
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel feathery P/S Co-doped graphitic carbon nitride for highly efficient synergistic photocatalytic H2O2 generation and tetracycline degradation†
Lian Qina,
Zhongli Zhao a,
Ning Fua,
Xiangyu Lia,
Lei Hub,
Xingang Lic and
Chun Zhang*a
a,
Ning Fua,
Xiangyu Lia,
Lei Hub,
Xingang Lic and
Chun Zhang*a
aSchool of Chemistry and Chemical Engineering, Lanzhou Jiaotong University, Lanzhou 730070, PR China. E-mail: zh-chun@163.com
bChongqing Titanium Industry Co. Ltd of Pangang Group, Chongqing 400055, PR China
cLaboratory of Applied Catalysis Science and Engineering, Tianjin University, Tianjin 300072, PR China
First published on 4th December 2024
Abstract
Graphitic carbon nitride (g-C3N4) has garnered significant attention in photocatalytic pollutant degradation for its non-toxicity and cost-effectiveness. However, its limited photocatalytic performance has hindered its applications. Addressing this, we successfully synthesized a novel feathery multifunctional catalyst, phosphorus and sulfur co-doped g-C3N4 (P0.3S0.2-CN), with an enlarged pore network through a hydrothermal method. This catalyst exhibits remarkable photocatalytic performance under visible light, achieving a hydrogen peroxide (H2O2) production rate of 28.6 mg L−1 h−1 and an efficiency of 87.3% in degrading tetracycline (TC). Comparative studies demonstrate that P0.3S0.2-CN outperforms singly doped catalysts P0.5-CN and S0.4-CN by increasing H2O2 yield by 28.67% and 53.28% and improving TC degradation by 15.2% and 11.5%, respectively. These improvements can be attributed to the synergetic effects of P and S co-doping and the high number of active sites provided by its peculiar morphology, which enhance charge transfer and photocatalytic activity, and a more pronounced conjugation effect, resulting in a high electrostatic potential surface conducive to adsorption and activation, as confirmed by density-functional theory calculations. Our findings propose a mechanism for the synergistic photocatalytic-Fenton degradation (PSF) of TC using P0.3S0.2-CN. This present research contributes to the advancement of g-C3N4-based photocatalysts and promotes the exploration of more efficient carbon-based catalysts for environmental remediation.
1. Introduction
The rapid development of industry and agriculture has inevitably resulted in a series of environmental issues, with various types of domestic sewage, industrial and agricultural wastewater, industrial soot, and automobile exhausts releasing harmful substances into the environment. Among these pollutants, antibiotics present in natural water sources poses a serious threat to both the ecological cycle and human health.1 Antibiotics are typically characterized by their high stability and resistance to biodegradation. Besides, traditional physical and chemical treatment methods may lead to secondary toxic contamination and generally require a long duration (at least 3 months). Moreover, the removal efficiency of physical-chemical treatment is usually insignificant, greatly limiting its application for organic pollutants. Tetracycline, one of the broad-spectrum antibiotics containing acylamino groups, dimethylamino groups, phenolic hydroxyl groups, and ketoenol conjugated double bonds, is extensively consumed and produced in China.2 Therefore, it is crucial to develop clean degradation methods that are free from secondary pollution and environmentally friendly. Advanced oxidation processes (AOPs) offer an excellent solution for oxidizing and mineralizing toxic and refractory organics in complex wastewater treatment compared to other conventional approaches.3,4 The main challenges associated with homogeneous Fenton systems include (1) high consumption cost of H2O2, (2) limited pH adaptability (pH ≤ 3), and (3) the low cyclic conversion rate of Fe3+/Fe2+.5–8 Interestingly, in situ generation and consumption of H2O2 within the Fenton system can effectively address these concerns related to costliness and safety risk.9 Therefore, the development of an in situ self-Fenton system is imperative for the efficient elimination of TC from the aqueous environment.The photocatalytic oxygen reduction reaction has emerged as a sustainable approach for the production of H2O2,10 offering significant advantages in terms of cost-effectiveness, environmental friendliness, safety and efficiency. The construction of PSF systems may also serve as an effective method for the degradation of TC.11,12 For instance, the CdS/rGO/Fe2+ composite in a PSF system achieved efficient degradation of various organic compounds by generating H2O2 in situ, exemplifying its effectiveness.13 However, limitations in the application of sulfides arise from their inherent instability and biotoxicity. In recent years, g-C3N4 materials have gained significant traction in the optoelectronics field due to their favorable energy band position, exceptional chemical stability, and cost-effectiveness.14,15 Nevertheless, the photocatalytic performance of g-C3N4 is greatly limited by its rapid carrier complexation and small specific surface area.16–20 Fortunately, the photocatalytic activity and efficiency of g-C3N4 can be enhanced through nanostructure engineering, ion doping, and the formation of composite materials. The desired morphology can be achieved through the process of hard template synthesis. However, during the removal of the template, nanocrystallinity inevitably appears as structural defects, which can be effectively addressed by employing self-assembly techniques. The supramolecular self-assembly process enables the formation of specialized nanostructures, including nanoribbons,21 nanorods,22 nanotubes23 and nanoflakes.24 These structures exhibit enhanced specific surface area and light absorption capacity, thereby facilitating superior light scattering ability, increased light utilization efficiency, larger specific surface area, and greater exposure of active sites.
