
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of different phase structure nano sized FePO4/C cathode material by ultrasonic-assisting and their electrochemical performance†
Chunmei Tanga,
Wei Maa,
Jichuan Huo*a and
Shuxin Liu *b
*b
aCollege of Materials and Chemistry, Southwest University of Science and Technology, Mianyang, Sichuan 621010, China. E-mail: huojichuan@163.com
bSchool of Chemistry and Chemical Engineering, Mianyang Techers' College, Mianyang, Sichuan 621000, China. E-mail: liushuxin88@126.com
First published on 25th February 2025
Abstract
As one of the cathode materials for phosphate-based sodium ion batteries, FePO4 has received extensive attention due to its excellent theoretical capacity and stability. However, the FePO4 cathode has the problem of low ionic conductivity and electronic conductivity, which limits its application in sodium-ion batteries. The phase composition and microstructure of FePO4 are crucial to ensure the excellent electrochemical properties. Therefore, in this paper, the nano-sized amorphous FePO4·2H2O/C with a particle size of only 50 nm was prepared by ultrasonic-assisted precipitation. The carbon black oxidized can be uniformly dispersed in samples and form a spatial network structure. Four kinds of crystalline cathode materials were successfully prepared by further treatment of FePO4·2H2O/C, including amorphous FePO4/C, hexagonal FePO4/C, monoclinic FePO4·2H2O/C and monoclinic/orthogonal FePO4·2H2O/C. The microstructure, phase composition, particle size distribution and specific surface area of the samples were characterized by XRD, SEM, TEM, EDS, Raman, and BET. The results show that the amorphous FePO4/C particle size is smaller, and the specific surface area is larger. The electrochemical properties of samples were analyzed by CV and EIS. The results show that the crystal structure affects the specific charge–discharge capacity, Na+ diffusion coefficient, and charge transfer resistance of the materials. The amorphous FePO4/C has excellent electrochemical performance, the specific discharge capacity is 149.8 mA h g−1, the Na+ diffusion coefficient is 2.71 × 10−16 cm2 s−1, and the charge transfer resistance is 139 Ω. The results show that the amorphous structure is effective for improving the electrochemical performance of FePO4 cathode materials.
1 Introduction
Lithium-ion batteries are currently one of the most common rechargeable batteries, and they are widely used in electronic devices, automobiles, power storage and so on. However, due to the limitation of lithium resources, and people's demand for low cost, strong safety, resource sustainability and higher energy density, the research and development of other secondary battery technologies, such as potassium ion batteries, aluminum ion batteries, and sodium-ion batteries, have gradually attracted attention.1 However, these batteries come with their own set of limitations and challenges. For example, aluminum ion batteries are prone to side reactions during charging and discharging, especially hydrogen evolution reactions, affecting their cycle stability and energy efficiency.2 Potassium ion batteries face challenges such as volume expansion and slow kinetics, resulting in poor performance during high-rate charge and discharge.3 And sodium-ion batteries have problems such as poor conductivity.4 For potassium ion batteries, Zhang et al.5 reported the ternary Bi0.4Sb1.6Te3 nanoparticles coupled with few-layered graphene hybrids, which exhibit a mitigated expansion of 28% during the potassiation/depotassiation process. At the same time, aiming at the problems existing in aluminum ion batteries, Zhang et al.6 designed a deep eutectic electrolyte named HEE30 to significantly improve the reversibility and electrochemical stability of aluminum ion batteries in a wide temperature range of −20 to 60 °C. For sodium-ion batteries, Hu et al.7 designed a series of Nasicon-type Na3+xMnTi1−xVx(PO4)3 cathode materials, which have multi-electron reactions, and have a high-voltage platform. Yang et al.8 proposed an interconnected microchip composed of carbon nanotubes and sulfur-doped TiO2(CNT/S-TiO2) as a high-performance anode material for SIBs, enhancing the conductivity and improving the ion transport kinetics. In a word, researchers are constantly exploring the technical improvement and development of various secondary batteries in terms of performance improvement, cost control, material selection and charge–discharge efficiency.In recent years, sodium-ion batteries have attracted more and more attention due to their low cost and abundant resource advantages, especially cathode materials. The cathode materials mainly include layered oxide, Prussian blue and polyanionic compounds. Among these promising cathode materials, FePO4 has become a research hotspot due to its simple preparation process, high structural stability, high safety, and one-dimensional Na+ channe.9–14 Although the FePO4 cathode material has many advantages, its low ionic and electronic conductivity limits its application.15
Many methods have been tried to improve the electrochemical performance of FePO4, including phase structure controlling, nanocrystallization and adding conductive agents, etc. The crystal phases of FePO4 are mainly amorphous, hexagonal, monoclinic, and orthogonal. Heterosite FePO4, as one of the orthogonal phases, is a metastable structure that cannot be directly prepared but is obtained by selective delithiation in LiFePO4.16 Most FePO4 crystal phases can be obtained by hydrothermal or direct precipitation, but the particle size is usually in the micron scale.17,18 To get a smaller particle size or nanocrystallization of FePO4, ball milling is the key technique, and to enhance the ionic and electronic conductivity, conductive agents were usually added, including graphene, carbon nanotubes, carbon black, etc. Wang et al.19 directly synthesized nanostructured amorphous FePO4–carbon nanotube (CNT) composites with high purity FePO4/C ratio controllable, the particles were refined to about 20 nm by ball milling, and showed a discharge specific capacity of 175.8 mA h g−1. However, the preparation process becomes more complicated and has higher energy consumption because of the implementation of the ball milling, so a new and simple method for nanoscale FePO4/C cathode materials with excellent performance is urgent.
