
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMorphology-driven electrochemical attributes of Cu-MOF: a high-performance anodic material for battery supercapacitor hybrids†
Noshaba Shakeelad,
Junaid Khan *bc and
Abdullah A. Al-Kahtanie
*bc and
Abdullah A. Al-Kahtanie
aDepartment of Physics, Abbottabad University of Science and Technology, Khyber Pakhtunkhwa, Pakistan
bDepartment of Physics, Government Postgraduate College No. 1, Abbottabad, Khyber Pakhtunkhwa, Pakistan. E-mail: junaidkhan.nanotech@gmail.com
cDepartment of Higher Education Achieves and Libraries, Government of Khyber Pakhtunkhwa, Pakistan
dDepartment of Chemical and Biological Engineering, Gachon University, 1342 Seongnam-daero, Seongnam 13120, Republic of Korea
eDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh-11451, Saudi Arabia
First published on 25th October 2024
Abstract
Metal–organic frameworks (MOFs) have garnered substantial attention as promising candidates for electrode materials due to their intriguing electrochemical properties. However, the quest for enhanced energy density and electrical conductivity persists. Manipulating surface morphology emerges as a pivotal strategy to modulate these attributes and unlock the full potential of MOFs in electrochemical applications. This research delves into a pioneering exploration of copper metal–organic framework synthesis employing pyridine-4-carboxylic acid via hydrothermal and sonochemical routes, focusing on sculpting its surface morphology. Through meticulous comparative analysis, we unveil the distinct morphological features between the bulk and thin flakes crafted via each method. Notably, our findings highlight the remarkable superiority of the sonochemical approach in delivering refined outcomes (594.2 C g−1 at 1 A g−1 to 331.0 C g−1 at 16 A g−1) over its hydrothermal counterpart. Furthermore, the application of the sono-synthesized sample in an asymmetric device reveals a specific energy of 74.92 W h kg−1 at 850 W kg−1, while it sustains an exceptional 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 765 W kg−1, maintaining a noteworthy specific energy of 34.4 W h kg−1. The pursuit of refining surface morphology stands as a critical avenue in the ongoing endeavor to optimize the electrochemical performance of MOFs, paving the way for their widespread utilization in advanced energy storage technologies.
765 W kg−1, maintaining a noteworthy specific energy of 34.4 W h kg−1. The pursuit of refining surface morphology stands as a critical avenue in the ongoing endeavor to optimize the electrochemical performance of MOFs, paving the way for their widespread utilization in advanced energy storage technologies.
1. Introduction
The intense focus on energy conversion and storage devices has surged, driven by mounting concerns regarding the persistent energy crisis linked to conventional fossil fuels.1 The growing demand for eco-friendly and sustainable energy solutions has significantly directed attention toward storage device technologies.2 In the realm of electrochemical energy storage, batteries and supercapacitors (SCs) stand out as promising technologies.3 SCs are regarded as one of the best choices for the next-generation of power sources because of their exceptional stability, high power density, and low cost.4 Their high electrochemical performance, quick charging, and long lifespan have attracted considerable attention.5 The storage process depends on ionic adsorption/desorption (EDLC) and redox reaction at the surface (pseudocapacitors).6 Carbon-rich materials like graphite, carbon nanotubes, graphene, activated carbon, and carbon black are frequently utilized in EDLC applications. Materials exhibiting pseudocapacitive properties have been reported for pseudocapacitors, such as metal oxides (MnO2, V2O5, RuO2·xH2O, NiO, Co3O4) and conductive polymers (PANI, PPy, PTh, PEDOT).7 Despite their impressive electrochemical qualities, SCs still have a long way to go to achieve stable, significant specific energy, which hinders their ability to address modern energy storage challenges.8 Rechargeable battery storage depends heavily on significant redox activities, especially at certain potentials.9 In light of their diminished specific power and cycle potential, these entities find reinforcement in their exceptional energy density attributes owing to the storage chemistry. Integrating both redox-dominant and electrostatic technologies into a single architecture, hybrid storage devices, also called battery-supercapacitor hybrids, have notably mitigated the concerns faced.10 Their ability to combine the advantages of both batteries and supercapacitors has made them promising candidates for various applications, including portable electronics, electric vehicles, and renewable energy storage systems.11Numerous studies have been focused on exploring the utilization of non-carbonaceous (battery-grade) materials as electrode components in hybrid configurations. A broad range of materials are included in this class comprising metal hydroxides (Zn(OH), Co(OH)2, and Ni(OH)2), oxides (NiO, Fe3O4, and TiO2), sulfides (Cu2S, CoS2, NiS, and WS2), phosphates (FePO4, NaMnPO4, and LiCoPO4), and MOFs.12 Metal–organic frameworks (MOFs) are emerging as a viable option to satisfy the requirements of next-generation energy storage systems. MOFs belong to a category of porous substances consisting of metallics nodes and organic linkers.13 Their modular characteristics enable extensive synthesis adaptability, providing opportunities for precise chemical adjustments as well as structural controls. Attributes including porosity, stability, conductivity, and particle shape can be optimized for specific uses via creative synthetic design. MOFs store charge via various mechanisms, including capacitive and redox reactions, contingent upon the specific organic framework used to accommodate the metal ions.14 The porous structure enables the creation of an electrostatic double layer at the electrode–electrolyte interface, thus facilitating the reversible adsorption/desorption of electrolyte ions.15 Specific MOFs demonstrate a quality comparable to batteries, engaging