The improvement of g-C3N4 materials for efficient photocatalysis through a single strategy may pose challenges, while elemental doping has been demonstrated as an effective approach to modulate both the photoelectric and chemical properties of g-C3N4.25–27 The heteroatom doping of g-C3N4 involves a robust coordination between carbon and nitrogen atoms, as well as other atoms, which benefits the delocalization of π-conjugated electrons for effective charge separation and modulation of the energy band structure.28–30 Theoretical studies based on first principles have shown a preferential substitution of S and P atoms for N and C atoms specifically at the edges of g-C3N4 within the gaps located at angular sites.31 Theoretical calculations revealed enhanced photocatalytic activity and stability in the sulfur-doped g-C3N4 (Sy-CN) heptazine-based framework.32 The presence of P-doped g-C3N4 (Px-CN) nanoribbons can effectively suppress the aggregation between NH2 groups, thereby promoting the degradation and hydrogenation of pollutants.33 However, the addition of a single element may not always be adequate to achieve significant improvements in performance. Co-doping of g-C3N4 with various elements can further accelerate the rate of electron–hole pair separation. For instance, the co-doping of S and Cl in g-C3N4 results in an augmentation of the specific surface area, modulation of the band gap, and facilitation of charge migration as well as an upward shift in the conduction band.34
Herein, a novel feathery P and S co-doped CN catalyst featuring an expanded network of pores P0.3S0.2-CN was successfully synthesized by hydrothermal procedure (Fig. 1). The incorporation of P and S elements not only enhances the efficiency of carrier separation, but also significantly boosts its redox ability compared to single doping. Experimental results illustrate that the active species ˙O2− plays a major role in driving the photocatalytic process. Under visible light irradiation, the photocatalytic H2O2 production rate of P0.3S0.2-CN reaches 28.6 mg L−1 h−1 and it degrades TC with a removal efficiency of 87.3%. Compared to the photocatalysts P0.5-CN and S0.4-CN, the H2O2 yield rate of P0.3S0.2-CN exhibited increases of 28.67% and 53.28%, respectively, and TC degradation rate increases of 15.2% and 11.5%, respectively. Moreover, we conducted a comprehensive analysis of differential charge and Bader charges to elucidate the exceptional co-doping performance through DFT calculations. The experimental and computational evidence both support P0.3S0.2-CN as an efficient catalytic material for synthetic H2O2 and TC degradation.
 | ||
| Fig. 1 Schematic depiction of the fabrication process of PxSy-CN. | ||
2. Experimental section
All reagents employed in this work were of analytical or chromatographic grade. The experiments were conducted using ultra-pure water with a conductivity of less than 0.2 μs cm−1. The pH adjustment was performed utilizing diluted aqueous solutions of NaOH and H2SO4. The synthesis process of doped carbon nitride and the subsequent photocatalytic performance testing are provided in the ESI.†3. Results and discussion
The information regarding the crystalline structure and phase composition of carbon nitride is presented in Fig. 2a. Two additional distinctive characteristic peaks are observed at approximately 13° and 27° in the XRD diffractograms, corresponding to the (100) and (002) crystallographic planes of carbon nitride, respectively. This observation suggests that the morphology control and doping have not altered its crystal structure. Interestingly, the diffraction peaks of the doped CN are shifted due to the difference in electronegativity between the different atoms. Doping reduces the attraction between the carbon and nitrogen layers, thereby increasing the spacing and ultimately causing a downward shift in the diffraction peaks.33 The broad absorption peaks observed in Fig. 2b, ranging from 3100 to 3600 cm−1, can be attributed to the stretching vibrations of –NH2, –NH and –OH groups. These peaks may arise from the residual amine groups and surface H2O molecules resulting from the incomplete condensation of the precursors.31 The absorption peak at 818 cm−1 is assigned to the bending vibration of the tri-s-triazine unit. No co-doping of P and S was observed, as these peaks overlapped with the C–N vibration peaks.35 The spectrum of P0.3S0.2-CN exhibits an additional peak at 534 cm−1, which can potentially be attributed to the P–S stretching mode.36 The N2 adsorption–desorption isotherms of the CN and P0.3S0.2-CN samples are depicted in Fig. S1a and b.† Both CN and P0.3S0.2-CN catalysts adhere to the IUPAC classification criteria for type IV adsorption isotherms, indicating the presence of mesoporous structures in the sample. The specific surface areas of CN and P0.3S0.2-CN are 33.45 and 48.23 m2 g−1, respectively, with corresponding pore sizes of 24.74 and 20.43 nm. The specific surface area of P0.3S0.2-CN is 5.84 times greater than that of the g-C3N4 prepared in the literature (8.25 m2 g−1).34 P0.3S0.2-CN exhibits a higher specific surface area and superior porosity, thus offering an increased number of reactive sites for photocatalytic reactions and facilitating adsorption of organic pollutants.37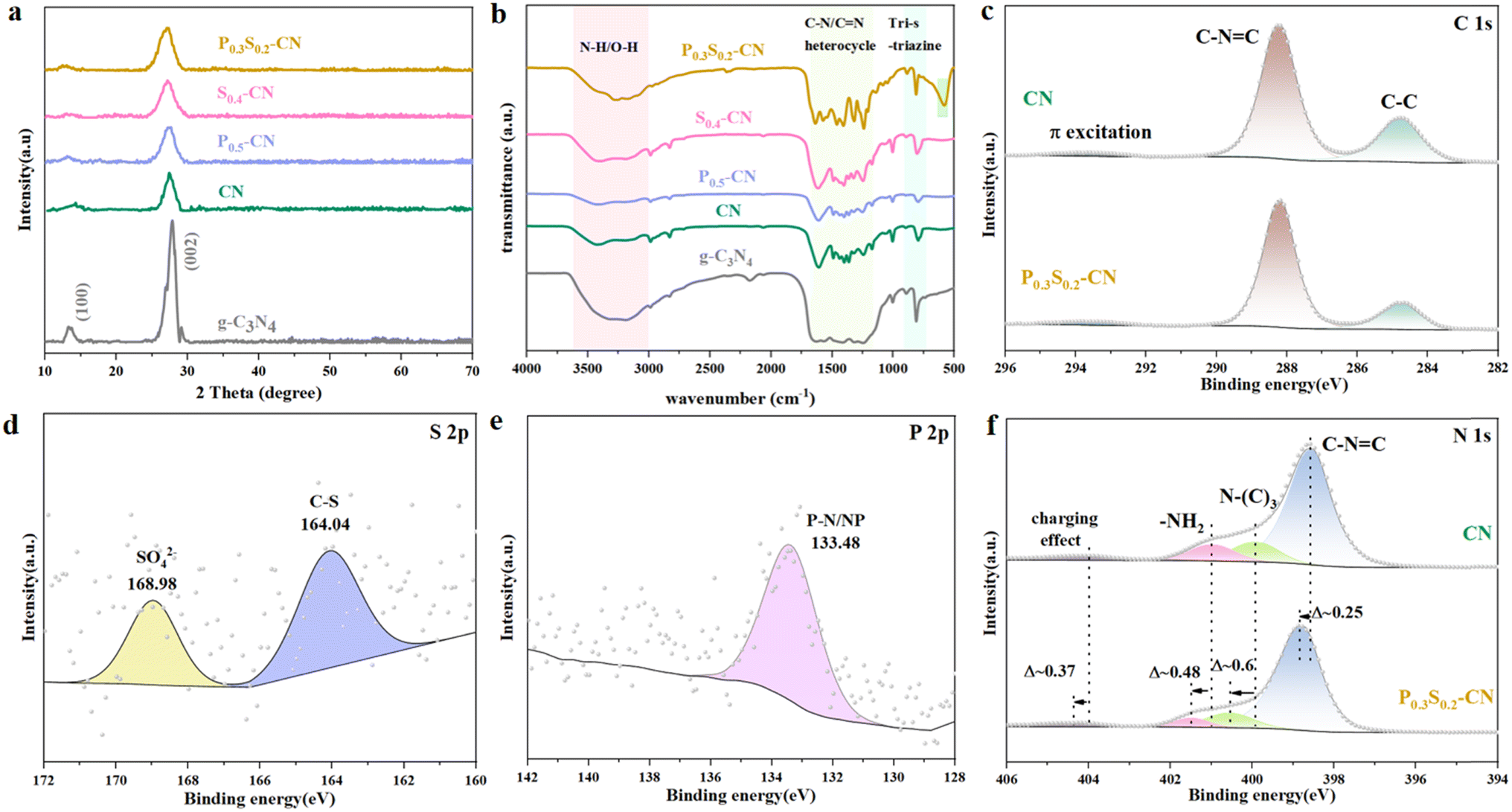 | ||
