
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative photoredox and N-heterocyclic carbene-catalyzed formal C–H acylation of cyclopropanes via a deconstruction–reconstruction strategy†
Fan
Gao
 ,
Tian
Wang
and
Xiaoyu
Yan
*
,
Tian
Wang
and
Xiaoyu
Yan
*
Key Laboratory of Advanced Light Conversion Materials and Biophotonics School of Chemistry and Life Resources, Renmin University of China, Beijing, 100872, China. E-mail: yanxy@ruc.edu.cn
First published on 22nd November 2024
Abstract
Cyclopropanes are ubiquitous and key structural motifs in commercially available drugs and bioactive molecules. Herein, we present regio-selective acylation of aryl cyclopropanes with cooperative photoredox and N-heterocyclic carbene catalysis. This approach involves a deconstruction–reconstruction strategy via γ-chloro-ketones as intermediates and fulfills the formal C(sp3)–H functionalization of cyclopropanes.
Cyclopropanes, as strained cycloalkanes, have gained significant attention due to their ubiquitous and key structural motif in commercially available pharmaceutical candidates and drugs as well as their promising bioactivities (Scheme 1).1 Additionally, compared with gem-dimethyl, isopropyl and phenyl groups, cyclopropane derivatives exhibit enhanced metabolic stability and reduced lipophilicity because of their structural characteristics involving high coplanarity of the ring-carbon atoms, enhanced π-character, relatively shorter C–C bonds, and shorter and stronger C–H bonds.2 However, the functionalization of strained cyclopropane frameworks represents an important challenge for chemical synthesis.
 | ||
| Scheme 1 Selected application examples of bioactive cyclopropane. | ||
Direct C(sp3)–H functionalization of strain cyclopropane frameworks has been recognized as an economic and simple strategy to access various cyclopropane scaffolds. Yu, Gaunt, Xu and others have reported powerful strategies to synthesize monofunctionalized cyclopropane derivatives3via the coordination interaction of transition-metals (Pd or Ir) and various directing groups (DGs) such as carboxylic acids,3f primary amines,3gN-aryl carboxamides,3iN-triflamides,3eN-aryl-aminomethyl,3h carboxamide,3c,d and ether3j (Scheme 2a).
 | ||
| Scheme 2 Functionalization of strained cyclopropanes. | ||
Those available protocols show good regioselectivity, introducing various functional groups to the β-position of DGs, while few directed C(sp3)–H functionalized examples at the α-position3l have been achieved. Besides requiring tedious processes to introduce appropriate directing groups (DGs) and costly transition-metals as catalysts, those aforesaid methodologies usually require harsh reaction conditions, like heating to keep high regio- and stereoselectivity.
Due to their high ring strain energies (ca. 27.5 kcal mol−1), cyclopropane derivatives easily undergo deconstruction of the coplanarity ring, and have been identified as versatile and powerful C3 units in synthesis. Diverse strategies catalyzed by Lewis acids,4 transition-metals (Rh, Ni, Pd, Fe),5 visible light6 and electricity7 have been developed to produce [3 + n] annulation products (Scheme 2b). Meanwhile, 1,3-difunctionalization of cyclopropanes has also been achieved, generating acyclic products with introduction of two distinct functional groups.8 We envisioned that, with suitable functional groups as leaving groups, 1,3-elimination reactions are viable, which will reconstruct the cyclopropane skeleton. The two-step strategy of deconstructive 1,3-difunctionalization and 1,3-elimination would fulfill the formal C(sp3)–H functionalization of cyclopropanes and avoid the preinstallation of directing groups. With the rapid development of radical N-heterocyclic carbene (NHC) catalysis9 and our contribution in this area,9,10 herein, we disclose a cooperative NHC and photoredox9h,11 catalyzed acylation of aryl cyclopropanes, which involves formal C–H functionalization that has been achieved via a deconstruction–reconstruction strategy with γ-chloro-ketones as intermediates. Either aromatic or aliphatic acyl groups can be selectively introduced to the α-position of aryl groups. Meanwhile, this method could be extended to esterification of cyclopropanes with carbonate esters.