in faradaic charge storage mechanisms rooted in reversible redox reactions. However, all the redox-active sites are not as readily available to electrolyte ions, causing the experimentally observed results to be much below the expected values.16 Passivation layers, surface reactions and other surface phenomena that impede ion transport are mostly to blame for the slow reaction kinetics that prevent the expected electrochemical outcomes.17 Additionally, problems like structural and surface imperfections leading to pore blocking that result in diffusion limitations cause the performance to further impede. Many efforts have been introduced to address these challenges. Y. Zhang synthesized MOF/CNT gels-infused configuration, resulting in 431.6 F g−1 storage capability. However, in Li-ion hybrid capacitor configuration, it could only gain a maximum specific energy of 20.2 W h kg−1 while preserving 89.4% capacitance (8000 cycles).18 Controllable multicomponent ZnO-ZnCo/MOF-PANI composites were reported by C. Yang with an energy density of 26 W h kg−1 and 75.3% retention after 5000 cycles.19 However, metal–organic frameworks (MOFs) are still in an early stage of development and require further advancement to narrow the disparity between demand and capabilities. The optimal selection of linkers and metal nodes from a diverse array remains a puzzle yet to be fully investigated.20 A comprehensive understanding of the charge storage mechanisms inherent to the respective complexes, along with strategies to mitigate the associated challenges, is imperative for elucidating the optimal utilization pathways within electrochemical systems.21 MOFs are usually fabricated using a variety of methods, including hydrothermal, solvothermal, hydrogel, sol–gel, chemical vapor deposition, microwave-assisted, and sonochemical processes. The choice of synthesis techniques significantly influences the morphology of the resulting sample, which has a significant influence on the final sample's morphology, including its size, shape, surface properties, and overall microscopic visual qualities.22 The final morphology of the sample is shaped by a number of critical parameters, including nucleation, growth rate, and surface contacts, all of which are influenced by the synthesis process selected. Refining the surface morphology can mitigate the abovementioned constraints.23 Surface modification can give MOF surfaces, and enhanced wettability promotes better electrolyte infiltration into the MOF pores, leading to improved ion accessibility and higher capacitance. This also reduces internal resistance and enables faster charge/discharge rates. This modification can alter the pseudocapacitive behavior in MOF electrodes, enabling additional charge storage mechanisms beyond diffusive insertions.24 Besides, surface modification can optimize pore size distribution, pore connectivity, and tortuosity, facilitating efficient ion transport within the MOF structure and improving the rate capability. By strategically choosing the synthesis approaches, the surface morphology of electrode materials can be tailored to meet the requirements of supercapacitor applications and the optimize performance metrics, ultimately advancing their potential for energy storage applications.
In this investigation, we delve into the synthesis of a copper metal–organic framework (Cu-MOF) employing pyridine-4-carboxylic acid via the hydrothermal and sonochemical routes, with a focus on shaping its surface morphology. Through meticulous comparative analysis, we unveil the distinct morphological nuances between the bulk flakes and thin flakes crafted via each method. Notably, our findings highlight the remarkable superiority of the sonochemical approach in delivering refined outcomes over its hydrothermal counterpart. Furthermore, the application of the sonochemically synthesized sample in an asymmetric device reveals unprecedented advancements in energy and power density characteristics.
2. Materials and methods
Copper chloride, pyridine-4-carboxylic acid, deionized water, ethanol, methanol, activated carbon, nickel foam, N-methyl-2-pyrrolidone, KOH, and PVDF were obtained from Sigma-Aldrich, all of which were of analytical grade, ensuring their suitability for high-precision experimental work.Cu-MOF was successfully synthesized using both hydrothermal and sonochemical techniques. Firstly, Cu-MOF was synthesized by the hydrothermal technique with pyridine-4-carboxylic acid serving as the organic ligand. The following is an overview of the synthesis process. In 10 mL of methanol, separate 0.1 M solutions of copper chloride (CuCl2) were made. A 0.1 M solution of pyridine-4-carboxylic acid was prepared by dissolving it in deionized (DI) water. To achieve homogeneity, the pyridine-4-carboxylic acid solution and the CuCl2 solution were extensively mixed and blended, resulting in a colloidal combination. After carefully pouring the colloidal liquid into a Teflon container, the container was put into an autoclave. For 12 hours, the autoclave containing the colloidal mixture was subjected to hydrothermal conditions in a furnace that was heated to 180 °C. The obtained Cu-MOF was purified after the hydrothermal reaction to get rid of any remaining contaminants. After that, the refined Cu-MOF was obtained. To eliminate any remaining solvent, the Cu-MOF underwent a drying procedure that involving vacuum drying or low heating and was ready for use as an active material after drying, demonstrating its adaptability for a range of uses, most notably as an electrode material (denoted as Q1).
Sonochemical synthesis was also used to create Cu-MOF using an organic linker of pyridine-4-carboxylic acid. A precursor solution was created by first dissolving 0.50 mol of CuCl2 and 0.50 mol of the organic linker in 100 milliliters of deionized water. The previously mentioned pyridine-4-carboxylic acid solution was added drop by drop and sonicated for 45 minutes. The synthesis was performed utilizing 40% amplitude and power of 600 Watts. The temperature was kept at 30 °C. The process was performed for 45 min with the condition of 2 s active and 1 s non-active sonication.
