
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, antiproliferative activity, and molecular dynamics simulation of pyrazoline-based derivatives as dual EGFR and HER-2 inhibitors†
Hani Mohamed Hafez a,
Basmat Amal M. Saidb,
Ahmed M. Sayedcd,
Eid Alatwie,
Bahaa G. M. Youssif*f,
Stefan Bräse*g and
Hany A. M. El-Sheriefh
a,
Basmat Amal M. Saidb,
Ahmed M. Sayedcd,
Eid Alatwie,
Bahaa G. M. Youssif*f,
Stefan Bräse*g and
Hany A. M. El-Sheriefh
aPharmaceutical Chemistry Branch, College of Pharmacy, Al-Esraa University College, Baghdad, Iraq
bCollege of Pharmacy, Al-Mustaqbal University, Babylon, 51001, Iraq
cDepartment of Pharmacognosy, Faculty of Pharmacy, Nahda University, 62513 Beni Suef, Egypt
dDepartment of Pharmacognosy, Collage of Pharmacy, Almaaqal University, 61014 Basrah, Iraq
eDepartment of Pharmacology, College of Pharmacy, Jouf University, Sakaka 72341, Aljouf, Saudi Arabia
fDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt. E-mail: bgyoussif2@gmail.com; Tel: +20-1098294419
gInstitute of Biological and Chemical Systems-Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
hDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Deraya University, Minia, Egypt
First published on 28th March 2025
Abstract
The dual targeting of EGFR and HER2 is an established anticancer strategy. A novel series including two distinct scaffolds, A (chalcone-based compounds, 4a–n) and B (pyrazoline-based compounds, 5a–n), was developed and synthesized. The antiproliferative efficacy of 4a–n and 5a–n was examined against a panel of four cancer cell lines. The findings indicated that pyrazoline derivatives 5a–n exhibited more efficacy than chalcone compounds 4a–n. Compounds 4n, 5d, and 5g were identified as the most effective antiproliferative derivatives. These compounds were further investigated as dual EGFR/Her2 inhibitors. Compound 5d inhibited EGFR-TK and HER2 significantly, with IC50 values of 0.126 and 0.061 μM, respectively. Moreover, compound 5d can induce a percentage of pre-G1 apoptosis by 78.53% in cell cycle analysis and cause early apoptosis with necrosis percent of 5.28. Docking and MD simulation illustrated the significant cytotoxic activity of the 5d compound and how it can be a promising scaffold with anticancer activity.
1. Introduction
Receptor tyrosine kinases (RTKs) are a class of tyrosine kinases that help transfer a phosphate group from ATP to a hydroxyl group on a tyrosine residue. These enzymes regulate various tasks in normal cells, including growth, motility, differentiation, and metabolism. Furthermore, they are crucial to oncogenesis.1,2 There are 58 known receptor tyrosine kinases (RTKs) in humans, which are divided into 20 subfamilies. These include the ERBB receptors, the insulin receptors, the platelet-derived growth factor (PDGF) receptors, and the vascular endothelial growth factor (VEGF) receptors.3,4 ERBB, a subclass I of the receptor tyrosine kinase superfamily, comprises four members: EGFR/ERBB1/HER1, ERBB2/HER2, ERBB3/HER3, and ERBB4/HER4. These members have a similar protein structure, with a tyrosine kinase-containing cytoplasmic domain, an extracellular ligand-binding domain, and a single transmembrane helix.5 According to reports, the epidermal growth factor receptor (EGFR) plays a crucial role in tumor growth and progression, including cell proliferation, apoptosis inhibition, metastasis, and angiogenesis.6,7 Moreover, HER2 functions as a significant cancer biomarker despite the absence of a ligand for it. HER2 must dimerize to be activated, and heterodimers formed between HER2, and other ERBB family members have gotten much attention because they are more stable and send signals more effectively. Oncological treatment, considered a significant target, typically generates EGFR/HER2 heterodimers.8Pyrazolines are five-membered heterocycles that consist of two adjacent nitrogen atoms and an endocyclic double bond in the ring structure. Of the three tautomeric structures of pyrazolines, 2-pyrazoline is the most prevalent.9 Researches reveal that diversely substituted pyrazolines exhibit various pharmacological actions, including anticancer effects. Certain pyrazoline-derived cytotoxic agents exhibit cancer chemopreventive characteristics.10–13
For example, Lv and colleagues demonstrated that the pyrazoline-based derivative I (Fig. 1) exhibited significant cytotoxicity against the breast cancer cell line (MCF-7), with an IC50 of 0.07 μM, through EGFR inhibition, which had an IC50 value of 0.06 μM.14 Al-Anazi et al.15 developed a novel series of pyrazoline-based compounds as EGFR inhibitors. The cytotoxic effects of these compounds were examined on hormonal and non-hormonal breast cancer cell lines. Most pyrazoline derivatives have shown significant cytotoxic effects against hormonal breast cancer. In vitro tests have shown compound II (Fig. 1) is the most potent pyrazoline derivative, blocking the EGFR kinase at a very low concentration of 0.19 μM.
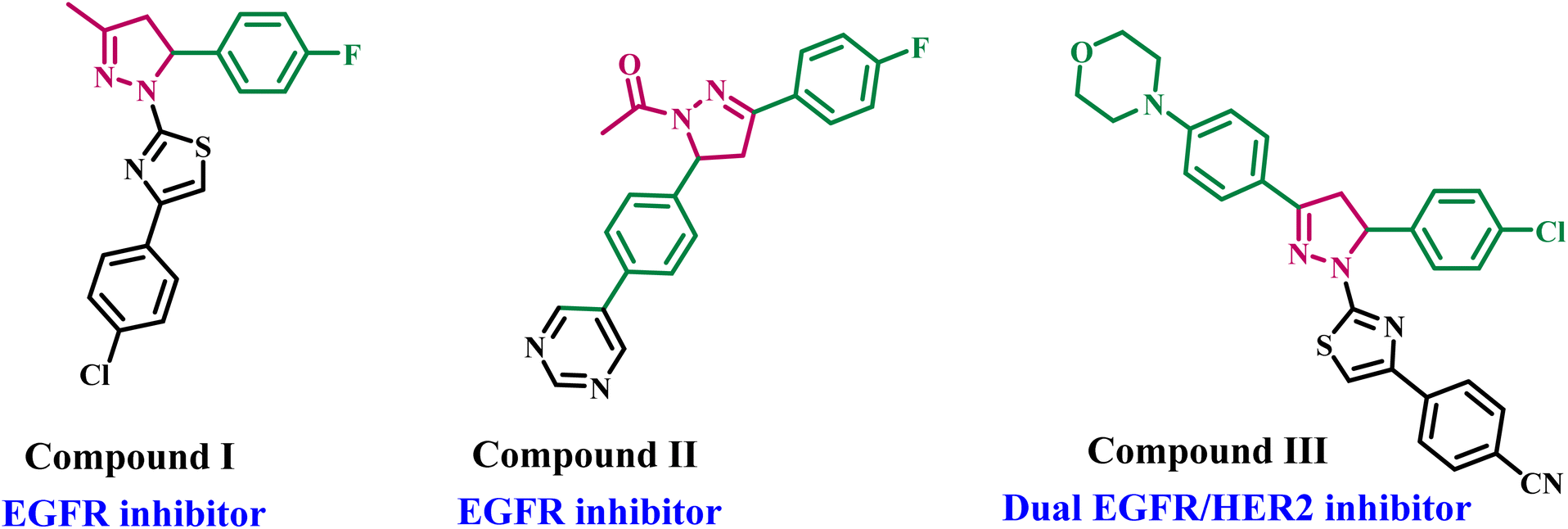 | ||
| Fig. 1 Structures of some pyrazoline-based derivatives as EGFR and/or HER2 inhibitors I–III. | ||
New pyrazoline derivatives were developed and evaluated for their cytotoxic activities against various cancer cell lines.16 The results indicated compound III (Fig. 1) is the most potent derivative. Compound III caused apoptosis and had a strong EGFR inhibitory effect, with an IC50 of 4.34 μM compared to erlotinib's IC50 of 0.05 μM. Compound III significantly suppressed HER2, with an IC50 value of 2.30 μM, so it functions as a dual EGFR and HER2 inhibitor.
On the other hand, chalcones are aromatic ketones that serve as the fundamental structure for numerous essential biological molecules.17 They exhibit diverse therapeutic properties and can be adaptable precursors for numerous complex compounds in medicinal chemistry.18 Recent research indicates that chalcones demonstrate diverse biological activities, with the most prominent anticancer properties.19,20 They may also reduce resistance to multiple chemotherapeutic agents.21 Chalcones selectively limit cancer formation and progression, indicating that they and their derivatives may serve as viable options for cancer treatment.22,23 Consequently, researchers are synthesizing various chalcones to assess their effectiveness in multiple therapeutic domains.
In a recent publication from our lab,19 we describe a new series of quinoline/chalcone hybrids that work as antiproliferative agents and block both EGFR and BRAFV600E. With an IC50 of 3.60 ± 0.60 μM for the MCF-7 breast cancer cell line, compound IV (Fig. 2) had the strongest antiproliferative effect against cancer cell proliferation of all the new hybrids. Furthermore, it demonstrated substantial inhibitory activity against EGFR and BRAFV600E, with IC50 values of 2.30 and 4.60 μM, respectively.
 | ||
| Fig. 2 Compounds IV and V as dual-targeting chalcone-based antiproliferative agents. | ||
In another publication,24 we describe a novel series of quinoline/chalcone-based compounds that stop cell growth by targeting EGFR and BRAFV600E. Compound V (Fig. 2) exhibited enhanced antiproliferative efficacy relative to doxorubicin (GI50 = 1.15 μM). It demonstrated a GI50 value of 3.30 μM against four human cancer cell lines. The compound showed significant inhibitory activity against EGFR and BRAFV600E, with IC50 values of 1.30 ± 0.12 μM and 3.80 ± 0.15 μM, respectively. In contrast, the reference erlotinib exhibited IC50 values of 0.08 ± 0.005 μM for EGFR and 0.06 ± 0.01 μM for BRAFV600E.