Studer's group has achieved the 1,3-difunctionalization of aryl cyclopropanes catalyzed by a cooperative NHC and organophotoredox catalyst, generating various γ-aroyloxy ketones.11d To facilitate reconstruction of cyclopropanes, we used acyl imidazole as the acyl source to avoid introducing the poor-leaving ester group. Hence, we first started our investigation by using 1-anisoylcyclopropane (1a) and N-anisoylimidazole (2a) as the model substrates in the presence of the triazolium-type NHC-1 as the organo-catalyst, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC-1) as the photocatalyst, LiCl as an additive, K2CO3 as the base and anhydrous DMF as the solvent at room temperature under an Ar atmosphere and the irradiation of 20 W blue LEDs. After 48 h of reaction, the desired tertiary cyclopropane product 3aa was obtained in 78% yield (Table 1, entry 1). When we reduced 2.0 equivalents of K2CO3 to 0.2 equivalents, a 50% yield of 3aa was obtained, accompanied by the 1,3-difunctionalization product 3aa′ in 26% yield (entry 2). This indicates that product 3aa′ was transformed into product 3aa after increasing the equivalent of base and that product 3aa′ was the key intermediate. Screening different organo-catalysts (entries 3–6) showed that the yields of 3aa decreased with other NHCs. Other photocatalysts (entries 7 and 8) like 4CzIPN also showed decreased yields. Lower yields were obtained in CH3CN and DMSO (entries 9 and 10). Subsequently, several bases and additives were explored (entries 11–15), and the results showed that LiCl and K2CO3 were the most suitable additive and base in this reaction system. Control experiments indicated that the NHC catalyst, photocatalyst and visible light were critical for this reaction (entries 16–18). Finally, a higher concentration for substrates gave a better yield of 85% (entry 19).
|
|
|||
|---|---|---|---|
| Entry | Variation from “standard conditions” | Yieldb3aa [%] | Yieldb3aa′ [%] |
| a All reactions were performed by using 1a (0.1 mmol), 2a (2.0 equiv.), PC-1 (2 mol%), NHC-1 (20 mol%), K2CO3 (2.0 equiv.), LiCl (2.0 equiv.), and anhydrous DMF (0.05 M) under blue LEDs (440 nm, 20 W), stirred at room temperature and in Ar for 48 h. b Yield was determined by 1H NMR. c Isolated yield was given in parentheses. | |||
| 1 | None | 78 | 0 |
| 2 | K2CO3 (0.2 equiv.) instead of K2CO3 (2.0 equiv.) | 50 | 26 |