After centrifuging the colloidal solution to extract the synthesized material, it was repeatedly washed with ethanol and DI water and then allowed to dry overnight at 80 °C (denoted as Q2). Both synthesis approaches are schematically illustrated in Fig. 1.
 | ||
| Fig. 1 Synthesis approach of Cu-MOF through the hydrothermal and sonochemical approach. | ||
2.1. Electrode characterization/fabrication
A mixture consisting of 80% synthesized Cu-MOF, 10% acetylene black, and 10% PVDF binder was used, with NMP (N-methyl-2-pyrrolidone) as the solvent to create a slurry. This slurry was stirred for 6 hours and then coated onto a 1 cm2 area of nickel foam that had been thoroughly cleaned and prepared as both the substrate and current collector. The electrode was then dried at 60 °C for 6 hours before undergoing electrochemical analyses.A 1 M KOH solution was used as the electrolyte in all electrochemical experiments. The specific capacity (Qs) in C g−1 was determined as follows.25
 | (1) |
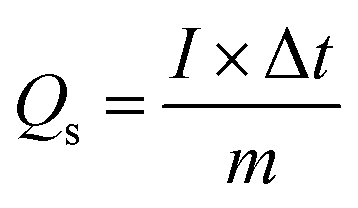 | (2) |
Here, m represents the active mass (g) deposited, ν is the potential sweep rate (V s−1), I is the current obtained, and Δt is the discharge time. The specific power (Ps) and specific energy (Es) were determined as follows.26
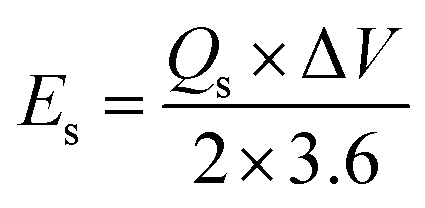 | (3) |
 | (4) |
3. Structural and surface morphology
The phase purity and crystallinity of the synthesized material were determined using X-ray diffraction (XRD). The findings and simulated XRD patterns (CCDC no. 226294) are also presented in Fig. 2(a). The lower strength of the peaks in the spectrum from the Q2 sample suggests that the generated material is amorphous as compared to Q1. This feature improves the electrochemical performance. The peaks are noted at 14.2, 18.7, 29.2, 44.7, 61.1, 68.9, and 79.3°, which correspond to (110), (200), (311), (400), (511), (420), and (600), respectively.27 The Q2 XRD spectrum reveals additional peaks at 8.4, 21.1, 27.5, 34.9, and 44.5°. It suggests that the additional sonochemical treatment of the MOF matrix caused the creation of an additional phase. The XRD results of both the samples exhibit a strong correlation with the simulated diffraction patterns, indicating that the experimental data closely matches the theoretical prediction of synthesis of Cu-MOFs. | ||
| Fig. 2 (a) XRD analysis with multiple peaks demonstrating the crystallinity of Cu-MOF synthesized via the hydrothermal and sonochemical approach, respectively, (b and c) SEM results unveiling the morphology of Q1 and (d and e) for Q2. | ||
The impact of employing different synthesis approaches on the morphology of the surface was examined using SEM. Fig. 2(b–e) demonstrates the results of Q1 and Q2 MOF at parallel resolutions and the results disclose noteworthy transformation. The bulky asymmetric flakes with a rough and coarse surface are obvious for Q1, leading to limited surface area and possible poor electrolyte accessibility (Fig. 2(b) and (c)).28 This can hinder the infiltration and diffusion of electrolyte ions inside the material, leading to slower ion transport kinetics and higher internal resistance.29 Long ion diffusion pathways, mechanical stresses during cycling (cause material degradation), particle agglomeration, and electrode delamination, which lead to performance degradation and reduced cycle life, are the properties of this bulk morphology.30 However, the incorporation of the sonochemical approach yielded a prominent transformation. Fig. 2(d) and (e) reveals a distinctive thin flake morphology, stacked in a radial arrangement, introducing a less textured and smoother surface. The alteration in the surface morphology entails notable ramifications for the electrochemical functionalities as an electrode material. The presence of a flaky structure results in an augmented surface area, casing potentially enhanced storage kinetics and ions accessibility.31 The increased surface area facilitates a greater abundance of active sites conducive to charge storage and electrochemical reactions at the interface (pseudocapacitive insertions). Moreover, the presence of flakes mitigates constraints linked with concentration polarization through the reduction of the diffusion path length, thereby resulting in enhanced ion transport efficiency and superior electrochemical performance.32 Enhanced electrolyte infiltration additionally diminishes internal impedance, thereby promoting superior ion transport and increasing the power output along with significant enhancement in the energy density.33 In brief, the implementation of the introductory sonochemical technique lead to a change in the surface morphology, thereby facilitating enhanced electrochemical efficacy, rendering it a favorable contender for diverse energy storage applications. Additionally, Q1 synthesis involves high temperature and pressure, which leads to more extensive crystal growth and the formation of bulk structures. The extended reaction time and elevated temperature accelerates the diffusion of reactants, promoting the growth of larger crystals. The fewer, more intense peaks in the XRD pattern also suggest well-defined, larger, and more ordered crystalline domains. Q2 synthesis uses ultrasonic waves to induce the rapid nucleation and growth of particles. The intense acoustic cavitation creates localized high temperatures and pressures, leading to the formation of smaller, thinner flakes. The shorter reaction times typically result in a more uniform and finer morphology. The XRD pattern showing multiple peaks with lower intensity also indicates a less crystalline material or smaller crystal sizes.
Energy-dispersive X-ray (EDX) spectroscopy was used to determine the elemental configuration of Cu-MOF (Fig. 3(a) and (b)), which reveals the existence of Cu, C, N, and O in both the synthesized samples, as expected for the synthesized MOF, thus confirming its successful synthesis. No additional peaks confirm the elemental purity of the synthesized samples. The elemental composition results obtained from the EDX analysis are presented in Table 1.
