
Ammonia electro-oxidation on nickel hydroxide: phases, pH and poisoning†
Inbal
Offen-Polak
 ,
Hilla
Ayali Aviram
,
Adan
Hijaze
,
Thierry K.
Slot
and
David
Eisenberg
*
,
Hilla
Ayali Aviram
,
Adan
Hijaze
,
Thierry K.
Slot
and
David
Eisenberg
*
Schulich Faculty of Chemistry, The Grand Technion Energy Program, and the Resnick Sustainability Center for Catalysis, Technion – Israel Institute of Technology, Technion City, 3200003 Haifa, Israel. E-mail: eisenberg@technion.ac.il
First published on 2nd December 2024
Abstract
Nickel hydroxide is a leading alternative to platinum group metals for electrocatalysis of the ammonia oxidation reaction (AOR), an important process for energy conversion and environmental remediation. Nevertheless, the dependence of AOR electrocatalysis on the different crystalline phases at the electrode surface (α-Ni(OH)2/γ-NiOOH vs. β-Ni(OH)2/β-NiOOH) has never been investigated. Herein, the crystalline β-Ni(OH)2 and the disordered α-Ni(OH)2 were synthesized and characterized by XRD, HRSEM, and Raman and FTIR spectroscopies. The respective electrocatalytic activity of the two phases was analysed at a broad range of ammonia concentrations (0.01–2 M) and pH values (11–13). Both phases electrocatalyze the oxidation of NH3 to N2, as proven by online mass spectrometry, but the α-Ni(OH)2/γ-NiOOH couple is more active. At high ammonia concentrations (>1 M), surface poisoning by adsorbed NH3 prevents access to OH−, leading to less NiOOH formation, lower AOR currents, and suppression of the OER side reaction. The poisoning is strong and irreversible on α-Ni(OH)2, as confirmed by soaking experiments. The difference in ammonia adsorption and electrocatalytic activity between the α-Ni(OH)2 and β-Ni(OH)2 emphasizes the importance of understanding the phase space of nickel hydroxide electrodes when designing low-cost electrocatalysts for the nitrogen cycle.
Introduction
The electro-oxidation of ammonia is important in the fields of energy and environmental remediation.1–5 Ammonia is a common pollutant present in wastewater, due to its extensive exploitation in industry and agriculture. The electro-oxidation of ammonia is used in electrochemical sensors6–8 and in wastewater remediation.9 In the energy sector, the (alkaline) ammonia oxidation reaction (AOR, reaction (I); E° for the opposite reduction reaction = −0.77 V vs. SHE) is used in direct ammonia fuel cells,10–12 or to produce hydrogen fuel from ammonia-containing water for energy storage applications.5,13–16 The production of hydrogen by ammonia electrolysis is theoretically more cost-effective and less energy-consuming than water electrolysis. Furthermore, wastewater remediation can be coupled with hydrogen production.9,11 Ammonia offers high energy density (3000 W h kg−1) and higher hydrogen storage capacity (17.7 wt%) than other hydrogen carriers, without releasing CO2.9 Moreover, infrastructure for ammonia synthesis and distribution already exists, due to its role as a fertilizer.| 2NH3 + 6OH− → N2 + 6H2O + 6e− | (I) |
Nickel is a fascinating and complex electrode material, first proposed for batteries in 1899.48,49 Despite over a century of electrochemical studies and broad applications in Ni–MH batteries,50 Ni–Cd batteries,51 and supercapacitors,52 the electrochemistry of the Ni(OH)2 surface remains elusive. The reason lies in the variety of crystalline phases of Ni(OH)2 and NiOOH, which show different degrees of hydration and intercalation,53 and interconvert through multiple routes (Fig. 1) and in different locations inside the electrode. The nickel hydroxide polymorphs range from crystalline brucite-structured β-Ni(OH)2 to a family of badly crystalline structures denoted under the umbrella term α-Ni(OH)2.54 The latter phase can intercalate water and anions, be turbostratically disordered (imperfect layer orientation), and can contain hydrogen bonding and point defects.55,56 Upon oxidation, the crystalline β-Ni(OH)2 transforms into crystalline β-NiOOH (reaction (II)), while the α-Ni(OH)2 becomes γ-NiOOH (reaction (III)).
| β-NiII(OH)2 + OH− → β-NiIIIOOH + e− + H2O | (II) |
| α-NiII(OH)2 + OH− → γ-NiIII/IVOOH + e− + H2O | (III) |
 | ||
| Fig. 1 Bode diagram of the limiting cases of divalent and trivalent NiOxHy phases.55–57 | ||
The α/γ couple has larger interlayer distances than the β/β couple, allowing ion intercalation and better ionic conductivity. Moreover, the γ-NiOOH phase can have higher average oxidation than 3+ on the Ni,56 leading to higher electronic conductivity than β-NiOOH.58 The α/γ couple is more catalytic towards the urea oxidation reaction (UOR),59 while the β/β couple is considered better for the oxygen evolution reaction (OER)60 and the hydrogen evolution reaction (HER).61 For the AOR, Łuczak and Lieder have hypothesized that the α/γ couple could be more catalytic towards the AOR.35 Importantly, the α/γ couple is less stable thermodynamically, aging to the β/β couple over time, or by the effects of high temperature, alkalinity or continuous electrochemical cycling.55,56
Several studies focused on the mechanism of the AOR, mostly for Pt and other noble metals,19,25,26,62–65 though only a few examined Ni(OH)2/NiOOH surfaces. Kapalka et al. demonstrated that ammonia oxidation on Ni/Ni(OH)2 occurs only after the surface has been oxidized to nickel oxyhydroxide,66 as was later supported by in situ Raman studies.67–69 The chemical oxidation of ammonia on NiOOH was further found to be spontaneous, as demonstrated by detecting nitrite (NO2−) formed on NiOOH under no bias.70 Two possible mechanisms were proposed for the AOR (in analogy for the oxidation of organic molecules): a direct electron transfer from the ammonia to the oxyhydroxide film (reactions (IV) and (V)), or an indirect oxidation where the formed oxyhydroxide is chemically reduced back to the hydroxide by an ammonia molecule (reaction (VI)).66
| Ni(OH)2 + OH− → NiOOH + H2O + e− | (IV) |
| NiOOH + NH3 → NiOOH(NH3)ads → NiOOH + products + ne− | (V) |
| NiOOH + NH3 + mOH− → Ni(OH)2 + products | (VI) |
We now report a detailed investigation of AOR electrocatalysis and its dependence on the phase of nickel hydroxide. For the first time, the electrochemistry of α-Ni(OH)2 and β-Ni(OH)2 are compared directly, over a broad range of conditions relevant to energy and environmental applications, namely ammonia concentrations from 0.01 to 2 M, and pH values from 11 to 13.13,72,73 We found that the α-Ni(OH)2/γ-NiOOH couple is more active towards the AOR throughout the concentration range, although the β-Ni(OH)2/β-NiOOH is also AOR-active. Both hydroxide phases are poisoned at high ammonia concentrations, leading to lower currents and delays in the onset potentials. The poisoning is stronger and irreversible in α-Ni(OH)2, probably due to stronger adsorption energies. The effects of pH on AOR further confirm the poisoning, as do ammonia soaking experiments.
Experimental procedures
Material synthesis
α-Ni(OH)2 and β-Ni(OH)2 were synthesized according to Chakrabarty, Offen-Polak et al.59 NiSO4·6H2O (Alfa Aesar, 98%) and NaOH (Alfa Aesar, 98%) were mixed in 35 mL of DI water in either 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio to give β-Ni(OH)2 (9.8 and 19.6 mmol, respectively) or a 3:1 molar ratio for α-Ni(OH)2 (9.8 and 3.3 mmol, respectively). For β-Ni(OH)2, the mixture was stirred for 30 min until it became homogeneous, transferred to a 50-mL Teflon-lined stainless-steel autoclave and placed in an oven at 120 °C for 24 hours. For α-Ni(OH)2, the mixture was stirred at room temperature for 3 hours. The resulting green powders were collected by centrifugation, washed with DI water repeatedly (until pH reached 7–8), and air-dried at 50 °C.
:2 molar ratio to give β-Ni(OH)2 (9.8 and 19.6 mmol, respectively) or a 3:1 molar ratio for α-Ni(OH)2 (9.8 and 3.3 mmol, respectively). For β-Ni(OH)2, the mixture was stirred for 30 min until it became homogeneous, transferred to a 50-mL Teflon-lined stainless-steel autoclave and placed in an oven at 120 °C for 24 hours. For α-Ni(OH)2, the mixture was stirred at room temperature for 3 hours. The resulting green powders were collected by centrifugation, washed with DI water repeatedly (until pH reached 7–8), and air-dried at 50 °C.