| 3 | NHC-2 instead of NHC-1 | 11 | 12 |
| 4 | NHC-3 instead of NHC-1 | 8 | 26 |
| 5 | NHC-4 instead of NHC-1 | 24 | 0 |
| 6 | NHC-5 instead of NHC-1 | 0 | 0 |
| 7 | PC-2 (5 mol%) instead of PC-1 | 32 | 0 |
| 8 | PC-3 instead of PC-1 | 11 | 0 |
| 9 | MeCN instead of DMF | 21 | 20 |
| 10 | DMSO instead of DMF | 42 | 0 |
| 11 | Cs2CO3 instead of K2CO3 | 57 | 0 |
| 12 | K3PO4 instead of K2CO3 | 29 | 22 |
| 13 | Na2CO3 instead of K2CO3 | 37 | 0 |
| 14 | Me4NCl instead of LiCl | 50 | 0 |
| 15 | KCl instead of LiCl | 15 | 0 |
| 16 | Without NHC-1 | 0 | 0 |
| 17 | Without PC-1 | 0 | 0 |
| 18 | In the dark | 0 | 0 |
| 19 | DMF (0.1 M) instead of DMF (0.05 M) | 85(79)c | 0 |

|
|||

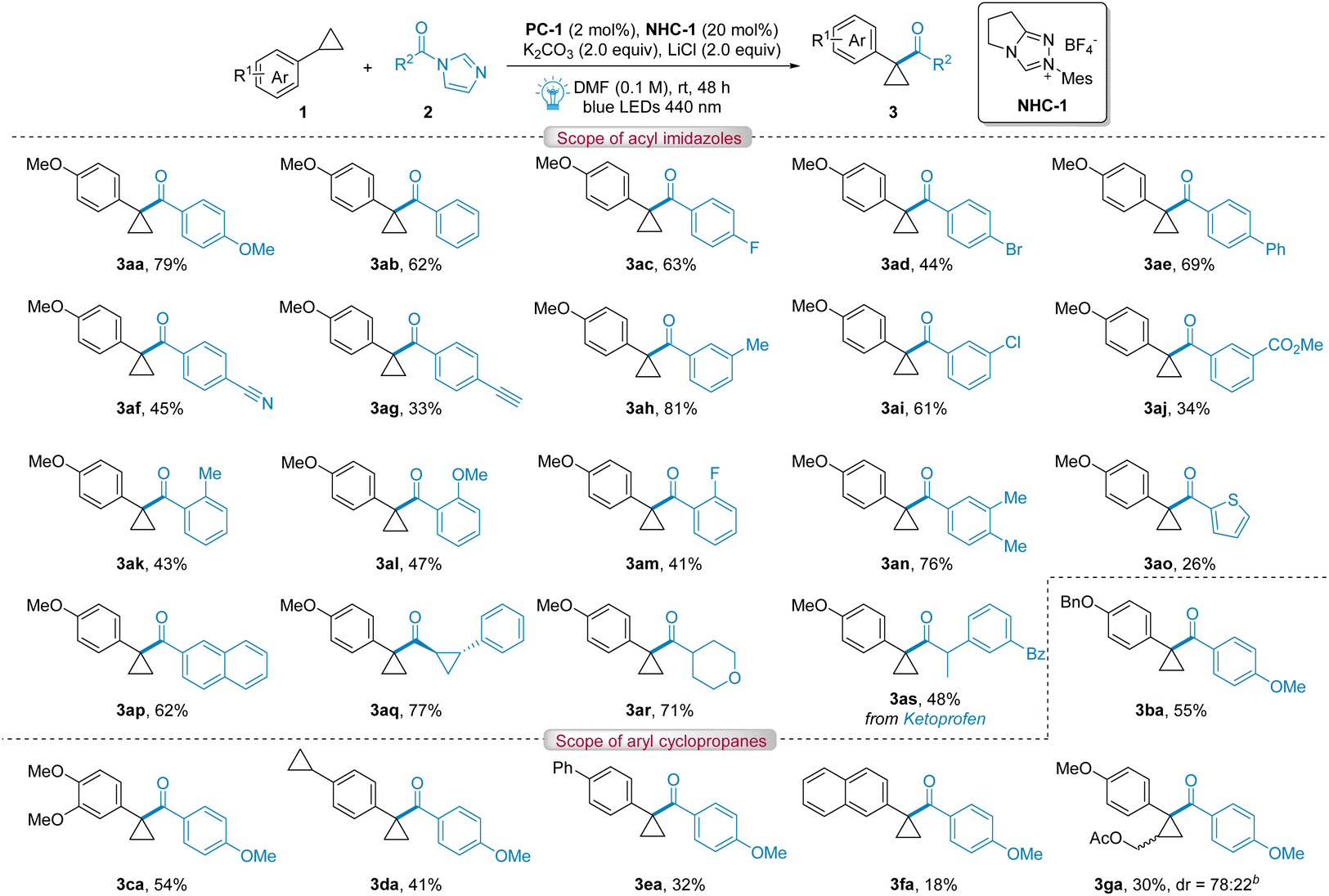
With the optimized reaction conditions in hand, we explored a series of acyl imidazoles 2 (Table 2). Benzoyl imidazoles with different substituents bearing electron-donating or electron-withdrawing groups at the para- or meta-position proceeded smoothly to afford the desired tertiary cyclopropanes 3aa–3aj in moderate to good yields (33–81%). In general, higher yields were obtained for benzoyl imidazoles with electron-donating groups while lower yields were obtained for benzoyl imidazoles with strong electron-withdrawing groups. A low yield was obtained for 3ad, which is due to dehalogenation. For benzoyl imidazoles with ortho-substituents, corresponding products were obtained in 41–47% yields due to the effect of steric hindrance. Different aromatic substituted acyl imidazoles like 2-thienyl and 2-naphthyl were also found to be suitable in this reaction, affording corresponding products 3ao and 3ap in 26% and 62% yields, respectively. We speculate that the oxidation of thienyl under reaction conditions results in the low yield for 3ao. NHC-catalyzed radical acylations were usually limited to aryl acyl substrates, while aliphatic substrates were challenged.11e Gratifyingly, we found that aliphatic acyl imidazoles could also react smoothly with cyclopropane 1a, leading to the formation of desired products 3aq–3ar in high yields (71–77%). To demonstrate the high functional group tolerance and broad substrate scope of acyl imidazoles, late-stage functionalization of a bioactive molecule derived from ketoprofen was explored, generating the corresponding α-acylated cyclopropane derivative 3as in moderate yield (48%).