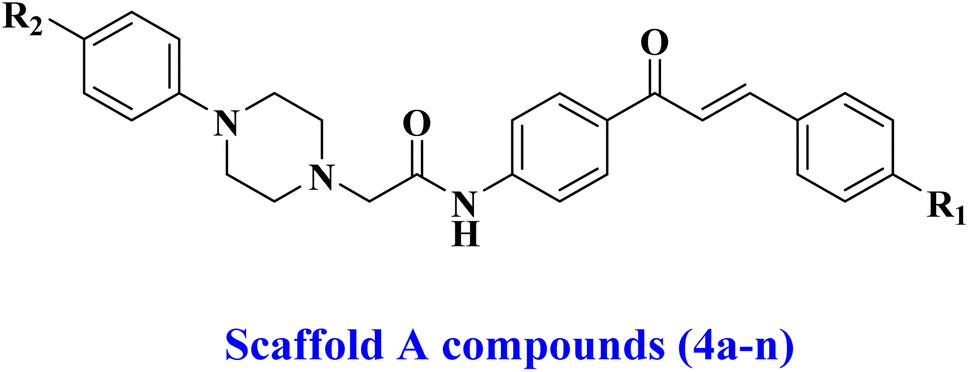
Building upon previously reported data and as part of our ongoing effort to develop a dual- or multi-targeted antiproliferative agent,12,13,25–28 this work details the development, synthesis, and biological evaluation of novel dual inhibitors aimed at EGFR and HER-2. The newly developed compounds are made up of two distinct scaffolds. Fig. 3 indicates that the scaffold A compounds (4a–n) are chalcone-based derivatives. The compounds in scaffold B are pyrazoline derivatives known as 5a–n (Fig. 3). The newly synthesized compounds 4a–n and 5a–n will be tested against a panel of four cancer cell lines for antiproliferative activity. The most promising compounds will be further tested as dual EGFR/HER2 inhibitors. Furthermore, we will study the apoptotic potential of the most potent compound. Finally, we will use molecular docking and dynamic simulations to investigate these compounds' probable binding mechanisms and interactions with receptor sites.
 | ||
| Fig. 3 Structures of new targets 4a–n and 5a–n. | ||
2. Experimental
2.1. Chemistry
General details: see ESI Appendix A.†2.1.1.1. N-(4-Cinnamoylphenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (4a). Yellow powder (70% yield, 0.32 g), mp 163–165 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.17 (d, J = 8.2 Hz, 2H), 7.95 (d, J = 15.6 Hz, 1H), 7.91–7.83 (m, 4H), 7.73 (d, J = 15.6 Hz, 1H), 7.52–7.40 (m, 3H), 6.89 (d, J = 8.9 Hz, 2H), 6.81 (d, J = 8.8 Hz, 2H), 3.67 (s, 3H), 3.36 (s, 2H), 3.08 (t, J = 4.8 Hz, 4H), 2.68 (t, J = 4.9 Hz, 4H); 13C NMR (101 MHz, DMSO) δ 187.56, 168.91, 152.89, 145.37, 143.51, 143.40, 143.10, 142.90, 134.77, 132.81, 132.44, 130.59, 130.49, 129.96, 129.84, 128.90, 128.81, 121.97, 118.94, 118.81, 117.38, 114.23, 61.75, 55.15, 52.79, 49.46. Anal. calcd for C28H29N3O3 (455.56): C, 73.82; H, 6.42; N, 9.22 found: C, 73.91; H, 6.58; N, 9.46.
2.1.1.2. 2-(4-(4-Methoxyphenyl)piperazin-1-yl)-N-(4-(3-(p-tolyl)acryloyl)phenyl)acetamide (4b). Yellow powder (82% yield, 0.38 g), mp 158–160 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.15 (d, J = 8 Hz, 2H), 8.00–7.62 (m, 6H), 7.25 (d, J = 8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.8 Hz, 2H), 3.66 (s, 3H), 3.36 (s, 2H), 3.08 (t, J = 4.8 Hz, 4H), 2.68 (t, J = 4.9 Hz, 4H). 2.33 (s, 3H); 13C NMR (101 MHz, DMSO) δ 187.56, 187.53, 168.94, 152.89, 145.38, 143.47, 143.43, 143.05, 142.99, 140.54, 132.67, 132.55, 132.07, 129.89, 129.76, 129.71, 129.53, 128.85, 120.92, 120.89, 118.93, 118.81, 118.66, 118.15, 117.38, 114.23, 62.06, 61.76, 55.14, 52.80, 49.48, 21.08. Anal. calcd for C29H31N3O3 (469.59): C, 74.18; H, 6.65; N, 8.95 found: C, 73.89; H, 6.79; N, 9.21.
2.1.1.3. N-(4-(3-(4-Methoxyphenyl)acryloyl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (4c). Yellow powder (73% yield, 0.35 g), mp 123–125 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 8.13 (d, J = 12 Hz, 2H), 7.89–7.82 (m, 3H), 7.78 (d, J = 12 Hz, 2H), 7.70 (d, J = 15.5 Hz, 1H), 7.01 (d, J = 12 Hz, 2H), 6.88 (d, J = 9.2 Hz, 2H), 6.81 (d, J = 9.2 Hz, 2H), 3.82 (s, 3H), 3.67 (s, 3H), 3.25 (s, 2H), 3.08 (t, J = 6.5 Hz, 4H), 2.68 (t, J = 5.0 Hz, 4H); 13C NMR (101 MHz, DMSO) δ 187.46, 187.43, 168.91, 161.28, 152.89, 145.39, 143.51, 143.39, 143.35, 142.90, 142.82, 142.69, 133.10, 132.72, 130.71, 129.81, 129.68, 129.62, 127.43, 127.40, 119.45, 118.91, 118.78, 118.74, 117.39, 114.40, 114.24, 62.01, 61.77, 55.37, 55.16, 52.81, 49.48. Anal. calcd for C29H31N3O4 (485.58): C, 71.73; H, 6.44; N, 8.65 found: C, 71.95; H, 6.63; N, 8.84.
2.1.1.4. 2-(4-(4-Methoxyphenyl)piperazin-1-yl)-N-(4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)acetamide (4d). Yellow powder (80% yield, 0.43 g), mp 162–164 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.17 (d, J = 8.7 Hz, 2H), 7.91–7.83 (m, 3H), 7.66 (d, J = 15.6 Hz, 1H), 7.23 (s, 2H), 6.89 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 9.1 Hz, 2H), 3.87 (s, 6H), 3.72 (s, 3H), 3.68 (s, 3H), 3.25 (s, 2H), 3.09 (q, J = 4.9 Hz, 4H), 2.67 (t, J = 4.9 Hz, 4H); 13C NMR (101 MHz, DMSO) δ 187.50, 168.94, 153.11, 152.90, 145.39, 143.94, 143.06, 139.67, 132.55, 130.34, 129.83, 121.12, 118.77, 117.39, 114.24, 106.49, 61.80, 60.14, 55.16, 52.81, 49.48. Anal. calcd for C31H35N3O6 (545.64): C, 68.24; H, 6.47; N, 7.70 found: C, 68.40; H, 6.55; N, 7.89.
2.1.1.5. N-(4-(3-(4-Bromophenyl)acryloyl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (4e). Yellow powder (81% yield, 0.43 g), mp 193–195 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.78–7.74 (m, 4H), 7.59–7.52 (m, 6H), 6.96 (d, J = 8 Hz, 2H), 6.88 (d, J = 8 Hz, 2H), 3.81 (s, 3H), 3.31 (s, 2H), 3.22 (t, J = 4.8 Hz, 4H), 2.88 (t, J = 4.9 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 187.42, 168.96, 152.89, 145.38, 143.18, 142.01, 134.10, 132.32, 131.86, 130.73, 129.89, 123.83, 122.77, 118.79, 117.39, 114.24, 61.78, 55.17, 52.80, 49.47. Anal. calcd for C28H28BrN3O3 (534.45): C, 62.93; H, 5.28; N, 7.86 found: C, 63.15; H, 5.47; N, 8.05.
2.1.1.6. N-(4-(3-(2,4-Dichlorophenyl)acryloyl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (4f). Yellow powder (74% yield, 0.38 g), mp 142–145 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 8.25 (d, J = 15.6 Hz, 1H), 8.16 (d, J = 8.5 Hz, 2H), 8.06–7.92 (m, 2H), 7.87 (d, J = 8.5 Hz, 2H), 7.77–7.75 (m, 1H), 7.55 (d, J = 15.6 Hz, 1H), 6.89 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 3.68 (s, 3H), 3.25 (s, 2H), 3.08 (t, J = 4.9 Hz, 4H), 2.68 (t, J = 4.9 Hz, 4H); 13C NMR (101 MHz, DMSO) δ 187.17, 169.01, 152.89, 145.39, 143.42, 136.68, 136.64, 135.49, 135.08, 132.01, 131.48, 130.16, 130.04, 129.98, 129.81, 129.48, 129.41, 127.93, 127.83, 125.29, 118.94, 118.81, 118.67, 118.16, 117.39, 114.24, 61.75, 55.17, 52.79, 49.48. Anal. calcd for C28H27C12N3O3 (524.44): C, 64.13; H, 5.19; N, 8.01 found: C, 64.39; H, 5.33; N, 8.29.
2.1.1.7. N-(4-(3-(Furan-2-yl)acryloyl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (4g). Yellow powder (69% yield, 0.31 g), mp 187–189 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.10–8.02 (m, 2H), 7.94–7.88 (m, 2H), 7.88–7.82 (m, 2H), 7.55–7.54 (m, 2H), 7.09–7.08 (m, 1H), 6.89 (d, J = 9.2 Hz, 2H), 6.81 (d, J = 9.2 Hz, 2H), 6.69 (m, 1H), 3.68 (s, 3H), 3.24 (s, 2H), 3.07 (d, J = 5.6 Hz, 3H), 2.67 (t, J = 4.9 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 187.04, 186.99, 168.94, 152.89, 151.23, 146.06, 145.39, 143.05, 143.00, 132.36, 129.96, 129.94, 129.59, 129.53, 119.00, 118.87, 118.70, 118.66, 117.39, 116.76, 116.71, 114.23, 113.09, 62.04, 61.75, 55.16, 52.79, 49.57, 49.48. Anal. calcd for C26H27N3O4 (445.52): C, 70.09; H, 6.11; N, 9.43 found: C, 70.25; H, 6.29; N, 9.64.
