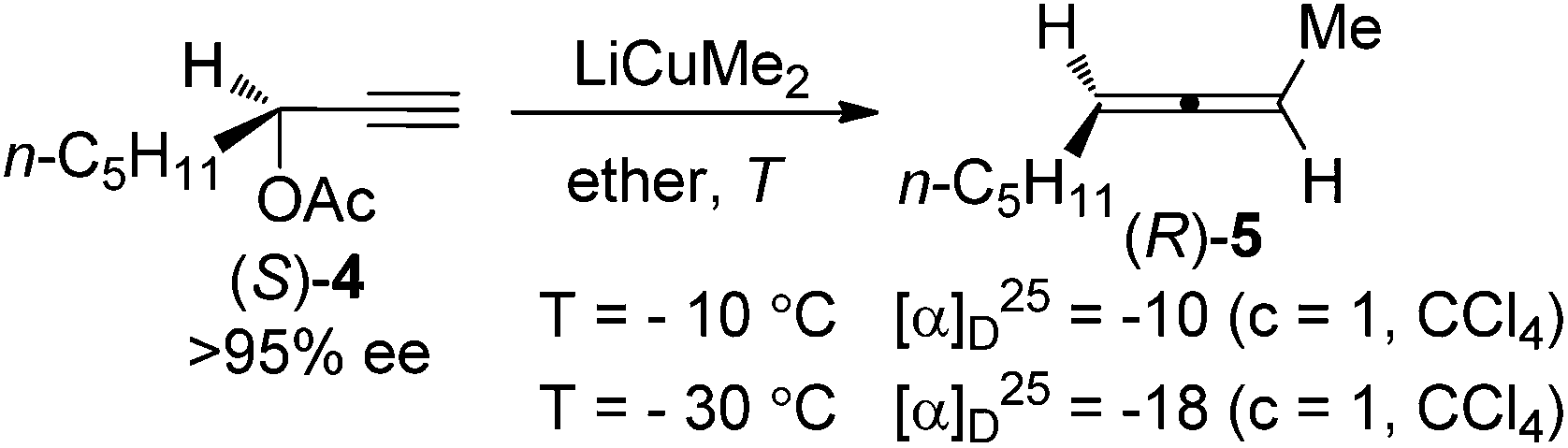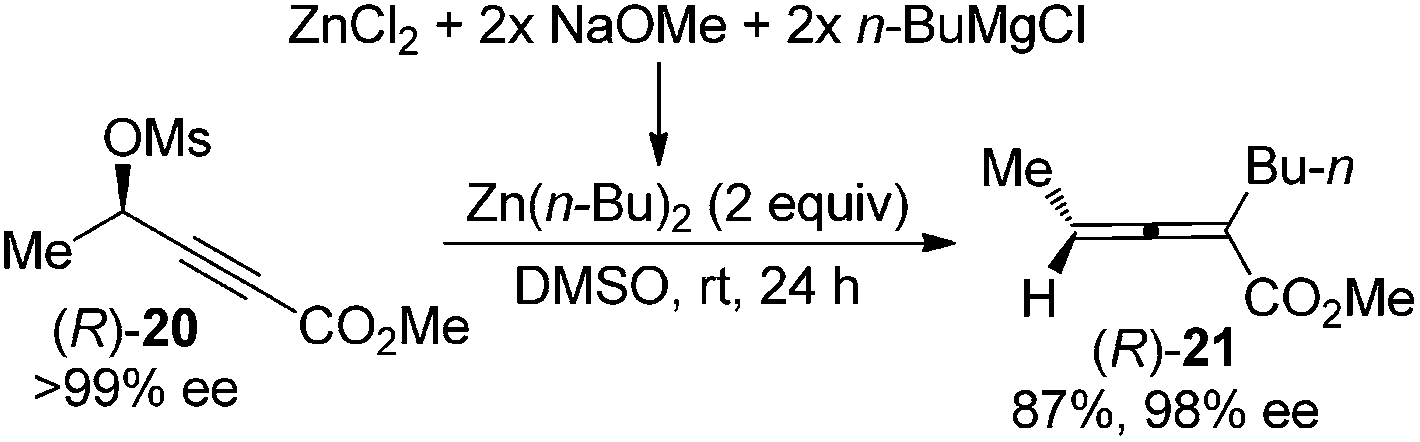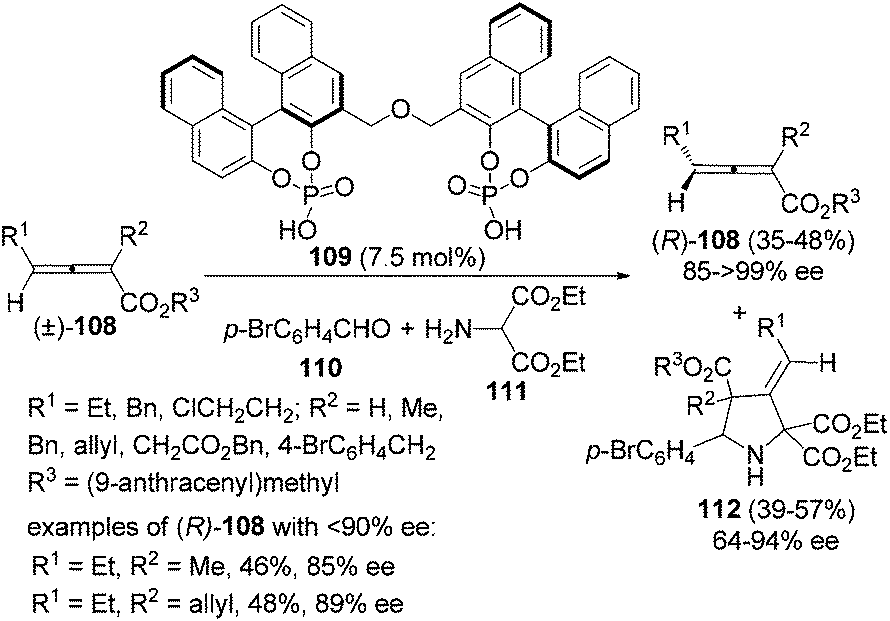DOI:
10.1039/C4QO00208C
(Review Article)
Org. Chem. Front., 2014,
1, 1210-1224
Conquering three-carbon axial chirality of allenes†
Received
25th July 2014
, Accepted 5th September 2014
First published on 10th September 2014
Abstract
While one-carbon central chirality of organic molecules has been recognized and extensively studied for more than a century, far less attention has been paid to three-carbon axial chirality of allenes, although they exist in nature with interesting biological activity and have been demonstrated with great synthetic potentials. However, remarkable progress has been made in this field in recent years, giving rise to axially chiral allenes with a wide range of functionalities with practical enantioselectivity. This review provides a concise account of enantioselective syntheses of axially chiral allenes with a selection of published protocols.
 Juntao Ye | Juntao Ye was born in 1986 in Hubei, China. He received his B.S. degree from Huazhong University of Science and Technology (HUST) in 2008. He was then admitted to Shanghai Institute of Organic Chemistry (SIOC) and obtained his Ph.D. degree in 2013 under the supervision of Prof. Shengming Ma. His doctoral research was focused on the synthesis and cyclization reactions of allenes. Currently, he is a postdoctoral fellow in Prof. Mark Lautens’ group at the University of Toronto. |
 Shengming Ma | Shengming Ma was born in 1965 in Zhejiang, China. He received his Ph.D. from Shanghai Institute of Organic Chemistry (SIOC) and became an assistant professor there in 1991. After postdoctoral research at the ETH with Prof. Venanzi and Purdue University with Prof. Negishi, he returned to SIOC in 1997. From February 2003 to September 2007, he was jointly appointed by SIOC and Zhejiang University (ZJU). In October 2007, he moved to East China Normal University to help build the research program in organic chemistry. After finishing his duty there, he moved his research activity in ECNU to Fudan University in September 2014. Currently he is also a research professor at SIOC and Qiu Shi Adjunct Professor at ZJU and CUHK. He received the Mr & Mrs Sun Chan Memorial Award in Organic Chemistry (2004), OMCOS Springer Award (2005), National Award for Research in Natural Science in China (Second-Class, 2006), and Natural Science Award of Shanghai (First-Class, 2010). |
1. Introduction
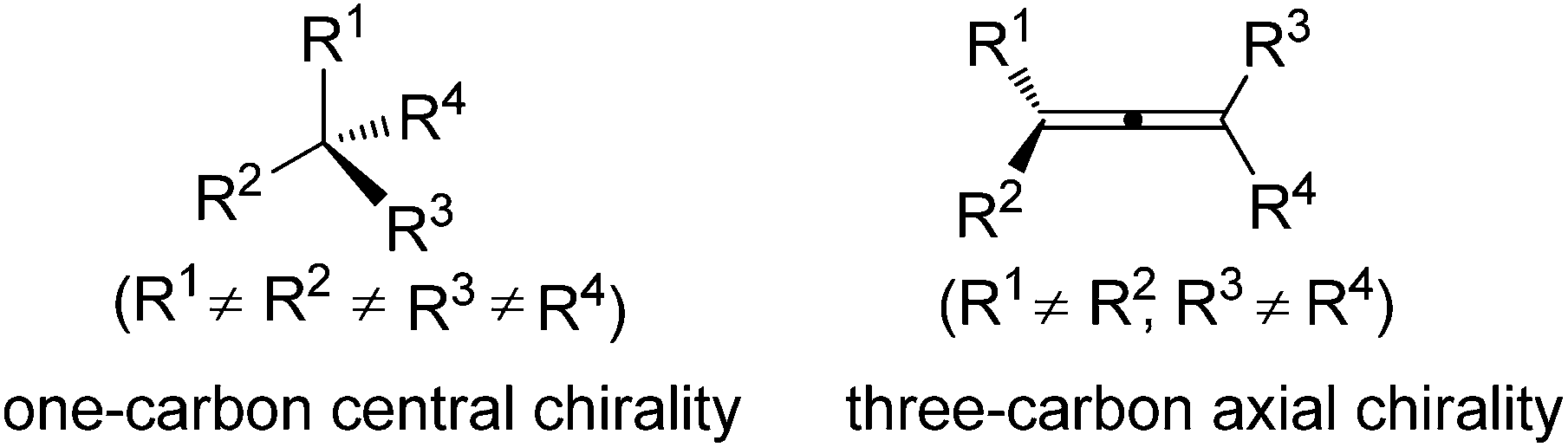
Chirality is an interesting phenomenon commonly existing in nature. A typical example is the hands of human beings: the left hand is a non-superimposable mirror image of the right hand. Nature also endows organic molecules with various types of chiralities such as central, axial, planar, and helical chiralities,1 among which one-carbon central chirality is the most frequently encountered one closely connected to biological activity and thus has been extensively studied ever since Jacobus van't Hoff and Joseph Le Bel's prediction in as early as 1874 (Fig. 1).2 Another type of chirality that has recently attracted broad interest of chemists is the axial chirality of allenes owing to the rapid development of allene chemistry in the last few decades as well as their occurrence in nature.3 However, construction of the axial chirality of allenes proved to be much more challenging,4 simply due to the fact that such an axial chirality spreads over a linear three-carbon atom unit.