 | ||
| Fig. 3 (a and b) EDX outcomes to depict the elemental analysis of hydrothermal and sonochemical approaches, respectively, (c and d) Color-mapping of Cu hydrothermal and sonochemical approach. | ||
| Q1 | Q2 | ||
|---|---|---|---|
| Element | Weight (%) | Element | Weight (%) |
| Copper (Cu) | 33.73% | Copper (Cu) | 39.70% |
| Carbon (C) | 18.60% | Carbon (C) | 16.72% |
| Oxygen (O) | 24.57% | Oxygen (O) | 25.68% |
| Nitrogen (N) | 23.10% | Nitrogen (N) | 17.90% |
EDX color-mapping elemental analysis was also performed to scrutinize the presence of Cu on the MOF surface. The analysis offers valuable insights into the structural and compositional differences between the two synthesis methods. Fig. 3(c) corresponds to the Q1 sample. The slower kinetics of this process compared to Q2 led to changes in the morphology as well as Cu (the redox center) concentration on the surface. Fig. 3(d) represents the EDX color-mapping outcomes obtained for the Q2 sample. The sonochemical approach leads to the rapid and efficient nucleation and growth of MOF crystals due to the cavitation effect induced by ultrasound waves. This resulted in a greater concentration of Cu on the surface of the electrode material, elucidating its potential redox behavior.34 Additionally, the uniformity in distribution is another factor supporting the significant electrochemical attributes. The analysis indicates that the sonochemically synthesized Cu-MOF, characterized by its thin, flaky morphology and higher surface concentration of copper compared to the bulkier hydrothermal counterpart, is a dominant contender for electrochemical applications.
The nitrogen adsorption–desorption isotherm of the Cu-MOF measured at 77 K is presented in Fig. S1.† The isotherm exhibits a characteristic type I loop indicative of the porous material. The BET calculated surface area from the isotherm is 1750 and 2100 m2 g−1, and the pore size distribution analysis confirms the presence of mesopores with a pore volume of 0.8 and 1.0 cm3 g−1 for Q1 and Q2, respectively.
4. Three-electrode configurations
Cyclic voltammetry (CV) tests were performed with a three-electrode setup to assess the electrochemical effectiveness of the synthesized nanomaterials. In the electrochemical setup, the working electrode, Cu-MOF, was submerged in an electrolyte solution of 1 M KOH. The CV scan was carried out in a specified potential range from 0 to 0.7 V at room temperature using different sweep rates. Fig. 4(a) and (b) show the CV curves for Q1 and Q2, respectively. The CV profile distinctly shows oxidation and reduction peaks, which are suggestive of faradaic processes involving the electrolyte species and the active electrode. These findings offer substantial evidence that the electrode is appropriate for use in battery-grade systems. The oxidation peaks gradually move to a higher potential range as the scan rates increase, and this is followed by an increase in the current. This modification can be ascribed to both faster reaction kinetics and the restricted accessibility of the electrolyte ions in the electrode materials. A stable state pattern in the CV spectrum, even at high scan rates, highlights the mechanism's reversibility and the electrode's excellent rate capabilities. The CV curves for Q1 and Q2, recorded at a scan rate of 3 mV s−1, are presented in Fig. 4(c) to facilitate a comparative analysis. With greater peak current and surface area under the CV scan graph, the dominant behavior of Q2 MOF is obvious. This is because of the predominant surface morphological aspects that are advantageous for the electrochemical activities. Eqn (1) was used to compute the electrode's specific capacity or its capacity to store charge. The synthesized MOF using the hydrothermal approach shows a specific capacity of 511.6 C g−1 at a scan rate of 3 mV s−1, which was boosted to a promising 695.7 C g−1 after tuning the surface morphology through the sonochemical approach (Fig. 4(d)). The depiction of storage chemistry behind the obtained outcomes was also carried out. A fitting approach (the b-value fitting) was used to support the battery-grade attributes regarding the observed redox peaks, as shown in Fig. 4(e). The battery-grade characteristics of the resultant materials were validated by the measured b-values of 0.50 and 0.47 obtained for Q1 and Q2, respectively. Separating the capacitive (faster kinetics) and diffusion-driven (slower kinetics) processes that contribute to charge storage chemistry has significantly deepened our understanding. The obtained current is the result of either a capacitive or diffusive current. The resulting current (v) is the sum of these faster kinetics (Ic(v)) and slower (Id(v)) phenomena.9| (v) = Ic(v) + Id(v) | (5) |
| (v) = k1v + k2v1/2 | (6) |
 | ||
| Fig. 4 (a and b) CV curves of Q1 and Q2 bare electrode, (c) CV comparison at 3 mV s−1, (d) Qs at different scan rates, (e) b-value fitting, and (f) diffusive contribution at multiple scan rates. | ||
Galvanostatic charge/discharge (GCD) characterization was carried out using a 1 M KOH electrolyte to fully assess the electrochemical performance of the developed Cu-MOF over a variety of current densities. Fig. 5(a) and (b) demonstrates the GCD profiles for Q1 and Q2 conducted at varying current densities while maintaining a constant voltage range of 0.6 V. Pointing to similarities with battery-type materials, the non-linear GCD curves in both scenarios were obtained. An in-depth evaluation of the relative performance of the two electrodes while charging and discharging at a rate of 2.0 A g−1 is given in Fig. 5(c). Q2 exhibits greater durability over a comparable period at 1 A g−1, signifying improved storage capacity. From the GCD curves, the specific capacity was calculated using eqn (2). Based on the discharge patterns of both, a comparison of their specific capacities is shown in Fig. 5(d). As it is consistent with the corresponding current densities, this method is considered more reliable for practical applications. Remarkably, the MOF produced by the sonochemical approach (Q2) showed a higher specific capacity of 594.2 C g−1 than the one obtained by the hydrothermal method (Q1), which produced a specific capacity of 462.0 C g−1. The shortened time available for electrolyte ions to interact with the electrode material is the reason for the drop in the specific capacity (Qs) with higher specific current levels. However, the Q2 electrode was able to sustain 55.70% of maximum storage capability even at 10 A g−1. This confirms that the strong rate capability of the Q2 electrode is driven by the improved surface morphology.
