DOI:
10.1039/D4RA06253A
(Paper)
RSC Adv., 2024,
14, 33952-33967
A doxorubicin loaded chitosan–poloxamer in situ implant for the treatment of breast cancer†
Received
29th August 2024
, Accepted 14th October 2024
First published on 25th October 2024
Abstract
Breast cancer is a serious concern for many women worldwide. Drug-loaded implants have shown several benefits over systemic administrations. To provide anti-cancer drugs with controlled release and reduced systemic toxicity, biodegradable in situ implants have attracted a lot of attention. In the present study, we aimed to design and optimize a doxorubicin-loaded chitosan–poloxamer in situ implant for breast cancer treatment. Utilizing Box–Behnken Design and a Quality-by-Design (QbD) methodology, the in situ implant was prepared with chitosan (X1), poloxamer 407 concentration (X2), and stirring time (X3) as the independent variables. It was characterized for its in vitro gelation time, pH, rheology, and morphology, and evaluated based on drug release profile, in vitro cytotoxicity activities, in vitro anti-inflammatory potential, in vitro cellular uptake, and in vivo anti-inflammatory and pharmacokinetics to ensure their therapeutic outcomes. The results revealed that the prepared formulation showed a gelation time of 26 ± 0.2 s with a viscosity of 8312.6 ± 114.2 cPs at 37 °C. The developed formulation showed better cytotoxic activity in MCF-7 cell lines compared to the free drug solution. It demonstrated reduced levels of pro-inflammatory cytokines in RAW 264.7 macrophages. Further, the prepared in situ implant increases the intracellular accumulation of DOX in the MCF-7 cells. The in vivo pharmacokinetic investigations depicted an increase in t1/2 and a decrease in AUC of the developed formulation resulting in prolonged drug release and there could be a lower drug concentration in the bloodstream than for the free drug. Therefore, the developed in situ implant may offer a viable option for breast cancer treatment.
1. Introduction
One of the most common and dangerous health issues that women face globally is breast cancer. Every year, over a million new cases are diagnosed. According to the World Health Organization, 670![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 individuals worldwide lost their lives to breast cancer in 2022, while 2.3 million women received a diagnosis.1 As a result, one of the greatest hazards to humanity is cancer, a global health issue with no known bounds in terms of death and incidence rates. The primary cause of death for patients with breast cancer is metastasis.2 The prognosis for metastatic patients remains unsatisfactory and dismal despite advancements in breast cancer treatment.
000 individuals worldwide lost their lives to breast cancer in 2022, while 2.3 million women received a diagnosis.1 As a result, one of the greatest hazards to humanity is cancer, a global health issue with no known bounds in terms of death and incidence rates. The primary cause of death for patients with breast cancer is metastasis.2 The prognosis for metastatic patients remains unsatisfactory and dismal despite advancements in breast cancer treatment.
Systemic treatments like chemotherapy involve intravenous delivery of anti-cancer drugs at maximum tolerable doses that cause serious toxicities in healthy tissues, such as cardiomyopathy and neutropenia3,4 and the drug reaches the tumor site in low concentrations, which decreases the tumor treatment's effectiveness.5,6 Further, surgery is a non-aggressive procedure that is regarded as the primary strategy in the early stages of breast cancer.7 Even with the advancements in surgical methods, there is still a chance that residual tumor cells will stay in the surgical margins or the circulation following the surgical interventions, increasing the risk of metastasis and cancer recurrence.8,9 As a result, alternative approaches are required to reduce the chance of distant metastasis, stop the tumor from growing back, and increase the effectiveness of tumor-targeted drugs.10 The use of targeted drug delivery has the potential to achieve therapeutic concentrations within the tumor site, which would reduce systemic levels and, in turn, reduce the likelihood of unwanted side effects.11
Implantable drug delivery devices are the better approaches for local drug administration to the tumor site.12 These implants use biopolymer components that are both biodegradable and non-biodegradable. The non-biodegradable implants require surgical procedures twice, as they need to be inserted and removed. Using biodegradable polymers in implantable devices could be a better approach as the body can naturally remove it after use, eliminating the need for surgical extraction and increasing the patient's acceptance.13 In recent decades, many biomaterials have been widely used to develop implantable drug-delivery devices against cancer.14 Particularly biopolymers have attracted more attention because of their beneficial qualities, which include biocompatibility, biodegradability, and ease of processing.15 In the past decade, drug delivery systems that are implanted have advanced significantly and have shown to be an extremely effective way to deliver drugs to specified areas.16 They also reduce the total amount of drug in blood circulation by providing accurate spatial control, recurrence probability can be reduced, enhancing the rate of overall survival, and shielding healthy cells from harm.17 By maintaining therapeutically relevant drug levels within the tumor for extended durations, they can overcome the limitations of short half-lives, dose-dependent systemic toxicity, and rapid clearance associated with conventional chemotherapy agents.18 Implantable drug delivery systems include hydrogel, nanofibers, in situ gel, and osmotic pumps used for local chemotherapeutics delivery.19 As an injectable drug delivery technique, in situ implants (ISI) have garnered a lot of interest as these systems are injectable into the body with a syringe, and after injection, they solidify to form a semisolid depot.20 When a stimulus (such as ions, temperature, or pH) is applied, the liquid combination precipitates, cross-links, or polymerizes to solidify into an “implant”.21 Among these, temperature-responsive in situ gel has been widely used over the past few years.
Temperature changes cause temperature-sensitive in situ gel to shift from a solution to a gel.
Poloxamer 407, a commercially accessible and standardized excipient approved for parenteral use by the FDA, is a well-known thermosensitive component focused by many researchers.22 Poloxamer (PO), being a biocompatible polymer composed of polyoxy ethylene (PEO), polyoxy propylene (PPO), and polyoxy ethylene chains (PEOn–PPOn–PEOn) to form nonionic triblock polymers.23 It provides flexibility and control in drug delivery system design, allowing for forming formulations with customized release profiles and improved therapeutic efficacy.24 Additionally, the literature suggests that chitosan (CH) has good biodegradability, low toxicity, and suitable biocompatibility.25 Furthermore, chitosan itself has significant effects on several tumor types that are anti-metastatic anti-proliferative, anti-angiogenetic, and pro-apoptotic.26,27 A thermosensitive chitosan-based injectable hydrogel was developed by Ahsan et al. showing an inhibiting effect on tumor development and used as an effective anticancer drug carrier.28 Hence, we aimed to develop in situ gel as an implant comprising chitosan and poloxamer 407 for treating breast cancer.
Here, we have used doxorubicin (DOX) as a model drug. It is a powerful cytotoxic anthracycline antibiotic that damages DNA and induces cancer cells to undergo apoptosis. This study highlights the potential role of the in situ implant incorporating chitosan and poloxamer 407 using the QbD approach in the treatment of breast cancer by assuring improved cytotoxicity activity, anti-inflammatory activity, and improvised therapeutic potential which would reduce the systemic toxicity of chemotherapy and cancer recurrence after surgical procedure.
2. Materials and methods
2.1. Materials
Chitosan (molecular weight 190–310 kDa, deacetylation degree 75–85%) was obtained from Loba Chemie Pvt Ltd, poloxamer 407 was purchased from Hi Media Pvt Ltd. Doxorubicin was procured as a gift sample from Sisco Research Laboratory Pvt. Ltd (Mumbai). Glacial acetic acid, 3-(4,5-dimethylthiazol2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Sigma), and Dulbecco's modified Eagle's medium (DMEM), dimethyl sulfoxide (DMSO) were obtained from Hi Media Pvt Ltd MCF-7 cells and RAW264.7 cells were bought from National Centre for Cell Science, Pune. All the remaining substances were of analytical grade.
2.2. Optimization
Using Design Expert® Software (version 13, Stat Ease Inc., Minneapolis, USA), the Box–Behnken design was chosen to optimize the in situ gel formulation using 33 factorial design with 3 factors, 3 levels, and 17 runs. In this study, chitosan concentration (X1), poloxamer 407 concentration (X2), and stirring time (X3) were selected as independent variables. The three factorial levels for these variables were designated as follows: −1, 0, and +1 for low, medium, and high levels, respectively as shown in Table 1. The viscosity at 25 °C (Y1), viscosity at 37 °C (Y2), and gelation time (Y3) were chosen as the dependent variables. Using response surface analysis, it was possible to understand the relationship and interaction terms between the parameters as well as the predicted points of the independent variables that would comprise the optimum formulation. A one-way ANOVA was used for the statistical study to ascertain how formulation parameters affected the response variables. Following the software's recommendation, we created one optimal formulation in triplicate. After identifying the range that the ideal conditions might fall into using overlay plots and the desirability function, we measured the dependent variables. To verify the accuracy of the model, we conducted a comparison between the predicted viscosity at 25 °C, viscosity at 37 °C, and gelation time with the observed viscosity at 25 °C, viscosity at 37 °C, and the gelation time.