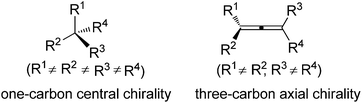 |
| | Fig. 1 One-carbon central chirality vs. three-carbon axial chirality. | |
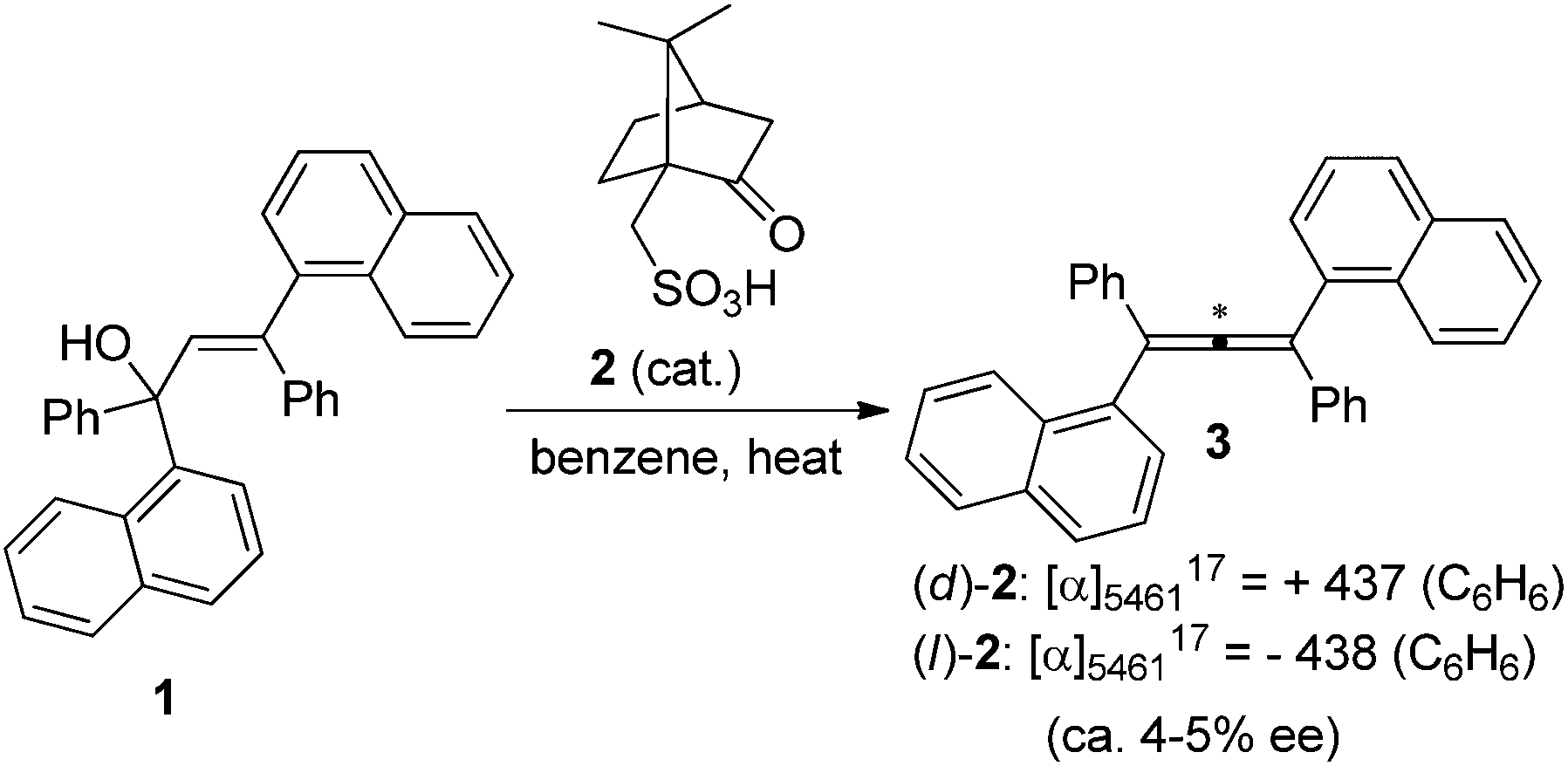
Traditionally, axially chiral allenes are prepared via central-to-axial chirality transfer of enantioenriched propargylic alcohols or their derivatives, kinetic resolution of racemic allenes, or olefination of ketenes with chiral ylide reagents.4 However, all these methods require a stoichiometric amount of chiral sources. Loss of enantiopurity during the chirality transfer and lack of generality are the major problems. Accordingly, more attention has been paid to catalytic asymmetric synthesis of axially chiral allenes, which in fact has a long history as the first synthesized axially chiral allene was prepared via asymmetric catalytic dehydration by Maitland and Mills in 1935: in the presence of a catalytic amount of (D)- or (L)-camphorsulphonic acid 2, allylic alcohol 1 was dehydrated to the corresponding optically active allenes 3 (Scheme 1),5a thereby experimentally verifying van't Hoff's early prediction6 that unsymmetrically substituted allenes would exhibit enantiomorphism for the first time. However, the vast potential of this early finding was not recognized at that time, probably due to the low enantioselectivity observed.5b
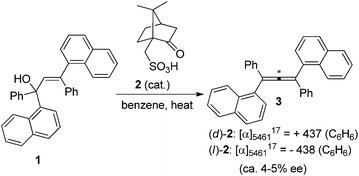 |
| | Scheme 1 Camphorsulphonic acid-catalyzed dehydration to axially chiral allene 3. | |
Fortunately, with the rapid development of new protocols to form racemic allenes as well as the emergence of numerous types of easily available commercialized chiral ligand skeletons, chemists around the world have witnessed remarkable progress in this area over the last few years by identifying new approaches and/or new chiral ligands from the well-established chiral ligand skeletons. In this review, we present a critical (rather than exhaustive) account of enantioselective synthesis of axially chiral allenes, with a focus on the most privileged ones.4