 | ||
| Fig. 5 (a and b) GCD results recorded at different current densities (1–10 A g−1) for Q1 and Q2 electrodes, respectively, (c) Comparison of GCD at 1 A g−1 for comparative analysis, (d) Qs calculated from the GCD results, showing the dominant performance of Q2. | ||
Electrochemical impedance spectroscopy (EIS) was performed to investigate the processes of interface and charge transfer resistance between the electrode and the electrolyte. A frequency ranges of 0 to 0.1 MHz and an amplitude of 10 millivolts were used in the measurement. The EIS measurements obtained for both are displayed in the Nyquist plot shown in Fig. 6. The high-frequency regions are shown in Fig. 6(b) as the zoomed part. The intercept aids as a measure of the ESR. The resistance at the current collector interface resistance, electrode resistance, electrolyte–electrode interface, and electrolytes are the several parts that make up the ESR. The ESR value of 0.78 Ω for Q2 indicates higher conductivity than that of the Q1 electrode, which is 0.98 Ω. As seen in Fig. 6(a), the slight semicircular pattern in the high-frequency zone points to low charge transfer resistances (Rct) for sonochemical approaches. Additionally, the low-frequency region's spectral line magnitude is directly correlated with the active materials' ion transport channel. The Q2-reformed surface morphology lowers the ion diffusion path, which speeds up the material's ability to transport electrolyte ions. It shows better electrical conductivity. The EIS measurement results are consistent with the GCD and CV analysis results, indicating that the Q2 electrode in the three-electrode assembly performs superiorly overall.
 | ||
| Fig. 6 (a) Nyquist plot of EIS. (b) Zoomed part of the high-frequency region. | ||
5. Battery-supercapacitor hybrid configuration
To scrutinize the real-time performance, the sono-electrode underwent testing in a hybrid configuration combining battery (Q2) and supercapacitor elements (activated carbon) in order to examine the asymmetric device performance. To allow ions to go through the channel it formed, a thin, porous membrane called a polymeric separator was positioned in between the two electrodes. A schematic representation of each component used in the device's design can be seen in Fig. 7(a). The ability of the capacitive electrode to promote electrolyte ion absorption and desorption is mostly in a negative potential range from 0 to −1 V. The voltage range from 0 to 0.7 V is ideal for the best operation of the Q2 electrode. This suggests 0 to 1.7 V as the operational potential range for the real device. The device was subjected to CV, with the potential sweep rates varied between 3 and 100 mV s−1 while keeping the designated potential window. The final results are shown in Fig. 7(b); the semi-rectangular shape including humps implies that both electrostatic and faradaic phenomena are involved in the storage mechanism. Moreover, the CV curve forms remain consistent at increasing scan rates, particularly up to 100 mV s−1. This finding highlights the system's exceptional potential for stability and rate capacity. The device was subjected to GCD testing over a similar potential range, covering current densities ranging from 1.0 A g−1 to 16 A g−1. The discharge curves that originated using the acquired GCD results are shown in Fig. 7(c). The non-linear and analogous symmetry displayed by the GCD profiles emphasizes the device's hybrid and reversible charge storage. The consistency of the GCD curve suggests that the designed device is stable when operated at different current densities.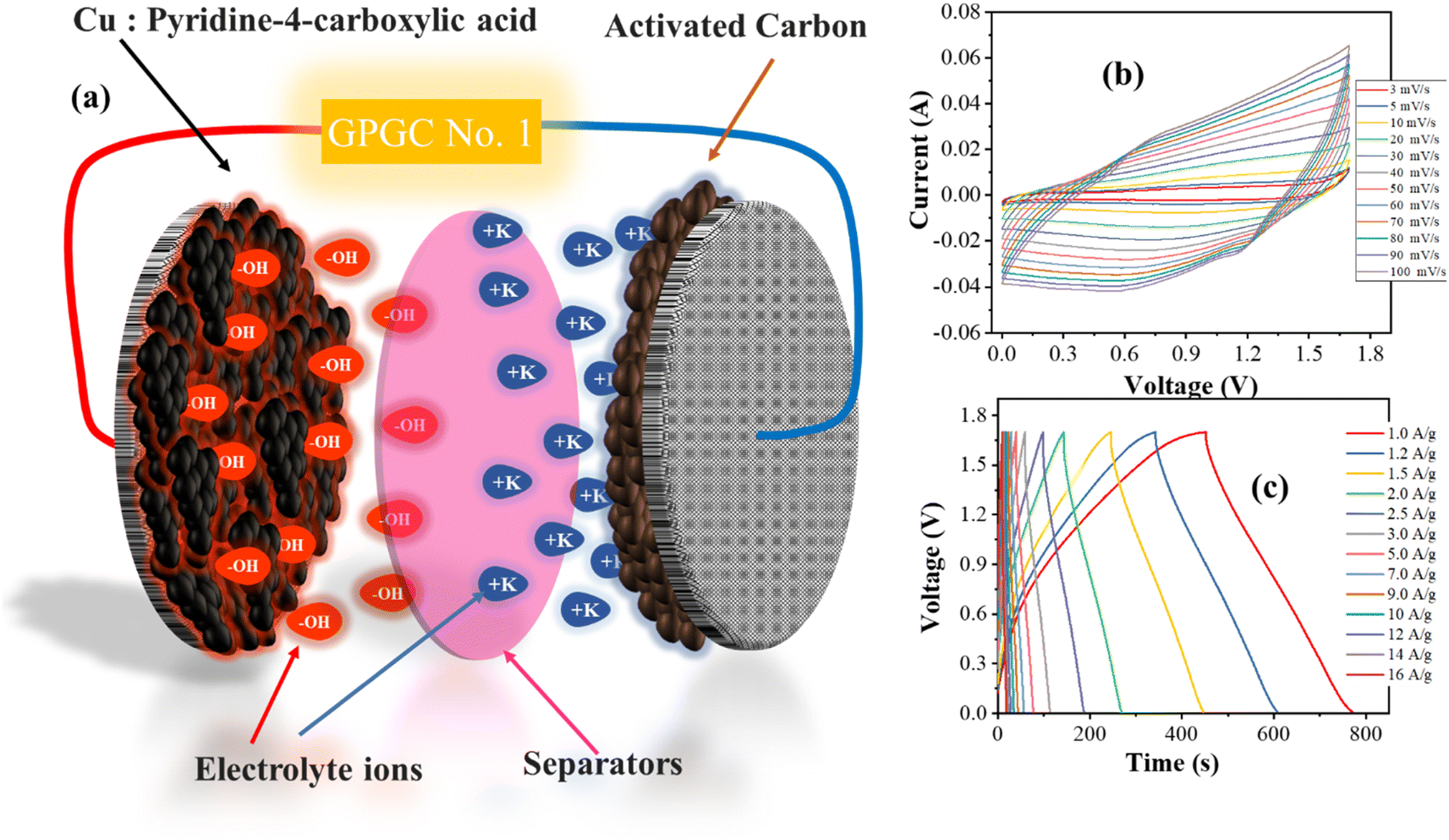 | ||
| Fig. 7 (a) Fabricated hybrid supercapacitor, (b) CV at 3–100 mV s−1, (c) GCD results. | ||
The specific capacity under various current density settings, as determined by the GCD results, is displayed in Fig. 8(a). At 1 A g−1, the device reached its maximal specific capacity (Qs) of 317.3 C g−1. The slow decline in the specific capacity that is seen at higher current densities can be explained by the little time that the electrolyte ions have to interact with the material when kinetics are fast. Particularly, the device maintains 145.7 C g−1 at a high current density of 16 A g−1. This remarkable preservation is the consequence of ions interaction with maximum active sites and rapid diffusion, made possible by the tuned morphological aspects. In Fig. 8(b), the device's energy density at the corresponding power density is displayed for comparison with previously published