
Synthesis and biological evaluation of novel D-ring fused steroidal N(2)-substituted-1,2,3-triazoles†
Branislava
Tenjović
 a,
Sofija
Bekić
*a,
Andjelka
Ćelić
b,
Edward
Petri
b,
Julia
Scholda
c,
Florian
Kopp
*c,
Marija
Sakač
a and
Andrea
Nikolić
a
a,
Sofija
Bekić
*a,
Andjelka
Ćelić
b,
Edward
Petri
b,
Julia
Scholda
c,
Florian
Kopp
*c,
Marija
Sakač
a and
Andrea
Nikolić
a
aDepartment of Chemistry, Biochemistry and Environmental Protection, Faculty of Sciences, University of Novi Sad, Trg Dositeja Obradovića 3, 21000 Novi Sad, Serbia. E-mail: sofija.bekic@dh.uns.ac.rs
bDepartment of Biology and Ecology, Faculty of Sciences, University of Novi Sad, Trg Dositeja Obradovića 2, 21000 Novi Sad, Serbia
cFaculty of Life Sciences, Department of Pharmaceutical Sciences, Clinical Pharmacy Group, University of Vienna, Josef-Holaubek-Platz 2, 1090, Vienna, Austria. E-mail: florian.kopp@univie.ac.at
First published on 18th October 2024
Abstract
In this study, a series of 13 new D-ring fused steroidal N(2)-substituted-1,2,3-triazoles were synthesized, characterized and evaluated for their biological activities. The relative binding affinities of the synthesized compounds for the ligand-binding domains of estrogen receptors α and β, androgen receptor and glucocorticoid receptor demonstrated that androstane derivatives 3a and 3h and estratriene derivative 4e showed highly specific and strong binding affinity for estrogen receptor β, while 3b, 3e, 4a and 4b displayed high binding affinity for the glucocorticoid receptor. The synthesized compounds were tested for their ability to inhibit aldo–keto reductases 1C3 and 1C4 in vitro by monitoring NADPH consumption using fluorescence spectroscopy. The most potent aldo–keto reductase 1C3 inhibitors were compounds 3h (71.17%) and 3f (69.9%). Moreover, a molecular docking study was carried out for compounds 3f and 3h against aldo–keto reductase 1C3 and results showed that compounds 3h and 3f could bind in the same site and orientation as EM1404. However, polar atoms in the triazole group enable additional hydrogen bonding deeper in SP1 with Tyr319, Tyr216 and the NADP+ cofactor, which are not visible in the AKR1C3-EM1404 crystal structure. The synthesized compounds were screened for their anticancer activity against four cancer cell lines. Compound 3f demonstrated moderate toxic effects across various cancer types, while displaying lower toxicity towards the healthy cell line. In summary, our findings indicate that N(2)-substituted-1,2,3-triazoles are high-affinity ligands for estrogen receptor β and glucocorticoid receptor, inhibitors of aldo–keto reductase 1C3 enzyme, and exhibit antiproliferative effects against cancer cells, suggesting that they could serve as scaffolds for anticancer drug development.
1. Introduction
Cancer is a leading cause of death globally, and the number of cancer patients is constantly increasing, exerting tremendous health, social and economic problems. Hormone-dependent cancers, such as breast and prostate cancer, are associated with the abnormal activities and/or expression of enzymes and receptors that participate in androgen or estrogen biosynthesis and signaling.1–4 Design of modified steroid hormones capable of modulating these targets has been shown to be a promising research strategy in the field of hormone-dependent cancer treatment. For example, steroidal anticancer agents such as abiraterone, dutasteride and exemestane have been clinically approved for cancer therapy.5–7 However, despite advances in the diagnosis, prevention and treatment of cancers, the need for safer, more effective anticancer drugs remains a challenge for medicinal chemists.Numerous reports in medicinal chemistry have focused on nitrogen-containing heterocyclic derivatives.8–13 These modifications enable the formation of non-covalent interactions, such as hydrogen bonding, π–π interactions, ion–dipole interactions, hydrophobic interactions, and van der Waals forces with various receptors and enzymes, and are capable of providing a wide spectrum of pharmacological properties, such as anticancer,14,15 antimalarial,16 antioxidant,17,18 antimicrobial,19 antiviral,20,21 antibacterial,22,23 anti-inflammatory,24,25 and antitubercular activities.26 As evidence of their pharmacological and medicinal significance, triazoles are present in many lead structures for the discovery of new drugs.27,28 The general procedure for obtaining 1,2,3-triazoles includes the reaction of azides with activated alkenes, alkynes, and carbonyl compounds, resulting in N(1)-substituted triazole derivatives.29,30N-Substituted triazoles can be obtained by reaction of NH-triazoles with electrophiles, for example by alkylation, arylation and acylation. All three nitrogen atoms in triazoles can participate in electrophilic substitution. However, due to greater thermodynamic stability and steric hindrance because of the presence of substituents on the C4 and C5 atoms, the probability of obtaining N(2)-substituted triazoles is higher than N(1)- and N(3)-substituted derivatives.31–33
Two common strategies for treatment of hormone-dependent cancers are inhibition of steroidogenic enzymes involved in hormone biosynthesis and development of hormone receptor antagonists that compete with endogenous steroid hormones and block their action.34 In the present study we focus on steroid-converting aldo–keto reductases (AKRs) and steroid receptors, both targets for anticancer therapeutics. AKRs are involved in the biosynthesis and metabolism of steroids by catalyzing NADPH-dependent reduction of endogenous and xenobiotic carbonyl-containing substrates.35 AKRs are overexpressed in hormone-related cancers and leukemias, and AKR enzymatic activity has been implicated in the development of resistance to chemotherapeutic drugs.36 Moreover, inhibition of AKR1C enzymes is believed to restore the chemosensitivity of cancer cells.37 Thus, aldo–keto reductase 1C3 (AKR1C3) has been proposed as a drug target for the treatment of hormone-dependent malignancies.
Prostate and breast cancers often express high levels of androgen receptor or estrogen receptor α, as well as androgen- or estrogen-regulated genes, respectively. Thus, antagonists that inhibit androgen or estrogen receptor function represent a gold standard in the treatment of hormone-sensitive cancers.38 However, hormone receptor-negative cancer subtypes do not respond to hormonal therapies and are associated with poorer prognosis. Increasing evidence also suggests that glucocorticoid receptor signaling may be involved in the progression and development of chemoresistance in ovarian cancer patients;39 glucocorticoid receptor ligands suppress growth of lymphoid cancer.40
In our previous work, we reported the synthesis of D-ring fused 1,2,3-triazoles of dehydroepiandrosterone and estra-1,3,5(10)-triene.41 To investigate the influence of N(2)-substituents on the biological activity of androstane and estratriene D-ring fused 1,2,3-triazoles, in the present study we synthesized a series of 13 compounds and screened their anticancer activity against human cancer cell lines. As a continuation of our previous biological activity evaluation studies of steroid derivatives,42–44 we evaluated the relative binding affinities of a series of new D-ring fused steroidal N(2)-substituted-1,2,3-triazoles for the ligand-binding domains (LBDs) of estrogen receptor α (ERα), estrogen receptor β (ERβ), androgen receptor (AR) and glucocorticoid receptor (GR) using a fluorescent yeast screen in vitro in order to identify potential anti-hormonal effects. Furthermore, inhibition potential against human AKR1C3 and selectivity over a homologous isoform, AKR1C4, were also investigated in order to identify selective AKR1C3 inhibitors.
2. Results and discussion
2.1. Synthesis
Previously synthesized steroid 1,2,3-triazoles 1 and 2 showed moderate antiproliferative acitvity.34 In order to improve this activity, substitution of the hydrogen atom of the triazole ring was performed. Although structurally similar, N(2)-substituted triazole has different properties than the N(1) and N(3) isomers, so they have a specific application. Considering the above discussion, the aim of this work was to synthesize N(2)-substituted derivatives.The strategy for the synthesis of N(2)-substituted 1,2,3-triazole derivatives can be the construction of the 1,2,3-triazole skeleton already functionalized in N(2) or the N(2) functionalization of the 1,2,3-triazole skeleton. In the present study, we synthesized N(2)-substituted 1,2,3-triazoles of an androstane (3a–h) and estratriene (4a–e) series (Scheme 1 and Table 1) starting from previously synthesized steroid triazoles 1 and 2.34 Alkyl groups of different lengths were chosen as substituents, as well as groups of two carbon atoms containing polar functions.
 | ||
| Scheme 1 Synthesis of steroidal N(2)-substituted-1,2,3-triazoles 3a–h and 4a–e. Reagents and reaction time are given in Table 1. | ||
| Entry | Starting comp. | Reagents | Reaction time | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1 | CH3I, KOH, MeOH | 75 min | 3a | 31 |
| 2 | 1 | CH3CH2Br, KOH, MeOH | 8 h | 3b | 33 |
| 3 | 1 | CH3(CH2)3Br, KOH, MeOH | 5 h | 3c | 27 |
| 4 | 1 | CH3(CH2)7Br, KOH, MeOH | 4 h | 3d | 28 |
| 5 | 1 | C6H5CH2Br, KOH, MeOH | 6.5 h | 3e | 21 |
| 6 | 1 | ClCH2CH2NH2, K2CO3, 18-crown-6, THF | 6 h | 3f | 30 |
| 7 | 1 | ClCH2COOC2H5, K2CO3, 18-crown-6, THF | 2.5 h | 3g | 18 |
| 8 | 1 | Ac2O, Py | 2 h | 3h | 32 |
| 9 | 2 | CH3I, KOH, MeOH | 75 min | 4a | 25 |
| 10 | 2 | CH3CH2Br, KOH, MeOH | 2 h | 4b | 20 |
| 11 | 2 | CH3(CH2)3Br, KOH, MeOH | 5 h | 4c | 21 |
| 12 | 2 | CH3(CH2)7Br, KOH, MeOH | 4 h | 4d | 22 |
| 13 | 2 | C6H5CH2Br, KOH, MeOH | 90 min | 4e | 19 |
Basic conditions were used for the reaction. Namely, a mixture of compound 1 or 2 and potassium hydroxide in methanol was stirred at room temperature for 20 min to deprotonate the triazole ring. After that, the corresponding haloalkane was added and the reaction mixture was refluxed for 75 min–8 h. At the end of the reaction, the resulting product was evaporated and purified by flash chromatography: from compound 1, N(2)-alkyl derivatives 3a–e (Table 1, entries 1–5) and from compound 2, 4a–e (Table 1, entries 9–13) were obtained. N(2)-derivatives 3f (Table 1, entry 6) and 3g (Table 1, entry 7) were synthesized by reaction of compound 1 with 2-chloroethylamine or ethyl chloroacetate in tetrahydrofuran (THF), in the presence of potassium carbonate and 18-crown-6 at boiling point THF for 6 h and 2.5 h, respectively. Acetylation of the N2 atom of the triazole ring of compound 1 was carried out with acetic anhydride in pyridine to provide compound 3h (Table 1, entry 8). By monitoring the reaction by thin-layer chromatography (TLC), it was observed that the hydroxyl group is acetylated first, followed by the triazole.
