
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning high-order multiexciton properties of colloidal CdSe quantum dots via size and surface modification†
Raktim
Baruah‡
ab,
Krishan
Kumar§
ab,
Jan
Dellith
b and
Maria
Wächtler‡
 *a
*a
aChemistry Department and State Research Center OPTIMAS, RPTU Kaiserslautern-Landau, Erwin-Schrödinger-Str. 52, 67663 Kaiserslautern, Germany
bDepartment Functional Interfaces, Leibniz Institute of Photonic Technology Jena, Albert-Einstein-Straße 9, 07745 Jena, Germany
First published on 14th May 2025
Abstract
Excitonic properties of colloidal quantum dots are strongly dependent on size and surface properties. The same is predicted to be valid for the properties of multiexcitons. To realize applications exploiting the generation of multiexcitons in colloidal nanocrystals, a comprehensive understanding of size and surface influence, e.g., on the lifetimes of multiexciton species, is required. In this study, we employ intensity-dependent transient absorption spectroscopy to probe multiexcitons in colloidal CdSe quantum dots of four different sizes capped with long-chained organic (majorly trioctylphosphine oxide) or short inorganic (sulfide, S2−) ligands. To analyze the intensity-dependent transient absorption data, a global fit method based on Markov Chain Monte Carlo sampling was employed. Applying a simple Auger recombination model, the lifetimes and spectra of multiexciton species were analyzed. The spectra obtained for different multiexciton species exhibit both size and surface functionalization dependent features, allowing us to distinguish between different species. Independent of the surface modification, we find that the multiexciton lifetimes follow the volume scaling laws established earlier. Owing to the strong surface hole trapping induced by the S2− ligands, S2−-capped QDs show prolonged multiexciton lifetimes compared to the QDs capped with the native organic ligands with long alkyl chains. Deconvoluting contributions of bleach, stimulated emission, and photoinduced absorption in the species spectra enables us to determine multiexciton binding energies. We observe a decrease of binding energies with increasing size and a clear reduction in multiexciton binding energies for the S2−-capped QDs. The observed trends can be explained by changes in the overlap of electron and hole wave functions depending on the QDs’ diameter and the charge carrier localization, which can be induced by trapping in surface defect sites.
Introduction
Semiconductor nanocrystals (NCs) have garnered considerable attention over the past few decades as prospective materials for light-harvesting applications,1–3 light-emitting diodes (LEDs)4–6 and NC lasers,7,8 among other potential uses. Owing to their distinctive size-dependent optoelectronic properties caused by the quantum confinement effect, NCs stand out as one of the most promising materials for such applications.9,10 One of the highly interesting phenomena exhibited by semiconductor NCs is the formation of multiple excitons or multiexcitons, a coherent state involving multiple excitations. Multiexcitons are either formed upon excitation with radiation of high photon density photoexcitation11 or via a process called multiexciton generation (MEG),12 which takes place at very high energy excitation with a photon energy of at least twice the band gap of the NC. Multiexcitons in NCs are particularly interesting for applications such as optical amplification.8,13 The formation of multiexcitons increases the population inversion probability enhancing the optical gain in NC lasers.8 Furthermore, multiexcitons are considered to potentially increase performance in photovoltaics14 as well as photocatalysis15,16 involving multielectron mechanisms. For example, photovoltaic materials implementing MEG hold the potential to surpass the so-called Shockley–Queisser limit and enhance the conversion efficiency in photovoltaics.12 In all these applications, the multiexciton lifetime is crucial. In photocatalysis and photovoltaics, long multiexciton lifetimes are a precondition for efficient separation and utilization of charge carriers before recombination.17 However, in contrast to bulk semiconductors, multiexcitons in NCs have short lifetimes of only a few tens to hundreds of ps.11,15,18,19 This is one of the major challenges for applications involving multiexcitons. The short multiexciton lifetime in NCs is due to the strong spatial confinement of charge carriers, which leads to strong Coulombic interactions between electrons and holes, and lowers the barrier for momentum conservation.18,20 Multiexciton recombination majorly occurs via Auger recombination, which is a three-carrier process, where the electron–hole exchange energy is transferred to a third carrier by promoting it to higher energy levels.18,21 The probability of Auger recombination and hence the multiexciton lifetimes depend on the NC's structural parameters such as size, shape, and composition. Exemplarily, a linear scaling of multiexciton lifetime with NC volume is typically found.21 On the other hand, heterostructures having unique band alignment can reduce carrier wavefunction overlap that can result in prolonged multiexciton lifetimes.15,22 Similarly, the surface properties of NCs also impact multiexciton lifetimes.17 In the past few years, a substantial number of theoretical and experimental studies have been conducted to unfold the mechanism of Auger recombination of multiexcitons in semiconductor NCs.20,21,23–25A particular emphasis has been placed on the investigation of Cd-chalcogenide-based materials, with a specific focus on CdSe quantum dots (QDs).26–28 The established electronic structure of CdSe QDs enables a comprehensive understanding of multiexciton states.29–31 Colloidal CdSe QDs, which can be synthesized via the hot injection method with narrow size distribution and high crystallinity,32 contain organic surface ligands (e.g., phosphonic acids, carboxylic acids, and amines with long aliphatic chains) that provide colloidal stability and prevent the QDs from aggregation and degradation by lowering the surface energy.33 With the help of surface modification, the parent ligands can be replaced with surface ligands containing different functional groups impacting the dispersibility of QDs to a wide range of solvents.34,35 Additionally, the type of surface ligand can also alter the optoelectronic properties of QDs by modifying the electronic structure. For example, for thiol-capped CdSe QDs, strong photoluminescence quenching is observed originating from surface hole localization at thiolate introduced trapping sites. In contrast, for surface ligands with different anchoring groups such as amines, the QD surface is passivated and photoluminescence can be increased.36 Besides organic surface ligands, inorganic single atom ligands, e.g., halides or sulfides (S2−), are extensively studied.37–39 S2−-capped QDs exhibit strong photoluminescence quenching caused by the enhanced hole trapping.38,40 The strong dependence of the monoexciton (X) processes, here reflected in the discussed changes in photoluminescence, on the QD surface properties raises the question on the impact of surface functionalization on multiexciton behaviour in these structures and whether the size of the QDs influences the sensitivity towards changes in surface properties. Multiexciton lifetimes in CdSe QDs have been shown to scale with the volume.21,41 Similar dependencies were observed in PbSe,21 HgTe,42 and InAs43 QDs and perovskite NCs,44,45 and hence this relation seems to be a universal trend. Trapping of charge carriers at surface trapping sites is discussed to decouple charge carriers from the QD core, significantly impacting multiexciton lifetimes.46 For example, Yan et al.47 demonstrated the effect of hole trapping via phenothiazine resulting in an increased biexciton (BX) lifetime of CdSe QDs. This implies that different surface functionalities could have a strong influence on the multiexciton properties. The surface volume ratio changes with the size of QDs and the decreasing charge carrier interaction extending multiexciton lifetimes provoke the question of whether the sensitivity of multiexciton properties to surface properties is dependent on the particle size.