2.1.1.8. 2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(4-cinnamoylphenyl)acetamide (4h). Yellow powder (76% yield, 0.35 g), mp 188–190 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 15.7 Hz, 1H), 7.88 (t, J = 6.5 Hz, 2H), 7.74 (d, J = 15.6 Hz, 1H), 7.49–7.44 (m, 1H), 7.24 (d, J = 8.3 Hz, 1H), 6.96 (d, J = 8.8 Hz, 1H), 3.35 (s, 1H), 3.21 (d, J = 5.3 Hz, 2H), 2.69 (t, J = 4.9 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 187.55, 168.90, 149.79, 143.40, 143.32, 143.22, 143.11, 134.77, 132.43, 132.35, 130.50, 129.83, 129.76, 128.90, 128.81, 128.60, 61.63, 52.47, 47.91. Anal. calcd for C27H26ClN3O2 (459.97): C, 70.50; H, 5.70; N, 9.14 found: C, 70.63; H, 5.81; N, 9.39.
2.1.1.9. 2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(4-(3-(p-tolyl)acryloyl)phenyl)acetamide (4i). Yellow powder (70% yield, 0.33 g), mp 175–177 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 8.15 (d, J = 8.2 Hz, 1H), 7.95–7.83 (m, 2H), 7.80–7.66 (m, 2H), 7.24 (dd, J = 19.3, 8.2 Hz, 3H), 6.94 (d, J = 8.7 Hz, 1H), 3.28–3.15 (m, 3H), 2.67 (t, J = 5.0 Hz, 2H), 2.35 (s, 2H); 13C NMR (101 MHz, DMSO) δ 187.57, 187.53, 168.89, 149.80, 143.48, 143.44, 143.02, 142.96, 140.56, 140.55, 132.56, 132.07, 129.90, 129.77, 129.71, 129.54, 128.86, 128.70, 128.61, 122.31, 120.92, 120.89, 118.94, 118.82, 117.42, 116.85, 61.64, 52.48, 47.91, 21.09. Anal. calcd for C28H28ClN3O2 (474.00): C, 70.95; H, 5.95; N, 8.87 found: C, 71.12; H, 6.07; N, 9.12.
2.1.1.10. 2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(4-(3-(4-methoxyphenyl)acryloyl)phenyl)acetamide (4j). Yellow powder (87% yield, 0.43 g), mp 158–160 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.13 (d, J = 12 Hz, 2H), 7.85–7.79 (m, 5H), 7.70 (d, J = 15.5 Hz, 1H), 7.22 (d, J = 9.2 Hz, 2H), 7.02 (d, J = 12 Hz, 2H), 6.94 (d, J = 9.2 Hz, 2H), 3.82 (s, 3H), 3.34 (s, 2H), 3.20 (t, J = 5.1 Hz, 3H), 2.67 (d, J = 9.9 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 187.42, 168.84, 161.27, 149.79, 143.38, 143.34, 142.87, 142.80, 132.72, 130.70, 129.80, 129.67, 129.61, 128.60, 127.42, 122.29, 119.44, 118.91, 118.78, 116.84, 114.39, 62.00, 61.64, 55.37, 52.47, 47.90. Anal. calcd for C28H28ClN3O3 (490.00): C, 68.63; H, 5.76; N, 8.58 found: C, 68.85; H, 5.69; N, 8.77.
2.1.1.11. 2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(4-(3-(3,4,5-trimethoxyphenyl)acryloyl)phenyl)acetamide (4k). Yellow powder (77% yield, 0.42 g), mp 185–187 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.93–7.85 (m, 2H), 7.68 (d, J = 15.5 Hz, 1H), 7.22 (d, J = 6.7 Hz, 2H), 6.96–6.92 (m, 1H), 3.87 (s, 3H), 3.72 (s, 2H), 3.34 (s, 1H), 3.19 (d, J = 5.3 Hz, 2H), 2.67 (d, J = 5.2 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 187.50, 168.89, 153.12, 149.79, 143.94, 143.05, 139.67, 132.56, 130.34, 129.83, 128.61, 122.31, 121.12, 118.91, 118.78, 116.84, 106.49, 61.67, 60.14, 56.14, 52.49, 47.91. Anal. calcd for C30H32ClN3O5 (550.05): C, 65.51; H, 5.86; N, 7.64 found: C, 65.34; H, 5.98; N, 7.90.
2.1.1.12. N-(4-(3-(4-Bromophenyl)acryloyl)phenyl)-2-(4-(4-chlorophenyl)piperazin-1-yl)acetamide (4l). Yellow powder (81% yield, 0.44 g), mp 200–202 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.98 (d, J = 15.6 Hz, 1H), 7.86 (t, J = 9.1 Hz, 2H), 7.71–7.63 (m, 2H), 7.21 (d, J = 8.7 Hz, 1H), 6.93 (d, J = 8.6 Hz, 1H), 3.26 (s, 1H), 3.19 (t, J = 5.0 Hz, 2H), 2.67 (t, J = 4.8 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 187.42, 168.92, 149.78, 143.23, 141.99, 134.09, 132.30, 131.85, 130.72, 129.87, 128.68, 128.59, 123.82, 122.76, 122.30, 118.81, 117.40, 116.83, 61.60, 52.46, 47.91. Anal. calcd for C27H25BrClN3O2 (538.87): C, 60.18; H, 4.68; N, 7.80 found: C, 60.36; H, 4.90; N, 8.04.
2.1.1.13. 2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(4-(3-(2,4dichlorophenyl)acryloyl)phenyl)acetamide (4m). Yellow powder (73% yield, 0.38 g), mp 180–182 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.26 (d, J = 8.6 Hz, 4H), 8.17 (dd, J = 8.7, 3.0 Hz, 7H), 8.03 (d, J = 15.6 Hz, 4H), 7.98–7.84 (m, 11H), 7.74 (d, J = 2.2 Hz, 4H), 7.55 (dd, J = 8.5, 2.1 Hz, 4H), 7.26–7.17 (m, 7H), 6.98–6.90 (m, 6H), 3.26 (s, 6H), 3.20 (t, J = 5.1 Hz, 12H), 2.67 (t, J = 5.0 Hz, 11H); 13C NMR (101 MHz, DMSO) δ 187.16, 168.96, 149.79, 143.42, 136.68, 136.64, 135.49, 135.08, 132.02, 131.47, 130.15, 130.03, 129.98, 129.80, 129.48, 128.69, 128.60, 127.93, 125.28, 122.30, 118.94, 118.82, 117.41, 116.84, 61.61, 52.46, 48.02, 47.91. Anal. calcd for C27H24Cl3N3O2 (528.86): C, 61.32; H, 4.57; N, 7.95 found: C, 61.45; H, 4.68; N, 8.16.
2.1.1.14. 2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(4-(3-(furan-2-yl)acryloyl)phenyl)acetamide (4n). Yellow powder (70% yield, 0.31 g), mp 207–209 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.10–8.06 (m, 2H), 7.92 (d, J = 1.8 Hz, 1H), 7.85 (d, J = 8.5 Hz, 2H), 7.56 (s, 2H), 7.25–7.22 (m, 2H), 7.10 (d, J = 3.4 Hz, 1H), 6.98–6.93 (m, 2H), 6.70 (dd, J = 3.4, 1.8 Hz, 1H), 3.33–3.16 (m, 8H), 2.68 (d, J = 10.0 Hz, 5H); 13C NMR (101 MHz, DMSO) δ 186.99, 168.87, 151.22, 149.80, 146.07, 143.02, 132.38, 129.97, 129.59, 129.53, 128.60, 122.30, 119.01, 118.89, 118.69, 118.65, 116.85, 116.78, 116.72, 113.09, 61.64, 52.47, 47.91. Anal. calcd for C25H24ClN3O3 (449.94): C, 66.74; H, 5.38; N, 9.34 found: C, 66.53; H, 5.60; N, 9.45.
2.1.2.1. N-(4-(1-Acetyl-5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (5a). White powder (83% yield, 0.42 g), mp 135–137 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 7.78–7.65 (m, 4H), 7.33–7.22 (m, 5H), 6.90 (d, J = 9.1 Hz, 2H), 6.83 (d, J = 9 Hz, 2H), 5.54 (dd, J = 17.9, 3.6 Hz, 1H), 3.84 (dd, J = 18.0, 11.9, 1H), 3.69 (s, 3H), 3.22 (s, 2H), 3.09 (m, 5H), 2.68 (t, J = 4.9 Hz, 4H), 2.31 (s, 3H); 13C NMR (101 MHz, DMSO) δ 168.54, 167.22, 153.84, 149.80, 142.49, 140.51, 128.64, 128.60, 127.33, 127.15, 125.39, 122.28, 119.32, 119.25, 116.84, 61.59, 59.76, 55.65, 52.48, 50.20, 42.64, 22.15. Anal. calcd for C30H33N5O3 (511.26): C, 70.43; H, 6.50; N, 13.69 found: C, 70.29; H, 6.63; N, 13.91.
2.1.2.2. N-(4-(1-Acetyl-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxy-phenyl)piperazin-1-yl)acetamide (5b). Yellowish white powder (75% yield, 0.39 g), mp 130–132 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 7.79–7.62 (m, 4H), 7.12 (d, J = 7.9 Hz, 2H), 7.05 (d, J = 8.2 Hz, 2H), 6.89 (d, J = 8 Hz, 2H), 6.82 (d, J = 8 Hz, 2H), 5.52 (dd, J = 17.9, 3.6 Hz, 1H), 3.79 (dd, J = 17.9, 11.6 Hz, 1H), 3.68 (s, 3H), 3.39 (dd, J = 17.9, 3.6 Hz, 1H), 3.21 (s, 2H), 3.08 (t, J = 6.1 Hz, 4H), 2.67 (t, J = 6.1 Hz, 4H), 2.28 (s, 3H), 2.25 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.07, 167.67, 154.34, 153.36, 145.84, 140.93, 140.00, 136.76, 129.59, 127.76, 125.81, 119.73, 119.24, 117.85, 114.70, 62.16, 59.55, 55.62, 53.26, 49.95, 42.57, 22.15, 21.06. Anal. calcd for C31H35N5O3 (525.65): C, 70.83; H, 6.71; N, 13.32 found: C, 70.98; H, 6.82; N, 13.59.