| Fig. 2 (a) XRD diffraction patterns, (b) FT-IR spectra, and XPS (c) C 1s spectra, (d) S 2p spectra, (e) P 2p spectra, and (f) N 1s spectra of various samples. | ||
The chemical composition and atomic state information of the samples were analyzed using X-ray photoelectron spectroscopy (XPS). Fig. S2† displays the complete spectra of CN and P0.3S0.2-CN, revealing the presence of the elements C, N, and O. High resolution XPS was employed to further elucidate the chemical state of each element in the samples. The binding energy of C 1s, as shown in Fig. 2c, is deconvoluted into three distinct peaks at 284.8 eV, 288.2 eV and 293.5 eV corresponding to the C–C bond, C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and π excitation bond of CN, respectively. Four discernible peaks are observed in the N 1s spectrum of CN: the C–NC bond (398.17 eV), N–(C)3 bond (399.93 eV), amino functional group (400.90 eV) and charging effect (403.75 eV).38,39 The binding energies of the four peaks of in the N 1s spectrum of P0.3S0.2-CN are shifted to varying degrees towards higher binding energies. This shift is due to the formation of new coordination bonds between N and P atoms which lead to the extra electron redistribution because of the nonmetal-doping. Simultaneously, the electron density around the N atoms decreases, leading to a shift of the four N 1s peaks in P0.3S0.2-CN to higher binding energies.40 The S 2p spectrum primarily shows binding energies at 164.04 eV and 168.98 eV, corresponding to the C–S bond41 within the nitrogen aromatic ring and the sulfur oxides released during thermal polymerization, respectively. The presence of a prominent peak at 133.48 eV in the P 2p spectrum, attributed to P–N coordination, signifies the substitution of C with P in the heptazine ring.42 Based on the aforementioned XPS analysis, successful replacement of C and N atoms by P and S atoms respectively occurred in the carbon nitride.
C bond and π excitation bond of CN, respectively. Four discernible peaks are observed in the N 1s spectrum of CN: the C–NC bond (398.17 eV), N–(C)3 bond (399.93 eV), amino functional group (400.90 eV) and charging effect (403.75 eV).38,39 The binding energies of the four peaks of in the N 1s spectrum of P0.3S0.2-CN are shifted to varying degrees towards higher binding energies. This shift is due to the formation of new coordination bonds between N and P atoms which lead to the extra electron redistribution because of the nonmetal-doping. Simultaneously, the electron density around the N atoms decreases, leading to a shift of the four N 1s peaks in P0.3S0.2-CN to higher binding energies.40 The S 2p spectrum primarily shows binding energies at 164.04 eV and 168.98 eV, corresponding to the C–S bond41 within the nitrogen aromatic ring and the sulfur oxides released during thermal polymerization, respectively. The presence of a prominent peak at 133.48 eV in the P 2p spectrum, attributed to P–N coordination, signifies the substitution of C with P in the heptazine ring.42 Based on the aforementioned XPS analysis, successful replacement of C and N atoms by P and S atoms respectively occurred in the carbon nitride.
The morphology and surface characteristics of the obtained samples are examined using scanning electron microscopy (SEM) and transmission electron microscopy (TEM), respectively. As depicted in Fig. S3,† the introduction of ammonium dihydrogen phosphate did not alter the rod-like morphology of MCA, thereby providing a certain acidic environment conductive to promoting the hydrolysis of MA into partial CA and serving as a source for phosphorus-doped carbon nitride. Conversely, when thiourea was added to the MCA supramolecule, it induced a flat and uniform orientation, resulting in the transformation of MCA rods into a planar stick-like shape.43 The stable hydrogen bond structure in MCA allows the vertical outflow of NH3 and other gases during calcination, resulting in the formation of a specific hollow CN structure. Fig. S4a and b† demonstrates that, as the phosphate content increases, Px-CN undergoes a transformation from fragmented hollow tubes to flat tubes.44 The morphology of Sy-CN undergoes changes at elevated temperatures (Fig. S4c†). With incorporation of thiourea, the morphology continuously changes as NH3 and other gases are released from the interior to the exterior during calcination. The diverse modes of phosphate binding, along with thiourea and MA-CA, lead to apparent alterations in the morphology of the final P0.3S0.2-CN. As shown in Fig. 3a–c, P0.3S0.2-CN exhibits a planar feather-like structure and a 2D irregular planar structure with an enlarged pore network. The selected area electron diffraction (SAED) image in Fig. 3d reveals that the diffraction rings are unclear and have blurred edges, indicating the poor crystallinity of the P0.3S0.2-CN.45–47 Elemental mapping (Fig. 3e) confirms the presence of the elements C, N, S, and P in P0.3S0.2-CN. However, it is noted that under high-temperature conditions, elemental S tends to volatilize with the gas, resulting in a reduced content.