N(2)-substituted derivatives 3a–h and 4a–e were successfully isolated from the reaction mixtures in a yield of 18–33%. Due to the complexity of the mixtures, no pure side products were isolated. The obtained synthesis results suggest that the yield of N(2)-substituted triazoles is not affected by the strength of the base and the type of solvent (protic or aprotic). Product yields of alkyl substituted triazoles are slightly higher in androstane than in the estrane series.
2.2. Spectral characterization
The identities of the synthesized compounds 3a–h and 4a–e were confirmed using high-resolution mass spectrometry (HRMS) and 1D (1H,13C{1H} and 15N{1H}) and 2D (1H-1H COSY, HSQC, 1H–13C and 1H–15N HMBC) NMR spectroscopy (ESI,† Fig. S1–S56). In the 1H–15N HMBC spectrum of the synthesized compounds, we observed a correlation between all three triazole nitrogen atoms and the hydrogen atoms of the methylene group attached to the triazole ring. In the 1H–13C HMBC spectra, there was no correlation of these hydrogens with C-16 and C-17 atoms. These facts indicate that the substituent on the triazole is attached to nitrogen N(2). The molecular formulas of the synthesized compounds were confirmed by HRMS measurements.2.3. Relative binding affinities of D-ring fused steroidal N(2)-substituted-1,2,3-triazoles for the ligand-binding domains of ERα, ERβ, AR and GR
Antagonists that compete with endogenous steroid hormones for binding sites on steroid receptors, together with inhibitors of steroid hormone synthetic pathways, remain a cornerstone of first-line endocrine therapy for hormone-sensitive cancers.45 However, there is a constant need for new therapeutic routes and more specific and effective drugs with minimum side effects for the management of hormone-sensitive cancers.46 In the present study, synthesized steroid triazoles were evaluated for relative binding affinities for the ligand binding domains (LBDs) of human recombinant estrogen receptor α (ERα-LBD), estrogen receptor β (ERβ-LBD), androgen receptor (AR-LBD) and rat recombinant glucocorticoid receptor (GR-LBD) using a fluorescent yeast-based biosensor as described.42,43,47,48 Modified yeast cells have been used as biosensors for drug discovery, as well as investigation of mechanisms of disease pathogenesis, biomolecular interactions and mitochondrial function.49LBDs of ERα, ERβ, AR or GR were expressed in-frame with yellow fluorescent protein (YFP) in Sacharomyces cerevisiae. The fluorescence level of LBD–YFP expression was assessed by using fluorimetry and fluorescence microscopy. The relative binding affinities were expressed as fold fluorescence change between recombinant cells treated with test compounds or DMSO solvent only. As a control, addition of positive control ligands resulted in increased fluorescence intensity, whereas treatment with low-affinity ligands resulted in low fluorescence signals under the same conditions. As can be seen in Fig. 1, none of the test compounds displayed affinity for ERα- and AR-LBD, suggesting a lack of estrogenic and androgenic activities. In our experience, introduction of modifications in the structure of steroid hormones often results in lower binding affinity of these steroid analogues for AR than their parent compounds.42,43 Among the tested compounds, androstane derivatives 3a and 3h and estratriene derivative 4e showed highly specific and strong binding affinity for ERβ-LBD, with similar fold fluorescence enhancements as a natural ligand, estrone. In agreement with these experimental findings, molecular docking simulations also suggest that 4e, 3h and 3a have affinity for ERβ-LBD (see ESI† Fig. S59). Although ERα is the predominant form in breast cancer and triggers proliferation, ERβ is believed to play a suppressive role. Thus, highly selective ERβ agonists without ERα activity are considered to be therapeutically useful agents.50–52 Furthermore, comparison of the relative ERβ binding affinities of compounds 3a–c for ERβ-LBD suggests that the longer N(2)-alkyl chain led to a decrease in binding affinity (Table 3). These results suggest the importance of alkyl chain length at the D-ring for the interaction with the receptor. Other steroid derivatives had no apparent affinity for the ERβ subtype. Moreover, steroid triazoles 3b, 3e, 4a and 4b displayed high binding affinity for GR-LBD with fold fluorescence enhancements ∼2, similar to synthetic corticosteroid drug prednisolone, suggesting they could be used as potential scaffolds for the development of improved immunomodulators with fewer side effects. Findings from this screen are in agreement with the GR-mediated activities of a series of steroid compounds.44,53 Comparing the structures of N-2-alkyl triazole derivatives, we noticed that the most favorable group for binding to GR-LBD is the ethyl group, and that increasing alkyl chain length attached to the triazole ring leads to decreased binding affinity (Table 3). Synthetic GR ligands are often used as palliative agents to reduce the side effects of chemotherapy54 and in low doses may suppress breast cancer growth.55 Many studies have described the antiproliferative effects of steroid triazoles against various cancers,15,56 but there is a lack of information about their potential to modulate the action of steroid hormone receptors, which could help improve the understanding of the mechanism of anticancer action and provide new suggestions for drug design. As can be seen in Fig. 2 treatment of yeast cells expressing GR-LBD with compounds 3b, 3e, 4a, and 4b and yeast cells expressing ERβ-LBD with compounds 3a, 3h and 4e resulted in a strong increase and relocalization of fluorescence intensity compared to the negative control. These findings are in agreement with results from fluorimetric screening in a 96-well format.
 | ||
| Fig. 1 Relative binding affinities of compounds 3a–h and 4a–e for the LBDs of ERα, ERβ, AR or GR measured using a fluorescent biosensor in yeast. Receptor LBDs were fused in frame with YFP and expressed in S. cerevisiae. Yeast cells were exposed to test compounds over 14–16 h and fluorescence was measured by fluorimetry in a 96-well format. Relative binding affinities were expressed as fold fluorescence change between ligand-treated cells and control cells in the absence of a ligand (normalized to 1). Higher values correspond to stronger affinity. Estrone and androstenedione were positive and negative control ligands, respectively, in ERα- and ERβ-binding assays, while for yeast cells expressing AR-LBD androstenedione and estrone were positive and negative control ligands, respectively. Estradiol served as a negative control and prednisolone as a positive control in the GR-binding assay. Error bars indicate propagated standard errors of the mean. | ||
 | ||
| Fig. 2 Yeast cells expressing GR-LBD treated with estradiol (− control), prednisolone (+ control) or compounds 3b, 3e, 4a and 4b, at a final concentration of 100 μM and yeast cells expressing ERβ-LBD treated with androstenedione (− control), estrone (+ control) or compounds 3a, 3h and 4e at a final concentration of 10 μM were observed by fluorescence microscopy. | ||
2.4. Inhibition potential of D-ring fused steroidal N(2)-substituted-1,2,3-triazoles against human AKR1C3 or AKR1C4
In the present study, we tested the ability of the synthesized D-ring fused steroidal N(2)-substituted-1,2,3-triazoles (compounds 3a–h and 4a–e) to inhibit AKR1C3 and AKR1C4 in vitro by monitoring NADPH consumption using fluorescence spectroscopy (Table 2 and Fig. 3–5). The most potent AKR1C3 inhibitors were compounds 3h (71.17%) and 3f (69.9%), while compounds 4e (43.74%) and 3e (43.53%) had moderate inhibition potential. These findings are consistent with previous research showing that AKR1C enzymes can be strongly inhibited by steroid analogues.43,44,57,58 Compounds possessing non-polar chains attached to the triazole ring (3a–3c and 4a–4d) do not exhibit inhibition of the AKR1C3 enzyme (Table 3), indicating that the presence of polar groups at this position is likely essential for inhibitory effects, as seen in derivatives 3f and 3h. Compound 3h, which inhibited AKR1C3 by more than 70%, (more than a known inhibitor ibuprofen) was selected for IC50 measurements. Based on an inhibition potential vs. concentration plot (Fig. 4), 3h displayed an IC50 value ∼25 μM, in the range of reported values for ibuprofen59 indicating potential therapeutic relevance. AKR1C3 inhibitors have been the subject of biomedical research and patent applications in recent years, but have not yet been approved for clinical use. To the best of our knowledge, this is the first study where N(2)-substituted-1,2,3-triazoles have been identified as AKR1C3 inhibitors. None of the tested steroid compounds showed inhibition against AKR1C4 (Fig. 5). The AKR1C4 isoform acts as 3-alpha hydroxysteroid dehydrogenase and plays a role in steroidogenic pathways by preventing accumulation of steroid hormones. Therefore, inhibition is often undesirable under physiological conditions. On the other hand, AKR1C3 inhibitors have been proposed for treatment of hormone-dependent cancers and overcoming resistance to chemotherapy. Results from the present study suggest that this series of steroid triazoles are promising scaffolds for structure-guided lead optimization and development of AKR1C3 inhibitors with high potency and selectivity. | ||