2.1.2.3. N-(4-(1-Acetyl-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (5c). White powder (71% yield, 0.37 g), mp 210–212 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 7.80–7.64 (m, 4H), 7.09 (d, J = 8 Hz, 2H), 6.81–6.90 (m, 4H), 6.82 (d, J = 8 Hz, 2H), 5.51 (dd, J = 17.9, 3.6 Hz, 1H), 3.84 (dd, J = 17.9, 11.6 Hz, 1H), 3.72 (dd, J = 17.9, 3.6 Hz, 1H), 3.71 (s, 3H), 3.68 (s, 3H), 3.22 (s, 2H), 3.08 (t, J = 4.9 Hz, 4H), 2.67 (t, J = 4.9 Hz, 4H), 2.27 (s, 3H); 13C NMR (101 MHz, DMSO) δ 168.53, 167.66, 162.82, 158.84, 154.34, 153.36, 145.85, 134.98, 127.76, 127.19, 126.50, 119.73, 119.25, 117.85, 114.71, 114.42, 62.15, 59.26, 55.63, 55.53, 53.26, 49.95, 42.54, 20.89. Anal. calcd for C31H35N5O4 (541.65): C, 68.74; H, 6.51; N, 12.93 found: C, 69.01; H, 6.67; N, 13.15.
2.1.2.4. N-(4-(1-Acetyl-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (5d). Whitish brown powder (80% yield, 0.48 g), mp 117–119 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 7.79–7.65 (m, 4H), 6.90 (d, J = 9.1 Hz, 2H), 6.82 (d, J = 9.1 Hz, 2H), 6.45 (s, 2H), 5.45 (dd, J = 17.9, 3.6 Hz, 1H), 3.78 (dd, J = 17.9, 11.6 Hz, 1H), 3.72 (dd, J = 17.9, 3.6 Hz, 1H), 3.72 (s, 6H), 3.68 (s, 3H), 3.62 (s, 3H), 3.22 (s, 2H), 3.07 (t, J = 4.9 Hz, 4H), 2.66 (t, J = 4.9 Hz, 4H), 2.32 (s, 3H); 13C NMR (101 MHz, DMSO) δ 168.48, 167.97, 153.51, 153.36, 145.85, 138.73, 136.98, 127.82, 119.74, 119.25, 117.85, 114.71, 102.94, 60.37, 59.99, 56.31, 55.63, 53.25, 49.95, 42.67, 20.90. Anal. calcd for C33H39N5O6 (601.70): C, 65.87; H, 6.53; N, 11.64 found: C, 66.09; H, 6.62; N, 11.91.
2.1.2.5. N-(4-(1-Acetyl-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (5e). White powder (72% yield, 0.42 g), mp 118–120 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.98 (s, 1H), 7.80–7.68 (m, 4H), 7.51 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 9.1 Hz, 2H), 5.51 (dd, J = 11.8, 4.6 Hz, 1H), 3.86 (dd, J = 18.0, 11.8 Hz, 1H), 3.82 (dd, J = 18.0, 4.6 Hz, 1H), 3.68 (s, 3H), 3.21 (s, 2H), 3.04 (t, J = 4.9 Hz, 4H), 2.67 (t, J = 4.9 Hz, 4H), 2.29 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.42, 167.91, 153.37, 145.83, 142.25, 140.92, 131.98, 128.29, 127.85, 125.41, 120.76, 119.75, 117.88, 114.72, 62.16, 55.64, 53.25, 49.94, 43.98, 22.11. Anal. calcd for C30H32BrN5O3 (590.52): C, 61.02; H, 5.46; N, 11.86 found: C, 60.97; H, 5.65; N, 12.04.
2.1.2.6. N-(4-(1-Acetyl-5-(2,4-dichlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamide (5f). Yellow powder (73% yield, 0.41 g), mp 160–162 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 7.77–7.63 (m, 3H), 7.37 (d, J = 8.3, Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 5.70 (dd, J = 12.1, 5.1 Hz, 1H), 3.91 (dd, J = 17.9, 12.0 Hz, 1H), 3.87 (dd, J = 12.1, 5.1 Hz, 1H), 3.68 (s, 3H), 3.21 (s, 2H), 3.08 (t, J = 3.3 Hz, 4H), 2.64 (t, J = 3.3 Hz, 4H), 2.34 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.58, 167.95, 154.70, 150.25, 144.14, 138.64, 132.96, 132.24, 129.57, 129.06, 128.23, 127.86, 122.76, 119.71, 119.18, 117.31, 62.06, 58.53, 52.94, 48.37, 41.24, 22.05. Anal. calcd for C30H31C12N5O3 (580.51): C, 62.07; H, 5.38; N, 12.06 found: C, 62.32; H, 5.49; N, 12.32.
2.1.2.7. N-(4-(1-Acetyl-5-(furan-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-methoxy-phenyl)piperazin-1-yl)acetamide (5g). Whitish gray powder (79% yield, 0.39 g), mp 130–132 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 7.79–7.70 (m, 4H), 7.57–7.52 (m, 1H), 6.89 (d, J = 9.1 Hz, 2H), 6.81 (d, J = 9.1 Hz, 2H), 6.37–6.35 (m, 1H), 6.30–6.29 (m, 1H), 5.61 (dd, J = 11.9, 4.5 Hz, 1H), 3.91 (dd, J = 17.9, 12.0 Hz, 1H), 3.87 (dd, J = 11.9, 4.5 Hz, 1H), 3.68 (s, 3H), 3.22 (s, 2H), 3.08 (t, J = 4.9 Hz, 4H), 2.67 (t, J = 4.9 Hz, 4H), 2.25 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.12, 167.82, 154.47, 153.36, 153.30, 145.85, 142.70, 141.01, 127.76, 126.30, 119.75, 117.86, 114.71, 110.91, 107.34, 62.15, 55.64, 53.38, 53.26, 49.95, 38.87, 22.15. Anal. calcd for C30H31C12N5O3 (580.51): C, 67.07; H, 5.38; N, 13.06 found: C, 62.29; H, 5.92; N, 13.48.
2.1.2.8. N-(4-(1-Acetyl-5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chloro-phenyl)piperazin-1-yl)acetamide (5h). White powder (83% yield, 0.43 g), mp 186–188 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 7.80–7.69 (m, 4H), 7.36–7.23 (m, 5H), 7.27–7.17 (d, J = 8 Hz, 2H), 6.95 (d, J = 9.1 Hz, 2H), 5.52 (dd, J = 11.8, 4.5 Hz, 1H), 3.86 (dd, J = 17.9, 11.6 Hz, 1H), 3.23–3.16 (m, 6H), 3.10 (dd, J = 18.0, 4.6 Hz, 1H), 2.66 (t, J = 5.0 Hz, 4H), 2.30 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.02, 167.74, 154.31, 150.25, 142.94, 140.97, 129.10, 129.06, 127.78, 127.61, 126.44, 125.85, 122.77, 119.74, 117.30, 62.04, 59.79, 52.94, 48.38, 42.63, 22.16. Anal. calcd for C29H30ClN5O2 (516.04): C, 67.50; H, 5.86; N, 13.57 found: C, 67.73; H, 5.92; N, 13.70.
2.1.2.9. N-(4-(1-Acetyl-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chloro-phenyl)piperazin-1-yl)acetamide (5i). White powder (66% yield, 0.35 g), mp 150–152 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 5H), 7.79–7.64 (m, 4H) 7.23 (d, J = 8 Hz, 2H), 7.12 (d, J = 7.9 Hz, 2H), 7.05 (d, J = 7.8 Hz, 2H), 6.95 (d, J = 8 Hz, 2H), 5.47 (dd, J = 11.6, 3.9 Hz, 1H), 3.79 (dd, J = 17.9, 11.7 Hz, 1H), 3.20–3.18 (m, 6H), 3.06 (dd, J = 17.9, 4.8 Hz, 1H), 2.66 (t, J = 6.1 Hz, 4H), 2.28 (s, 3H), 2.25 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.02, 167.70, 154.35, 150.24, 141.57, 140.86, 139.98, 136.78, 129.60, 129.06, 127.80, 127.76, 126.53, 125.80, 122.78, 119.76, 119.24, 117.31, 62.06, 59.54, 52.94, 48.37, 42.57, 22.15, 21.06. Anal. calcd for C30H32ClN5O2 (530.07): C, 67.98; H, 6.09; N, 13.21 found: C, 68.15; H, 6.21; N, 13.48.
2.1.2.10. N-(4-(1-Acetyl-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chlorophenyl)piperazin-1-yl)acetamide (5j). Yellow powder (67% yield, 0.36 g), mp 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 7.76–7.66 (m, 4H), 7.22 (d, J = 9.0 Hz, 2H), 7.09 (d, J = 8.5 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 5.52 (dd, J = 18.0, 4.9 Hz, 1H), 3.78 (dd, J = 18.0, 11.4 Hz, 1H), 3.71 (s, 3H), 3.24–3.16 (m, 6H), 3.08 (dd, J = 17.9, 4.9 Hz, 1H), 2.66 (t, J = 5.0 Hz, 4H), 2.27 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.04, 167.74, 158.83, 154.35, 150.23, 140.85, 134.95, 129.06, 127.79, 127.76, 127.18, 126.55, 122.79, 119.77, 119.26, 117.30, 114.41, 62.04, 59.27, 55.52, 52.93, 48.37, 42.53, 22.16. Anal. calcd for C30H32ClN5O2 (530.07): C, 67.98; H, 6.09; N, 13.21 found: C, 68.16; H, 6.07; N, 12.95.