The spectroscopic techniques employed for the investigation of multiexciton properties primarily encompass transient absorption23,48 (TA) spectroscopy and time-resolved photoluminescence49–51 (tr-PL) spectroscopy. In addition to the temporal evolution, TA spectra with complex contributions of state-filling and Stark effects to the spectral features offer a comprehensive representation of the electronic population and (multi)exciton states.20,52,53 Because of the Pauli exclusion principle, the filling of quantized energy levels leads to the bleach of the corresponding transition in the transient spectra.23,54 This effect enables to determine the relative carrier population by analysing the relative changes of bleach amplitudes with respect to the pump intensity. Additionally, local fields of the pump generated carriers cause energetic shifts in optical transitions as well as changes in oscillator strengths.20 These effects result in characteristic changes in the TA spectrum that are unique for multiexcitons of varying order. Utilizing the advantage of distinguishing multiexciton species with their unique spectral features, TA spectroscopy has the potential to detect high-order multiexcitons beyond BX in excitation intensity-dependent experiments. Typically, the intensity-dependent TA data are analysed at a single probe wavelength of the band-edge bleach.19 The fast-decaying components of the band-edge bleach appearing at high pump intensities are attributed to multiexciton annihilation due to Auger recombination.19 This method works well for the evaluation of BX in CdSe QDs. With higher-order multiexciton population at high pump intensities, the evaluation of single wavelength kinetics can be challenging. This is because of the relatively weak contribution of higher-order multiexcitons (higher than BX) to the signal at the band-edge as well as the presence of ultrafast trapping,55 hot carrier cooling,56etc., components occurring on a similar time range as the decay of higher-order multiexcitons that could be misleading in assigning multiexciton recombination decay components. The spectral features of different excitonic states tend to overlap to form a complex signature that needs careful analysis and can only be reliably disentangled by global analysis of a broad spectral range.26,57 Ashner et al.57 and Shulenberger et al.27 have determined the spectra and lifetimes of X and BX in CsPbBr3 NCs and CdSe QDs, respectively, using a Markov Chain Monte Carlo (MCMC) sampling method. Applying this technique, the detection of multiexcitons should not be limited to BX.58
In this work, we present a comparative investigation of multiexciton properties in dependence on the size and properties of the NC surface. To this end, we perform intensity-dependent TA spectroscopy on four different sizes of CdSe QDs, ranging from 2.6 nm to 4.8 nm diameters, and surface functionalization with a long-chain aliphatic organic ligand shell (majorly containing trioctylphosphine oxide, TOPO and phosphonic acids octadecylphosphonic acid) or short inorganic ligand (S2−). The TOPO- and S2−-capped QDs provide the two extremes of weak and strong surface trapping, respectively. To analyse the intensity-dependent TA data, we adopt the global fitting method based on MCMC sampling established by Ashner et al.57 to simultaneously determine the spectra and lifetime of multiexciton species. Analysing the spectral profiles of different multiexciton species, we also determine multiexciton binding energies using a multi-Gaussian fit method, which particularly complements the understanding of the influence of surface functionalization on multiexciton properties.
Experimental
Chemicals
Trioctylphosphine oxide (TOPO, 99%), trioctylphosphine (TOP, 97%), cadmium oxide (CdO, 99.99%), selenium (Se, 99.99%), sodium sulfide (Na2S, 98%), toluene (99.8% anhydrous), N-methylformamide (NMF, 99%), hexane (95% anhydrous), acetonitrile (MeCN, 99.8% anhydrous), acetone (99.5%) and methanol (MeOH, 99.8% anhydrous) were purchased from Sigma Aldrich and octadecylphosphonic acid (ODPA, 97%) was purchased from Carl Roth.Synthesis of colloidal CdSe QDs
CdSe QDs were synthesized following the well-established hot-injection method.59 Briefly, 60 mg of CdO, 0.28 g of ODPA and 3.0 g of TOPO were mixed in a 25 mL three-necked flask. The mixture was heated to 80 °C under an N2 atmosphere followed by evacuation until gas release stopped. Under nitrogen, the mixture was heated to 150 °C and evacuated for 1 h. After that, the reaction vessel was filled with an N2 atmosphere again and further heated up. The mixture became optically clear and colourless at around 300 °C, and at 320 °C, 1.5 g of TOP was injected into the mixture. When the temperature reached 380 °C, a solution of TOP:Se (0.058 g Se dissolved in 0.36 g TOP) was injected and the reaction mixture was immediately cooled by removing the heating mantel, followed by additional cooling with cold water. At 60 °C, 10 mL of toluene were injected into the mixture. The QDs were precipitated by adding 10 mL of methanol to the reaction mixture and centrifugation at 5300 rpm. The precipitate was redispersed in toluene and the same step was repeated three more times for purification. Finally, the QDs were dispersed in 10 mL of toluene and stored in a glovebox. Following this protocol resulted in QDs with a 2.6 nm diameter. To synthesize 3.4 nm QDs, TOP:Se was injected at 370 °C and the reaction mixture was cooled in a similar manner as described for the 2.6 nm QD synthesis. 4 nm and 4.8 nm QDs were obtained by injecting TOP:Se at 370 °C. However, instead of immediate cooling, the reaction mixture was held at 370 °C for 3 min (for 4 nm) and 5 min (for 4.8 nm).The synthesized QDs contain a mixture of ligands such as TOPO, ODPA, and TOP, which are denoted as TOPO-capped QDs in the subsequent discussion.
Ligand exchange
The original ligands were exchanged with S2− ligands using a biphasic method.37 1 mL of the QD solution was precipitated with 1 mL of MeOH followed by centrifugation at 5300 rpm for 10 min. The supernatant was discarded, and the precipitate was redispersed in 1 mL of hexane. 1 mL of Na2S (1 M, NMF) solution was added to the QD solution in hexane and two separate phases of NMF (transparent) and hexane (coloured) were observed. After 2 hours of stirring, the QDs were transferred to the NMF phase, indicating a successful ligand exchange. Then, the clear hexane phase was removed. Furthermore, 2 mL of hexane was added to the QD solution, which was again removed after 5 minutes of stirring. Then, 2 mL MeCN was added to precipitate the QDs in NMF, followed by centrifugation at 5300 rpm for 10 min. Finally, the QDs were dispersed in 1 mL NMF. The QDs obtained after the ligand exchange are denoted as S2−-capped QDs in the subsequent discussion.UV-vis absorption spectroscopy
Steady-state UV-vis absorption spectra were collected using a Jasco V-600 spectrophotometer in the wavelength range from 200 nm to 800 nm. 1 cm quartz cuvettes were used for QD solutions (in toluene for TOPO-capped QDs and NMF for S2−-capped QDs).Photoluminescence spectroscopy
Photoluminescence spectra of QD solutions, prepared in 1 cm quartz cuvettes, were recorded using an FLS 980 Fluorometer from Edinburgh Instruments with a 400 nm excitation wavelength.Transmission electron microscopy (TEM)
TEM images were collected using a Hitachi HT7820 instrument with an acceleration voltage of 100 kV. For this, a drop of QD solution was drop cast on carbon film-supported copper grids followed by vacuum drying.Fourier transform infrared spectroscopy (FT-IR)
FTIR spectra were collected using a Bruker Tensor 27 spectrometer in attenuated total reflection (ATR) mode. Samples were prepared by dropping colloidal QD solutions on CaF2 substrates.Transient absorption spectroscopy (TA)