| a All reactions were performed by using 1a (0.1 mmol), 2 (2.0 equiv.), PC-1 (2 mol%), NHC-1 (20 mol%), K2CO3 (2.0 equiv.), LiCl (2.0 equiv.), and anhydrous DMF (0.1 M) under blue LEDs (440 nm, 20 W), stirred at room temperature and in Ar for 48 h, isolated yields were given. b dr was determined by 1H NMR. |
|---|
|
The scope of aryl cyclopropanes was also investigated (Table 2). For aryl cyclopropanes bearing various substituents at the para-positions of the aryl moiety such as benzyloxy, disubstituted methoxy, cyclopropyl and phenyl, the corresponding products 3ba–3ea were obtained in moderate yields (32–55%). Notably, it could selectively afford mono-acylated cyclopropane derivatives 3da from a substrate bearing two cyclopropyl groups at 1,4-positions of the benzene ring. A low yield was obtained for 3fa, which is due to acylation at the naphthalene ring.12 Finally, to address the limitation of regioselectivity and diastereoselectivity, an unsymmetric cyclopropane, 1-ethoxy-2-phenylcyclopropane was explored to produce 3ga in 30% yield, showing excellent regioselectivity albeit moderate diastereoselectivity (dr = 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22).
:22).
To demonstrate the ease and practicality of this method, a “one-pot, two-step” process was performed starting from carboxylic acid 4, generating 3aa in 80% yield (compared to 79% when starting from N-anisoylimidazole 2a) (Scheme 3). NHC-catalyzed radical esterification reactions have been developed recently.11g We also extended this deconstruction–reconstruction strategy for the formal esterification of cyclopropanes. With diethyl dicarbonate 5 as the esterification reagent, 1a was converted to α-esterified cyclopropanes 6 in 74% yield. Furthermore, 6 can be easily converted to amide product 7, which has the skeleton of lumacaftor (Scheme 4).
 | ||
| Scheme 3 “One-pot, two-step” acylation reaction from carboxylic acid. | ||
 | ||
| Scheme 4 Esterification of cyclopropane 1a and further transformation. | ||
To gain a deep insight into the mechanism for this transformation, several control experiments were subsequently conducted. Initially, radical scavengers (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) were employed respectively, which clearly inhibited the reaction (Scheme 5a). These results indicated that a radical process might be involved. Moreover, the intermediates A and B were successfully detected by high-resolution mass spectrometry (HRMS), implying that the NHC-derived ketyl radical and alkyl radical were generated in this transformation. Subsequently, Stern–Volmer fluorescence quenching experiments were carried out (for details see ESI†). As shown in Scheme 5b, the obvious linear relationships and different slopes between the fluorescence intensities and the concentrations of 1a and 9 suggested that the excited photocatalyst was more favorable to be quenched by acyl azolium 9.
 | ||
| Scheme 5 Control experiments and linear relationship between I0/I and the concentration of 1a and 9. | ||
Based on these results of control experiments and previous investigations, we proposed the following mechanism (Scheme 6). The excited photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC*) (Eox = −0.89 V vs. SCE)13 was oxidatively quenched by acyl azolium 9 (E1/2 = −0.81 V vs. SCE)14 to generate NHC-derived ketyl radical intermediate III and PC˙+. Then, the substrate 1a (E1/2 = + 1.35 V vs. SCE) was oxidized by PC˙+ (EPC˙+/PC = + 1.69 V vs. SCE) to regenerate ground photocatalyst PC and intermediate I. Subsequently, chloride ions reacted with intermediate I to produce alkyl radical intermediate II, which coupled with the NHC-derived ketyl radical intermediate III to produce γ-chloro-ketones as intermediates IV. Finally, the targeted product V was afforded via nucleophilic substitution in the presence of a base.
