
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time in situ monitoring as a tool for comparison of electrochemical advanced oxidation processes for the decolourisation of azo and indigoid dyes†
Chelsea M. Schroeder‡
 ,
Taylor M. Koehler‡ and
Nicholas E. Leadbeater*
,
Taylor M. Koehler‡ and
Nicholas E. Leadbeater*
Department of Chemistry, University of Connecticut, 55 North Eagleville Road, Storrs, Connecticut 06269, USA. E-mail: nicholas.leadbeater@uconn.edu
First published on 4th December 2024
Abstract
The widespread use of synthetic dyes has led to the release of substantial amounts of dye-contaminated wastewater, posing significant environmental and health concerns. This study focuses on the use of anodic and electrochemically activated persulfate oxidation for the degradation of organic contaminants. Specifically, the structural variations of nine dyes in the indigoid and azo families, and their impact on the efficiency of electrochemical oxidation were analysed. An in situ continuous monitoring apparatus with a UV-visible detector was employed to collect data in real-time. The electrochemically activated persulfate system demonstrated higher efficiency compared to the anodic oxidation approach. In both systems the efficiency of decolourisation was highly dependent on the structure of the pollutant. Electron-withdrawing substituents in direct conjugation with the chromophore, bulky auxochromes, and extended aromatic systems significantly decreased the decolourisation efficiency. Conversely, changing the location of electron-withdrawing groups and adding electron-donating substituents increased the decolourisation efficiency, even overcoming the detrimental effects of bulky groups and extended conjugation. This type of systematic structural comparison study is essential for highlighting the interconnected nature of pollutant structure and degradation speed so that efficient electrochemical oxidation systems can be designed for the treatment of genuine wastewater effluent containing more than one pollutant.
Introduction
Bright colours are a staple of modern society, where nearly every industry relies on synthetic dyes to enhance the appearance of their products.1,2 This is particularly the case in the textile industry, which releases 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of dye-contaminated wastewater every year.3 Approximately 80% of this effluent is discharged directly into watersheds where it can be used for irrigating farmland without treatment.4,5 Major concerns arise as most dyes are resistant to traditional water treatment methods,6,7 and their mutagenic and genotoxic properties can devastate entire ecosystems, reduce crop yields, and bioaccumulate in all living things.4,7
000 tons of dye-contaminated wastewater every year.3 Approximately 80% of this effluent is discharged directly into watersheds where it can be used for irrigating farmland without treatment.4,5 Major concerns arise as most dyes are resistant to traditional water treatment methods,6,7 and their mutagenic and genotoxic properties can devastate entire ecosystems, reduce crop yields, and bioaccumulate in all living things.4,7
Electrochemical advanced oxidation processes (EAOPs) have emerged as one of the most innovative alternative wastewater treatment methods.2,3,8–10 EAOPs are promising due to their high efficiency, small physical footprint, simple operation, and capability for automation. They work by generating reactive oxygen species (ROS) in situ, which eliminates the need for chemical additives.1,8,10,11 The ROS can break recalcitrant pollutants into smaller organic components like small carboxylic acids, or mineralise them into carbon dioxide and water.1,12
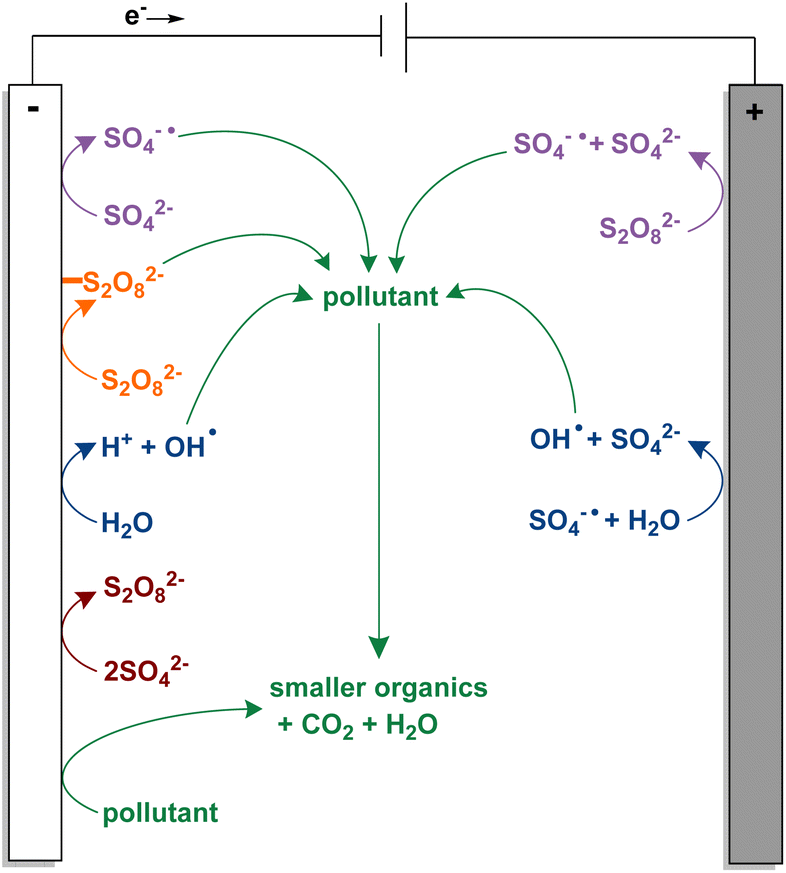