2. From propargylic alcohols or their derivatives
2.1 From propargylic alcohol derivatives
2.1.1 Copper-catalyzed or -mediated transformations.
Since the pioneering work of Crabbé and co-workers in 1968,7 SN2′ substitution of propargylic derivatives such as acetates, carbonates, sulfinates, sulfonates, phosphates, halides, and ethers with organocopper or cuprate reagents has become one of the most popular methods for the synthesis of allenes.4 Crabbé et al. also observed that central chirality of the enantioenriched propargylic acetate (S)-4 could be transferred to axial chirality of the allene product; however, the efficiency of chirality transfer was not clear due to the fact that the enantiomeric excess (ee) of the allene (R)-5 was not determined (Scheme 2).8a
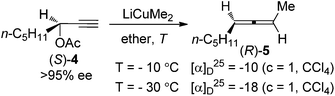 |
| | Scheme 2 Central-to-axial chirality transfer of enantioenriched propargylic acetate in the presence of LiCuMe2. | |
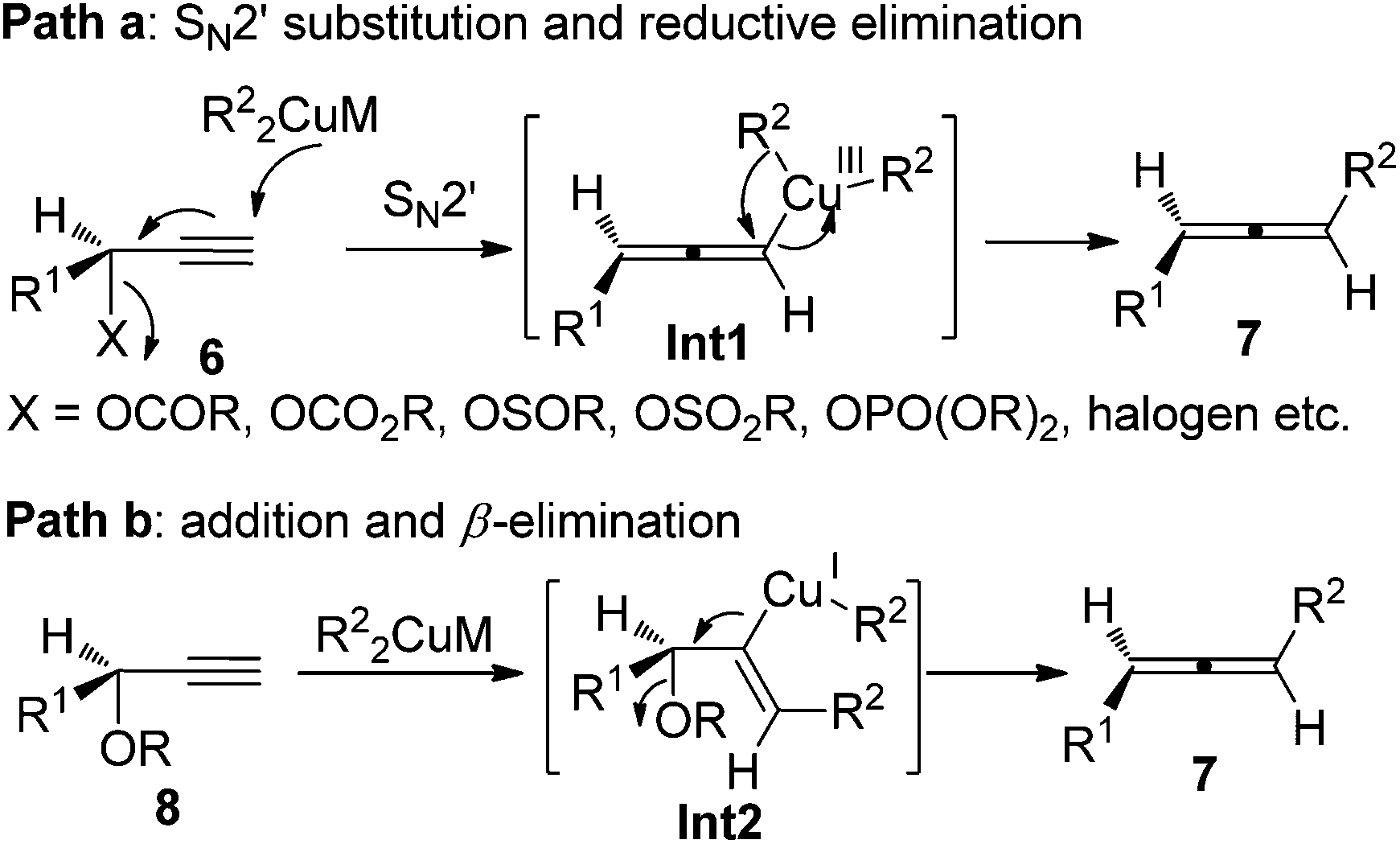
Given the high potential of this methodology, great effort has been made by Crabbé,8a,b Claesson,8c,9a Elsevier,8d,e and Alexakis et al.8f–h to broaden the substrate scope, improve the efficiency of chirality transfer, and elucidate the reaction mechanism. It turned out that the chirality transfer process is affected by a series of factors such as the nature of electrophiles, nucleophiles, leaving groups, ligands, solvent, temperature, and even reaction time.8,9 In addition, in situ racemization of the allene products caused by organocopper or cuprate reagents that are derived from Grignard or organolithium reagents has also been observed.9 Generally speaking, there are two possible reaction pathways for these reactions:8f–h (1) for propargylic derivatives with a good leaving group such as acetates, carbamates, sulfinates, sulfonates, phosphates, and halides, the reaction is more likely to proceed via SN2′ substitution followed by reductive elimination from the Cu(III) intermediate Int1 (path a, Scheme 3); (2) for those with a poorer leaving group such as propargylic ethers or epoxides, a syn-insertion followed by anti-β-elimination may be operative (path b, Scheme 3). In both cases, an overall anti displacement was observed.
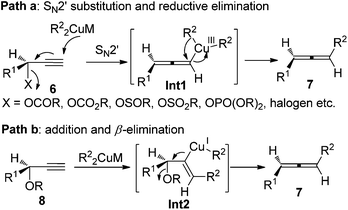 |
| | Scheme 3 Two possible pathways of the chirality transfer process. | |
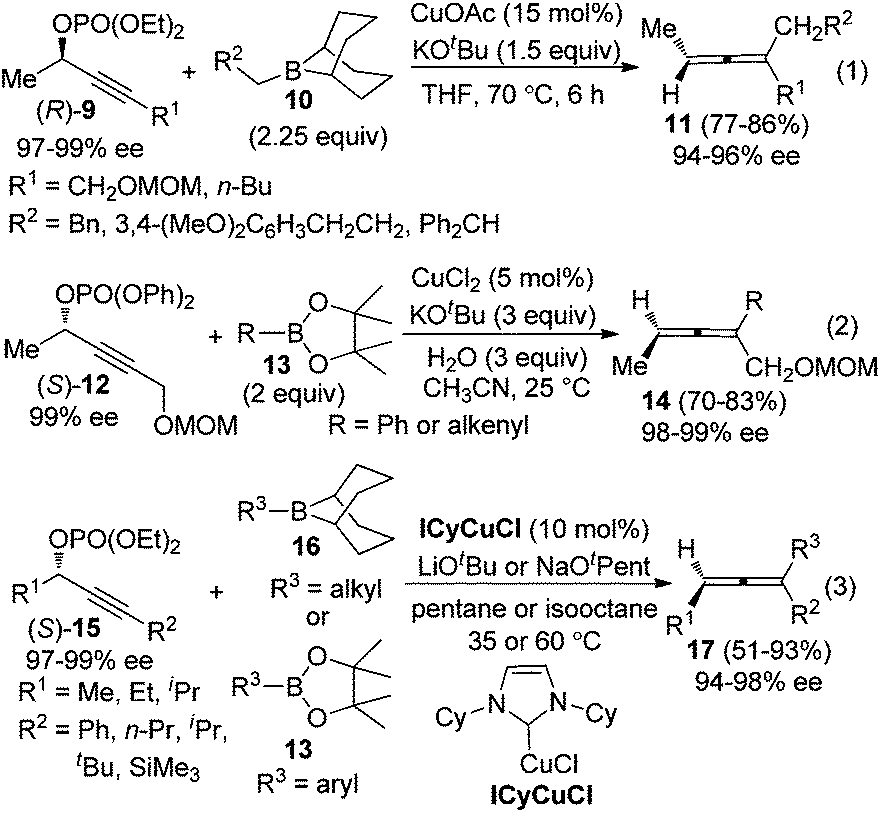
To avoid racemization in the traditional cuprate-based methods, Sawamura et al. employed alkylboranes 1010a or phenyl- and alkenylboronates 1310b to couple with optically active propargylic phosphates 9 or 12, under the catalysis of CuOAc or CuCl2, furnishing trisubstituted allenes in good yields with excellent central-to-axial chirality transfer (eqn (1) and (2), Scheme 4). Almost at the same time, Lalic and co-workers independently demonstrated that, in the presence of N-heterocyclic carbene (NHC)-ligated copper complexes (ICyCuCl), propargylic phosphates 15 could be coupled with alkyl boranes or arylboronic esters to give trisubstituted axially chiral allenes in moderate to good yields without obvious loss of enantiomeric purity (eqn (3), Scheme 4).11
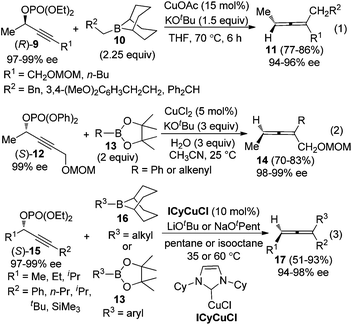 |
| | Scheme 4 Chirality transfer of propargylic phosphates with alkylboranes or aryl- and alkenylboronates. | |
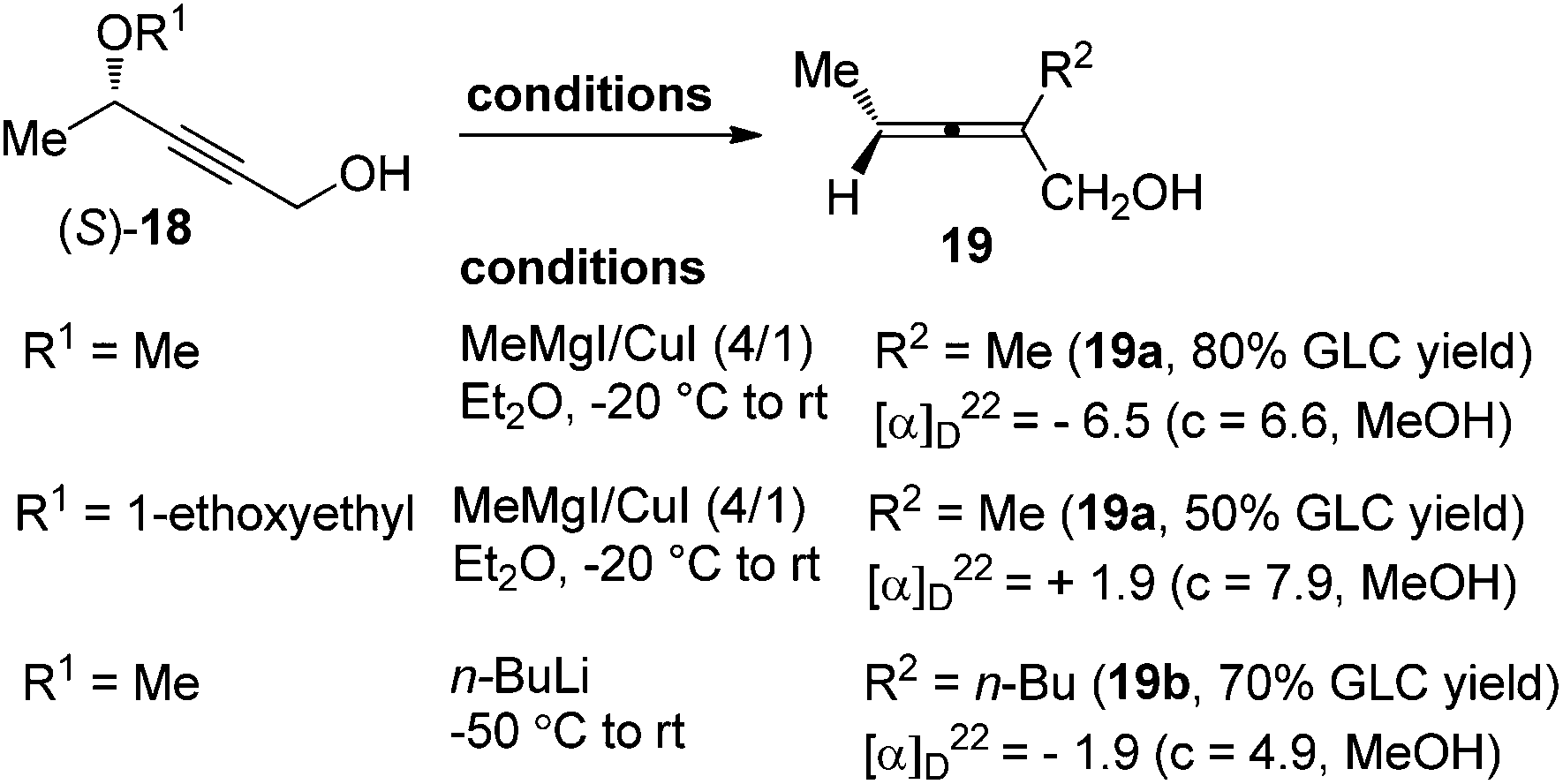