 | ||
| Fig. 3 (a) SEM, (b) and (c) TEM, (d) selected area electron diffraction and (e) EDS mapping images of P0.3S0.2-CN. | ||
The UV-vis DRS are presented in Fig. S5.† For g-C3N4, it exhibits significant absorption within the range of 200–450 nm with an absorption edge value of approximately 450 nm. In contrast, the light absorption of P0.3S0.2-CN is significantly enhanced by the co-doping of P and S elements, resulting in an extended edge range from 450 to 750 nm. The introduction of P and S elements along with the unique morphological structure accelerates the light trapping ability, leading to the generation of photo-induced carriers. As depicted in Fig. 4a, the band gap energy for P0.3S0.2-CN was calculated to be 2.45 eV. The VB position ratio of CN to the normal hydrogen electrode was determined at pH = 7.48 The CB positions were obtained through combined band gap and EVB-XPS analysis. Notably, P0.3S0.2-CN possesses the highest negative CB position (ranging from 1.52 to 0.93) and the narrowest band gap, as shown in Fig. 4b and c, indicating that electrons can be more easily excited into the CB and promoting a stronger reduction reaction involving photo-generated e− and h+. The extent of complexation between photo-generated e− and h+ can be evaluated by PL spectroscopy. The PL emission spectra were excited under a wavelength of 360 nm, and the peaks were centered at around 457 nm. As depicted in Fig. 7, P0.3S0.2-CN shows the lowest PL peak intensity which indicates that neither P nor S acts as a recombination center for photo-generated electrons and holes in CN. Fewer electrons are recombining with holes and leaving the conduction band, resulting in lower photon energy. This suggests that the recombination rate of electrons and holes is suppressed, meaning the separation efficiency of the electron–hole pairs is higher.49,50 The blue shift observed in doped CN's PL spectrum may be attributed to changes in its band gap caused by doping.51 TPC curves are displayed in Fig. 4e; remarkably, the photocurrent response of P0.3S0.2-CN is superior to those of the other samples which suggests a prolonged lifetime for photo-generated carriers. Furthermore, the Nyquist diagram reveals that the arc radius for P0.3S0.2-CN photocatalyst is smaller than those of the others (Fig. 4f), suggesting that doping with both P and S elements reduces charge transfer resistance, thereby facilitating rapid separation and transfer of photoinduced charges.
 | ||
| Fig. 4 (a) Band gap values, (b) VB-XPS, (c) energy band structure, (d) PL spectra, (e) TPC spectrograms, (f) EIS plots of various species. | ||
The photocatalytic H2O2 generation performance of various carbon nitrides under visible light irradiation was investigated under natural pH conditions. As shown in Fig. 5a, the H2O2 yield of hydrothermal-calcined CN is 3.8 times higher than that of directly calcined g-C3N4, which can be attributed to the formation of a partially tubular structure and hydrophilic functional groups on the surface of CN.52 By adjusting the ratio of ammonium dihydrogen phosphate (NH4H2PO4), the optimum P0.5-CN was found to produce H2O2 at a concentration of 1.308 mM (Fig. S6a†). Varying the amount of thiourea resulted in S0.4-CN producing H2O2 at a concentration of 1.098 mM (Fig. S6b†). These results indicate that the appropriate amounts of P and S doping could significantly enhance the production performance of photocatalytic H2O2. As depicted in Fig. S6c,† four PxSy-CN photocatalysts with different P and S contents were prepared by varying the amounts of NH4H2PO4 and thiourea, all exhibiting higher H2O2 generation performance compared to the mono-doped P or S CN catalysts. After 120 min of photocatalytic reaction, P0.3S0.2-CN accumulated an H2O2 concentration as high as 1.683 mM under visible light irradiation, surpassing the other catalysts’ performance in terms of H2O2 generation. The above findings indicate that co-doping plays a facilitating role and the appropriate amounts of P and S doping can significantly improve the photocatalytic production of H2O2. The yield of H2O2 produced by P0.3S0.2-CN is 28.6 mg L−1 h−1, which exceeds the yield reported in previous literature (Table S1†).
 | ||
| Fig. 5 (a) Photocatalytic production of H2O2 by various photocatalysts, (b) 50 mg photocatalyst for 4 consecutive cycles of H2O2 production, (c) photocatalytic oxidative degradation of TC (20 mg L−1) by multifarious catalysts (30 mg), and (d) 30 mg photocatalyst employed for 4 consecutive cycles of TC degradation. | ||
Decomposition experiments were conducted to investigate the proportion of P0.3S0.2-CN that decomposes H2O2 during the photocatalytic reaction. As shown in Fig. S6e,† a slight reduction in H2O2 concentration (0.591 mM) was observed after 120 min of light exposure. The control experiment without P0.3S0.2-CN demonstrated H2O2 production at a concentration of 0.011 mM (Fig. S6f†), yielding an H2O2 decomposition efficiency of 3.33%.
TC was selected as a representative pollutant to evaluate the photocatalytic properties of the various carbon nitrides under visible light. All photocatalytic reactions were carried out until adsorption–desorption equilibrium was reached under dark stirring conditions. As shown in Fig. 5c, the TC concentration remained essentially unchanged in the absence of light. With the exception of g-C3N4, the other CN photocatalysts exhibited higher adsorption capacity for TC, which can be attributed to their relatively larger specific surface area. For the photocatalytic degradation experiments with 30 mg photocatalysts and an initial TC concentration of 20 mg L−1, P0.3S0.2-CN, P0.5-CN, S0.4-CN, CN, and g-C3N4 achieved degradation capacities of 80.6%, 72.1%, 75.8%, 67.4%, and 43.3%, respectively (Fig. S7a†), with pseudo primary kinetic rate constants of 0.0163 min−1, 0.0126 min−1, 0.0140 min−1, 0.0110 min−1, and 0.00567 min−1, respectively (Fig. S7a†). The kinetics analysis revealed that P0.3S0.2-CN exhibited superior photocatalytic degradation efficiency compared to the other carbon nitride materials due to its co-doping effect and presence of more reactive sites. After each experiment, the photocatalysts were washed with deionized water and vacuum dried for subsequent use. The recycling test showed a gradual decrease in both TC removal rate and H2O2 yield compared to their initial values (Fig. 5b and d). The slight decrease in photocatalytic performance may be ascribed to the loss of some active sites caused by adsorption from reaction products. We conducted Fourier transform infrared spectroscopy (Fig. S7e†) testing on the P0.3S0.2 photocatalyst after four cycles of photocatalytic activity. The characteristic peaks of the original functional groups remained unchanged, indicating that the photocatalyst exhibits good stability.