The simplest EAOP is anodic oxidation (AO) which generates hydroxyl radicals from water at the anode (Fig. 1). Organic contaminants can be degraded through interactions with these highly oxidising hydroxyl radicals (E° 2.8 V vs. SHE) or direct electron transfer with the anode.11,13–15 This process is controlled by mass transfer, and the efficiency is limited by the oxygen evolution potential (OEP) of the anode.16–18 Active anodes like graphite, platinum, IrO2, and RuO2 have a low OEP, and generate chemisorbed hydroxyl radicals. Inactive anodes like boron-doped diamond (BDD), PbO2, and Ti4O7 have a higher OEP, leading to physiosorbed hydroxyl radicals that are more reactive due to the greater distance from the electrode surface.13,14,17 Of the inactive anodes, BDD has the highest stability, corrosion resistance, and operable pH range, as well as the widest electrochemical window, and superior catalytic activity.8 When combined with sulfate-, chloride-, carbonate-, or phosphate-based electrolytes, BDD anodes can generate highly oxidising species from the electrolyte in the bulk solution. These species can react with and degrade pollutants more efficiently than hydroxyl radicals because they are not limited to the area around the anode.8,15,17 Fortunately, textile effluent is often high in both chloride and sulfate ions, obviating the need for additional reagents. Electrolytes with chloride are the most studied but can lead to toxic halogenated intermediates that are more difficult to remove than the original pollutant.14,19,20 As such, an electrochemically activated persulfate (EAP) indirect oxidation system, like the one depicted in Fig. 2, is more desired.15,17,21,22 Besides anode material, EAOPs can be influenced by a large number of variables, one of the most important being the identity of the pollutant.
 | ||
| Fig. 1 Anodic oxidation. | ||
 | ||
| Fig. 2 Electrochemically activated persulfate. | ||
It is very common for literature methods to report the use of a single dye as a model pollutant to investigate an EAOP. However, this leaves the impact of dye structure on the efficiency of EAOPs largely unknown.1,23 With over 10000 different dyes, it is essential to understand how pollutant structure impacts the efficiency of oxidation.6 All dyes have a chromophore that determines their color, and auxochromes that control the intensity of the color and its solubility. Two of the most commonly used dye types are azo and indigoid (Fig. 3). Over 70% of dyes used are azo dyes. Coveted for their diverse colors and ease of use, they contain a –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– moiety in their chromophore, with sulfate, nitro, and amine auxochromes being common. Indigoid dyes have a polymethine chromophore and are most commonly used to dye jeans.1,6,7,9 The dose that causes death in 50% (LD50) of mice for both dye types is well within the concentration found in some wastewater.3
N– moiety in their chromophore, with sulfate, nitro, and amine auxochromes being common. Indigoid dyes have a polymethine chromophore and are most commonly used to dye jeans.1,6,7,9 The dose that causes death in 50% (LD50) of mice for both dye types is well within the concentration found in some wastewater.3
 | ||
| Fig. 3 Structural motif of two common classes of dye. | ||
Our group has previously developed an apparatus for real-time in situ continuous monitoring of electrochemical decolourisation processes (Fig. 4). This system utilises a 3D-printed vessel holder to block excess light from entering the electrochemical cell and assuring the appropriate alignment of other components. The apparatus contains an LED light source on one side, aligned with an optical fiber that connects to a UV-visible (UV-vis) spectrophotometer on the other side. The design enables highly reproducible absorbance measurements to be taken in situ, eliminating the need for time-consuming sample collection and machine operation. Decolourisation in EAOPs has been shown to continue for several hours after the electricity is turned off, so any delay between sample preparation and analysis could potentially inflate values. As a result, continuous monitoring systems like the one utilized in these studies are essential.24,25 Related approaches in the literature include an automated UV-vis system that pumps solution out of an electrochemical cell to fill a cuvette,26 as well as online HPLC systems to detect intermediates.27–29 However, our system has the key advantage of being capable of taking real-time measurements in situ.