However, in spite of all the advances mentioned above, direct access of highly enantioenriched 2-substituted α-allenols, which are versatile building blocks in organic synthesis,12 was yet to be well established, probably due to the presence of a free hydroxyl group. Claesson et al. reported that 2-substituted α-allenols 19 could be obtained by reacting propargylic ethers 18 with methylmagnesium iodide–copper(I) iodide (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or n-butyllithium; however, the efficiency of chirality transfer is very low (Scheme 5).8c Thus, further explorations in this area are still highly desirable.
:1) or n-butyllithium; however, the efficiency of chirality transfer is very low (Scheme 5).8c Thus, further explorations in this area are still highly desirable.
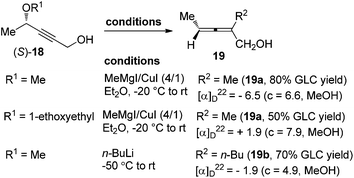 |
| | Scheme 5 Synthesis of optically active 2-substituted α-allenols via chirality transfer. | |
An alternative approach to circumvent the propensity of organocopper or cuprates to racemize enantioenriched allenes is to utilize organozinc reagents as the nucleophile, as demonstrated by Kondo and co-workers in the highly stereospecific SN2′ reaction of propargylic mesylate (R)-20 (Scheme 6).13 A dramatic solvent effect of DMSO was observed for achieving higher reactivity.
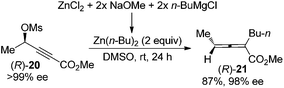 |
| | Scheme 6 Chirality transfer of propargylic mesylate with organozinc reagents without a catalyst. | |
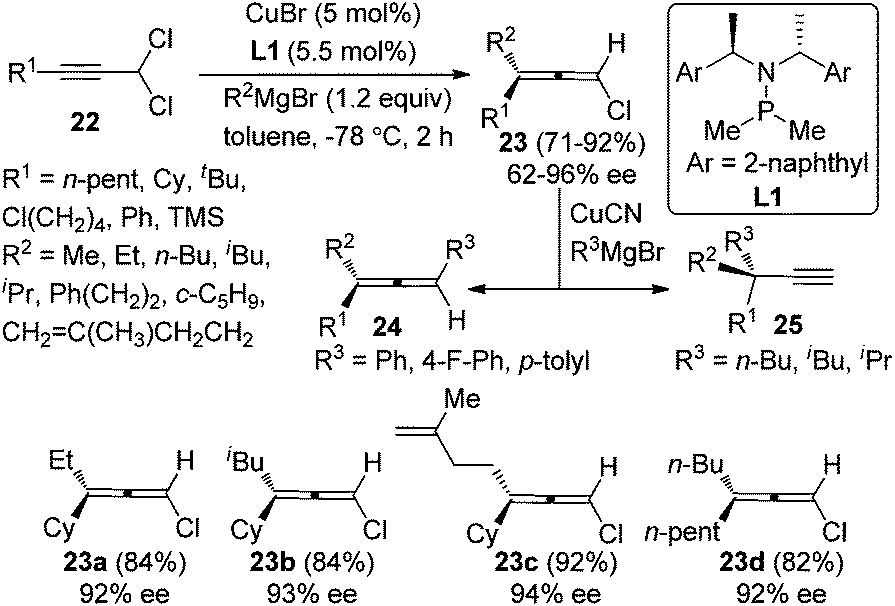
While Cu-catalyzed chirality transfer of enantioenriched propargylic alcohol derivatives with Grignard reagents to synthesize axially chiral allenes has met with considerable success, an asymmetric variant of this reaction is yet to be realized. A major breakthrough in this area was made by Alexakis et al. in 2012.14 By employing the chiral ligand L1 developed in their own group, chloroallenes 23 were obtained in high yields with exclusive regioselectivity and moderate to good enantioselectivity from 1,1-dichloropropargylic compound 22 and alkyl Grignard reagents (Scheme 7). Notably, the chloroallene products could be easily transformed into trisubstituted allenes 24 or terminal alkynes 25 with a propargylic quaternary carbon center without appreciable loss of enantiopurity in the presence of aryl or alkyl Grignard reagents, respectively.
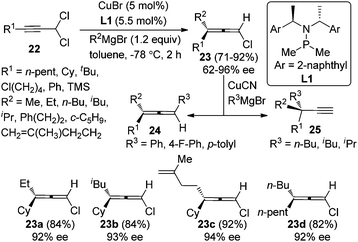 |
| | Scheme 7 Catalytic asymmetric synthesis of chloroallenes from 1,1-dichloropropargylic compounds. | |
2.1.2 Palladium-catalyzed transformations.
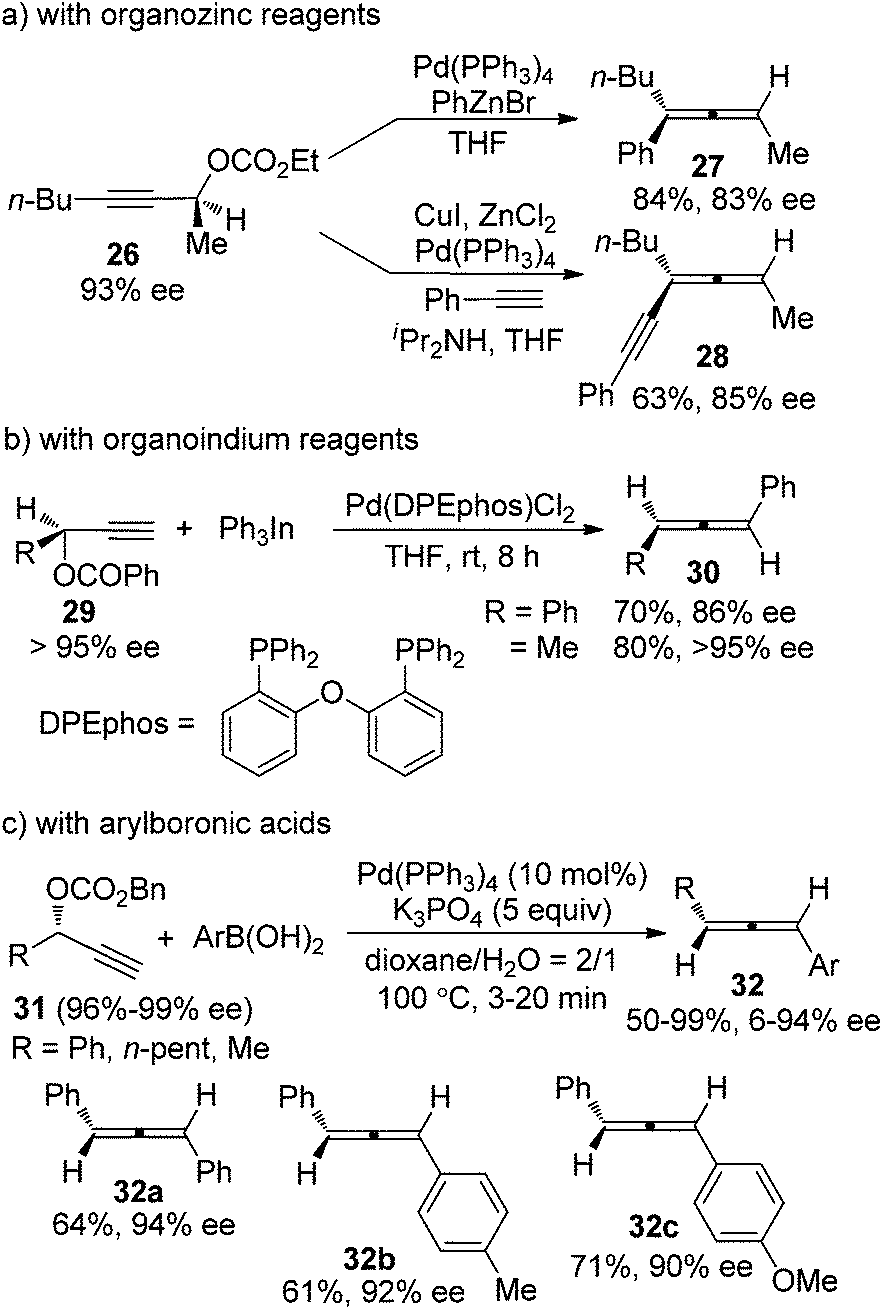
Palladium-catalyzed coupling reaction of propargylic compounds with various nucleophiles is another straightforward approach for the synthesis of allenes.4 However, deterioration of enantiopurity was also often observed in the chirality transfer process, as demonstrated in the reactions where organozinc15a or organoindium15b reagents or arylboronic acids15c were utilized as the coupling partner of propargylic electrophiles (Scheme 8). For example, in the reactions of propargylic carbonates 31 with arylboronic acids, only three products 32a–c were obtained with ≥90% ee, while the other arylboronic acids or alkyl-substituted propargylic carbonates all gave products with low efficiency of chirality transfer (c, Scheme 8). Isomerization between η1-propargyl and η1-allenylpalladium intermediates, which were generated from oxidative addition of the propargylic electrophiles with Pd(0), was believed to be responsible for the loss of optical activity. Thus, while loss of enantiopurity was observed in both Cu- and Pd-catalyzed transformations, the reason behind is different as a result of the different mechanisms of these reactions.