In this work, different phase compositions, microstructures, and nano-sized FePO4·2H2O/C and FePO4/C cathode materials were prepared by ultrasonic-assisting technique with carbon black preoxidation technique. The particle size reached about 50 nm and the carbon black was evenly distributed. Meanwhile, it showed excellent electrochemical performance.
2 Materials and methods
2.1 Materials preparation
The amorphous FePO4·2H2O/C was prepared by ultrasonic-assisted precipitating with carbon black preoxidation technique. Firstly, carbon black was oxidized in an oil bath at 80 °C for 5 h by H2O2 and oxidized carbon black with strong hydrophilicity was obtained, next, the oxidized carbon black (5% wt) was dispersed into NH4H2PO4 (1 M, 60 ml) solution under strongly stirring, and then the NH4H2PO4/oxidized carbon black solution was slowly dropped into Fe(NO3)3·9H2O (1 M, 60 ml) solution to obtain a mixed solution. The mixed solution was heated to 80 °C in the mechanical-ultrasonic assisted instrument with ultrasonic power 300 W and stirring speed 300 rpm, and an appropriate amount of NH4·H2O was slowly dripped to adjust pH, then the reaction was continued for 4 h and stand for 1 h. Finally, the amorphous FePO4·2H2O/C was obtained. The preparation mechanism is shown in Fig. 1. | ||
| Fig. 1 Preparation mechanism diagram. | ||
Other four kinds of cathode materials were prepared by further treatment of amorphous FePO4·2H2O/C: (1) amorphous FePO4·2H2O/C was calcined at different temperature in a nitrogen atmosphere for a certain time, the amorphous and hexagonal FePO4/C without crystal water were separately obtained. (2) FePO4·2H2O/C was dispersed in 5 mol L−1 H3PO4 solution, then heated and aged at different temperature in the closed reactor for different time, the monoclinic FePO4·2H2O/C and orthogonal/monoclinic FePO4/C·2H2O were separately obtained.
2.2 Materials characterization
X-ray diffractometer (XRD) was used to collect the diffraction pattern of the powder to obtain the crystal structure information. The microstructure of the samples was measured by field emission scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM). The stretching vibration of carbon materials and main functional groups was analyzed using Raman spectroscopy. The particle size and specific surface area of the samples were analyzed by nitrogen adsorption/desorption testing.2.3 Battery assembling and electrochemical measurements
The active material was mixed with conductive agent (Super P) and binder (PVDF) according to the mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 into a uniform slurry, coated on aluminum foil, and baked in a vacuum drying oven at 80 °C for 20 h. After that, the positive electrode sheet required to assemble the half battery was obtained. The active material was used as the positive electrode, the sodium sheet as negative electrode, the electrolyte was 1 mol L−1 NaPF6 (EC:DEC:EMC = 1:1:1 vol%) solution, and the separator was glass fiber (GF/D Whatman).
:1:1 into a uniform slurry, coated on aluminum foil, and baked in a vacuum drying oven at 80 °C for 20 h. After that, the positive electrode sheet required to assemble the half battery was obtained. The active material was used as the positive electrode, the sodium sheet as negative electrode, the electrolyte was 1 mol L−1 NaPF6 (EC:DEC:EMC = 1:1:1 vol%) solution, and the separator was glass fiber (GF/D Whatman).
The constant current charge–discharge and cycle performance of the half-cell were tested by the LAND C2001A test system, the test voltage range was 1.5–4.5 V (vs. Na/Na+), and the charge–discharge current density was 0.1C, which defined 1C = 178 mA g−1. The electrochemical impedance spectroscopy (EIS) was employed using a CHI660E electrochemical workstation over the frequency range from 0.01 to 100 kHz with an amplitude of 5 mV. The cyclic voltammetry of the half-cell was tested using an electrochemical workstation (CHI660E) at 0.1 mV s−1.