According to Fig. 6a, the introduction of Fe2+ before the reaction resulted in a higher photocatalytic degradation efficiency of P0.3S0.2-CN compared to P0.3S0.2-CN without treatment after 60 min of photocatalytic oxidation. However, adding Fe2+ before light did not enhance the degradation performance. Thus, the combination of photocatalytic oxidation and Fenton oxidation (two-step oxidation) accelerated the photocatalytic degradation process. The effect of P0.3S0.2-CN dosage (10–70 mg) on TC removal efficiency was investigated under visible light photocatalytic oxidation-Fenton oxidation (Fig. 6b and S7b†). It was found that when an excessive amount of carbon nitride catalyst was utilized, there was a marked decrease in photoactivation volume due to the increase solution turbidity.53 Fig. 6c and S7c† display the effect of different initial TC concentrations on P0.3S0.2-CN catalyzed TC degradation efficiency and its corresponding pseudo primary reaction kinetics. The final photodegradation efficiency remained essentially unchanged when increasing TC concentration from 10 mg L−1 to 20 mg L−1. Nevertheless, when TC concentration was further increased from 20 mg L−1 to 80 mg L−1, there was an apparent suppression in photocatalytic degradation efficiency with a corresponding decrease in kinetic constant kpfo from 0.0201 min−1 to 0.0089 min−1. The decrease in the photodegradation efficiency can be attributed to both high TC concentration and limited activity of intermediate products competing for active sites on the photocatalyst. As shown in Fig. 6d and S7d,† reducing the initial pH from 5.3 to 3.6 led to an increase in reaction rate constant from 0.0201 min−1 to 0.0229 min−1, implying the excellent performance of the photocatalytic oxidation-Fenton oxidation system under acidic conditions. However, adjusting the pH to 10.7 brought about a notable decrease in the reaction rate constant (0.0115 min−1). The observations suggest that P0.3S0.2-CN has the ability to generate a greater amount of reactive substances in acidic conditions.
 | ||
| Fig. 6 Under visible light irradiation, effect of (a) Fe2+ addition on the photocatalytic degradation of TC by P0.3S0.2-CN, (b) dosage of the P0.3S0.2-CN catalyst, (c) initial concentration of TC, (d) different pH values on photocatalytic oxidation-Fenton oxidative degradation. | ||
To investigate the effect of P and S doping on catalytic activity, we performed DFT analog calculations on the CN and P0.3S0.2-CN. Fig. S8† illustrates the optimized structures of the top and side of the CN, P–CN, S–CN, and P0.3S0.2-CN catalytic surfaces. Fig. S9† presents the theoretical calculated band structures for CN and P0.3S0.2-CN. It is observed that the bandgap value decreases significantly after doping CN with P and S elements, and a smaller bandgap is more favorable for electron migration, improving the photoelectrochemical performance of the material. However, the calculated bandgap values for semiconductors are often greatly underestimated; therefore, relying solely on theoretically calculated bandgap values to analyze trends is not recommended.54 We computed the atomic charges near the doping sites of CN and P0.3S0.2-CN to gain further insights. The optimized top surface structures of CN and P0.3S0.2-CN catalytic surfaces are presented in Fig. 7a and b, respectively. As shown in Fig. 7d, the atomic charge of the C-bound S atom is −0.015 eV, while that of the N-bound P atom exhibits a larger positive value. Specifically, within the P–N bond, we observed atomic charges for the P, N2, and N3 atoms of +0.842, −0.405, and −0.476, respectively. Our findings display that doping leads to a reduction in the charges of the C1 and C2 atoms in CN, while S exhibits higher positivity compared to N1. The charges of the N2 and N3 atoms in CN decrease, with the P atoms carrying larger positive charges. Notably, the positive charge of P surpasses that of C3, indicating the replacement of N1 by S and C3 by P through covalent bonding with neighboring N. This formation of a conjugated system exhibits a more pronounced conjugation effect than an induced effect, resulting in a high electrostatic potential surface conducive to adsorption and activation of the adsorbent. The differential charge densities on the surfaces of CN and P0.3S0.2-CN are depicted in Fig. 7e and f, respectively. Upon S substitution doping, the original N1 of CN undergoes a transition from electron acquisition to electron loss, while the corresponding C3 becomes more prominent in its original state of electron loss after P doping. The DOS of CN and P0.3S0.2-CN surfaces were calculated and are shown in Fig. 7g and h, respectively. We found that after doping CN with P and S elements, the P 2p and S 2p orbital energy levels appear near the Fermi level. In P0.3S0.2-CN, the conduction band edge moves closer to the Fermi level, resulting in a reduced bandgap. A smaller bandgap allows electrons to more easily transition from the valence band to the conduction band, thereby enhancing the material's photoelectrochemical performance.
 | ||
| Fig. 7 The optimized structures of the (a) CN and (b) P0.3S0.2-CN; the DFT-computed atomic charges at the (c) CN and (d) P0.3S0.2-CN doping sites are provided near each atom. The differential charge density plots of (e) CN and (f) P0.3S0.2-CN. DOS plots of (g) CN and (h) P0.3S0.2-CN. | ||
Importantly, the potential photodegradation pathways and intermediates of TC proposed by LC-MS are shown in Fig. 8. Mass spectrometry inferred seventeen possible intermediates involved in the oxidation process of TC, and their molecular and structural formulas are present in Table S3.† In pathway I, two demethylations yield intermediate P2 (m/z = 416), while deamidation reactions lead to the formation of intermediate P3 (m/z = 358). The CC bond breaking generates intermediate product P4 (m/z = 322). Notably, P6 can be oxidized by the resulting ˙O2− to form small molecule P7 (m/z = 150) through a series of C–C bonding, CC bond breaking and ring opening reactions.55 Pathway II involves conversion to P8 (m/z = 411) via dehydroxylation and cyclo-decomposition followed by C–C bond breakage and sequential oxidation resulting in P11 (m/z = 136). In pathway III, C–C bond breaking and demethylation produce intermediate P13 (m/z = 418), while atomic rearrangement allows for dehydroxylation and deamidation leading to the formation of P14 (m/z = 357). Oxidation of double bonds within TC triggers ring opening reactions yielding P15 (m/z = 331), after which radical decomposition oxidizes CC bonds, amide groups, –CH3 groups, and –N(CH3)2 groups, forming smaller molecules. Finally, these intermediates undergo further oxidation, causing compounds such as CO2, H2O, and so on.