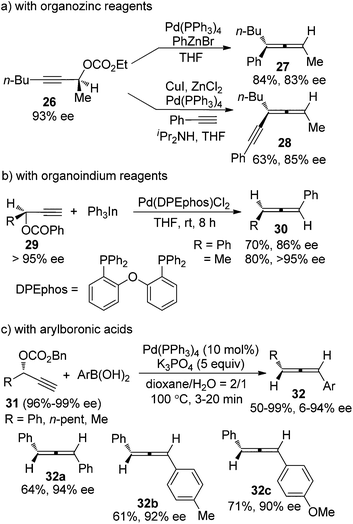 |
| | Scheme 8 Chirality transfer of propargylic electrophiles with a variety of organometallic reagents. | |
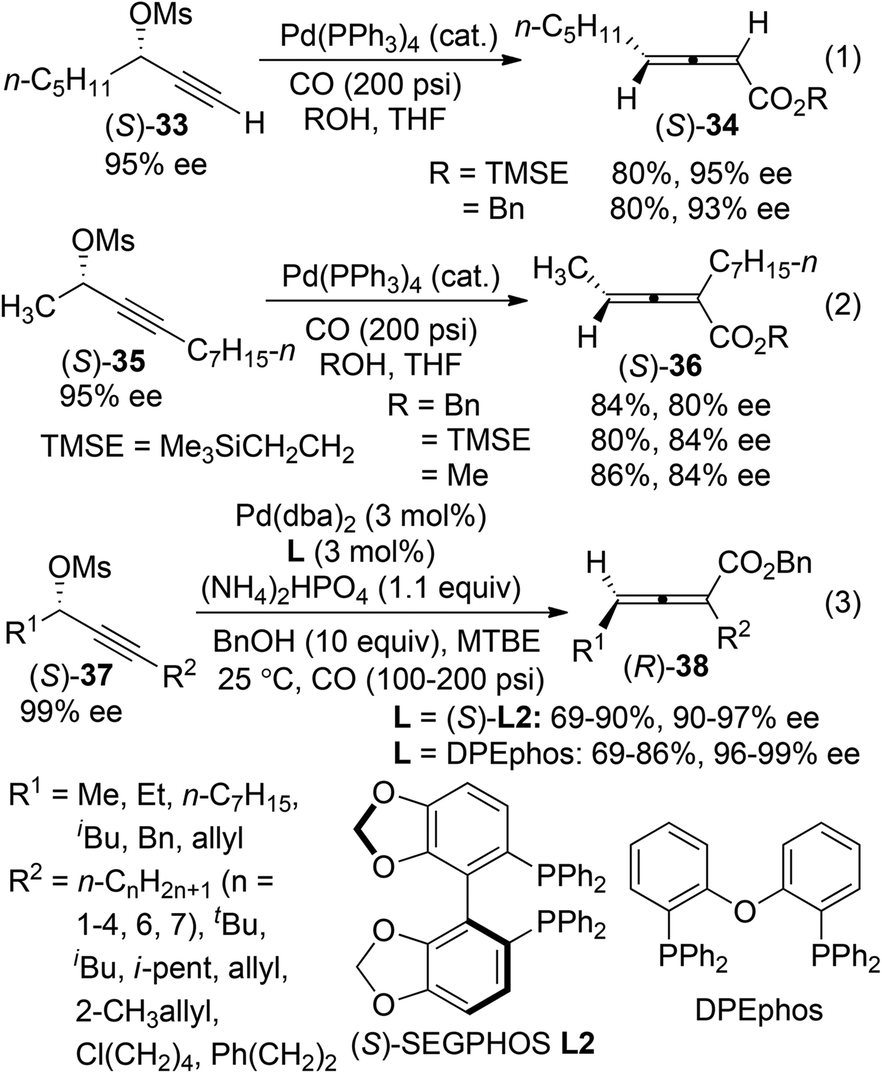
2,3-Allenoates are versatile building blocks in organic synthesis due to the synthetic potentials of the ester functionality.3h One of the most widely used methods for the synthesis of axially chiral 2,3-allenoates is based on Pd-catalyzed carbonylation reactions of optically active propargylic alcohol derivatives under a carbon monoxide atmosphere.4 In Pd-catalyzed carbonylation reactions of optically active propargylic mesylates reported by Marshall et al.,16 retention of enantiomeric purity was observed in the case of terminal alkyne (S)-33 (eqn (1), Scheme 9). In contrast, an obvious loss of enantiopurity was encountered when internal alkyne derivative (S)-35 was utilized (eqn (2), Scheme 9). To address such a concern referring to the internal C–C triple bond, two modified procedures were recently developed, in which biphosphine ligand (S)-SEGPHOS L2 or DPEphos was applied to minimize racemization during the chirality transfer process of the internal propargyl mesylates (S)-37 (eqn (3), Scheme 9).17
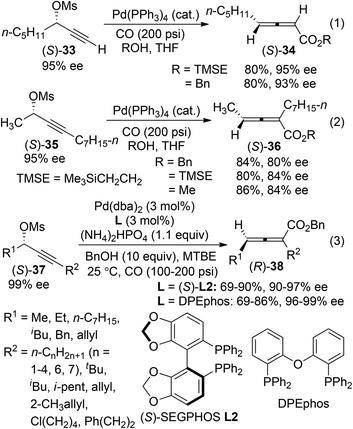 |
| | Scheme 9 Synthesis of axially chiral 2,3-allenoates via carbonylation reactions of propargylic mesylates. | |
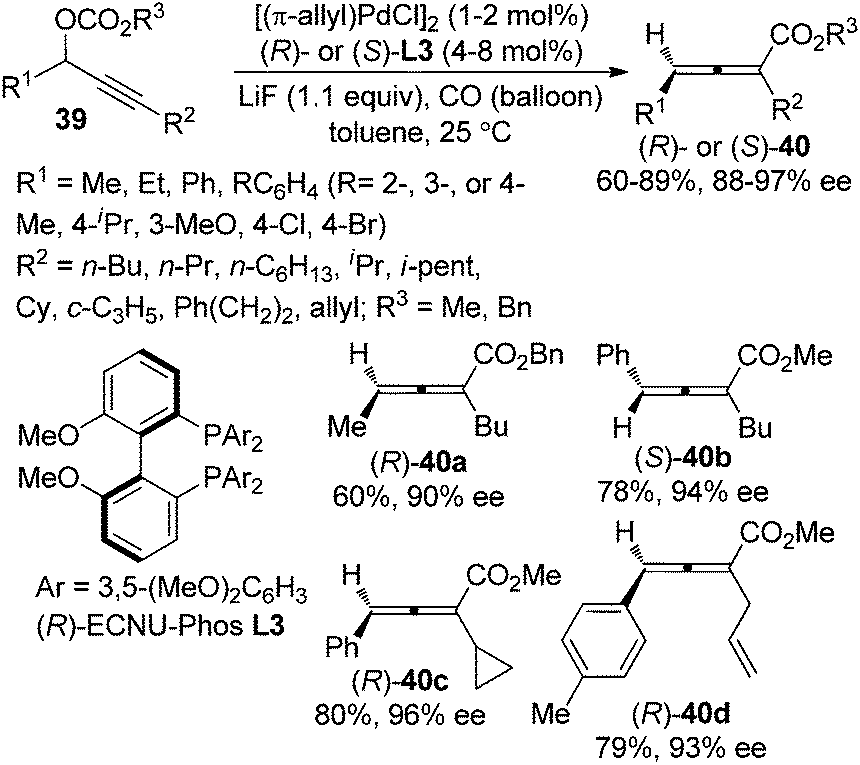
Very recently, we further developed a highly enantioselective protocol for the synthesis of 2,3-allenoates 40 directly from racemic propargylic carbonates 39 by utilizing the newly developed biphenyl biphosphine ligand (R)- or (S)-ECNU-Phos L3 (Scheme 10).18 The 3,5-dimethoxy substituents on the phenyl group linked to the phosphorus atom (Ar) may provide the required steric and electronic environments for achieving a high enantioselectivity at room temperature, which is of critical importance for this type of reaction due to the temperature-sensitive nature of electron-deficient axially chiral allenes. However, terminal propargylic carbonates (R2 = H) are not compatible with this procedure and further efforts are needed.