research. The hybrid device exhibits a striking specific energy of 74.9 W h kg−1 as well as outstanding power characteristics, with 850 W kg−1 at 1 A g−1. In addition, it sustains an exceptional specific power of 13765 W kg−1 at a current density of 16 A g−1 while maintaining a noteworthy specific energy of 34.4 W h kg−1. The reduced diffusion path length and improved conductivity along with pseudocapacitive involvement via morphology tuning have made it possible for device to boost the specific power to about 16 times while sustaining 45.91% specific energy. Increased electrolyte penetration facilitated more effective ion transport by further lowering the internal resistance. These results show a notable improvement over previously published results in the literature on batteries and supercapacitors. To scrutinize the device potential for its practical utilization, it was tested for 5000 consecutive GCD cycles at 16 A g−1. An exceptional 97.95% capacity retention was observed at the end of the test, showing excellent stability and durability (Fig. 8(c)). The Nyquist plot obtained from EIS measurements carried out before and after, followed by a stability test, is shown in Fig. 8(d). Before and after the stability evaluation, the X-intercepts showed an equivalent series resistance (ESR) value of 0.41 Ω and 0.42 Ω, respectively (inset). The slight decline in the slope of the EIS spectrum suggests a decrease in the Warburg resistance. This is because consecutive charge discharge activated the maximum redox channels, leading to efficient ion transport. The EIS spectra show no discernible changes, which shows that the device is reliable under the testing circumstances and retains its electrical properties. The equivalent electric circuit used for EIS fitting is also presented in the inset. R defines the combined series resistance and Rct is the charge transfer resistance. W is the Warburg component while CPC and CPD correspond to the constant phase element capacitive and diffusive storage, respectively.
 | ||
| Fig. 8 (a and b) Qs, Es, and Ps for the real device, (c) GCD cyclic test results, (d) EIS results in the Nyquist plot. | ||
The performance of the fabricated device is also presented in Table 2 in comparison with similar reported literature.
Morphology-driven better electrochemical yields overall, longer lifespans, enhanced durability, and greater efficiency are all attributed to the superior conductive qualities of Cu-MOF for high-performance energy storage applications.
6. Conclusion
In conclusion, our research has demonstrated the potential of Cu-MOF as electrode materials in electrochemical applications, with a particular focus on enhancing the energy density and electrical conductivity through the manipulation of surface morphology. By synthesizing copper MOFs using pyridine-4-carboxylic acid via both hydrothermal and sonochemical methods, we elucidated the distinct morphological variations from bulk flakes to thin flakes. Significantly, our findings underscore the superior performance of the sonochemical method, yielding refined outcomes surpassing those of the hydrothermal route. Remarkably, it showed a higher specific capacity of 594.2 C g−1 than the one obtained by the hydrothermal method, which produced a specific capacity of 462.0 C g−1. The sonochemical electrode was also able to sustain 55.70% of the maximum storage capability even at 10 A g−1. Additionally, it exhibited a notable specific energy of 74.9 W h kg−1 at 850 W kg−1, along with an exceptional power density of 13765 W kg−1, maintaining a noteworthy specific energy of 34.4 W h kg−1 when subjected to a battery-supercapacitor hybrid configuration. Besides, the exceptional 97.95% capacity conservation was observed after 5k cycles. Our study underscores the importance of refining the surface morphology to optimize the electrochemical performance of MOFs, thereby opening avenues for their widespread utilization in advanced energy storage technologies.
Data availability
The data will be made available on request.Conflicts of interest
The authors declare that there are no conflicts of interest regarding the publication of this article.Acknowledgements
The authors thank the “researchers supporting project number (RSP2024R266), King Saud university, Riyadh, Saudi Arabia” for their financial support.References
- J. Khan, N. Shakeel and S. Alam, Unlocking the potential of sulfurized electrode materials for next-generation supercapacitor technology, Int. J. Hydrogen Energy, 2024, 88, 1163–1185 CrossRef CAS.