Material characterization
Powder X-ray diffraction (XRD) was performed in a Rigaku SmartLab X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å), scan rate of 1° min−1. A Raman spectrometer (LabRam, 1800 grating) was used with 532 nm laser excitation. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) transmittance spectra were collected on a Bruker Tensor 27 spectrometer. High-resolution scanning electron microscopy (HRSEM) was performed in secondary electrons mode on a Zeiss Ultra+ microscope, at a 4 kV acceleration voltage. The Ni concentration in the solution was measured by ICP-OES 5100 (Agilent Technologies).Electrochemical testing
The ink solution was prepared with 1 mg of the catalyst (either α-Ni(OH)2 or β-Ni(OH)2) dispersed by 30 min sonication in a solution containing 300 μL deionized water and 180 μL ethanol and 20 μL of 20 μL Nafion® 5 wt% dispersion (Alfa Aesar). 10 μL of the ink solution was applied by drop-cast on a polished glassy carbon electrode (5 mm diameter) and dried at 50 °C. The catalyst loading was 0.02 mg or 0.1 mg cm−2. Electrochemical measurements were performed by using a Biologic VSP multichannel potentiostat. Reversible hydrogen electrode (RHE) was used as reference electrode and a graphite rod as counter electrode. The electrolyte contained 0.1 M KOH and ammonia water solution (25% v/v) at the desired concentrations, along with 0.5 M K2SO4, required to maintain high ionic conductivity. The pH was monitored before and after the addition of ammonia, and pH was not changed by more than 0.2 pH units, except for 2 M ammonia, where it deviated by 0.4 units. For measurements at lower pH, (NH4)2SO4 was used instead for ammonia source, and the pH was monitored and adjusted accordingly by addition of KOH. Before a measurement, the electrolyte was purged for 30 min by Ar at 25.0 ± 0.1 °C and flowed continuously above the solution during the experiments. For the Fe-free experiments, electrolytes were cleaned by the Trotochaud procedure74 in a 0.1 M KOH solution. Then, the K2SO4 and ammonia were added to the Fe-free solution. Cyclic voltammetry (CV) was recorded at a scan rate of 10 mV s−1. Before measurements, each electrode was cycled in the non-faradaic potential range to improve wetting (0.8 V to 1.2 V vs. RHE, 10 cycles, 100 mV s−1). Additionally, each electrode went through 10 activation cycles before ammonia addition (0.8 V to 1.55 V vs. RHE, 10 cycles, 10 mV s−1). Measurements were repeated at least 3–6 times for each concentration and pH combination, for each material, and all presented trends were shown to be consistent. Rotating ring-disk electrode (RRDE) experiments were performed at each ammonia concentration, where CVs were measured (after the initial activation) at 1600 rpm rotation, Edisk = 0.8–1.9 V vs. RHE and Ering = 0.6 V vs. RHE. A small and constant baseline ring current was subtracted.Results and discussion
Phase characterization
To investigate the AOR electrocatalysis on the different phases of nickel hydroxide, both α-Ni(OH)2 and β-Ni(OH)2 were synthesized. The synthetic procedure and the characterization are fully described in our previous study for a broad range of crystallinities.59 The crystallinity of the nickel hydroxide materials was determined by X-ray diffraction (XRD). Indeed, the sharp peaks of the β-Ni(OH)2 match the diffraction pattern of this phase (JCPDS 14-0117, Fig. 2a). Moreover, the XRD confirms a crystalline and ordered β-Ni(OH)2, while the α-phase is amorphous, with barely visible broad peaks. The morphologies of the catalysts were characterized by high resolution scanning electron microscopy (HR-SEM, Fig. 2b and c). The α-Ni(OH)2 phase consists of irregular agglomerates (hundreds of nanometers in size), while the β-Ni(OH)2 powder is composed of uniform hexagonal platelets (∼60 nm). | ||
| Fig. 2 Material characterization of the α-Ni(OH)2 and β-Ni(OH)2 catalysts, as prepared: (a) XRD, with the β-Ni(OH)2 lines marked (JCPDS 14-0117). (b) and (c) HRSEM micrographs of the two materials. (d) Raman spectra and (e) ATR-FTIR spectra of the two materials. | ||
The structures of the α-Ni(OH)2 and β-Ni(OH)2 phases were further characterized by Raman spectroscopy (Fig. 2d) and Fourier transform infrared (FTIR) spectroscopy (Fig. 2e). Raman peaks at 318, 445, 872 and 3590 cm−1 are typical for β-Ni(OH)2, and at 460 and 3656 for α-Ni(OH)2. In the latter, a broad band at 3000–3600 cm−1 arises from the high hydration of the structure.56,77 Furthermore, strong sulphate intercalation peaks appear for α-Ni(OH)2 at 603, 985 and 1060 cm−1, indicating a disordered and open structure allowing intercalation.78–80 The FTIR spectra confirm the phase assignments, with fingerprint peaks of β-Ni(OH)2 at 340, 418, 506 and at 3630 cm−1, and for α-Ni(OH)2 at 374, 455 and 673 cm−1.78,81 The hydration of the α-Ni(OH)2 phase appear as a broad band at about ∼3400 cm−1. In contrast, the sharp peak at 3630 cm−1 corresponds to free hydroxyl groups in β-Ni(OH)2, as the closed-packed crystalline structure does not permit hydrogen bonding with intercalated water.81
Ammonia oxidation electrocatalysis
The AOR electrocatalytic activity of α-Ni(OH)2 and β-Ni(OH)2 was investigated by CV, before and after the addition of ammonia. Without ammonia (Fig. 3a), both α-Ni(OH)2 and β-Ni(OH)2 show oxidation and reduction peaks related to the reversible nickel hydroxide ⇌ nickel oxyhydroxide reaction (reaction (VI)). The voltametric waves on crystalline and well-defined β-Ni(OH)2 are sharper than the CV wave on the disordered α-Ni(OH)2 material. Furthermore, the α/γ couple has an earlier onset potential than the β/β couple (1.38 V and 1.42 V vs. RHE, respectively), and its oxidation produces a larger total charge. When ammonia is added (0.75 M, Fig. 3b), both catalysts show higher current densities for oxidation, and a different shape of the wave, suggesting that ammonia is indeed oxidized. The oxidation onset potentials are also delayed, to 1.39 V for α-Ni(OH)2 and to 1.45 V for β-Ni(OH)2. The electrocatalytic activity of nickel hydroxide towards the AOR is further confirmed by detection of N2(g) by differential scanning mass spectrometry (DEMS, Fig. 3c, d and Fig. S1, ESI†). In this experiment, the DEMS probe is placed above the electrode, measuring the gaseous species emitted during cell operation. Note that the DEMS experiment only provides qualitative information on the evolution of gaseous species and could be used to determine faradaic efficiencies. The rise in N2 production follows the voltametric ammonia oxidation peak, for both phases, taking into account a 5 second delay due to the bubble formation, release, and detection time (Fig. 3d). This proves, for the first time, that both NiOOH surface phases (γ and β) electrocatalyze the oxidation of NH3 to N2. The fact that the AOR starts only after the NiOOH formed confirms the hypothesized role of Ni3+ in the AOR electrocatalysis.66 This is analogous to UOR and OER, where the catalytic reaction follows the transition to NiOOH.60,82 Consequently, the use of pure nickel hydroxide as an anode in a direct ammonia fuel cell is impractical due to this high overpotential. This distinction is crucial, as was recently stressed for direct urea or alcohol fuel cells.83 The activity of nickel hydroxides towards the AOR is about 5 times inferior to its UOR activity.59 The poor performance of the pure nickel hydroxides explains the extensive focus on alloying and doping studies.4,29,35–42 Nevertheless, the fundamental differences between the nickel hydroxide phases are crucial to explore for the future design of doped catalysts, and for general understanding of the nitrogen cycle. | ||
| Fig. 3 Cyclic voltammetry (CV) of α-Ni(OH)2 (red) and β-Ni(OH)2 (black) in (a) 0.1 M KOH + 0.5 M K2SO4 electrolyte and (b) with 0.75 M NH3 added. (c) Differential electrochemical mass spectrometry (DEMS) of the N2 signal produced (black, top) during CV at 5 mV s−1 from 0.8 V to 1.55 V (red, bottom) with 1 M NH3 added. (d) Enlarged portion of the DEMS experiment, focusing on the onset of N2 evolution; the traces are averaged over 7 measurements, to improve signal-to noise ratio. | ||
A side-reaction of O2 evolution could contribute to the anodic wave. However, there is no OER peak in the CV up to 1.55 V, in the absence of ammonia (Fig. 3a). When ammonia is added, the catalytic wave becomes sharp and harder to deconvolute (Fig. 3b), so to rule out the OER, we performed RRDE for both α-Ni(OH)2 and β-Ni(OH)2 (Fig. 4). The disk-deposited Ni(OH)2 electrodes were cycled up to a high potential (1.9 V), where OER is expected (reaction (VII)), while the ring potential was held at 0.6 Vvs.RHE, to detect the produced O2 by reducing it (reaction (VIII)). The RRDE experiments were conducted at 1600 rpm at four ammonia concentrations of 0, 0.1, 0.75 and 1.5 M.