3 Results and discussion
3.1 Characterization of materials
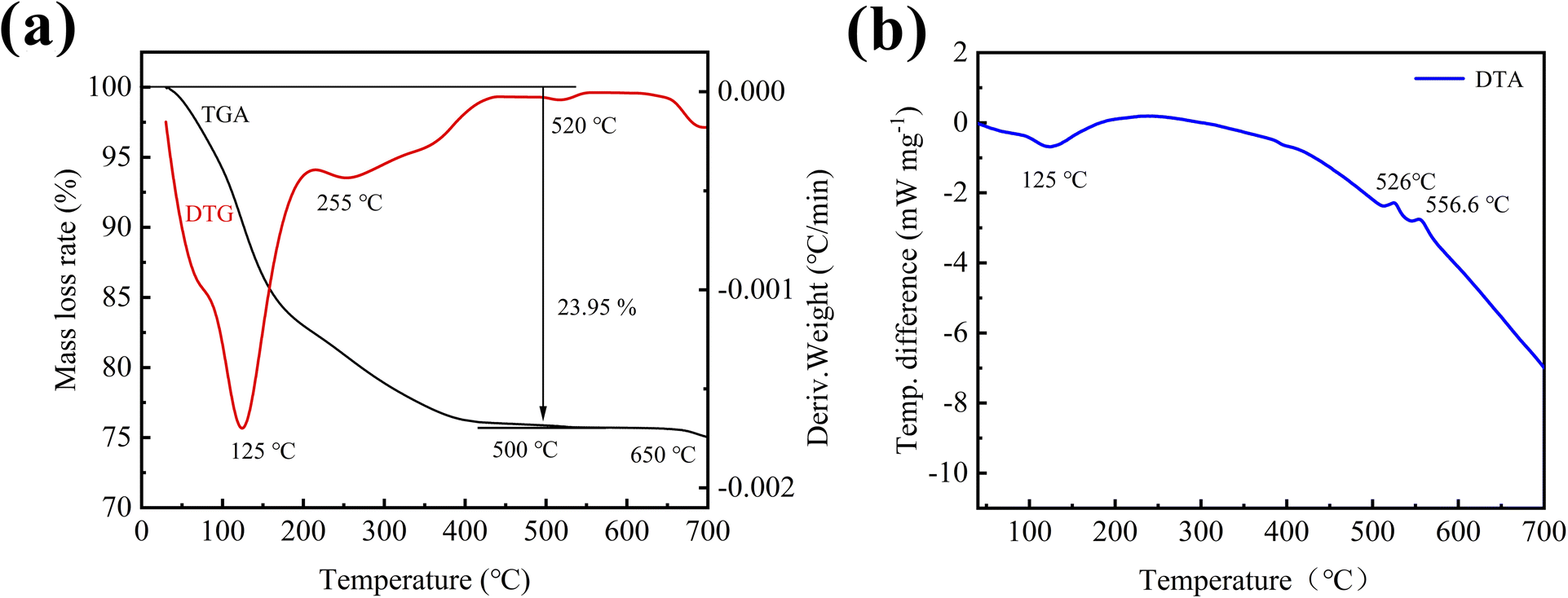
To analyze the phase structure and the amount of crystal water in the initial sample without aging and calcining, the XRD, TGA, DTG and DTA of the sample were tested. Fig. 2 shows XRD patterns of the initial sample, and its calcined sample. As can be seen from the figure, the XRD diffraction pattern of the initial sample only has a wide dispersion peak at about 25°, which is an amorphous structure, and its phase composition (including the phase of carbon) cannot be determined. To further determine the composition of the initial sample, the sample was calcined at 600 °C, and its diffraction pattern had strong diffraction peaks at 20.3°, 25.8°, 37.9°, etc., which were found to be consistent with quartz-type iron phosphate (COD number: 96-901-2513) standard card has a high degree of coincidence, indicating that the main phase of the initial sample prepared by ultrasonic assisted precipitation is iron phosphate. Meanwhile, thermal analysis was performed on the initial sample to further determine the crystal water content of the synthesized iron phosphate by precipitation. | ||
| Fig. 2 XRD patterns of the initial sample and its calcined sample. | ||
Fig. 3 shows the thermal analysis curves of the initial sample at a heating rate of 10 °C min−1. It can be seen from the TGA curve (Fig. 3a) that the quality of the sample begins to decline at about 40 °C until 500 °C, the weight of the sample is reduced by 23.95%. Combined with the analysis of the DTG curve, it can be found that the weight loss rate of the sample is the highest at 125 °C, followed by 255 °C, corresponding to the wide endothermic peak of the DTA curve (Fig. 3b) around 125 °C, it is speculated that the decomposition of crystal water and adsorbed water occurs in this temperature range. And after 500 °C, the TGA/DTG curves remain level, and the DTA curve is smooth and no difference. No significant mass loss or other physical/chemical reactions occurred in the sample, indicating that the crystal water and adsorbed water were completely removed. According to thermal analysis, the weight percentage of the dehydrated sample is 76.05%, and the sample contains 5% carbon. By calculating the actual weight percentage of the dehydrated sample is 74.79%, and according to formula (1), the number of crystal water is 2.83. Since the sample contains part of adsorbed water, there should be two crystal water. Therefore, it can be concluded that the initial sample prepared in this experiment is amorphous FePO4·2H2O/C cathode material.
 | (1) |
 | ||
| Fig. 3 TGA/DTG (a) and DTA (b) curves of the initial sample. | ||
In addition to determining the amount of crystal water in the sample, it can also be found through the thermal analysis that there are exothermic peaks in the DTA curve at about 520 °C and 556.6 °C, while the corresponding TGA/DTG curve does not change significantly. It is inferred that the crystal transformation of iron phosphate from amorphous to quartz type occurs at this time.