TA spectroscopy was performed using a home-built setup. The output of a 1 kHz Ti:Sapphire laser (Astrella, Coherent, USA) with a pulse duration of ∼100 fs at an 800 nm central wavelength was split into two parts, one to generate a 400 nm pump pulse through an optical parametric amplifier (TOPAS, Light Conversion, Lithuania) and the other to focus the beam into a rotating CaF2 crystal to generate a spectrally broad supercontinuum probe pulse. The probe pulses travel through an optical delay line with a delay of maximum 2 ns and were then split into a probe and a reference beam. Pump and probe beams were spatially and temporally overlapped at the sample position. The probe and reference pulses were spectrally resolved with a Czerny–Turner spectrograph and the spectra were detected with a CCD detector. The relative polarization of the pump and probe was set to the magic angle, 54.7°. The repetition rate of the pump pulse was reduced to 500 Hz using a mechanical chopper and the consecutive probe pulses were used to calculate the differential signals. The pump power was varied with a rotating neutral density filter wheel and measured using a power meter. Five power points between 15 μW and 1500 μW were recorded for each sample. The Gaussian profile of the pump beam was recorded using a beam profiler to estimate the area to determine the power density (intensity). The QD solutions were measured in 1 mm inert quartz cuvettes (TOPO-capped QDs in toluene and S2−-capped QDs in NMF). The chirp correction of the TA data was done by using a Python-based package, KiMoPack.60 The chirp-corrected data were then cut to the window of 2 ps to 1900 ps and 430 nm to 700 nm to perform the MCMC fit using a MATLAB package.57Results and discussion
TOPO-capped CdSe QDs of four different sizes were synthesized via hot injection, as described in the experimental section in detail. The QD sizes were estimated from the lowest energy band edge transition in the steady-state absorption spectra using an empirical formula described by Yu et al.61 and further validated by TEM image analysis (see the ESI,† Fig. S1 and Table S1). The four QDs are denoted according to the diameters determined from the absorption spectra, as 2.6 nm, 3.4 nm, 4.0 nm, and 4.8 nm QDs. The steady-state absorption spectra exhibit the characteristic distinct features of the three lowest energy 1S3/2–1Se (1S), 2S3/2–1Se (2S), and 1P3/2–1Pe (1P) excitonic transitions (Fig. 1).29 With increasing the QD size, the 1S peak position is redshifted as a consequence of the quantum confinement effect.62 To prepare the respective S2−-capped QDs, a biphasic ligand exchange39 protocol was adapted. The FT-IR spectra confirm efficient ligand exchange with disappearing vibrational signatures of the original ligand sphere (Fig. S2, ESI†). The steady-state absorption spectra of the four QDs show no significant changes upon ligand exchange (Fig. 1a), indicating that size distributions are unchanged. In contrast, strong changes are observed in the photoluminescence spectra (Fig. S3, ESI†). A strong quenching of the band edge photoluminescence and enhancement of the trap state photoluminescence occurs for S2−-capped QDs (Fig. S3, ESI†), originating from hole trapping in surface sites introduced by the S2− ligands.38–40 | ||
| Fig. 1 (a) Steady-state absorption spectra of TOPO-capped (solid lines) and S2−-capped (dashed lines) QDs of four different sizes. (b) Schematic of 1S, 2S and 1P electronic transitions from the valence band levels to conduction band levels of CdSe QDs. (c) Steady-state absorption spectrum (top panel) of 2.6 nm TOPO-capped QDs with marked 1S, 2S and 1P transitions, and the corresponding bleach features of these transitions marked in the TA spectrum collected at 2 ps delay (bottom panel) as B1, B2, and B3. A1 and A3 are the photoinduced absorption signatures. | ||
To investigate exciton and multiexciton properties, intensity-dependent TA spectroscopy was performed with pump pulse intensities between 1.0 × 104 μW cm−2 and 1.0 × 106 μW cm−2 (400 nm central wavelength and 100 fs pulse duration). A comparison of the 2 ps TA spectrum collected at the lowest excitation intensity with the steady-state absorption spectrum for the TOPO-capped 2.6 nm QDs is shown in Fig. 1c. The comparison allows us to assign the characteristic bleach signals B1, B2, and B3, to the 1S, 2S, and 1P transitions, respectively (Fig. 1c). These transitions occur in the other three QDs in the respective different spectral positions according to the size-dependent shift of the spectral signatures (Fig. S4, ESI†). The intensity-dependent TA spectra of the 2.6 nm TOPO- and S2−-capped QDs at a delay of 2 ps show a gradual increase of B1, B2, and B3 amplitudes with the excitation intensity (Fig. 2c, g and Fig. S5, ESI†). Additionally, the 2 ps TA spectra of 2.6 nm TOPO-capped QDs exhibit pronounced photoinduced absorption (PIA) bands, A1 and A3, redshifted from the B1 and B3 features (Fig. 2c). With increasing the excitation intensity, the A1 and A3 features gradually disappear in the spectra at 2 ps. However, irrespective of the excitation intensity, these features reappear during signal decay after a delay of around 100 ps. In contrast, for the S2−-capped QDs the PIA features are missing. The intensity-dependent TA spectra of the 3.4 nm, 4.0 nm, and 4.8 nm TOPO- and S2−-capped QDs are shown in the ESI† (Fig. S6–S11). The 3.4 nm QDs exhibit similar features to the 2.6 nm QDs. In the case of the TOPO-capped 4.0 nm and 4.8 nm QDs, the B3 component dominates the spectrum over B1 and B2 at 2 ps at high intensities, whereas at lower intensities, B1 and B2 are more pronounced compared to B3 (Fig. S9 and S11, ESI†). The A1 and A3 features are similarly observed for the TOPO-capped 3.4 nm, 4.0 nm, and 4.8 nm QDs (Fig. S6–S11, ESI†), while the A1 features are again absent in the corresponding S2−-capped QDs regardless of the excitation intensity and delay time (between 2 ps and 1900 ps). Furthermore, the A3 features are weaker in comparison to the TOPO-capped QDs.
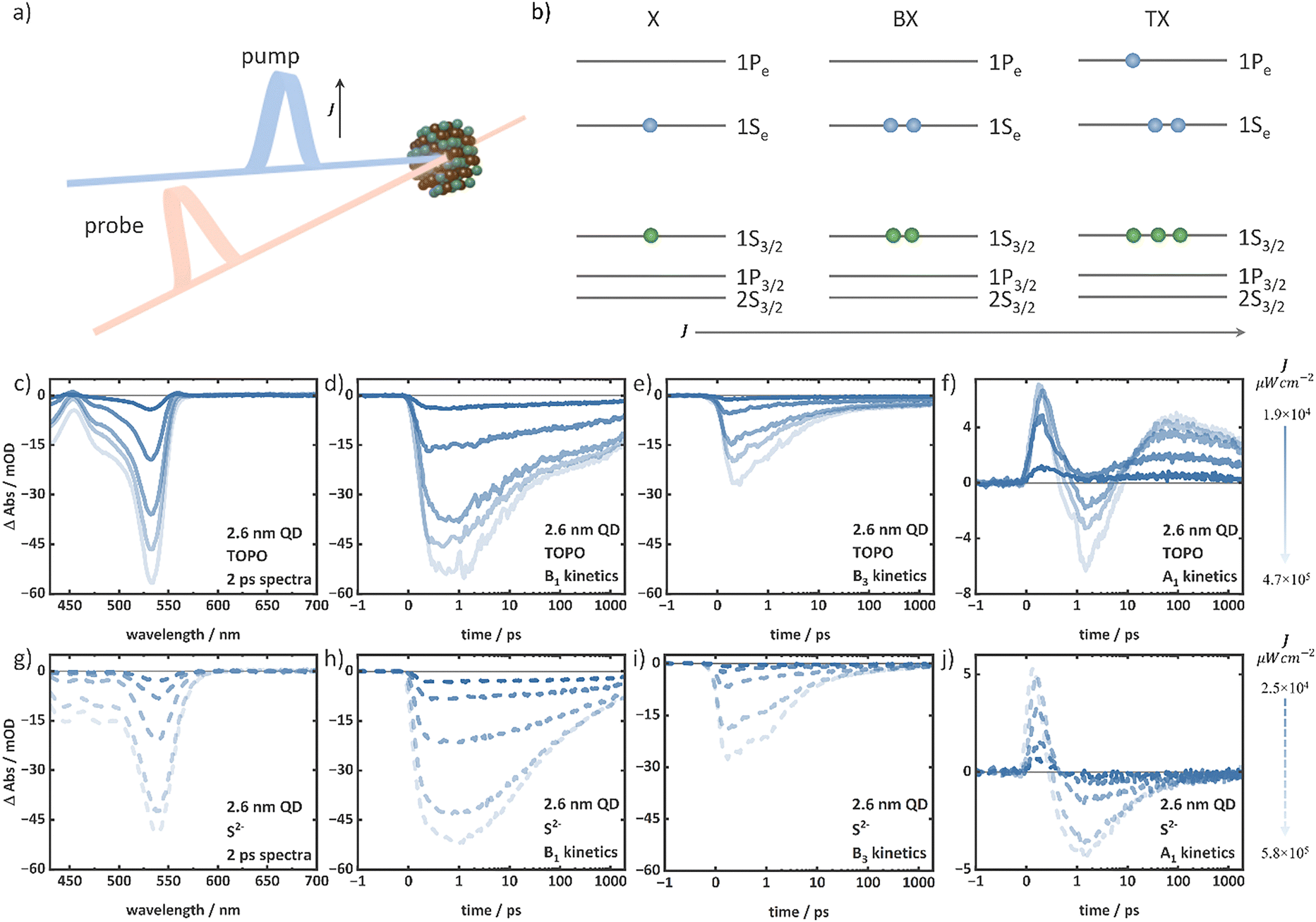 | ||
| Fig. 2 (a) Schematic of TA spectroscopy. J indicates the pump intensity. (b) Schematic of the energy level diagram of CdSe QDs representing increasing carrier population (exemplarily forming X, BX, and TX) with the intensity (J). The intensity-dependent TA data of the TOPO-capped 2.6 nm QD depicting the (c) spectra at 2 ps, (d) B1 kinetics, (e) B3 kinetics, and (f) A1 kinetics. The intensity-dependent TA data of the S2−-capped 2.6 nm QD depicting the (g) spectra at 2 ps, (h) B1 kinetics, (i) B3 kinetics, and (j) A1 kinetics. Increasing intensity is represented by the lines on the right side. | ||
An increase in the excitation intensity not only modifies the spectral shapes but also the temporal signal development. The decay of the B1 and B3 features of 2.6 nm QDs exhibit arising additional fast components between 2 ps and 500 ps with increasing excitation intensity (Fig. 2d, e, h and i). As the excitation intensity is increased, the A1 features of the TOPO- and S2−-capped 2.6 nm QDs disappear and the signal at this position at ∼1.5 ps gradually turns negative (Fig. 2f and j). In the case of the 2.6 nm TOPO-capped QDs, the A1 feature reappears with increasing delay time and shows similar decay independent of the excitation intensity. In the case of the S2−-capped 2.6 nm QD, however, the signal remains negative up to 1900 ps (Fig. 2f and j) at the typical A1 position. The B1, B3, and A1 kinetics of the TOPO- and S2−-capped 3.4 nm, 4.0 nm, and 4.8 nm QDs also exhibit similar behaviour (see the ESI,† Fig. S6–S11). At the latest recorded delay time of 1900 ps, the TA spectra at different intensities (Fig. S12, ESI†) appear identical when normalized to the B1 peak position.