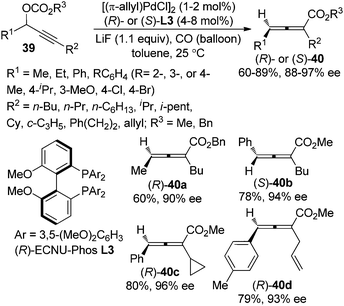 |
| | Scheme 10 Catalytic asymmetric synthesis of 2,3-allenoates from racemic propargylic carbonates. | |
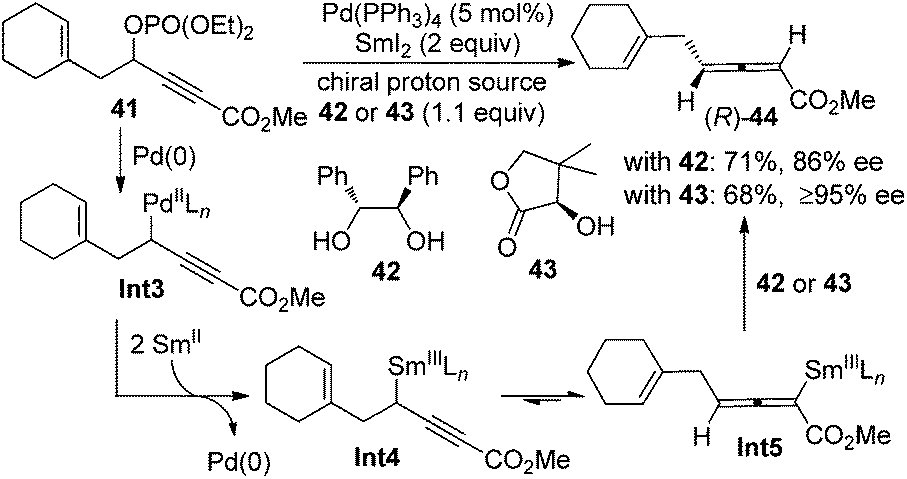
Besides the chirality transfer approach and asymmetric catalysis, 2,3-allenoates may also be prepared by the enantioselective protonation using a stoichiometric amount of chiral reagents. Mikami et al. reported that in the presence of chiral proton source 42 or 43, allenylsamarium(III) intermediates Int5 generated from the corresponding palladium species Int3 could be asymmetrically protodemetallated to afford axially chiral allene (R)-44 in practical yields with a reasonably high enantiopurity (Scheme 11).19 However, the generality of this approach has not been explored, as only a single example was reported.
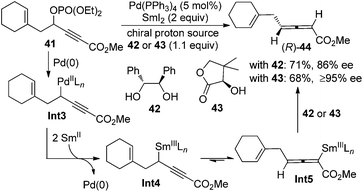 |
| | Scheme 11 Synthesis of 2,3-allenoates using stoichiometric amounts of chiral proton sources. | |
2.2 From propargylic alcohols
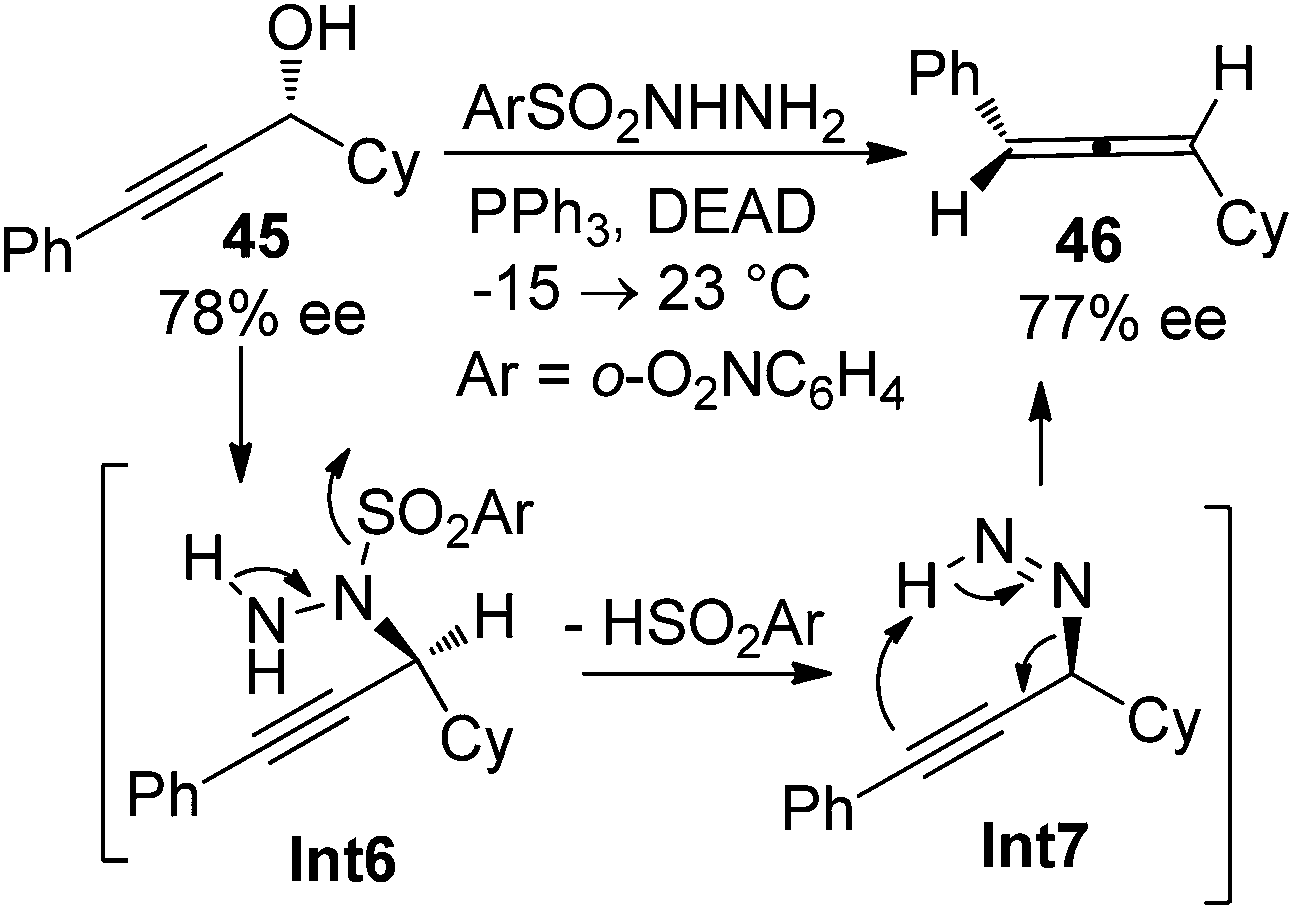
While the aforementioned methodologies all rely on the transformations of propargylic alcohol derivatives with a suitable leaving group, propargylic alcohols themselves have also been utilized to prepare allenes. Myers et al. reported such an example by using enantioenriched propargylic alcohol 45 (78% ee) with o-nitrobenzenesulfonylhydrazine to give axially chiral allene 46 with complete retention of enantiopurity (Scheme 12).20 The reaction is believed to proceed via the formation of propargylic hydrazine intermediate Int6 followed by intramolecular [1,5]-H transfer.