 | ||
| Fig. 4 Real-time in situ continuous UV-vis monitoring apparatus for monitoring electrochemical reactions. | ||
Results and discussion
In this work, we have employed our apparatus and previously optimised reaction conditions to study the performance of two different EAOPs; namely AO and EAP.24,25 These two systems have been used in the decolourisation of nine different textile dyes of the azo and indigoid families with the objective of determining how overall dye structure, number of carbons atoms, steric hindrance, and electron-withdrawing groups impact the rate and extent of decolourisation. The dyes selected for the study are shown in Fig. 5. From the indigoid class, we chose indigo carmine (IC) and potassium indigotrisulfonate (KIT). Due to their wide structural variation, the azo dye class was split into two families, phenyl- and naphthyl-based examples. From the phenyl-azo family, we selected food yellow, 6 (FY6), methyl orange (MO), ethyl orange (EO), and acid yellow 36 (AY36). From the naphthyl-azo family, we chose acid red 29 (AR29), acid red 176 (AR176), and sulfanilic acid azochromotrop (SPADNS). | ||
| Fig. 5 Structures of indigoid and azo dyes compared in this study. | ||
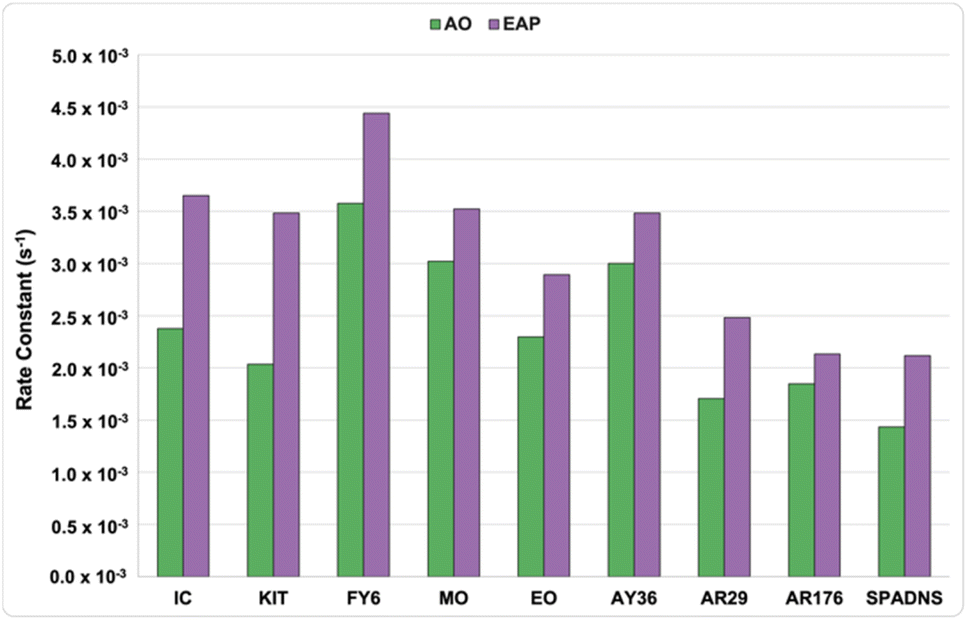
First-order rate constants at 20 min are presented in Fig. 6, and further graphical visualisation is provided in the ESI.† Overall, FY6, with a first-order rate constant of 4.44 × 10−3 s−1, was the easiest to decolourise, and SPADNS, with a first-order rate constant of 1.43 × 10−3 s−1, was the hardest to decolourise. Both AO and EAP showed the same overall trend in decolourisation speed, where phenyl-azo dyes are decolourised the fastest, followed by indigoid dyes, and naphthyl-azo dyes were the slowest. For all dyes studied, the rate of decolourisation with EAP was faster than decolourisation with AO. This was expected, as EAP uses a BDD anode to generate reactive species in the bulk solution, whereas AO relies on pollutants interacting with the anode to initiate decolourisation.14
 | ||
| Fig. 6 First order rate constants of nine dyes under AO and EAP conditions. | ||
In the indigoid category, the structure of IC and KIT differ in the number and location of sulfonate groups, but show similar decolourisation trends (Fig. 7). In both systems, KIT (AO: 2.04 × 10−3 s−1, EAP: 3.48 × 10−3 s−1) decolourised slightly more slowly than IC (AO: 2.38 × 10−3 s−1, EAP: 3.65 × 10−3 s−1), indicating that the more electron-deficient a pollutant is, the slower it decolourises. Of the dyes included in this study, the indigoid class show a larger difference between the first-order rate constants of AO and EAP than any of the azo dyes. Previous literature reports show that EAP is significantly more efficient at decolourising electron deficient pollutants than AO because of the nucleophilic nature of the sulfate radical compared to the hydroxyl radical.25 Within the phenyl-azo category, the structure of FY6, MO, and EO differs by the