The observed spectral features can be understood according to the previous reports in the literature.20,23,63 The primary contribution to the TA bleach signal in NCs such as CdSe QDs is the state-filling, i.e., the population of conduction band levels with electrons.48 The increasing bleach contributions with an increasing intensity indicate an increasing electron population in the conduction band levels. Fig. 2b illustrates the energy level diagram of CdSe QDs with increasing carrier population with the excitation intensity (exemplarily for X, BX, and triexciton (TX) formation). The 1Se and 1Pe levels in the conduction band exhibit different spin degeneracies of 2-fold and 6-fold, respectively.29 When the 1Se level is fully occupied with two electrons, the 1Pe level starts to fill. Therefore, the increasing B1 bleach amplitudes in all TOPO- and S2−-capped QDs (in the 2 ps TA spectra) indicate the presence of BX and the substantial influence in the B3 feature for 4 nm and 4.8 nm QDs (Fig. S8–S11, ESI†) suggest the presence of even higher-order multiexcitons than BX. The B3 feature is observable but weak at a low intensity regime (irrespective of the delay time and excitation intensity) when the population is primarily of the X and BX character, i.e., when only the 1Se level is populated (see Fig. 2c and g exemplarily for the 2 ps TA spectra of 2.6 nm TOPO- and S2−-capped QDs). In this case, the B3 bleach signals do not originate from the state-filling effect, but rather from the carrier-induced Stark effect.20 The local fields of the carriers in the X and BX states modulate the oscillator strengths of the electronic transition, which result in the bleaching of optical transitions, such as the B3 feature, without any state-filling effect.20,52 The A1 feature is attributed to the biexciton effect.20,26 At low excitation intensities, when the 1Se level is singly occupied, the probe pulse generates another exciton (due to the 2-fold degeneracy of the 1Se level) and the attractive interaction between the two excitons lowers the B1 transition energy, resulting in a red-shifted PIA (i.e., the A1). Therefore, the energetic difference between the A1 and B1 transitions is a measure of the biexciton binding energy. For the TOPO-capped QDs, the prominent A1 feature at 2 ps (see Fig. 2c exemplarily for the 2.6 nm TOPO-capped QD) at low excitation intensities and at 1900 ps irrespective of the excitation intensities indicates the presence of the X species that allows the biexciton effect to occur. Consequently, the reduction of A1 amplitudes with increasing excitation intensity signifies the formation of multiexcitons (atleast BX). Similar to the 2.6 nm TOPO-capped QDs, the 3.4 nm, 4.0 nm, and 4.8 nm TOPO-capped QDs also exhibit A1 features, but with different energetic shifts relative to the B1 bleach position indicating different biexciton binding energies due to the different QD sizes.64 On the other hand, the absent A1 features in the S2−-capped QDs regardless of the intensity and delay time (exemplarily see 2 ps and 1900 ps TA spectra of 2.6 nm S2−-capped QDs, Fig. 2g and Fig. S12b, ESI†) indicate smaller biexciton binding energies. A reduction in the biexciton binding energy may result in a significant degree of overlap between the A1 and B1 features, which could potentially lead to even complete absence of the A1 positive signal. Similar to the A1 feature, the interaction at the 1Pe level gives rise to A3 redshifted from the B3 feature. As the excitation intensity increases, the 1Pe level gradually fills due to the formation of higher-order multiexcitons compared to BX, as illustrated in Fig. 2b. This results in a decrease in the amplitude of the A3. At high intensity, A3 is also superimposed with B2 and B3, with substantial negative contributions.
Similar temporal developments of the discussed features with increasing excitation intensity have been previously reported in the literature.20,23,63 Excitation at 400 nm, which is above the 1Pe level, results in the formation of hot excitons that subsequently cool to the 1Se level via the 1Pe level in a timescale of hundreds of fs.23,65,66 This cooling process is reflected in the initial rise of the B1 and B3 signals. As the hot excitons begin to cool, the 1Pe levels are filled faster than the 1Se, resulting in an early bleach maximum at 0.5 ps of B3 (Fig. 2e and i), followed by the B1 at 1.5 ps (Fig. 2d and h). At low excitation intensities, the B3 decay in the sub-ps regime can be attributed to the cooling of hot electrons from the 1Pe to the 1Se level. The hot exciton formation is also reflected in the initial positive amplitude formation ∼0.2 ps for the A1 feature (Fig. 2f and j, exemplarily for 2.6 nm TOPO- and S2−-capped QDs).66 When the carriers are at higher energy levels, the Stark effect dominates over the state-filling effect in the TA signal. The hot carriers, which occupy high energy levels and are subjected to a strong local field, modulate the oscillator strength of the optical transitions, thereby giving rise to a pronounced A1 feature. The further decay of A1 up to ∼1.5 ps indicates the cooling process of the hot carriers (Fig. 2f and j, exemplarily for 2.6 nm TOPO- and S2−-capped QDs). However, the dynamics before 1.5 ps in the bleach features also contains contributions of, e.g., ultrafast carrier trapping.66 The band relaxation (i.e., the cooling of hot carriers to the lowest energy levels) is completed within ∼1.5 ps, as indicated by the B1 bleach maximum (Fig. 2d and h). On the time scales exceeding 2 ps, the signals undergo decay due to the recombination of band edge excitonic states. At high intensities, the presence of multiexcitons gives rise to additional components in the decay kinetics, namely those associated with multiexciton recombination. The additional fast-decaying components in B1 kinetics (Fig. 2d and h) with increasing excitation intensities can be attributed to Auger recombination on a timescale of up to a few hundred picoseconds until only X remains.20,23 Ultimately, X recombines via both radiative and non-radiative processes on timescales of up to tens of nanoseconds.67,68 The identical decay kinetics independent of the excitation intensity of B1 after approximately 500 ps as illustrated in Fig. 2d and h (exemplarily for 2.6 nm TOPO- and S2−-capped QDs) predominantly comprise contributions from X decay, which is identical independent of the initial excitation intensity. At low excitation intensities, the A1 feature shows a remaining positive amplitude after ∼1.5 ps following cooling, which is caused by the biexciton effect as previously discussed. However, at high excitation intensities, the A1 amplitude turns negative. This is due to the formation of multiexcitons that do not allow the biexciton effect to occur with the filled 1Se level. In the TOPO-capped QDs (Fig. 2f, exemplarily for the 2.6 nm TOPO-capped QD), the A1 reappears after different delay times with varying excitation intensities. This reappearance of the biexciton effect originates from the X species formed by multiexciton relaxation. According to the discussion of the spectral features, the A1 features in the S2−-capped QDs (Fig. 2j, exemplarily for the 2.6 nm S2−-capped QDs) remain absent and only negative amplitudes are observed in the region of the A1 feature irrespective of all excitation intensities.