 | ||
| Fig. 8 Photocatalytic degradation pathway of TC in aqueous solution by P0.3S0.2-CN under visible light irradiation. | ||
The main active species of responsible for TC degradation in P0.3S0.2-CN were investigated by radical trapping experiments, as depicted in Fig. 9a. Isopropyl alcohol (IPA), p-benzoquinone (BQ), and EDTA-2Na were employed as scavengers of hydroxyl radicals (˙OH), superoxide radicals (˙O2−), and h+. The addition of IPA and EDTA-2Na brought about a decrease in the removal rate of TC from 79.4% to 72.8% and 63.9%, respectively, suggesting that the impact of ˙OH and h+ on the degradation efficiency was limited. However, when BQ was introduced, the removal rate of TC dropped to only 38.2%, with a corresponding decrease in kinetic rate from 0.007 min−1 to 0.0034 min−1 (Fig. 9b). This indicates that ˙O2− played a crucial role in the TC degradation process. To further validate the generation of ˙O2− and ˙OH in P0.3S0.2-CN, electron spin resonance (ESR) spectroscopy employing DMPO (5,5-dimethyl-1-pyrroline-N oxide) as a spin trap was utilized. As shown in Fig. 9c and d, under dark conditions, the intensity of DMPO-˙OH and DMPO-˙O2− was negligible. However, under light irradiation, DMPO-˙OH (intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1) and DMPO-˙O2− (intensity ratio of 1:1:1:1) exhibited increased intensity with prolonged irradiation time.56,57 The results indicate that P0.3S0.2-CN can continuously generate ˙OH and ˙O2− under visible light irradiation. In the photocatalytic system involving P0.3S0.2-CN, O2 can be reduced via two-electron transfer to produce ˙O2−, some portion of which is further reduced to H2O2, while another part of ˙O2− plays a dominant role in the photocatalytic degradation. Furthermore, the involvement of ˙OH is observed during this degradation process.
:2:2:1) and DMPO-˙O2− (intensity ratio of 1:1:1:1) exhibited increased intensity with prolonged irradiation time.56,57 The results indicate that P0.3S0.2-CN can continuously generate ˙OH and ˙O2− under visible light irradiation. In the photocatalytic system involving P0.3S0.2-CN, O2 can be reduced via two-electron transfer to produce ˙O2−, some portion of which is further reduced to H2O2, while another part of ˙O2− plays a dominant role in the photocatalytic degradation. Furthermore, the involvement of ˙OH is observed during this degradation process.
 | ||
| Fig. 9 Under visible light irradiation and added scavenger conditions, (a) TC and (b) corresponding kinetic constants of P0.3S0.2-CN degradation and EPR spectra of P0.3S0.2-CN in the dark and under visible light irradiation with (c) DMPO-˙OH and (d) DMPO-˙O2−. | ||
Based on the aforementioned results, a proposed mechanism diagram of P0.3S0.2-CN during the photocatalytic two-step oxidation is shown in Fig. 10. Upon visible light irradiation, photo-generated electrons migrate from the VB to the CB, while h+ is produced on the VB.58,59 Because of the more negative potential of P0.3S0.2-CN (−0.93 eV) compared to O2/˙O2− (−0.33 eV), the photoinduced e− reduces O2 to ˙O2− on the CB. A portion of ˙O2− can directly oxidize TC, leading to the formation of some organic intermediates,60 while others can be reduced to H2O2. Since the energy level of h+ in the photocatalyst VB (1.52 eV) is lower than E(˙OH/H2O = 2.68 eV vs. NHE), it suggests that h+ lacks sufficient energy for oxidizing H2O and producing ˙OH.61,62 Nevertheless, h+ can capture photogenerated e− and use them for oxidizing TC as well as its organic intermediates. During photocatalytic degradation, H2O2 activates e− to generate ˙OH, which further accelerates the TC degradation process through the Fenton reaction. Eventually, TC is degraded into small molecules and harmless substances such as CO2 and H2O.63
 | ||
| Fig. 10 Mechanism diagram of the photocatalytic two-step oxidation by P0.3S0.2-CN. | ||
4. Conclusions
In this study, we successfully synthesized a feathery multifunctional P0.3S0.2-CN photocatalyst with an enlarged pore network using a hydrothermal technique. Demonstrating a notable photocatalytic H2O2 production rate of 28.6 mg L−1 h−1 under visible light, P0.3S0.2-CN also achieves an impressive 87.3% efficiency in degrading TC. When compared to the singly doped photocatalysts P0.5-CN and S0.4-CN, P0.3S0.2-CN shows a significant enhancement in H2O2 yield by 28.67% and 53.28%, respectively, and TC degradation by 15.2% and 11.5%. The catalyst's exceptional performance is attributed to the synergistic effects of phosphorus and sulfur co-doping, which not only enhances charge carrier separation but also improves the redox capabilities more effectively than single-doping strategies. Density functional theory calculations reveal that co-doping leads to electron clustering near heteroatoms and enhances carrier migration and separation on the catalytic surface of graphitic carbon nitride. This research provides a pioneering approach in designing high-efficiency catalysts for the degradation of organic pollutants, showcasing significant advancements in photocatalyst development by optimizing elemental doping strategies to enhance photocatalytic activity.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Lian Qin: writing –review & editing, conceptualization, supervision, methodology; Zhongli Zhao: Project conceptualization, experimentation, formal analysis, modify; Ning Fu: material synthesis and characterization, supervision, data arrangement; Xiangyu Li: investigation, formal analysis; Lei Hu: software, laboratory aids, methodology, formal analysis. Xingang Li: conceptualization, investigation; Chun Zhang: writing – review & editing, resources funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Key Research and Development Plan of Gansu Province (No. 22YF7GA148).References
- Z. Wei, J. Liu and W. Shangguan, Chin. J. Catal., 2020, 41, 1440–1450 CrossRef CAS.