| 4OH− → O2 + 2H2O + 4e− | (VII) |
| O2 + 2H2O + 4e− → 4OH− | (VIII) |
 | ||
| Fig. 4 RRDE experiments at 1600 rpm, with different added ammonia concentrations (0, 0.1, 0.75 and 1.5 M). The disk potential was scanned between 0.8 to 1.9 V, while the Pt ring potential was set to 0.6 V to detect O2. The disk and ring currents are presented vs. the disk potential for (a) α-Ni(OH)2 and (b) β-Ni(OH)2. | ||
AOR at different ammonia concentrations and pH values
To understand how the Ni(OH)2/NiOOH phases behave at different ammonia concentrations, we cycled both α-Ni(OH)2 and β-Ni(OH)2 electrodes in 0.01 to 2 M NH3 (Fig. 5a). This broad range covers the ammonia concentration expected in different applications, from waste-water remediation (0.01–0.07 M)72 to direct ammonia fuel cells and ammonia electrolyzers for H2 production (0.5–5 M).13,73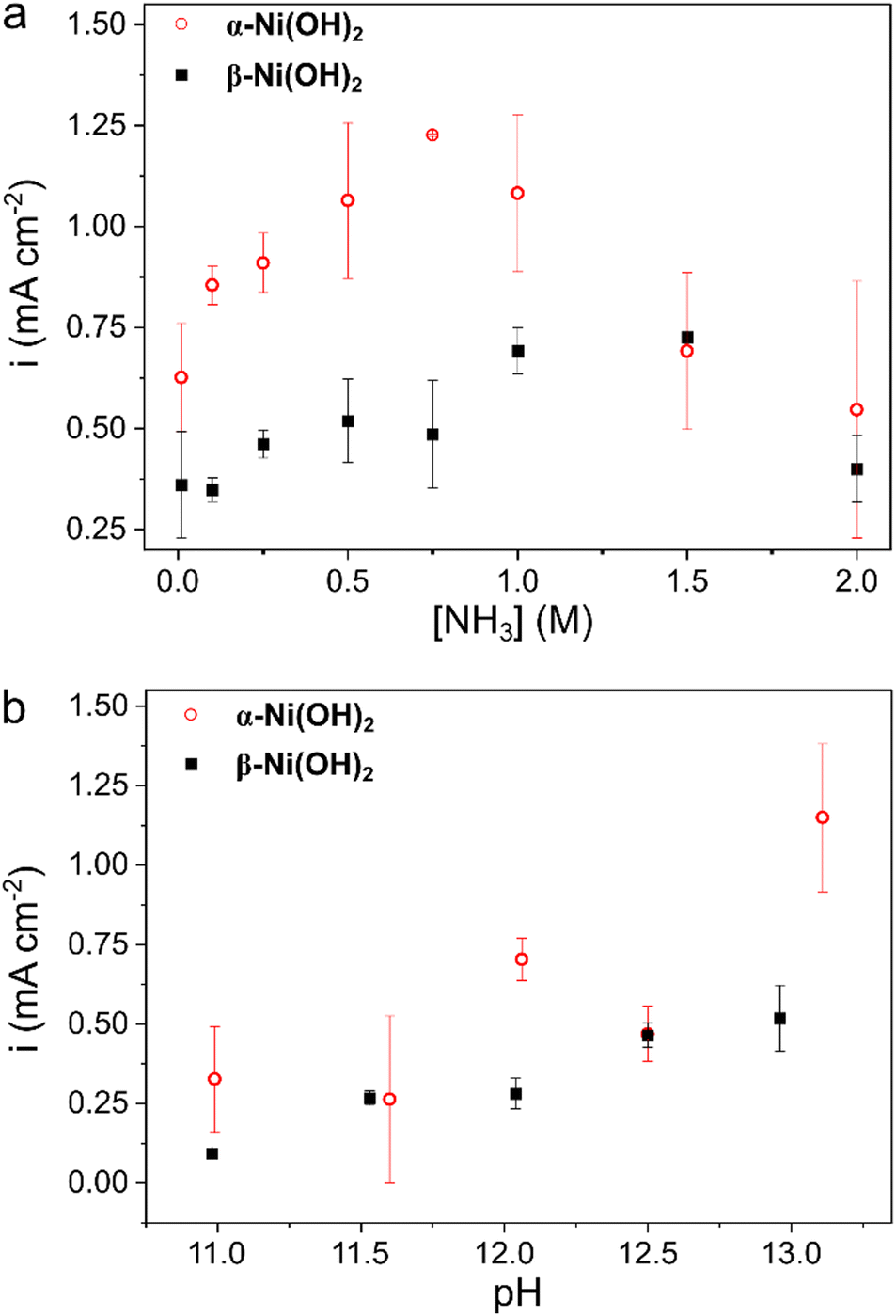 | ||
| Fig. 5 The oxidation current density at E = 1.54 V for both α-Ni(OH)2 (red circles) and β-Ni(OH)2 (black squares) over a wide range of (a) ammonia concentrations, and (b) pH of the electrolyte. Each data point is an average of at least 3 measurements. A representative CV for each data point appears in Fig. S2–S5 (ESI†). | ||
With the increase in ammonia concentration, the current rises linearly, up until 0.75 M, for both α-Ni(OH)2 and β-Ni(OH)2. This confirms that both phases can drive AOR electrocatalysis. Comparing the performance of the two catalysts, α-Ni(OH)2 is clearly a more active electrocatalyst than β-Ni(OH)2, with higher current densities for most of the range of ammonia concentrations studied, by up to 250%. This difference is too high to arise simply from enhanced electronic conductivity (due to Ni3+ defects58) or better ionic conductivity and mass transport through its open structure. After all, this difference in current with ammonia is quite larger than the ∼30% difference without ammonia, where the peak current varies between 0.75 ± 0.16 mA cm−2 for α-Ni(OH)2, and 0.53 ± 0.15 mA cm−2 for β-Ni(OH)2. This reveals that the higher AOR currents on α-Ni(OH)2 do not arise from inherent electrochemical differences between the phases, but rather due to better electrocatalytic activity of the α/γ couple. We believe that our findings can explain recent observations in the literature,84 where a naturally oxidized nickel foam (probably consisting of amorphous α-Ni(OH)2, as identified by earlier onsets of the NF5 and NF10 samples) exhibited higher AOR currents than the aged samples (probably consisting of β-Ni(OH)2, as identified by belated onsets of the NF50 sample). Interestingly, at ammonia concentrations above 1 M, the AOR current density starts to decay. This will be discussed in the next section.
To investigate the effect of pH on AOR, the two catalysts were cycled with 0.5 M ammonia, and the pH was adjusted in the range 11 to 13 (Fig. 5b). This range was chosen to produce the electrochemically active NH3 form (rather than NH4+), which is expected to be present at pH > 11 in over 98% fraction (NH4+ pKa = 9.24).70,85 Moreover, the electro-dissolution of Ni2+ in the presence of ammonia, to form a [Ni(NH3)6]2+ complex, is expected to decrease at alkaline pH.86,87 In our experiments, the oxidation currents increase with pH, on both α-Ni(OH)2 and β-Ni(OH)2. This is so, because the hydroxide ion is a reactant in both the Ni(OH)2 oxidation (reaction (IV)) and the AOR (reaction (I)). Nevertheless, the α-Ni(OH)2 maintains higher current densities than β-Ni(OH)2, in the entire pH range. This demonstrates that the advantage of α-Ni(OH)2 as an electrocatalyst is independent of the pH. Interestingly, the voltametric waves change their shapes with ammonia concentration and pH, depending on the Ni(OH)2 phases (Fig. S2 and S3, ESI†).