nature of the groups attached to the amine functionality. FY6 contains a simple NH2 moiety, MO has two methyl groups, and EO has two ethyl groups. For these three dyes, the rate of decolourisation is directly proportional to the length of the carbon chain where FY6 is fastest (AO: 3.58 × 10−3 s−1, EAP: 4.44 × 10−3 s−1) and EO is slowest (AO: 2.30 × 10−3 s−1, EAP: 2.89 × 10−3 s−1) (Fig. 8 and 9). This suggests that increasing the number of carbon atoms on the amine auxochrome increases the time needed for electrochemical oxidation. Following this logic, AY36, which contains an N-phenyl group, should have the slowest decolourisation rate of the dyes studied in this category. However, it shows an intermediate rate (AO: 3.00 × 10−3 s−1, EAP: 3.48 × 10−3 s−1), likely due to the location of the electron-withdrawing sulfonate group. FY6, MO, and EO have a sulfonate group in the para position, while AY36 has a sulfonate group in the meta position, meaning it is not directly conjugated to the azo bond. The secondary amine of AY36 is also more electron-donating than the tertiary amine functionalities of MO and EO. This means that the azo bond of AY36 to has a higher electron density than the other dyes in this category; an important feature as azo dye degradation occurs by cleavage of the azo bond.30 Consequently, both the number of carbon atoms on the amine group and the overall electronic nature of the dye play significant and interconnected roles in the efficiency of EAOP.
 | ||
| Fig. 7 Dye remaining vs. time for the AO and EAP decolourisation of IC and KIT. | ||
 | ||
| Fig. 8 Dye remaining vs. time for the AO and EAP decolourisation of FY6 and MO. | ||
 | ||
| Fig. 9 Dye remaining vs. time for the AO and EAP decolourisation of EO and AY36. | ||
Analysis of the first-order rate constants of the selected naphthyl-azo dyes indicates that they are harder to decolourise than the phenyl-azo examples, likely due to their more extensive aromatic systems. AR29, AR176, and SPADNS differ by the identity of a para electron-withdrawing group. AR29 does not have a substituent, AR176 bears a nitro group, and SPADNS has a sulfonate group. The first-order rate constants at 20 min suggest that this structural change has little impact on the rate. However, a plot of the percent dye remaining vs. time provides more information about their long-term behavior (Fig. 10). The extent of decolourisation of AR29, AR176, and SPADNS was 97%, 98%, and 97%, respectively after 90 min AO. However, better efficiency of >99%, 98%, and >99% decolourisation was achieved using EAP in half the time. Since extended aromatic systems are more difficult to decolourise, the oxidising effect of the longer-lived (30–40 μs) nucleophilic sulfate radical compared to the shorter (1 μs) lifetime of the unselective hydroxyl radical is more apparent.4 As a consequence, the efficiency of EAP for highly electron-deficient extended aromatic systems is much higher than that of AO for the same compounds.