Table 1 Independent variables and their corresponding levels for DOX–CH–PO in situ implant
| Independent variables |
Symbol |
Coded levels |
| −1 |
0 |
1 |
| Chitosan concentration (%) |
X1 |
0.3 |
0.5 |
0.7 |
| Poloxamer 407 concentration (%) |
X2 |
10 |
20 |
30 |
| Stirring time (second) |
X3 |
60 |
105 |
150 |
2.3. Validation of data analysis and response surface technique
Design Expert software from Stat-Ease was modified to utilize the gathered results. By employing Analysis of Variance (ANOVA), the proposed models were verified. The program generated primary and interaction effects of the variables in the form of “3D response surface plots, 2D perturbation curves, and contour plots.” We selected the combination that predicted the highest degree of desirability, and we compared the projected values with the experiment results.
3. Preparation and physiochemical characterization of DOX–CH–PO in situ implant
Poloxamer 407 solutions were prepared by the cold method process. The optimized amount of polymer was dispersed in distilled, deionized water and stirred continuously for one hour at room temperature. The poloxamer solutions that had partially dissolved were kept in the refrigerator at 4 °C until the polymer had entirely dissolved, which took around 24 hours. The chitosan solution was made by adding the optimized amount in a 2% w/v acetic acid solution and stirring continuously until the chitosan was completely dissolved. Aqueous solution of doxorubicin was added to the chitosan solution with continuous stirring. Finally, this drug solution was added to the poloxamer solution at room temperature and evaluated for different parameters.29
3.1. Fourier transform infrared spectroscopy (FTIR)
The drug and polymers in the prepared formulation and solid mixture were examined for possible interactions using infrared spectroscopy (FT/IR-4600, Jasco, Japan). The metal structure of the device was loaded with roughly 10 mg of powdered samples, and FT-IR readings were taken. The spectra of poloxamer 407, pure doxorubicin, chitosan, and the prepared formulation were compared at 400 to 4000 cm−1. A reference spectrum was compared to identify distinctive peaks representing any drug–polymer interaction.
3.2. Differential scanning colorimetry (DSC)
Using reference material as a sample, this approach estimates the temperature-dependent difference in heat required to raise the sample's temperature. Using a differential scanning calorimeter (DSC-60, Shimadzu, Kyoto, Japan), thermograms of poloxamer 407, pure doxorubicin, chitosan, and formulation were recorded. The sample weight ranged from 2 to 3 mg, and the DSC measurements were carried out in a nitrogen-purging atmosphere with a scanning rate of 10 °C min−1. The samples underwent a heating cycle of 10 °C min−1, ranging from 0 to 350 °C.
3.3. X-ray diffraction (XRD)
XRD is an essential analytical tool for determining the crystalline or amorphous solid state in which drugs may occur. X-ray diffraction investigation of the pure drug, polymers, and formulation was carried out using a Rigaku X-ray diffractometer. The glass slide containing the powdered materials under evaluation was put on the X-ray diffractometer. Within a 2θ range of 10 to 90°, the scanning rate was 10 minutes.
3.4. Pharmaceutical characterization of DOX–CH–PO in situ implant
3.4.1. In vitro gelation time. The behavior of the sol–gel transition was determined using the test tube inversion technique.A 5 mL stoppered test tube with an inner diameter of 10 mm was filled with 1 mL of each prepared solution. Following that, the test tubes were submerged in a water bath with a thermostat set to 37 °C. By flipping the tube once every second until no flow was seen, the sol–gel transition was tracked.30
3.4.2. Investigation of the rheological properties. The measurement of the in situ gel's viscosity, which was quantified in centipoises (cPs), was the most significant rheological investigation. A thermo-responsive in situ gel transforms from a liquid at room temperature to a gel at physiological temperature. The formulation should have an optimum viscosity, which would allow easy instillation at the tumor site as a solution, but also facilitate the formulation to undergo rapid sol-to-gel transition. Viscosity at 25 ± 2 °C and 37 ± 2 °C temperatures of the developed formulation was measured with a viscometer (DV2T Brookfield Viscometer) equipped with spindle number 63 at 50 rpm.
3.4.3. pH. The ideal pH for an in situ gel formulation will allow the formulation to remain stable while also preventing patient irritation upon injection. The pH buffers 4 and 7 were used to calibrate the digital pH meter. A glass electrode was thoroughly dipped into the formulation samples, and twenty milliliters of the optimal formulation were placed in a beaker. Then, the pH of the solution was determined in triplicate.31
3.4.4. Morphological study of the prepared formulation. Scanning electron microscopy (ZEISS, Evo 18, Thornwood, NY, United States) was used to assess the morphological characteristics of the in situ gel. After the gel formation, the optimized formulation was frozen at −20 °C for 24 h and then lyophilized for 48 h. The SEM was used to investigate the cross-section of the lyophilized sample after it was shattered in liquid nitrogen and created a photomicrograph image by scanning an object with an electron beam in a raster scan pattern at 1000 and 2000 magnification. The sample that had been gold-sputter-coated was held in a tiny area using the SEM sample holder. With an accelerating voltage of 15 kV, the SEM images were taken.
3.4.5. In vitro biodegradation analysis. The degradation was measured as a percentage of weight loss (WL) by taking fresh in situ implants weighing 0.1 g in 1 milliliter of PBS at two pH values (5.5 and 7.4) and 37 °C. It was then followed with the addition of 13 mg L−1 lysozyme solutions (same as those found in human serum). The mixtures were agitated at 60 rpm for various durations (1, 3, 5, 7, 9, and 11 days) at 37 °C. Every day, a new lysozyme solution was maintained via replacement. Following the conclusion of every degradation phase, the samples were removed from the medium, cleaned with distilled water, and dried in a vacuum oven at 50 °C until their weight remained constant. The following equation was used to compute the weight loss (%) after the experiment was run in triplicate.32
where Wi denotes the sample's starting weight and Wf its final weight following degradation.
3.4.6. In vitro drug release studies. The paddle method was used to monitor drug release with some modest adjustments. Pre-soaked in PBS 7.4 for 12 hours, the dialysis bag has a molecular weight cut-off of 12000 g mol−1 (6 cm long). Before being put in a dissolution tester, each dialysis bag was fastened with two clamps on either end. Before sealing and placing the dialysis bags in a dissolution tester, DOX–CH–PO solution and free DOX solution were added. Using 500 mL of PBS as a release medium, the test condition involved 50 rpm stirring speed at 37 ± 0.5 °C. Fresh medium was added to the release medium at predetermined intervals. Similarly, it was performed at 0.1 M acetate buffer pH 5.5. The amount of drug released was measured using HPLC at a maximum wavelength of 254 nm.
3.5. Biological characterization of DOX–CH–PO in situ implant
3.5.1. Cytotoxicity assay. Using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) technique, the in vitro cytotoxicity of free DOX, and DOX–CH–PO ISI was assessed against MCF-7 cells. In a 96-well culture plate (Tarsons, India), cells in the logarithmic phase of culture were plated at a density of 5 × 104 cells per well and allowed to adhere for 24 hours. The entire media was changed to DMEM (Dulbecco's Modified Eagle media) after a day. Then the control cells were treated with free DOX, and DOX–CH–PO IFI (10 to 50 μg mL−1) and incubated for 24 h. After adding the MTT solution (0.5 mg mL−1) and letting it sit for three hours, 100 μL of DMSO was added to dissolve the formazan crystals. A microplate reader was then used to measure the absorbance at 540 nm. The values were presented as mean ± SD and the tests were run in triplicate.33
3.5.2. In vitro anti-inflammatory study. The anti-inflammatory activity of the proposed DOX–CH–PO ISI was determined in RAW 264.7 macrophage cell lines. These cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% Pen-strep (a combination of streptomycin and penicillin) from a 100 U mL−1 Pen-strep stock solution, and 5% supplied CO2 at 37 °C. The RPMI 1640 medium containing 100 g mL−1 of the DOX–CH–PO sample completely dissolved. The cells were divided into three groups, i.e., the negative control (NC) group, a disease control group (LPS-treated), and the DOX–CH–PO group. In the next step, 50 μL of LPS (1 g mL−1) was administered to the DC group and DOX–CH–PO group, while 50 μL of RPMI 1640 medium was given to the negative control group. TNF-α, IL-1β, and IL-6 expression levels were measured using ELISA kits, in accordance with the manufacturer's recommendations. The supernatants were subjected to antigen–antibody reactions following the user's handbook that came with the ELISA kits, and the degree of cytokine expression was assessed. After adding 50 μL of stop buffer to each well, the cytokine level was measured using a microplate reader, and absorbances were recorded at 450 nm.34
3.5.3. Cellular uptake. Fluorescent microscopic imaging was used to assess the cellular absorption of DOX–CH–PO ISI. To be more specific, a coverslip was placed in each well of six-well plates. In 6-well plates, 3 × 105 cells per well were planted on the coverslips with 2 milliliters of culture media. Cells were treated with DOX–CH–PO ISI at equivalent doses of 200 μg mL−1 DOX after a 24 hour incubation period. Following incubation for 4 hours, the drug-containing medium was disposed of. The cells underwent three PBS washes, were fixed for thirty minutes in 75% ice-cold ethanol, and were examined using a fluorescent microscope (Cilika, BT-P-FL). The fluorescence intensity was obtained by using the software ImageJ 1.53.35
3.5.4. In vivo anti-inflammatory study. An in vivo assessment of the DOX–CH–PO ISI's anti-inflammatory action was conducted to appraise its potential. For this, following the 20th day of treatment, breast tissue samples were taken from recently sacrificed animals. We separated the breast tissues and homogenized them to perform the biochemical study, yielding a 10% homogenate in phosphate buffer. The homogenates underwent a 15 minute centrifugation at 10000 rpm and 4 °C in an Eppendorf 5425 R G ultracentrifuge. The ELISA kits' instructions for performing antigen–antibody reactions on supernatants were followed to measure the levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β).36 Two groups were assessed for total protein (pg mL−1): group II, which received treatment, and group I, which did not get treatment (disease control).