 | ||
| Scheme 6 Plausible reaction mechanism. | ||
Conclusions
Directed C(sp3)–H functionalization of strained cyclopropanes usually requires the coordination of transition-metals (Pd or Ir), ligands and various directing groups (DGs) to obtain high regio- and stereoselectivity cyclopropane scaffolds. In this paper, we present a deconstruction–reconstruction strategy for the regio-selective acylation of aryl cyclopropanes with cooperative photoredox and N-heterocyclic carbene catalysis, fulfilling the formal C(sp3)–H acylation of cyclopropanes. Besides that aromatic and aliphatic acyl groups can be selectively introduced to the α-position of aryl groups, this method could be extended to esterification of cyclopropanes with carbonate esters. α-Esterified cyclopropanes subsequently transformed into amide products, undoubtedly providing a far superior alternative for the preparation of analogous derivatives of commercially available pharmaceutical candidates and available drugs. Further studies on the applications of this deconstruction–reconstruction strategy will be reported in due course.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
F. G. and X. Y. conceived and designed the study. F. G. and T. W. performed the synthetic experiments with input from X. Y. The mechanistic investigations were performed by F. G. The manuscript was prepared by F. G. and X. Y. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22171284 and 22471288), the Fundamental Research Funds for the Central Universities and the Research Funds of Renmin University of China (Program 20XNLG20), and Public Computing Cloud Platform, Renmin University of China.Notes and references
- (a) T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed; (b) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS; (c) M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC; (d) M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046–14128 CrossRef CAS PubMed; (e) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1648 CrossRef CAS PubMed; (f) D. Y. K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- K. B. Wiberg, Angew. Chem., Int. Ed., 1986, 25, 312–322 CrossRef.
- (a) Y. Shi, Q. Gao and S. Xu, J. Am. Chem. Soc., 2019, 141, 10599–10604 CrossRef CAS; (b) M. Yasui, R. Ota, C. Tsukano and Y. Takemoto, Org. Lett., 2018, 20, 7656–7660 CrossRef CAS; (c) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598–19601 CrossRef CAS; (d) S. Jerhaoui, J.-P. Djukic, J. Wencel-Delord and F. Colobert, ACS Catal., 2019, 9, 2532–2542 CrossRef CAS; (e) K. S. L. Chan, H.-Y. Fu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 2042–2046 CrossRef CAS; (f) P.-X. Shen, L. Hu, Q. Shao, K. Hong and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545–6549 CrossRef CAS PubMed; (g) Z. Zhuang and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 12015–12019 CrossRef CAS PubMed; (h) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 12842–12845 CrossRef CAS PubMed; (i) R. Parella, B. Gopalakrishnan and S. A. Babu, Org. Lett., 2013, 15, 3238–3241 CrossRef CAS; (j) T. Xie, L. Chen, Z. Shen and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202300199 CrossRef CAS; (k) J. Rodrigalvarez, L. A. Reeve, J. Miró and M. J. Gaunt, J. Am. Chem. Soc., 2022, 144, 3939–3948 CrossRef CAS; (l) Q. Gao and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202218025 CrossRef CAS; (m) Y. Shi, Y. Yang and S. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201463 CrossRef CAS.
- (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS; (c) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804–818 RSC; (d) A. U. Augustin and D. B. Werz, Acc. Chem. Res., 2021, 54, 1528–1541 CrossRef CAS PubMed; (e) Y. Xia, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 9192–9204 CrossRef CAS.
- (a) S. Lv, W.-F. Xu, T.-Y. Yang, M.-X. Lan, R.-X. Xiao, X.-Q. Mou, Y.-Z. Chen and B.-D. Cui, Org. Lett., 2024, 26, 3151–3157 CrossRef CAS PubMed; (b) L. Liu and J. Montgomery, J. Am. Chem. Soc., 2006, 128, 5348–5349 CrossRef CAS; (c) M. H. Shaw, E. Y. Melikhova, D. P. Kloer, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2013, 135, 4992–4995 CrossRef CAS; (d) L. Jiao, S. Ye and Z.-X. Yu, J. Am. Chem. Soc., 2008, 130, 7178–7179 CrossRef CAS PubMed; (e) G. Bhargava, B. Trillo, M. Araya, F. López, L. Castedo and J. L. Mascareñas, Chem. Commun., 2010, 46, 270–272 RSC.
- (a) Y. Xu, H.-X. Gao, C. Pan, Y. Shi, C. Zhang, G. Huang and C. Feng, Angew. Chem., Int. Ed., 2023, 62, e202310671 CrossRef CAS; (b) S. Li and L. Zhou, Org. Lett., 2024, 26, 3294–3298 CrossRef CAS; (c) M.-M. Wang, T. V. T. Nguyen and J. Waser, Chem. Soc. Rev., 2022, 51, 7344–7357 RSC; (d) A. S. Harmata, B. J. Roldan and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2023, 62, e202213003 CrossRef CAS.