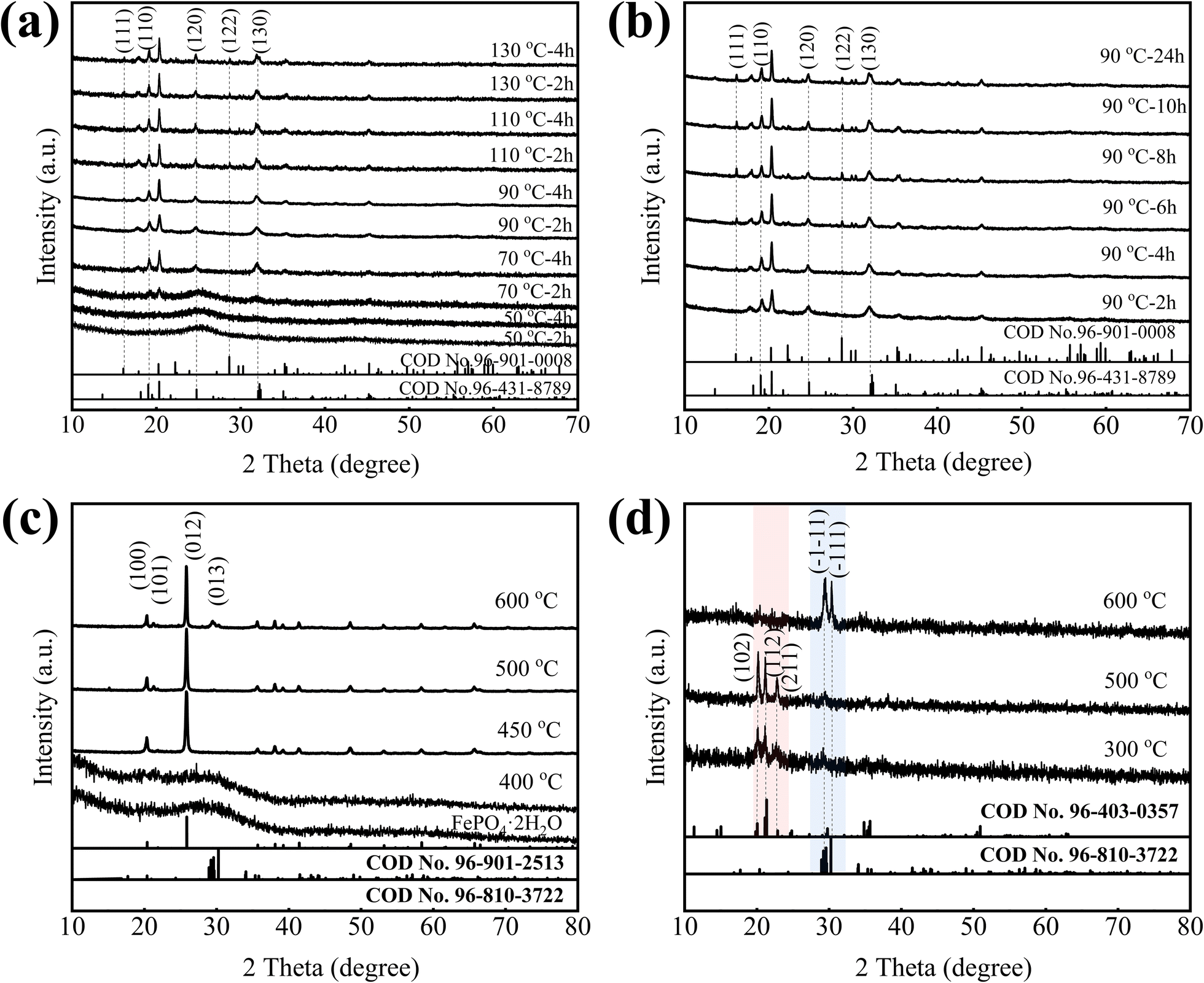
To get different phase structures FePO4·2H2O/C, the amorphous FePO4·2H2O/C was aged at different temperature in the closed reactor for different time. Fig. 4a shows the XRD patterns of aged samples. First of all, we know that the FePO4·2H2O/C without aging is an amorphous structure, and with the increase of aging temperature, the amorphous structure gradually transforms into a crystal structure. Further analysis find that the samples aged at 50 °C are amorphous structures. When the aging temperature is 70 °C and the time is 2 h, some weaker diffraction peaks appear at around 19°, 20.5°, 24.5° and 32°, which belong to monoclinic structure (COD no. 96-431-8789), and the aging time is extended to 4 h, these diffraction peaks gradually increase. When the aging temperature is 90 °C and the time is 2 h or 4 h, the phase structure of samples still remains in monoclinic structure. When the temperature rises further, some new diffraction peaks appear at around 16°, 22° and 29° which belong to orthogonal structure (COD no. 96-901-0008), but the diffraction peaks of monoclinic structure do not diminish, it indicates that the orthogonal structure transforms from amorphous structure, not monoclinic structure, and the transform process is slower and more difficult than monoclinic structure. All of the samples, including amorphous, monoclinic and monoclinic/orthogonal structures contain crystal water.
 | ||
| Fig. 4 The XRD pattern of samples aged at different temperatures for a-FPH/C (a), aged at 90 °C in different time for a-FPH/C (b), calcined at different temperature for a-FPH/C (c), and calcined at different temperature for om-FPH/C (d). | ||
In order to study the phase transform process of the FePO4·2H2O/C, amorphous FePO4·2H2O/C was aged at 90 °C for different time. From Fig. 4b, it can be found that the orthogonal diffraction peaks of FePO4·2H2O (COD no. 96-901-0008) increase gradually with aging time, and the monoclinic diffraction peaks did not diminish, the orthogonal and monoclinic structure coexist.
To obtain different phase structures of FePO4/C without crystal water, the amorphous FePO4·2H2O/C was calcined at 400 °C, 450 °C, 500 °C and 600 °C in a nitrogen atmosphere for 2 h. Fig. 4c is the XRD patterns of FePO4/C prepared at different calcination temperature, it can be found that the calcination temperature has a major impact on the phase structure of FePO4, when the calcination temperature is 400 °C, the phase structure of FePO4 is amorphous structure, which indicates that when FePO4·2H2O/C is calcined below 400 °C, the phase structure is amorphous structure. With the calcination temperature rising, a rise of only 50 °C, the amorphous structure transforms into hexagonal structure, it has a high matching degree with the diffraction peaks of hexagonal FePO4 indexed in the standard card (COD no. 96-901-2513), and there are no other peaks, indicating that the FePO4 crystal can be synthesized at 450 °C. As temperature calcination continues to rise, the phase structure does not change. When the temperature rises to 600 °C, the anorthic Fe2P2O7 phase (COD no. 96-810-3722) appears at nearly 30°, which indicates that Fe3+ is reduced to Fe2+ by carbon black, the reaction mechanism could be: 2FePO4 + C = Fe2P2O7 + CO↑, 2FePO4 + CO = Fe2P2O7 + CO2↑. The above analysis indicates that the amorphous and hexagonal FePO4/C without crystal water can be only obtained by calcining amorphous FePO4·2H2O/C, and other phase structure FePO4/C without crystal water cannot be obtained.