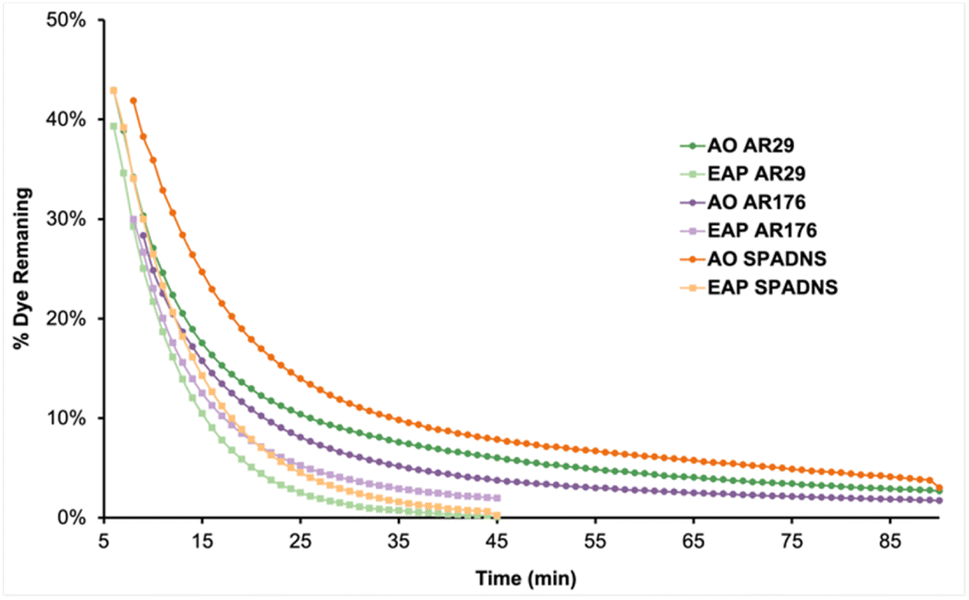 | ||
| Fig. 10 Dye remaining vs. time for the AO and EAP decolourisation of AR29, AR176, and SPADNS. | ||
Experimental section
General considerations
An IKA ElectraSyn 2.0 with a standard 10 mL vial was interfaced with an Ocean Insight SR-2UVV400-25 spectrophotometer with a range of 183–909 nm, optical resolution of 1.33 nm, and signal to noise ratio of 380:1. The spectrophotometer was attached to an Ocean Insight P400-1-SR 400 μm diameter optical fiber (200 nm to 1.1 μm) with a 74-UV collimating lens (185 nm to 2.5 μm). A diffuse white LED with a range of 420–700 nm was employed as the light source. Data was collected using OceanView 2.0 from Ocean Insight. Graphite and boron-doped diamond electrodes were obtained from IKA. The 3D-printed interface (STL file available in the ESI†) was fabricated from black polylactic acid using an Encina Tina2S 3D-printer.