As ammonia is added, the CV shows: (i) a delay in the onset potential for oxidation, (ii) a rise in anodic current, especially in higher overpotentials, and (iii) a decline in the cathodic peak current in the back scan. We quantitatively analyzed these changes using three parameters (Fig. 6): Δi to mark the change in current at E = 1.535 V vs. RHE; ΔEonset to mark the delay in oxidation onset potential, relative to the ammonia-free electrolyte; and ic,NH3/ic,none (%) to mark the relative change in the cathodic back peak, when comparing electrolytes with and without NH3. These changes are reported in comparison to the redox peaks without ammonia, to exclude any differences in conductivity. The additional current at higher ammonia concentrations (Δi, Fig. 6b) is initially higher in α-Ni(OH)2 than in β-Ni(OH)2. However, when [NH3] surpasses 0.75 M, Δi plummets in both, even becoming negative in α-Ni(OH)2. The decline in Δi in β-Ni(OH)2 is smaller and only starts above 1 M NH3. These observations suggests that NH3 saturates and blocks the surface both towards the AOR (reaction (I)), and then even towards the OH− needed to oxidize Ni(OH)2 to NiOOH (reaction (VI)). This is in contrast to the UOR, where rising urea concentrations give rise to surface saturation, but no decline in the current even at 2 M urea.82 Moreover, such a delay in AOR onset on Ni(OH)2 was recently observed with rising [NH3].84
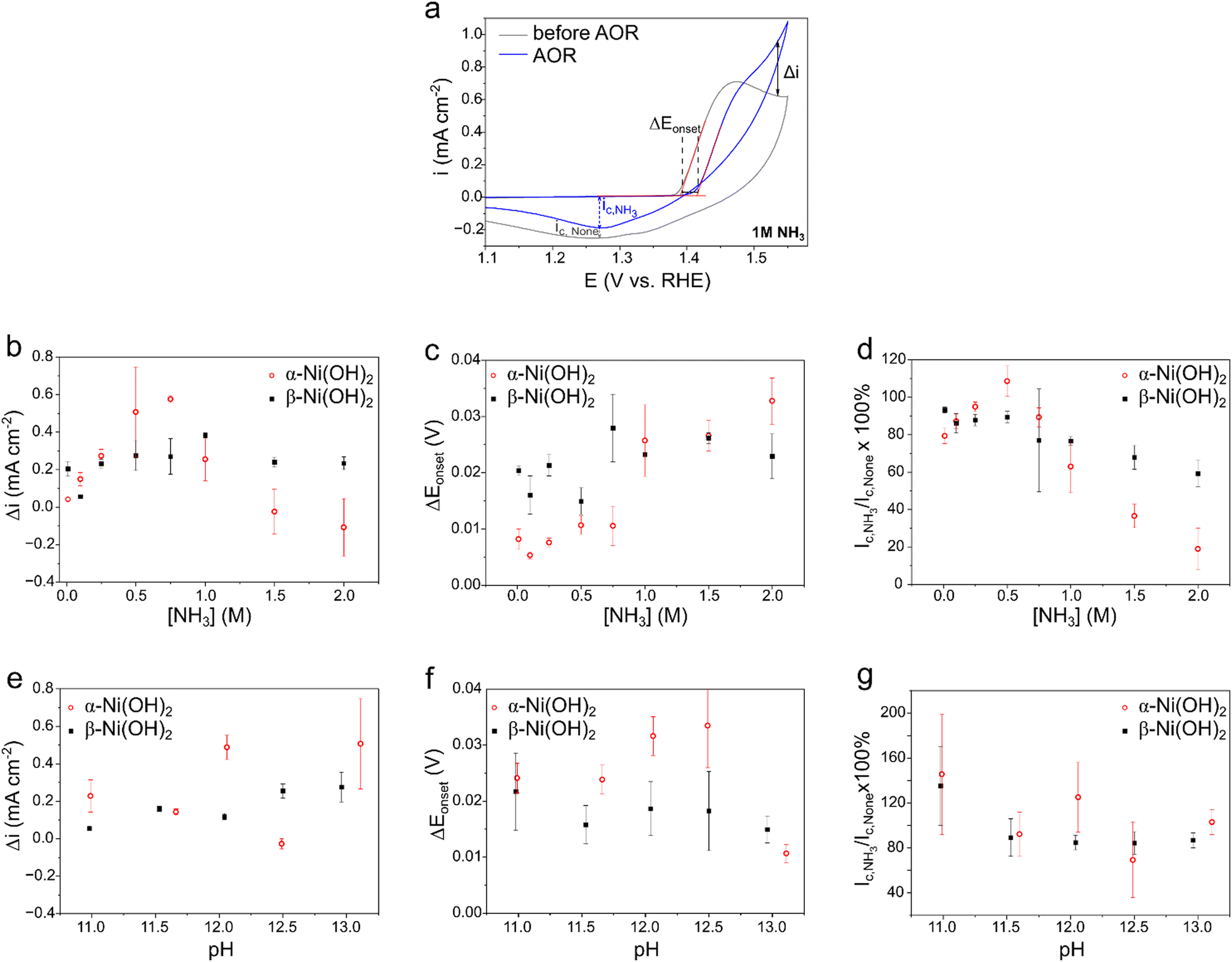 | ||
| Fig. 6 Analysis of the changes in the voltametric waves. (a) Definitions of change parameters (see text for further explanations), comparing cycle 10 without NH3 and cycle 1 with 1 M NH3, on α-Ni(OH)2, in 1 M KOH + 0.5 M K2SO4. (b)–(d) The effect of ammonia concentration on the (b) additional oxidation current at 1.535 V, (c) delay in onset potential, and (d) the relative change in back reduction current, for α-Ni(OH)2 (red circles) and β-Ni(OH)2 (blue squares). (e)–(g) The effect of pH on the same parameters. | ||
The onset potential for oxidation (as defined by the intersection of tangent lines, Fig. 6a) is delayed on both phases, even in the lowest concentrations (ΔEonset > 0, Fig. 6c). This is in contrast to other reaction systems, where the catalytic oxidation either coincides with the NiII/NiIII transition potential (like in the UOR88) or follows after it (like in OER89) – but does not affect the NiII/NiIII redox potential itself. Here, ΔEonset is quite constant on both phases in low ammonia concentrations, increasing dramatically at [NH3] > 0.75 M. The delay in onset probably arises from binding of NHx species to Ni(OH)2, stronger here than the binding of urea, for example (where the effect is not observed). In the literature, the adsorption energies of NHx species on Ni(OH)2 were calculated for the case of gaseous ammonia or urea on the (0001) facet of β-Ni(OH)2, showing comparable values around 310–340 kJ mol−1.70,90 However, calculations on the surface of α-Ni(OH)2 are challenging, due to the ill-defined structure of this phase. When the ammonia concentration is further increased, the OH− can no longer react with the Ni(OH)2 surface to oxidize it to NiOOH (reaction (VI)), thus delaying the NiII/NiIII transition.
The suppression of the transition can be further studied by analyzing the cathodic peak variation (ic,NH3/ic,none, Fig. 6d). Note that ic represents only the NiIII → NiII reduction, since the ammonia oxidation is irreversible. The value of ic,NH3/ic,none initially increases on both phases, then decreasing when [NH3] > 0.5 M. Notably, the decline in cathodic currents at higher ammonia concentrations is more steep for α-Ni(OH)2 than β-Ni(OH)2, with almost an entirely irreversible process at 2 M. The decline in ic,NH3/ic,none after [NH3] > 0.5 M indicates that there is less and less NiOOH available to be reduced, as the NH3 concentration rises. This could happen if NH3 molecules block OH− from accessing surface NiII sites, thus generating less NiOOH to begin with. Alternatively, the NiOOH could be blocked by NH3 molecules, if the AOR is slow enough, decreasing the effective surface available for the back reduction. Overall, the surface of Ni(OH)2 and/or NiOOH could be blocked by NHx species (x = 0–3), effectively poisoned similarly to poisoning of Pt by Nads during AOR electrocatalysis.62 In fact, ammonia adsorption by breaking Ni–O bonds, followed by dehydrogenation, were proposed as the first AOR steps.70
Interestingly, a decline in the cathodic current following urea oxidation on Ni(OH)2/NiOOH was previously proposed as evidence for an indirect AOR mechanism (reaction (IV)). The reason is that the chemical reduction of NiIII back to NiII by ammonia would leave less NiOOH to undergo electrochemical reduction in the back scan, resulting in a lower cathodic current.82 However, this is hardly the case here: the decline in AOR at higher [NH3] – which should revive the cathodic currents, as less NiOOH is chemically back-reduced – actually leads to lower, not higher, cathodic currents. Thus, a mechanism switch is hard to confirm here, and surface poisoning remains the most viable hypothesis. Furthermore, one must remember that the Ni(OH)2 electrodes can contain a mosaic of phases that are ever changing at the timescale of the CV experiment, complicating a possible discussion of a mechanism shift.