2.1.2.11. N-(4-(1-Acetyl-5-(3,4,5-trimethoxyphenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chlorophenyl)piperazin-1-yl)acetamide (5k). White powder (63% yield, 0.34 g), mp 162–164 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 7.79–7.62 (m, 4H), 7.22 (d, J = 9.0 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 6.45 (s, 2H), 5.52 (dd, J = 18.0, 5.3 Hz, 1H), 3.86 (dd, J = 17.9, 11.6 Hz, 1H), 3.72 (s, 6H), 3.62 (s, 3H), 3.22–3.19 (m, 6H), 3.12 (dd, J = 18.0, 5.3 Hz, 1H), 2.66 (t, J = 4.9 Hz, 4H), 2.32 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.04, 168.05, 154.45, 153.51, 150.23, 140.85, 138.70, 136.98, 129.05, 127.81, 126.51, 122.79, 119.77, 117.30, 102.92, 62.04, 60.37, 60.00, 56.29, 52.93, 48.36, 42.67, 22.16. Anal. calcd for C32H36ClN5O5 (606.12): C, 63.41; H, 5.99; N, 11.55 found: C, 63.59; H, 6.14; N, 11.79.
2.1.2.12. N-(4-(1-Acetyl-5-(4-bromophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chlorophenyl)piperazin-1-yl)acetamide (5l). White powder (83% yield, 0.42 g), mp 110–112 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 1H), 7.79–7.64 (m, 4H), 7.51 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 5.51 (dd, J = 11.9, 4.5 Hz, 1H), 3.83 (dd, J = 18.1, 11.5 Hz, 1H), 3.21 (m, 6H), 3.11 (dd, J = 18.1, 4.9 Hz, 1H), 2.66 (t, J = 5.0 Hz, 4H), 2.29 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.13, 169.01, 167.83, 154.31, 150.24, 142.26, 140.97, 131.97, 129.06, 128.29, 127.82, 126.35, 122.79, 120.63, 119.74, 119.21, 117.30, 62.07, 59.29, 52.95, 48.38, 42.35, 22.13. Anal. calcd for C29H29BrClN5O2 (594.94): C, 58.55; H, 4.91; N, 11.77 found: C, 58.34; H, 5.12; N, 11.95.
2.1.2.13. N-(4-(1-Acetyl-5-(2,4-dichlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chlorophenyl)piperazin-1-yl)acetamide (5m). Yellowish white powder (73% yield, 0.38 g), mp 155–157 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 7.79–7.63 (m, 3H), 7.37 (dd, J = 8.4, 2.2 Hz, 2H), 7.22 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 5.70 (dd, J = 12.1, 5.1 Hz, 1H), 3.90 (dd, J = 18.0, 12.0 Hz, 1H), 3.21–3.18 (m, 6H), 3.05 (dd, J = 17.9, 5.3 Hz, 1H), 2.66 (t, J = 5.0 Hz, 3H), 2.34 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.05, 167.94, 154.52, 154.49, 153.37, 145.84, 141.74, 141.03, 138.63, 132.97, 132.24, 129.56, 128.21, 127.85, 126.18, 119.68, 119.17, 117.84, 114.70, 62.19, 57.29, 55.62, 53.28, 49.95, 41.23, 22.04. Anal. calcd for C29H28Cl3N5O2 (584.93): C, 59.55; H, 4.83; N, 11.97 found: C, 59.76; H, 4.97; N, 12.86.
2.1.2.14. N-(4-(1-Acetyl-5-(furan-2-yl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)-2-(4-(4-chlorophenyl)piperazin-1-yl)acetamide (5n). Whitish gray powder (79% yield, 0.39 g), mp 124–126 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 7.80–7.65 (m, 4H), 7.54 (d, J = 3.3 Hz, 1H), 7.23 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.38 (t, J = 2.5 Hz, 1H), 6.30 (d, J = 3.3 Hz, 1H), 5.61 (dd, J = 11.8, 4.4 Hz, 1H), 3.69 (dd, J = 15.3, 11.8 Hz, 1H), 3.22–3.19 (m, 7H), 2.67 (t, J = 5.0 Hz, 4H), 2.25 (s, 3H); 13C NMR (101 MHz, DMSO) δ 169.01, 167.79, 154.42, 153.31, 150.25, 142.69, 140.94, 129.06, 127.81, 127.77, 126.36, 122.78, 119.76, 117.31, 110.91, 107.33, 62.08, 53.39, 52.96, 48.38, 38.87, 22.16. Anal. calcd for C27H28ClN5O3 (506.00): C, 64.09; H, 5.58; N, 13.84 found: C, 63.87; H, 5.74; N, 14.09.
2.2. Biological evaluation
2.3. Molecular docking study and molecular dynamics simulation
All experimental methods, data, and tools for molecular docking and MD simulation were submitted in ESI Appendix A.†3. Results and discussion
3.1. Chemistry
Scheme 1 details the synthesis of the target compounds 4a–n and 5a–n. Chalcones 1a–g and their chloroacetamide derivatives 2a–g were synthesized using the typical base-catalyzed Claisen–Schmidt condensation of various aromatic aldehydes with 4-aminoacetophenone,32 then acetylated the free amino group using chloroacetyl chloride.33 Acetylation was an effective approach for incorporating a spacer group into the chalcone molecule. Compounds 4a–n were prepared by alkylating substituted N-phenylpiperazines 3a–b with compounds 2a–g in DMF, which contains K2CO3 as a base,34 Scheme 1. | ||
| Scheme 1 Synthesis of targeted compounds 4a–n and 5a–n. | ||
Compounds 4a–n were validated using 1H NMR, 13C NMR, and elemental analysis. Compounds 4a–n exhibited distinct and significant signals at various chemical shifts in their 1H NMR spectra. We observe the NH-group at approximately δ 9.50–10.60 ppm. Also, the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH bond, which is known to be an important part of chalcones, showed clear doublet peaks in the chemical shift range of δ 6.8–6.9 ppm, along with a coupling constant of 15.6–16 Hz, which shows that the chalcone double bond is in a trans configuration. Two protons corresponding to the CH2 group of the acetamide spacer were identified through a singlet signal at δ 3.25–3.36 ppm. Additionally, piperazine protons exhibited two prominent triplet peaks at δ 2.60–2.67 and δ 3.18–3.21 ppm. The 13C NMR of 4a–n showed two prominent signals at δ 167 and 187 ppm, corresponding to the amidic carbonyl and ketonic carbonyl of the chalcone moiety, respectively.
CH bond, which is known to be an important part of chalcones, showed clear doublet peaks in the chemical shift range of δ 6.8–6.9 ppm, along with a coupling constant of 15.6–16 Hz, which shows that the chalcone double bond is in a trans configuration. Two protons corresponding to the CH2 group of the acetamide spacer were identified through a singlet signal at δ 3.25–3.36 ppm. Additionally, piperazine protons exhibited two prominent triplet peaks at δ 2.60–2.67 and δ 3.18–3.21 ppm. The 13C NMR of 4a–n showed two prominent signals at δ 167 and 187 ppm, corresponding to the amidic carbonyl and ketonic carbonyl of the chalcone moiety, respectively.
Scheme 1 illustrates the reaction of compound 4a–n with hydrazine hydrate to synthesize compounds 5a–n.35 The 1H NMR spectra of N-acetyl pyrazolines 5a–n showed different peaks that were the protons of the cyclized forms CH and CH2. These peaks showed up as doublets of doublets in three different areas at δ 3.08–3.10, 3.84–3.90, and 5.52–5.55 ppm. The 13C NMR spectrum of 5a–n showed the absence of chalcone's ketonic carbonyl and the presence of amidic carbonyl at δ 168 ppm.