Chemicals
Potassium indigotrisulfonate, methyl orange, acid yellow 36, acid red 29, acid red 176, and sulfanilic acid azochromotrop were purchased from Sigma-Aldrich. Ethyl orange was purchased from Thermo Fisher Scientific. Food yellow 6 was purchased from TCI America. Indigo carmine was purchased from Spectrum Chemical. Sodium persulfate was purchased from Oakwood Chemicals.General procedure
The 3D-printed sleeve was placed on the stage of the IKA ElectraSyn 2.0 single-vial holder. An optical fiber was attached to the front cavity in the 3D-printed sleeve by means of a collimating lens. The other end of the optical fiber was attached to the spectrophotometer. A standard 5 mm diffuse white LED was placed in the slot at the back of the 3D-printed sleeve and connected to a bench power supply using alligator clips. The power supply was set to 2.7 V and allowed to warm up for 10 min until the absorbance spectrum of the LED remained at a constant intensity. To calibrate the spectrophotometer, a standard 10 mL capacity ElectraSyn 2.0 vial containing deionised water (8 mL) was placed inside of the assembled electrochemical/spectrophotometer interface, and the resulting absorbance spectrum was set to zero.To prepare a reaction, an ElectraSyn 2.0 vial cap was equipped with the desired anode and cathode, a 10 mL capacity vial was charged with 8 mL of a 100 μM dye solution in deionised water, sodium persulfate (14 mM), and a magnetic stir bar. For AO conditions, a graphite anode and cathode were used while EAP processes employed a BDD anode and graphite cathode. The reaction vessel was placed inside the assembled electrochemical/spectrophotometer interface, and the contents electrolysed at a constant current of 5 mA while stirring the reaction mixture at 1000 rpm. Absorbance spectra were collected every minute with an automated clicker until the oxidation was deemed complete by in situ UV-visible spectrophotometer detection. Cartesian points of absorbance vs. time at the λmax were transferred to a Microsoft Excel workbook. All reactions were performed in triplicate, absorbances were averaged and then converted to μM values by employing a calibration curve. Finally, μM values were converted to percentage dye remaining and plotted as a function of time.
To clean the boron-doped diamond electrodes between trials, they were rinsed with water, then acetone, and then dried with lab air; the graphite electrodes were rinsed with water and acetone then polished on 2000 grit sandpaper.
Conclusions
In summary, we have investigated the significant role that pollutant structure has on the decolourisation efficiency of AO and EAP using real-time in situ UV-vis monitoring. We screened two indigoid dyes and seven azo dyes. EAP proves to be more efficient than AO for all the dyes studied, especially when they comprise of extended aromatic systems and highly electron deficient substituents. As a result, phenyl functionalised azo dyes exhibit faster decolourisation as compared to indigoid and naphthyl-based azo dyes. Electron-deficient dyes decolourise slower than their more electron-rich counterparts. Increasing the number of carbon atoms attached to the amine functionality of azo dyes has a detrimental effect on decolourisation efficiency. However, the slowing effect of increasing chain length can be ameliorated by the electronic properties of the pollutant. Overall, our findings highlight the importance of considering both the chemical structure and electronic properties of dyes when designing efficient electrochemical oxidation systems for wastewater treatment. The results of this study will aid the design and understanding of how EAOP would operate in wastewater where multiple pollutants are present.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Connecticut Research Excellence Program, Office of Undergraduate Research (TMK), and Bobbitt Chou fund (CMS) are thanked for funding.Notes and references
- A. B. Isaev, N. S. Shabanov, A. G. Magomedova, P. V. Nidheesh and M. A. Oturan, Environ. Chem. Lett., 2023, 21, 2863–2911 CrossRef CAS.
- H. Jiang, H. Chen, K. Wei, L. Liu, M. Sun and M. Zhou, Chemosphere, 2023, 341, 140083 CrossRef CAS PubMed.
- L. D. Paquini, L. T. Marconsini, L. P. R. Profeti, O. S. Campos, D. Profeti and J. Ribeiro, Braz. J. Chem. Eng., 2023, 40, 623–653 CrossRef CAS.
- J. Lin, W. Ye, M. Xie, D. H. Seo, J. Luo, Y. Wan and B. Van Der Bruggen, Nat. Rev. Earth Environ., 2023, 4, 785–803 CrossRef CAS.
- H. M. Solayman, Md. A. Hossen, A. Abd Aziz, N. Y. Yahya, K. H. Leong, L. C. Sim, M. U. Monir and K.-D. Zoh, J. Environ. Chem. Eng., 2023, 11, 109610 CrossRef CAS.
- F. Deng and E. Brillas, Sep. Purif. Technol., 2023, 316, 123764 CrossRef CAS.
- R. Al-Tohamy, S. S. Ali, F. Li, K. M. Okasha, Y. A.-G. Mahmoud, T. Elsamahy, H. Jiao, Y. Fu and J. Sun, Ecotoxicol. Environ. Saf., 2022, 231, 113160 CrossRef CAS.
- P. Brosler, A. V. Girão, R. F. Silva, J. Tedim and F. J. Oliveira, Environments, 2023, 10, 15 CrossRef.