| Fig. 3 Change in NADPH fluorescence over time during in vitro reduction of 9,10-phenanthrenequinone by AKR1C3 in the absence (reaction) or presence of either test compounds (3a–h and 4a–e) or known inhibitor ibuprofen (IBU) at a final concentration of 33 μM. Enzyme activity was measured by monitoring the decrease in NADPH fluorescence at ex/em 340/460 nm. Blank represents a control probe in the absence of enzyme. | ||
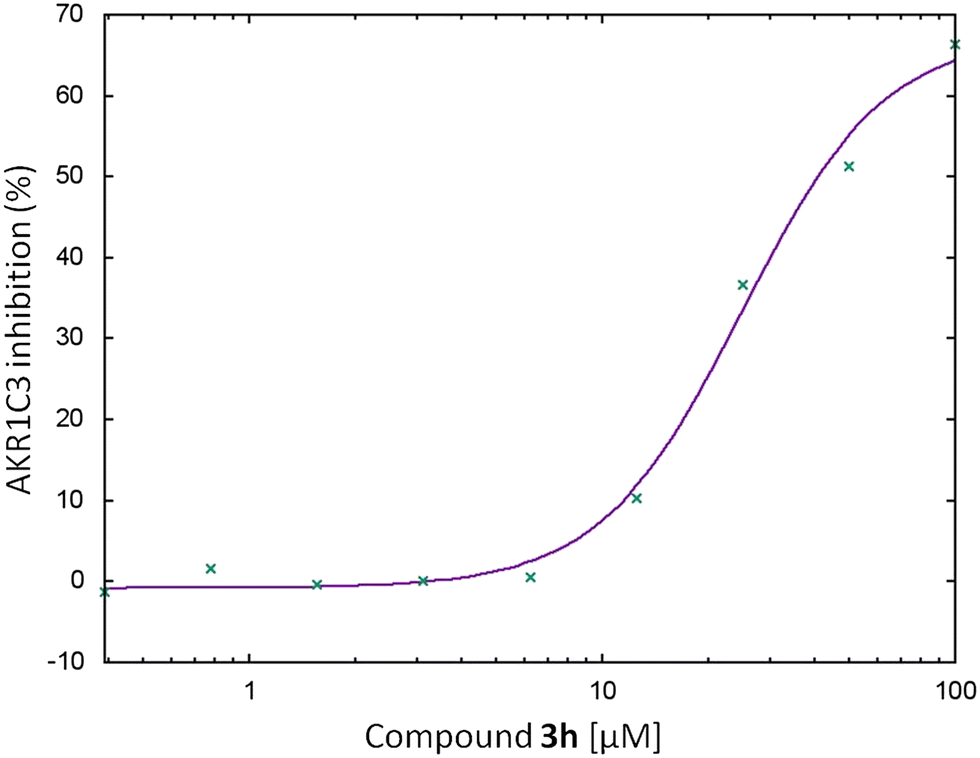 | ||
| Fig. 4 Sigmoidal dose-dependence of AKR1C3 inhibition by the N(2)-substituted 1,2,3-triazole derivative of androstane 3h. The inhibitory effect of increasing concentrations of compound 3h (0, 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50, 100, and 133 μM) on AKR1C3-catalyzed reduction of 9,10-phenanthrenequinone. NADPH consumption during reaction was monitored by fluorescence spectroscopy. | ||
 | ||
| Fig. 5 Change in NADPH fluorescence over time during in vitro reduction of 9,10-phenanthrenequinone by AKR1C4 in the absence (reaction) or presence of either test compounds (3a–h and 4a–e) or known inhibitor ibuprofen (IBU) at a final concentration of 33 μM. Enzyme activity was measured by monitoring the decrease in NADPH fluorescence at ex/em 340/460 nm. Blank represents a control probe in the absence of enzyme. | ||
| Compound | R2 | Cytotoxicity (>50%) | Binding (fold fluorescence > 1.4) | AKR1C3 inhibition (>50%) | |
|---|---|---|---|---|---|
| ERβ | GR | ||||
| 3a | –CH3 | + | |||
| 3b | –CH2CH3 | + | |||
| 3c | –(CH2)3CH3 | ||||
| 3e | –CH2C6H5 | + | |||
| 3f | –CH2CH2NH2 | + | + | ||
| 3g | –CH2COOCH2CH3 | ||||
| 3h | CH3CO– | + | + | ||
| 4a | –CH3 | + | |||
| 4b | –CH2CH3 | + | |||
| 4d | –(CH2)7CH3 | ||||
| 4e | –CH2C6H5 | + | |||
2.5. Molecular docking of compounds 3f and 3h against AKR1C3
To model the molecular basis of AKR1C3 inhibition by D-ring fused steroidal N(2)-substituted-1,2,3-triazole compounds, we conducted molecular docking simulations with Autodock Vina using the PyRx platform.60,61 Autodock Vina has been shown to successfully predict molecular interactions and binding energies between bioactive ‘ligand’ compounds and their protein ‘receptors’.60,61 Compounds 3h and 3f were chosen for molecular docking analysis because they represent the strongest AKR1C3 inhibitors identified in the present study.For protein ‘receptor’, the structure of AKR1C3 in complex with another steroidal inhibitor compound known as EM1404 was selected (PDB 1ZQ5).62 EM1404 is a steroidal spiro-δ-lactone compound which displays strong, specific inhibition of AKR1C3.62 This particular structure of AKR1C3 was used for molecular docking because the overall shape and dimensions of compounds 3h and 3f are qualitatively similar to EM1404. In particular, the test compounds contain a D-ring fused triazole, whereas EM104 has a spiro lactone group attached to the D-ring. In the X-ray structure (PDB 1ZQ5), the steroidal core of EM1404 is bound in the conserved steroid channel of AKR1C3 through hydrophobic interactions with gatekeeper residues Leu54 and Trp227; while the spiro lactone functional group is bound via interactions with Ser118, Asn167, Phe306, Phe311, and Tyr319 in AKR1C3. These residues form a subpocket in the AKR1C3 ligand binding site known as SP1 that is involved in binding all AKR1C3 ligands known to date.59 The binding orientation of EM1404 depends on hydrogen bonding interactions between the spiro lactone and Ser118 at the D-ring and between a polar group at C3 and Arg226 and Ser129. To validate the parameters that we used for molecular docking, EM1404 was redocked into the structure of AKR1C3 with this ligand removed, and Autodock Vina was able to accurately reproduce the binding geometry of EM1404 seen in the crystal structure. Using these same parameters, Autodock Vina predicts that compounds 3h and 3f could bind to AKR1C3 in the same site and orientation as EM1404. Binding energies predicted using Autodock Vina for compound 3h (−10.1 kcal mol−1) and 3f (−8.8 kcal ml−1) are energetically favorable, and are comparable to that predicted for EM1404 (−14.1 kcal mol−1). As can be seen in Fig. 6, the steroidal core of 3h and 3f superimposes well with EM1404 and is also predicted to bind in the steroidal channel via interactions with Trp227 and Leu54. In addition, the triazole derivatives of compounds 3h and 3f occupy the SP1 subpocket in a similar position to the lactone group of EM1404. Moreover, polar modifications at the C3 position in 3h, 3f and EM1404 seem to play similar roles in binding AKR1C3 by participating in hydrogen bonding with Arg226. Thus, in general, the binding orientation of all three inhibitors seems to depend on polar interactions at the D-ring and A-ring side of the steroidal core. However, polar atoms in the triazole group enable additional hydrogen bonds with Tyr319, Tyr216 and the NADP+ cofactor deeper in SP1 that are not visible in the structure of AKR1C3 in complex with EM1404.
 | ||
| Fig. 6 Molecular docking of compounds 3h (panel A) and 3f (panel B) against AKR1C3. Top ranking docking poses (magenta sticks) are shown superimposed onto the structure of AKR1C3 in complex with EM1404 (green sticks). Predicted hydrogen bonds between docked compounds and AKR1C3 residues (blue dashes) are shown in comparison with hydrogen bonds between EM1404 and AKR1C3 (red dashes). | ||
To visualize molecular interactions between compound 3h and AKR1C3, a two dimensional plot was generated using the program LigPlot+ using coordinates for the top ranking docking pose (Fig. 7).63 Based on these analyses of our molecular docking results, the D-ring fused triazole modification is important for interactions with the SP1 site of AKR1C3, while polar modifications at the C3 position could orient the inhibitor compound in the AKR1C3 binding pocket.
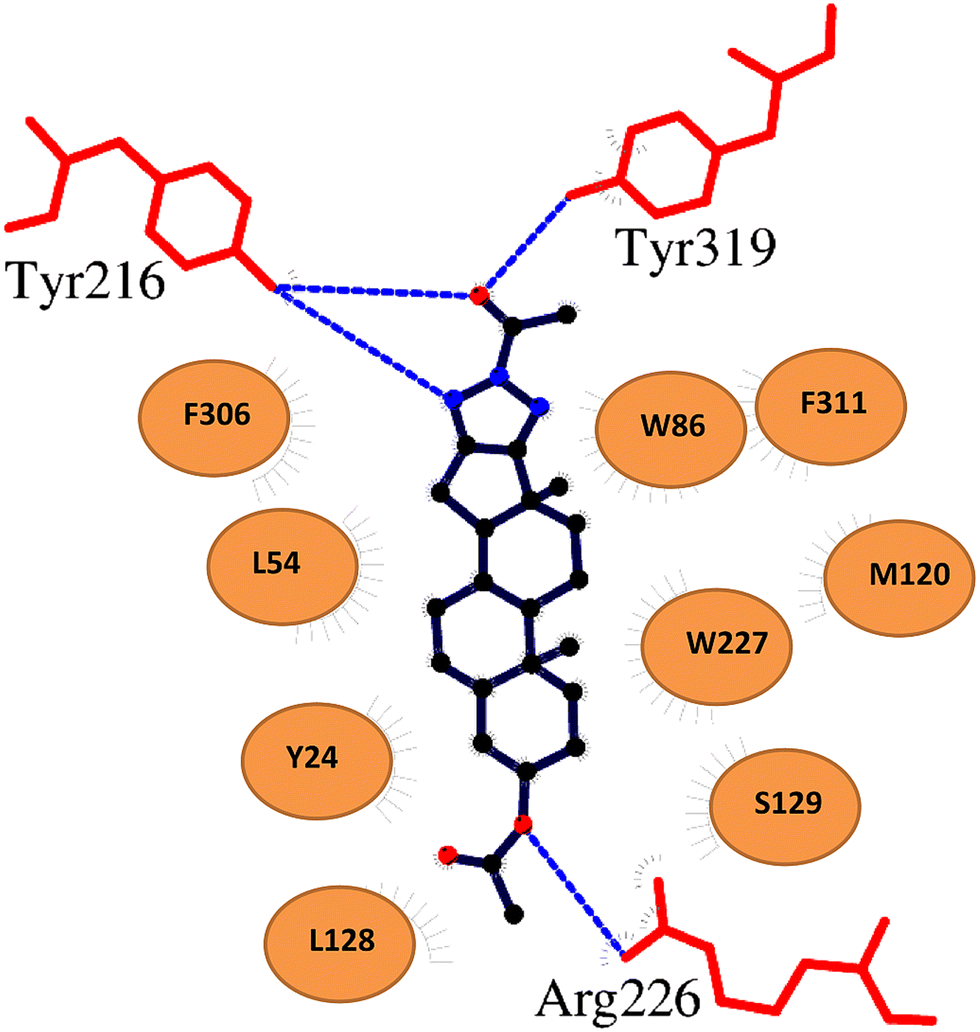 | ||
| Fig. 7 Two dimensional plot of molecular interactions between compound 3h and AKR1C3. Molecular interactions predicted by docking were visualized using LigPlot+. Hydrogen bonds are shown as blue dashes between 3h and specific residues in AKR1C3 (shown as red lines). Other AKR1C3 residues predicted to be involved in binding compound 3h are shown as colored circles. | ||
2.6. Cytotoxic properties
To assess the cytotoxic properties of novel steroid triazoles, we conducted a comprehensive screening across four distinct cancer cell lines representing various cancer types: HCT116 (human colon cancer), Huh7 (human hepatoma), MCF7 (human estrogen receptor-positive breast cancer) and MDA-MB-231 (human triple-negative breast cancer). Initially, each compound (3a–c, 3e–h, 4a and b and 4d and e) was administered to each cell line at a broad concentration range (0.01–100 μM), followed by evaluation using a resazurin assay to observe metabolic activity (ESI,† Fig. S57 and S58), which is indicative of cell viability and cytotoxicity.64,65 Our criterion for potential cytotoxicity was set at a minimum of 50% reduction in cell viability across at least three of the four tested cancer cell lines, which was met by compound 3f. Subsequently, we refined our analysis by subjecting this compound to a narrower concentration range (1–100 μM) in a repeated cell viability assay, which confirmed its cytotoxic potential with IC50 values ranging between 22.3 and 93.6 μM (Fig. 8 and ESI,† Table S1). | ||
| Fig. 8 Cell viability assays with the toxic compound 3f in indicated human cancer cell lines. Statistical significance was determined by one-way ANOVA Dunnett's test, where each treatment group (n = 3) was compared to the control. * P < 0.05, ** P < 0.001, *** P < 0.0001. | ||
To explore the selectivity of the observed decrease in cell viability towards cancer cells, we extended our investigation and included a noncancerous, hTERT-immortalized human fibroblast cell line (BJ-5ta). Intriguingly, compound 3f exhibited lower cytotoxicity against these untransformed fibroblasts compared to the previously tested cancer cell lines (Fig. 9).
 | ||
| Fig. 9 Cell viability assay with the cytotoxic compound 3f in human BJ-5ta fibroblasts. Statistical significance was determined by one-way ANOVA Dunnett tests, where each treatment group (n = 3) was compared to the control. *** P < 0.0001. | ||
Therefore, compound 3f shows potential as an anticancer drug candidate, demonstrating moderate cytotoxicity across various cancer types while displaying lower toxicity towards healthy, untransformed cells. The initial screening indicates that this compound is not specific for any particular cancer type or steroid receptor expression, such as the estrogen receptor. Notably, comparable cytotoxic effects were observed in both estrogen receptor-positive human breast cancer cells (MCF7) and triple-negative human breast cancer cells (MDA-MB-231), suggesting a potential mechanism independent of estrogen or other steroid receptors. Further investigation is warranted to elucidate the precise mechanisms of action and associated biological implications of this lead compound. Additionally, structural optimization of this lead compound may enhance its anticancer efficacy while reducing its toxicity against untransformed, healthy cells. The summary of in vitro biological activities of the tested compounds in this study including their cytotoxicity, steroid receptor binding affinities, and AKR1C3 enzyme inhibition properties is presented in Table 3.
3. Experimental section
3.1. Chemistry
NMR spectra were recorded on a Bruker AVANCE III 400 (BRUKER, Rheinstetten, Germany) spectrometer operating at 400 MHz (1H) and 100 MHz (13C) and are reported in ppm (δ-scale) downfield from the tetramethylsilane internal standard; coupling constants (J) are given in Hz. Melting points were determined using a Stuart melting point apparatus digital SMP10 and are reported uncorrected. ESI-HRMS mass spectrometry was performed on an Orbitrap Exploris 240 instrument and Exactive Orbitrap, Thermo Scientific instrument. TLC analysis was carried out using Al-backed silica-gel sheets (TLC Silica gel 60 F254, Merck) and after elution, plates were visualized under UV radiation (254 nm) with a UV lamp (UVGL-58 Handheld UV Lamp). Then a development step with sulfuric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), followed by heating at 120 °C was performed. Chromatographic separations were performed on silica gel columns (Kieselgel 60, 0.04–0.063 mm, Merck). All solutions were dried over anhydrous sodium sulfate.
:1), followed by heating at 120 °C was performed. Chromatographic separations were performed on silica gel columns (Kieselgel 60, 0.04–0.063 mm, Merck). All solutions were dried over anhydrous sodium sulfate.
3β-Hydroxy-2′-methyl-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3a). White crystals (eluent: petroleum ether–ethyl acetate, 2
:1, yield 31%); mp 229–230 °C; 1H NMR (CDCl3, 400 MHz) δ: 1.03 (s, 3H, 3H-18); 1.09 (s, 3H, 3 H-19); 1.11–1.18 (overlapping signals, 2H, H-1a, H-9); 1.49–1.56 (m, 1H, H-2a); 1.60–1.79 (overlapping signals, 4H, H-7a, 2 H-11, H-12a); 1.82–1.91 (overlapping signals, 3H, H-1b, H-2b, H-8); 1.92–2.02 (m, 1H, H-14); 2.06–2.14 (m, 1H, H-7b); 2.15–2.36 (overlapping signals, 3H, 2 H-4, H-12b); 2.39 (dd, 1H, Jgem = 14.4 Hz, J15b,14 = 2.4 Hz, H-15b); 2.69 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 3.49–3.60 (m, 1H, H-3); 4.11 (s, 3H, N-CH3); 5.37–5.42 (m, 1H, H-6). 13C NMR (CDCl3, 101 MHz) δ: 18.05 (C-18); 19.37 (C-19); 20.39 (C-11); 24.31 (C-15); 30.67 (C-8); 31.43 (C-7); 31.60 (C-2); 33.93 (C-12); 36.82 (C-10); 37.10 (C-1); 39.80 (C-13); 41.23 (N-CH3); 42.26 (C-4); 50.54 (C-9); 61.70 (C-14); 71.66 (C-3); 120.98 (C-6); 141.16 (C-5); 152.50 (C-16); 162.50 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H30N3O 328.23834; found 328.23867.
2′-Ethyl-3β-hydroxy-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3b). White crystals (eluent: petroleum ether–acetone, 6
:1, yield 33%); mp 146 °C; 1H NMR (CDCl3, 400 MHz) δ: 1.01 (s, 3H, 3 H-18); 1.07 (s, 3H, 3 H-19); 1.08–1.16 (overlapping signals, 2H, H-1a, H-9); (m, 1H, H-2a); 1.47–1.54 (overlapping signals, 4H, H-2a, N-CH2CH3); 1.63–1.74 (overlapping signals, 4H, H-7a, 2 H-11, H-12a); 1.80–1.90 (overlapping signals, 3H, H-1b, H-2b, H-8); 1.92–2.01 (m, 1H, H-14); −2.04-2.12 (m, 1H, H-7b); 2.18–2.34 (overlapping signals, 3H, 2 H-4, H-12b); 2.37 (dd, 1H, Jgem = 14.4 Hz, J15b,14 = 2.4 Hz, H-15b); 2.67 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 3.46–3.57 (m, 1H, H-3); 4.36 (q, 2H, J = 7.2 Hz, N-CH2CH3); 5.34–3.39 (m, 1H, H-6). 13C NMR (CDCl3, 101 MHz) δ: 15.20 (N-CH2CH3); 18.03 (C-18); 19.37 (C-19); 20.39 (C-11); 24.27 (C-15); 30.66 (C-8); 31.43 (C-7); 31.57 (C-2); 33.93 (C-12); 36.82 (C-10); 37.11 (C-1); 39.79 (C-13); 42.25 (C-4); 49.62 (N-CH2CH3); 50.54 (C-9); 61.66 (C-14); 71.54 (C-3); 120.91 (C-6); 141.22 (C-5); 152.11 (C-16); 162.06 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H32N3O 342.25454; found 342.25350.
2′-Butyl-3β-hydroxy-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3c). White crystals (eluent: petroleum ether – ethyl-acetate, 2:1, yield 27%); mp 109 °C; 1H NMR (CDCl3, 400 MHz) δ: 0.94 (t, 3H, J = 7.4 Hz, N-CH2CH2CH2CH3); 1.07 (s, 3H, H-18); 1.09 (s, 3H, H-19); 1.10–1.18 (overlapping signals, 2H, H-1a, H-9); 1.29–1.39 (m, 2H, N-CH2CH2CH2CH3); 1.45–1.76 (overlapping signals, 7H, H-2a, H-7a, 2 H-11, H-12a, N-CH2CH2CH2CH3); 1.82–1.93 (overlapping signals, 3H, H-1b, H-2b, H-8); 1.98 (m, 1H, H-14); 2.05–2.014 (m, 1H, H-7b); 2.20–2.41 (overlapping signals, 4H, 2 H-4, H-12b, H-15b); 2.69 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 3.50–3.59 (m, 1H, H-3); 4.33 (t, 3H, J = 7.2 Hz, N-CH2CH2CH2CH3); 5.37–5.42 (m, 1H, H-6). 13C NMR (CDCl3, 101 MHz) δ: 13.60 (N-CH2CH2CH2CH3), 18.05 (C-18); 19.39 (C-19); 19.87 (N-CH2CH2CH2CH3); 20.41 (C-11); 24.29 (C-15); 30.68 (C-8); 31.44 (C-7); 31.61 (C-2); 32.18 (N-CH2CH2CH2CH3); 33.95 (C-12); 36.83 (C-10); 37.11 (C-1); 39.79 (C-13); 42.27 (C-4); 50.56 (C-9); 54.49 (N-CH2CH2CH2CH3); 61.65 (C-14); 71.64 (C-3); 121.00 (C-6); 141.16 (C-5); 152.03 (C-16); 162.01 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H36N3O 370.28584; found 370.28534.
3β-Hydroxy-2′-octyl-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3d). Colorless oil (eluent: petroleum ether–acetone, 8
:1, yield 28%); 1H NMR (CDCl3, 400 MHz) δ: 0.86 (t, 3H, J = 7.2 Hz, N-(CH2)7CH3); 1.02 (s, 3H, H-18); 1.08 (s, 3H, H-19); 1.02–1.15 (overlapping signals, 2H, H-1a, H-9); 1.22–1.36 (overlapping signals, 10H, CH2 from N-CH2(CH2)6CH3); 1.46–1.56 (m, 1H, H-2a); 1.60–1.76 (overlapping signals, 4H, H-7a, 2 H-11, H-12a); 1.82–1.97 (overlapping signals, 5H, H-1b, H-2b, H-8, CH2 from N-CH2(CH2)6CH3); 1.94–2.02 (m, 1H, H-14); 2.02–2.40 (overlapping signals, 5H, 2 H-4, H-7b, H-12b, H-15b); 2.68 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.2 Hz, H-15a); 3.50–3.58 (m, 1H, H-3); 4.31 (t, 2H, J = 7.2 Hz, N-CH2(CH2)6CH3); 5.37–5.41 (m, 1H, H-6). 13C NMR (CDCl3, 101 MHz) δ: 14.10 (N-(CH2)7CH3); 18.04 (C-18); 19.38 (C-19); 20.40 (C-11); 22.62 (CH2 from octyl group); 24.29 (C-15); 26.61 (CH2 from octyl group); 29.03 (CH2 from octyl group); 29.09 (CH2 from octyl group); 30.13 (CH2 from octyl group); 30.68 (C-8); 31.44 (C-7); 31.60 (C-2); 31.74 (CH2 from octyl group); 33.94 (C-12); 36.83 (C-10); 37.11 (C-1); 39.79 (C-13); 42.27 (C-4); 50.56 (C-9); 54.79 (N-CH2(CH2)6CH3); 61.65 (C-14); 71.61 (C-3); 120.98 (C-6); 141.18 (C-5); 152.02 (C-16); 162.00 (C-17).
2′-Benzyl-3β-hydroxy-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3e). White crystals (eluent: petroleum ether–acetone, 4
:1, yield 21%); mp 142–143 °C; 1H NMR (CDCl3, 400 MHz) δ: 1.04 (s, 3H, H-18); 1.09 (s, 3H, H-19); 1.10–1.18 (overlapping signals, 2H, H-1a, H-9); 1.46–1.76 (overlapping signals, 5H, H-2a, H-7a, 2 H-11, H-12a); 1.81–1.94 (overlapping signals, 3H, H-1b, H-2b, H-8); 1.98–2.39 (overlapping signals, 6H, 2 H-4, H-7b, H-12b, H-14, H-15b); 2.69 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 3.49–3.59 (m, 1H, H-3); 5.37–5.41 (m, 1H, H-6); 5.50 (s, 2H, N-CH2C6H5); 7.27–7.32 (overlapping signals, 5H, C6H5). 13C NMR (CDCl3, 101 MHz) δ: 18.04 (C-18); 19.38 (C-19); 20.39 (C-11); 24.36 (C-15); 30.68 (C-8); 31.42 (C-7); 31.60 (C-2); 33.89 (C-12); 36.82 (C-10); 37.10 (C-1); 39.87 (C-13); 42.26 (C-4); 50.53 (C-9); 58.36 (N-CH2C6H5); 61.53 (C-14); 71.64 (C-3); 120.97 (C-6); 127.91 (Ph); 128.01 (Ph); 128.70 (Ph); 136.17 (Ph); 141.16 (C-5); 153.02 (C-16); 162.68 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H34N3O 404.26964; found 404.26998.
3-Benzyloxy-2′-methyl- 2′H-[1,2,3]triazolo[4′,5′:16,17]estra-1,3,5(10)-triene (4a). White crystals (eluent: petroleum ether–ethyl acetate, 6
:1, yield 25%); mp 118 °C; 1H NMR (CDCl3, 400 MHz) δ: 1.05 (s, 3H, H-18); 1.46–1.57 (m, 1H, H-7a); 1.66–1.92 (overlapping signals, 4H, H-8, 2 H-11, H-12a); 1.94–2.03 (m, 1H, H-7b); 2.25–2.30 (m, 1H, H-14); 2.31–2.52 (overlapping signals, 3H, H-9, H-12b, H-15b); 2.80 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 2.90–3.00 (m, 2H, H-6); 4.15 (s, 3H, N-CH3); 5.07 (s, 2H, CH2C6H5); 6.77 (d, 1H, J = 2.5, H-4); 6.82 (dd, 1H, J1,2 = 8.8, J2,4 = 2.7, H-2); 7.24 (d, 1H, J = 8.4, H-1); 7.39–7.47 (overlapping signals, 5H, C6H5). 13C NMR (CDCl3, 101 MHz) δ: 18.39 (C-18); 24.14 (C-15); 26.09 (C-11); 27.58 (C-7); 29.62 (C-6); 34.04 (C-12); 37.39 (C-8); 40.29 (C-13); 41.27 (N-CH3); 44.43 (C-9); 60.82 (C-14); 69.98 (CH2C6H5); 112.39 (C-2); 114.92 (C-4); 126.11 (C-1); 127.46 (C-2 and C-6 from C6H5); 127.89 (C-4 from C6H5); 128.57 (C-3 and C-5 from C6H5); 132.58 (C-5); 137.28 (C-1 from C6H5); 137.75 (C-10); 152.42 (C-16); 156.89 (C-3); 162.57 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H30N3O 400.23889; found 400.23787.
3-Benzyloxy-2′-ethyl-2′H-[1,2,3]triazolo[4′,5′:16,17]estra-1,3,5(10)-triene (4b). White crystals (eluent: petroleum ether–ethyl acetate, 8
:1, yield 20%); mp 134 °C; 1H NMR (CDCl3, 400 MHz) δ: 1.04 (s, 3H, H-18); 1.54 (t, 3H, J = 7.2 Hz, N-CH2CH3); 1.56–1.83 (overlapping signals, 4H, H-7a, H-8, 2 H-11); 1.84–1.93 (m, 1H, H-12a); 1.96–2.05 (m, 1H, H-7b);; 2.16–2.26 (m, 1H, H-14); 2.30–2.52 (overlapping signals, 3H, H-9, H-12b, H-15b); 2.80 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.8 Hz, H-15a); 2.85–3.01 (m, 2H, H-6); 4.41 (q, 2H,J = 7.2 Hz, N-CH2CH3); 5.05 (s, 2H, CH2C6H5); 6.76 (d, 1H, J = 2.8 Hz, H-4); 6.81 (dd, 1H, J2,1 = 8.8 Hz, J2,4 = 2.8 Hz, H-2); 7.23 (d, 1H, J = 8.4 Hz, H-1); 7.33–7.44 (overlapping signals, 5H, C6H5). 13C NMR (CDCl3, 101 MHz) δ: 15.22 (N-CH2CH3); 18.36 (C-18); 24.12 (C-15); 26.10 (C-11); 27.58 (C-7); 29.62 (C-6); 34.05 (C-12); 37.39 (C-8); 40.30 (C-13); 44.44 (C-9); 49.68 (N-CH2CH3); 60.80 (C-14); 69.98 (CH2C6H5); 112.38 (C-2); 114.91 (C-4); 126.11 (C-1); 127.46 (C-2 and C-6 from C6H5); 127.89 (C-4 from C6H5); 128.57 (C-3 and C-5 from C6H5); 132.61 (C-5); 137.28 (C-1 from C6H5); 137.76 (C-10); 152.07 (C-16); 156.88 (C-3); 162.17 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H32N3O 414.25454; found 414.25331.
3-Benzyloxy-2′-butyl-2′H-[1,2,3]triazolo[4′,5′:16,17]estra-1,3,5(10)-triene (4c). Colorless oil (eluent: petroleum ether–ethyl acetate, 6
:1, yield 21%); 1H NMR (CDCl3, 400 MHz) δ: 0.94 (t, 3H, J = 7.2 Hz, N-(CH2)3CH3); 1.02 (s, 3H, H-18); 1.29–1.39 (m, 2H, N-CH2CH2CH2CH3); 1.45–1.58 (m, 1H, H-7a); 1.62–1.84 (overlapping signals, 5H, H-8, 2 H-11, N-CH2CH2CH2CH3); 1.88–2.04 (overlapping signals, 2H, H-7b, H-12a); 2.14–2.24 (m, 1H, H-14); 2.30–2.52 (overlapping signals, 3H, H-9, H-12b, H-15b); 2.78 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 2.84–3.00 (m, 2H, H-6); 4.34 (t, 2H, J= 7.2 Hz, N-CH2(CH2)2CH3); 5.04 (s, 2H, CH2C6H5); 6.74 (d, 1H, J = 2.0 Hz, H-4); 6.80 (dd, 1H, J2,1 = 8.4 Hz, J2,4 = 2.4 Hz, H-2); 7.21 (d, 1H, J = 8.4 Hz, H-1); 7.30–7.38 (overlapping signals, 5H, C6H5). 13C NMR (CDCl3, 101 MHz) δ: 13.61 (N-(CH2)3CH3); 18.38 (C-18); 19.89 (CH2 from butyl group); 24.13 (C-15); 26.11 (C-11); 27.58 (C-7); 29.62 (C-6); 32.19 (CH2 from butyl group); 34.06 (C-12); 37.40 (C-8); 40.29 (C-13); 44.44 (C-9); 54.52 (N-CH2(CH2)2CH3); 60.78 (C-14); 69.98 (CH2C6H5); 112.38 (C-2); 114.92 (C-4); 126.12 (C-1); 127.46 (C-2 and C-6 from C6H5); 127.89 (C-4 from C6H5); 128.57 (C-3 and C-5 from C6H5); 132.63 (C-5); 137.29 (C-1 from C6H5); 137.76 (C-10); 151.96 (C-16); 156.88 (C-3); 162.10 (C-17).
3-Benzyloxy-2′-octyl-2′H-[1,2,3]triazolo[4′,5′:16,17]estra-1,3,5(10)-triene (4d). Colorless oil (eluent: petroleum ether–ethyl acetate, 10
:1, yield 22%); 1H NMR (CDCl3, 400 MHz) δ: 0.89 (t, 3H, J = 6.8 Hz, N-(CH2)7CH3); 1.04 (s, 3H, H-18); 1.22–1.36 (overlapping signals, 10H, CH2 from N-CH2(CH2)6CH3); 1.53 (m, 1H, H-7a); 1.62–1.84 (overlapping signals, 3H, H-8, 2 H-11); 1.88–2.04 (overlapping signals, 4H, H-7b, H-12a, CH2 from N-CH2(CH2)6CH3); 2.17–2.27 (m, 1H, H-14); 2.28–2.52 (overlapping signals, 3H, H-9, H-12b, H-15b); 2.80 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 2.87–3.01 (m, 2H, H-6); 4.34 (t, 2H, J = 7.2 Hz, N-CH2(CH2)6CH3); 5.06 (s, 2H, CH2C6H5); 6.76 (d, 1H, J = 2.8 Hz, H-4); 6.81 (dd, 1H, J2,1 = 8.4 Hz, J2,4 = 2.8 Hz, H-2); 7.23 (d, 1H, J = 8.4 Hz, H-1); 7.33–7.46 (overlapping signals, 5H, C6H5). 13C NMR (CDCl3, 101 MHz) δ: 14.12 (N-(CH2)7CH3); 18.38 (C-18); 22.64 (CH2 from octyl group); 24.13 (C-15); 26.11 (C-11); 26.64 (CH2 from octyl group); 27.59 (C-7); 29.06 (CH2 from octyl group); 29.12 (CH2 from octyl group); 29.63 (C-6); 30.16 (CH2 from octyl group); 31.76 (CH2 from octyl group); 34.07 (C-12); 37.41 (C-8); 40.29 (C-13); 44.44 (C-9); 54.83 (N-CH2(CH2)6CH3); 60.78 (C-14); 69.98 (CH2C6H5); 112.39 (C-2); 114.92 (C-4); 126.12 (C-1); 127.46 (C-2 and C-6 from C6H5); 127.89 (C-4 from C6H5); 128.57 (C-3 and C-5 from C6H5); 132.62 (C-10); 137.29 (C-1 from C6H5); 137.76 (C-5); 151.96 (C-16); 156.89 (C-3); 162.09 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C33H44N3O 498.34844; found 498.34717.
3-Benzyloxy-2′-benzyl-2′H- [1,2,3]triazolo[4′,5′:16,17]estra-1,3,5(10)-triene (4e). Colorless oil (eluent: petroleum ether–ethyl acetate, 5
:1, yield 19%); 1H NMR (CDCl3, 400 MHz) δ: 1.03 (s, 3H, H-18); 1.51–2.02 (overlapping signals, 6H, H-8, H-7a, H-7b, H-12a, 2 H-11); 2.14–2.24 (m, 1H, H-14); 2.25–2.52 (overlapping signals, 3H, H-9, H-12b, H-15b); 2.79 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 2.84–3.00 (m, 2H, H-6); 5.04 (s, 2H, O-CH2C6H5); 5.51 (s, 2H, N-CH2C6H5); 6.74 (d, 1H, J = 2.4 Hz, H-4); 6.79 (dd, 1H, J2,1 = 8.4 Hz, J2,4 = 2.4 Hz, H-2); 7.21 (d, 1H, J = 8.8 Hz, H-1); 7.25–7.40 (overlapping signals, 10H, 2× C6H5). 13C NMR (CDCl3, 101 MHz) δ: 18.37 (C-18); 24.20 (C-15); 26.09 (C-11); 27.57 (C-7); 29.61 (C-6); 34.01 (C-12); 37.40 (C-8); 40.36 (C-13); 44.40 (C-9); 58.40 (N-CH2C6H5); 60.65 (C-14); 69.98 (CH2C6H5); 112.39 (C-2); 114.91 (C-4); 126.11 (C-1); 127.46 (C-2 and C-6 from C6H5); 127.90 (C-4 from C6H5); 128.02 (aromatic C from N-Bn); 128.57 (C-3 and C-5 from C6H5); 128.71 (aromatic C from N-Bn); 132.58 (C-5); 136.20 (aromatic C from N-Bn); 137.28 (C-1 from C6H5); 137.74 (C-10); 152.95 (C-16); 156.89 (C-3); 162.76 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C32H34N3O 476.26964; found 476.27012.
2′-(2′′-Aminoethyl)-3β-hydroxy-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3f). Colorless oil (eluent: dichloromethane–methanol, 20
:1, yield 30%); 1H NMR (DMSO-d6, 400 MHz) δ: 0.92 (s, 3H, H-18); 1.00 (s, 3H, H-19); 1.02–1.10 (overlapping signals, 2H, H-1a, H-9); 1.35–2.20 (overlapping signals, 13H, H-1b, 2 H-2, 2 H-4, 2 H-7, H-8, 2 H-11, 2 H-12, H-14); 2.32 (dd, 1H, Jgem = 14.4 Hz, J15b,14 = 2.3 Hz, H-15b); 2.58 (dd, 1H, Jgem = 14.4 Hz, J15a,14 = 6.4 Hz, H-15a); 2.94 (t, 2H, J = 6.4 Hz, –CH2CH2NH2); 4.24 (t, 2H, J = 6.4 Hz, –CH2CH2NH2); 5.26–5.33 (m, 1H, H-6). 13C NMR (DMSO-d6, 101 MHz) δ: 18.41 (C-18); 19.56 (C-19); 20.42 (C-11); 24.07 (C-15); 30.64 (C-8); 31.23 (C-7); 31.86 (C-2); 34.08 (C-12); 36.84 (C-10); 37.20 (C-1); 39.85 (C-13); 42.03 (–CH2CH2NH2); 42.68 (C-4); 50.41 (C-9); 57.37 (–CH2CH2NH2); 61.42 (C-14); 70.44 (C-3); 120.48 (C-6); 142.04 (C-5); 152.05 (C-16); 161.95 (C-17). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H33N4O 357.26489; found 357.26489.
2′-Ethoxycarbonylmethyl-3β-hydroxy-2′H-[1,2,3]triazolo[4′,5′:16,17]androst-5-ene (3g). Colorless oil (eluent: petroleum ether–acetone, 10
:1, yield 18%); 1H NMR (CDCl3, 400 MHz) δ: 1.05 (s, 3H, H-18); 1.09 (s, 3H, H-19); 1.10–1.18 (overlapping signals, 2H, H-1a, H-9); 1.27 (t, 3H, J = 7.2 Hz, COOCH2CH3); 1.46–1.77 (overlapping signals, 5H, H-2a, H-7a, 2 H-11, H-12a); 1.83–1.95 (overlapping signals, 3H, H-1b, H-2b, H-8); 1.97–2.44 (overlapping signals, 6H, 2 H-4, H-7b, H-12b, 2 H-14, H-15b); 2.73 (dd, 1H, Jgem = 14.8 Hz, J15a,14 = 6.4 Hz, H-15a); 3.49–3.59 (m, 1H, H-3); 4.23 (q, 2H, J = 7.2 Hz, COOCH2CH3); 5.12 (s, 2H, CH2COOCH2CH3); 5.37–5.42 (m, 1H, H-6). 13C NMR (CDCl3, 101 MHz) δ: 14.09 (COOCH2CH3); 17.98 (C-18); 19.38 (C-19); 20.37 (C-11); 24.35 (C-15); 30.70 (C-8); 31.41 (C-7); 31.58 (C-2); 33.75 (C-12); 36.81 (C-10); 37.09 (C-1); 39.85 (C-13); 42.24 (C-4); 50.49 (C-9); 55.11 (CH2COOCH2CH3); 61.42 (C-14); 61.98 (COOCH2CH3); 71.64 (C-3); 120.94 (C-6); 141.15 (C-5); 153.82 (C-16); 163.49 (C-17); 167.56 (COOCH2CH3). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H34N3O3 400.25947; found 400.25943.
:1) was added to pH 1, and extracted with dichloromethane (3 × 20 mL). The combined organic extracts were dried and evaporated. The crude product was purified by column chromatography (petroleum ether–ethyl acetate, 8:1) affording pure compound 3h at a yield of 32%. Mp 204–205 °C.
1H NMR (CDCl3, 400 MHz) δ: 1.11 (s, 6H, H-18 and H-19); 1.12–1.19 (overlapping signals, 2H, H-1a, H-9); 1.56–1.79 (overlapping signals, 5H, H-2a, H-7a, 2 H-11, H-12a); 1.85–2.13 (overlapping signals, 6H, H-1b, H-2b, H-7b, H-8, 2 H-14); 2.04 (s, 3H, CH3CO-O); 2.24–2.50 (overlapping signals, 4H, H-4, H-12b, H-15b); 2.73 (s, 3H, CH3CO-N); 2.79 (dd, 1H, Jgem = 15.6 Hz, J15a,14 = 6 Hz, H-15a); 4.56–4.68 (m, 1H, H-3); 5.39–5.45 (m, 1H, H-6). 13C NMR (CDCl3, 101 MHz) δ: 17.49 (C-18); 19.30 (C-19); 20.16 (C-11); 21.42 (CH3CO-O); 22.35 (CH3CO-N); 24.03 (C-15); 27.67 (C-2); 30.86 (C-8); 31.32 (C-7); 33.14 (C-2); 36.80 (C-1); 36.87 (C-10); 38.06 (C-4); 39.66 (C-13); 50.16 (C-9); 60.85 (C-14); 73.66 (C-3); 121.55 (C-6); 141.15 (C-5); 158.54 (C-16); 166.54 (CH3CO-N); 167.11 (C-17); 170.59 (CH3CO-O). HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C23H31N3O3Na 420.22576; found 420.22597.
3.2. Fluorescent yeast-based biosensor
Relative binding affinities of steroid derivatives 3a–h and 4a–e for ERα-LBD, ERβ-LBD, AR-LBD and GR-LBD were assessed using a fluorescent yeast-based biosensor as previously described.42,43,47,48S. cerevisiae FY250 strain (MATα, ura3–52, his3Δ200, leu2Δ1, trp1Δ63) and plasmid constructs pRF4–6-ERα LBD-EYFP, pRF4–6-ERβ LBD-EYFP, pRF4–6-AR LBD-EYFP and pRF4–6-GR LBD-EYFP used in the present study were provided by Dr. Blake Peterson, University of Kansas.66 Yeast transformations were performed using a lithium acetate/polyethylene glycol procedure and cells were plated on synthetic dropout medium containing 6.7 g L−1 yeast nitrogen base w/o amino acids and ammonium sulfate, 1.92 g L−1 yeast synthetic drop-out medium supplement lacking tryptophan, 2% agar and 2% glucose and incubated at 30 °C. Transformed yeast cells were preinoculated in the same drop-out liquid medium containing 2% raffinose and grown to saturation in a Biosan orbital shaker-incubator ES-20/60 at 28 °C, with an orbital shaking speed of 140 rpm. After overnight incubation, cells were diluted to achieve an initial optical density at 600 nm (OD600nm) of 0.15 and allowed to grow until the logarithmic phase (OD600nm of ∼0.6). Production of receptor-LBD–YFP fusions was induced with 2% galactose. Cells were exposed to steroid compounds 3a–h and 4a–e at a final concentration of 10 μM in ER- and AR- and 100 μM in GR-binding assays for 14–16 h at 25 °C in the dark. Cells treated with dimethyl sulfoxide (DMSO)-only served as a solvent control. Non-induced cells grown in the absence of galactose were used to determine non-specific fluorescence background effects. Positive control ligands for yeast cells expressing ERα/β-, AR- and GR-LBD were estrone (negative control AR ligand), androstenedione (negative control ER ligand) and prednisolone, respectively, while estradiol was used as a negative control in the GR-binding assay. Fluorescence levels were measured in a 96-well microplate (Greiner bio-one). All experiments were performed in triplicate at a controlled temperature of 25 °C. Fluorescence readings were conducted using a Fluoroskan Ascent FL fluorometer setting excitation and emission parameters at 485 nm and 538 nm, respectively. Relative binding affinities of ligands were expressed as fold fluorescence enhancements relative to the DMSO control (normalized to 1) and were calculated as described.47 Histograms were created using Origin Pro 9 (Origin Lab, Northampton, MA, USA). Error bars indicate propagated standard errors of the mean. Yeast cells selected on the basis of fluorimetric signal were observed under a fluorescence microscope Olympus BX51 using a FITC filter.3.3. Heterologous expression, purification and enzymatic activity assay of human AKR1C3 and AKR1C4
Escherichia coli strain BL21(DE3) was obtained from Novagen (Merck, Germany). Plasmid constructs pET28b(+)-AKR1C3 and pET28b(+)-AKR1C4 were provided by Prof. Dr. Chris Bunce (University of Birmingham).67 Protocols for expression and purification of active human AKR1C3 and AKR1C4 from Escherichia coli were identical and performed as stated in our previous work.44,58E. coli cells harboring pET28b(+)-AKR1C3 or pET28b(+)-AKR1C4 plasmids were grown in 1 L of LB medium (10 g L−1 tryptone, 5 g L−1 yeast extract, 5 g L−1 NaCl) supplemented with 50 μg mL−1 kanamycin at 37 °C to an OD600nm of 0.4–0.6. Protein expression was then induced with 0.5 mM isopropyl 1-thio-D-galactopyranoside (IPTG) and cells were incubated at 25 °C for an additional 20 h. The induced cells were harvested by centrifugation at 5000×g for 10 minutes, and resuspended in 20 mM Tris-HCl pH 8.0, 5 mM imidazole. AKR1C3 and AKR1C4 were produced as N-terminally His-tagged proteins and purified by immobilized Ni-affinity chromatography using HisTrap IMAC column and size-exclusion chromatography using Bio-Gel P-10. Cells were lysed using a combination of three freeze–thaw cycles with 1 mg mL−1 lysozyme and 7 cycles of sonication treatment (Soniprep 150) on ice. Following lysis, the lysate was centrifuged for 45 minutes at 13400×g, 4 °C and a clear supernatant was loaded onto a HisTrap column previously equilibrated in 20 mM Tris-HCl pH 8.0, 0.5 M NaCl. The column was then washed in the same buffer containing 20 mM imidazole (10 column volumes) and eluted using 400 mM imidazole (5 column volumes). Eluted fractions with the highest AKR content were pooled and desalted on a size-exclusion column and residual imidazole was removed. The protein was frozen in aliquots and stored in 20 mM Tris-HCl pH 8.0, 0.1 M NaCl, 10% glycerol and 1 mM DTT at −80 °C until use. The protein concentration was determined by the Bradford method,68 using bovine serum albumin (BSA) as a standard. The purity of the protein samples was verified by SDS-PAGE. To test the potential of steroid derivatives 3a–h and 4a–e to inhibit the AKR1C3 and AKR1C4 activity, a standard enzymatic assay based on monitoring the decrease of NADPH cofactor fluorescence was performed, as previously described.42,58 Compounds 3a–h and 4a–e were screened for their ability to inhibit the reduction of a general AKR1C substrate, 9,10-phenanthrenequinone (PQ). The assay mixture (total volume 300 μL) containing 4 μM PQ for AKR1C3 and 17 μM PQ for AKR1C4 assay in 100 mM potassium phosphate buffer pH 6.0 was pipetted in a 96-well microplate in duplicate. The concentration of NADPH was maintained at 250 μM. The reaction was initiated by adding the enzyme (80 μg mL−1 AKR1C3; 160 μg mL−1 AKR1C4) and conducted at 37 °C. The fluorescence intensity (excitation 340 nm, emission 460 nm) was continuously measured in kinetic mode over 10 min at 30 s intervals using a Fluoroskan Ascent FL fluorometer. Inhibition potentials of steroid triazoles and ibuprofen (a known AKR1C inhibitor, control) were tested at a final concentration of 33 μM. A decrease in NADPH fluorescence in the reaction mixture (reaction) compared to the control in the absence of enzyme (blank) indicated AKR activity. The normalized values of fluorescence intensities at appropriate wavelengths were plotted as a function of reaction time and analyzed by linear regression using Origin Pro 9 (Origin Lab, Northampton, MA, USA). The inhibition potential of the tested compounds and ibuprofen was calculated from the slope of the linear part of the curve. The enzyme activity without added inhibitor was considered to represent 100% activity. For compound 3h, a sigmoidal dose–response curve for AKR1C3 inhibition was obtained by measuring the effect of increasing inhibitor concentrations (0, 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50, 100, and 133 μM). IC50 defined as the concentration of compound required for 50% of maximum enzyme inhibition in vitro was estimated using an online tool (http://www.ic50.org/).
3.4. Molecular docking
Compounds that inhibited >50% of AKR1C3 activity were chosen for further analysis by molecular docking. Compounds 3h (71.1% inhibition) and 3f (69.9%) were modeled using the X-ray structure of 3a as a template in the program AVOGADRO.69 The geometry of three-dimensional models of 3h and 3f was energy minimized using AVOGADRO using the conjugate gradient algorithm in an MMF94 force-field with a convergence setting of 10e−7. Gasteiger partial charges were calculated for ligand atoms and ligand coordinate files were converted to the PDBQT format for molecular docking using the script, ‘ligand.c’, in the program VEGA ZZ (version 3.2.1.33). For molecular docking, the ‘receptor’ used was an X-ray structure of AKR1C3 in complex with EM1404, a steroidal inhibitor that has a lactone ring attached to the D-ring of the steroid core (PDB 1ZQ5).62 Using the script ‘receptor.c’ in VEGA ZZ, hydrogen atoms were added to the receptor; Gasteiger partial charges were calculated and the coordinate file was converted to the PDBQT format.70 Molecular docking simulations were conducted for 3h and 3f with Autodock Vina using the program PyRx.60,61 Docking simulations were centered on the AKR1C3 active site (x = 31.935, y = 27.429, z = 15.149) with a search space of 25 × 25 × 25 Å and an exhaustiveness setting of 32. As a positive control, the ligand present in the X-ray structure of AKR1C3 (EM1404) was redocked into the active site using the same parameters. Top ranking docking poses for 3h, 3f and EM1404 were selected based on the predicted binding energy (kcal mol−1) and further analyzed using the program PyMol. Docking results were analyzed by superposition onto the experimental X-ray structure of AKR1C3 in complex with EM1404. 3D plots of the docking results were created using PyMol.71 2D plots of the docking results were created using the program LigPlot+ and labeled using PowerPoint.63 Molecular docking was also used to model interactions between 3a, 3h and 4e and ERβ-LBD (ESI† Fig. S59). Autodock Vina was used as described for AKR1C3, with the following modifications: coordinates from a structure of ERβ in complex with a triazine compound (PDB 1NDE) were used as the receptor. Simulations were centered on the ERβ active site (x = 110.144, y = 8.060, z = −108.215) with an exhaustiveness setting of 16.3.5. Cell culture
For the cell viability experiments, the following cancer cell lines were utilized: HCT116, Huh7, MCF7 and MDA-MB-231. All cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with high glucose, 10% fetal bovine serum (FBS), 100 units per mL penicillin and 100 μg mL−1 streptomycin and were cultured in a humidified atmosphere with 5% CO2 at 37 °C. Additionally, the noncancerous hTERT-immortalized human foreskin fibroblasts BJ-5ta were cultured in DMEM supplemented with high glucose, 20% Medium 199, 10% FBS, 100 units per mL penicillin and 100 μg mL−1 streptomycin.3.6. Cell viability assay
In the initial phase of cytotoxicity screening, each of the four cancer cell lines was seeded in 96-well plates and treated with varying concentrations of the test compounds, ranging from 0.01 to 100 μM for a duration of 72 h. The grade of compound solubility determined their dissolution in DMSO at stock concentrations of 10, 50 or 100 mM. Corresponding controls were selected based on the stock concentration of each compound, resulting in a final DMSO concentration of 0.1%, 0.2% or 1% in growth medium. Following a 72 h treatment period, cells were incubated with 0.01 mg mL−1 resazurin for an additional 2 h, after which fluorescence was quantified at 584 nm using a plate reader. The protocol was adapted from Kumar et al. 2018.72 Compounds eliciting a minimum 50% reduction in cell viability across at least three of the four tested cell lines were classified as toxic and subjected to further evaluation via another round of resazurin cell viability assay. In this subsequent phase, all cell lines were exposed to a narrower concentration range spanning from 1 to 100 μM. Additionally, the substance identified as cytotoxic was also tested on BJ-5ta cells, employing concentrations ranging from 0.01 to 100 μM over a 72 h period. For statistical analysis, a one-way ANOVA with Dunnett's test was performed. This test is used to identify which treatment groups differ significantly from the control group.4. Conclusions
In the present study, new steroid triazoles were synthesized. We identified three compounds, 3a, 3h and 4e that showed specific binding affinity for ERβ-LBD and four compounds, 3b, 3e, 4a and 4b that displayed high binding affinity for GR-LBD. Compounds 3h and 3f were potent and selective AKR1C3 inhibitors and 3f was cytotoxic against human cancer cell lines. These results suggest that 3f could be a potential candidate for the development of anticancer steroidal triazoles.Data availability
Data are available upon request from the authors.Author contributions
Branislava Tenjović – investigation; Sofija Bekić – investigation, formal analysis, methodology, writing original draft, visualization; Andjelka Ćelić – formal analysis, writing original draft, visualization; Edward Petri – investigation, formal analysis, writing original draft, visualization; Julia Scholda – investigation, formal analysis, writing original draft, visualization; Florian Kopp – formal analysis, writing original draft, visualization; Marija Sakač – supervision, writing – review & editing, visualization; Andrea Nikolić – investigation, formal analysis, methodology, writing original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support of the Ministry of Science, Technological Development and Innovation of the Republic of Serbia (Grants No. 451-03-66/2024-03/ 200125 & 451-03-65/2024-03/200125) and the financial support of the Provincial Secretariat for Higher Education and Scientific Research of the Autonomous Province of Vojvodina (Project No. 142-451-3463/2023-01). Results on cancer cells were obtained within the framework of the bilateral cooperation between the Republic of Austria and the Republic of Serbia (Project No. OeAD RS15/2022 and 337-00-577/2021-09/27).References
- R. Minorics and I. Zupko, Anti-Cancer Agents Med. Chem., 2018, 18(5), 652 CrossRef CAS.
- M. Merlani, N. Nadaraia, L. Amiranashvili, A. Petrou, A. Geronikaki, A. Ciric, J. Glamoclija and M. Sokovic, Molecules, 2023, 28, 1167 CrossRef CAS.
- L. Amiranashvili, N. Nadaraia, M. Merlani, C. Kamoutsis, A. Petrou, A. Geronikaki, P. Pogodin, D. Druzhilovskiy, V. Poroikov, A. Ciric, J. Glamočlija and M. Sokovic, Antibiotics, 2020, 9(5), 224 CrossRef CAS.
- A. Ansari, A. Ali, M. Asif, M. A. Rauf, M. Owais and Shamsuzzaman, Steroids, 2018, 134, 22–36 CrossRef CAS.
- C. M. Zobniw, A. Causebrook and M. K. Fong, Res. Rep. Urol., 2014, 6, 97 CAS.
- M. Shiota, R. Inoue, K. Tashiro, K. Kobayashi, S. Horiyama, H. Kanji, M. Eto, S. Egawa, J. Haginaka and H. Matsuyama, J. Clin. Pharmacol., 2023, 63(4), 445 CrossRef CAS.
- Y. Wang, F. Jing and H. Wang, Adv. Ther., 2022, 39(2), 862 CrossRef CAS PubMed.
- K. M. Elattar and A. El-Mekabaty, J. Heterocyclic Chem., 2021, 58(2), 389 CrossRef CAS.
- M. Monier, A. El-Mekabaty, D. Abdel-Latif, B. Doğru Mert and K. M. Elattar, Steroids, 2020, 154, 108548 CrossRef CAS.
- K. Sharma, H. Kumar and Priyanka, Steroids, 2023, 191, 109171 CrossRef CAS.
- Y. Hou, C. Shang, T. Meng and W. Lou, Steroids, 2021, 171, 108852 CrossRef CAS.
- A. Bayazeed, R. B. Alnoman, K. Alatawi, O. M. Alatawi, A. M. Alqahtani, M. Mojally, N. A. Alenazi and N. M. El-Metwaly, J. Saudi Chem. Soc., 2023, 27(6), 101754 CrossRef CAS.
- K. Taruneshwar, A. Shome, Chahat and P. A. Chawla, Bioorg. Chem., 2023, 138, 106680 CrossRef.
- M. M. Alam, Arch. Pharm., 2022, 355(1), e2100158 CrossRef PubMed.
- D. S. Agarwal, R. Sakhuja, R. M. Beteck and L. J. Legoabe, Steroids, 2023, 197, 109258 CrossRef CAS.
- X. M. Chu, C. Wang, W. L. Wang, L. L. Liang, W. Liu, K. K. Gong and K. L. Sun, Eur. J. Med. Chem., 2019, 166, 206 CrossRef CAS.
- A. Pachuta-Stec, Mini-Rev. Med. Chem., 2022, 22(7), 1081 CrossRef CAS PubMed.
- D. Hadjipavlou-Litina, I. E. Głowacka, J. Marco-Contelles and D. G. Piotrowska, Antioxidants, 2023, 12(1), 36 CrossRef CAS.
- R. Matta, J. Pochampally, B. N. Dhoddi, S. Bhookya, S. Bitla and A. G. Akkiraju, BMC Chem., 2023, 17, 61 CrossRef CAS PubMed.
- S. A. El-Sebaey, ChemistrySelect, 2020, 5(37), 11654 CrossRef CAS.
- I. A. Seliem, S. S. Panda, A. S. Girgis, Y. Moatasim, A. Kandeil, A. Mostafa, M. A. Ali, E. S. Nossier, F. Rasslan, A. M. Srour, R. Sakhuja, T. S. Ibrahim, Z. K. M. Abdel-samii and A. M. M. Al-Mahmoudy, Bioorg. Chem., 2021, 114, 105117 CrossRef CAS.
- M. Strzelecka and P. Świątek, Pharmaceuticals, 2021, 14(3), 224 CrossRef CAS PubMed.
- F. Gao, T. Wang, J. Xiao and G. Huang, Eur. J. Med. Chem., 2019, 173, 274 CrossRef CAS PubMed.
- N. Kuntala, J. Mareddy, J. Rani Telu, V. Banothu, S. Pal and J. Shree Anireddy, J. Heterocyclic Chem., 2021, 58(10), 2018 CrossRef CAS.
- T. Azim, M. Wasim, M. S. Akhtar and I. Akram, BMC Complementary Med. Ther., 2021, 21, 304 CrossRef CAS.
- A. Sharma, A. K. Agrahari, S. Rajkhowa and V. K. Tiwari, Eur. J. Med. Chem., 2022, 238, 114454 CrossRef CAS PubMed.
- K. Bozorov, J. Zhao and H. A. Aisa, Bioorg. Med. Chem., 2019, 27(16), 3511 CrossRef CAS PubMed.
- M. A. Abdalah, H. Salah, S. M. Gomha and H. Rashdan, Egypt. J. Chem., 2024, 67(13), 287 CrossRef.
- M. Marzi, M. Farjam, Z. Kazeminejad, A. Shiroudi and A. Kouhpayeh, J. Chem., 2022, 2022, 7884316 Search PubMed.
- D. P. Vala, R. M. Vala and H. M. Patel, ACS Omega, 2022, 7(42), 36945 CrossRef CAS PubMed.
- S. Gupta, N. Chandna, A. K. Singh and N. Jain, J. Org. Chem., 2018, 83(6), 3226 CrossRef CAS.
- G. Luo, C. Sun, Y. Li, X. Li and Z. Zhao, RSC Adv., 2018, 8(49), 27610 RSC.
- K. D. Khalil, S. M. Riyadh, S. M. Gomha and I. Ali, Int. J. Biol. Macromol., 2019, 130, 928 CrossRef CAS PubMed.
- A. E. Kulmany, B. E. Herman, I. Zupko, M. Sinreih, T. Lanisnik Rizner, M. Savić, A. Oklješa, A. Nikolić, V. Nagy, I. Ocsovszki, M. Szecsi and S. Jovanović-Šanta, J. Steroid Biochem. Mol. Biol., 2021, 214, 105997 CrossRef CAS PubMed.
- T. M. Penning, P. Wangtrakuldee and R. J. Auchus, Endocr. Rev., 2019, 40(2), 447 CrossRef PubMed.
- T. M. Penning, S. Jonnalagadda, C. P. Trippier and T. L. Rižner, Pharmacol. Rev., 2021, 73(3), 1150 CrossRef CAS PubMed.
- S. He, X. Chu, Y. Wu, J. Jiang, P. Fang, Y. Chen, Y. Liu, Z. Qiu, Y. Xiao, Z. Li, D. Pan, Q. Zhang, H. Xie, S. Xing, F. Feng, W. Liu, Q. Guo, L. Zhao, P. Yang and H. Sun, J. Med. Chem., 2023, 66(14), 9537 CrossRef CAS PubMed.
- Y. J. Ko and S. P. Balk, Curr. Pharm. Biotechnol., 2004, 5(5), 459 CAS.
- R. Buonaiuto, G. Neola, S. C. Cecere, A. Caltavituro, A. Cefaliello, E. Pietroluongo, P. De Placido, M. Giuliano, G. Arpino and C. De Angelis, Biomolecules, 2023, 13(4), 653 CrossRef CAS.
- K. T. Lin and L. H. Wang, Steroids, 2016, 111, 84 CrossRef CAS PubMed.
- M. N. Sakač, A. R. Gaković, J. J. Csanadi, E. A. Djurendić, O. Klisurić, V. Kojić, G. Bogdanović and K. M. Penov Gaši, Tetrahedron Lett., 2009, 50, 4107 CrossRef.
- I. Z. Kuzminac, D. S. Jakimov, S. S. Bekić, A. S. Ćelić, M. A. Marinović, M. P. Savić, V. N. Raičević, V. V. Kojić and M. N. Sakač, Bioorg. Med. Chem., 2021, 30, 115935 CrossRef CAS.
- M. P. Savić, J. J. Ajduković, J. J. Plavša, S. S. Bekić, A. S. Ćelić, O. R. Klisurić, D. S. Jakimov, E. T. Petri and E. A. Djurendić, MedChemComm, 2018, 9(6), 969 RSC.
- T. L. Šestić, J. J. Ajduković, S. S. Bekić, A. S. Ćelić, S. T. Stojanović, S. J. Najman, M. A. Marinović, E. T. Petri, D. Đ. Škorić and M. P. Savić, J. Steroid Biochem. Mol. Biol., 2023, 233, 106362 CrossRef.
- G. Chaput and N. Sumar, Can. Fam. Physician, 2022, 68(4), 271 CrossRef PubMed.
- A. E. Harris, V. M. Metzler, J. Lothion-Roy, D. Varun, C. L. Woodcock, D. B. Haigh, C. Endeley, M. Haque, M. S. Toss, M. Alsaleem, J. L. Persson, L. J. Gudas, E. Rakha, B. D. Robinson, F. Khani, L. M. Martin, J. E. Moyer, J. Brownlie, S. Madhusudan, C. Allegrucci, V. H. James, C. S. Rutland, R. G. Fray, A. Ntekim, S. de Brot, N. P. Mongan and J. N. Jeyapalan, Front. Endocrinol., 2022, 13, 1006101 CrossRef PubMed.
- S. S. Bekić, M. A. Marinović, E. T. Petri, M. N. Sakač, A. R. Nikolić, V. V. Kojić and A. S. Ćelić, Steroids, 2018, 130, 22 CrossRef PubMed.
- S. Bjedov, S. Bekić, M. Marinović, D. Škorić, K. Pavlović, A. Ćelić, E. Petri and M. Sakač, J. Serb. Chem. Soc., 2023, 88(2), 123 CrossRef CAS.
- S. Dhakal and I. Macreadie, Microorganisms, 2022, 10(9), 1772 CrossRef CAS.
- B. S. Katzenellenbogen and J. A. Katzenellenbogen, Breast Cancer Res., 2000, 2, 1 Search PubMed.
- Y. Zhou and X. Liu, Biomark. Res., 2020, 8(1), 39 Search PubMed.
- J. Datta, N. Willingham, J. M. Manouchehri, P. Schnell, M. Sheth, J. J. David, M. Kassem, T. A. Wilson, H. S. Radomska, C. C. Coss, C. E. Bennett, R. K. Ganju, S. D. Sardesai, M. Lustberg, B. Ramaswamy, D. G. Stover and M. A. Cherian, Front. Oncol., 2022, 12, 857590 Search PubMed.
- Z. Jin, H. Lin, S. Srinivasan, J. C. Nwachukwu, N. Bruno, P. R. Griffin, K. W. Nettles and T. M. Kamenecka, Bioorg. Med. Chem. Lett., 2017, 27(2), 347 CrossRef CAS PubMed.
- T. Diab, S. S. AlKafaas, T. I. Shalaby and M. Hessien, Bioorg. Chem., 2020, 99, 103792 CrossRef CAS PubMed.
- J. M. Pang, Y. C. Huang, S. P. Sun, Y. R. Pan, C. Y. Shen, M. C. Kao, R. H. Wang, L. H. Wang and K. T. Lin, Steroids, 2020, 164, 108738 CrossRef CAS PubMed.
- T. Ostlund, F. Alotaibi, J. Kyeremateng, H. Halaweish, A. Kasten, S. Iram and F. Halaweish, Steroids, 2022, 177, 108950 CrossRef CAS PubMed.
- P. Brozic, S. Turk, T. Lanisnik Rizner and S. Gobec, Curr. Med. Chem., 2011, 18(17), 2554 CrossRef CAS.
- M. A. Marinović, S. S. Bekić, M. Kugler, J. Brynda, J. Škerlová, D. Đ. Škorić, P. Řezáčová and A. S. Ćelić, RSC Med. Chem., 2023, 14(2), 341 RSC.
- M. C. Byrns, Y. Jin and T. M. Penning, J. Steroid Biochem. Mol. Biol., 2011, 125(1–2), 95 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455 CrossRef CAS.
- S. Dallakyan and A. J. Olson, Methods Mol. Biol., 2015, 1263, 243 CrossRef CAS.
- W. Qiu, M. Zhou, M. Mazumdar, A. Azzi, D. Ghanmi, F. Labrie and S. X. Lin, J. Biol. Chem., 2007, 282(11), 8368 CrossRef CAS PubMed.
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51(10), 2778 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2003, 267(17), 5421 CrossRef PubMed.
- R. Hamid, Y. Rotshteyn, L. Rabadi, R. Parikh and P. Bullock, Toxicol. In Vitro, 2004, 18(5), 703 CrossRef CAS PubMed.
- S. S. Muddana and B. R. Peterson, ChemBioChem, 2003, 4(9), 848 CrossRef CAS PubMed.
- N. J. Davies, R. E. Hayden, P. J. Simpson, J. Birtwistle, K. Mayer, J. P. Ride and C. M. Bunce, Cancer Res., 2009, 69(11), 4769 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72(1–2), 248 CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4(1), 1 Search PubMed.
- A. Pedretti, A. Mazzolari, S. Gervasoni, L. Fumagalli and G. Vistoli, Bioinformatics, 2021, 37(8), 1174 CrossRef CAS PubMed.
- W. L. DeLano, Protein Crystallography, 2002, 40(1), 82 Search PubMed.
- P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harb. Protoc., 2018, 462 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00297k |
| This journal is © The Royal Society of Chemistry 2025 |