3.5.5. In vivo pharmacokinetics study. To examine the absolute bioavailability and pharmacokinetic parameters of three groups of female Wistar rats, an equal dosage of free DOX and DOX–CH–PO ISI (5 mg kg−1 DOX) was intravenously and intramuscularly injected into each group respectively. Rat retro-orbital plexus blood samples (200 μL) were drawn at predetermined intervals (6 h, 12 h, 24 h, 48 h, 72 h), and the plasma was separated by centrifuging the samples for 10 minutes at 5000 rpm. Before analysis, all plasma samples were kept at −20 °C. 100 μL of plasma samples were vortexed for three minutes after adding 1 mL of acetonitrile and water mixture solution. After being collected, the supernatant was evaporated in nitrogen-filled conditions. Following centrifugation and a vortex, the residue was resolved using 100 μL of mobile phase, and the supernatant was subjected to HPLC analysis. A C18-HL reversed-phase column (150 4.6 mm, 3.5 m; Zorbax Ltd) was used to evaluate the samples. At ambient temperature (25–28 °C), a binary gradient solvent system was used for the elution, with a flow rate of 1.00 mL min−1, with acetonitrile (ACN): sodium dodecyl sulphate and orthophosphoric acid as the mobile phase. Plasma from untreated rats was used to prepare standard calibration curves of concentrations 0.2, 0.4, 0.6, 0.8, and 1 μg mL−1. The PK Solver program computed the pharmacokinetic parameters using the linear trapezoidal approach and non-compartmental studies.37
3.6. Statistical analysis
A statistical analysis was performed on the data using version 9 of GraphPad Prism. One-way analysis of variance (ANOVA) was used to compare group variances, with a significance level of P = 0.05. The sample sizes range from n = 3 to n = 6, and the data is shown as mean ± standard deviation (SD).
4. Results and discussion
4.1. Statistical optimization of DOX–CH–PO in situ implant
Based on the Box–Behnken factorial design, the design expert projected a total of 17 runs for three components, which were modified at three distinct levels: chitosan concentration (X1), poloxamer 407 concentration (X2), and stirring time (X3). These runs were varied at three distinct levels (coded as −1, 0, and +1). The viscosity at 25 °C (Y1), viscosity at 37 °C (Y2), and gelation time (Y3) were studied as responses shown in Table 2.
Table 2 Response values of experimental runs
| Formulation code |
X1 |
X2 |
X3 |
Y1 |
Y2 |
Y3 |
| Chitosan (%) |
Poloxamer 407 (%) |
Stirring time (s) |
Viscosity at 25 °C (cPs) |
Viscosity at 37 °C (cPs) |
Gelation time (s) |
| F1 |
0.3 |
20 |
150 |
141.5 |
7563.2 |
23 |
| F2 |
0.5 |
20 |
105 |
151.8 |
8245.3 |
30 |
| F3 |
0.7 |
20 |
150 |
145.3 |
7845.6 |
25 |
| F4 |
0.7 |
10 |
105 |
138.6 |
6945.6 |
35 |
| F5 |
0.3 |
20 |
60 |
149.3 |
8015.4 |
23 |
| F6 |
0.5 |
20 |
105 |
153.6 |
8612.3 |
27 |
| F7 |
0.5 |
30 |
150 |
176.5 |
9784.6 |
10 |
| F8 |
0.3 |
30 |
105 |
172.1 |
9542.1 |
15 |
| F9 |
0.7 |
30 |
105 |
183.6 |
10453.3 |
12 |
| F10 |
0.3 |
10 |
105 |
140.2 |
7398.8 |
37 |
| F11 |
0.7 |
20 |
60 |
153.5 |
8512.4 |
27 |
| F12 |
0.5 |
10 |
150 |
135.6 |
6712.2 |
40 |
| F13 |
0.5 |
20 |
105 |
149.2 |
8098.2 |
28 |
| F14 |
0.5 |
20 |
105 |
152.2 |
8412.3 |
26 |
| F15 |
0.5 |
20 |
105 |
150.7 |
8145.9 |
28 |
| F16 |
0.5 |
30 |
60 |
183.2 |
10356.9 |
11 |
| F17 |
0.5 |
10 |
60 |
144.6 |
7789.8 |
35 |
4.1.1. Effect of independent variables on viscosity at 25 °C (Y1). The polynomial equation (eqn (1)) for response Y1, on viscosity at 25 °C, was generated by the model. The primary and secondary effects of each response might be ascertained using factor coefficients. Predicting the reaction for levels of each element is feasible by using an equation stated in terms of coded factors. By default, factors with high levels are coded as +1, while those with low levels are coded as −1. By comparing the factor coefficients in the coded equation, we can ascertain the elements' respective relative importance.| | |
Y1 = 151.5 + 2.2375X1 + 19.55 X2 − 3.9625 X3 + 3.275 X1X2 − 0.1 X1X3 + 0.575 X2X3 − 2.725 X12 + 9.85 X22 − 1.375 X32
| (1) |
The equation showed that chitosan (X1) and poloxamer 407 (X2) have a favorable impact on the in situ implant while stirring time (X3) has a negative impact on it. The fact that X3's magnitude was most readily apparent led to the conclusion that its detrimental effects were most severe. The combined interaction effect of X1X2 and X2X3 was also found to be synergistic, with X1X3 having the largest detrimental effect on the gel. As a result, it can be inferred that increasing the stirring time (X3), there is a decrease of viscosity at 25 °C and as the concentration of poloxamer 407 (X2) increases, there is an increase in the viscosity at 25 °C of the in situ implant.
With a p-value of less than 0.05 (<0.0001), the ANOVA for the quadratic response surface model of viscosity at 25 °C was determined to be significant. With a p-value of 0.0020, factor A (chitosan concentration) was found to have a significant impact on viscosity at 25 °C. On the other hand, factor C and B's p-values of less than 0.0001 indicate that they significantly affect viscosity at 25 °C. There was a significant correlation between the actual and expected results, as evidenced by the variations of less than 0.2 between the adjusted R2 (0.9925) and predicted R2 (0.9898) for the viscosity at 25 °C quadratic model. It is recommended to utilize this model to navigate the design space. The contour plot of the reaction and response surface for Y1 are shown in Fig. 1.