The detailed comparative analysis of the B1, B3, and A1 spectral features and kinetics of the intensity-dependent TA data of the TOPO- and S2−-capped QDs clearly indicates the presence of contributions of high-order multiexcitons and significant differences in the dependence on surface functionalization. To evaluate the differences quantitatively, we employ a global analysis routine based on the MCMC-based target analysis established by Ashner et al.57 for the analysis of large data-sets of intensity-dependent TA, enabling us to determine multiple fit parameters with high precision. It provides means to assess the quality of the selected kinetic model and the uncertainty of the determined parameters, expressed through posterior probability distributions. The MCMC method utilizes random sampling guided by the target distribution, thereby ensuring that the samples are representative of the true posterior distribution. To ensure the precise estimation of the fit parameters, the process undergoes a chain of convergence toward the true distribution. Using this method, the cascade Auger recombination kinetic model of multiexciton recombination is solved.21 It is assumed that the Nth-order multiexciton relaxes stepwise to the respective lower order exciton.
| N → N − 1 → N − 2 → N − 3… → 2 → 1 | (1) |
Hence, to extract multiexciton lifetime and spectra, the development of concentrations of the contributing species is modelled by the following differential equations
 | (2) |
 | (3) |
Here, 〈N〉 is the average number of excitons generated by pump excitation, σ is an absorption-cross-section parameter, that can be converted to the cross-sectional area α (see the ESI†),27,58,69 and J is the excitation intensity of the pump. The TA data are treated as a sum of the spectra of the contributing species (SN(λ)) corresponding to each multiexciton species, N. In the modelling routine, the spectral shape of SN is assumed to be independent of the excitation intensity and only its contribution to the overall spectrum varies with the co-efficient AN(t, J), which is dependent on the excitation intensity and time, and represents the relative concentration of the species.
 | (4) |
The initial amplitude (AN(0, J)) of the contributing X and multiexciton species spectra (SN(λ)) corresponds to PN,0, which is the known initial concentration determined by eqn (3). In the MCMC fit, the intensity-dependent TA data are simultaneously sampled to determine SN, τN, and σ as fit parameters.
In our analysis, the timeframe below 2 ps was neglected due to the strong superposition with signatures from cooling and trapping. Hence, the contributions of higher-order multiexcitons with much shorter lifetimes, below 2 ps cannot be determined. The longest probe delay time point in our data is 1900 ps, which does not include the complete X recombination regime, but covers the complete expected timescale of multiexciton dynamics. The goodness-of-fit and accuracy of the kinetic model are reflected in the kinetics along with the fits and the contour plots of the fitted parameters (see the ESI†). Depending on the size of the QDs, kinetic models comprising TX or even tetraexciton (QX) species were used (Fig. 3a). It should be noted that, the spectrum of the highest-order exciton modelled most probably is not a pure spectrum and can contain contributions of even higher-order multiexcitons.
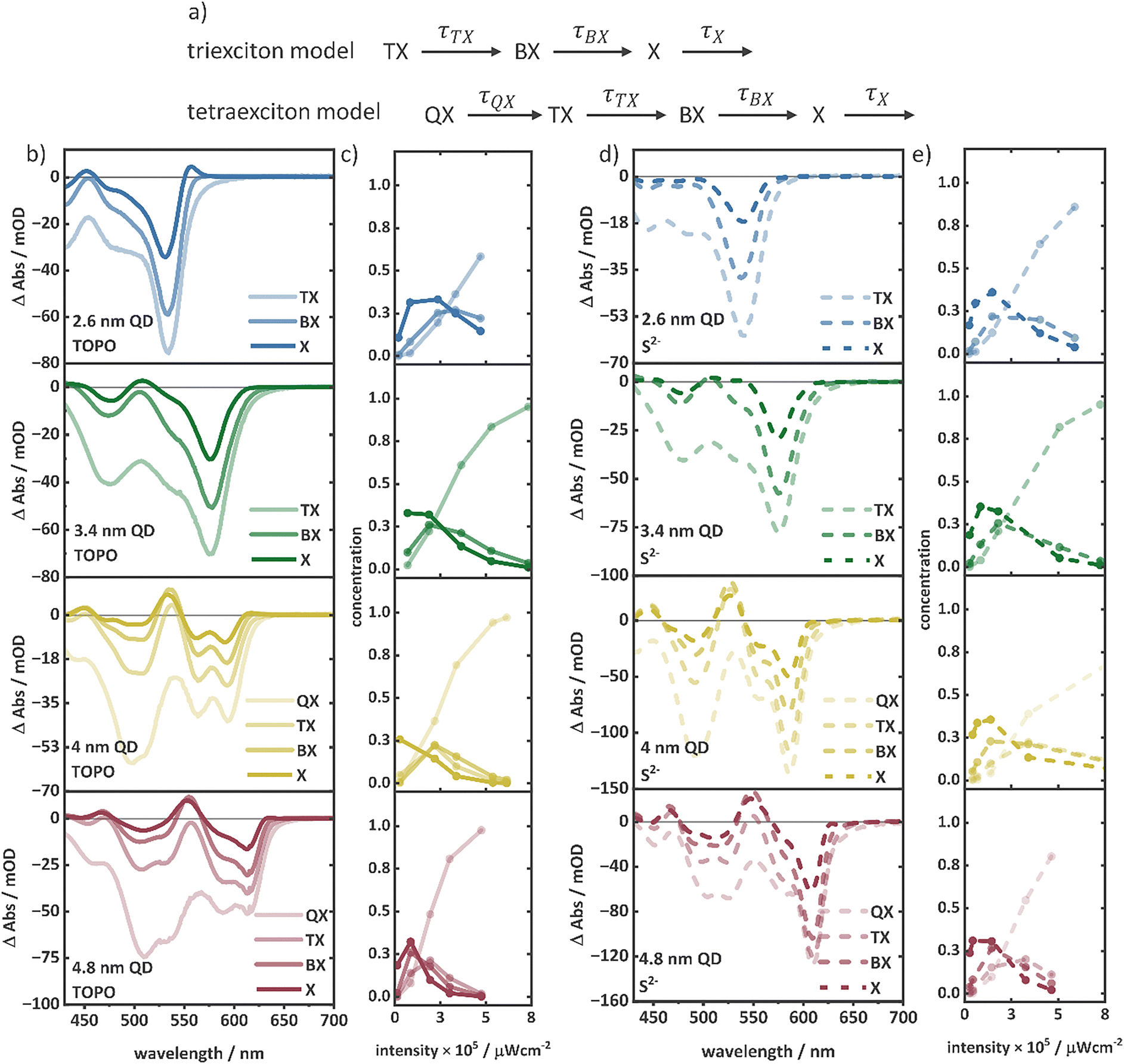 | ||
| Fig. 3 (a) Illustration of the TX and QX kinetic models used for the MCMC fit. (b) Multiexciton species spectra and the (c) initial concentration of multiexciton species over the pump intensities in TOPO-capped QDs of four different sizes. (d) Multiexciton species spectra and the (e) initial concentration of multiexciton species over the pump intensities in S2−-capped QDs of four different sizes. | ||
Fig. 3b and d illustrate the multiexciton species spectra obtained by this procedure along with the initial concentrations for each species for the series of TOPO- and S2−-capped QDs. As expected, at low excitation intensities, the X concentration dominates the transient spectra, while at high excitation intensities, multiexciton species are the dominating species determining the spectral features. For instance, the A1 feature observed at low excitation intensities in the TOPO-capped QDs (Fig. 3b) is consistent with the X spectrum. The BX, TX, and QX spectra do not exhibit the A1 feature, which is also similarly not observed in the TA spectra at high excitation intensities. The determined spectral shapes of the species are also in agreement with the proposed energy level structure, as shown in Fig. 2b. The increasing amplitudes of B1, B2, and B3 features from X to QX (Fig. 3a and c) are caused by the increasing population in the conduction band levels (1Se and 1Pe). The A1 feature is observed only in the X spectra of the TOPO-capped QDs. This is because only in X, the 1Se level is singly occupied, allowing the biexciton effect to occur. As the 1Se level is fully occupied from BX to higher multiexcitons, the biexciton effect cannot take place resulting in the absence of the A1 feature. High-order multiexcitons above BX show a negative signal in the same wavelength region as the A1 feature in X (e.g., at 560 nm of the 2.6 nm QD), which is attributed to the stimulated emission (SE).70 The A1 feature in the X spectra of S2−-capped QDs are absent (Fig. 3c). The A3 feature is on the other hand a consequence of the Stark effect induced shift of the B3 transition occurring at the 1Pe level. As the 1Pe level starts to fill from TX to QX, the B3 feature appears with significantly high amplitude resulting in decreased A3 amplitude due to the spectral superposition of B3 and A3. The multiexciton lifetimes estimated from the MCMC fit of the TOPO- and S2−-capped QDs are shown in Table 1. Multiexciton lifetime decreases with the increasing QD size. Due to the small volume in QDs, fast Auger recombination plays a dominant role because of the strong overlap of charge carrier wavefunctions.20 Therefore, multiexciton lifetimes are shorter in small QDs.21 In good agreement with previous reports, the multiexciton lifetimes (in particular τBX and τTX) of the series of TOPO- and S2−-capped QDs exhibit nearly cubic dependency with the QD radius.21 The lifetimes follow the relation, τMX ∝ Rn, where τMX is the multiexciton lifetime, and R is the QD radius (Fig. 4a). For the TOPO-capped QDs, n = 3.0 ± 0.6 for τBX and n = 2.9 ± 0.2 for τTX is determined. The S2−-capped QDs also exhibit similar dependency with n = 3.0 ± 0.7 for τBX and n = 3.8 ± 0.9 for τTX.