3.2. Biological evaluation
|
||||||
|---|---|---|---|---|---|---|
| Compd | Cell viability% | Antiproliferative activity IC50 ± SEM (nM) | GI50 (average IC50) | |||
| HCT-116 | MCF-7 | MDAMB-231 | PC-3 | |||
| a —: Not determined. | ||||||
| 4a | 89 | 68.64 ± 3.7 | 72.43 ± 3.7 | 58.74 ± 3.1 | 78.23 ± 4.0 | 70 |
| 4b | 90 | 79.64 ± 3.8 | 65.48 ± 3.5 | 77.25 ± 3.9 | 31.98 ± 2.1 | 64 |
| 4c | 92 | 18.09 ± 1.4 | 28.32 ± 1.8 | 23.12 ± 1.7 | 34.51 ± 2.1 | 26 |
| 4d | 87 | 29.11 ± 1.9 | 24.23 ± 1.7 | 15.46 ± 1.3 | 61.42 ± 3.5 | 32 |
| 4e | 90 | 64.81 ± 3.5 | 52.45 ± 2.9 | 43.63 ± 2.5 | 73.93 ± 3.8 | 59 |
| 4f | 86 | >100 | >100 | 88.64 ± 4.4 | >100 | 100 |
| 4g | 82 | 17.31 ± 1.4 | 9.37 ± 0.7 | 8.51 ± 0.7 | 14.15 ± 1.2 | 12 |
| 4h | 85 | 49.56 ± 2.8 | 41.34 ± 2.4 | 32.83 ± 2.3 | 20.37 ± 1.5 | 36 |
| 4i | 89 | 86.09 ± 4.3 | 91.60 ± 4.6 | 81.58 ± 4.1 | >100 | 90 |
| 4j | 91 | 27.44 ± 1.9 | 42.93 ± 2.5 | 38.72 ± 2.3 | 66.85 ± 3.5 | 44 |
| 4k | 89 | 34.76 ± 2.2 | 17.86 ± 1.4 | 19.95 ± 1.6 | 42.92 ± 2.5 | 29 |
| 4l | 87 | 75.35 ± 4.1 | 80.78 ± 3.9 | 63.41 ± 3.4 | 86.36 ± 4.3 | 76 |
| 4m | 86 | 44.79 ± 2.6 | 35.81 ± 2.2 | 26.36 ± 1.8 | 55.24 ± 3.2 | 41 |
| 4n | 90 | 8.67 ± 0.9 | 6.94 ± 0.4 | 4.80 ± 0.3 | 9.08 ± 0.8 | 7 |
| Doxorubicin | — | 5.23 ± 0.3 | 4.17 ± 0.2 | 3.18 ± 0.1 | 8.87 ± 0.6 | 5 |
| Sorafenib | — | 5.47 ± 0.3 | 7.26 ± 0.3 | 7.64 ± 0.4 | 11.53 ± 0.9 | 8 |
|
||||||
|---|---|---|---|---|---|---|
| Comp. | Cell viability% | Antiproliferative activity IC50 ± SEM (nM) | GI50 (average IC50) | |||
| HCT-116 | MCF-7 | MDAMB-231 | PC-3 | |||
| a —: Not determined. | ||||||
| 5a | 87 | 32.08 ± 2.1 | 19.56 ± 1.4 | 25.70 ± 1.7 | 46.29 ± 2.5 | 31 |
| 5b | 89 | 24.93 ± 1.8 | 13.76 ± 1.1 | 10.36 ± 0.9 | 21.92 ± 1.6 | 18 |
| 5c | 92 | 18.63 ± 1.5 | 9.68 ± 0.8 | 8.21 ± 0.7 | 16.22 ± 1.3 | 13 |
| 5d | 91 | 6.47 ± 0.4 | 5.05 ± 0.4 | 2.89 ± 0.1 | 12.95 ± 1.0 | 7 |
| 5e | 86 | 26.75 ± 2.0 | 15.19 ± 1.3 | 19.33 ± 1.5 | 29.57 ± 1.9 | 23 |
| 5f | 89 | 53.98 ± 3.2 | 47.52 ± 2.6 | 51.56 ± 2.8 | 57.80 ± 3.3 | 53 |
| 5g | 85 | 8.06 ± 0.7 | 3.78 ± 0.2 | 5.97 ± 0.3 | 7.48 ± 0.6 | 6 |
| 5h | 90 | 74.94 ± 4.1 | 85.42 ± 4.6 | 68.27 ± 3.7 | 87.61 ± 4.3 | 79 |
| 5i | 89 | 62.51 ± 3.4 | 54.68 ± 3.1 | 56.13 ± 3.2 | 67.76 ± 3.6 | 60 |
| 5j | 86 | >100 | 94.05 ± 4.9 | 89.44 ± 4.3 | >100 | 100 |
| 5k | 90 | 52.83 ± 3.0 | 40.76 ± 2.4 | 33.82 ± 2.1 | 45.69 ± 2.6 | 43 |
| 5l | 93 | 62.51 ± 3.4 | 54.68 ± 3.1 | 56.13 ± 3.2 | 67.76 ± 3.6 | 60 |
| 5m | 91 | 69.88 ± 3.7 | 71.26 ± 3.6 | 64.66 ± 3.5 | 76.68 ± 3.9 | 70 |
| 5n | 84 | 39.62 ± 2.3 | 23.89 ± 1.7 | 27.75 ± 1.8 | 36.82 ± 2.2 | 32 |
| Doxorubicin | — | 5.23 ± 0.3 | 4.17 ± 0.2 | 3.18 ± 0.1 | 8.87 ± 0.6 | 5 |
| Sorafenib | — | 5.47 ± 0.3 | 7.26 ± 0.3 | 7.64 ± 0.4 | 11.53 ± 0.9 | 7 |

The antiproliferative activity of scaffold A (chalcone-based) compounds 4a–n was generally weak to moderate, with GI50 values ranging from 7 to 100 μM against the four examined cancer cell lines. In comparison, the standard doxorubicin had a GI50 value of 5 μM, while sorafenib had a GI50 value of 8 μM. Compounds 4c, 4d, 4g and 4n exhibited the highest potency among the derivatives, with GI50 values ranging from 7 to 32 μM. Compounds 4g and 4n (GI50 = 12 and 7 μM, respectively) demonstrated equivalent potency to the reference drugs doxorubicin and sorafenib. Among the newly synthesized derivatives 4a–n, compound 4n (R1 = furan-2-yl, R2 = Cl, scaffold A) had the highest potency with a GI50 value of 7 μM. It showed similar potency to doxorubicin (GI50 = 5 μM) and sorafenib (GI50 = 8 μM). Compound 4n outperformed the reference drug sorafenib against the two types of breast cancer cells, MCF-7 and MDAMB-231, with IC50 values of 6.94 and 4.80 μM, respectively. Sorafenib had IC50 values of 7.26 and 7.64 μM for the same cell lines. These results showed that compound 4n was 1.6 times more potent than sorafenib against the breast cancer MDAMB-231 cell line, nevertheless, compound 4n exhibits lower potency than doxorubicin against all cancer cell types.
Compound 4g (R1 = furan-2-yl, R2 = OMe, scaffold A), which shares the same structure as compound 4n but with a methoxy group at the para position of N-phenyl piperazine moiety, exhibited a GI50 of 12 μM (1.7-fold less potent than compound 4n). This indicates that a chlorine atom at the para position of N-phenyl piperazine moiety is more tolerated for the antiproliferative activity than a methoxy group. Compound 4g was 2-fold less potent than 4n against the two breast cancer cell lines MCF-7 and MDAMB-231.
Moreover, substituting the para-position of the phenyl group of the chalcone moiety (R1) is crucial for the antiproliferative action of compounds 4a–n. Compounds 4h–m share the same structure as compound 4n but with different substituents at the para-position of the phenyl group of the chalcone moiety. The GI50 values of 4h–m ranged from 29 to 90 μM, making them at least four times less potent than compound 4n (GI50 = 7 μM). The data suggest that the furan group at the para-position of the phenyl group of the chalcone moiety is crucial for activity. Still, other substituents such as H atom, methyl group, methoxy group, and halogen atoms are less well-tolerated.
Compounds 4i (R1 = Me, R2 = Cl, scaffold A) and 4l (R1 = Br, R2 = Cl, scaffold A) had the highest GI50 values, indicating the lowest potency of the compounds 4h–m. Their GI50 values were 90 and 76 μM, 13 and 11 times less potent than compound 4n (R1 = furan-2-yl, R2 = Cl, scaffold A). This indicates that a methyl group and a bromine atom (R1) are the least tolerated for antiproliferative activity.
The antiproliferative activity of scaffold B compounds, 5a–n, was moderate to high compared to the open-ring counterparts, 4a–n (scaffold A compounds). The compounds 5a–n had improved GI50 values ranging from 6 to 79 μM, except for 5j, which had a GI50 value of 100 μM (the least potent one). Compounds 5b, 5c, 5d, and 5g were found to be the most efficient derivatives with GI50 values of 18, 13, 7, and 6 μM, respectively, making compounds 5d and 5g equipotent to the references doxorubicin and sorafenib (GI50 values 5 and 7 μM, respectively). Compound 5d (R1 = 3,4,5-tri-OMe, R2 = OMe, and scaffold B) had the highest potency among the derivatives tested against the MDAMB-231 breast cancer cell line, with an IC50 value of 2.89 ± 0.1 μM. It was more potent than both doxorubicin and sorafenib. Compound 5d exhibited 2.7 times greater potency than sorafenib against the MDAMB-231 breast cancer cell line, as seen in Table 2. Compound 5g (R1 = furan-2-yl, R2 = OMe, and scaffold B) demonstrated the highest efficiency against the MCF-7 breast cancer cell line. Its IC50 value was measured at 3.78 ± 0.2 μM, which is lower than the IC50 values of doxorubicin (4.17 ± 0.2 μM) and sorafenib (7.26 ± 0.3 μM). This indicates that compound 5g is more potent than doxorubicin and sorafenib against the MCF-7 breast cancer cell line. These in vitro assays suggest that compounds 5d, 5g, and 4n can be anti-breast cancer agents. However, further structural modifications are necessary to enhance their potency.
Compounds 5b (R1 = CH3, R2 = OMe, and scaffold B) and 5c (R1 = OMe, R2 = OMe, and scaffold B) demonstrated GI50 values of 18 and 13 μM, respectively being 2-folds less potent than compound 5d and 5g, these data indicate that the presence of furan moiety or tri-methoxy group at the para-position of the phenyl group of the chalcone moiety is essential for activity. Replacement of these groups (furan or tri-methoxy group) with other substituents like methyl or methoxy group resulted in a marketed decrease in the antiproliferative activity.
| Comp. | EGFR IC50 ± SEM (μM) | HER2 IC50 ± SEM (μM mL−1) |
|---|---|---|
| 4n | 0.799 ± 0.026 | 0.227 ± 0.007 |
| 5d | 0.126 ± 0.004 | 0.061 ± 0.002 |
| 5g | 1.192 ± 0.039 | 0.771 ± 0.025 |
| Erlotinib | 0.076 ± 0.002 | — |
| Lapatinib | — | 0.026 ± 0.001 |
The results of this in vitro assay test are consistent with the results of the antiproliferative assay. Compound 5d (R1 = 3,4,5-tri-OMe, R2 = OMe, scaffold B), the most effective antiproliferative agent, was also the most effective EGFR inhibitor with an IC50 value of 0.126 ± 0.004 μM. It was equally potent as the reference erlotinib (IC50 value = 0.076 ± 0.002 μM). Compounds 4n and 5g had promising EGFR inhibitory effects, with IC50 values of 0.799 and 1.192 μM, respectively. This means they were at least 10 times less effective as EGFR inhibitors than erlotinib. The data indicate that compound 5d is an efficacious antiproliferative agent that may function as an EGFR inhibitor.
| DNA content | Comment | |||
|---|---|---|---|---|
| Code | % G0–G1 | % S | % G2/M | |
| 5d | 78.53 | 18.11 | 3.36 | Cell cycle arrest @ G1 |
| Control | 64.15 | 24.38 | 11.47 | — |
 | ||
| Fig. 4 Cell cycle analysis of MDA-MB 231 cells treated with (A) vehicle (control), (B) compound 5d (0.250 μM). | ||
| Code | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 5d | 29.59 | 17.13 | 8.89 | 3.57 |
| Control | 2.21 | 0.34 | 0.15 | 1.72 |
 | ||
| Fig. 5 Detection of apoptosis in MDA-MB 231 cells. (A): vehicle control, (B) compound 5d (0.250 μM). | ||
3.3. Molecular docking study and molecular dynamics simulation
To gain insight into the potential interaction mechanisms of both 4n and 5d with EGFR or HER2, we modeled the structures of these compounds and then used Autodock Vina37 to dock them into the active sites of the kinase domains of each receptor (PDB IDs 1M17 and 3PP0, respectively).38 We used PyMOL software39 to edit and visualize the resulting binding poses. In our docking experiments, we used the modeled structures of both 5d enantiomers (R and S) as a racemic combination. The docking scores and binding patterns of compound 4n and the R enantiomer of 5d were similar in the active site of the EGFR kinase domain (−10.94 and −10.16 kcal mol−1, respectively). However, the docking scores of 4n were higher than those of 5d (i.e., the R enantiomer) in the active site of the HER2 kinase domain (−12.34 and −9.11 kcal mol−1, respectively). The 5d S enantiomer exhibited significantly worse compatibility inside the active regions of both kinase domains, with docking scores of −7.3 and −7.1 kcal mol−1 for EGFR and HER2, respectively.To better understand how each compound interacts with the active parts of the kinase domains of both receptors, we simulated their docking positions for 200 ns34–36,40,41. After that, we looked at each MD trajectory to find the chemical's main binding conformation, its RMSD profile, and its absolute binding free energies (ΔGBind). Fig. 6A–C illustrate that the MD simulation-derived predominant binding modes of compounds 4n and 5d (R enantiomer) when they were combined with the co-crystallized inhibitor inside the EGFR kinase domain. These interactions revealed several important details about their binding affinities and how they work.