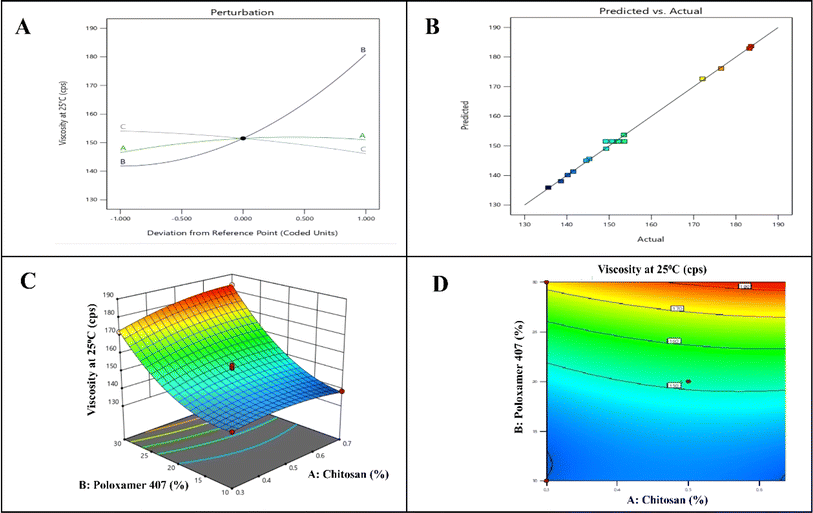 |
| | Fig. 1 QBD model graph for viscosity at 25 °C. (A) Perturbation curve, (B) linear correlation plot, (C) response surface plot, (D) contour plot. | |
4.1.2. Effect of independent variables on viscosity at 37 °C (Y2). The 2D and 3D graphs illustrate the likely impacts of changing independent variables on the degree of viscosity at 37 °C deviation. The viscosity at 37 °C deviation is shown in the actual equation (eqn (2)).| | |
Y2 = 8302.8 + 154.675 X1 + 1411.31 X2 − 346.112 X3 + 341.1 X1X2 − 53.65 X1X3 + 126.325 X2X3 − 197.288 X12 + 479.437 X22 − 121.362 X32
| (2) |
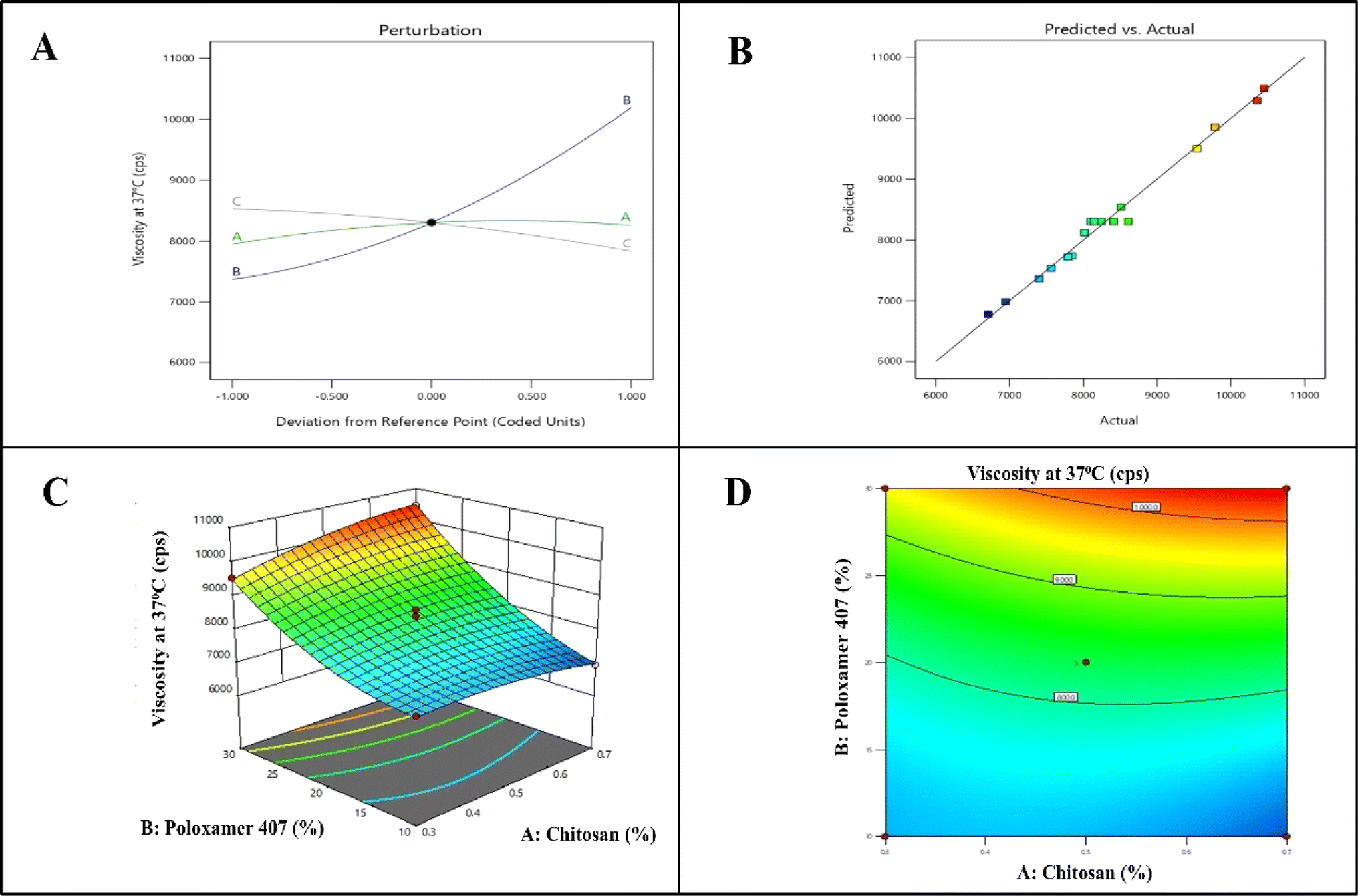
The equation showed that the chitosan (X1) and poloxamer 407 (X2) have a positive impact on the viscosity at 37 °C, whereas the stirring time (X3) has a negative impact. Similarly, it was found that the interaction between X1X2 and X2X3 was synergistic, while X1X3 had a detrimental effect on viscosity at 37 °C. It indicates that as the concentration of poloxamer 407 (X2) and chitosan (X1) increases, there is an increase in the viscosity at 37 °C. Micelle production and micellar aggregation occur in response to an increase in poloxamer 407 concentration. The concentration must be higher than the micellar concentration for the gel phase to occur. Hydrophobic parts of the poloxamer 407 are kept apart in cold water by hydrogen bonding between POP chains and water. With an increase in temperature, the hydrogen bonding is broken and hydrophobic interactions are forming the gel.
The p-value for the model, which is less than 0.05, is less than 0.0001, indicating that the ANOVA for the quadratic response surface model for viscosity at 37 °C is significant. With a p-value of 0.0450, X1 was found to have a significant impact on viscosity at 37 °C. X2 and X3 had p-values of less than 0.0001 and 0.0010, respectively, indicating a substantial impact on viscosity at 37 °C. The adjusted R2 (0.9728) and projected R2 (0.9448), which displayed discrepancies of less than 0.2, demonstrated a strong correlation between the expected and actual findings for the viscosity at 37 °C quadratic model. Fig. 2 displays the response surface plot and response contour plot for Y2.
 |
| | Fig. 2 QBD model graph for viscosity at 37 °C. (A) Perturbation curve, (B) linear correlation plot, (C) response surface plot, (D) contour plot. | |
4.1.3. Effect of independent variables on gelation time (Y3). Fig. 3's representation of a 2D and 3D response surface plot showed that gelation time has a positive association with X1 and X3. The inverse relationship between poloxamer 407 and gelation time was also demonstrated by a negative value for the linear coefficient of X2. With increase in the concentration of poloxamer 407 (X2), gelation time decreases.| | |
Y3 = 25.4118 + 0.125 X1 − 12.375 X2 + 0.25 X3
| (3) |
 |
| | Fig. 3 QBD model graph for gelation time (A) Perturbation curve, (B) linear correlation plot, (C) response surface plot, (D) contour plot. | |
Since the model's p-value is less than 0.05 (<0.0001), the ANOVA for the quadratic response surface model of gelation time was determined to be significant. X1 has p-value of 0.8954 showing that factor A had significant effect on the gelation time. While p-values for X2 and X3 were <0.0001 and 0.7928 showing that it has significant effect on the gelation time. The projected R2 (0.8836) and adjusted R2 (0.9155) for the gelation time linear model both showed discrepancies smaller than 0.2, showing a significant correlation between the results of the experiment and those expected.
4.1.4. Optimization and validation of the model. Based on the desired range of response values, independent factors were obtained using statistical and graphical analysis. An optimal in situ implant formulation was chosen using the Design Expert Software point prediction based on the desirability factor's proximity to 1. It is determined that the optimized batch with the highest desirability of 1 was chosen, with X1 = 0.5% (chitosan concentration), X2 = 20% (poloxamer 407), and X3 = 105 s (stirring time). The model was validated by observing the responses of an optimal formulation that was developed. The projected and obtained results (viscosity at 25 °C-152.6 cPs, viscosity at 37 °C-8312.6 cPs, and gelation time-26 s) are in good agreement, demonstrating the optimized in situ implant formulation's rationality.