To obtain monoclinic or orthogonal FePO4/C without crystal water, the monoclinic/orthogonal FePO4·2H2O/C were calcined at 300 °C, 500 °C, 600 °C for 8 h, the XRD patterns shown in Fig. 4d. When the calcination temperature is 300 °C and 500 °C, the main phase is the orthorhombic Fe3P4O14 (COD no. 96-403-0357) and little anorthic Fe2P2O7 (COD no. 96-810-3722), it indicates that some of Fe3+ are reduced. When the calcination temperature is 600 °C, the main phase is the anorthic Fe2P2O7 indicating that most of Fe3+ are reduced, this can be attributed to the fact that Fe3+ is reduced by carbon black. These experiments indicate that the monoclinic or orthogonal FePO4/C without crystal water cannot be synthesized by calcination due to the presence of carbon black.
Based on the above analysis, the samples of the initial amorphous FePO4·2H2O/C without aging and calcining (a-FPH/C), monoclinic FePO4·2H2O/C (m-FPH/C) and orthogonal/monoclinic FePO4·2H2O/C (om-FPH/C) prepared by aging a-FPH/C at 90 °C for 2 h and 8 h, and amorphous FePO4/C (a-FP/C) and hexagonal FePO4/C (h-FP/C) prepared by calcining a-FPH/C at 400 °C for 2 h and at 600 °C for 2 h are used as research objects.
Fig. 5 is the Raman spectrum of the samples, and the characteristic Raman peaks belonging to carbon can be detected. The G band corresponds to the E2g phonon at the Brillouin zone center, which indicates sp2 hybridized carbon atoms arranged in a graphitic structure, the D band arises from defects and disorder within this structure. By comparing the peak areas of the G band and D band, the large ratio of peak area (Table 1) of the G band relative to the D band exhibits a high degree of graphitization, and highly graphitized carbon materials are known to possess enhanced electrical conductivity.16,20–26 Meanwhile, all of the ratios of IG/ID are basically equal, which indicates that the oxidizing, calcining and aging processes do no effect carbon black. Furthermore, it can be observed that a broad band centered at 1064 cm−1 is present in the Raman spectrum of m-FPH/C, om-FPH/C and h-FP/C, it is the characteristic of iron being tetrahedrally coordinated attributed to the formation of tetrahedrally-coordinated iron atoms to the enhanced surface energy kinetics at lower crystallite sizes,27–29 whereas this band is not present in a-FPH/C and a-FP/C, further demonstrating their amorphous structure.
 | ||
| Fig. 5 Raman spectrum of the samples. | ||
| Samples | Peak D | Peak G | IG/ID | ||
|---|---|---|---|---|---|
| Area | Position (cm−1) | Area | Position (cm−1) | ||
| a-FPH/C | 247988.48 |
1370.1 | 182993.54 |
1584.2 | 0.738 |
| m-FPH/C | 236596.62 |
1370.6 | 166715.26 |
1592.9 | 0.705 |
| om-FPH/C | 203405.07 |
1363.4 | 145787.86 |
1586.3 | 0.718 |
| a-FP/C | 170298.07 |
1363.4 | 120221.00 |
1584.2 | 0.706 |
| h-FP/C | 161927.08 |
1363.9 | 119317.61 |
1582.3 | 0.737 |
Fig. 6 is the SEM and TEM of the samples. In the figures, the amorphous FePO4·2H2O/C (a-FPH/C) particles are globular and uniform in size, about 50 nm (Fig. 6b). The monoclinic m-FPH/C and orthogonal/monoclinic om-FPH/C are flake (Fig. 6c–f), and there are some small flocculent particles in these samples, they should be carbon black, and the flake of m-FPH/C is bigger than om-FPH/C, the thickness of flakes is about 20 nm. Both amorphous a-FP/C and hexagonal h-FP/C have near-spherical shapes (Fig. 6g–j), the particle dispersion is more uniform, and the particle size is about 50 nm (Fig. 6h and j), and in contrast to the a-FPH/C, the calcination temperature and phase changing are almost no effects on particles size. At the same time, because the samples of a-FPH/C, a-FP/C and h-FP/C are spherical and the particle size is small, carbon black is not easy to identify.
 | ||
| Fig. 6 SEM and TEM of samples. (a) and (b) are SEM and TEM of a-FPH/C, (c) and (d) are SEM and TEM of m-FPH/C, (e) and (f) are SEM and TEM of om-FPH/C, (g) and (h) are SEM and TEM of a-FP/C, (i) and (j) are SEM and TEM of h-FP/C. | ||
In order to further prove the existence of carbon and the distribution of each element, elemental analysis of amorphous FePO4·2H2O/C (a-FPH/C) is carried out by energy dispersion spectroscopy (EDS), and the results are shown in Fig. 7. As shown in the figures, all elements are evenly distributed, the molar ratio of Fe/P is close to 1:1, and the content of carbon black is 5 wt%. In summary, the nano-size FePO4·2H2O/C cathode materials with uniform particle distribution were successfully prepared by ultrasonic-assisted preparation, and carbon black to form a conducting network structure in samples.