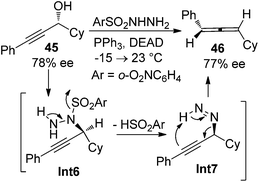 |
| | Scheme 12 Chirality transfer of propargylic alcohol in the presence of o-nitrobenzenesulfonylhydrazine. | |
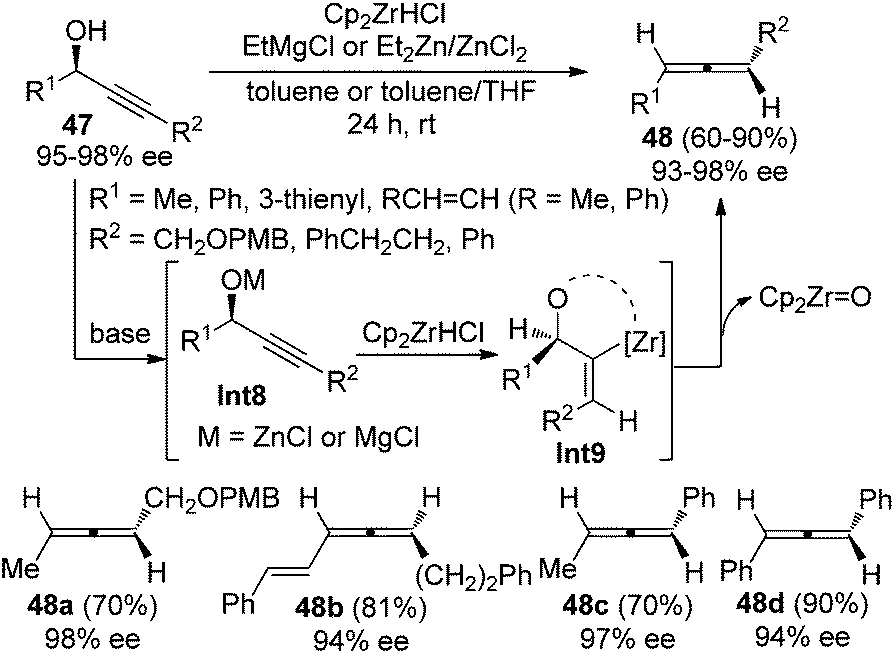
Ready and co-workers demonstrated an alternative approach to achieve this goal (Scheme 13).21 With EtMgCl or the combination of Et2Zn and ZnCl2 as the base, highly enantioenriched propargylic alcohols 47 were converted to the corresponding disubstituted allenes in good yields and with high stereochemical purity in the presence of the Schwartz reagent (Cp2ZrHCl). Strict syn-hydrozirconation followed by syn-elimination of Cp2ZrO accounts for the high efficiency of the chirality transfer process.
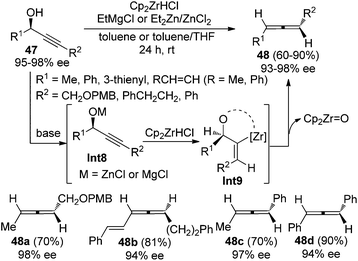 |
| | Scheme 13 Chirality transfer of propargylic alcohols in the presence of the Schwartz reagent. | |
3. From terminal alkynes, aldehydes, and amines – allenylation of terminal alkynes (ATA) reaction
3.1 Chiral amine approach
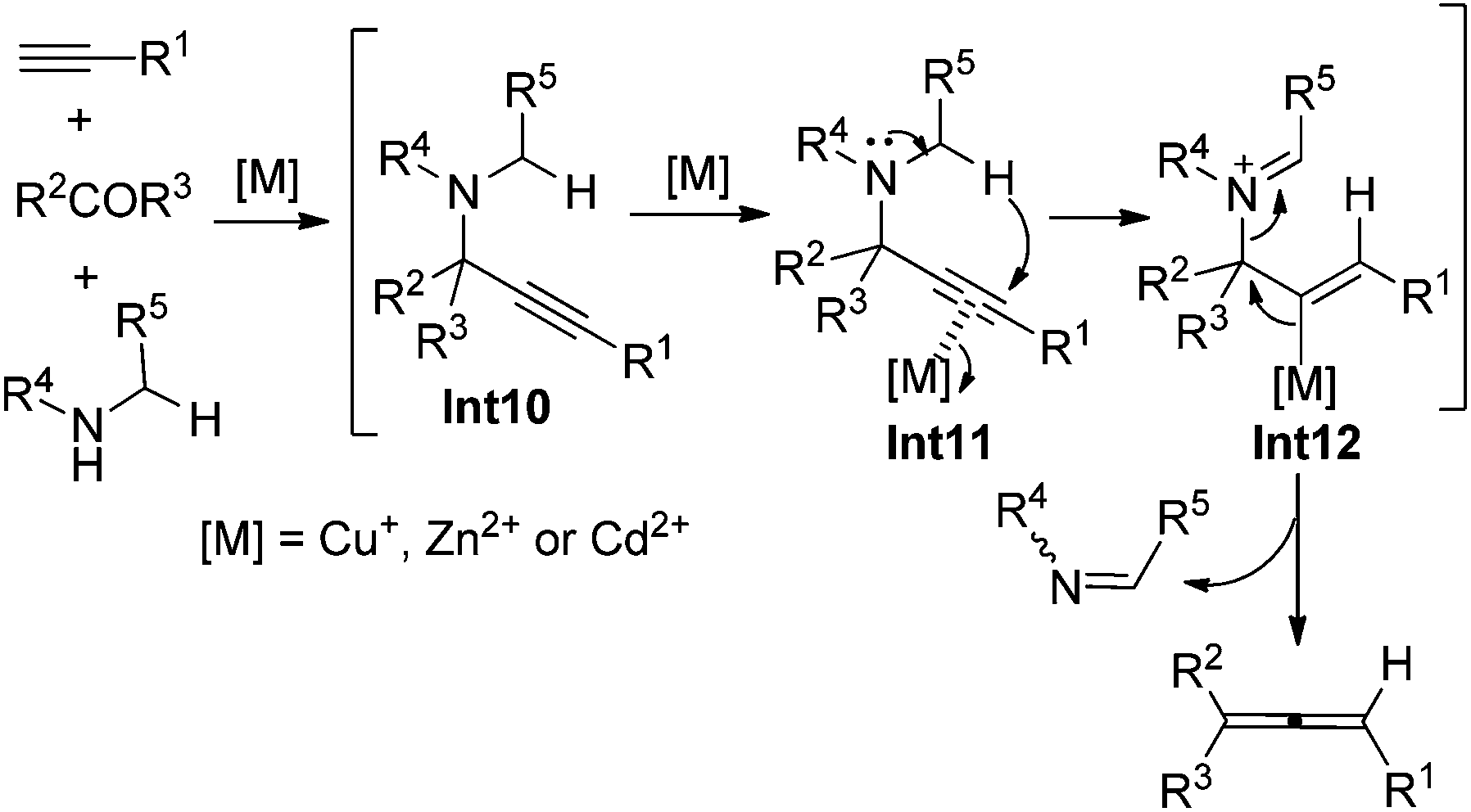
In 1979, Crabbé et al. developed an efficient synthesis of monosubstituted allenes directly from terminal alkynes in the presence of paraformaldehyde and diisopropylamine, albeit with low yields and a limited scope only workable with paraformaldehyde.22 We have later modified this procedure for higher yields with CuI and Cy2NH23a and established efficient ZnI2- or CdI2-mediated synthesis of 1,3-disubstituted23b or 1,1,3-trisubstituted23c allenes from terminal alkynes and aldehydes or ketones in the presence of secondary amines. The ATA reaction is believed to proceed via an initial formation of propargylic amine intermediate Int10 followed by metal-mediated intramolecular [1,5]-H shift and β-elimination (Scheme 14).22,23
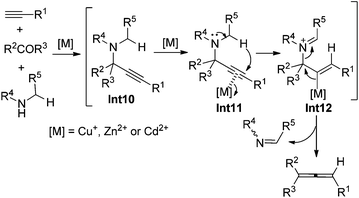 |
| | Scheme 14 A possible mechanism of the ATA reaction. | |
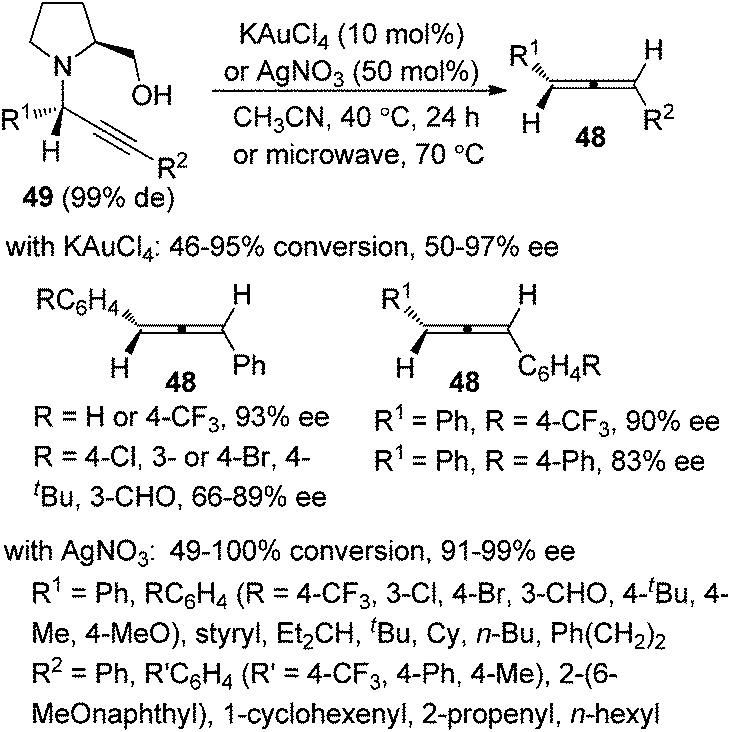
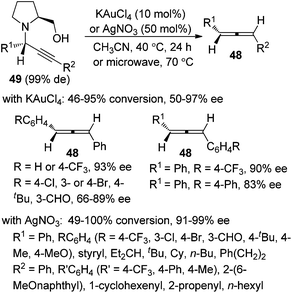
It should be noted that Che and co-workers developed the Au(III)-catalyzed24a or Ag(I)-mediated24b enantioselective synthesis of axially chiral allenes from enantioenriched propargylic amines 49 (Scheme 15), which, in turn, were prepared via Au(III) salen complex-catalyzed three-component coupling reactions of terminal alkynes, aldehydes, and chiral prolinol.24c Although the Au(III)-catalyzed approach is confined to 1,3-diarylallenes and electron-poor propargylic amines, the efficiency of the chirality transfer process is generally good in the Ag(I)-mediated protocol. However, the conversions in both cases are unsatisfactory as most of the substrates failed to reach full conversion under the reaction conditions.