4.2. Preparation and physiochemical characterization of DOX–CH–PO in situ implant
The in situ gel was prepared by optimizing the parameters. The in situ gel was formulated using poloxamer 407 (20% w/v) dispersed in distilled water which was kept in the refrigerator at 4 °C for 24 hours for completely dissolving. Chitosan (0.5% w/v) was dissolved in a 2% w/v acetic acid solution. Aqueous solution of doxorubicin was added to the chitosan solution with continuous stirring. Finally, at room temperature, this drug solution was added to the poloxamer solution, it provides ease of injection and in situ gelation at the injection site.
4.2.1. FTIR. FT-IR analysis was performed to ascertain whether there is an interaction between poloxamer 407, chitosan, and doxorubicin in the in situ gel or not. The FT-IR spectra of poloxamer 407, chitosan, doxorubicin, and formulation are displayed in Fig. 4A. The characteristic peaks of doxorubicin of FTIR spectra at 3747.98 cm−1 (O–H stretch), 3313.11 cm−1 (N–H stretch), 2894.63 cm−1 (C–H stretch), 1729.83 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch), and 1412.62 cm−1 (CC ring stretch). The peak pattern in the formulation spectrum was precisely the same as that of the pure drug. Considerable surface modification is depicted by a small alteration in the fingerprint region's peak intensity. The formulation had several distinct peaks at approximately 1100 cm−1, which corresponded to the typical peaks of poloxamer 407. The spectra of the in situ implant formulation show all of the doxorubicin's distinctive peaks, indicating that there has been no chemical alteration.
O stretch), and 1412.62 cm−1 (CC ring stretch). The peak pattern in the formulation spectrum was precisely the same as that of the pure drug. Considerable surface modification is depicted by a small alteration in the fingerprint region's peak intensity. The formulation had several distinct peaks at approximately 1100 cm−1, which corresponded to the typical peaks of poloxamer 407. The spectra of the in situ implant formulation show all of the doxorubicin's distinctive peaks, indicating that there has been no chemical alteration.
 |
| | Fig. 4 (A) FT-IR spectrum, (B) DSC thermogram, and (C) XRD spectrum depicting peaks of doxorubicin, poloxamer 407, chitosan, and formulation (D) SEM images of optimized DOX–CH–PO ISI at different magnification. | |
4.2.2. DSC. The DSC thermogram of pure doxorubicin showed an endothermic peak at 225.48 °C. Due to the amorphous nature of chitosan, it does not show any peak (Fig. 4B). So, the thermogram of the formulation has not shifted, indicating no change in the physical state between the drug and polymers. It revealed that the drug is in crystalline form in the formulation.
4.2.3. XRD. Fig. 4C display XRD data of poloxamer 407, chitosan, doxorubicin, and formulation. XRD spectrum of doxorubicin indicates peak position (2θ) at 16.49°, 21.79°, 22.43°, and 24.92°, which is crystalline whereas chitosan and poloxamer 407 showed amorphous nature. The XRD results for the formulation showed a peak intensity that was noticeably close to the plain drug. It indicates that the drug retains its property in the formulation and has no change in the physical state.
4.3. Pharmaceutical characterization of DOX–CH–PO in situ implant
4.3.1. In vitro gelation time. For in situ gel systems, gelation time is an essential feature. In general, a shorter gelation period would be preferable to reduce the amount of time needed for the implanted dosage to solidify into a thick gel. Using the test tube inversion method, the behavior of the sol–gel transition was found. At 37 °C, the optimum preparation in the vial took <26 ± 0.2 s to stop flowing, indicating rapid gelation which would help to develop a depot drug delivery system. The thermoresponsive behavior of poloxamer 407 and the interaction with chitosan leads to a depot formation at physiological temperatures.38 Z. Fathalla et al. reported that a combination of poloxamer 407 with chitosan showed a gelation time of 17.9 ± 3.5 s for ophthalmic delivery systems.39 The optimized formulation was further characterized for various physicochemical parameters.
4.3.2. pH. A pH meter calibrated using standard buffers of pH 4 and pH 7 under the protocol was used to check the optimized batch's pH. The developed formulation was measured using a digital pH meter and was found to be 6.81. This pH value is compatible with the site and there would be no irritation as the pH of solid tumors is typically between 6.5 to 7.2, indicating a slightly acidic environment.40
4.3.3. Investigation of rheological properties. Viscosity has a great influence on thermosensitive in situ gels. As indicated, a formulation should ideally have a low viscosity at application and a high viscosity afterward to enable it to remain at the application site. The prepared formulations F1–F17 of the temperature-triggered in situ implant were used to measure the viscosity of the preparation by using Brookfield Viscometer (DV-II + Pro). After raising the temperature from 25 to 37 °C, the viscosity of the prepared formulation was measured in the range of 6712.26 to 10453.3 cPs. The optimized batch showed a viscosity of around 8312.6 ± 114.2 cPs at 37 °C. When chitosan interacts with poloxamer 407 through hydrogen bonding and electrostatic interactions, it enhances the formulation viscosity.41 Upon increasing the temperature, poloxamer 407 forms micelles due to the hydrophobic nature of poly(propylene oxide) blocks, and these micelles aggregate into a gel network at body temperature.42
4.3.4. Morphological study of the prepared in situ implant. The morphology study of the optimized batch was carried out in SEM (Hitachi S300N) at two different magnifications. At 1000× magnification, the gel matrix surface gave the impression of being rough and corrugated as displayed in Fig. 4D. At 2000× magnification, it showed cylindrical crystal surface morphology but a structure with a multilayer. Phase separation or polymer separation was not evident.
4.3.5. In vitro degradation analysis. Fig. 5 shows the biodegradability of the optimized formulation in the presence of a lysozyme enzyme. The findings showed that the formulation underwent biodegradation due to the reduced sugar released from the samples. Maximum weight loss values of 74.24 ± 2.2% (pH = 5.5) and 67.58 ± 1.7% (pH = 7.4) were observed when the degradation rate was progressively raised for up to 11 days due to the presence of chitosan and poloxamer 407. The existence of all the hydrophilic water-soluble components and their great biodegradability supports the current finding. This gel's ability to adsorb lysosomes is explained by several functional hydrophilic NH2, OH– and COOH groups in these substances. At pH 5.5, chitosan is in a more protonated and soluble form, leading to a faster degradation process than at pH 7.4.43,44
 |