 | ||
| Fig. 7 Element distribution mappings of a-FPH/C. | ||
Fig. 8 shows the N2 adsorption–desorption isotherm and pore size distribution of the samples. The adsorption–desorption curves of the five samples display IV-type isotherms with H1 hysteresis loop, H3 hysteresis loop, and H4 hysteresis loop, H1 hysteresis loop, H3 hysteresis loop, respectively. The type of hysteresis loop corresponds to the specific pore structure information. Among them, H1 is a uniform pore model, which can be considered as a cylindrical pore, while H3 and H4 have a large adsorption amount under high pressure, which can be considered as a narrow pore formed by the accumulation of flake particles or a large pore formed by the accumulation of big particles. At the same time, the pore size distribution also further verifies that the pore of samples is mainly mesoporous. Table 2 shows the specific surface area and pore size of the samples. It can be seen that the amorphous structure a-FPH/C and a-FP/C have a larger specific surface area of 85.021 m2 g−1 and 70.289 m2 g−1. This is because the calcining changes particle size and porosity, and heating and aging change particle size and shape.
 | ||
| Fig. 8 N2 adsorption–desorption isotherm and pore size distribution of a-FPH/C (a), m-FPH/C (b), om-FPH/C (c), a-FP/C (d), and h-FP/C (e). | ||
| Sample | SBET (m2 g−1) | Pore volume (cm3 g−1) | Average pore size (nm) |
|---|---|---|---|
| a-FPH/C | 85.021 | 0.598 | 24.314 |
| m-FPH/C | 22.303 | 0.106 | 19.649 |
| om-FPH/C | 46.255 | 0.086 | 19.640 |
| a-FP/C | 70.289 | 0.341 | 14.845 |
| h-FP/C | 14.579 | 0.066 | 17.513 |
3.2 Electrochemical performance analysis
Fig. 9 shows the electrochemical properties of samples prepared by ultrasonic-assisted preparation. Fig. 9a illustrates the Nyquist plot of samples, all of the curves comprise a straight line at low frequencies and a semicircle at high to medium frequencies, where the linear portion signifies sodium ion diffusion within the electrode, while the semicircular part denotes charge transfer occurring on the cathode surface.30,31 The corresponding equivalent circuit model is presented in Fig. 9a, the charge transfer resistance (Rct) values of a-FPH/C, m-FPH/C, om-FPH/C, a-FP/C and h-FP/C after fitting are 205, 833, 618, 139, 1020 Ω, respectively. It can be found that the Rct of a-FP/C is much lower than that of other phase structures, indicating that the amorphous FePO4/C materials have better electronic conductivity. The reason is that a larger specific surface area of a-FP/C improves the point contact between the active particles and the carbon black, which is conducive to bipolar diffusion,18 and the isotropic and defectless properties of amorphous FePO4 provides a large number of continuous channels for Na+.32,33 | ||
| Fig. 9 Electrochemical impedance spectroscopy of samples (a). The relationship between the resistance (Z′) and the inverse square root of the angular speed (b). The slope (δ) and Na+ diffusion coefficient of samples (c). Cyclic voltammetry of samples at a scan rate of 0.1 mV s−1 in the voltage range of 1.5–4.5 V at room temperature (d). | ||
Since there is a good linear relationship between the resistance (Z′) and the angular velocity (ω−1/2) (Fig. 9b), the specific Na+ diffusion rate (DNa+) can be calculated according to this relationship. The sodium diffusion coefficient in Fig. 9c is calculated using formula (2).34–38
 | (2) |
The calculated results (Fig. 9c) show that the diffusion coefficients of Na+ are 2.34 × 10−16, 2.24 × 10−16, 2.40 × 10−16, 2.71 × 10−16 cm2 s−1 and 1.81 × 10−17 cm2 s−1 for a-FPH/C, m-FPH/C, om-FPH/C, a-FP/C, and h-FP/C, respectively. The maximum Na+ diffusion coefficient of a-FP/C further proves that the amorphous structure without crystal water is favorable to the electrochemical performance. Fig. 9d is the cyclic voltammetry of samples at a scan rate of 0.1 mV s−1 in the voltage range of 1.5–4.5 V at room temperature, one pair of oxidation–reduction current peaks of Fe3+/Fe2+ can be seen located near 3.1 V, the wide oxidation–reduction peak proves that the sodiation/desodiation process is a continuous one-phase oxidation–reduction reaction. Meanwhile, a-FP/C has the smallest potential difference, indicating the lowest degree of electrode polarization, which also shows that the amorphous structure, nano-sized spherical particles, large specific surface area, spatial network structure of carbon black enhance the sodiation/desodiation kinetics of cathode material.