- P. Tenorio-Chávez, G. A. Elizalde-Velázquez, L. M. Gómez-Oliván and M. D. Hernández-Navarro, Sci. Total Environ., 2023, 859, 159731 CrossRef PubMed.
- Y. Lee, D. L. Sedlak and U. von Gunten, Environ. Sci. Technol., 2023, 57, 18391–18392 CrossRef CAS PubMed.
- Q. Zou, B. Wang, B. Gao, T. Jiang, Q. Feng, M. Chen, J. Zhang and X. Zhang, Biochar, 2023, 5, 1–24 CrossRef.
- J. Zhang, G. Zhang, Q. Ji, H. Lan, J. Qu and H. Liu, Appl. Catal., B, 2020, 266, 118665 CrossRef CAS.
- J. Xu, Q. Zhang, X. Gao, P. Wang, H. Che, C. Tang and Y. Ao, Angew. Chem., Int. Ed., 2023, 62, e202307018 CrossRef CAS PubMed.
- X. Yang, X. Zeng, H. Chen, L. Xin, J. Pan, H. Ji and K. Cheng, Chem. Eng. J., 2024, 483, 148697 CrossRef CAS.
- X. Zhang, J. Tang, L. Wang, C. Wang, L. Chen, X. Chen, J. Qian and B. Pan, Nat. Commun., 2024, 15, 917 CrossRef CAS PubMed.
- M. Wu, X. Guo, Y. Cao, H. Yu, Z. Hu, Y. Yang, T. Yao and J. Wu, Chem. Eng. J., 2024, 489, 151091 CrossRef CAS.
- A. Torres-Pinto, H. Boumeriame, C. G. Silva, J. L. Faria and A. M. T. Silva, ACS Sustainable Chem. Eng., 2023, 11, 894–909 CrossRef CAS.
- L. Xie, T. Zhang, X. Wang, W. Zhu, Z. Liu, M. Liu, J. Wang, L. Zhang, T. Du, C. Yang, M. Zhu and J. Wang, J. Cleaner Prod., 2022, 359, 131808 CrossRef CAS.
- M. Yang, W. Wang, H. Ma, X. Lu, L. Chen, Y. Li and H. Ma, Chem. Eng. J., 2024, 479, 147882 CrossRef CAS.
- Z. Jiang, L. Wang, J. Lei, Y. Liu and J. Zhang, Appl. Catal., B, 2019, 241, 367–374 CrossRef CAS.
- S. R. Nagella, R. Vijitha, B. Ramesh Naidu, K. S. V. Krishna Rao, C. Ha and K. Venkateswarlu, Nano Energy, 2023, 111, 108402 CrossRef CAS.
- H. Liang, A. Wang, R. Cheng, F. Chen, P. Kannan, C. Molochas and P. Tsiakaras, Chem. Eng. J., 2024, 489, 151145 CrossRef CAS.
- Y. Li, M. Gu, X. Zhang, J. Fan, K. Lv, S. A. C. Carabineiro and F. Dong, Mater. Today, 2020, 41, 270–303 CrossRef CAS.
- Y. Li, D. Zhang, J. Fan and Q. Xiang, Chin. J. Catal., 2021, 42, 627–636 CrossRef CAS.
- Q. Gu, P. P. Jiang, Y. Shen, K. Zhang, P. T. Wai and A. Haryono, Mol. Catal., 2021, 504, 111441 CrossRef CAS.
- L. Chen, X. Liu, D. Wang, Y. Xia, G. Yan, X. Huang and X. Wang, Dalton Trans., 2023, 52, 2845–2852 RSC.
- L. Chen, H. Huang, Y. Zheng, W. Sun, Y. Zhao, P. S. Francis and X. Wang, Dalton Trans., 2018, 47, 12188–12196 RSC.
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew Chem. Int. Ed. Engl., 2019, 58, 16644–16650 CrossRef CAS PubMed.
- X. Zhao, Y. Zhang, F. Li, Y. Wang, W. Pan and D. Y. C. Leung, Carbon, 2021, 179, 42–52 CrossRef CAS.
- B. Zhu, G. Jiang, S. Chen, F. Liu, Y. Wang and C. Zhao, Chem. Eng. J., 2022, 430, 132843 CrossRef CAS.
- Z. Liu, J. Huang, B. Shao, H. Zhong, Q. Liang, Q. He, T. Wu, Y. Pan, Z. Peng, X. Yuan, Y. Liu and C. Zhao, Chem. Eng. J., 2021, 426, 130767 CrossRef CAS.
- Q. Liu, J. Shen, X. Yu, X. Yang, W. Liu, J. Yang, H. Tang, H. Xu, H. Li, Y. Li and J. Xu, Appl. Catal., B, 2019, 248, 84–94 CrossRef CAS.
- P. Sun, Z. Chen, J. Zhang, G. Wu, Y. Song, Z. Miao, K. Zhong, L. Huang, Z. Mo and H. Xu, Appl. Catal., B, 2024, 342, 123337 CrossRef CAS.
- G. Wu, Z. He, Q. Wang, H. Wang, Z. Wang, P. Sun, Z. Mo, H. Liu and H. Xu, J. Mater. Sci. Technol., 2024, 195, 1–8 CrossRef CAS.
- K. Huang, C. Li, X. Zhang, L. Wang, W. Wang and X. Meng, Green Energy Environ., 2023, 8, 233–245 CrossRef CAS.
- L. Chen, S. Ning, R. Liang, Y. Xia, R. Huang, G. Yan and X. Wang, Int. J. Hydrogen Energy, 2022, 47, 14044–14052 CrossRef CAS.
- Z. Mo, Z. Miao, P. Yan, P. Sun, G. Wu, X. Zhu, C. Ding, Q. Zhu, Y. Lei and H. Xu, J. Colloid Interface Sci., 2023, 645, 525–532 CrossRef CAS PubMed.