| τ QX/ps | τ TX/ps | τ BX/ps | α × 10−15 cm−2 | τ QX/ps | τ TX/ps | τ BX/ps | α × 10−15 cm−2 | |
|---|---|---|---|---|---|---|---|---|
| TOPO | S2− | |||||||
| 2.6 nm | — | 2.242.252.23 | 12.7212.7312.68 | 1.6011.6021.601 | — | 5.495.505.48 | 61.8661.9861.78 | 2.0422.0432.041 |
| 3.4 nm | — | 5.075.085.06 | 47.1247.1547.10 | 2.1492.1492.148 | — | 6.866.876.86 | 76.3376.3576.31 | 2.1762.1772.176 |
| 4.0 nm | 1.841.851.83 | 8.978.988.96 | 79.5279.6779.48 | 3.4683.4693.467 | 3.893.903.88 | 25.5325.5725.49 | 107.92108.04107.79 | 2.3072.3082.307 |
| 4.8 nm | 2.032.032.04 | 13.5813.5913.57 | 104.13104.24104.07 | 4.6094.6104.608 | 5.405.425.39 | 38.8138.9138.75 | 275.83276.59275.00 | 2.9712.9732.970 |
 | ||
| Fig. 4 (a) Dependence of TX and BX lifetimes on the radius of TOPO- and S2−-capped QDs. (b) Dependence of lifetimes on the exciton order of TOPO- and S2−-capped 4 nm and 4.8 nm QDs. (c) Illustration of TX, in a TOPO-capped QD with the carrier density concentrated in the CdSe core and in a S2−-capped QD with a trapped hole on the surface site. | ||
For 4.0 nm and 4.8 nm QDs (having atleast three multiexciton lifetimes, Fig. 4b), the lifetimes of multiexcitons in dependence on the order (N) can be described by τMX ∝ Nn. The TOPO-capped 4.0 nm QDs show n = 5.4 ± 0.03, and the 4.8 nm QDs show n = 5.5 ± 0.5. Similarly, for the S2−-capped 4.0 nm QDs, n = 4.8 ± 0.6, and for the 4.8 nm QDs, n = 5.6 ± 0.6. Clearly, with both the surface ligands, TOPO and S2−, the 4.0 nm and the 4.8 nm QD exhibit similar dependence of the multiexciton lifetime on the exciton order of around the 5th order. The dependency on the mulitexciton order describes the type of kinetic process involved in Auger recombination. In previous reports, several mechanisms have been discussed.21 A cubic dependency stands for a three-carrier model, where e.g., an electron and a hole annihilate via exciting a third charge carrier (electrons or holes). Theoretically, strongly confined charge carriers in materials like QDs exhibit cubic dependency.20,21 The quadratic model is observed in larger particles where Auger recombination occurs via exciton–exciton interactions.20 For higher-order multiexcitons, these two models become complicated, because the manifold degeneracy of higher energy levels creates different possibilities of carrier wave function mixing.21 For instance, when there is a TX state, the electron population is distributed over 1Se and 1Pe levels of the conduction band, and the hole population is distributed over 1S3/2 and 1P3/2 before ultrafast cooling and located in the 1S3/2 level after cooling. In such a situation, when the charge carriers are distributed over levels with manifold degeneracy, multiple possibilities of transitions in Auger recombination can occur influencing the dependency of the Auger lifetime in N. Taking this into account, multiexciton lifetimes are scaled following a statistical model as described by Klimov et al.21
However, according to our fit, the 5th-order kinetic model does not match with any of the previous reports. In this context, it has to be pointed out that the QDs possess significant surface carrier traps, which become more pronounced with S2− functionalization as indicated by the analysis of the photoluminescence spectra (Fig. S3, ESI†). The kinetic models discussed above, e.g., the statistical models, do not consider the population of surface trap states. It is possible that the presence of hole trapping sites further increases the number of recombination channels and consequently the Auger process shows high-order mechanisms. To verify the participation of surface trap states in Auger recombination, the statistical models need to be modified to take the additional recombination channels into account, which is challenging due to the undefined electronic structure of surface trap states.
A general trend can be observed comparing the multiexciton lifetimes of TOPO- and S2−-capped QDs. The multiexciton lifetimes of the S2−-capped QDs show a significant increase in comparison to those of the respective TOPO-capped QDs (Table 1). The increase in multiexciton lifetimes can be attributed to a decrease in the carrier wavefunction overlap induced by carrier localization in surface trap states, which suppresses the Auger process.22,47,71 The S2−-capped QDs, with higher surface hole trapping sites (compared to TOPO-capped QDs), therefore show prolonged multiexciton lifetimes. To further validate this effect of surface hole trapping on multiexciton lifetimes, determination of multiexciton binding energies can be useful. This is because the multiexciton binding energy and the extent of charge carrier wavefunction overlap influencing the Coulomb interactions between charge carriers are connected. Particularly, the change in spectral profiles for the X species, with a diminished A1 feature in S2−-capped QDs indicates a change in binding energies. Therefore, taking advantage of the MCMC determined multiexciton spectra, we employ a quantitative Gaussian fit analysis to deconvolute the ground state bleach (GSB), PIA, and SE contributions in the species spectra and to determine the shift between the B1 and A1 features exactly to derive the BX binding energies (ΔBX).26,72,73 Similarly, tri- (ΔTX), tetra- (ΔQX), and penta- (ΔPX) exciton binding energies can be estimated from the shifts between B3 and A3 in the BX, TX, and QX spectra, respectively.26,74 The Gaussian functions used to describe the species spectra are described in eqn (S3)–(S6) (ESI†). A detailed description of the fitting procedure is given in the ESI.† The obtained fit results are shown in Fig. S22–S26 (ESI†) and Tables S10–S14 (ESI†). The multiexciton binding energies of the TOPO-capped QDs increase with a decreasing size and increasing exciton order (Table 2).
| Δ BX/meV | Δ TX/meV | Δ QX/meV | Δ PX/meV | |
|---|---|---|---|---|
| TOPO | ||||
| 2.6 nm | 35.937.834.0 | 114.4125.292.3 | 203.6228.8191.6 | — |
| 3.4 nm | 24.226.921.6 | 75.096.166.1 | 111.0127.0102.3 | — |
| 4.0 nm | 22.723.422.1 | 66.275.055.0 | 109.6127.787.9 | 155.6208.891.7 |
| 4.8 nm | 18.119.916.2 | 49.658.325.9 | 70.891.947.7 | 102.1127.883.7 |
| S2− | ||||
| 4.8 nm | 12.918.57.4 | 37.039.432.2 | 58.073.257.2 | 64.670.651.4 |
To determine the dependency with the QD radius, we fit the function, ΔMX ∝ Rn, where ΔMX is the multiexciton binding energy, and R is the QD radius. For BX, TX, and QX, n = 1.2 ± 0.4, n = 1.4 ± 0.2, and n = 1.7 ± 0.1, respectively. Multiexciton binding energies show weaker dependence on the QD radius than the lifetimes. These observations match with previous reports.64 Reducing the QD size enhances the wavefunction overlap leading to an increased multiexciton binding energy as well as the Auger rate as discussed above. Similarly, with an increasing exciton order, the carrier density increases resulting in the increased multiexciton binding energy. Multiexciton binding energies in CdSe QDs are described as bound, i.e., with negative binding energy. Strandell et al.28 report negative multiexciton binding energy up to TX for CdSe QDs of different sizes. Utilizing the Gaussian fit method, we obtained binding energies up to pentaexciton (ΔPX). Here, this is to be clarified that the highest order multiexcitons obtained for each QD may contain contributions of higher order multiexciton states. The ΔPX for the 4.0 nm and 4.8 nm QDs, and ΔQX for the 2.6 nm and 3.4 nm QDs are determined from the highest detected multiexciton species, and thereby may contain a certain degree of error.