| | Fig. 5 Degradation percentage of DOX–CH–PO ISI at pH = 5.5 and pH = 7.4. Values represent mean ± SD (n = 3). | |
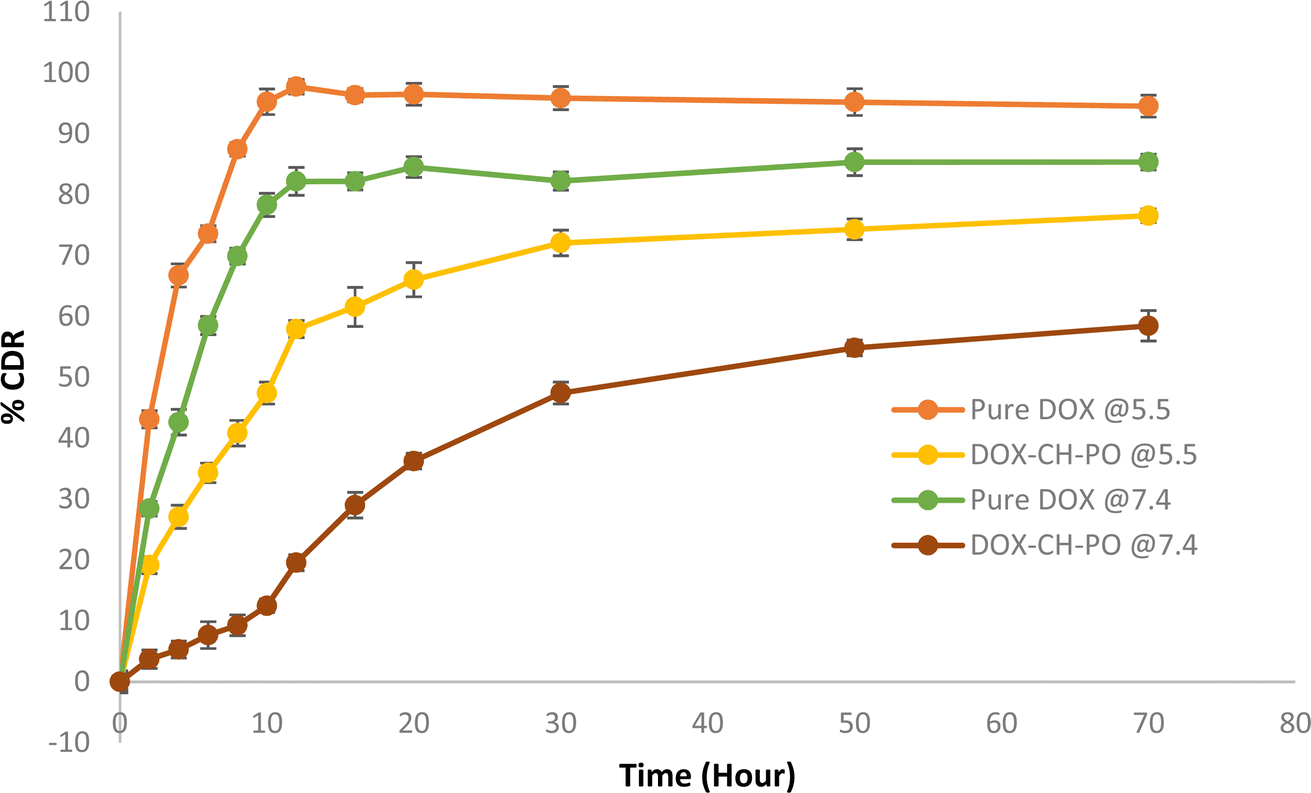
4.3.6. In vitro drug release studies. Fig. 6 displays the cumulative percent of DOX release versus time profiles for pure DOX, and DOX–CH–PO ISI. At pH 7.4, it was observed that about 85.32 ± 1.2% of DOX was released from pure DOX over 72 hours whereas 29.01 ± 2.1% of DOX was released from the gel within 16 h and 58.44 ± 2.5% after 72 hours displaying a slow–release profile. In contrast, there was 76.52 ± 1.3% of DOX release over 72 hours from the gel at pH 5.5. A controlled release of doxorubicin was seen in both pH conditions. The drug doxorubicin exhibits a notable increase in release at pH 5.5, owing to its improved solubility under slightly acidic pH circumstances. In pure DOX, the release pattern depends upon the intrinsic physicochemical nature of the drug. Additionally, the developed formulation formed a rigid matrix, slowing the drug's diffusion out of the gel. The combination of the thermosensitivity of poloxamer 407 and the pH sensitivity of chitosan affects the delayed drug release pattern. The gelation helps to localize the drug at the tumor site, creating a depot that slowly releases the drug over time, ensuring a sustained therapeutic effect.45 This delayed release helps maintain the effective concentration for a longer period, reduce dose dependency, and increase drug efficacy. J. Varshosaz et al. reported similar results in the combination of chitosan with poloxamer 407 showing sustained release of the drug for the ocular delivery of ciprofloxacin.46
 |
| | Fig. 6 In vitro drug release of pure DOX and DOX–CH–PO ISI. Results are presented as the mean ± SD, n = 3. | |
4.4. Biological characterization of DOX–CH–PO in situ implant
4.4.1. Cytotoxicity assay. In vitro cell viability or cytotoxicity of prepared DOX–CH–PO ISI and free DOX in MCF-7 cells were investigated. The cell growth inhibition activity of free DOX and DOX–CH–PO against breast cancer is shown in Fig. 7A. The free DOX and DOX–CH–PO ISI killed 49.09 ± 3.9%, and 26.47 ± 2.8% of the MCF-7 cells respectively. When chitosan interacts with the negatively charged cell membranes of cancer cells, it may be more likely to accumulate in tumor cells rather than in healthy tissues, reducing off-target effects. Poloxamer enhances the penetration of the drug into the cancer cells due to its ability to disrupt cell membranes, facilitating drug uptake in cells. This data demonstrates that DOX–CH–PO ISI exhibited better anticancer cytotoxicity as combining the drug with poloxamer and chitosan has a synergistic effect of killing the MCF-7 cells compared to free DOX. According to microscopic photographs, live cells are shaped differently from dead cells, which are bright and refractile. Typical images of living and dying cells taken under a microscope (Fig. 7C and D). Y. I. Cho et al. reported that the thermosensitive hydrogels made of chitosan–doxorubicin conjugates showed that chitosan–doxorubicin also had significant cytotoxicity comparable to free doxorubicin.47 Previous reports also indicated that a combination of P407 and doxorubicin showed the synergistic killing of MC-38 tumor cells and reduced tumor cell proliferation.48
 |
| | Fig. 7 (A) Effect of free DOX and DOX–CH–PO ISI on the viability of the MCF-7 cells (B) microscopical images of cells of control (C) microscopical images of cells of free DOX 5 μg mL−1 (D) microscopical images of cells of DOX–CH–PO ISI 5 μg mL; 200× magnification data represent mean ± SD, n = 3 (***p < 0.001). | |
4.4.2. In vitro anti-inflammatory study. The in vitro anti-inflammatory study of the DOX–CH–PO ISI was investigated using RAW264.7 cells. The present investigation involved pre-treating RAW264.7 cells' LPS-induced inflammatory response with DOX–CH–PO ISI. Fig. 8A indicates that the DOX–CH–PO ISI has anti-inflammatory properties by downregulating pro-inflammatory cytokines in RAW264.7 cell lines including TNF-α, IL-6, and IL-1β. Chitosan has excellent anti-inflammatory properties inhibiting pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α. Previously, it has been demonstrated chitosan attenuates inflammation through multiple mechanisms, such as altering cytokine expression, scavenging free radicals, and regulating immunological responses.49 It has been earlier reported on the impact of chitosan and its oligosaccharides on LPS-stimulated RAW 264.7 cells, the researchers found that exposure to chitosan and its oligosaccharides resulted in a reduction of LPS-induced TNF-α and IL-6 release in the culture medium.50 The results indicated that DOX–CH–PO ISI pretreated LPS-induced RAW264.7 cells may serve anti-inflammatory properties and lower inflammatory factor levels.
 |
| | Fig. 8 (A) In vitro anti-inflammatory effect of normal cells, disease control cells, and DOX–CH–PO ISI. (B) In vivo anti-inflammatory study of DOX–CH–PO ISI on different cytokines levels. Data represent mean ± SD, n = 3 (**p < 0.01, ***p < 0.001). | |
4.4.3. Cellular uptake. The MCF-7 cell lines were used to study the cellular uptake of DOX–CH–PO ISI. As displayed in Fig. 9B, the fluorescence microscopy image demonstrated that the cell lines incorporated DOX–CH–PO ISI (represented by the red dots). After 4–6 hours of observation, the formulation group showed a higher mean fluorescence intensity of 23.2 (red channel) than the control group (2.1) (Fig. 9C and D). The DOX–CH–PO ISI conjugates exhibited higher fluorescence intensity, suggesting a higher amount of cellular uptake compared with the control MCF-7 cell line. The positively charged chitosan makes it easier for it to interact with the negatively charged cell membranes, which improves cell adhesion and uptake.51 Additionally, poloxamer interacts with the lipid bilayer of the cell membrane to increase permeability, which amplifies this impact even further. This combination may improve the drug's ability to penetrate cells. Recently, it has been reported that doxorubicin-loaded poly (methacrylamide) based copolymeric nanoparticles functionalized with chitosan showed significant cellular internalization.52
 |
| | Fig. 9 In vitro cellular uptake of (A) negative control (B) DOX–CH–PO ISI in MCF-7 cells; 200× magnification (C) fluorescence intensity of control group (D) fluorescence intensity of formulation group. | |
4.4.4. In vivo anti-inflammatory study. Published research indicates that chitosan can reduce the production of TNF-α, IL-1β, and IL-6 in LPS-stimulated RAW264.7 cells. In the current study, DOX–CH–PO ISI significantly reduced the levels of cytokines, including IL-6, TNF-α, and IL-1β (Fig. 8B). The study suggests that DOX–CH–PO ISI may lessen the generation of pro-inflammatory cytokines when used in anticancer treatment as there was a decrease in the infiltration of inflammatory cells in the treated groups.