Since a competition between ammonia and hydroxide ions seems to play a key role in the observed trends, Δi, ΔEonset and then changes to the cathodic back-reduction peak were analyzed for α-Ni(OH)2 and β-Ni(OH)2 at different electrolyte pH (Fig. 6e, f and g). The ammonia concentration was set to 0.5 M, to maximize the difference between the phases. Interestingly, the effect of pH was found to be much weaker than that of the ammonia concentration. There is a small increase in Δi with pH; since OH− is a reactant for both the nickel hydroxide oxidation and the AOR (reactions (VI) and (I)), the current is indeed expected to rise with increasing concentration of hydroxide ions. The near-constant ΔEonset at pH > 11, suggests that the oxidation is no longer limited by [OH−], indicating that surface blocking by NH3 becomes key. Note that by defining the onset delay as the reviewed parameter, rather than the onset potential, we can focus solely on the differences originating from the NH3/OH− ratios on the pH-insensitive RHE.
The value of ic,NH3/ic,none is somewhat higher at lower pH, reaching higher than 100% values. This could be due to some electrodissolution of Ni in ammonia to form nickel ammonium complexes, which intensifies in lower pH;86,87 the reverse redeposition of Ni could be the side reduction process. However, dissolution (at open circuit) is minimal (see below). Nickel hydroxide can extract Fe traces from the electrolyte, leading to iron doping which affects its electrochemistry,74,91 including for electrocatalysis of the UOR92 and the OER.93 To rule out the effect of Fe impurities on the observed trends in AOR, we repeated the CV cycling in Fe-free solutions at several ammonia concentrations (Fig. S6 and S7, ESI†). The resulting voltammograms present the same trends as observed in Fig. 6 for Δi and ΔEonset, differing somewhat in the current densities, and showing a 30 mV earlier onset for α-Ni(OH)2. Hence, the trends in onset delays and current variations vs. [NH3] are unrelated to any iron incorporated in the Ni(OH)2.
Surface poisoning by ammonia
To investigate the poisoning of Ni(OH)2/NiOOH surfaces in high ammonia concentrations, the electrodes were either soaked or voltammetrically cycled in 2 M ammonia, and then transferred to an ammonia-free electrolyte. If the ammonia is weakly adsorbed to the surface, it should desorb (at least partially) in the NH3-free electrolyte. Then, the hydroxide ions would again be able to oxidize the surface and the nickel peak current density would recover. However, the nickel redox peaks are never fully restored after switching to the ammonia-free electrolyte, for neither α-Ni(OH)2 or β-Ni(OH)2 (Fig. 7 and Fig. S8, ESI†). Notably, the surface adsorption is stronger on α-Ni(OH)2: the currents do not recover at all, and the Ni peaks are almost eliminated. On β-Ni(OH)2, most of the current is restored after soaking in NH3 (Fig. 7b), but less is restored after CV cycling (Fig. S8, ESI†). Overall, these observations suggest that α-Ni(OH)2 binds NH3 (or a NHx intermediate) more strongly than β-Ni(OH)2, and the surface remains poisoned even in the ammonia-free electrolyte. To gauge the possibility of Ni electro-dissolution, which could be enhanced by the presence of NH3,86,87 we measured the Ni content in the electrolyte by ICP-MS following soaking in 1 mL of 2 M ammonia solution for 10 minutes, as the duration of an electrochemical measurement in ammonia. The results show that only about 1% of the α-Ni(OH)2 electrode mass has been dissolved. Therefore, the dissolution of nickel does not explain the huge decay in activity after soaking in ammonia solution, appearing at Fig. 7a. | ||
| Fig. 7 CV of nickel hydroxide in an ammonia free solution before (10th cycle, in black) and after (2nd cycle, blue) soaking the electrode in 2 M NH3 solution for 10 minutes (same as the duration of an AOR CV experiment) for (a) α-Ni(OH)2 and (b) β-Ni(OH)2. | ||
The difference in NH3 adsorption energy between α-Ni(OH)2 and β-Ni(OH)2 corroborates the trends in AOR electrocatalysis (Fig. 8). At low NH3 concentrations, the AOR currents are higher on α-Ni(OH)2 since it adsorbs NH3 more strongly, promoting dehydrogenation steps. Meanwhile, at high NH3 concentrations, strongly adsorbed ammonia poisons the sites and decreases the local pH, thus causing large onset delays and the drop in peak current. Furthermore, the fact that NH3 adsorbs strongly on Ni(OH)2 explains the delay in the onset potential for oxidation to NiOOH, which was first observed here. To the best of our knowledge, this is the first time that irreversible poisoning of α-Ni(OH)2 by ammonia is reported. The weaker adsorption to β-Ni(OH)2 explains the lower currents and the low dependance on ammonia concentrations for the trends of Δi and ΔE in Fig. 6. Moreover, reexamining the RRDE experiment in Fig. 4, we notice that the AOR currents almost disappear at high ammonia concentrations, despite the rotation eliminating mass transfer limitations. Then, at very high potentials (E > 1.75 V vs. RHE), the current increases, while O2 is not produced, according to the ring currents. Possibly, the strongly adsorbed NHx species are getting released by forming NOx species, similarly to Nads on Pt.62,94
 | ||
| Fig. 8 Schematic of the AOR on Ni(OH)2/NiOOH, comparing the α/γ vs. the β/β couple as a function of ammonia concentration. Ammonia binds more strongly to α-Ni(OH)2, leading to better electrocatalysis at intermediate [NH3], and to irreversible poisoning at high [NH3]. | ||
Interestingly, the poisoning related phenomena are more pronounced for α-Ni(OH)2 than for β-Ni(OH)2, i.e. the current decline and the onset delay at ammonia saturation. This further suggests a stronger adsorption energy of ammonia for the surface of α-Ni(OH)2 than β-Ni(OH)2. This is supported by how the currents can be recovered on β-Ni(OH)2 in the desorption experiments (Fig. 7), while α-Ni(OH)2 remains blocked.
Could the different responses of α-Ni(OH)2 and β-Ni(OH)2 to surface poisoning arise from different electrochemical surface areas (ECSAs)? Unfortunately, the ECSA of nickel hydroxide phases is hard to determine, due to interfering contributions from intercalation, overcharging, and pseudocapacitance.75 Herein, we measured the ECSA by two methods: (1) double layer capacitance measured by CV in the non-faradaic region, and (2) electrochemical impedance spectroscopy (EIS), as proposed by Watzele et al.76 (Fig. S9, ESI†). In the EIS method, the adsorption capacitance (Ca) of oxygenated intermediates, formed at the onset of the OER, is extracted from EIS spectra by fitting them to an equivalent circuit (Fig. S9, ESI†). At potentials where these species reach full surface coverage, this value reaches a plateau, and can be correlated to the electrochemically active surface area. The double-layer capacitance values measured by the first method were similar for the two phases: 6 μF for α-Ni(OH)2 and 7 μF for β-Ni(OH)2. Typically, ECSA values are calculated from the double-layer capacitance by dividing it by the specific capacitance of the material. However, this value is also debated in the literature, with reported values ranging from 40 to 300 μF cm−2.95,96 Therefore, we can examine the capacitance values comparatively. The EIS method yielded capacitance values of 80 μF for α-Ni(OH)2 and 9 μF for β-Ni(OH)2 (Fig. S9, ESI†). Each method has a priori limitations: double-layer capacitance by CV may include processes such as intercalation, adding non-faradaic currents, while the new EIS approach shows inconsistent dependency of Ca on potential, similarly to what we observe (Fig. S9, ESI†). Regardless if the ECSA of α-Ni(OH)2 is equal or even 10 times higher than that of β-Ni(OH)2, it is clear that it is not lower, and thus the α-Ni(OH)2 poisoning effect is not related to surface area limitations, but rather due to stronger adsorption.