- (a) Q. Wang, Q. Wang, Y. Zhang, Y. M. Mohamed, C. Pacheco, N. Zheng, R. N. Zare and H. Chen, Chem. Sci., 2021, 12, 969–975 RSC; (b) D. Saha, I. M. Taily, N. Banerjee and P. Banerjee, Chem. Commun., 2022, 58, 5459–5462 RSC.
- (a) S. M. Banik, K. M. Mennie and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 9152–9155 CrossRef CAS PubMed; (b) L. Ge, D.-X. Wang, R. Xing, D. Ma, P. J. Walsh and C. Feng, Nat. Commun., 2019, 10, 4367 CrossRef PubMed; (c) D. Petzold, P. Singh, F. Almqvist and B. König, Angew. Chem., Int. Ed., 2019, 58, 8577–8580 CrossRef CAS PubMed; (d) S. Kolb, M. Petzold, F. Brandt, P. G. Jones, C. R. Jacob and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 15928–15934 CrossRef CAS; (e) H. Zhang, H. Xiao, F. Jiang, Y. Fang, L. Zhu and C. Li, Org. Lett., 2021, 23, 2268–2272 CrossRef CAS PubMed; (f) W. Sheng, X. Huang, J. Cai, Y. Zheng, Y. Wen, C. Song and J. Li, Org. Lett., 2023, 25, 6178–6183 CrossRef CAS; (g) Y. Yue, Y. Song, S. Zhao, C. Zhang, C. Zhu and C. Feng, Org. Lett., 2023, 25, 7385–7389 CrossRef CAS PubMed; (h) H. Huang, X. Luan and Z. Zuo, Angew. Chem., Int. Ed., 2024, 63, e202401579 CrossRef CAS; (i) M. Li, Y. Wu, X. Song, J. Sun, Z. Zhang, G. Zheng and Q. Zhang, Nat. Commun., 2024, 15, 8930 CrossRef CAS PubMed.
- (a) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS; (b) J. J. Chen, Y. Zhang and H. M. Huang, Catal. Sci. Technol., 2022, 12, 5241–5251 RSC; (c) Q.-Z. Li, X.-X. Kou, T. Qi and J.-L. Li, ChemCatChem, 2022, 15, e202201320 CrossRef; (d) K. Liu, M. Schwenzer and A. Studer, ACS Catal., 2022, 12, 11984–11999 CrossRef CAS; (e) K.-Q. Chen, H. Sheng, Q. Liu, P.-L. Shao and X.-Y. Chen, Sci. China:Chem., 2021, 64, 7–16 CrossRef CAS; (f) L. Dai and S. Ye, Chin. Chem. Lett., 2021, 32, 660–667 CrossRef CAS; (g) A. V. Bay and K. A. Scheidt, Trends Chem., 2022, 4, 277–290 CrossRef CAS; (h) X. Wang, S. Wu, R. Yang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2023, 14, 13367–13383 RSC; (i) Z.-Z. Zhang, R. Zeng, Y.-Q. Liu and J.-L. Li, ChemCatChem, 2024, 16, e202400063 CrossRef CAS; (j) X.-Y. Ye, Y. Xie and Y. R. Chi, Trends Chem., 2024, 6, 504–509 Search PubMed; (k) T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630–5636 RSC; (l) X. Chen, H. Wang, Z. Jin and Y. R. Chi, Chin. J. Chem., 2020, 38, 1167–1202 CrossRef CAS.
- (a) F. Gao, Z. Zhang and X. Yan, ChemCatChem, 2024, 16, e202301331 Search PubMed; (b) C. Liu, Z. Zhang, L.-L. Zhao, G. Bertrand and X. Yan, Angew. Chem., Int. Ed., 2023, 62, e202303478 Search PubMed; (c) Z. Zhang, S. Huang, C.-Y. Li, L.-L. Zhao, W. Liu, M. Melaimi, G. Bertrand and X. Yan, Chem Catal., 2022, 2, 3517–3527 Search PubMed; (d) W. Liu, A. Vianna, Z. Zhang, S. Huang, L. Huang, M. Melaimi, G. Bertrand and X. Yan, Chem Catal., 2021, 1, 196–206 Search PubMed; (e) T. Wang, Z. Zhang, F. Gao and X. Yan, Org. Lett., 2024, 26, 6915–6920 CrossRef CAS PubMed.