5. Conclusion
In the present study, a biodegradable and sustained-release DOX–CH–PO in situ implant was successfully prepared for the treatment of breast cancer. The in situ implant's physicochemical, pharmacological, and biological potential for treating breast cancer has been effectively developed and characterized. The QbD method was used to prepare seventeen formulations (F1–F17) from 33 factorial designs and optimize the amounts of chitosan, poloxamer 407, and stirring time. The results showed that a liquid formulation that gels upon injection at 37 °C in 26 ± 0.2 s was successfully produced using 0.5% w/v chitosan and 20% w/v poloxamer 407 (optimized DOX–CH–PO ISI). The optimized formulation showed good rheological properties and extended drug release. The prepared formulation showed better cytotoxicity due to the synergistic potential of chitosan and poloxamer against the MCF-7 cells compared to the free drug. The DOX–CH–PO ISI exhibits anti-inflammatory characteristics by inhibiting pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, in RAW264.7 cell lines. It has been observed that DOX–CH–PO in situ implant increases the cellular uptake in the MCF-7 cells. The in vivo pharmacokinetic the developed formulation results showed an increase in the plasma half-life, a decrease in the peak plasma concentration, and AUC which may indicate maintaining high concentrations of the drug within the tumor and less exposure of the drug to the bloodstream. Therefore, it is possible to enhance the effectiveness and decrease the systemic toxicity of chemotherapy drugs by using the sustained-release DOX–CH–PO in situ implant which may be a viable option for the treatment of breast cancer. Further, the intrinsic toxicological and repeated dose kinetics profiling is key to predict the clinical outcomes.
Data availability
The authors declare that the data supporting the findings of this study entitled “Doxorubicin loaded chitosan–poloxamer in situ implant for the treatment of breast cancer” available within the paper. Should any raw data files be needed in another format, they are available from the corresponding author upon reasonable request. Source data are provided in this paper.
Author contributions
Guru Prasanna Sahoo: manuscript writing, data collection and proof reading. Vineet Kumar Rai: data collection, experimental work, compilation, and manuscript writing. Deepak Pradhan: proofreading, software and language editing. Jitu Halder: data analysis, language editing and critical analysis. Tushar Kanti Rajwar: compilation, manuscript writing. Ritu Mahanty: data collection and experimental work. Ivy Saha: data collection, experimentation and statistics. Ajit Mishra: data collection and experimental work. Priyanka Dash: animal experimentation and proof reading. Chandan Dash: software analysis, data collection and proof reading. Jameel Al-Tamimi: data analysis, language editing. Salim Manoharadas: data analysis, language editing and funding support. Biswakanth Kar: animal experiment and data interpretation. Goutam Ghosh: conceptualization of the topics and critical analysis. Goutam Rath: conceptualization of the topic and design of the table of content, supervision, data analysis and paper writing, editing and proof reading.
Conflicts of interest
The authors declare no conflict of interest
Acknowledgements
The DBT Builder project funded the research with order no (BT/INF/22/SP45078/2022). The authors would also like to thank the Researchers Supporting Project Number (RSPD2024R1082), King Saud University, Riyadh, Saudi Arabia. King Saud University, Riyadh, Saudi Arabia for funding this research work. The authors also acknowledge the financial support of the Department of Biotechnology (DBT), Govt. India.
References
- Breast Cancer, World Health Organization (WHO), available from: https://www.who.int/news-room/fact-sheets/detail/breast-cancer Search PubMed.
- S. Nardin, E. Mora, F. M. Varughese, F. D'Avanzo, A. R. Vachanaram and V. Rossi, et al., Breast Cancer Survivorship, Quality of Life, and Late Toxicities, Front. Oncol., 2020, 10, 864 CrossRef PubMed.
- R. De Souza, P. Zahedi, C. J. Allen and M. Piquette-Miller, Polymeric drug delivery systems for localized cancer chemotherapy, Drug Delivery, 2010, 17(6), 365–375 CrossRef CAS PubMed.
- O. G. Scharovsky, L. E. Mainetti and V. R. Rozados, Metronomic Chemotherapy: Changing the Paradigm That More Is Better, Curr. Oncol., 2009, 16(2), 7–15 CrossRef CAS PubMed.
- S. Talebian, J. Foroughi, S. J. Wade, K. L. Vine, A. Dolatshahi-Pirouz and M. Mehrali, et al., Biopolymers for Antitumor Implantable Drug Delivery Systems: Recent Advances and Future Outlook, Adv. Mater., 2018, 30(31), 1706665 CrossRef PubMed.
- S. H. Jang, M. G. Wientjes, D. Lu and J. L. -S. Au, Drug Delivery and Transport to Solid Tumors, Pharm. Res., 2003, 20(9), 1337–1350 CrossRef CAS PubMed.
- J. M. Binkley, S. R. Harris, P. K. Levangie, M. Pearl, J. Guglielmino and V. Kraus, et al., Patient perspectives on breast cancer treatment side effects and the prospective surveillance model for physical rehabilitation for women with breast cancer, Cancer, 2012, 118(S8), 2207–2216 CrossRef PubMed.
- A. I. Riggio, K. E. Varley and A. L. Welm, The lingering mysteries of metastatic recurrence in breast cancer, Br. J. Cancer, 2021, 124(1), 13–26 CrossRef PubMed.
- A. Ahmad, Pathways to Breast Cancer Recurrence, ISRN Oncol., 2013, 1–16 Search PubMed.
- C. Pacheco, A. Baião, T. Ding, W. Cui and B. Sarmento, Recent advances in long-acting drug delivery systems for anticancer drug, Adv. Drug Delivery Rev., 2023, 194, 114724 CrossRef CAS PubMed.
- A. Banerjee, S. Pathak, V. D. Subramanium, G. Dharanivasan, R. Murugesan and R. S. Verma, Strategies for targeted drug delivery in treatment of colon cancer: current trends and future perspectives, Drug Discovery Today, 2017, 22(8), 1224–1232 CrossRef CAS PubMed.
- A. Kumar and J. Pillai. Implantable drug delivery systems, in Nanostructures for the Engineering of Cells, Tissues and Organs. Elsevier; 2018. p. 473–511. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128136652000132 Search PubMed.
- J. A. Camargo, A. Sapin, C. Nouvel, D. Daloz, M. Leonard and F. Bonneaux, et al., Injectable PLA-based in situ forming implants for controlled release of Ivermectin a BCS Class II drug: solvent selection based on physico-chemical characterization, Drug Dev. Ind. Pharm., 2013, 39(1), 146–155 CrossRef CAS PubMed.
- S. A. Chew and S. Danti, Biomaterial-Based Implantable Devices for Cancer Therapy, Adv. Healthcare Mater., 2017, 6(2), 1600766 CrossRef PubMed.
- B. D. Weinberg, E. Blanco and J. Gao, Polymer Implants for Intratumoral Drug Delivery and Cancer Therapy, J. Pharm. Sci., 2008, 97(5), 1681–1702 CrossRef CAS PubMed.
- A. A. Exner and G. M. Saidel, Drug-eluting polymer implants in cancer therapy, Expert Opin. Drug Delivery, 2008, 5(7), 775–788 CrossRef CAS PubMed.
- J. G. Hiremath, N. S. Khamar, S. G. Palavalli, C. G. Rudani, R. Aitha and P. Mura, Paclitaxel loaded carrier based biodegradable polymeric implants: preparation and in vitro characterization, Saudi Pharm. J., 2013, 21(1), 85–91 CrossRef PubMed.
- M. Ebrahimnia, S. Alavi, H. Vaezi, M. Karamat-Iradmousa and A. Haeri, Exploring the vast potentials and probable limitations of novel and nanostructured implantable drug delivery systems for cancer treatment, EXCLI J., 2024, 23, 143–179 Search PubMed.
- M. Souri, S. Elahi and M. Soltani, Intratumoral implantable drug delivery system for targeted localized chemotherapy in breast cancer, J. Drug Delivery Sci. Technol., 2024, 94, 105519 CrossRef CAS.
- S. Kempe and K. Mäder, In situ forming implants—an attractive formulation principle for parenteral depot formulations, J. Controlled Release, 2012, 161(2), 668–679 CrossRef CAS PubMed.
- M. Parent, C. Nouvel, M. Koerber, A. Sapin, P. Maincent and A. Boudier, PLGA in situ implants formed by phase inversion: critical physicochemical parameters to modulate drug release, J. Controlled Release, 2013, 172(1), 292–304 CrossRef CAS PubMed.
- Y. Chen, J. H. Lee, M. Meng, N. Cui, C. Y. Dai and Q. Jia, et al., An Overview on Thermosensitive Oral Gel Based on Poloxamer 407, Materials, 2021, 14(16), 4522 CrossRef CAS PubMed.
- M. Almeida, M. Magalhães, F. Veiga and A. Figueiras, Poloxamers, poloxamines and polymeric micelles: definition, structure and therapeutic applications in cancer, J. Polym. Res., 2018, 25(1), 31 CrossRef.
- E. Giuliano, D. Paolino, M. Fresta and D. Cosco, Mucosal Applications of Poloxamer 407-Based Hydrogels: An Overview, Pharmaceutics, 2018, 10(3), 159 CrossRef CAS PubMed.