A similar treatment of the species spectra allowed us to determine the multiexciton binding energies of the 4.8 nm S2−-capped QD (Table 2). The multiexciton binding energies for the S2−-capped 4.8 nm QD are smaller in comparison to those for the TOPO-capped 4.8 nm QD. The reduction of the multiexciton binding energy can be explained by the reduced carrier wavefunction overlapping due to the surface hole trapping reducing the electron–hole attractive interaction.49 The surface hole trapping results in a relative increase of the electron–electron repulsion shifting the multiexciton states more towards an unbound state resulting in lowering the multiexciton binding energy.49,75 This results in a strong spectral overlap of the PIA feature and the respective bleach.73Fig. 5a and c show the individual Gaussians obtained for the B1 and A1 of the X spectra of TOPO- and S2−-capped 4.8 nm QDs. It can clearly be seen that the stronger overlap of B1 and A1 components leads to the nearly vanishing of the overall A1 feature to the red of the B1 bleach in the spectra of the S2−-capped QDs. With the weak PIA features in the species spectra of 2.6 nm, 3.4 nm and 4.8 nm QDs, reliable fits of eqn (S3)–(S6) (ESI†) were challenging. We assume, due to the smaller QD size with a higher carrier density, the electron–electron repulsion gets more and more dominating further reducing the binding energy and the spectral shift, or even resulting in an unbound multiexciton species with a positive binding energy. In this case, the A1 feature can be expected to blue shift from B1 resulting in a more complex superposition of multiple spectral features, e.g., with B2 and A3. Therefore, the Gaussian fit of the species spectra of S2−-capped QDs with eqn (S3)–(S6) (ESI†) might be misleading with overparameterization of the fit parameters and was not performed for the smaller S2−-capped QDs.
 | ||
| Fig. 5 The A1 and B1 peaks obtained from the multi-Gaussian fitting of the X spectra with eqn (S3) (ESI†) of 4.8 nm (a) TOPO-capped and (c) S2−-capped QDs. The figures demonstrate the extent of the A1 and B1 overlap, which determines the ΔBX. (b) Excitonic state diagram representing the optical transitions corresponding to A1 and B1 in the X species. (d) Radius dependency (ΔMX ∝ Rn) of ΔMX of BX, TX and QX obtained from the multi-Gaussian fitting of eqn (S3)–(S6) (ESI†). | ||
Conclusions
To summarize, multiexciton lifetimes and spectral shapes for four different sizes of colloidal CdSe QDs with two surface ligands (long chain organic capping or short inorganic strong hole trapping ligands) have been analysed by utilizing global fitting of the intensity-dependent TA spectra. The multiexciton lifetimes exhibit the ‘universal volume scaling’ independent of the surface functionalization. Upon introduction of the S2− ligand, the multiexciton lifetimes significantly increased. This is attributed to the reduction of the carrier wavefunction overlap due to strong hole trapping by S2− surface traps resulting in the suppression of the Auger process. A quantitative spectral evaluation was performed to determine multiexciton binding energies. A weak scaling with the volume in good agreement with the previous literature was observed. The reduction of the multiexciton binding energy in S2−-capped QDs indicates a relative decrease in electron–hole Coulomb attraction. This behaviour can be expected due to the hole localization in the surface trap states of S2−-capped QDs. Hence, this study exemplifies the potential of applying spectral analysis of excitation intensity-dependent TA-data to access details in multiexciton properties and advance the understanding of the impact of the structural change, here in this study changes in size and surface functionalization, to influence multiexciton dynamics, which lays the foundation for exploiting multiexciton effects in applications such as photocatalysis, photovoltaics, NC lasing, etc.Author contributions
Raktim Baruah: conceptualization, data curation, formal analysis, investigation, verification, and writing – original draft and review & editing. Krishan Kumar: writing – review & editing. Jan Dellith: data curation and formal analysis. Maria Wächtler: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review & editing.Data availability
The supporting data for this study are shown in the ESI.† The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Mathias Micheel for the valuable discussion. We acknowledge the German Research Foundation (DFG) for supporting this work under project number 39881677–CRC 1375 NOA, C04, project number 364549901–TRR234 CATALIGHT, TP B04 and project number 468735112 – KU 4220/1-1.References
- X. L. Xiang, L. X. Wang, J. J. Zhang, B. Cheng, J. G. Yu and W. Macyk, Adv. Photonics Res., 2022, 3, 2200065 CrossRef CAS.
- M. A. Holmes, T. K. Townsend and F. E. Osterloh, Chem. Commun., 2012, 48, 371–373 RSC.
- K. F. Wu and T. Q. Lian, Chem. Soc. Rev., 2016, 45, 3781–3810 RSC.
- V. Singh, Priyanka, P. V. More, E. Hemmer, Y. K. Mishra and P. K. Khanna, Mater. Adv., 2021, 2, 1204–1228 RSC.
- Y. Shu, X. Lin, H. Qin, Z. Hu, Y. Jin and X. Peng, Angew. Chem., Int. Ed., 2020, 59, 22312–22323 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulovic, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- Y. S. Park, J. Roh, B. T. Diroll, R. D. Schaller and V. I. Klimov, Nat. Rev. Mater., 2021, 6, 382–401 CrossRef CAS.
- N. Ahn, C. Livache, V. Pinchetti and V. I. Klimov, Chem. Rev., 2023, 123, 8251–8296 CrossRef CAS PubMed.
- R. Koole, E. Groeneveld, D. Vanmaekelbergh, A. Meijerink and C. de Mello Donegá, in Nanoparticles: Workhorses of Nanoscience, ed. C. de Mello Donegá, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 13–51 Search PubMed.
- M. G. Bawendi, M. L. Steigerwald and L. E. Brus, Annu. Rev. Phys. Chem., 1990, 41, 477–496 CrossRef CAS.
- V. I. Klimov, J. Phys. Chem. B, 2006, 110, 16827–16845 CrossRef CAS PubMed.
- H. Eshet, R. Baer, D. Neuhauser and E. Rabani, Nat. Commun., 2016, 7, 13178 CrossRef CAS PubMed.
- F. García-Santamaría, Y. F. Chen, J. Vela, R. D. Schaller, J. A. Hollingsworth and V. I. Klimov, Nano Lett., 2009, 9, 3482–3488 CrossRef PubMed.
- M. C. Beard, J. Phys. Chem. Lett., 2011, 2, 1282–1288 Search PubMed.
- H. M. Zhu, Y. Yang and T. Q. Lian, Acc. Chem. Res., 2013, 46, 1270–1279 Search PubMed.
- Y. Ben-Shahar, J. P. Philbin, F. Scotognella, L. Ganzer, G. Cerullo, E. Rabani and U. Banin, Nano Lett., 2018, 18, 5211–5216 CrossRef CAS PubMed.
- Y. W. Liu, D. A. Cullen and T. Q. Lian, J. Am. Chem. Soc., 2021, 143, 20264–20273 Search PubMed.
- C. Melnychuk and P. Guyot-Sionnest, Chem. Rev., 2021, 121, 2325–2372 CrossRef CAS PubMed.
- V. I. Klimov, A. A. Mikhailovsky, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Science, 2000, 287, 1011–1013 Search PubMed.
- V. I. Klimov, Annu. Rev. Phys. Chem., 2007, 58, 635–673 CrossRef CAS PubMed.
- V. I. Klimov, J. A. McGuire, R. D. Schaller and V. I. Rupasov, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195324 Search PubMed.
- D. G. Kong, Y. Y. Jia, Y. P. Ren, Z. X. Xie, K. F. Wu and T. Q. Lian, J. Phys. Chem. C, 2018, 122, 14091–14098 CrossRef CAS.
- V. I. Klimov, J. Phys. Chem. B, 2000, 104, 6112–6123 Search PubMed.