Conclusions
The two phases of nickel hydroxide, disordered α-Ni(OH)2 and crystalline β-Ni(OH)2, were synthesized, characterized and comparatively evaluated for their electrocatalytic performance towards the ammonia oxidation reaction. Both γ-NiOOH and β-NiOOH (resulting from oxidation of α-Ni(OH)2 and β-Ni(OH)2, respectively) can catalyze the AOR. Both yield N2, as determined by differential electrochemical mass spectrometry. The α-Ni(OH)2 electrocatalyst is more reactive towards ammonia oxidation, throughout most of the concentration range between 0.01 M and 2 M. Its catalytic activity drops at high ammonia concentrations, with lower currents and belated onset potentials when [NH3] > 1 M. Soaking experiments confirm a surface poisoning effect, where NH3 and/or other NHx intermediates adsorb to the Ni(OH)2 surface, blocking access for the OH− ions required for the Ni(OH)2 → NiOOH oxidation and for the AOR. The surface poisoning is completely irreversible for α-Ni(OH)2, and partially reversible on β-Ni(OH)2, suggesting stronger adsorption energies on the disordered, intercalating α-Ni(OH)2 phase. Overall, our findings demonstrate the key role of the phases of the Ni(OH)2/NiOOH electrocatalysts on ammonia oxidation, as well as the strong effects of poisoning and competition with hydroxide-consuming reactions. We hope that these findings will broaden our understanding of electrocatalysis of nitrogen-based fuels, and of metal oxide electrocatalysts in general.Author contributions
I. O. P. performed the electrochemical experiments (with H. A. A. and A. H.), the DEMS experiments (with T. K. S.) and all characterizations. I. O. P. wrote the original draft. D. E. coordinated the project and edited the manuscript.Data availability
Additional electrochemical measurements and analysis have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Israel Ministry of Science and Technology (grant 1001556424) and the Israel Ministry of Energy (grant 222-11-059) for partial funding, and Dr Thomas Hartman for graphic design. I. O. P. thanks the Israeli Smart Transportation Research Center (ISTRC) for a graduate fellowship. T. K. S. thanks the Azrieli Foundation and the Israel Academy of Science and Humanities for post-doctoral fellowships.Notes and references
- X. Ding, Y. Ji, H. Huang, J. Huang, S. Chen, C. Yang, F. Li and M. Luo, Electrocatalysis of the ammonia oxidation reaction, Chem. Catal., 2024, 4(6), 100932 CrossRef CAS.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, A Roadmap to the Ammonia Economy, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- Y. Zhao, B. P. Setzler, J. Wang, J. Nash, T. Wang, B. Xu and Y. Yan, An Efficient Direct Ammonia Fuel Cell for Affordable Carbon-Neutral Transportation, Joule, 2019, 3, 2472–2484 CrossRef CAS.
- L. Jiang and X. Fu, An Ammonia-Hydrogen Energy Roadmap for Carbon Neutrality: Opportunity and Challenges in China, Engineering, 2021, 7(12), 1688–1691 CrossRef.
- A. Boretti, Reviewing the progress toward an ammonia energy storage ecosystem, Int. J. Hydrogen Energy, 2024, 63, 472–479 CrossRef CAS.
- B. A. L. de Mishima, D. Lescano, T. M. Holgado and H. T. Mishima, Electrochemical oxidation of ammonia in alkaline solutions: its application to an amperometric sensor, Electrochim. Acta, 1998, 43, 395–404 CrossRef.
- N. Mayo, R. Harth, U. Mor, D. Marouani, J. Hayon and A. Bettelheim, Electrochemical response to H2, O2, CO2 and NH3 of a solid-state cell based on a cation- or anion-exchange membrane serving as a solid polymer electrolyte, Anal. Chim. Acta, 1995, 310, 139–144 CrossRef CAS.
- B. Timmer, W. Olthuis and A. van den Berg, Ammonia sensors and their applications—a review, Sens. Actuators, B, 2005, 107, 666–677 CrossRef CAS.
- C. Zhong, W. B. Hu and Y. F. Cheng, Recent advances in electrocatalysts for electro-oxidation of ammonia, J. Mater. Chem. A, 2013, 1, 3216–3238 RSC.
- D. K. Bora and A. Faik, Direct Ammonia Fuel Cells for Power Generation: Recent Technological Advances and Mobility Applications, Curr. Opin. Green Sustainable Chem., 2024, 100944 CrossRef.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, Review—Ammonia Oxidation Electrocatalysis for Hydrogen Generation and Fuel Cells, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- H. Shi, J. Tang, W. Yu, M. O. Tadé and Z. Shao, Advances in power generation from ammonia via electrocatalytic oxidation in direct ammonia fuel cells, Chem. Eng. J., 2024, 488, 150896 CrossRef CAS.
- F. Vitse, M. Cooper and G. G. Botte, On the use of ammonia electrolysis for hydrogen production, J. Power Sources, 2005, 142, 18–26 CrossRef CAS.
- H. Liu, X. Xu, D. Guan and Z. Shao, Minireview on the Electrocatalytic Ammonia Oxidation Reaction for Hydrogen Production and Sewage Treatment, Energy Fuels, 2024, 38, 919–931 CrossRef CAS.
- Y. Feng, L. Huang, Z. Xiao, X. Zhuang, T. S. Aslam, X. Zhang, Y.-X. Tan and Y. Wang, Temporally Decoupled Ammonia Splitting by a Zn–NH3 Battery with an Ammonia Oxidation/Hydrogen Evolution Bifunctional Electrocatalyst as a Cathode, J. Am. Chem. Soc., 2024, 146, 7771–7778 CrossRef CAS PubMed.
- P. Babar and G. G. Botte, Recent Advances in Ammonia Electrolysis for Sustainable Hydrogen Generation, ACS Sustainable Chem. Eng., 2024, 12(35), 13030–13047 CrossRef CAS.
- G. Jeerh, M. Zhang and S. Tao, Recent progress in ammonia fuel cells and their potential applications, J. Mater. Chem. A, 2021, 9, 727–752 RSC.
- K. Siddharth, Y. Chan, L. Wang and M. Shao, Ammonia electro-oxidation reaction: Recent development in mechanistic understanding and electrocatalyst design, Curr. Opin. Electrochem., 2018, 9, 151–157 CrossRef CAS.
- S. I. Venturini, D. R. Martins de Godoi and J. Perez, Challenges in Electrocatalysis of Ammonia Oxidation on Platinum Surfaces: Discovering Reaction Pathways, ACS Catal., 2023, 13, 10835–10845 CrossRef CAS.
- V. Rosca and M. T. M. Koper, Electrocatalytic oxidation of ammonia on Pt(111) and Pt(100) surfaces, Phys. Chem. Chem. Phys., 2006, 8, 2513–2524 RSC.
- Z. Gong, H. Wang, C. Li, Q. Sang, Y. Xie, X. Zhang and Y. Liu, Progress in the design and performance evaluation of catalysts for low-temperature direct ammonia fuel cells, Green Chem. Eng., 2025, 6(1), 54–67 CrossRef.
- G. G. Botte, Electrolysis of ammonia towards sustainability, Bol. Acad. Cienc. Fis., Mat. Nat., 2024, 84(1), 1–22 Search PubMed.
- H. Kim, S. Hong, H. Kim, Y. Jun, S. Y. Kim and S. H. Ahn, Recent progress in Pt-based electrocatalysts for ammonia oxidation reaction, Appl. Mater. Today, 2022, 29, 101640 CrossRef.
- H. Gerischer and A. Mauerer, Untersuchungen Zur anodischen Oxidation von Ammoniak an Platin-Elektroden, J. Electroanal. Chem. Interfacial Electrochem., 1970, 25, 421–433 CrossRef CAS.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, The role of adsorbates in the electrochemical oxidation of ammonia on noble and transition metal electrodes, J. Electroanal. Chem., 2001, 506, 127–137 CrossRef CAS.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Nitrogen Cycle Electrocatalysis, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- B. Achrai, Y. Zhao, T. Wang, G. Tamir, R. Abbasi, B. P. Setzler, M. Page, Y. Yan and S. Gottesfeld, A Direct Ammonia Fuel Cell with a KOH-Free Anode Feed Generating 180 mW cm−2 at 120 °C, J. Electrochem. Soc., 2020, 167, 134518 CrossRef CAS.
- S. Johnston, L. Kemp, B. Turay, A. N. Simonov, B. H. R. Suryanto and D. R. MacFarlane, Copper-Catalyzed Electrosynthesis of Nitrite and Nitrate from Ammonia: Tuning the Selectivity via an Interplay Between Homogeneous and Heterogeneous Catalysis, ChemSusChem, 2021, 14, 4793–4801 CrossRef CAS PubMed.
- S. Johnston, S. Cohen, C. K. Nguyen, K. N. Dinh, T. D. Nguyen, S. Giddey, C. Munnings, A. N. Simonov and D. R. MacFarlane, A Survey of Catalytic Materials for Ammonia Electrooxidation to Nitrite and Nitrate, ChemSusChem, 2022, 15, e202200614 CrossRef CAS PubMed.
- X. Jiang, D. Ying, X. Liu, M. Liu, S. Zhou, C. Guo, G. Zhao, Y. Wang and J. Jia, Identification of the role of Cu site in Ni–Cu hydroxide for robust and high selective electrochemical ammonia oxidation to nitrite, Electrochim. Acta, 2020, 345, 136157 CrossRef CAS.
- A. Cleetus, H. Teller and A. Schechter, CuCr Bimetallic Catalyst for Selective Electrooxidation of Ammonia at Room Temperature, ChemCatChem, 2023, 15, e202300035 CrossRef CAS.
- T. M. Vu, S. Johnston, D. Simondson, C. K. Nguyen, T. D. Nguyen, D. V. Zeil, R. K. Hocking, D. R. Macfarlane and A. N. Simonov, High-Rate, High-Selectivity Electrochemical Oxidation of Ammonia to Nitrite with a Silver-Based Catalyst, ACS Catal., 2024, 10974–10986 CrossRef CAS.