 | ||
| Fig. 6 Dynamic binding modes of 4n (yellow structure), 5d (R enantiomer; brick red structure), and the co-crystallized inhibitors (blue structures) inside the active sites of kinase domains of both EGFR ((A)–(C), respectively; PDB ID 1M17) and HER2 ((D)–(F), respectively; PDB ID 3PP0). The depicted binding poses were retrieved from the MD simulation trajectories as the most populated binding pose. | ||
For compound 4n, we detected a stable hydrogen bond with the THR-766 residue (89% of simulation duration), which acts as an anchoring point within the binding pocket. Hydrophobic residues, such as LEU-820, LEU-834, VAL-702, and PHE-699, enhance the stability of 4n via van der Waals interactions. The closeness of extra residues like ASP-831 and MET-742 supports the idea that they bind through non-polar interactions even more. The aromatic rings of compound 4n indicate potential pi–pi stacking interactions with PHE-699, which occurred for 57% of the simulation duration, thereby increasing its binding affinity. Furthermore, the terminal electron-deficient chlorophenyl ring engaged in stable π-anion interactions with the negatively charged residue ASP-831 (85% of the simulation duration).
Regarding 5d (R configuration), its structure exhibited a distinct binding profile, characterized by a stable hydrogen bond with PRO-770 (76% of simulation time), which helps establish its orientation within the kinase pocket. Hydrophobic interactions involving residues such as LEU-820, VAL-702, and LEU-694 provide additional stabilization. The presence of PHE-771 PRO-717 and PRO-770 in the vicinity suggests further HER hydrophobic interactions, strengthening the binding of compound 5d. Moreover, the p-methoxy aromatic ring of 5d potentially engages in T-shaped pi–pi stacking with PHE-771 (55% of simulation time). In comparison, the electron-rich trimethoxy one formed a stable π-cation interaction with LYS-721 (89% of simulation time), enhancing its affinity for the kinase domain.
The dynamic binding modes of both 4n and 5d (R enantiomer) shared a common interaction with the co-crystallized inhibitor (i.e., erlotinib), particularly the hydrophobic ones with LEU-820, LEU-694, VAL-702, and PHE-771. The presence of aromatic rings in all three molecules also allows for potential pi–pi stacking with pivotal residues like PHE-699 and PHE-771, contributing to their binding affinity and specificity. These interactions highlight the compounds' capabilities to fit within the hydrophobic pocket of the kinase domain, thereby informing their potential as EGFR inhibitors.
These binding behaviors of both 4n and 5d (R enantiomer) were translated into steady RMSD profiles throughout MD simulations comparable to that of the co-crystalized inhibitor (average RMSD = 2.44 Å; Fig. 7A). Hence, their estimated ΔGBind values were convergent (−9.24, −9.48, −9.39, respectively).
 | ||
| Fig. 7 RMSD profiles of 4n, 5d, and the co-crystallized inhibitors inside the active sites of kinase domains of both EGFR (A) and HER2 (B) throughout 200 ns MD simulation. | ||
As shown in Fig. 7A, compound 4n demonstrates the highest structural stability within the binding pocket, as reflected by its consistently lower RMSD values, ranging between 1.0 and 3.0 Å. Its RMSD profile stabilizes around 2.0 Å relatively early in the simulation (after approximately 20 ns) and exhibits minimal fluctuations afterward. This behavior suggests compound 4n forms a rigid and stable complex with the EGFR kinase domain, potentially indicating a strong binding affinity and specificity.
In contrast, compound 5d (R enantiomer) displays the highest fluctuation in its RMSD values, oscillating between 1.5 and 4.0 Å throughout the simulation period. This high level of variability suggests that compound 5d undergoes significant conformational changes, indicating a more flexible or adaptable binding mode within the kinase pocket. Such flexibility may imply weaker interactions with EGFR, potentially resulting in lower binding affinity or specificity than compound 4n and the co-crystallized inhibitor.
The co-crystallized inhibitor shows intermediate behavior, with RMSD values fluctuating between 1.5 and 3.5 Å. While the inhibitor exhibits more flexibility than compound 4n, it is notably more stable than compound 5d. The RMSD pattern suggests that the co-crystallized inhibitor maintains a relatively stable interaction within the EGFR kinase domain while allowing some conformational flexibility. This balance between stability and flexibility may reflect the inhibitor's ability to achieve effective binding while accommodating necessary structural adjustments during the simulation.
Regarding HER2's kinase domain, Fig. 6D–F (representing the most populated poses of 4n and 5d alongside the co-crystallized inhibitor) demonstrated that both 4n and 5d (R enantiomer) exhibited comparable interactions inside the active site of HER2's kinase domain that were also concurrent to that of the co-crystallized inhibitor. 4n forms three stable hydrogen bonds with the main chain of LEU-726 and LEU-796, alongside the side chain of ASP-863 (78%, 75%, and 71% of the simulation time, respectively) that serve as key anchoring interactions in the kinase domain's active site. The compound's proximity to ASP-808 suggests the presence of π-anion interaction (88% of simulation time) with the terminal electron deficient chlorophenyl ring that further stabilizes the binding conformation. Hydrophobic residues, including LEU-726, LEU-796, VAL-734, and LEU-852, likely engage in van der Waals interactions with the hydrophobic regions of compound 4n, reinforcing its affinity for the binding site. Furthermore, MET-774 and PHE-864 stabilize the compound within the kinase domain through hydrophobic contacts.
For compound 5d (R enantiomer), the docking pose reveals stable hydrogen bonds with LYS-753, THR-798, and ARG-849 (66%, 53%, and 61% of simulation time, respectively), facilitating its precise orientation and interaction within the binding pocket. This hydrogen-bonding network, particularly with LYS-753, mirrors typical interactions observed in effective HER2 inhibitors.38 The surrounding hydrophobic environment, composed of residues such as LEU-726, VAL-734, LEU-785, and LEU-852, plays a crucial role in stabilizing compound 5d. The involvement of residues like MET-774 and PHE-864 provides further hydrophobic contacts, contributing to the compound's overall stability. The aromatic rings of compound 5d also likely engage in T-shaped pi–pi stacking interactions with PHE-864, reinforcing its binding affinity within the HER2 kinase domain.
The co-crystallized inhibitor demonstrates a well-defined binding profile characterized by a network of hydrogen bonds involving ASP-863, THR-862, and LYS-753. These interactions are vital for the inhibitor's specificity and robust binding within the kinase domain. The hydrophobic surroundings, including LEU-726, LEU-852, and MET-801, enhance the inhibitor's stability through van der Waals interactions. Potential pi–pi stacking with PHE-864 further strengthens the inhibitor's binding affinity.
Collectively, these results illustrate that compounds 4n and 5d (R enantiomer) share key binding features with the co-crystallized inhibitor, such as hydrogen bonding, hydrophobic contacts, and pi–pi stacking interactions. These compounds' ability to occupy and stabilize within the HER2 kinase pocket underscores their potential as HER2 inhibitors, providing valuable insights for further optimization and development.
The RMSD profiles and ΔGBind values of both compounds were also convergent and comparable to those of the co-crystallized inhibitor, indicating stable binding modes (average RMSD = 2.88 Å; ΔGBind values = −9.22, −8.29, −9.89 kcal mol−1, respectively; Fig. 7B).
Among the three compounds, 4n displays the lowest and most consistent RMSD values, predominantly fluctuating between 1.0 and 2.5 Å throughout the 200 ns simulation. This low range and steady fluctuation indicate that compound 4n maintains a rigid and stable interaction within the HER2 binding pocket. The minimal structural deviations from the initial conformation suggest that 4n fits snugly within the binding site, potentially reflecting a strong binding affinity and a well-defined interaction with the kinase domain (Fig. 7B).
In contrast, 5d (R enantiomer) shows higher RMSD fluctuations, ranging between 1.5 and 4.0 Å, indicating a more dynamic interaction with the HER2 kinase domain. These larger oscillations suggest that compound 5d undergoes notable conformational adjustments throughout the simulation, implying a more flexible binding mode. Such flexibility might indicate weaker binding affinity or a less optimized fit within the binding pocket, as the compound seems to require continuous adjustments to accommodate its position (Fig. 7B).
The co-crystallized inhibitor exhibits intermediate RMSD values, generally fluctuating between 2.0 and 3.5 Å. While it displays more variability than compound 4n, it remains more stable than compound 5d. This intermediate pattern suggests that the co-crystallized inhibitor maintains a relatively stable conformation within the kinase domain, similar to compound 4n, but with a degree of flexibility that could be advantageous for effective binding. The balance of moderate flexibility and overall stability aligns with its role as a known inhibitor of HER2.