- J. Khan, A. Ahmed, M. I. Saleem and A. A. Al-Kahtani, Benzene-1, 4-dicarboxylic acid-based Ni-MOF for efficient battery-supercapacitor hybrids: Electrochemical behavior and mechanistic insights, J. Energy Storage, 2024, 100, 113455 CrossRef.
- J. Khan and N. Shakeel, Copper-based Para-Phthalic Acid Metal-Organic Framework: An efficient anodic material for super-capattery hybrid systems, Mater. Chem. Phys., 2024, 326, 129763 CrossRef CAS.
- J. Khan, Experimental and theoretical insights into benzene-1, 4-dicarboxylic acid based Co-MOFs: an anodic material for expedient battery-supercapacitor hybrids, Sustainable Energy Fuels, 2024, 8, 4355–4364 RSC.
- S. S. Shah, F. Niaz, M. A. Ehsan, H. T. Das, M. Younas, A. S. Khan, H. U. Rahman, S. A. Nayem, M. Oyama and M. A. Aziz, Advanced strategies in electrode engineering and nanomaterial modifications for supercapacitor performance enhancement: A comprehensive review, J. Energy Storage, 2024, 79, 110152 CrossRef.
- I. Shaheen, I. Hussain, T. Zahra, M. S. Javed, S. S. A. Shah, K. Khan, M. B. Hanif, M. A. Assiri, Z. Said and W. U. Arifeen, Recent advancements in metal oxides for energy storage materials: design, classification, and electrodes configuration of supercapacitor, J. Energy Storage, 2023, 72, 108719 CrossRef.
- I. Melkiyur, Y. Rathinam, P. S. Kumar, A. Sankaiya, S. Pitchaiya, R. Ganesan and D. Velauthapillai, A comprehensive review on novel quaternary metal oxide and sulphide electrode materials for supercapacitor: Origin, fundamentals, present perspectives and future aspects, Renewable Sustainable Energy Rev., 2023, 173, 113106 CrossRef CAS.
- K. O. Oyedotun, J. O. Ighalo, J. F. Amaku, C. Olisah, A. O. Adeola, K. O. Iwuozor, K. G. Akpomie, J. Conradie and K. A. Adegoke, Advances in supercapacitor development: materials, processes, and applications, J. Electron. Mater., 2023, 52(1), 96–129 CrossRef CAS.
- A. G. Olabi, Q. Abbas, P. A. Shinde and M. A. Abdelkareem, Rechargeable batteries: Technological advancement, challenges, current and emerging applications, Energy, 2023, 266, 126408 CrossRef CAS.
- J. Khan, Zincronization-induced surface modification of CoMn phosphate for improved electrochemical performance in battery-supercapacitor hybrid systems, J. Energy Storage, 2024, 81, 110432 CrossRef.
- H. Guo, M. Qiao, J. Yan, L. Jiang, J. Yu, J. Li, S. Deng and L. Qu, Fabrication of Hybrid Supercapacitor by MoCl5 Precursor-Assisted Carbonization with Ultrafast Laser for Improved Capacitance Performance, Adv. Funct. Mater., 2023, 33(23), 2213514 CrossRef CAS.
- A. Afif, S. M. Rahman, A. T. Azad, J. Zaini, M. A. Islan and A. K. Azad, Advanced materials and technologies for hybrid supercapacitors for energy storage–A review, J. Energy Storage, 2019, 25, 100852 CrossRef.
- M. Z. Iqbal, M. Shaheen, M. W. Khan, S. Siddique, S. Aftab, S. M. Wabaidur and M. J. Iqbal, Exploring MOF-199 composites as redox-active materials for hybrid battery-supercapacitor devices, RSC Adv., 2023, 13(5), 2860–2870 RSC.
- C. N. Hong, A. B. Crom, J. I. Feldblyum and M. R. Lukatskaya, Metal-organic frameworks for fast electrochemical energy storage: Mechanisms and opportunities, Chem, 2023, 9(4), 798–822 CAS.
- S. J. Shin, J. W. Gittins, C. J. Balhatchet, A. Walsh and A. C. Forse, Metal–organic framework supercapacitors: challenges and opportunities, Adv. Funct. Mater., 2023, 2308497 Search PubMed.
- M. Saleem, F. Ahmad, M. Fatima, A. Shahzad, M. S. Javed, S. Atiq, M. A. Khan, M. Danish, O. Munir and S. M. B. Arif, Exploring new frontiers in supercapacitor electrodes through MOF advancements, J. Energy Storage, 2024, 76, 109822 CrossRef.
- M. I. Anwar, M. Asad, L. Ma, W. Zhang, A. Abbas, M. Y. Khan, M. Zeeshan, A. Khatoon, R. Gao and S. Manzoor, Nitrogenous MOFs and their composites as high-performance electrode material for supercapacitors: Recent advances and perspectives, Coord. Chem. Rev., 2023, 478, 214967 CrossRef CAS.
- Y. Zhang, Q. Xie, R. Shao, J. Ding, J. Liu, W. Xu and Y. Wang, Green synthesis of MOF/CNT gels via in-situ physical mixing strategy toward quasi-solid-state Li-ion hybrid capacitor, J. Energy Storage, 2024, 86, 111156 CrossRef.
- D. Qin, B. Zhou, Z. Li and C. Yang, Construction of controllable multicomponent ZnO-ZnCo/MOF-PANI composites for supercapacitor applications, J. Mol. Struct., 2024, 1309, 138140 CrossRef CAS.