Fig. 10a is the initial charge–discharge curves of samples at 0.1C at room temperature. The discharge specific capacity of a-FPH/C, m-FPH/C, om-FPH/C, a-FP/C and h-FP/C is 124.1, 94.9, 114.6, 149.8, 68.1 mA h g−1, respectively. From the comparison, whether or not containing crystal water, the amorphous structure materials have higher discharge specific capacity, the reason is the amorphous structure can provide more Na+ transport channels, meanwhile, the amorphous structure has a larger specific surface area, which means that there are more contact surfaces with conductive carbon black and electrolytes. Moreover, the amorphous a-FP/C without crystal water has the maximum discharge specific capacity, this may be the crystal water may hinder Na+ transport channel and may react with electrolyte, reducing the reversibility of sodiation/desodiation during charge and discharge. In the calcining amorphous FePO4·2H2O/C process, with the increase of calcination temperature, the amorphous structure is transformed into hexagonal structure which has high stability, but the more stable structure usually has lower electrochemical activity,39 so it has the minimum discharge specific capacity. Comparing m-FPH/C and om-FPH/C, the discharge specific capacity of om-FPH/C containing partially orthogonal structure is higher than m-FPH/C, it shows that orthogonal structure has better electrochemical performance than monoclinic structure. Fig. 10b is the cycling performance of samples at 0.1C at room temperature, the cycling performance of a-FP/C is excellent, and with the increase in the number of cycles, the capacity retention rate is 94.06%. It also has been found that the discharge specific capacity of the samples with crystal water (a-FPH/C, m-FPH/C and om-FPH/C) decreases seriously with the increase of the number of cycles, it further shows that the crystal water will be detrimental to charge and discharge process. Based on the above experimental results, compared with the existing literature,11,14,15,25,40 this work adopts a relatively simple experimental process to synthesize amorphous structural FePO4/C cathode materials, and obtains similar or better experimental results.
 | ||
| Fig. 10 Charge–discharge curves (a) of samples and cycling performance (b) of samples at 0.1C at room temperature. | ||
4 Conclusion
To study the electrochemical performance of FePO4 with different crystal phases, ultrasonic-assisting technique was applied to the precipitation reaction process of FePO4/C for sodium ion battery, and the different phase composition and microstructure cathode materials including amorphous and hexagonal structure FePO4/C without crystal water, and the amorphous, monoclinic and orthogonal/monoclinic FePO4/C with crystal water were prepared. The peroxidation for carbon black and ultrasonic application in the preparation process obtain the high dispersity and ultrafining cathode materials. Furthermore, the peroxidation doesn't change the structure of carbon black. Because of the highly nano-sized particles, higher specific surface area, and a continuous and uniform carbon spatial network structure, all the samples showed a certain improvement in electrochemical performance, especially a-FP/C. Moreover, the amorphous structure can provide more Na+ transport channels than other phase structures, and the crystal water was removed by calcining which is more conducive to ion transport, so, a-FP/C shows an excellent discharge specific capacity, the maximum Na+ diffusion coefficient, the minimum charge transfer resistance and good cycle performance. This work provides a new insight into the preparation of the enhanced electrochemical performance for FePO4/C, and is helpful in developing new battery materials through the ultrasonic technique.Data availability
Data will be made available on request.Author contributions
Chunmei Tang: methodology, investigation, writing – original draft. Wei Ma: data curation. Jichuan Huo: conceptualization, supervision. Shuxin Liu: writing – review, conceptualization, resources & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research and Innovation Team Project of Mianyang Teachers' College (CXTD2023PY06).References
- C. Zhang, S. Chou, Z. Guo and S. Dou, Adv. Funct. Mater., 2024, 34, 2308001 CrossRef CAS.
- T. Dong, K. L. Ng, Y. Wang, O. Voznyy and G. Azimi, Adv. Energy Mater., 2021, 11, 2100077 CrossRef CAS.
- Z. Xiao, F. Xia, L. Xu, X. Wang, J. Meng, H. Wang, X. Zhang, L. Geng, J. Wu and L. Mai, Adv. Funct. Mater., 2022, 32, 2108244 CrossRef CAS.
- D. Wang, Y. Wu, J. Lv, R. Wang and S. Xu, Colloids Surf., A, 2019, 583, 123957 CrossRef CAS.
- L. Zhang, J. Liu, Y. Zhai, S. Zhang, W. Wang, G. Li, L. Sun, H. Li, S. Qi, S. Chen, R. Wang, Q. Ma, J. Just and C. Zhang, Adv. Mater., 2024, 36, 2313835 CrossRef CAS PubMed.
- X. Zhang, R. Wang, Z. Liu, Q. Ma, H. Li, Y. Liu, J. Hao, S. Zhang, J. Mao and C. Zhang, Adv. Energy Mater., 2024, 14, 2400314 CrossRef CAS.
- P. Hu, T. Zhu, C. Cai, X. Wang, L. Zhang, L. Mai and L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202219304 CrossRef CAS PubMed.
- C. Wang, Q. Yao, M. Wang, C. Zheng, N. Wang, Z. Bai, J. Yang, S. Dou and H. Liu, Adv. Funct. Mater., 2024, 34, 2301996 CrossRef CAS.
- X. Zhao, S. Yang, X. Song, Y. Wang, H. Zhang, M. Li and Y. Wang, Adv. Sci., 2024, 11, 2405176 CrossRef CAS PubMed.
- Y. Wang, Z. Feng, D. Laul, W. Zhu, M. Provencher, M. L. Trudeau, A. Guerfi and K. Zaghib, J. Power Sources, 2018, 374, 211–216 CrossRef CAS.
- W. Wang, S. Wang, H. Jiao, P. Zhan and S. Jiao, Phys. Chem. Chem. Phys., 2015, 17, 4551–4557 RSC.