- D. J. Little, D. O. Edwards, M. R. I. Smith and T. W. Hamann, As Precious as Platinum: Iron Nitride for Electrocatalytic Oxidation of Liquid Ammonia, ACS Appl. Mater. Interfaces, 2017, 9, 16228–16235 CrossRef CAS PubMed.
- Y.-J. Shih and C.-H. Hsu, Kinetics and highly selective N2 conversion of direct electrochemical ammonia oxidation in an undivided cell using NiCo oxide nanoparticle as the anode and metallic Cu/Ni foam as the cathode, Chem. Eng. J., 2021, 409, 128024 CrossRef CAS.
- J. Łuczak and M. Lieder, Nickel-based catalysts for electrolytic decomposition of ammonia towards hydrogen production, Adv. Colloid Interface Sci., 2023, 319, 102963 CrossRef PubMed.
- Z. Mao, Y. Tian, B. Guo, R. Chen, Y. Zeng, F. Hou, X. Yan and J. Liang, Modulation of charge distribution enabling CuNi nano-alloys for efficient ammonia oxidation reaction to nitrite production, Chem. Eng. J., 2024, 484, 149570 CrossRef CAS.
- S. Cohen, S. Johnston, C. K. Nguyen, T. D. Nguyen, D. A. Hoogeveen, D. V. Zeil, S. Giddey, A. N. Simonov and D. R. MacFarlane, A CoOxHy/β-NiOOH electrocatalyst for robust ammonia oxidation to nitrite and nitrate, Green Chem., 2023, 25, 7157–7165 RSC.
- W. Xu, D. Du, R. Lan, J. Humphreys, D. N. Miller, M. Walker, Z. Wu, J. T. S. Irvine and S. Tao, Electrodeposited NiCu bimetal on carbon paper as stable non-noble anode for efficient electrooxidation of ammonia, Appl. Catal., B, 2018, 237, 1101–1109 CrossRef CAS.
- H. Zhang, Y. Wang, Z. Wu and D. Y. C. Leung, An ammonia electrolytic cell with NiCu/C as anode catalyst for hydrogen production, Energy Proc., 2017, 142, 1539–1544 CrossRef CAS.
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and M. Tawalbeh, Electrochemical oxidation of ammonia on nickel oxide nanoparticles, Int. J. Hydrogen Energy, 2020, 45, 10398–10408 CrossRef CAS.
- J. Huang, J. Cai and J. Wang, Nanostructured Wire-in-Plate Electrocatalyst for High-Durability Production of Hydrogen and Nitrogen from Alkaline Ammonia Solution, ACS Appl. Energy Mater., 2020, 3, 4108–4113 CrossRef CAS.
- S. Kitano, M. L. Ooi, T. Yamamoto, S. Matsumura and M. Yamauchi, Catalytic Roles and Synergetic Effects of Iron-Group Elements on Monometals and Alloys for Electrochemical Oxidation of Ammonia, Bull. Chem. Soc. Jpn., 2021, 94, 1292–1299 CrossRef CAS.
- M. Zhu, Y. Yang, S. Xi, C. Diao, Z. Yu, W. S. V. Lee and J. Xue, Deciphering NH3 Adsorption Kinetics in Ternary Ni–Cu–Fe Oxyhydroxide toward Efficient Ammonia Oxidation Reaction, Small, 2021, 17, 2005616 CrossRef CAS PubMed.
- M. Gonzalez-Reyna, M. S. Luna-Martínez and J. F. Perez-Robles, Nickel supported on carbon nanotubes and carbon nanospheres for ammonia oxidation reaction, Nanotechnology, 2020, 31, 235706 CrossRef CAS PubMed.
- P. D. Tanwade, A. V. Munde, B. B. Mulik, A. Adhikari, R. Patel and B. R. Sathe, NiO-Nanoparticle-Embedded Polyaniline for Enhanced Ammonia and Water Oxidation Reactions, Energy Fuels, 2023, 37, 19959–19970 CrossRef CAS.
- H. K. Vu, T. Mahvelati-Shamsabadi, T. T. Dang, S. H. Hur, S. G. Kang and J. S. Chung, Synergistic Effects of Ni and Cu in Morphology-Controlled NiCu Electrocatalysts for Ammonia Electro-oxidation, ACS Appl. Nano Mater., 2023, 6(22), 20688–20699 CrossRef CAS.
- W. Xu, R. Lan, D. Du, J. Humphreys, M. Walker, Z. Wu, H. Wang and S. Tao, Directly growing hierarchical nickel-copper hydroxide nanowires on carbon fibre cloth for efficient electrooxidation of ammonia, Appl. Catal., B, 2017, 218, 470–479 CrossRef CAS.
- E. W. Jungner, Primärt eller sekundärt elektriskt element, Swedish Pat., 10177, 1899 Search PubMed.
- T. A. Edison, US Pat., US701804A, 1902 Search PubMed.
- S. Arya and S. Verma, Rechargeable Batteries, John Wiley & Sons, Ltd, 2020, pp. 131–175 Search PubMed.
- C. Jeyaseelan, A. Jain, P. Khurana, D. Kumar and S. Thatai, Rechargeable Batteries, John Wiley & Sons, Ltd, 2020, pp. 177–194 Search PubMed.
- L. Zhang, D. Shi, T. Liu, M. Jaroniec and J. Yu, Nickel-based materials for supercapacitors, Mater. Today, 2019, 25, 35–65 CrossRef CAS.
- M. E. G. Lyons, R. L. Doyle, I. Godwin, M. O’Brien and L. Russell, Hydrous Nickel Oxide: Redox Switching and the Oxygen Evolution Reaction in Aqueous Alkaline Solution, J. Electrochem. Soc., 2012, 159, H932–H944 CrossRef CAS.
- A. Delahaye-Vidal and M. Figlarz, Textural and structural studies on nickel hydroxide electrodes. II. Turbostratic nickel(II) hydroxide submitted to electrochemical redox cycling, J. Appl. Electrochem., 1987, 17, 589–599 CrossRef CAS.
- P. Oliva, J. Leonardi, J. F. Laurent, C. Delmas, J. J. Braconnier, M. Figlarz, F. Fievet and A. de Guibert, Review of the structure and the electrochemistry of nickel hydroxides and oxy-hydroxides, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Nickel hydroxides and related materials: A review of their structures, synthesis and properties, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- S. Fujita, S. Baranton, C. Coutanceau and G. Jerkiewicz, Design, Synthesis, and Characterization of Carbon-Supported β-Ni(OH)2 Nanosheets for Miniaturized Nickel–Metal Hydride Batteries, Energy Technol., 2024, 12, 2301268 CrossRef CAS.
- S. Deabate, F. Henn, S. Devautour and J. C. Giuntini, Conductivity and Dielectric Relaxation in Various Ni(OH)2 Samples, J. Electrochem. Soc., 2003, 150, J23 CrossRef CAS.
- S. Chakrabarty, I. Offen-Polak, T. Y. Burshtein, E. M. Farber, L. Kornblum and D. Eisenberg, Urea oxidation electrocatalysis on nickel hydroxide: the role of disorder, J. Solid State Electrochem., 2021, 25, 159–171 CrossRef CAS.
- Y. Chen, K. Rui, J. Zhu, S. X. Dou and W. Sun, Recent Progress on Nickel-Based Oxide/(Oxy)Hydroxide Electrocatalysts for the Oxygen Evolution Reaction, Chem. – Eur. J., 2019, 25, 703–713 CrossRef CAS PubMed.
- X. Yu, J. Zhao, L.-R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, Hydrogen Evolution Reaction in Alkaline Media: Alpha- or Beta-Nickel Hydroxide on the Surface of Platinum?, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- I. Katsounaros, M. C. Figueiredo, F. Calle-Vallejo, H. Li, A. A. Gewirth, N. M. Markovic and M. T. M. Koper, On the mechanism of the electrochemical conversion of ammonia to dinitrogen on Pt(100) in alkaline environment, J. Catal., 2018, 359, 82–91 CrossRef CAS.
- I. Katsounaros, T. Chen, A. A. Gewirth, N. M. Markovic and M. T. M. Koper, Evidence for Decoupled Electron and Proton Transfer in the Electrochemical Oxidation of Ammonia on Pt(100), J. Phys. Chem. Lett., 2016, 7, 387–392 CrossRef CAS PubMed.
- H. Wang, D. R. Dekel and H. D. Abruña, Unraveling the Mechanism of Ammonia Electrooxidation by Coupled Differential Electrochemical Mass Spectrometry and Surface-Enhanced Infrared Absorption Spectroscopic Studies, J. Am. Chem. Soc., 2024, 146, 15926–15940 CrossRef CAS PubMed.