- Y. Guo, K. Wang, Y. Hong, H. Wu and Q. Zhang, Recent progress on pristine two-dimensional metal–organic frameworks as active components in supercapacitors, Dalton Trans., 2021, 50(33), 11331–11346 RSC.
- V. Hegde, U. Uthappa, M. Suneetha, T. Altalhi, S. S. Han and M. D. Kurkuri, Functional porous Ce-UiO-66 MOF@ Keratin composites for the efficient adsorption of trypan blue dye from wastewater: A step towards practical implementations, Chem. Eng. J., 2023, 461, 142103 CrossRef CAS.
- X. Hou, J. Sun, M. Lian, Y. Peng, D. Jiang, M. Xu, B. Li and Q. Xu, Emerging Synthetic Methods and Applications of MOF-Based Gels in Supercapacitors, Water Treatment, Catalysis, Adsorption, and Energy Storage, Macromol. Mater. Eng., 2023, 308(2), 2200469 CrossRef CAS.
- A. A. Kulkarni, N. K. Gaikwad, A. P. Salunkhe, R. M. Dahotre and T. S. Bhat, Transition Metal Phosphates: A Paradigm for Electrochemical Supercapacitors, J. Electroanal. Chem., 2023, 117795 CrossRef CAS.
- G. Chaturvedi, R. Jaiswal, S. Ilangovan, S. Sujatha, K. Ajeesh and S. S. V. Tatiparti, A systematic approach for selecting suitable morphologies for supercapacitor applications through morphological stability map–A case of Ni-MOF, Ceram. Int., 2023, 49(6), 9382–9394 CrossRef CAS.
- A. Khan, F. Shaheen, M. Roman, R. Ahmad, K. Mehboob and M. H. Aziz, Highly redox active mesoporous Ni/Co-organic framework as a potential battery type electrode material for high energy density supercapattery, J. Energy Storage, 2023, 58, 106317 CrossRef.
- J. Khan, N. Shakeel, M. Z. Iqbal and A. A. Al-Kahtani, Zincronization-induced surface modification of CoMn phosphate for improved electrochemical performance in battery-supercapacitor hybrid systems, J. Energy Storage, 2024, 81, 110432 CrossRef.
- N. He, X. Yang, L. Shi, X. Yang, Y. Lu, G. Tong and W. Wu, Chemical conversion of Cu2O/PPy core-shell nanowires (CSNWs): a surface/interface adjustment method for high-quality Cu/Fe/C and Cu/Fe3O4/C CSNWs with superior microwave absorption capabilities, Carbon, 2020, 166, 205–217 CrossRef CAS.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, All pseudocapacitive MXene-RuO2 asymmetric supercapacitors, Adv. Energy Mater., 2018, 8(13), 1703043 CrossRef.
- T. Zhang and F. Ran, Design strategies of 3D carbon-based electrodes for charge/ion transport in lithium ion battery and sodium ion battery, Adv. Funct. Mater., 2021, 31(17), 2010041 CrossRef CAS.
- R. Guo, K. Zhang, W. Zhao, Z. Hu, S. Li, Y. Zhong, R. Yang, X. Wang, J. Wang and C. Wu, Interfacial challenges and strategies toward practical sulfide-based solid-state lithium batteries, Energy Mater. Adv., 2023, 4, 0022 CrossRef CAS.
- G. J. Lim, X. Liu, C. Guan and J. Wang, Co/Zn bimetallic oxides derived from metal organic frameworks for high performance electrochemical energy storage, Electrochim. Acta, 2018, 291, 177–187 CrossRef CAS.
- J. Yang, B. Yin, Y. Sun, H. Pan, W. Sun, B. Jia, S. Zhang and T. Ma, Zinc anode for mild aqueous zinc-ion batteries: challenges, strategies, and perspectives, Nano-Micro Lett., 2022, 14, 1–47 CrossRef PubMed.
- E. Muchuweni, E. T. Mombeshora, C. M. Muiva and T. S. Sathiaraj, Lithium-ion batteries: Recent progress in improving the cycling and rate performances of transition metal oxide anodes by incorporating graphene-based materials, J. Energy Storage, 2023, 73, 109013 CrossRef.
- M. Shaheen, M. Z. Iqbal, M. W. Khan, S. Siddique, S. Aftab and S. M. Wabaidur, Evaluation of a redox-active Cu-MOF and Co-MOF as electrode materials for battery–supercapacitor-type hybrid energy storage devices, Energy Fuels, 2023, 37(5), 4000–4009 CrossRef CAS.
- Z. Maliha, M. Rani, R. Neffati, A. Mahmood, M. Z. Iqbal and A. Shah, Investigation of copper/cobalt MOFs nanocomposite as an electrode material in supercapacitors, Int. J. Energy Res., 2022, 46(12), 17404–17415 CrossRef CAS.
- M. K. Singh, S. Krishnan, K. Singh and D. K. Rai, CNT Interwoven Cu-MOF: A Synergistic Electrochemical Approach for Solid-State Supercapacitor and Hydrogen Evolution Reaction, Energy Fuels, 2024, 38(13), 12098–12110 CrossRef CAS.
- Y. Ji, W. Li, Y. You and G. Xu, In situ synthesis of M (Fe, Cu, Co and Ni)-MOF@ MXene composites for enhanced specific capacitance and cyclic stability in supercapacitor electrodes, Chem. Eng. J., 2024, 496, 154009 CrossRef CAS.
- M. Z. Iqbal, U. Aziz, S. Aftab, S. M. Wabaidur, S. Siddique and M. J. Iqbal, A hydrothermally prepared lithium and copper MOF composite as anode material for hybrid supercapacitor applications, ChemistrySelect, 2023, 8(6), e202204554 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05790b |
| This journal is © The Royal Society of Chemistry 2024 |