- Q. Fan, L. Lei, G. Yin, Y. Chen and Y. Sun, Electrochem. Commun., 2014, 38, 120–123 CrossRef CAS.
- M. R. Cerón, M. Izquierdo, N. Alegret, J. A. Valdez, A. Rodríguez-Fortea, M. M. Olmstead, A. L. Balch, J. M. Poblet and L. Echegoyen, Chem. Commun., 2016, 52, 64–67 RSC.
- S.-Y. Duan, J.-Y. Piao, T.-Q. Zhang, Y.-G. Sun, X.-C. Liu, A.-M. Cao and L.-J. Wan, NPG Asia Mater., 2017, 9, e414 CrossRef CAS.
- Y. Wang, M. Deng, X. Zhang, J. Zhang, Y. Sui, K. Sun, K. Rao and L. Wu, J. Colloid Interface Sci., 2024, 661, 23–32 CrossRef CAS PubMed.
- A. Banday, R. Shahid, M. Cupta and S. Murugavel, RSC Adv., 2023, 13, 18332 RSC.
- J. Ma, L. Wang, F. Yu and X. Dai, Chem. Eng. J., 2019, 370, 938–943 CrossRef CAS.
- C. Li, X. Wang, J. Li and H. Wang, Chem. Commun., 2018, 54, 4349–4352 RSC.
- Z. Wang and Y. Lu, ACS Omega, 2019, 4, 14790–14799 CrossRef CAS PubMed.
- K. He, Z. Xu, X. Zhang, Q. Li and F. Wang, Spectrochim. Acta, Part A, 2023, 289, 122249 CrossRef CAS PubMed.
- N. Hassanzadeh, S. K. Sadrnezhaad and G. Chen, Electrochim. Acta, 2016, 208, 188–194 CrossRef CAS.
- I. P. Vali, B. S. Anusha, M. Pruthvija, S. Savitha, S. Ravindra, M. Nagaveni, P. S. Poojitha and N. Swathi, Mater. Chem. Phys., 2024, 318, 129240 CrossRef CAS.
- M. Shi, D. Bao, S. Li, B. Wulan, J. Yan and Q. Jiang, Adv. Energy Mater., 2018, 8, 1800124 CrossRef.
- C. M. Burba, J. M. Palmer and B. S. Holinsworth, J. Raman Spectrosc., 2009, 40, 225–228 CrossRef CAS.
- Z. Wang, J. Liu, X. Zhang, Y. Wang, D. Wang, W. Shi, H. Li, P. Zhang, J. Man and L. Liu, J. Non-Cryst. Solids, 2024, 634, 122983 CrossRef CAS.
- A. Banday, R. Shahid, S. S. Meena, S. M. Yusuf and S. Murugavel, Phys. Chem. Chem. Phys., 2020, 22, 15478–15487 RSC.
- L. Popovi, D. De Waal and J. C. A. Boeyens, J. Raman Spectrosc., 2005, 36, 2–11 CrossRef.
- J. Haines, O. Cambon and S. Hull, Z. Kristallogr. – Cryst. Mater., 2003, 218, 193–200 Search PubMed.
- P. Tarte, Solid State Ionics, 1990, 42, 177–196 CrossRef CAS.
- W. Lou, Y. Zhang, Y. Zhang, S. Zheng, P. Sun, X. Wang, S. Qiao, J. Li, Y. Zhang, D. Liu, M. Wenzel and J. J. Weigan, J. Alloys Compd., 2021, 856, 158148 Search PubMed.
- W. Wang, S. Wang, H. Jiao, P. Zhan and S. Jiao, Phys. Chem. Chem. Phys., 2015, 17, 4551–4557 RSC.
- C. X. Guo, Y. Q. Shen, Z. L. Dong, X. D. Chen, X. W. Lou and C. M. Li, Energy Environ. Sci., 2012, 5, 6919 RSC.
- Y. Yin, Y. Hu, P. Wu, H. Zhang and C. Cai, Chem. Commun., 2012, 48, 2137 RSC.
- X. Ren, Z. Li, J. Cao, S. Tian, K. Zhang, J. Guo, L. Wen and G. Liang, J. Alloys Compd., 2021, 867, 158776 CrossRef CAS.
- Y. Zuo, J. Yue, Z. Ma and Z. Zuo, J. Phys. Chem. Solids, 2022, 160, 110354 CrossRef CAS.
- H. Xiao, J. P. Pender, M. A. Meece-Rayle, J. P. De Souza, K. C. Klavetter, H. Ha, J. Lin, A. Heller, C. J. Ellison and C. B. Mullins, ACS Appl. Mater. Interfaces, 2017, 9, 22641–22651 CrossRef CAS PubMed.
- Y. Zhang, H. Jin, H. Tu, Y. Zuo, Q. Luo, P. Li, Z. Chen, J. Jia and L. Zhang, J. Ind. Text., 2023, 53, 1–16 CrossRef.
- X. Tian, Y. Zhou, X. Tu, Z. Zhang and G. Du, J. Power Sources, 2017, 340, 40–50 Search PubMed.
- Y. Pei, C. Liu, Z. Han, Z. G. Neale, W. Qian, S. Xiong, Z. Jiang and G. Cao, J. Power Sources, 2019, 431, 75–83 CrossRef CAS.
- B. Pandit, B. Fraisse, L. Stievano, L. Monconduit and M. T. Sougrati, Electrochim. Acta, 2022, 409, 139997 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00107b |
| This journal is © The Royal Society of Chemistry 2025 |