- C. Hu, W. Hung, M. Wang and P. Lu, Carbon, 2018, 127, 374–383 CrossRef CAS.
- A. Mohammad, P. Chandra, M. E. Khan, C. Choi and T. Yoon, Adv. Colloid Interface Sci., 2023, 322, 103048 CrossRef CAS PubMed.
- S. Wang, F. He, X. Zhao, J. Zhang, Z. Ao, H. Wu, Y. Yin, L. Shi, X. Xu, C. Zhao, S. Wang and H. Sun, Appl. Catal., B, 2019, 257, 117931 CrossRef CAS.
- X. Li, J. Zhang, Y. Huo, K. Dai, S. Li and S. Chen, Appl. Catal., B, 2021, 280, 119452 CrossRef CAS.
- J. Huang, D. Li, R. Li, Q. Zhang, T. Chen, H. Liu, Y. Liu, W. Lv and G. Liu, Chem. Eng. J., 2019, 374, 242–253 CrossRef CAS.
- M. Bellardita, E. I. Garcia-Lopez, G. Marci, I. Krivtsov, J. R. Garcia and L. Palmisano, Appl. Catal., B, 2018, 220, 222–233 CrossRef CAS.
- F. Yi, H. Gan, H. Jin, W. Zhao, K. Zhang, H. Jin, H. Zhang, Y. Qian and J. Ma, Sep. Purif. Technol., 2020, 233, 115997 CrossRef CAS.
- Q. Zheng, D. P. Durkin, J. E. Elenewski, Y. Sun, N. A. Banek, L. Hua, H. Chen, M. J. Wagner, W. Zhang and D. Shuai, Environ. Sci. Technol., 2016, 50, 12938–12948 CrossRef CAS PubMed.
- J. Wang, L. Tang, G. Zeng, Y. Liu, Y. Zhou, Y. Deng, J. Wang and B. Peng, ACS Sustainable Chem. Eng., 2017, 5, 1062–1072 CrossRef CAS.
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, X. Li, B. Li, W. Dong and Y. Zhou, Appl. Catal., B, 2020, 272, 118970 CrossRef CAS.
- Z. Chen, T. Fan, M. Shao, X. Yu, Q. Wu, J. Li, W. Fang and X. Yi, Appl. Catal., B, 2019, 242, 40–50 CrossRef CAS.
- N. Rafique, A. H. Asif, R. A. K. Hirani, H. Wu, L. Shi, S. Zhang, S. Wang, Y. Yin, S. Wang and H. Sun, J. Colloid Interface Sci., 2023, 632, 117–128 CrossRef CAS PubMed.
- D. Guo, Y. Wang, C. Chen, J. He, M. Zhu, J. Chen and C. Zhang, Chem. Eng. J., 2021, 422, 130035 CrossRef CAS.
- G. Moon, M. Fujitsuka, S. Kim, T. Majima, X. Wang and W. Choi, ACS Catal., 2017, 7, 2886–2895 CrossRef CAS.
- S. Bach, V. R. Celinski, M. Dietzsch, M. Panthoefer, R. Bienert, F. Emmerling, J. S. A. Der Guenne and W. Tremel, J. Am. Chem. Soc., 2015, 137, 2285–2294 CrossRef CAS PubMed.
- L. E. Franken, E. J. Boekema and M. C. A. Stuart, Adv. Sci., 2017, 4, 1600476 CrossRef PubMed.
- P. Moeck and P. Fraundorf, Physics, 2006, 2, 533–543 Search PubMed.
- D. M. Zhao, C. L. Dong, B. Wang, C. Chen, Y. C. Huang, Z. D. Diao, S. Z. Li, L. J. Guo and S. H. Shen, Adv. Mater., 2019, 31, 1903545 CrossRef CAS PubMed.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 16850–16856 CrossRef CAS PubMed.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176, 44–52 CrossRef.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- Q. You, Q. Zhang, M. Gu, R. Du, P. Chen, J. Huang, Y. Wang, S. Deng and G. Yu, Chem. Eng. J., 2022, 431, 133927 CrossRef CAS.
- Y. Liu, X. Wang, Q. Sun, M. Yuan, Z. Sun, S. Xia and J. Zhao, J. Hazard. Mater., 2022, 424, 127387 CrossRef CAS PubMed.
- M. Achehboune, M. Khenfouch, I. Boukhoubza, I. Derkaoui, B. M. Mothudi, I. Zorkani and A. Jorio, Comput. Condens. Matter, 2022, 31, e00627 CrossRef.
- X. Ma, G. Liu, L. Wang, S. Xin, B. Zhang, Q. Chen, Q. Yan, G. Zhang, Y. Zhang and Y. Xin, J. Environ. Chem. Eng., 2023, 11, 110483 CrossRef CAS.
- M. Razavi-Esfali, T. Mahvelati-Shamsabadi, H. Fattahimoghaddam and B. Lee, Chem. Eng. J., 2021, 419, 129503 CrossRef CAS.
- Y. Zeng, X. Zhan, B. Hong, Y. Xia, Y. Ding, T. Cai, K. Yin, X. Wang, L. Yang and S. Luo, Chem. Eng. J., 2023, 452, 139434 CrossRef CAS.
- S. Li, C. Wang, M. Cai, Y. Liu, K. Dong and J. Zhang, J. Colloid Interface Sci., 2022, 624, 219–232 CrossRef CAS PubMed.
- Y. Ran, Y. Cui, Y. Zhang, Y. Fang, W. Zhang, X. Yu, H. Lan and X. An, Chem. Eng. J., 2022, 431, 133348 CrossRef CAS.
- T. Zhao, D. Li, Y. Zhang and G. Chen, J. Colloid Interface Sci., 2022, 628, 966–974 CrossRef CAS PubMed.
- K. Saravanakumar and C. M. Park, Chem. Eng. J., 2021, 423, 130076 CrossRef CAS.
- R. Shen, K. He, A. Zhang, N. Li, Y. H. Ng, P. Zhang, J. Hu and X. Li, Appl. Catal., B, 2021, 291, 120104 CrossRef CAS.
- O. Altan, E. Altintas, S. Alemdar and O. Metin, Chem. Eng. J., 2022, 441, 136047 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05879h |
| This journal is © The Royal Society of Chemistry 2024 |