- J. P. Philbin and E. Rabani, Nano Lett., 2018, 18, 7889–7895 CrossRef CAS PubMed.
- R. Vaxenburg, A. Rodina, A. Shabaev, E. Lifshitz and A. L. Efros, Nano Lett., 2015, 15, 2092–2098 CrossRef CAS PubMed.
- T. Labrador and G. Dukovic, J. Phys. Chem. C, 2020, 124, 8439–8447 CrossRef CAS.
- K. E. Shulenberger, S. J. Sherman, M. R. Jilek, H. R. Keller, L. M. Pellows and G. Dukovic, J. Chem. Phys., 2024, 160, 014708 Search PubMed.
- D. P. Strandell, A. Ghosh, D. Zenatti, P. Nagpal and P. Kambhampati, J. Phys. Chem. Lett., 2023, 14, 6904–6911 Search PubMed.
- D. J. Norris and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16338–16346 CrossRef CAS PubMed.
- J. M. Pietryga, Y. S. Park, J. H. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Chem. Rev., 2016, 116, 10513–10622 CrossRef CAS PubMed.
- A. L. Efros and M. Rosen, Annu. Rev. Mater. Res., 2000, 30, 475–521 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- M. Liu, G. Tang, Y. Liu and F. L. Jiang, J. Phys. Chem. Lett., 2024, 15, 1975–1984 CrossRef CAS PubMed.
- M. Green, J. Mater. Chem., 2010, 20, 5797–5809 RSC.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS PubMed.
- K. J. Schnitzenbaumer and G. Dukovic, J. Phys. Chem. C, 2014, 118, 28170–28178 CrossRef CAS.
- K. J. Schnitzenbaumer, T. Labrador and G. Dukovic, J. Phys. Chem. C, 2015, 119, 13314–13324 CrossRef.
- A. Nag, M. V. Kovalenko, J. S. Lee, W. Y. Liu, B. Spokoyny and D. V. Talapin, J. Am. Chem. Soc., 2011, 133, 10612–10620 CrossRef CAS PubMed.
- A. Nag, D. S. Chung, D. S. Dolzhnikov, N. M. Dimitrijevic, S. Chattopadhyay, T. Shibata and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 13604–13615 CrossRef CAS PubMed.
- A. Pandey and P. Guyot-Sionnest, J. Chem. Phys., 2007, 127, 111104 CrossRef PubMed.
- C. Melnychuk and P. Guyot-Sionnest, J. Phys. Chem. Lett., 2018, 9, 2208–2211 CrossRef CAS PubMed.
- I. Robel, R. Gresback, U. Kortshagen, R. D. Schaller and V. I. Klimov, Phys. Rev. Lett., 2009, 102, 177404 CrossRef PubMed.
- Y. L. Li, X. Luo, T. Ding, X. Lu and K. F. Wu, Angew. Chem., Int. Ed., 2020, 59, 14292–14295 CrossRef CAS PubMed.
- J. A. Castañeda, G. Nagamine, E. Yassitepe, L. G. Bonato, O. Voznyy, S. Hoogland, A. F. Nogueira, E. H. Sargent, C. H. B. Cruz and L. A. Padilha, ACS Nano, 2016, 10, 8603–8609 CrossRef PubMed.
- M. Califano, J. Phys. Chem. Lett., 2018, 9, 2098–2104 CrossRef CAS PubMed.
- C. Yan, D. Weinberg, D. Jasrasaria, M. A. Kolaczkowski, Z. J. Liu, J. P. Philbin, A. D. Balan, Y. Liu, A. M. Schwartzberg, E. Rabani and A. P. Alivisatos, ACS Nano, 2021, 15, 2281–2291 CrossRef CAS PubMed.
- V. I. Klimov, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 13740–13749 CrossRef CAS.
- M. Achermann, J. A. Hollingsworth and V. I. Klimov, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 245302 CrossRef.
- H. Utzat, K. E. Shulenberger, O. B. Achorn, M. Nasilowski, T. S. Sinclair and M. G. Bawendi, Nano Lett., 2017, 17, 6838–6846 CrossRef CAS PubMed.
- K. E. Shulenberger, T. S. Bischof, J. R. Caram, H. Utzat, I. Coropceanu, L. Nienhaus and M. G. Bawendi, Nano Lett., 2018, 18, 5153–5158 CrossRef CAS PubMed.
- V. Klimov, S. Hunsche and H. Kurz, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 8110–8113 CrossRef CAS PubMed.
- V. Klimov, S. Hunsche and H. Kurz, Phys. Status Solidi, 1995, 188, 259–267 CrossRef CAS.
- R. D. Schaller and V. I. Klimov, Phys. Rev. Lett., 2006, 96, 097402 CrossRef PubMed.
- P. Tyagi and P. Kambhampati, J. Chem. Phys., 2011, 134, 094706 CrossRef PubMed.
- E. Hendry, M. Koeberg, F. Wang, H. Zhang, C. D. Donegá, D. Vanmaekelbergh and M. Bonn, Phys. Rev. Lett., 2006, 96, 057408 CrossRef CAS PubMed.
- M. N. Ashner, S. W. Winslow, J. W. Swan and W. A. Tisdale, J. Phys. Chem. A, 2019, 123, 3893–3902 CrossRef CAS PubMed.
- K. Kumar and M. Wächtler, Nanoscale, 2025, 17, 7141–7152 RSC.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- C. Müller, T. Pascher, A. Eriksson, P. Chabera and J. Uhlig, J. Phys. Chem. A, 2022, 126, 4087–4099 CrossRef PubMed.
- L. Q. W. William Yu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef.
- A. I. Ekimov, A. L. Efros and A. A. Onushchenko, Solid State Commun., 1985, 56, 921–924 CrossRef CAS.
- D. P. Morgan and D. F. Kelley, J. Phys. Chem. C, 2020, 124, 8448–8455 CrossRef CAS.
- S. L. Sewall, R. R. Cooney, K. E. H. Anderson, E. A. Dias, D. M. Sagar and P. Kambhampati, J. Chem. Phys., 2008, 129, 084701 CrossRef PubMed.
- A. J. Nozik, Annu. Rev. Phys. Chem., 2001, 52, 193–231 CrossRef CAS PubMed.
- P. Kambhampati, J. Phys. Chem. C, 2011, 115, 22089–22109 CrossRef CAS.
- K. E. Knowles, E. A. McArthur and E. A. Weiss, ACS Nano, 2011, 5, 2026–2035 CrossRef CAS PubMed.
- E. A. McArthur, A. J. Morris-Cohen, K. E. Knowles and E. A. Weiss, J. Phys. Chem. B, 2010, 114, 14514–14520 CrossRef CAS PubMed.
- M. N. Ashner, S. W. Winslow, J. W. Swan and W. A. Tisdale, J. Phys. Chem. A, 2019, 123, 3893–3902 CrossRef CAS PubMed.
- S. L. Sewall, R. R. Cooney, E. A. Dias, P. Tyagi and P. Kambhampati, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235304 CrossRef.
- W. X. Yang, Y. W. Yang, A. L. Kaledin, S. He, T. Jin, J. R. McBride and T. Q. Lian, Chem. Sci., 2020, 11, 5779–5789 RSC.
- C. Zhang, T. N. Do, X. W. Ong, Y. T. Chan and H. S. Tan, Chem. Phys., 2016, 481, 157–164 CrossRef CAS.
- S. Palato, H. Seiler, H. Baker, C. Sonnichsen, P. Brosseau and P. Kambhampati, J. Chem. Phys., 2020, 152, 104710 CrossRef CAS PubMed.
- Y. Kobayashi and N. Tamai, J. Phys. Chem. C, 2010, 114, 17550–17556 CrossRef CAS.
- M. E. Reimer, D. Dalacu, P. J. Poole and R. L. Williams, J. Phys.: Conf. Ser., 2010, 210, 012019 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00608b |
| ‡ Current address: Institute of Physical Chemistry and Kiel Nano, Surface and Interface Science (KiNSIS), Kiel University, Max-Eyth-Straße 1, 24118, Kiel, Germany. E-mail: waechtler@phc.uni-kiel.de |
| § Institut für Physik, Carl von Ossietzky Universität Oldenburg, 26129 Oldenburg, Germany. |
| This journal is © the Owner Societies 2025 |