- N. N. Inamdar and V. Mourya, Chitosan and Low Molecular Weight Chitosan: Biological and Biomedical Applications, in Advanced Biomaterials and Biodevices, ed. Tiwari A. and Nordin A. N., Wiley, 1st edn, 2014, pp. 183–242. available from: https://onlinelibrary.wiley.com/doi/10.1002/9781118774052.ch6 Search PubMed.
- H. S. Adhikari and P. N. Yadav, Anticancer Activity of Chitosan, Chitosan Derivatives, and Their Mechanism of Action, Int. J. Biomater., 2018, 2018, 1–29 CrossRef PubMed.
- C. Qin, Y. Du, L. Xiao, Z. Li and X. Gao, Enzymic preparation of water-soluble chitosan and their antitumor activity, Int. J. Biol. Macromol., 2002, 31(1–3), 111–117 CrossRef CAS PubMed.
- A. Ahsan, M. A. Farooq and A. Parveen, Thermosensitive Chitosan-Based Injectable Hydrogel as an Efficient Anticancer Drug Carrier, ACS Omega, 2020, 5(32), 20450–20460 CrossRef CAS PubMed.
- T. Gratieri, G. M. Gelfuso, E. M. Rocha, V. H. Sarmento, O. De Freitas and R. F. V. Lopez, A poloxamer/chitosan in situ forming gel with prolonged retention time for ocular delivery, Eur. J. Pharm. Biopharm., 2010, 75(2), 186–193 CrossRef CAS PubMed.
- L. Liu, X. Tang, Y. Wang and S. Guo, Smart gelation of chitosan solution in the presence of NaHCO3 for injectable drug delivery system, Int. J. Pharm., 2011, 414(1–2), 6–15 CrossRef CAS PubMed.
- S. Gupta, Carbopol/Chitosan Based pH Triggered In Situ Gelling System for Ocular Delivery of Timolol Maleate, Sci. Pharm., 2010, 78(4), 959–976 CrossRef CAS PubMed.
- E. Kenawy, A. M. Omer, T. M. Tamer, M. A. Elmeligy and M. S. M. Eldin, Fabrication of biodegradable gelatin/chitosan/cinnamaldehyde crosslinked membranes for antibacterial wound dressing applications, Int. J. Biol. Macromol., 2019, 139, 440–448 CrossRef CAS PubMed.
- M. A. Fathalla Z, A. Vangala, M. Longman, K. A. Khaled, A. K. Hussein and O. H. El-Garhy, et al., Poloxamer-based thermoresponsive ketorolac tromethamine in situ gel preparations: Design, characterisation, toxicity and transcorneal permeation studies, Eur. J. Pharm. Biopharm., 2017, 114, 119–134 CrossRef PubMed.
- İ. Erol, N. Üstündağ Okur, D. Orak, H. Sipahi, A. Aydın and Ö. Özer, Tazarotene-loaded in situ gels for potential management of psoriasis: biocompatibility, anti-inflammatory and analgesic effect, Pharm. Dev. Technol., 2020, 25(8), 909–918 CrossRef PubMed.
- B. Xu, L. Yuan, Y. Hu, Z. Xu, J. J. Qin and X. D. Cheng, Synthesis, Characterization, Cellular Uptake, and In Vitro Anticancer Activity of Fullerenol–Doxorubicin Conjugates, Front. Pharmacol, 2021, 11, 598155 CrossRef PubMed.
- R. Jayaganesh, P. Pugalendhi and R. Murali, Effect of citronellol on NF-kB inflammatory signaling molecules in chemical carcinogen-induced mammary cancer in the rat model, J. Biochem. Mol. Toxicol., 2020, 34(3), e22441 CrossRef CAS PubMed.
- H. S. Mahajan and S. Gattani, In situ gels of Metoclopramide Hydrochloride for intranasal delivery: in vitro evaluation and in vivo pharmacokinetic study in rabbits, Drug Delivery, 2010, 17(1), 19–27 CrossRef CAS PubMed.
- H. Abdeltawab, D. Svirskis and M. Sharma, Formulation strategies to modulate drug release from poloxamer based in situ gelling systems, Expert Opin. Drug Delivery, 2020, 17(4), 495–509 CrossRef CAS PubMed.
- Z. Fathalla, W. W. Mustafa, H. Abdelkader, H. Moharram, A. M. Sabry and R. G. Alany, Hybrid thermosensitive-mucoadhesive in situ forming gels for enhanced corneal wound healing effect of L-carnosine, Drug Delivery, 2022, 29(1), 374–385 CrossRef CAS PubMed.
- S. Lee and A. Shanti, Effect of Exogenous pH on Cell Growth of Breast Cancer Cells, Int. J. Mol. Sci., 2021, 22(18), 9910 CrossRef CAS PubMed.
- K. H. Ullah, F. Rasheed, I. Naz, N. Ul Haq, H. Fatima and N. Kanwal, et al., Chitosan Nanoparticles Loaded Poloxamer 407 Gel for Transungual Delivery of Terbinafine HCl, Pharmaceutics, 2022, 14(11), 2353 CrossRef CAS PubMed.
- S. Kajdič, F. Vrečer and P. Kocbek, Preparation of poloxamer-based nanofibers for enhanced dissolution of carvedilol, Eur. J. Pharm. Sci., 2018, 117, 331–340 CrossRef PubMed.
- A. M. Omer, T. M. Tamer, M. A. Hassan, P. Rychter, M. S. Mohy Eldin and N. Koseva, Development of amphoteric alginate/aminated chitosan coated microbeads for oral protein delivery, Int. J. Biol. Macromol., 2016, 92, 362–370 CrossRef CAS PubMed.
- C. Galocha-León, C. Antich, A. Voltes-Martínez, J. A. Marchal, M. Mallandrich and L. Halbaut, et al., Development and characterization of a poloxamer hydrogel composed of human mesenchymal stromal cells (hMSCs) for reepithelization of skin injuries, Int. J. Pharm., 2023, 647, 123535 CrossRef PubMed.
- T. Kojarunchitt, S. Baldursdottir, Y. D. Dong, B. J. Boyd, T. Rades and S. Hook, Modified thermoresponsive poloxamer 407 and chitosan sol–gels as potential sustained-release vaccine delivery systems, Eur. J. Pharm. Biopharm., 2015, 89, 74–81 CrossRef CAS PubMed.
- J. Varshosaz, M. Tabbakhian and Z. Salmani, Designing of a Thermosensitive Chitosan/Poloxamer In Situ Gel for Ocular Delivery of Ciprofloxacin, Open Drug Deliv. J., 2008, 2(1), 61–70 CrossRef CAS.
- Y. I. Cho, S. Park, S. Y. Jeong and H. S. Yoo, In vivo and in vitro anti-cancer activity of thermo-sensitive and photo-crosslinkable doxorubicin hydrogels composed of chitosan–doxorubicin conjugates, Eur. J. Pharm. Biopharm., 2009, 73(1), 59–65 CrossRef CAS PubMed.
- Z. Gu, C. Da Silva, K. Van Der Maaden, F. Ossendorp and L. Cruz, Liposome-Based Drug Delivery Systems in Cancer Immunotherapy, Pharmaceutics, 2020, 12(11), 1054 CrossRef CAS PubMed.
- M. Oliveira, S. Santos, M. Oliveira, A. Torres and M. Barbosa, Chitosan drives anti-inflammatory macrophage polarisation and pro-inflammatory dendritic cell stimulation, Eur. Cells Mater., 2012, 24, 136–153 CrossRef CAS PubMed.
- K. Azuma, T. Osaki, S. Minami and Y. Okamoto, Anticancer and Anti-Inflammatory Properties of Chitin and Chitosan Oligosaccharides, J. Funct. Biomater., 2015, 6(1), 33–49 CrossRef PubMed.
- Y. Zaiki, A. Iskandar and T. W. Wong, Functionalized chitosan for cancer nano drug delivery, Biotechnol. Adv., 2023, 67, 108200 CrossRef CAS PubMed.
- P. Manhas, C. Cokca, R. Sharma, K. Peneva, N. Wangoo and D. Sharma, et al., Chitosan functionalized doxorubicin loaded poly(methacrylamide) based copolymeric nanoparticles for enhanced cellular internalization and in vitro anticancer evaluation, Int. J. Biol. Macromol., 2024, 259, 129242 CrossRef CAS PubMed.
- X. Cao, J. Geng, S. Su, L. Zhang, Q. Xu and L. Zhang, et al., Doxorubicin-Loaded Zein in Situ Gel for Interstitial Chemotherapy, Chem. Pharm. Bull., 2012, 60(10), 1227–1233 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Biswakanth Kara,
Goutam Ghosha and
Goutam Rath
b,
Biswakanth Kara,
Goutam Ghosha and
Goutam Rath