- A. Iqbal, A. Yusaf, M. Usman, T. Hussain Bokhari and A. Mansha, Int. J. Environ. Anal. Chem., 2023, 1–35 CAS.
- J. Mukherjee, B. K. Lodh, R. Sharma, N. Mahata, M. P. Shah, S. Mandal, S. Ghanta and B. Bhunia, Chemosphere, 2023, 345, 140473 CrossRef CAS.
- D. Gümüş, Water Air Soil Pollut., 2023, 234, 301 CrossRef.
- A. L. Magdaleno, E. Brillas, S. Garcia-Segura and A. J. Dos Santos, Sep. Purif. Technol., 2024, 345, 127295 CrossRef CAS.
- Y. Jiang, H. Zhao, J. Liang, L. Yue, T. Li, Y. Luo, Q. Liu, S. Lu, A. M. Asiri, Z. Gong and X. Sun, Electrochem. Commun., 2021, 123, 106912 CrossRef CAS.
- Y. Bashir, R. Raj, M. M. Ghangrekar, A. K. Nema and S. Das, RSC Sustain., 2023, 1, 1912–1931 RSC.
- R. Fu, P.-S. Zhang, Y.-X. Jiang, L. Sun and X.-H. Sun, Chemosphere, 2023, 311, 136993 CrossRef CAS.
- D. Zhi, Y. Lin, L. Jiang, Y. Zhou, A. Huang, J. Yang and L. Luo, J. Environ. Manage., 2020, 260, 110125 CrossRef CAS.
- A. Fernandes, M. J. Nunes, A. S. Rodrigues, M. J. Pacheco, L. Ciríaco and A. Lopes, Molecules, 2021, 26, 4821 CrossRef CAS.
- G. Kuchtová, L. Hojová, A. V. Staňová, M. Marton, M. Vrška, M. Behúl, P. Michniak, M. Vojs and L. Dušek, Electrochim. Acta, 2023, 464, 142924 CrossRef.
- J. Alagesan, M. Jaisankar, S. Muthuramalingam, E. Mousset and P. V. Chellam, Chemosphere, 2021, 262, 128381 CrossRef CAS.
- C. Gutiérrez-Bouzán and M. Pepió, Ind. Eng. Chem. Res., 2014, 53, 18993–19000 CrossRef.
- L. W. Matzek and K. E. Carter, Chemosphere, 2016, 151, 178–188 CrossRef CAS.
- W. Li, G. Liu, D. Miao, Z. Li, Y. Chen, X. Gao, T. Liu, Q. Wei, L. Ma, K. Zhou and Z. Yu, J. Environ. Chem. Eng., 2020, 8, 103997 CrossRef CAS.
- C. K. C. Araújo, G. R. Oliveira, N. S. Fernandes, C. L. P. S. Zanta, S. S. L. Castro, D. R. Da Silva and C. A. Martínez-Huitle, Environ. Sci. Pollut. Res., 2014, 21, 9777–9784 CrossRef.
- C. M. Schroeder, A. León Sandoval, K. K. Ohlhorst and N. E. Leadbeater, Chem.:Methods, 2023, e202300014 CAS.
- C. M. Schroeder, T. M. Koehler, K. K. Ohlhorst and N. E. Leadbeater, RSC Adv., 2023, 13, 33559–33565 RSC.
- G. Kuchtová, P. Mikulášek and L. Dušek, Monatsh. Chem., 2022, 153, 237–243 CrossRef.
- A. B. De Souza, J. Mielcke, I. Ali, R. Dewil, T. Van De Goor and D. Cabooter, J. Environ. Chem. Eng., 2023, 11, 109993 CrossRef CAS.
- M. A. Sandoval, N. Zúñiga-Mallea, L. C. Espinoza, J. Vidal, P. Jara-Ulloa and R. Salazar, ChemistrySelect, 2019, 4, 13856–13866 CrossRef CAS.
- E. Kusmierek, E. Chrzescijanska, M. Szadkowska-Nicze and J. Kaluzna-Czaplinska, J. Appl. Electrochem., 2011, 41, 51–62 CrossRef CAS.
- M. Nashat, M. Mossad, H. K. El-Etriby and M. Gar Alalm, Chemosphere, 2022, 286, 131579 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07657e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |