
Design, synthesis, and evaluation of benzhydrylpiperazine-based novel dual COX-2/5-LOX inhibitors with anti-inflammatory and anti-cancer activity†
Poorvi
Saraf
a,
Bhagwati
Bhardwaj
a,
Akash
Verma
a,
Mohammad Aquib
Siddiqui
b,
Himanshu
Verma
b,
Pradeep
Kumar
c,
Samridhi
Srivastava
a,
Sairam
Krishnamurthy
b,
Saripella
Srikrishna
c and
Sushant Kumar
Shrivastava
 *a
*a
aPharmaceutical Chemistry Research Laboratory, Department of Pharmaceutical Engineering & Technology, Indian Institute of Technology (Banaras Hindu University), Varanasi-221005, India. E-mail: skshrivastava.phe@itbhu.ac.in; Tel: +91 945 2156 527
bPharmacology Research Laboratory, Department of Pharmaceutical Engineering & Technology, Indian Institute of Technology (Banaras Hindu University), Varanasi-221005, India
cDepartment of Biochemistry, Institute of Science, Banaras Hindu University, Varanasi-221005, India
First published on 17th October 2024
Abstract
Piperazine derivatives were screened using the ChEMBL database, paving the way for the design, synthesis, and evaluation of a novel series of dual COX-2/5-LOX inhibitors and identifying their role in mitigating cancer cell proliferation. Compound 9d with 4-Cl substitution at the terminal phenyl ring showed promising inhibition of COX-2 (IC50 = 0.25 ± 0.03 μM) and 5-LOX (IC50 = 7.87 ± 0.33 μM), outperforming the standards celecoxib (IC50 = 0.36 ± 0.023 μM) and zileuton (IC50 = 14.29 ± 0.173 μM), respectively. The two most active derivatives 9d and 9g indicated a significant anti-inflammatory response in a paw edema model by inhibiting PGE2, IL-6, and TNF-α and an increase in IL-10 concentrations. Interestingly, 9d effectively reduced pain by 55.78%, closely comparable to the 59.09% exhibited by the standard indomethacin, and was also devoid of GI, liver, kidney, and cardiac toxicity. Furthermore, 9d demonstrated anti-cancer potential against in vitro A549, COLO-205, and MIA-PA-CA-2 human cancer cell lines and an in vivo Drosophila cancer model. The pharmacokinetic investigations revealed that 9d has good oral absorption characteristics.
1. Introduction
Inflammation is a biological defence mechanism against harmful stimuli controlled by the body's immune system. Inflammation-related disorders may develop due to an aberrantly extended host's defensive response when tissue loses homeostasis.1 Inflammation is a multifaceted condition generally identified by an outbreak of biochemical mediators such as prostaglandins (PGs), leukotrienes (LTs), cytokines, platelet-activating factor (PAF), histamine, and reactive oxygen species (ROS).2 A specific inflammatory mechanism can trigger inflammation but is more likely to manifest with other inflammatory pathways.3 The involvement of the mediators of arachidonic acid (AA) biosynthetic pathways in the pathogenesis of cardiovascular and cancer diseases has sparked considerable interest in their inflammatory pathways.4,5 Non-steroidal anti-inflammatory medicines (NSAIDs), primarily used to treat inflammatory disorders, have also played an essential role in anti-inflammatory function in conditions such as cardiovascular and cancer diseases. Also, NSAIDs confirm their place in the WHO Essential Medicines Model 23rd List.6Cyclooxygenase (COX) and lipoxygenase (LOX) enzymes contribute significantly to AA metabolism. The COX enzyme produces prostanoids i.e., PGs and thromboxane A2 (TXA2), and the lipoxygenase (5-LOX) enzyme generates LTs.7 NSAIDs exert action by inhibiting the COX enzymes. COX-1, the constitutive form, is responsible for the body's defence mechanism, while COX-2 manifests during the inflammatory condition.8 The inhibition of constitutive COX-1 is believed to be the trigger of NSAID-induced gastrointestinal (GI) adverse effects and therefore, selective COX-2 inhibitors have been developed.9,10 Selective COX-2 inhibitors typically display bulkier moieties that interact with the COX-2 specific active site and preclude the molecule from fitting within the relatively smaller COX-1 channel.11 Although selective COX-2 inhibitors were devoid of GI toxicity, they were implicated in serious cardiovascular disease (CVD) risks, stroke, and cardiac arrest.12
The LOX pathway, which is parallel to the COX pathway in AA metabolism, can access more AA due to COX inhibition.13 The 5-lipoxygenase activating protein (FLAP) activates the liberated AA to trigger the 5-LOX enzyme and prominently shunts LT synthesis, which mediates inflammatory reactions by promoting bronchoconstriction, and edema indicating adverse effects in pathological illnesses like CVD, asthma, inflammatory bowel disease, and psoriasis.14 Dual COX-2/5-LOX inhibition is a proclaiming strategy that focuses on retaining the fundamental activity of NSAIDs, culminating in safer and efficacious anti-inflammatory drugs.15 Additionally, some studies suggested that inhibiting 5-LOX indirectly reduces the levels of inflammatory cytokines and dual inhibition has neuroprotective action in neurodegenerative diseases.16
Licofelone and tepoxalin were developed as dual COX/LOX inhibitors but could not reach the market due to mixed clinical results.17 Still, there is an unavailability of safer multifunctional anti-inflammatory medications. Thus, the prevailing therapeutic emphasis is on the design and development of novel compounds with impressive safety profiles instead of seeking higher potency variants of currently available drugs. The modern approach of simultaneously inhibiting multiple enzymes with a single inhibitor molecule has been adopted on the notion that inflammation is a multifactorial disease that is associated with numerous targets. This strategy might effectively combat the progression of multifaceted inflammatory diseases.
The piperazine framework represents a vital core having a medicinal significance which has shown potential for drug discovery and development of novel lead compounds.18,19 The piperazine framework has been reported to aid in the improvement of anti-inflammatory activity.20 Furthermore, several investigations have identified similarities between NSAIDS and benzhydrylpiperazine containing antihistamines (e.g. cetirizine, hydroxyzine) that might be explored to develop novel molecular hybrids with promising pharmacological properties.21,22 Based on these presumptions, initially, we have performed virtual screening (VS) of all piperazine-containing molecules obtained from the ChEMBL database against COX-2 enzymes. The structure-based drug design approach was utilized and 59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 090 piperazine-containing ligands from the ChEMBL database (https://www.ebi.ac.uk/chembl/, accessed in April 2022) were screened using the crystal structure of COX-2 (PDB code: 3LN1). The identified hit was employed to rationally design novel multifunctional anti-inflammatory compounds. The top two hits, ChEMBL342253 and ChEMBL4794855, displayed the common presence of benzhydrylpiperazine and displayed favourable docking results for the COX-2 enzyme, but they did not demonstrate potential binding against the 5-LOX enzyme. They failed to exhibit acceptable molecular stability in dynamic simulation studies (Fig. S1–S4, ESI†).
090 piperazine-containing ligands from the ChEMBL database (https://www.ebi.ac.uk/chembl/, accessed in April 2022) were screened using the crystal structure of COX-2 (PDB code: 3LN1). The identified hit was employed to rationally design novel multifunctional anti-inflammatory compounds. The top two hits, ChEMBL342253 and ChEMBL4794855, displayed the common presence of benzhydrylpiperazine and displayed favourable docking results for the COX-2 enzyme, but they did not demonstrate potential binding against the 5-LOX enzyme. They failed to exhibit acceptable molecular stability in dynamic simulation studies (Fig. S1–S4, ESI†).
The molecular hybridization approach was further employed to develop multifunctional ligands with improved molecular binding and stability against both COX-2 and 5-LOX enzymes by incorporating the pharmacophoric features of some known compounds, including hit molecules. Biphenyl rings are usually evident in coxibs, such as celecoxib, as they are necessary to bind to the active COX-2 pocket. This characteristic is also evident in licofelone, a dual COX/LOX inhibitor. The hit compounds from the ChEMBL database shared similarities with the scaffold found in licofelone and antihistamines, such as levocetirizine and hydroxyzine. Thus, considering several therapeutic benefits of the scaffold, it is retained in the designed molecules. Subsequently, 1,3,4-oxadiazole was selected as an auxiliary scaffold considering its multifunctional potential.23,24 We identified dual COX-2/5-LOX inhibitory potential in recently developed molecular hybrids containing 1,3,4-oxadiazole (A).9 Also, the experimental drug zibotentan, containing 1,3,4-oxadiazole, is under investigation for its anti-cancer activity.25,26 Based on the aforementioned information, a novel series of molecular hybrids have been developed (Fig. 1), and their molecular binding and stability were confirmed through in silico studies. The envisioned compounds have been successfully synthesized, characterized, and assessed biologically.
 | ||
| Fig. 1 Design strategy for novel benzhydrylpiperazine-based molecular hybrids. | ||
2. Results and discussion
2.1. Chemistry
The series of designed novel molecular hybrids (9a–u) were synthesized by connecting benzhydrylpiperazine and substituted phenyl oxadiazoles with a –CH2 linker as depicted in Scheme 1. In brief, Boc-piperazine first reacted with 1-chloro-4-(chloro(phenyl)methyl)benzene (3) to obtain benzhydrylpiperazine (5). In another reaction, benzoic acid esters were obtained by reacting substituted benzoic acids (6a–u) with hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) in acetonitrile (ACN), which eventually reacted with hydrazine hydrate to yield benzoic acid hydrazides (7a–u). Thereafter, benzoic acid hydrazides (7a–u) were converted into oxadiazole derivatives (8a–u) in a cyclization reaction. The 1H NMR spectra confirmed the formation of 8a–u by showing the presence of a methylene proton singlet in the δH = 4.01–5.11 ppm range, which is also confirmed by the presence of signals in the range of δc = 3.91–4.21 ppm in the 13C NMR spectra. In the final reaction, 8a–u reacted with benzhydrylpiperazine (5) with potassium hydroxide (KOH) in solvent dimethylformamide (DMF) via nucleophilic substitution reaction to yield the corresponding target compounds (9a–u). In the 1H NMR spectra, the disappearance of the –NH proton of benzhydrylpiperazine (5) confirmed the final product formation (9a–u) and was verified by the 13C NMR spectra. The 13C NMR spectra displayed a signal in the range of δc = 52.90–55.48 ppm for N–CH2. The high-resolution mass spectrometry (HRMS) results also confirmed the molecular weights of all synthesized derivatives. Additionally, the purity of the synthesized derivatives has also been verified by high-performance liquid chromatography (HPLC). | ||
| Scheme 1 Reagents and conditions: (a) DCM, Boc2O, stirring at 0–5 °C for 1–2 h; (b) toluene, stirring at 80–90 °C for 6–8 h; (c) HOBt, EDC, ACN, stirring for 2–4 h; hydrazine hydrate, 0–5 °C to RT; (d) POCl3, chloroacetic acid, reflux for 4–6 h; (e) DCM, CF3CO2H, 0 °C to RT; (f) DMF, KOH, RT, stirring for 4 h. | ||
2.2. Pharmacology/biology
2.2.1.1. COX-1 and COX-2 inhibition activities. The colorimetric inhibitor screening assay method was utilized to evaluate the inhibitory potential of the synthesized compounds on COX-1 and COX-2 enzymes.27 Indomethacin and celecoxib were utilized as the reference drugs. A range of submicromolar to low micromolar IC50 values were observed for the compounds (Table 1). All the compounds displayed COX-2 selectivity, and the most active compounds against COX-2 were 9d, 9e, 9g, and 9l, with IC50 values of 0.25 μM, 0.47 μM, 0.49 μM, and 0.48 μM, respectively. The calculated selectivity index (SI) confirmed the COX-2 selectivity of celecoxib and the synthesized compounds. Compound 9d with a SI of 133.59 emerged as the most potent COX-2 inhibitor, while indomethacin and celecoxib demonstrated SI values of 0.91 and 82.04, respectively.
| CMP | –R | IC50 COX-2 | IC50 COX-1 | Selectivity indexb | IC50 5-LOX |
|---|---|---|---|---|---|
| (μM)a | (μM)a | (μM)a | |||
| a All values are presented as the mean ± SD. b COX-2 selectivity index = IC50 of COX-1/IC50 of COX-2. nd: not determined. | |||||
| 9a | 3-OCH3 | 0.66 ± 0.076 | 36.26 ± 0.993 | 54.94 | 15.28 ± 0.219 |
| 9b | 4-OCH3 | 0.54 ± 0.074 | 34.46 ± 0.755 | 65.6 | 10.35 ± 0.110 |
| 9c | 3-Cl | 0.53 ± 0.061 | 32.03 ± 0.196 | 60.43 | 16.23 ± 0.018 |
| 9d | 4-Cl | 0.25 ± 0.030 | 34.07 ± 0.828 | 133.59 | 7.87 ± 0.334 |
| 9e | 2,4-diCl | 0.47 ± 0.014 | 30.32 ± 0.414 | 64.52 | 14.38 ± 0.630 |
| 9f | 4-Cl-3-NO2 | 0.72 ± 0.021 | 38.54 ± 0.854 | 55.11 | 11.21 ± 0.221 |
| 9g | 4-NO2 | 0.49 ± 0.012 | 41.06 ± 0.214 | 87.63 | 9.16 ± 0.210 |
| 9h | 3,5-diNO2 | 0.60 ± 0.117 | 37.73 ± 0.096 | 62.42 | 18.04 ± 0.041 |
| 9i | 2,4-diF | 0.58 ± 0.071 | 34.76 ± 0.078 | 59.58 | 9.33 ± 0.132 |
| 9j | 2,4,5-triF | 0.52 ± 0.053 | 32.71 ± 0.209 | 62.72 | 19.94 ± 0.259 |
| 9k | 4-OCF3 | 0.63 ± 0.075 | 27.88 ± 0.773 | 44.6 | 9.40 ± 0.032 |
| 9l | 4-F | 0.48 ± 0.081 | 34.19 ± 0.105 | 71.23 | 11.64 ± 0.676 |
| 9m | 2-NO2 | 0.74 ± 0.054 | 29.89 ± 0.046 | 40.61 | 13.53 ± 0.043 |
| 9n | 3-CN | 0.63 ± 0.088 | 23.03 ± 0.429 | 36.67 | 15.02 ± 0.146 |
| 9o | 4-CN | 0.59 ± 0.024 | 25.49 ± 0.131 | 43.35 | 13.49 ± 0.202 |
| 9p | H | 0.61 ± 0.071 | 38.42 ± 0.398 | 63.08 | 9.81 ± 0.201 |
| 9q | 3-OH | 0.73 ± 0.050 | 36.30 ± 0.015 | 50.03 | 20.26 ± 0.105 |
| 9r | 4-OH | 0.66 ± 0.127 | 36.34 ± 0.447 | 54.89 | 10.78 ± 0.277 |
| 9s | 4-CF3 | 0.57 ± 0.027 | 26.95 ± 0.108 | 47.03 | 13.70 ± 0.236 |
| 9t | 3-CH3 | 0.68 ± 0.031 | 30.71 ± 0.177 | 44.9 | 9.89 ± 0.018 |
| 9u | 4-CH3 | 0.70 ± 0.132 | 32.05 ± 0.267 | 45.78 | 18.10 ± 0.270 |
| Celecoxib | 0.36 ± 0.023 | 29.99 ± 0.060 | 82.04 | nd | |
| Zileuton | nd | nd | nd | 14.29 ± 0.173 | |
| Indomethacin | 0.35 ± 0.013 | 0.32 ± 0.008 | 0.91 | nd | |
2.2.1.2. 5-LOX inhibition activity. The 5-LOX inhibitory potential of the synthesized compounds was also investigated. The results revealed that most of the evaluated derivatives were 5-LOX enzyme inhibitors with comparable potency to standard zileuton. Among all the examined derivatives, 9d and 9g demonstrated the highest inhibitory potential with IC50 values of 7.87 μM and 9.16 μM, respectively, while zileuton displayed an IC50 value of 14.29 μM. Upon a closer examination of Table 1, noteworthy characteristics of the dual COX-2/5-LOX inhibitory potential of the synthesized compounds become evident.
 | ||
| Fig. 2 Enzyme kinetics of compound 9d against COX-2: [A] the Lineweaver–Burk displays competitive inhibition; [B] Dixon plot with Ki at the negative x-axis intersection point. | ||
2.2.4.1. Acute oral toxicity. Acute oral toxicity assessment was performed on healthy female Wistar rats following the OECD 423 guidelines. Compounds 9d and 9g were well-tolerated up to a dose of 500 mg kg−1 and exhibited no detrimental effects. Also, no mortality was observed even after 14 days, indicating a significant safety margin. Thus, the compounds can be examined by additional in vivo investigations. The efficacy of compounds 9d and 9g was further validated through the histological study of the kidneys, liver, lungs, and heart.
2.2.4.2. Carrageenan-induced rat paw edema model. Carrageenan-induced paw edema is a standard exploratory model for assessing the anti-inflammatory effect of different compounds. Carrageenan injected locally into the sub-plantar region of the rat paw causes a severe inflammatory response visible within 30 minutes. Carrageenan-induced paw edema occurs in two stages, wherein the pro-inflammatory histamine, kinins, and serotonin are released in the early stage (around 1 h), followed by the release of prostaglandins, free radicals and pro-inflammatory cytokines in the delayed stage (3 h post-treatment).28 The results of the anti-inflammatory effects of compounds 9d and 9g are presented in Table 2. In comparison with the standard drug indomethacin (Indo 10), the investigated compounds markedly reduce paw edema. The anti-inflammatory effect peaked at 5 h, indicating the compounds' ability to inhibit prostaglandin release in the late stage.
| Swelling thickness in mm | |||||||
|---|---|---|---|---|---|---|---|
| 0 h | 1 h | 2 h | 3 h | 4 h | 5 h | 6 h | |
| The statistics are displayed as mean ± SD; the control group was administered with 0.3% Na CMC solution (10 ml kg−1, p.o.) in distilled water. The standard group was given indomethacin (Indo 10) at a dose of 10 mg kg−1, p.o. in 0.3% Na CMC. The test compounds 9d and 9g were given in doses of 5 mg, 10 mg, and 20 mg kg−1, p.o. in 0.3% Na CMC.a p < 0.05 vs. control.b p < 0.05 vs. Indo 10. | |||||||
| Control | 3.48 ± 0.21 | 8.47 ± 0.25 | 8.65 ± 0.53 | 8.56 ± 0.45 | 8.36 ± 0.78 | 8.47 ± 0.32 | 8.33 ± 0.42 |
| Indo 10 | 3.37 ± 0.23 | 3.74 ± 0.23a | 3.73 ± 0.26a | 3.38 ± 0.19a | 3.14 ± 0.26a | 3.17 ± 0.26a | 3.29 ± 0.11a |
| 9d (5 mg kg−1) | 3.51 ± 0.19 | 6.18 ± 0.54a,b | 5.63 ± 0.48a,b | 4.67 ± 0.38a,b | 4.18 ± 0.45a | 3.98 ± 0.45a | 4.14 ± 0.35a |
| 9d (10 mg kg−1) | 3.21 ± 0.14 | 5.15 ± 0.49a,b | 3.96 ± 0.51a,b | 3.47 ± 0.29a,b | 3.32 ± 0.47a | 3.29 ± 0.26a | 3.37 ± 0.31a |
| 9d (20 mg kg−1) | 3.43 ± 0.18 | 5.12 ± 0.32a,b | 4.11 ± 0.23a,b | 3.41 ± 0.35a,b | 3.22 ± 0.39a | 3.24 ± 0.19a | 3.41 ± 0.35a |
| 9g (5 mg kg−1) | 3.59 ± 0.24 | 5.74 ± 0.53a,b | 5.25 ± 0.47a,b | 4.87 ± 0.41a,b | 4.57 ± 0.43a,b | 4.22 ± 0.31a | 4.14 ± 0.31a |
| 9g (10 mg kg−1) | 3.44 ± 0.27 | 5.32 ± 0.41a,b | 4.55 ± 0.36a,b | 4.41 ± 0.22a,b | 3.88 ± 0.56a,b | 3.76 ± 0.42a | 3.89 ± 0.28a |
| 9g (20 mg kg−1) | 3.65 ± 0.18 | 5.16 ± 0.52a,b | 4.78 ± 0.43a,b | 4.54 ± 0.34a,b | 4.32 ± 0.49a,b | 3.96 ± 0.23a | 3.91 ± 0.31a |
2.2.4.3. Effect on prostaglandin E2 (PGE2). PGE2 inhibition is reported to be a successful approach to inflammation therapy.29 NSAIDs suppress inflammation by decreasing the production of prostanoids, mainly PGE2. PGE2 activates more during inflammatory conditions, thus the inhibition of specific PGE2 is another approach to subside the adverse effects of NSAIDs. Herein, we have performed an assay to evaluate the PGE2 inhibition efficiency of the promising derivatives, 9d and 9g. The results (Fig. 3) indicated that 9d and 9g markedly inhibit PGE2 in a dose-dependent manner.
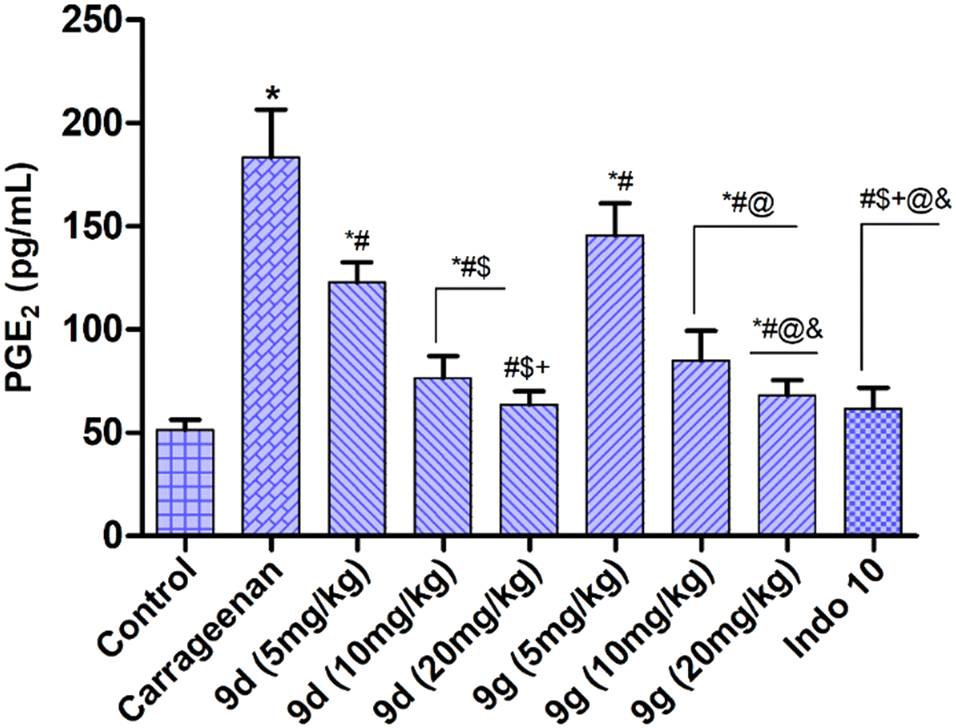 | ||
| Fig. 3 Rat paw tissue concentration of PGE2 for compounds 9d and 9g at 5 mg, 10 mg, and 20 mg kg−1, p.o.; data presented in the mean ± SD. *p < 0.05 vs. control; #p < 0.05 vs. carrageenan; $p < 0.05 vs.9d (5 mg kg−1); +p < 0.05 vs.9d (10 mg kg−1); @p < 0.05 vs.9g (5 mg kg−1); &p < 0.05 vs. (9g 10 mg kg−1); Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
2.2.4.4. Effect on cytokine levels. Pro-inflammatory cytokines have been linked to inflammation and are implicated in chronic inflammatory diseases. The decrease in the levels of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) lowers the chances of cardiovascular toxicity and an increase in the level of interleukin-10 (IL-10) presents promising anti-inflammatory activity.30–32 The ongoing investigation involved the measurement of IL-6, TNF-α, and IL-10 concentrations from paw tissue samples collected from rats that were treated with indomethacin and test compounds 9d and 9g, respectively (Fig. 4). The examined compounds showed a substantial decrease in concentrations of IL-6 and TNF-α. Compound 9d was the most active among the tested compounds with IL-6 and TNF-α inhibition in a manner comparable with the standard drug indomethacin. Also, there is an increase in IL-10 levels indicating the anti-inflammatory activity of the tested compounds.
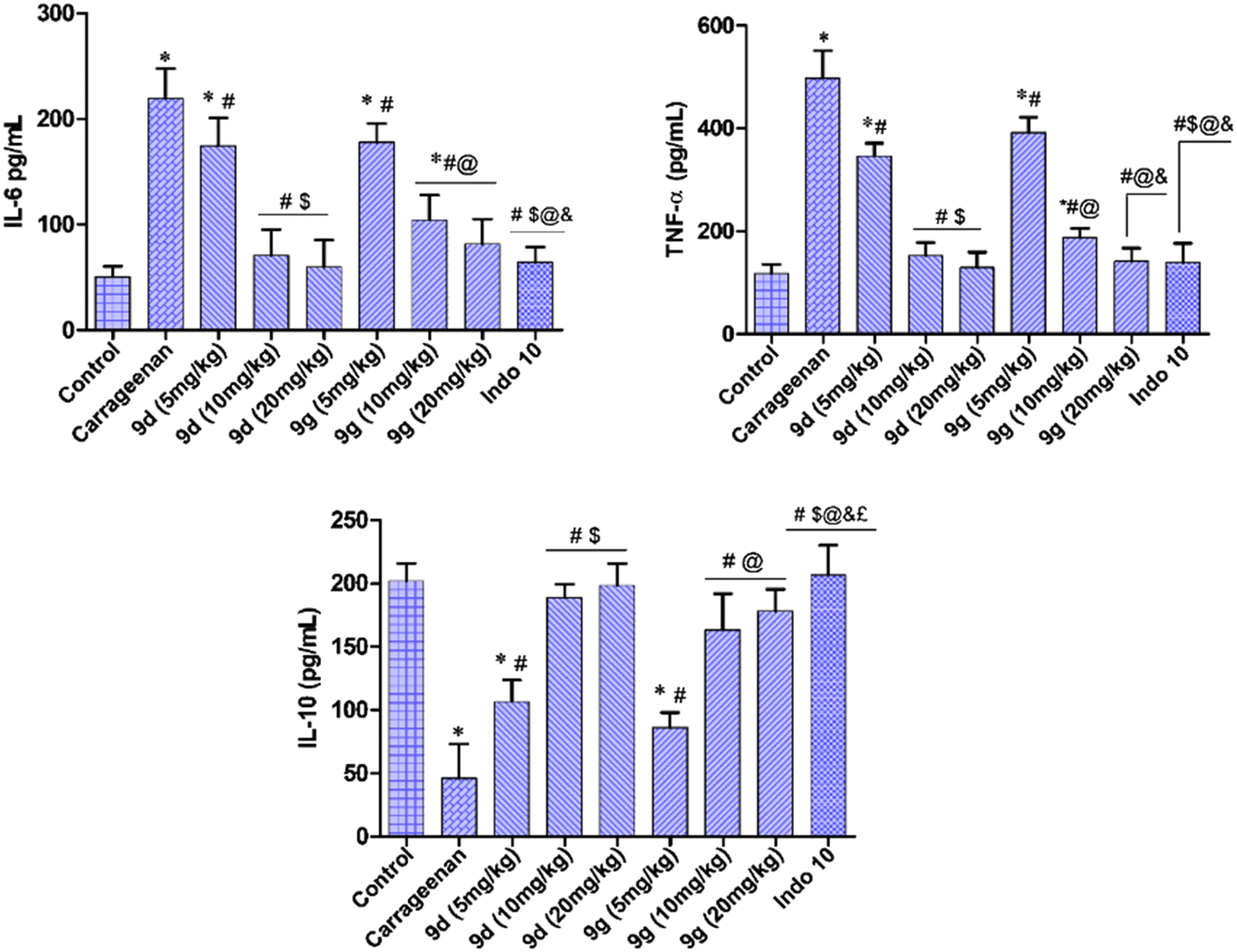 | ||
| Fig. 4 Rat paw tissue concentrations of IL-6, TNF-α, and IL-10 for compounds 9d and 9g at 5 mg, 10 mg, and 20 mg kg−1, p.o.; data presented as the mean ± SD. *p < 0.05 vs. control; #p < 0.05 vs. carrageenan; $p < 0.05 vs.9d (5 mg kg−1); @p < 0.05 vs.9g (5 mg kg−1); &p < 0.05 vs.9g (10 mg kg−1); £p < 0.05 vs.9g (20 mg kg−1); Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
2.2.4.5. Arachidonic acid-induced rat paw edema model. Arachidonic acid injection into the rat's hind paws causes an abrupt and persistent inflammatory response that is inhibited by dual COX/LOX inhibitors and corticosteroids, but not by selective COX inhibitors. The phlogistic activity of LTs renders this model valuable for investigating anti-inflammatory drugs with mechanisms different from those of COX inhibitors.33 A sub-plantar injection of AA produced substantial edema within 30 minutes and hit a peak at 60 minutes. The findings of this study (Table 3) suggested that compounds 9d and 9g mediate its effects by blocking both COX and LOX inflammatory pathways.
| Groups | Dose | Mean ± SD | %Protection |
|---|---|---|---|
| The statistics are displayed as mean ± SD; the control group was administered with 0.3% Na CMC solution (10 ml kg−1, p.o.) in distilled water. The standard groups were given zileuton and celecoxib at a dose of 10 mg kg−1, p.o. in 0.3% Na CMC. The test compounds 9d and 9g were given in doses of 5 mg, 10 mg, and 20 mg kg−1, p.o. in 0.3% Na CMC. | |||
| Control | 10 ml kg−1 | 6.51 ± 0.37 | 0 |
| Zileuton | 10 mg kg−1 | 1.79 ± 0.31 | 72.59 |
| Celecoxib | 10 mg kg−1 | 3.39 ± 0.18 | 47.95 |
| 9d | 5 mg kg−1 | 2.86 ± 0.26 | 56.12 |
| 10 mg kg−1 | 2.01 ± 0.20 | 69.22 | |
| 20 mg kg−1 | 1.94 ± 0.34 | 70.21 | |
| 9g | 5 mg kg−1 | 2.92 ± 0.20 | 55.22 |
| 10 mg kg−1 | 2.30 ± 0.33 | 62.33 | |
| 20 mg kg−1 | 2.24 ± 0.29 | 64.36 | |
2.2.4.6. Ulcerogenic risk assessment. Compounds 9d and 9g, which have significant anti-inflammatory properties, were investigated for ulcerogenic potential in the in vivo rat model. The experimental animals were sacrificed and their stomachs were examined for ulcerogenic effects (Table 4). The examination showed that the evaluated compounds depicted excellent GI safety profiles with ulcer indices of 1.87 and 9.37 for 9d and 9g, respectively, at a dosage of 20 mg kg−1, p.o. Conversely, the rats who received indomethacin suffered from severe stomach ulcers with an ulcer index of 304.16 and perforations across their stomachs. The indomethacin group displayed an ulcer score of 3.04 ± 0.42 indicating severe mucosal lesions. These findings, along with the histological evaluation of gastric mucosa, provide additional evidence of impressive gastric tolerance of the synthesized compounds in comparison with indomethacin (Fig. 5).
| Compound | Dose | Ulcer score | Ulcer index | Ulceration% |
|---|---|---|---|---|
| The statistics are displayed as mean ± SD; control group: 10 ml kg−1, p.o dose of 0.3% Na CMC in distilled water, indomethacin group: Indo 10, 10 mg kg−1, p.o dose in 0.3% Na CMC; the synthesized compounds 9d and 9g were administered in doses of 5 mg, 10 mg, and 20 mg kg−1, p.o. in 0.3% Na CMC. | ||||
| Control | 10 ml kg−1 | 0 | 0 | 0 |
| Indo 10 | 10 mg kg−1 | 3.041 ± 0.42 | 304.16 | 100 |
| 9d | 5 mg kg−1 | 0 | 0 | 0 |
| 10 mg kg−1 | 0.020 ± 0.01 | 2.083 | 1.38 | |
| 20 mg kg−1 | 0.018 ± 0.050 | 1.875 | 1.25 | |
| 9g | 5 mg kg−1 | 0.010 ± 0.005 | 1.041 | 0.667 |
| 10 mg kg−1 | 0.031 ± 0.009 | 3.125 | 4.85 | |
| 20 mg kg−1 | 0.093 ± 0.010 | 9.37 | 4.86 | |
 | ||
| Fig. 5 Gastric mucosa histology photomicrographs (10×): (A) the control rat gastric mucosa with naturally aligned gastric pits as indicated by the black arrow; (B) the indomethacin-treated rat group with ulceration-induced exudation of surface epithelium (red arrows); (C & D) the test compound 9d and 9g treated rats' gastric mucosa showing a normal mucosal architecture (black arrows). | ||
2.2.4.7. Biochemical analysis. Reactive oxygen species (ROS) possess a tendency to aggravate inflammatory responses.34 To investigate if oxidative stress contributes to gastric mucosal damage, the glutathione (GSH), superoxide dismutase (SOD), and nitrite levels have been examined and the results are presented in Fig. 6. A significant variation in oxidative stress was observed in the stomach tissues of indomethacin and the control groups. Compound 9d demonstrated substantial efficacy in enhancing antioxidant activity compared to 9g and the standard drug indomethacin.
 | ||
| Fig. 6 Gastric mucosal tissue concentrations of GSH, SOD, and nitrite for reference standard indomethacin (Indo 10), 9d, and 9g. The statistics are displayed as mean ± SD; rats (n = 6)/group; *p < 0.05 vs. control (0.3% sodium CMC mixture in distilled water; 10 ml kg−1 given orally); #p < 0.05 vs. Indo 10 (10 mg kg−1 given orally) in 0.3% sodium CMC solution; $p < 0.05 vs.9d (20 mg kg−1); Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
2.2.4.8. Assessment of liver and kidney functions. NSAIDs have been associated with drug-induced liver and kidney toxicity. Compounds 9d, 9g, and indomethacin were tested for their effects on the livers of rats using the serum biomarkers alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Both ALT and AST levels were increased in the indomethacin-treated group, while there was no significant difference between the control group and the synthesized derivative group. They were also checked for kidney function of rats; the indomethacin group showed higher levels of creatinine and urea while the synthesized derivatives had a non-significant difference from the control group, indicating the renal safety of the novel compounds The results of estimating these biochemical markers validated the relative safety of the synthesized derivatives in comparison with the standard drug (Fig. 7).
 | ||
| Fig. 7 The measurement of ALT, AST, creatinine, and urea in the serum of rats treated with standard indomethacin (Indo 10), 9d, and 9g. The statistics are displayed as mean ± SD; rats (n = 6)/group; *p < 0.05 vs. control (0.3% sodium CMC mixture in distilled water; 10 ml kg−1 given orally); #p < 0.05 vs. Indo 10 (10 mg kg−1 given orally) in 0.3% sodium CMC solution; Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
2.2.4.9. Effect on platelet aggregation. Inflammation induces vascular injury and recruits platelets by adhering to injured subendothelial cells resulting in the accumulation of platelets. Platelet activation releases multiple platelet aggregating agents, including adenosine diphosphate (ADP), which exacerbates platelet aggregation and causes secondary myocardial tissue damage.35 To discover the role of compounds 9d and 9g in the production of platelets, we conducted a platelet aggregation assay. The results of the study showed that the treatment with compounds 9d and 9g reduces platelet aggregation (Fig. 8).
 | ||
| Fig. 8 The measurement of platelet aggregation percent in rat groups treated with AA, 9d, 9g, and aspirin. The statistics are displayed as mean ± SD; rats (n = 6)/group; *p < 0.05 vs. AA; Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
2.2.4.10. Assessment of cardiotoxic liability.
2.2.4.10.1. Lactate dehydrogenase (LDH) and creatine kinase-MB (CK-MB). The present study was designed to investigate the cardiotoxic liabilities of compounds 9d and 9g in Wistar rats. The presence of enzymes LDH and CK-MB in serum indicates myocardial damage. An isoproterenol-induced myocardial infarction model was used to develop myocardial damage in rodents.36 The rats treated with isoproterenol (ISO) had significantly elevated levels of serum LDH and CK-MB, whereas the rats treated with celecoxib did not exhibit a significant difference in serum LDH and CK-MB levels. However, compounds 9d and 9g displayed substantially lower levels of both LDH and CK-MB (Fig. 9).
 | ||
| Fig. 9 The measurement of LDH and CK-MB in the serum of rats treated with isoproterenol (ISO), celecoxib, 9d, and 9g. The statistics are displayed as mean ± SD; n = 6 rats per group; *p < 0.05 vs. control (0.3% sodium CMC mixture in distilled water; 10 ml kg−1 given orally); #p < 0.05 vs. ISO (100 mg kg−1 given s.c.) in 0.3% sodium CMC solution; @p < 0.05 vs. ISO + celecoxib (10 mg kg−1 given orally); Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
2.2.4.10.2. Cardiac troponin-I (cTn-I). Cardiac troponin-I (cTn-I) is yet another marker of drug-induced cardiac cell rupture, leading to enzyme leakage into the bloodstream. The increased troponin level has been reported in the serum of celecoxib-treated groups,37 which is consistent with the results of the present findings. The loss of membrane integrity due to myocyte injury might be the reason for troponin release which is indicative of coronary diseases. When compared to the celecoxib group, both 9d and 9g showed lower levels of cTn-I, indicating that they did not exacerbate the existing condition, thus suggesting the absence of cardiotoxicity in the test compounds (Fig. 10).
 | ||
| Fig. 10 The measurement of cTn-I in the serum of rats treated with isoproterenol (ISO), celecoxib, 9d, and 9g. The statistics are displayed as mean ± SD; n = 6 rats per group; *p < 0.05 vs. control (0.3% sodium CMC mixture in distilled water; 10 ml kg−1 given orally); #p < 0.05 vs. ISO (100 mg kg−1 given s.c.) in 0.3% sodium CMC solution; @p < 0.05 vs. ISO + celecoxib (10 mg kg−1 given orally); Tukey's test is carried out following the repeated measure one-way ANOVA. | ||
Rats were randomly chosen from each experimental group to investigate cardiac tissue histology and sacrificed. The histopathology of compounds 9d and 9g is similar to that of the control group, confirming that the synthesized compounds do not cause toxicity in cardiac tissues (Fig. S8, ESI†).
2.2.4.11. Assessment of analgesic activity. The writhing test has been performed to evaluate the analgesic activity of the promising derivatives. Acetic acid is used to induce pain of peripheral origin. Acetic acid is believed to function indirectly by stimulating endogenous mediators, which activate nociceptive neurons susceptible to NSAIDs and narcotics.8 In this study (Table 5), compound 9d exhibited a greater percentage of pain inhibition than the standard group indomethacin. Compound 9d with a dose of 20 mg kg−1 had 17.83 ± 1.94 writhes and a percentage of pain inhibition of 55.78 ± 4.81%, which is close to the standard indomethacin producing 16.50 ± 1.87 writhes and 59.09 ± 4.63% pain inhibition. The test compound 9g produced 20.33 ± 2.16 writhes and 49.58 ± 5.35% pain inhibition.
| Group | No. of writhes (30 min) | %Pain inhibition |
|---|---|---|
| The statistics are displayed as mean ± SD; control group: 10 ml kg−1, p.o dose of 0.3% Na CMC in distilled water, indomethacin group: Indo 10, 10 mg kg−1, p.o dose in 0.3% Na CMC; the investigated compounds 9d and 9g were administered in doses of 20 mg, p.o. in 0.3% Na CMC. | ||
| Control | 40.33 ± 3.01 | 0 |
| Indo 10 | 16.50 ± 1.87 | 59.09 ± 4.63 |
| 9d | 17.83 ± 1.94 | 55.78 ± 4.81 |
| 9g | 20.33 ± 2.16 | 49.58 ± 5.35 |
 | ||
| Fig. 11 Cytotoxic potential of compounds in the A549 human lung cancer cell line (A–C), COLO-205 human colon cancer cell line (D–F) and MIA-PA-CA-2 (G–I) human pancreatic cancer cell line. The control group depicted untreated cancer cell lines, while the ADR group showed the treatment with the reference standard, and the 9d group showed test compound treatment. Red arrows indicate the cancer cells and black arrows indicate the decrease in the number of cancer cells. | ||
| Compound | Cell line | GI50 (μM) |
|---|---|---|
| A549 | 0.077 | |
| ADR | COLO-205 | 0.0999 |
| MIA-PA-CA-2 | 0.002 | |
| 9d | A549 | 0.731 |
| COLO-205 | 1.22 | |
| MIA-PA-CA-2 | 2.32 |
 | ||
| Fig. 12 Histogram represents the percent of flies eclosed in the untreated control group and 9d treated group with 1 μM, 10 μM, 50 μM, 100 μM, and 200 μM concentrations. Dunnett's multiple comparisons test was used to ascribe the significance values. Non significance (ns), p > 0.05, *p < 0.05, and ***p > 0.001. | ||
 | ||
| Fig. 13 Histogram represents the percent of different developmental stages of WT, diseased (repo-GAL4/UAS-RasV12), and diseased treated with 9d at 10 μM and 50 μM concentrations. Tukey's multiple comparisons test was used to ascribe the significance values. Non significance (ns), p > 0.05, *p < 0.05, **p < 0.01, and ***p > 0.001. | ||
2.2.6.1. Toxicity assessment. A comprehensive toxicity study in wild type Drosophila resulted in the LD50 dose of 100 μM for compound 9d. At doses of 1 μM and 10 μM, approximately 92% and 84% of flies effectively eclosed, exhibiting minimal toxicity. Also, at 50 μM dose, there was successful eclosion of nearly 71% of F1 flies into adults. However, at doses of 100 μM and 200 μM, the flies' eclosion was reduced to 41% and 24%, respectively (Fig. 12).
2.2.6.2. In vivo anti-cancer efficacy. The anti-cancer potential of test compound 9d was examined by treating RasV12-induced glioblastoma cancer. Compound 9d at 10 μM and 50 μM concentrations resulted in therapeutic efficacy in a dose-dependent manner (Fig. S9, ESI†). The study revealed that there was near absolute lethality at the larval stage in the development of cancer bearing larvae, whereas the 9d treated groups demonstrated significant improvement in larval development to pupal transition. Interestingly, at 10 μM dose, there was a statistically significant improvement in pupal development of the 9d treated group (p = 0.14). Notably, at 50 μM dosage, 9d showed a significant reduction in 3rd instar larval lethality, increased 3rd instar larval survivability, and transition to early pupal development (p = 0.006). The 9d treatment at both 10 μM and 50 μM dosages improved 3rd instar larval survivability (p = 0.12 and p = 0.006, respectively) and early pupal survivability (p = 0.007 and p < 0.001, respectively) compared to untreated glioblastoma cancer bearing larvae as illustrated in Fig. 13.
| Parameters | Effect of 9d |
|---|---|
| a All values are expressed as the mean ± SD (n = 3). | |
| C max (μg ml−1) | 10.32 ± 0.043 |
| T max (h) | 3.00 ± 0.554 |
| (AUC)0–24 (μg ml−1 h−1) | 32.06 ± 0.625 |
| t ½ (h) | 2.11 ± 0.053 |
| MRT (h) | 3.42 ± 0.0614 |
3. Computational studies
3.1. Molecular docking
The molecular docking studies were conducted on COX-2 (PDB id: 3LN1) and 5-LOX (PDB id: 6N2W), respectively. Initially, the grids were generated and validated by extracting and re-docking the co-crystallized ligands. The original co crystallized ligand and the re-docked ligand alignment were superimposed and the RMSD values were found to be 0.34 Å and 1.58 Å for the COX-2 and 5-LOX enzymes, respectively. The designed compounds exhibited encouraging glide and dock scores with improved molecular binding into the respective grids of COX-2 and 5-LOX. The docking results on the COX-2 grid indicated that the biphenyl rings of designed compound 9d best occupy the COX-2 active binding pocket and showed favorable hydrophobic interaction with the binding pocket residues such as Ala513, Tyr371, Val509, Met508, Phe504, and Val102, which are similar to the interaction of the selective COX-2 inhibitor, celecoxib (Fig. 14). Other active site residues were also observed which include π-cation interaction with Arg106, charged interaction with Arg499, and hydrogen bonding with Leu338 which confirms the active site binding of the synthesized compound. | ||
| Fig. 14 3D docking poses (PDB id-3LN1): (A) celecoxib with a docking score of −12.636 showing interactions with active site amino acid residues such as Arg106, Leu338, Val509, Phe504, Arg499, and Met508. (B) Compound 9d with a docking score of −10.830 also showing interactions with active site amino acid residues Ala513, Tyr371, Leu338, Arg106, Val509, Arg499, and Met508. | ||
The binding mode of docked compounds on the 5-LOX grid (PDB id: 6N2W) displayed that compound 9d is well occupied on the 5-LOX enzymatic cleft and showed interaction with active site residues such as His367, His372, Gln363, Gln557, and Asn407. His372 showed additional π–π stacking and π-cation interactions, and Ile406, Ile673, Leu607, and Gln363 indicated significant hydrophobic interactions (Fig. 15). Further, the molecular mechanics-generalized born surface area (MM-GBSA) assay of designed compounds identified the hit compound 9d with the minimum binding free energy of −36.628 and −48.307 kcal mol−1 for COX-2 and 5-LOX enzymes, respectively (Tables S2 and S3, ESI†).
 | ||
| Fig. 15 3D docking poses (PDB id- 6N2W): (A) zileuton with a docking score of −5.287 showing interactions with active site amino acid residues Gln363, His367, Gln557, His372, Leu607, and Ile406. (B) Compound 9d with a docking score of −5.859 also showing interactions with active site amino acid residues such as Gln363, Gln557, His372, Leu607, Ile673, and Asn407. | ||
3.2. Molecular dynamics simulations
The stability of the docked poses was evaluated through a 100 ns molecular dynamics (MD) simulation run for 9d on both the targets COX-2 and 5-LOX. The outcomes of molecular dynamics investigations on COX-2 revealed that derivative 9d showed favorable interactions with the key residues including hydrogen bonding interaction with Leu338. The hydrophobic interaction was observed for Phe504 and Ala513, which were crucial for the binding and stabilization of inhibitors in the COX-2 active site.44 The protein-ligand RMSD value was found within the acceptable limit of 1–3 Å for 100 ns simulated trajectory (Fig. 16).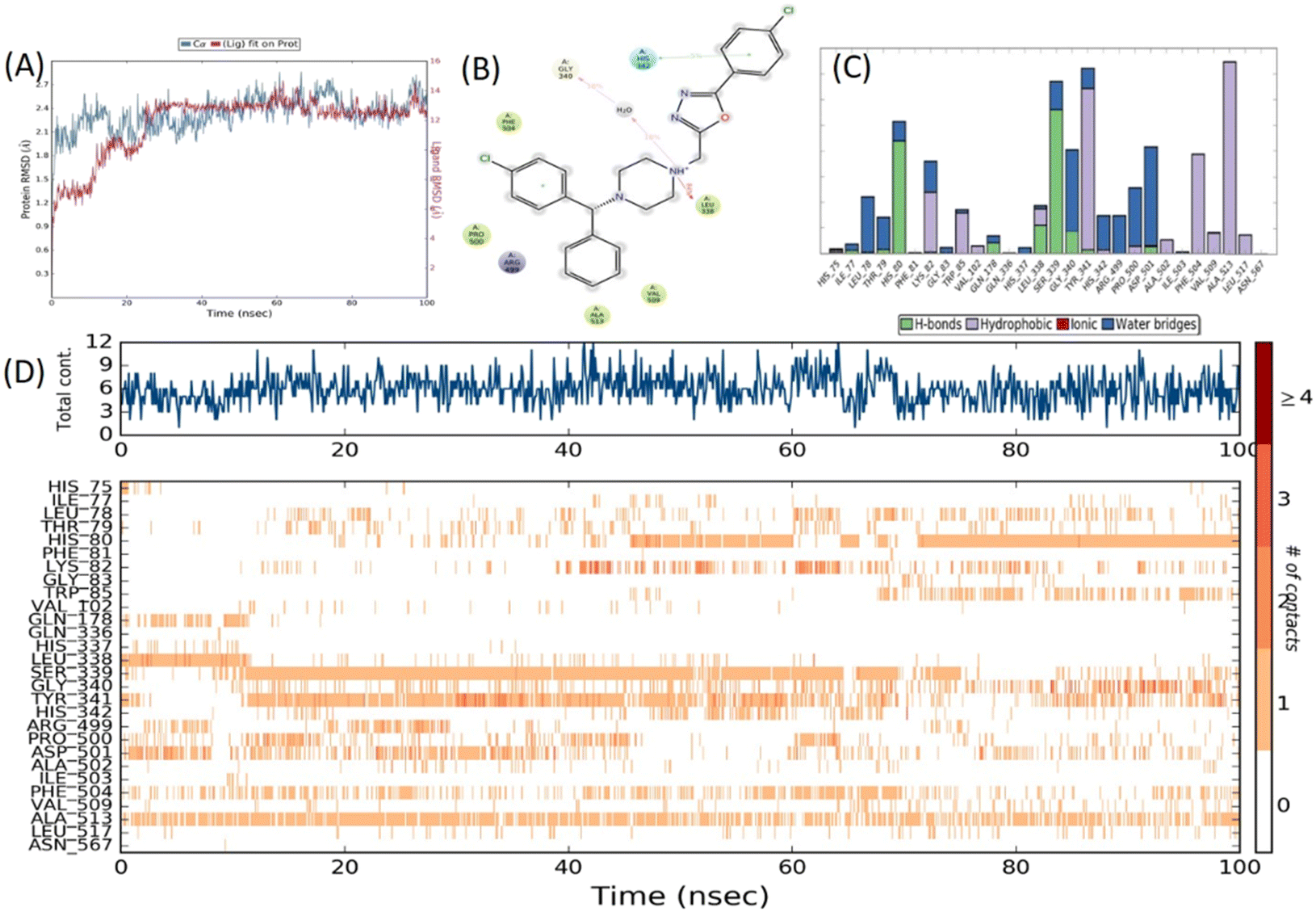 | ||
| Fig. 16 MD simulation studies of the 9d–COX-2 (3LN1) docked complex. [A] RMSD graph of 9d for the 100 ns run; [B] 2D-representation showing percent interaction with active site amino acid residues; [C] histogram depicting interaction between 9d and the protein; [D] timeline representation of interactions with all amino acid residues at each time frame. | ||
The molecular dynamics simulation of 9d on 5-LOX indicated significant hydrophobic interactions with the crucial amino acid residues His367, His372, Ile406, and Ala410. The protein-ligand depicted stable alignment throughout the 100 ns simulation run. Furthermore, the docked complex exhibited a strong hydrogen bonding interaction with Ile673, which is the main carboxylate of the C-terminus of the 5-LOX structural framework.45 The timeline representation also indicated that the interactions of important amino acid residues were constant throughout each time frame during the MD simulation run (Fig. 17). The results of molecular dynamic simulations confirm our belief that the designed compound could possess multitarget potential and might be explored in other inflammatory targets.
 | ||
| Fig. 17 MD simulation studies of the 9d–5-LOX (6N2W) docked complex. [A] RMSD graph of 9d for the 100 ns run; [B] 2D-representation showing percent interaction with active site amino acid residues; [C] histogram depicting interaction between 9d and the protein; [D] timeline representation of interactions with all amino acid residues at each time frame. | ||
4. Conclusions
The current strategy focuses on the synthesis of multitarget anti-inflammatory drugs rather than the conventional single-target based approach. The inflammatory pathways are interconnected, suggesting that stimulating one process might activate other pathways. Herein, we have designed, synthesized, and evaluated novel molecular hybrids of privileged moieties, benzhydrylpiperazine and 1,3,4-oxadiazole, with favorable results. A thorough pharmacological assessment revealed that the promising derivatives 9d and 9g have the anti-inflammatory and analgesic activity of classical NSAIDs while avoiding their primary drawbacks by curtailing the production of gastroprotective prostaglandins, thus devoid of GI, kidney, and liver toxicity. Through inhibition of the 5-LOX pathway, the promising compounds also suppress bronchoconstrictive leukotrienes. Moreover, unlike some selective COX-2 inhibitors, compounds 9d and 9g are cardioprotective and restrict platelet aggregation. Additionally, they displayed inflammatory cytokine inhibition and antioxidant properties. The most promising compound 9d also depicted anti-cancer activity in in vitro human cancer cell lines (A549, COLO-205, and MIA-PA-CA-2) and an in vivo Drosophila cancer model. Further research on these molecular hybrids of two physiologically active scaffolds could open up new avenues for the development of medicines with anti-inflammatory properties that are safer to use over a prolonged period than currently available therapies.5. Experimental
5.1. Chemistry
:hexane (50:50 v/v). This reaction mixture was then added dropwise into the solution of hydrazine hydrate in ACN at 0–5 °C. After the completion of the reaction, the reaction mixture was quenched with about 15–20 ml of water and extracted in a workup with EtOAc. The pure compounds 7a–u were obtained by concentrating the organic layer in a vacuum. Compounds 8a–u were obtained by dissolving the corresponding benzhydrazides (7a–u) and 1.2 equivalent chloroacetic acid in POCl3 (3–4 ml). The reaction was monitored by TLC in EtOAc:hexane (30:70 v/v). The reaction mixture was carefully added to crushed ice and the precipitate was collected as pure compounds after washing, filtering, and drying.48
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(3-methoxyphenyl)-1,3,4-oxadiazole (9a). Light brown solid, yield 74%; mp 258–259 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.38 (t, J = 7.6 Hz, 1H, Ar–H), 7.32–7.29 (m, 5H, Ar–H), 7.28–7.25 (m, 3H, Ar–H), 7.22 (tt, J = 7.7, 1.6 Hz, 3H, Ar–H), 6.95 (dt, J = 7.3, 1.4 Hz, 1H, Ar–H), 5.19 (s, 1H, Hchiral), 3.78 (s, 3H, OCH3), 3.54 (s, 2H, CH2), 2.75 (s, 4H, piperazine H), 2.54 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 166.14, 163.80, 162.11, 141.54, 139.07, 134.44, 129.22, 129.14, 128.96, 128.46, 128.41, 127.71, 127.26, 119.57, 117.36, 116.11, 75.37, 55.52, 52.96, 51.33, 51.12; HRMS [M + 1]+ calculated 475.1895, found 475.1884; HPLC purity: 96.71%, retention time: 4.39 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(4-methoxyphenyl)-1,3,4-oxadiazole (9b). Light brown solid, yield 76%; mp 257–258 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 8.9 Hz, 2H, Ar–H), 8.00 (d, J = 8.9 Hz, 2H, Ar–H), 7.34 (s, 4H, Ar–H), 7.19 (d, J = 7.2 Hz, 1H, Ar–H), 7.03 (d, J = 8.9 Hz, 2H, Ar–H), 7.00 (d, J = 8.9 Hz, 2H, Ar–H), 4.21 (s, 1H, Hchiral), 3.89 (s, 2H, CH2), 3.88 (s, 3H, OCH3), 2.66 (s, 4H, piperazine H), 2.44 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 165.32, 164.12, 162.74, 162.39, 162.23, 129.19, 128.94, 128.79, 128.71, 128.65, 128.60, 127.84, 127.26, 116.63, 116.32, 114.60, 114.50, 114.47, 75.33, 55.48, 52.91, 51.80, 51.56; HRMS [M + 1]+ calculated 475.1895, found 475.1886; HPLC purity: 97.58%, retention time: 4.36 min.
2-(3-Chlorophenyl)-5-((4-((4-chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazole (9c). Light brown solid, yield 81%; mp 262–264 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.05 (s, 1H, Ar–H), 7.96 (d, J = 7.8 Hz, 1H, Ar–H), 7.51 (d, J = 7.9 Hz, 1H, Ar–H), 7.44 (t, J = 8.0 Hz, 1H, Ar–H), 7.34 (dd, J = 7.0, 4.8 Hz, 5H, Ar–H), 7.25–7.17 (m, 4H, Ar–H), 4.21 (s, 1H, Hchiral), 3.90 (s, 2H, CH2), 2.65 (s, 4H, piperazine H), 2.40 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 164.26, 163.67, 141.93, 141.13, 135.22, 132.64, 131.84, 130.43, 129.23, 129.17, 128.70, 128.64, 127.89, 127.82, 127.25, 127.00, 125.44, 125.11, 75.29, 53.02, 51.84, 51.52; HRMS [M + 1]+ calculated 479.1400, found 479.1390; HPLC purity: 96.11%, retention time: 4.85 min.
2-(4-Chlorophenyl)-5-((4-((4-chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazole (9d). Light brown solid, yield 84%; mp 263–264 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 8.3 Hz, 2H, Ar–H), 7.49 (d, J = 8.3 Hz, 2H, Ar–H), 7.33 (d, J = 5.5 Hz, 5H, Ar–H), 7.21 (dd, J = 20.1, 7.8 Hz, 4H, Ar–H), 4.21 (s, 1H, Hchiral), 3.89 (s, 2H, CH2), 2.65 (s, 4H, piperazine H), 2.44 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 164.58, 163.50, 141.91, 141.11, 138.10, 132.65, 129.55, 129.44, 129.17, 128.69, 128.62, 128.29, 128.23, 127.82, 127.24, 122.32, 75.29, 53.03, 51.84, 51.53; HRMS [M + 1]+ calculated 479.1400, found 479.1407; HPLC purity: 96.81%, retention time: 4.88 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(2,4-dichlorophenyl)-1,3,4-oxadiazole (9e). Brown solid, yield 78%; mp 259–261 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 8.4 Hz, 1H, Ar–H), 7.60 (d, J = 1.2 Hz, 1H, Ar–H), 7.42 (dd, J = 8.4, 1.4 Hz, 1H, Ar–H), 7.36 (dd, J = 7.3, 4.8 Hz, 4H, Ar–H), 7.29 (d, J = 7.4 Hz, 3H, Ar–H), 7.21 (dd, J = 17.0, 9.7 Hz, 2H, Ar–H), 4.24 (s, 1H, Hchiral), 3.96 (s, 2H, CH2), 2.69 (s, 4H, piperazine H), 2.43 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 164.00, 163.02, 141.95, 141.14, 138.23, 133.96, 132.64, 131.97, 131.16, 129.16, 129.09, 128.89, 128.82, 128.71, 128.65, 127.81, 127.76, 127.64, 127.25, 121.71, 75.28, 52.92, 51.74, 51.56; HRMS [M + 1]+ calculated 513.1010, found 513.1051; HPLC purity: 96.24%, retention time: 4.96 min.
2-(4-Chloro-3-nitrophenyl)-5-((4-((4-chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazole (9f). Dark brown solid, yield 82%; mp 256–258 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.31 (d, J = 1.4 Hz, 1H, Ar–H), 7.85 (dd, J = 7.5, 1.4 Hz, 1H, Ar–H), 7.65 (d, J = 7.5 Hz, 1H, Ar–H), 7.33 (dd, J = 7.6, 1.5 Hz, 2H, Ar–H), 7.31–7.26 (m, 4H, Ar–H), 7.27–7.22 (m, 3H, Ar–H), 5.19 (s, 1H, Hchiral), 3.35 (s, 2H, CH2), 2.66 (s, 4H, piperazine H), 2.42 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 168.11, 165.41, 150.25, 142.88, 142.01, 132.69, 130.22, 129.27, 129.21, 129.16, 129.13, 128.91, 128.78, 128.64, 126.10, 124.83, 76.16, 52.88, 51.71, 51.49; HRMS [M + 1]+ calculated 524.1251, found 524.1203; HPLC purity: 95.16%, retention time: 5.15 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(4-nitrophenyl)-1,3,4-oxadiazole (9g). Dark brown solid, yield 86%; mp 258–259 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 8.6 Hz, 2H, Ar–H), 8.28 (d, J = 8.6 Hz, 2H, Ar–H), 7.36 (dd, J = 6.9, 4.8 Hz, 5H, Ar–H), 7.29 (s, 2H, Ar–H), 7.25–7.21 (m, 2H, Ar–H), 4.24 (s, 1H, Hchiral), 3.95 (s, 2H, CH2), 2.69 (s, 4H, piperazine H), 2.44 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 164.45, 163.68, 149.63, 141.86, 141.06, 132.67, 129.28, 129.20, 129.16, 128.72, 128.66, 128.12, 127.97, 127.85, 127.81, 127.28, 124.59, 124.38, 75.28, 53.13, 51.88, 51.50; HRMS [M + 1]+ calculated 490.1640, found 490.1640; HPLC purity: 98.65%, retention time: 4.30 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(3,5-dinitrophenyl)-1,3,4-oxadiazole (9h). Dark brown solid, yield 81%; mp 255–257 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 9.31 (t, J = 1.5 Hz, 1H, Ar–H), 8.96 (d, J = 1.6 Hz, 2H, Ar–H), 7.35–7.32 (m, 4H, Ar–H), 7.31 (s, 2H, Ar–H), 7.29 (d, J = 7.6 Hz, 2H, Ar–H), 7.27 (s, 1H, Ar–H), 4.40 (s, 1H, Hchiral), 3.85 (s, 2H, CH2), 2.66 (s, 4H, piperazine H), 2.49 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 169.74, 162.11, 149.44, 140.28, 139.64, 132.14, 129.85, 129.71, 129.43, 128.22, 128.01, 127.24, 126.17, 119.68, 76.11, 52.22, 51.78, 51.46; HRMS [M + 1]+ calculated 535.1491, found 535.1454; HPLC purity: 95.15%, retention time: 5.13 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(2,4-difluorophenyl)-1,3,4-oxadiazole (9i). Dark brown solid, yield 82%; mp 259–260 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.54 (dt, J = 7.1, 5.0 Hz, 1H, Ar–H), 7.32 (d, J = 3.0 Hz, 1H, Ar–H), 7.29 (d, J = 1.6 Hz, 2H, Ar–H), 7.28–7.25 (m, 4H, Ar–H), 7.24–7.20 (m, 2H, Ar–H), 6.95–6.90 (m, 2H, Ar–H), 4.37 (s, 1H, Hchiral), 3.48 (s, 2H, CH2), 2.63 (s, 4H, piperazine H), 2.46 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 169.38, 165.88, 165.47, 164.93, 164.88, 163.23, 163.11, 162.21, 162.14, 154.61, 154.12, 141.88, 141.81, 139.17, 132.16, 130.81, 130.79, 129.22, 129.18, 128.12, 128.09, 116.44, 116.41, 116.38, 116.36, 114.11, 114.04, 105.68, 105.66, 105.63, 75.33, 53.18, 51.55, 51.19; HRMS [M + 1]+ calculated 481.1601, found 481.1669; HPLC purity: 96.33%, retention time: 4.81 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(2,4,5-trifluorophenyl)-1,3,4-oxadiazole (9j). Brown solid, yield 84%; mp 257–259 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.37 (dd, J = 7.9, 5.6, 2.1 Hz, 1H, Ar–H), 7.32–7.29 (m, 2H, Ar–H), 7.30–7.26 (m, 4H, Ar–H), 7.25 (d, J = 5.4 Hz, 2H, Ar–H), 7.23–7.19 (m, 1H, Ar–H), 6.92 (td, J = 8.1, 5.0 Hz, 1H, Ar–H), 5.19 (s, 1H, Hchiral), 3.73 (s, 2H, CH2), 2.70 (s, 4H, piperazine H), 2.58 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 170.01, 168.94, 163.91, 163.18, 159.85, 157.85, 157.81, 157.79, 157.76, 153.78, 153.75, 152.93, 152.87, 150.81, 150.76, 148.81, 148.78, 148.59, 148.55, 146.71, 146.68, 140.56, 139.07, 132.44, 129.90, 129.74, 128.39, 126.73, 116.26, 116.01, 113.94, 113.63, 113.56, 113.32, 106.43, 106.38, 105.88, 105.81, 75.41, 52.81, 51.58, 51.23; HRMS [M + 1]+ calculated 499.1507, found 499.1438; HPLC purity: 94.93%, retention time: 4.90 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(4-(trifluoromethoxy) phenyl)-1,3,4-oxadiazole (9k). Dark brown solid, yield 81%; mp 259–260 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 7.5 Hz, 2H, Ar–H), 7.32–7.29 (m, 2H, Ar–H), 7.30–7.28 (m, 2H, Ar–H), 7.27 (s, 3H, Ar–H), 7.24 (s, 1H, Ar–H), 7.23–7.18 (m, 1H, Ar–H), 7.05 (d, J = 7.5 Hz, 2H, Ar–H), 5.16 (s, 1H, Hchiral), 3.35 (s, 2H, CH2), 2.62 (s, 4H, piperazine H), 2.39 (s, 4H, piperazine H). 13C NMR (125 MHz, CDCl3) δ 172.00, 164.09, 152.13, 152.06, 151.89, 151.63, 140.22, 138.06, 132.04, 129.88, 129.72, 129.31, 128.38, 126.69, 125.04, 123.61, 122.94, 120.73, 120.45, 120.31, 120.27, 120.25, 118.74, 75.58, 52.91, 52.64, 50.12; HRMS [M + 1]+ calculated 529.1613, found 529.1569; HPLC purity: 95.41%, retention time: 4.61 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(4-fluorophenyl)-1,3,4-oxadiazole (9l). Brown solid, yield 84%; mp 258–259 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.08 (dd, J = 8.7, 5.2 Hz, 2H, Ar–H), 7.36 (dd, J = 7.5, 5.2 Hz, 4H, Ar–H), 7.28 (d, J = 7.3 Hz, 2H, Ar–H), 7.25–7.21 (m, 3H, Ar–H), 7.21–7.18 (m, 2H, Ar–H), 4.24 (s, 1H, Hchiral), 3.91 (s, 2H, CH2), 2.67 (s, 4H, piperazine H), 2.46 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 165.83, 164.57, 163.82, 163.35, 141.93, 141.14, 132.63, 129.35, 129.28, 129.18, 128.69, 128.64, 127.83, 127.24, 120.17, 116.48, 116.30, 75.29, 53.01, 51.84, 51.54; HRMS [M + 1]+ calculated 463.1695, found 463.1733; HPLC purity: 96.47%, retention time: 4.44 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(2-nitrophenyl)-1,3,4-oxadiazole (9m). Brown solid, yield 82%; mp 256–258 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.31 (dd, J = 7.5, 1.4 Hz, 1H, Ar–H), 7.84 (dd, J = 7.5, 1.4 Hz, 1H, Ar–H), 7.81 (td, J = 7.4, 1.4 Hz, 1H, Ar–H), 7.77 (td, J = 7.5, 1.5 Hz, 1H, Ar–H), 7.76–7.61 (m, 3H, Ar–H), 7.59 (d, J = 7.4 Hz, 2H, Ar–H), 7.58 (dd, J = 9.4, 5.8 Hz, 3H, Ar–H), 7.59–7.58 (m, 1H, Ar–H), 4.28 (s, 1H, Hchiral), 3.98 (s, 2H, CH2), 2.73 (s, 4H, piperazine H), 2.47 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 165.38, 164.17, 150.59, 141.66, 141.04, 133.72, 130.99, 129.29, 129.18, 128.64, 128.61, 128.10, 127.96, 127.87, 127.82, 127.78, 124.39, 124.52, 75.44, 53.11, 51.82, 51.51; HRMS [M + 1]+ calculated 490.1640, found 490.1696; HPLC purity: 96.52%, retention time: 4.39 min.
3-(5-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazol-2-yl) benzonitrile (9n). Brown solid, yield 85%; mp 255–256 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 1.3 Hz, 2H, Ar–H), 7.64 (d, J = 5.9 Hz, 2H, Ar–H), 7.40–7.29 (m, 6H, Ar–H), 7.27–7.15 (m, 3H, Ar–H), 5.22 (s, 1H, Hchiral), 3.86 (s, 2H, CH2), 2.77 (s, 4H, piperazine H), 2.63 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 171.06, 163.11, 141.47, 138.21, 133.44, 133.18, 129.88, 129.83, 129.75, 129.46, 129.41, 128.32, 127.93, 127.71, 127.27, 120.19, 117.33, 75.49, 52.96, 51.16, 51.03; HRMS [M + 1]+ calculated 470.1742, found 470.1802; HPLC purity: 95.52%, retention time: 4.56 min.
4-(5-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazol-2-yl) benzonitrile (9o). Brown solid, yield 85%; mp 255–256 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 7.4 Hz, 2H, Ar–H), 7.68 (d, J = 7.5 Hz, 2H, Ar–H), 7.32 (dd, J = 14.7, 5.7 Hz, 6H, Ar–H), 7.29 (dd, J = 10.9, 4.5 Hz, 3H, Ar–H), 5.17 (s, 1H, Hchiral), 3.08 (s, 2H, CH2), 2.69 (s, 4H, piperazine H), 2.50 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 173.10, 163.42, 14.54, 139.07, 133.34, 123.98, 129.91, 129.73, 129.38, 129.32, 128.31, 127.33, 126.93, 120.16, 114.41, 75.44, 52.87, 51.16, 51.34; HRMS [M + 1]+ calculated 470.1742, found 470.1793; HPLC purity: 95.88%, retention time: 4.59 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-phenyl-1,3,4-oxadiazole (9p). Light brown solid, yield 82%; mp 257–259 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.55 (dd, J = 7.4, 1.4 Hz, 2H, Ar–H), 7.41 (t, J = 7.4 Hz, 2H, Ar–H), 7.35 (dt, J = 4.8, 2.0 Hz, 1H, Ar–H), 7.32–7.29 (m, 4H, Ar–H), 7.28–7.27 (m, 2H, Ar–H), 7.27–7.24 (m, 2H, Ar–H), 7.25–7.20 (m, 1H, Ar–H), 5.17 (s, 1H, Hchiral), 3.33 (s, 2H, CH2), 2.71 (s, 4H, piperazine H), 2.52 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 176.01, 168.06, 142.76, 140.77, 133.42, 132.56, 129.93, 129.71, 129.33, 129.02, 128.34, 127.17, 126.91, 126.18, 75.18, 52.82, 51.19, 51.31; HRMS [M + 1]+ calculated 445.1790, found 445.1850; HPLC purity: 96.12%, retention time: 4.11 min.
3-(5-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazol-2-yl) phenol (9q). Brown solid, yield 71%; mp 257–258 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 8.22 (s, 1H, OH), 7.30–7.27 (m, 4H, Ar–H), 7.27–7.25 (m, 2H, Ar–H), 7.24 (s, 1H, Ar–H), 7.24 (d, J = 2.2 Hz, 1H, Ar–H), 7.24–7.18 (m, 2H, Ar–H), 7.13–7.08 (m, 2H, Ar–H), 6.83 (dt, J = 7.5, 1.4 Hz, 1H, Ar–H), 5.20 (s, 1H, Hchiral), 3.55 (s, 2H, CH2), 2.76 (s, 4H, piperazine H), 2.56 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 174.00, 164.20, 159.61, 141.56, 139.07, 132.44, 130.91, 129.90, 129.74, 129.33, 128.39, 126.73, 126.15, 121.00, 118.30, 114.24, 76.58, 52.86, 51.18, 50.13; HRMS [M + 1]+ calculated 461.1739, found 461.1690; HPLC purity: 93.82%, retention time: 3.98 min.
4-(5-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-1,3,4-oxadiazol-2-yl) phenol (9r). Brown solid, yield 73%; mp 259–260 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 9.17 (s, 1H, OH), 7.43 (d, J = 7.5 Hz, 2H, Ar–H), 7.32–7.21 (m, 7H, Ar–H), 7.15 (d, J = 7.5 Hz, 2H, Ar–H), 7.00 (d, J = 7.5 Hz, 2H, Ar–H), 5.19 (s, 1H, Hchiral), 3.61 (s, 2H, CH2), 2.66 (s, 4H, piperazine H), 2.45 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 173.00, 163.07, 161.72, 142.56, 139.06, 132.41, 129.90, 129.74, 129.31, 128.39, 127.58, 126.73, 117.41, 115.81, 77.58, 52.84, 52.18, 50.33; HRMS [M + 1]+ calculated 461.1739, found 461.1696; HPLC purity: 94.24%, retention time: 4.21 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazole (9s). Dark brown solid, yield 84%; mp 259–260 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.66 (d, J = 7.5 Hz, 2H, Ar–H), 7.54 (d, J = 7.5 Hz, 2H, Ar–H), 7.29 (m, 5H, Ar–H), 7.28 (t, J = 6.5 Hz, 3H, Ar–H), 7.24–7.19 (m, 1H, Ar–H), 4.35 (s, 1H, Hchiral), 3.65 (s, 2H, CH2), 2.64 (s, 4H, piperazine H), 2.45 (s, 4H, piperazine H); 13C NMR (125 MHz, CDCl3) δ 173.01, 162.08, 141.56, 139.07, 134.27, 132.44, 132.37, 132.16, 129.90, 129.74, 129.33, 128.39, 127.60, 126.73, 126.67, 126.62, 126.57, 126.50, 126.43, 126.40, 126.37, 126.34, 125.51, 123.41, 121.32, 77.54, 52.83, 52.19, 49.39; HRMS [M + 1]+ calculated 513.1664, found 513.1718; HPLC purity: 96.46%, retention time: 4.63 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(m-tolyl)-1,3,4-oxadiazole (9t). Brown solid, yield 81%; mp 254–255 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.48 (t, J = 1.4 Hz, 1H, Ar–H), 7.43 (dt, J = 7.3, 1.4 Hz, 1H, Ar–H), 7.36 (t, J = 7.4 Hz, 1H, Ar–H), 7.31 (d, J = 7.5 Hz, 2H, Ar–H), 7.27 (m, 5H, Ar–H), 7.25–7.19 (m, 3H, Ar–H), 5.19 (s, 1H, Hchiral), 3.81 (s, 2H, CH2), 2.64 (s, 4H, piperazine H), 2.53 (s, 4H, piperazine H), 2.32 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 173.00, 162.20, 140.55, 139.02, 138.98, 132.44, 131.01, 129.93, 129.76, 128.41, 128.39, 127.89, 126.74, 126.70, 126.51, 77.58, 52.86, 52.18, 49.31, 21.36; HRMS [M + 1]+ calculated 459.1946, found 459.1898; HPLC purity: 95.41%, retention time: 5.55 min.
2-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-5-(p-tolyl)-1,3,4-oxadiazole (9u). Brown solid, yield 83%; mp 255–256 °C; TLC (hexane/EtOAc 80
:20 v/v); 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 7.5 Hz, 2H, Ar–H), 7.32–7.29 (m, 3H, Ar–H), 7.28 (d, J = 1.0 Hz, 2H, Ar–H), 7.26 (dd, J = 7.6, 3.6 Hz, 4H, Ar–H), 7.25–7.19 (m, 2H, Ar–H), 5.18 (s, 1H, Hchiral), 3.86 (s, 2H, CH2), 2.62 (s, 4H, piperazine H), 2.50 (s, 4H, piperazine H), 2.31 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 173.06, 162.07, 140.60, 140.56, 139.07, 132.44, 129.90, 129.74, 129.33, 129.09, 128.39, 126.98, 126.73, 125.47, 77.52, 52.85, 52.19, 49.33, 21.31; HRMS [M + 1]+ calculated 459.1946, found 459.1989; HPLC purity: 95.62%, retention time: 5.58 min.
5.2. Pharmacological/biological assays
5.2.1.1. COX-1 and COX-2 inhibition activity49. The colorimetric inhibitor screening assay method has been employed to identify the COX-1/COX-2 inhibition efficiency of the synthesized derivatives. All the reagents and enzymes were purchased from Cayman Chemicals, USA. Initially, assay buffer (item no. 760114) was used to prepare final concentrations of hemin (item no. 760116) and COX enzymes (item no. 760110 and 760108). The enzymes COX-1 (400 units per ml) and COX-2 (400 units per ml) were activated and kept on ice when thawed. The substrate arachidonic acid (item no. 760113) was prepared in a final concentration of 1.1 mM. While performing the assay, 160 μl of assay buffer and 10 μl of hemin were added to the background wells, and 100% initial activity wells were filled with 150 μl of assay buffer, 10 μl of hemin, and 10 μl of either COX-1 or COX-2. The synthesized inhibitors (10 μl) were added to the inhibitor well along with assay buffer, hemin, and enzyme COX-1 or COX-2. All the assay wells were activated by the addition of 20 μl colorimetric substrate N,N,N′,N′,-tetramethylbenzene-1,4-diamine (TMPD) and 20 μl arachidonic acid to initiate the reaction. A UV-spectrometer was used to measure absorbance at 590 nm. Percent inhibition graphs were plotted to determine the IC50 values.
5.2.1.2. 5-LOX inhibition activity50. A 5-LOX inhibitor screening assay kit developed by Cayman Chemicals, (catalog # 760700) was utilized to confirm the 5-LOX inhibition ability of the synthesized derivatives. The compounds were tested in duplicate as per the manufacturer's protocol. The blank well contained 100 μl assay buffer, while 90 μl enzyme and 10 μl were added to the positive control wells. The inhibitor wells were filled with 90 μl enzyme and 10 μl test inhibitor. After 5 minutes of incubation at room temperature, 10 μl AA substrate was added to each well, and the plate was shaken for 10 minutes. Finally, after adding 100 μl of chromogen to each well, the absorbance was determined with a UV spectrophotometer at 490 nm. The IC50 values were calculated by plotting percent inhibition graphs.
5.2.1.3. In vitro enzyme kinetic study51. The enzyme kinetic assay was performed to determine the type of COX-2 inhibition by compound 9d. The kinetic parameters Km and Vmax were calculated at a fixed concentration of the enzyme against six varied concentrations of the substrate (5–500 μM). Compound 9d was examined at three different concentrations (0.03, 0.15, and 0.30 μM), and the inhibitor concentration was tested against six varied concentrations of the substrate. The experiment was carried out in triplicate. The Lineweaver–Burk technique was executed to determine the inhibition kinetics. Furthermore, the relevant Ki value was obtained from a Dixon plot.
5.2.2.1. Animals. All of the research experiments were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the Committee for the Control and Supervision of Experiments on Animals (CCSEA), and approved by the Institutional Animal Ethics Committee (Approval no. IIT(BHU)/IAEC/2023/019). Healthy Wistar rats of either sex weighing 200–250 g were purchased from the Central Animal Facility, Banaras Hindu University. The rats were housed in cages and had unrestricted water and commercial food access. The rats were housed in a climate with controlled humidity and temperature (45–60% RH and 25 ± 2 °C) with a 12-hour light/dark cycle.
5.2.2.2. Acute oral toxicity. The study was authorized by the Institutional Animal Ethical Committee (IAEC) with approval number IIT(BHU)/IAEC/2023/019. The acute oral toxicity of the top two compounds, 9d and 9g, was studied in female Wistar rats. The animals had free water access and fasted for the entire night before the study. The drugs were given in gradually increasing dosages varying from 100 to 500 mg kg−1 after dissolving in 0.3 % w/v Na-carboxymethylcellulose (CMC). The animals were kept under observation for any unusual signs and indications such as seizures, lethargy, diarrhea, salivation, drowsiness, etc. for 24 hours and then every day for 14 days. Animals were sacrificed for histopathological evaluation of possible GI, hepatic, cardiovascular, and renal damage.52
5.2.2.3. Carrageenan-induced paw edema model. Acute inflammation was triggered in rats by sub-plantar injection of 0.1 ml of a 1% w/v carrageenan solution in the left hind paw. The control group received the vehicle, while the standard group was given indomethacin in 10 mg kg−1, p.o. dose, in 0.3% Na CMC. The test groups of animals (9d and 9g) were administered with compounds 9d and 9g orally in doses of 5 mg, 10 mg, and 20 mg kg−1 in 0.3% Na CMC, 1 hour before the administration of carrageenan. The paw volume was measured every hour for up to six hours after the injection of carrageenan, employing a digital vernier caliper.53
5.2.2.4. Effect on prostaglandin E2 (PGE2). Compounds 9d and 9g were tested for their effect on PGE2. Rat paw tissues were collected from different groups of the carrageenan-induced paw edema model and rinsed in PBS (pH 7.4) to remove excess blood.54 The tissues were minced, homogenized, filtered, and centrifuged at 2000 rpm for 20 minutes at 4 °C. The respective supernatant was collected carefully and analysis was performed as per the protocol mentioned in the ELISA kit (KLR0504) obtained from Krishgen Biosystems, USA.
5.2.2.5. Effects on the cytokine level. Rat paw tissue homogenates were prepared from different rat groups in the carrageenan-induced rat paw edema model. These homogenates were filtered and centrifuged for 20 minutes at 2000 rpm at 4 °C. The supernatant was collected, and IL-6, TNF-α, and IL-10 levels were measured using ELISA kits (Krishgen Biosystems, India) according to the manufacturer's instructions.
5.2.2.6. Arachidonic acid-induced paw edema model. A sub-plantar injection of 0.1 ml of 0.5% w/v arachidonic acid in 0.2 M carbonate buffer (pH 8.4) into the hind paw of rats caused significant edema within 5 minutes, which hit a peak in 1 h. The control group received 0.3% CMC (10 ml kg−1, p.o.), while the standard group animals were given zileuton (10 mg kg−1, p.o.). The other groups were administered with the test compounds 9d and 9g in doses of 5 mg, 10 mg, and 20 mg kg−1 in 0.3% Na CMC. A digital vernier caliper was used to measure the swelling thickness (mm) 1 h after the arachidonic acid injection.33
5.2.2.7. Ulcerogenic risk assessment. The ulcerogenic liability experiment was carried out on fasting rats, which were randomly divided into different groups, with each group having n = 6 rats. The rats in the control group received distilled water containing 0.3% CMC, while the rats in the standard group were given 10 mg kg−1 indomethacin. Compounds 9d and 9i have been administered to the test groups in doses of 5 mg, 10 mg, and 20 mg kg−1 in 0.3% Na CMC. All groups of animals were dosed via oral gavage once daily for three consecutive days and rats under anesthesia were sacrificed on the 3rd day after 6 h of dosing. The stomachs were quickly taken out and washed with saline to remove the excess material, and the stomach mucosa was examined using a hand-held lens. The percentage of stomach ulceration, ulcer index, and ulcer score were estimated following a previously reported scoring method.55 Some stomachs were randomly chosen from each group and preserved in 10% formalin solution. The slides were stained with hematoxylin and eosin dyes for histological inspection.
5.2.2.8. Biochemical analysis. The levels of oxidative stress biomarkers were determined in random stomachs from each animal group. The stomach tissues were homogenized in cold phosphate buffer saline (pH 7.4) and centrifuged at 1000 rpm for 10 minutes at 4 °C. The respective supernatant was collected and their protein content was estimated following the standard Bradford assay. The levels of GSH, SOD, and nitrate were measured to evaluate the antioxidant activity of the test substances as per the reported procedures.51
5.2.2.9. Assessment of liver and kidney functions. Blood samples from different rat groups were collected from the retro-orbital plexus and allowed to clot at room temperature. The serum was separated by centrifugation at 2000 rpm for 20 minutes at 4 °C. The serum biomarkers ALT, AST, creatinine, and urea were determined using commercial assay kits (Merck, Germany) according to the described protocol.
5.2.2.10. Effect on platelet aggregation. Platelet-rich plasma (PRP) was extracted from blood samples obtained from various rat groups during cardiotoxicity evaluation activities. Tyrode's buffer was used to adjust the platelet count to 4 × 108 cells per ml and the anti-platelet aggregation activity of test compound 9d was evaluated using 96-well plate aggregometry. Initially, the absorbance was recorded at 570 nm on a UV-spectrophotometer. Afterward, 10 μl of ADP and 5 μg ml−1 collagen were added to induce platelet aggregation. The plate was incubated for 5 min at 37 °C with shaking. Again, the absorbance was measured at 570 nm to observe the change in value. The platelet aggregation percent was calculated as per the previously reported method.56
5.2.2.11. Assessment of cardiotoxic liability. The rats were allocated into five different groups (n = 6 per group). Group I consisted of control animals receiving 0.3% CMC solution as a vehicle, group II received ISO (100 mg kg−1, s.c.), group III animals were administered with ISO (100 mg kg−1, s.c.), followed by the treatment with celecoxib in 0.3% CMC solution. Group IV and group V animals received the treatment with the oral administration of test compounds 9d and 9g, post ISO (100 mg kg−1, s.c.) administration. The blood samples were taken from the retro-orbital plexus and the serum was separated by centrifugation at 4000 rpm at 4 °C for 10 minutes. The levels of LDH, CKMB, and cTn-1 were determined using commercially available kits. For the estimation of LDH and CKMB, a Span diagnostic kit, India, was utilized in accordance with the manufacturer's protocol, and a rat cTn-I ELISA kit from Krishgen Biosystems, India, was used for the cTn-I estimation.
5.2.2.12. Assessment of analgesic activity57. The rats were randomly divided into four different groups (n = 6 per group). The control group received 0.3% CMC in distilled water as a vehicle, and the other groups received standard indomethacin and compounds 9d and 9g respectively. After 30 minutes of vehicle and drug administration, 0.6% v/v of acetic acid was injected subcutaneously into all groups. The writhing activity was observed and the numbers of stretching were carefully counted for the next 30 minutes. When compared to the control group, a reduction in the number of writhing responses demonstrated the pain-inhibitory response in both the standard and test groups.
The percent growth was expressed as the ratio of the average absorbance of the test well to the average absorbance of the control wells × 100. The images of the sample tested were taken using a Ti–S inverted research microscope – Nikon with a magnification of ×20, with Eclipse Image processing software NIS-Elements.
:H2O (85:15) as the mobile phase. The standard calibration curve was used to determine the plasma drug concentration in each sample at a detection wavelength of λ = 254 nm. The pharmacokinetic parameters, including Tmax, Cmax, (AUC)0–24, t1/2, and MRT, were determined using an extravascular non-compartmental model in Kinetica 5.0 (Thermo Scientific Kinetica, USA).60
5.3. Molecular docking
All in silico studies were performed using the Schrödinger Maestro 2018.1 glide XP software. The protein data bank (PDB) has been initially utilized to obtain the protein structures of COX-2 (PDB ID: 3LN1) and 5-LOX (PDB ID: 6N2W) for the purpose of the structure-based drug design approach. The protein error was addressed with the protein preparation wizard tool. Energy minimizations and structural refinements were conducted using the OPLS force field. Subsequently, grids were generated around the active sites of the co-crystallized ligands within the crystal structures using the protein grid generation module. The grids were validated by extracting and re-docking the co-crystallized ligands into the respective generated grids of COX-2 and 5-LOX.The virtual screening was conducted using Glide's structure-based virtual screening protocol. This method identified the compounds based on their interactions with the targeted active site. The screening process involves three hierarchical steps: high throughput virtual screening (HTVS), standard precision (SP) docking, and extra precision (XP) docking. During the screening, a 30% filter criterion was applied at each step of HTVS, SP, and XP docking to identify potential leads. The screened compound poses were then analyzed using the docking post-processing and pose filtration protocols in Maestro.
Using the Desmond module, molecular dynamics simulations validated the stability of the protein–ligand docked complex. The system builder tool was initially employed to build a system creating a virtual TI3P water environment and neutralizing it by the addition of counter atoms. The minimization tool was set at a convergence threshold of 1.0 kcal mol−1 Å−1 with a maximum of 2000 iterations. Docked complexes were subjected to a 100 ns molecular dynamic simulation run. The trajectory was recorded at intervals of 100 ns, resulting in approximately 1000 frames, while the energy threshold was maintained at 9.6 during the simulation run.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the Central Instrument Facility (CIF), Indian Institute of Technology (Banaras Hindu University), Varanasi for conducting the NMR experiments. The authors also express their gratitude to the anti-cancer drug screening facility (ACDSF) at ACTREC, Tata Memorial Centre, Navi Mumbai for conducting in vitro testing for anti-cancer activity evaluation of drugs.References
- P. Libby, Nutr. Rev., 2007, 65, S140–S146 CrossRef.
- K. S. Alharbi, S. K. Alenezi and G. Gupta, in Recent Developments in Anti-Inflammatory Therapy, Elsevier, 2023, pp. 1–9 Search PubMed.
- B. Wu, Q. H. Sodji and A. K. Oyelere, Cancers, 2022, 14, 552 CrossRef CAS.
- B. Wang, L. Wu, J. Chen, L. Dong, C. Chen, Z. Wen, J. Hu, I. Fleming and D. W. Wang, Signal Transduction Targeted Ther., 2021, 6, 1–30 CrossRef PubMed.
- A. Aliabadi, E. Khanniri, M. Mahboubi-Rabbani and M. Bayanati, Eur. J. Med. Chem., 2023, 115866 CrossRef CAS.
- W. H. Organization, WHO EML 23rd List, 2023, https://iris.who.int/bitstream/handle/10665/371090/WHO-MHP-HPS-EML-2023.02-eng.pdf?sequence=1 Search PubMed.
- J. Z. Haeggstrom and C. D. Funk, Chem. Rev., 2011, 111, 5866–5898 CrossRef.
- A. G. Banerjee, N. Das, S. A. Shengule, R. S. Srivastava and S. K. Shrivastava, Eur. J. Med. Chem., 2015, 101, 81–95 CrossRef CAS PubMed.
- P. Saraf, P. N. Tripathi, M. K. Tripathi, A. Tripathi, H. Verma, D. K. Waiker, R. Singh and S. K. Shrivastava, Bioorg. Chem., 2022, 129, 106147 CrossRef CAS.
- S. Shrivastava, D. Jain and P. Trivedi, Int. J. Pharm. Sci., 2003, 58, 389–391 CAS.
- P. Kumari, P. Singh, J. Kaur and R. Bhatti, J. Med. Chem., 2021, 64, 9550–9566 CrossRef CAS PubMed.
- C. D. Funk and G. A. FitzGerald, J. Cardiovasc. Pharmacol., 2007, 50, 470–479 CrossRef CAS.
- E. M. Gedawy, A. E. Kassab and A. M. El Kerdawy, Eur. J. Med. Chem., 2020, 189, 112066 CrossRef CAS.
- D. Poeckel and C. D. Funk, Cardiovasc. Res., 2010, 86, 243–253 CrossRef CAS PubMed.
- C. Charlier and C. Michaux, Eur. J. Med. Chem., 2003, 38, 645–659 CrossRef CAS PubMed.
- S. M. Razavi, D. Khayatan, Z. N. Arab, S. Momtaz, K. Zare, R. M. Jafari, A. R. Dehpour and A. H. Abdolghaffari, Prostaglandins Other Lipid Mediators, 2021, 157, 106587 CrossRef CAS PubMed.
- H. T. Nguyen, T.-Y. Vu, V. Chandi, H. Polimati and V. B. Tatipamula, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- A. Jain, J. Chaudhary, H. Khaira, B. Chopra and A. Dhingra, Drug Res., 2021, 71, 62–72 CrossRef CAS.
- A. Sharma, S. Wakode, F. Fayaz, S. Khasimbi, F. H. Pottoo and A. Kaur, Curr. Pharm. Des., 2020, 26, 4373–4385 CrossRef CAS PubMed.
- S. Koparde, K. M. Hosamani, V. Kulkarni and S. D. Joshi, Chem. Data Collect., 2018, 15, 197–206 CrossRef.
- R. Bartzatt, J. Adv. Med. Pharm. Sci., 2017, 1–18 Search PubMed.
- J. Tatarkiewicz, P. Rzodkiewicz, M. Żochowska, A. Staniszewska and M. Bujalska-Zadrożny, Arch. Med. Sci., 2019, 15, 537 CrossRef CAS.
- S. Bhati, V. Kumar, S. Singh and J. Singh, J. Mol. Struct., 2019, 1191, 197–205 CrossRef CAS.
- R. K. Srivastava, S. Shankar, S. K. Shrivastava and A. G. Banerjee, U.S. Pat., 11208389, 2021 Search PubMed.
- S. Valente, D. Trisciuoglio, T. De Luca, A. Nebbioso, D. Labella, A. Lenoci, C. Bigogno, G. Dondio, M. Miceli and G. Brosch, J. Med. Chem., 2014, 57, 6259–6265 CrossRef CAS.
- K. Miller, J. Moul, M. Gleave, K. Fizazi, J. Nelson, T. Morris, F. Nathan, S. McIntosh, K. Pemberton and C. Higano, Prostate Cancer Prostatic Dis., 2013, 16, 187–192 CrossRef CAS PubMed.
- S. Bano, K. Javed, S. Ahmad, I. Rathish, S. Singh and M. Alam, Eur. J. Med. Chem., 2011, 46, 5763–5768 CrossRef CAS PubMed.
- H. Sadeghi, V. Hajhashemi, M. Minaiyan, A. Movahedian and A. Talebi, Eur. J. Pharmacol., 2011, 667, 396–401 CrossRef CAS PubMed.
- A. M. Mohassab, H. A. Hassan, D. Abdelhamid, A. M. Gouda, H. A. Gomaa, B. G. Youssif, M. O. Radwan, M. Fujita, M. Otsuka and M. Abdel-Aziz, J. Mol. Struct., 2021, 1244, 130948 CrossRef CAS.
- T. Liebregts, B. Adam, C. Bredack, A. Röth, S. Heinzel, S. Lester, S. Downie–Doyle, E. Smith, P. Drew and N. J. Talley, Gastroenterology, 2007, 132, 913–920 CrossRef CAS PubMed.
- P. Stenvinkel, M. Ketteler, R. J. Johnson, B. Lindholm, R. Pecoits-Filho, M. Riella, O. Heimbürger, T. Cederholm and M. Girndt, Kidney Int., 2005, 67, 1216–1233 CrossRef CAS PubMed.
- X. Zhang, L. Cui, B. Chen, Q. Xiong, Y. Zhan, J. Ye and Q. Yin, Complement. Ther. Clin. Pract., 2021, 42, 101291 CrossRef PubMed.
- M. DiMartino, G. Campbell, C. Wolff and N. Hanna, Agents Actions, 1987, 21, 303–305 CrossRef CAS PubMed.
- Y. Ranneh, F. Ali, A. M. Akim, H. A. Hamid, H. Khazaai and A. Fadel, Appl. Biol. Chem., 2017, 60, 327–338 CrossRef CAS.
- K. Ramakrishna and S. Krishnamurthy, Int. J. Dev. Neurosci., 2023, 83, 31–43 CrossRef CAS.
- A. G. Banerjee, N. Das, S. A. Shengule, P. A. Sharma, R. S. Srivastava and S. K. Shrivastava, Bioorg. Chem., 2016, 69, 102–120 CrossRef CAS PubMed.
- S. Ahmad, B. P. Panda, M. Fahim, N. Dhyani and K. Dubey, Iran. J. Pharm. Res., 2018, 17, 155 CAS.
- X.-H. Che, C.-L. Chen, X.-L. Ye, G.-B. Weng, X.-Z. Guo, W.-Y. Yu, J. Tao, Y.-C. Chen and X. Chen, Oncol. Rep., 2016, 35, 1680–1688 CrossRef CAS PubMed.
- L. Goossens, N. Pommery and J. Pierre Henichart, Curr. Top. Med. Chem., 2007, 7, 283–296 CrossRef CAS PubMed.
- V. A. Pratyusha, G. S. Victoria, M. F. Khan, D. T. Haokip, B. Yadav, N. Pal, S. C. Sethi, P. Jain, S. L. Singh and S. Sen, Sci. Rep., 2018, 8, 5248 CrossRef.
- W. E. Kattan and J. F. Hancock, Biochem. J., 2020, 477, 2893–2919 CrossRef CAS.
- S. Mukhopadhyay, M. G. Vander Heiden and F. McCormick, Nat. Cancer, 2021, 2, 271–283 CrossRef CAS PubMed.
- H. Zhao, L. Wu, G. Yan, Y. Chen, M. Zhou, Y. Wu and Y. Li, Signal Transduction Targeted Ther., 2021, 6, 263 CrossRef CAS.
- S. H. Ullah, A. Khan, S. A. Halim, R. Khan, X.-D. Pan, R. Ullah, A. Wadood, A. Khalid, A. N. Abdalla and S. Khogeer, Bioorg. Chem., 2023, 140, 106760 CrossRef PubMed.
- N. C. Gilbert, S. G. Bartlett, M. T. Waight, D. B. Neau, W. E. Boeglin, A. R. Brash and M. E. Newcomer, Science, 2011, 331, 217–219 CrossRef CAS PubMed.
- Z. Benfodda, V. Fritz, C. Henriquet, C. Fattorusso, G. C. Cebrian-Torrejon, M. Persico, A. Di Dato, M. Menna, H. Blancou and L. Fajas, Med. Chem., 2017, 7, 257–267 CAS.
- C. A. Kumar, K. Vinaya, J. N. S. Chandra, N. Thimmegowda, S. B. Prasad, C. Sadashiva and K. Rangappa, J. Enzyme Inhib. Med. Chem., 2008, 23, 462–469 CrossRef.
- P. Sharma, A. Tripathi, P. N. Tripathi, S. S. Singh, S. P. Singh and S. K. Shrivastava, ACS Chem. Neurosci., 2019, 10, 4361–4384 CrossRef CAS.
- H. Fretz, A. Valdenaire, J. Pothier, K. Hilpert, C. Gnerre, O. Peter, X. Leroy and M. A. Riederer, J. Med. Chem., 2013, 56, 4899–4911 CrossRef CAS.
- K.-C. Hsu, W.-C. HuangFu, T. E. Lin, M.-W. Chao, T.-Y. Sung, Y.-Y. Chen, S.-L. Pan, J.-C. Lee, S.-C. Tzou and C.-M. Sun, Sci. Rep., 2020, 10, 10510 CrossRef CAS PubMed.
- D. K. Waiker, A. Verma, P. Saraf, G. Ta, S. Krishnamurthy, R. N. Chaurasia and S. K. Shrivastava, ACS Omega, 2023, 8, 9394–9414 CrossRef CAS.
- S. Manju, Bioorg. Chem., 2020, 100, 103882 CrossRef.
- S. K. Shrivastava, P. Srivastava, R. Bandresh, P. N. Tripathi and A. Tripathi, Bioorg. Med. Chem., 2017, 25, 4424–4432 CrossRef CAS.
- M. H. Abdelrahman, B. G. Youssif, A. H. Abdelazeem, H. M. Ibrahim, A. M. Abd El Ghany, L. Treamblu and S. N. A. Bukhari, Eur. J. Med. Chem., 2017, 127, 972–985 CrossRef CAS.
- A. A. Marzouk, E. S. Taher, M. S. A. Shaykoon, P. Lan, W. H. Abd-Allah, A. M. Aboregela and M. F. El-Behairy, Bioorg. Chem., 2021, 111, 104883 CrossRef CAS PubMed.
- K. Ramakrishna and S. Krishnamurthy, Nat. Prod. Res., 2022, 36, 6044–6049 CrossRef CAS.
- E. A. Ugbogu, H. Okoro, O. Emmanuel, O. C. Ugbogu, C. N. Ekweogu, M. Uche, E. D. Dike and S. N. Ijioma, J. Ethnopharmacol., 2024, 319, 117224 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- J. Kode, J. Kovvuri, B. Nagaraju, S. Jadhav, M. Barkume, S. Sen, N. K. Kasinathan, P. Chaudhari, B. S. Mohanty and J. Gour, Bioorg. Chem., 2020, 105, 104447 CrossRef CAS.
- A. Tripathi, P. K. Choubey, P. Sharma, A. Seth, P. N. Tripathi, M. K. Tripathi, S. K. Prajapati, S. Krishnamurthy and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 183, 111707 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00471j |
| This journal is © The Royal Society of Chemistry 2025 |