4. Conclusion
We synthesized a novel series of two scaffold compounds, chalcone/phenylpiperazine scaffold 4a–n and pyrazoline/phenylpiperazine scaffold 5a–n and evaluated their antiproliferative activity against EGFR and HER2. The results indicated that the compounds of scaffold B, 5a–n, exhibited more potency than those of scaffold A, 4a–n. Compounds 4n, 5d, and 5g exhibited the highest efficacy among all evaluated compounds against a panel of four cancer cell lines. These three derivatives were subsequently examined for mechanistic analysis against EGFR-TK and HER-2. Compound 5d was identified as an efficient antiproliferative agent that acts as a dual EGFR/HER2 inhibitor and demonstrated cell cycle arrest at the pre-G1 phase. The docking investigation and molecular dynamics simulation indicated that compounds 4n and 5d (R enantiomer) exhibit essential binding characteristics with the co-crystallized inhibitor, including hydrogen bonding, hydrophobic interactions, and pi–pi stacking interactions. All in vitro experiments demonstrated that 5d is a promising lead compound that requires more structural modification to give a more potent derivative.Data availability
Data will be available upon request from the authors.Author contributions
H. M. Hafez: formal analysis, methodology, software, writing–original draft. B. A. M. Said: investigation, validation, visualization, writing–original draft. A. M. Sayed: methodology, writing–original draft. E. Alatwi: writing–original draft. SB: investigation, validation, visualization, writing–review and editing. BY: conceptualization, data duration, formal analysis, investigation, methodology, resources, software, supervision, validation, visualization, writing–original draft, writing–review and editing. H. A. M. El-Sherief: methodology, resources, software, writing–original draft, writing–review and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.References
- T. Kovacs, F. Zakany and P. Nagy, Cancers, 2022, 14, 944 CAS.
- D. Kumar and M. I. Hassan, in Receptor Tyrosine Kinases in Neurodegenerative and Psychiatric Disorders, Elsevier, 2023, pp. 245–276 Search PubMed.
- Z. Batool, A. Azfal, L. Liaquat, S. Sadir, R. Nisar, A. Inamullah, A. U. F. Ghalib and S. Haider, in Receptor Tyrosine Kinases in Neurodegenerative and Psychiatric Disorders, Elsevier, 2023, pp. 117–185 Search PubMed.
- C. B. Pulivarthi, S. S. Choubey, S. K. Pandey, A. S. Gautam and R. K. Singh, Receptor Tyrosine Kinases in Neurodegenerative and Psychiatric Disorders, 2023, pp. 45–77 Search PubMed.
- Z. Batool, A. Azfal, L. Liaquat, S. Sadir, R. Nisar, A. Inamullah, A. U. F. Ghalib and S. Haider, in Receptor Tyrosine Kinases in Neurodegenerative and Psychiatric Disorders, Elsevier, 2023, pp. 117–185. Search PubMed.
- L. H. Al-Wahaibi, A. M. Elshamsy, T. F. Ali, B. G. Youssif, S. Bräse, M. Abdel-Aziz and N. A. El-Koussi, ACS Omega, 2024, 9, 34358–34369 CAS.
- L. H. Al-Wahaibi, B. G. Youssif, H. A. Abou-Zied, S. Bräse, A. B. Brown, H. N. Tawfeek and E. M. El-Sheref, RSC Med. Chem., 2024, 15, 2538–2552 CAS.
- J. Yoon and D.-Y. Oh, Nat. Rev. Clin. Oncol., 2024, 1–26 Search PubMed.
- D. Gandhi, A. Sethiya, N. Sahiba, D. K. Jangid and S. Agarwal, in Nanocatalysis, CRC Press, 2022, pp. 219–243 Search PubMed.
- D. O. Ozgun, H. I. Gul, C. Yamali, H. Sakagami, I. Gulcin, M. Sukuroglu and C. T. Supuran, Bioorg. Chem., 2019, 84, 511–517 Search PubMed.
- M. Karabacak, M. D. Altıntop, H. İ. Çiftçi, R. Koga, M. Otsuka, M. Fujita and A. Özdemir, Molecules, 2015, 20, 19066–19084 CAS.
- L. H. Al-Wahaibi, H. A. Abou-Zied, E. A. Beshr, B. G. Youssif, A. M. Hayallah and M. Abdel-Aziz, Int. J. Mol. Sci., 2023, 24, 9104 CAS.
- L. H. Al-Wahaibi, H. A. Abou-Zied, M. Hisham, E. A. Beshr, B. G. Youssif, S. Bräse, A. M. Hayallah and M. Abdel-Aziz, Molecules, 2023, 28, 6586 CAS.
- P.-C. Lv, H.-Q. Li, J. Sun, Y. Zhou and H.-L. Zhu, Bioorg. Med. Chem., 2010, 18, 4606–4614 CAS.
- M. Al-Anazi, M. Khairuddean, B. O. Al-Najjar, M. M. Alidmat, N. N. S. N. M. Kamal and M. Muhamad, Arab. J. Chem., 2022, 15, 103864 CrossRef CAS.
- B. Sever, M. D. Altıntop, M. O. Radwan, A. Özdemir, M. Otsuka, M. Fujita and H. I. Ciftci, Eur. J. Med. Chem., 2019, 182, 111648 CrossRef.
- H. A. Jasim, L. Nahar, M. A. Jasim, S. A. Moore, K. J. Ritchie and S. D. Sarker, Biomolecules, 2021, 11, 1203 CAS.
- A. Rammohan, J. S. Reddy, G. Sravya, C. N. Rao and G. V. Zyryanov, Environ. Chem. Lett., 2020, 18, 433–458 CrossRef CAS.
- M. Hisham, H. A. Hassan, H. A. Gomaa, B. G. Youssif, A. M. Hayalah and M. Abdel-Aziz, Anti-Cancer Agents Med. Chem., 2023, 23, 1932–1943 CrossRef CAS PubMed.
- M. Hisham, H. A. Hassan, H. A. Gomaa, B. G. Youssif, A. M. Hayallah and M. Abdel-Aziz, J. Mol. Struct., 2022, 1254, 132422 CrossRef CAS.
- J. Xiao, M. Gao, Q. Diao and F. Gao, Curr. Top. Med. Chem., 2021, 21, 348–362 CrossRef CAS PubMed.
- F. F. Leite, N. F. de Sousa, B. H. M. de Oliveira, G. D. Duarte, M. D. L. Ferreira, M. T. Scotti, J. M. B. Filho, L. C. Rodrigues, R. O. de Moura and F. J. B. Mendonça-Junior, Molecules, 2023, 28, 4009 CrossRef CAS PubMed.
- P. Kumar, R. Singh, D. Sharma, Q. P. Hassan, B. Gopu and J. M. H. Anal, Bioorg. Med. Chem. Lett., 2024, 107, 129795 CrossRef CAS PubMed.
- A. M. Mohassab, H. A. Hassan, D. Abdelhamid, A. M. Gouda, B. G. Youssif, H. Tateishi, M. Fujita, M. Otsuka and M. Abdel-Aziz, Bioorg. Chem., 2021, 106, 104510 CrossRef CAS.
- H. A. Abou-Zied, E. A. Beshr, H. A. Gomaa, Y. A. Mostafa, B. G. Youssif, A. M. Hayallah and M. Abdel-Aziz, Arch. Pharm., 2023, 356, 2200464 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, E. M. El-Sheref, A. A. Hassan, S. Bräse, M. Nieger, B. G. Youssif, M. A. Ibrahim and H. N. Tawfeek, Pharmaceuticals, 2023, 16, 1014 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, M. A. Mahmoud, Y. A. Mostafa, A. E. Raslan and B. G. Youssif, J. Enzyme Inhib. Med. Chem., 2023, 38, 376–386 CrossRef CAS.
- L. H. Al-Wahaibi, A. F. Mohammed, F. E.-Z. S. A. Rahman, M. H. Abdelrahman, X. Gu, L. Trembleau and B. G. Youssif, J. Enzym. Inhib. Med. Chem., 2023, 38, 2218602 Search PubMed.
- R. A. Mekheimer, S. M. Allam, M. A. Al-Sheikh, M. S. Moustafa, S. M. Al-Mousawi, Y. A. Mostafa, B. G. Youssif, H. A. Gomaa, A. M. Hayallah and M. Abdelaziz, Bioorg. Chem., 2022, 121, 105693 CrossRef CAS.
- S. E. Wang, A. Narasanna, M. Perez-Torres, B. Xiang, F. Y. Wu, S. Yang, G. Carpenter, A. F. Gazdar, S. K. Muthuswamy and C. L. Arteaga, Cancer Cell, 2006, 10, 25–38 CrossRef CAS PubMed.
- P. Smolewski, E. Bedner, L. Du, T. C. Hsieh, J. M. Wu, D. J. Phelps and Z. Darzynkiewicz, Cytometry: The Journal of the International Society for Analytical Cytology, 2001, 44, 73–82 CrossRef CAS.
- E. Brahmana, J. Kaban, G. Haro and M. Ginting, Rasayan J. Chem., 2022, 15, 3–11 Search PubMed.
- M. A. Mahmoud, A. F. Mohammed, O. I. Salem, H. A. Gomaa and B. G. Youssif, Arch. Pharm., 2022, 355, 2200009 CrossRef CAS.
- M. S. Mohamed, H. A. Elsherief, H. M. Hafez, O. A. Alsaidan, S. I. Alzarea and A. M. AboulMagd, Mol. Diversity, 2023, 27, 2133–2146 CrossRef CAS PubMed.
- M. Johnson, B. Younglove, L. Lee, R. LeBlanc, H. Holt Jr, P. Hills, H. Mackay, T. Brown, S. L. Mooberry and M. Lee, Bioorg. Med. Chem. Lett, 2007, 17, 5897–5901 CrossRef CAS PubMed.
- F. F. Hagar, S. H. Abbas, D. Abdelhamid, H. A. Gomaa, B. G. Youssif and M. Abdel-Aziz, Arch. Pharm., 2023, 356, 2200357 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS.
- K. Aertgeerts, R. Skene, J. Yano, B.-C. Sang, H. Zou, G. Snell, A. Jennings, K. Iwamoto, N. Habuka and A. Hirokawa, J. Biol. Chem., 2011, 286, 18756–18765 CrossRef CAS PubMed.
- S. Yuan, H. S. Chan and Z. Hu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1298 Search PubMed.
- J. V. Ribeiro, R. C. Bernardi, T. Rudack, K. Schulten and E. Tajkhorshid, Biophys. J., 2018, 114, 673a–674a CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01169h |
| This journal is © The Royal Society of Chemistry 2025 |