- (a) Q. Liu and X.-Y. Chen, Org. Chem. Front., 2020, 7, 2082–2087 RSC; (b) Q.-Z. Li, M.-H. He, R. Zeng, Y.-Y. Lei, Z.-Y. Yu, M. Jiang, X. Zhang and J.-L. Li, J. Am. Chem. Soc., 2024, 146, 22829–22839 Search PubMed; (c) H. Cai, X. Yang, S.-C. Ren and Y. R. Chi, ACS Catal., 2024, 14, 8270–8293 CrossRef CAS; (d) Z. Zuo, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 25252–25257 CrossRef CAS; (e) W.-C. Liu, X. Zhang, L. Chen, R. Zeng, Y.-H. Tian, E.-D. Ma, Y.-P. Wang, B. Zhang and J.-L. Li, ACS Catal., 2024, 14, 3181–3190 CrossRef CAS; (f) S. Li, C. Zhang, S. Wang, W. Yang, X. Fang, S. Fan, Q. Zhang, X.-X. Li and Y.-S. Feng, Org. Lett., 2024, 26, 1728–1733 CrossRef CAS; (g) N. Tanaka, J. L. Zhu, O. L. Valencia, C. R. Schull and K. A. Scheidt, J. Am. Chem. Soc., 2023, 145, 24486–24492 CAS; (h) S. Byun, M. U. Hwang, H. R. Wise, A. V. Bay, P. H. Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2023, 62, e202312829 CrossRef CAS PubMed; (i) A. V. Bay, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143–9148 CrossRef CAS; (j) A. V. Bay, K. P. Fitzpatrick, G. A. González-Montiel, A. O. Farah, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2021, 60, 17925–17931 CrossRef CAS; (k) J. Liu, X.-N. Xing, J.-H. Huang, L.-Q. Lu and W.-J. Xiao, Chem. Sci., 2020, 11, 10605–10613 RSC; (l) A. V. Bay, E. J. Farnam and K. A. Scheidt, J. Am. Chem. Soc., 2022, 144, 7030–7037 CrossRef CAS PubMed; (m) S.-C. Ren, X. Yang, B. Mondal, C. Mou, W. Tian, Z. Jin and Y. R. Chi, Nat. Commun., 2022, 13, 2846 CrossRef CAS PubMed; (n) Y. Sato, Y. Goto, K. Nakamura, Y. Miyamoto, Y. Sumida and H. Ohmiya, ACS Catal., 2021, 11, 12886–12892 CAS; (o) Q.-Y. Meng, N. Döben and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 19956–19960 CAS; (p) X. Yu, A. Maity and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202310288 CAS; (q) H. Huang, Q.-S. Dai, H.-J. Leng, Q.-Z. Li, S.-L. Yang, Y.-M. Tao, X. Zhang, T. Qi and J.-L. Li, Chem. Sci., 2022, 13, 2584–2590 RSC; (r) K. Liu and A. Studer, J. Am. Chem. Soc., 2021, 143, 4903–4909 CAS; (s) X. Yu, Q.-Y. Meng, C. G. Daniliuc and A. Studer, J. Am. Chem. Soc., 2022, 144, 7072–7079 CAS; (t) Q.-Y. Meng, L. Lezius and A. Studer, Nat. Commun., 2021, 12, 2068 CAS; (u) X. Wang, R. Yang, B. Zhu, Y. Liu, H. Song, J. Dong and Q. Wang, Nat. Commun., 2023, 14, 2951 CrossRef CAS PubMed.
- (a) Y. Goto, M. Sano, Y. Sumida and H. Ohmiya, Nat. Synth., 2023, 2, 1037–1045 CrossRef CAS; (b) J. Reimler, X. Y. Yu, N. Spreckelmeyer, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202303222 CrossRef CAS PubMed.
- K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed.
- L. Delfau, S. Nichilo, F. Molton, J. Broggi, E. Tomas-Mendivil and D. Martin, Angew. Chem., Int. Ed., 2021, 60, 26783–26789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06355d |
| This journal is © The Royal Society of Chemistry 2025 |