- K. Endo, Y. Katayama and T. Miura, A rotating disk electrode study on the ammonia oxidation, Electrochim. Acta, 2005, 50, 2181–2185 CrossRef CAS.
- A. Kapałka, A. Cally, S. Neodo, C. Comninellis, M. Wächter and K. M. Udert, Electrochemical behavior of ammonia at Ni/Ni(OH)2 electrode, Electrochem. Commun., 2010, 12, 18–21 CrossRef.
- R. Wang, H. Liu, K. Zhang, G. Zhang, H. Lan and J. Qu, Ni(II)/Ni(III) redox couple endows Ni foam-supported Ni2P with excellent capability for direct ammonia oxidation, Chem. Eng. J., 2021, 404, 126795 CrossRef CAS.
- L. Wang, K. Jiang, Z. Wang, T. Li, D. Wang and Y.-Q. Liu, Enabling surface reconstruction through a heterostructured Ni3S4@NiCo2O4/NF towards efficient ammonia oxidation reaction, Chem. Eng. J., 2024, 492, 152268 CrossRef CAS.
- A. Ashok Kashale, C.-T. Wu, H.-F. Hsu and I.-W. Peter Chen, In Situ monitoring intermediate stages in ammonia oxidation reaction via high performance NiCuBOx−1/NF electrocatalysts, Chem. Eng. J., 2023, 474, 145907 CrossRef CAS.
- J. J. Medvedev, Y. Tobolovskaya, X. V. Medvedeva, S. W. Tatarchuk, F. Li and A. Klinkova, Pathways of ammonia electrooxidation on nickel hydroxide anodes and an alternative route towards recycled fertilizers, Green Chem., 2022, 24, 1578–1589 RSC.
- A. Allagui, S. Sarfraz and E. A. Baranova, NixPd1−x (x = 0.98, 0.93, and 0.58) nanostructured catalysts for ammonia electrooxidation in alkaline media, Electrochim. Acta, 2013, 110, 253–259 CrossRef CAS.
- A. Estejab, D. A. Daramola and G. G. Botte, Mathematical model of a parallel plate ammonia electrolyzer for combined wastewater remediation and hydrogen production, Water Res., 2015, 77, 133–145 CrossRef CAS PubMed.
- G. G. Bottle and C. A. Feickert, Electro Decomposition of Ammonia into Hydrogen for Fuel Cell Use, Defense Technical Information Center, Fort Belvoir, VA, 2012 Search PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. D. Obradović and S. L. Gojković, Challenges in determining the electrochemically active surface area of Ni-oxides in the oxygen evolution reaction, J. Electroanal. Chem., 2022, 918, 116479 CrossRef.
- S. Watzele, P. Hauenstein, Y. Liang, S. Xue, J. Fichtner, B. Garlyyev, D. Scieszka, F. Claudel, F. Maillard and A. S. Bandarenka, Determination of Electroactive Surface Area of Ni-, Co-, Fe-, and Ir-Based Oxide Electrocatalysts, ACS Catal., 2019, 9, 9222–9230 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, Applications of in Situ Raman Spectroscopy for Identifying Nickel Hydroxide Materials and Surface Layers during Chemical Aging, ACS Appl. Mater. Interfaces, 2014, 6, 3141–3149 CrossRef CAS PubMed.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, Raman and infrared spectroscopy of α and β phases of thin nickel hydroxide films electrochemically formed on nickel, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- R. L. Frost, M. L. Weier and J. T. Kloprogge, Raman spectroscopy of some natural hydrotalcites with sulphate and carbonate in the interlayer, J. Raman Spectrosc., 2003, 34, 760–768 CrossRef CAS.
- C. Liu and Y. Li, Synthesis and characterization of amorphous α-nickel hydroxide, J. Alloys Compd., 2009, 478, 415–418 CrossRef CAS.
- P. V. Kamath and G. N. Subbanna, Electroless nickel hydroxide: synthesis and characterization, J. Appl. Electrochem., 1992, 22, 478–482 CrossRef CAS.
- V. Vedharathinam and G. G. Botte, Understanding the electro-catalytic oxidation mechanism of urea on nickel electrodes in alkaline medium, Electrochim. Acta, 2012, 81, 292–300 CrossRef CAS.
- G. A. Tsirlina, Organic fuels oxidation: a common misunderstanding related to non-noble fuel cell catalysts, Curr. Opin. Electrochem., 2024, 101567 CrossRef CAS.
- M. K. Adak, H. K. Basak, S. Kumar and B. Chakraborty, Detection of a Nickel-Oxide Nanolayer During Mild Acid Treatment of Nickel Foam and Its Effect on Alkaline Oxygen Evolution and Ammonia Oxidation Reactions, ACS Appl. Nano Mater., 2024, 7, 8329–8340 CrossRef CAS.
- K. Emerson, R. C. Russo, R. E. Lund and R. V. Thurston, Aqueous Ammonia Equilibrium Calculations: Effect of pH and Temperature, J. Fish. Res. Bd. Can., 1975, 32, 2379–2383 CrossRef CAS.
- M.-H. Tsai, T.-C. Chen, Y. Juang, L.-C. Hua and C. Huang, High catalytic performance of CuCo/nickel foam electrode for ammonia electrooxidation, Electrochem. Commun., 2020, 121, 106875 CrossRef CAS.
- E. Asselin, Thermochemistry of the Fe, Ni and Co-NH3-H2O systems as they relate to the Caron process: a review, Min., Metall., Explor., 2011, 28, 169–175 CAS.
- B. K. Boggs, R. L. King and G. G. Botte, Urea electrolysis: direct hydrogen production from urine, Chem. Commun., 2009, 4859–4861 RSC.
- G. A. Snook, N. W. Duffy and A. G. Pandolfo, Detection of Oxygen Evolution from Nickel Hydroxide Electrodes Using Scanning Electrochemical Microscopy, J. Electrochem. Soc., 2008, 155, A262 CrossRef CAS.
- S. W. Tatarchuk, R. M. Choueiri, A. J. MacKay, S. J. Johnston, W. M. Cooper, K. S. Snyder, J. J. Medvedev, A. Klinkova and L. D. Chen, Understanding the Mechanism of Urea Oxidation from First-Principles Calculations, ChemPhysChem, 2024, 25, e202300889 CrossRef CAS PubMed.
- Y. J. Son, S. Kim, V. Leung, K. Kawashima, J. Noh, K. Kim, R. A. Marquez, O. A. Carrasco-Jaim, L. A. Smith, H. Celio, D. J. Milliron, B. A. Korgel and C. B. Mullins, Effects of Electrochemical Conditioning on Nickel-Based Oxygen Evolution Electrocatalysts, ACS Catal., 2022, 12, 10384–10399 CrossRef CAS.
- V. M. Zemtsova, A. G. Oshchepkov and E. R. Savinova, Unveiling the Role of Iron in the Nickel-Catalyzed Urea Oxidation Reaction, ACS Catal., 2023, 13, 13466–13473 CrossRef CAS.
- R. A. Marquez, E. Kalokowski, M. Espinosa, J. T. Bender, Y. Jun Son, K. Kawashima, C. E. Chukwuneke, L. A. Smith, H. Celio, A. Dolocan, X. Zhan, N. Miller, D. J. Milliron, J. Resasco and C. Buddie Mullins, Transition metal incorporation: electrochemical, structure, and chemical composition effects on nickel oxyhydroxide oxygen-evolution electrocatalysts, Energy Environ. Sci., 2024, 17, 2028–2045 RSC.
- Y.-J. Shih, Y.-H. Huang and C. P. Huang, Electrocatalytic ammonia oxidation over a nickel foam electrode: Role of Ni(OH)2(s)–NiOOH(s) nanocatalysts, Electrochim. Acta, 2018, 263, 261–271 CrossRef CAS.
- A. G. Oshchepkov, G. Braesch, A. Bonnefont, E. R. Savinova and M. Chatenet, Recent Advances in the Understanding of Nickel-Based Catalysts for the Oxidation of Hydrogen-Containing Fuels in Alkaline Media, ACS Catal., 2020, 10, 7043–7068 CrossRef CAS.
- S. S. Jeon, P. W. Kang, M. Klingenhof, H. Lee, F. Dionigi and P. Strasser, Active Surface Area and Intrinsic Catalytic Oxygen Evolution Reactivity of NiFe LDH at Reactive Electrode Potentials Using Capacitances, ACS Catal., 2023, 13, 1186–1196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrochemical measurements and analysis. See DOI: https://doi.org/10.1039/d4cp02950j |
| This journal is © the Owner Societies 2025 |