 |
| | Scheme 15 Au(III)- or Ag(I)-promoted synthesis of axially chiral allenes from enantioenriched propargylic amines. | |
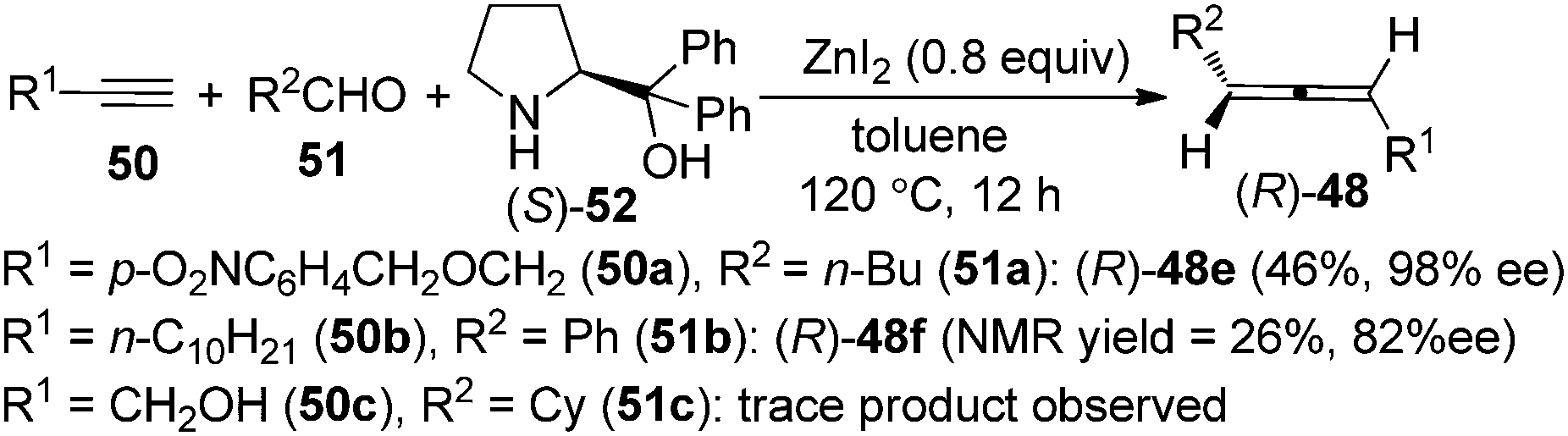
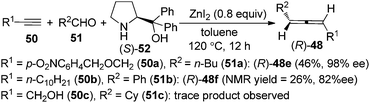
As the ZnI2-mediated protocol for aldehydes23b makes enantioselective synthesis of 1,3-disubstituted allenes possible by utilizing chiral amines as well as chiral ligands, we turned our attention to the enantioselective allenylation of terminal alkynes (EATA) via such an approach. Thus, a “chiral amine” approach was developed using commercially available and inexpensive (S)- or (R)-α,α-diphenylprolinol 52 as the chiral amine (Scheme 16).25,26 However, extensive studies showed that the scope of the reaction was quite limited as only terminal alkyne 50a with a sterically bulky group and aliphatic aldehydes are suitable substrates; simple alkynes such as 50b with a less sterically bulky alkyl group and propargyl alcohols such as 50c with a free hydroxyl group all gave very poor results.25
 |
| | Scheme 16 Initial observations on the chiral amine approach. | |
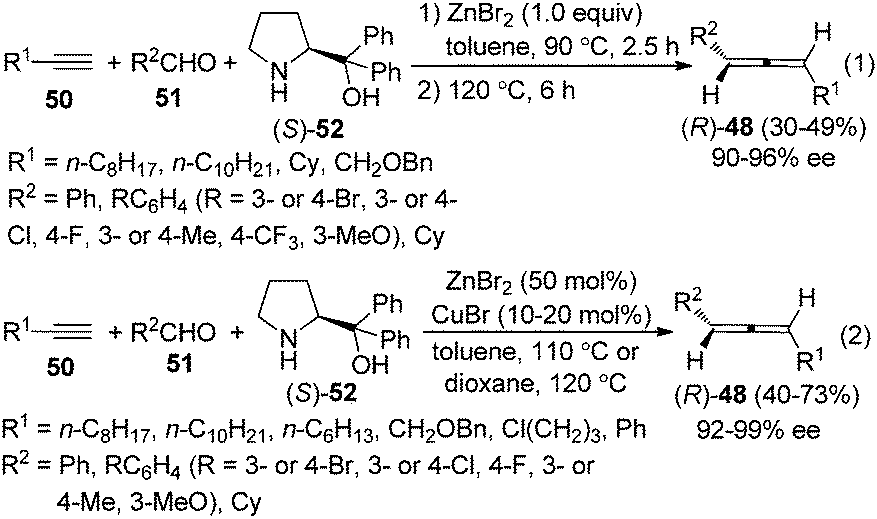
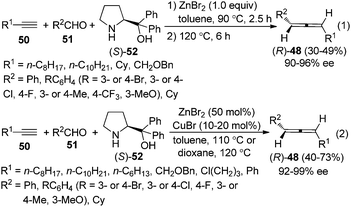
Thus, a “two-stage” procedure for the reactions of simple terminal alkynes with aromatic or aliphatic aldehydes was established to give the allene products with excellent enantioselectivity; however, the yields are still quite low (eqn (1), Scheme 17).26a Although Periasamy et al. recently reported the synthesis of axially chiral allenes from simple non-functionalized terminal alkynes using α,α-diphenylprolinol 52,27a we have not been able to reproduce their results in terms of ee and yield: the calibrated specific optical rotations of the same allenes with similar ee values from our study and the data in their report are different.26a To further improve the yield and enantioselectivity as well as broaden the scope of this reaction, a Cu+/Zn2+ bimetallic approach was developed, providing axially chiral allenes (R)-48 in somewhat higher yields with excellent enantioselectivity (eqn (2), Scheme 17).26b However, heteroaromatic and α,β-unsaturated aldehydes are still incompatible substrates for the reaction. Control experiments revealed that CuBr is responsible for the efficient formation of the propargylic amine intermediate while both CuBr and ZnBr2 play crucial roles in the propargylic amine-to-allene transformation.
 |
| | Scheme 17 Synthesis of simple axially chiral allenes via the chiral amine approach. | |
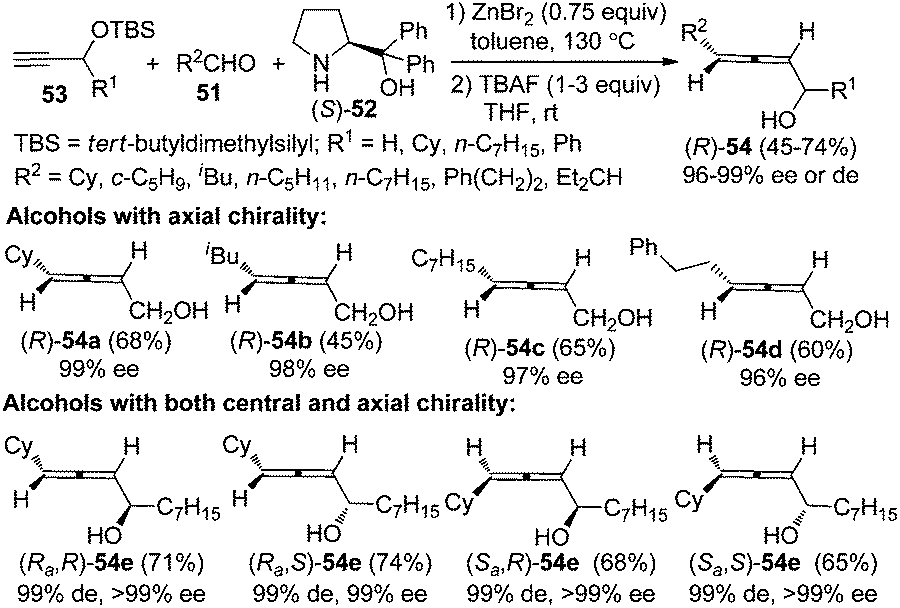
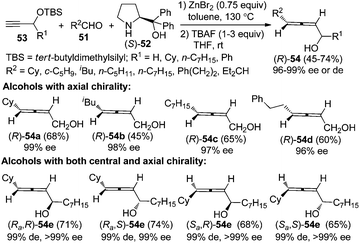
α-Allenols with axial chiralities are valuable building blocks in organic synthesis due to the synthetic potential of the hydroxyl group.12 The reactions of TBS-protected propargyl alcohol 53 (R1 = H) with various aliphatic aldehydes afforded primary α-allenols such as (R)-54a–d in practical yields with excellent enantioselectivity after deprotection of the TBS group (Scheme 18).26c Synthesis of α-allenols with both central and axial chiralities is challenging. Notably, all of the four diastereoisomers of secondary α-allenols 54e could be highly stereoselectively prepared simply by adjusting the absolute configurations of the central chiralities in the TBS-protected secondary propargylic alcohols 53 and α,α-diphenylprolinol 52. Control experiments revealed that the TBS group is acting not only as a protecting group but also as a steric-dictating group for the excellent enantioselectivity. However, this reaction is currently not applicable to aromatic and α,β-unsaturated aldehydes.
 |
| | Scheme 18 Synthesis of axially chiral α-allenols via the chiral amine approach. | |
3.2 Chiral ligand approach
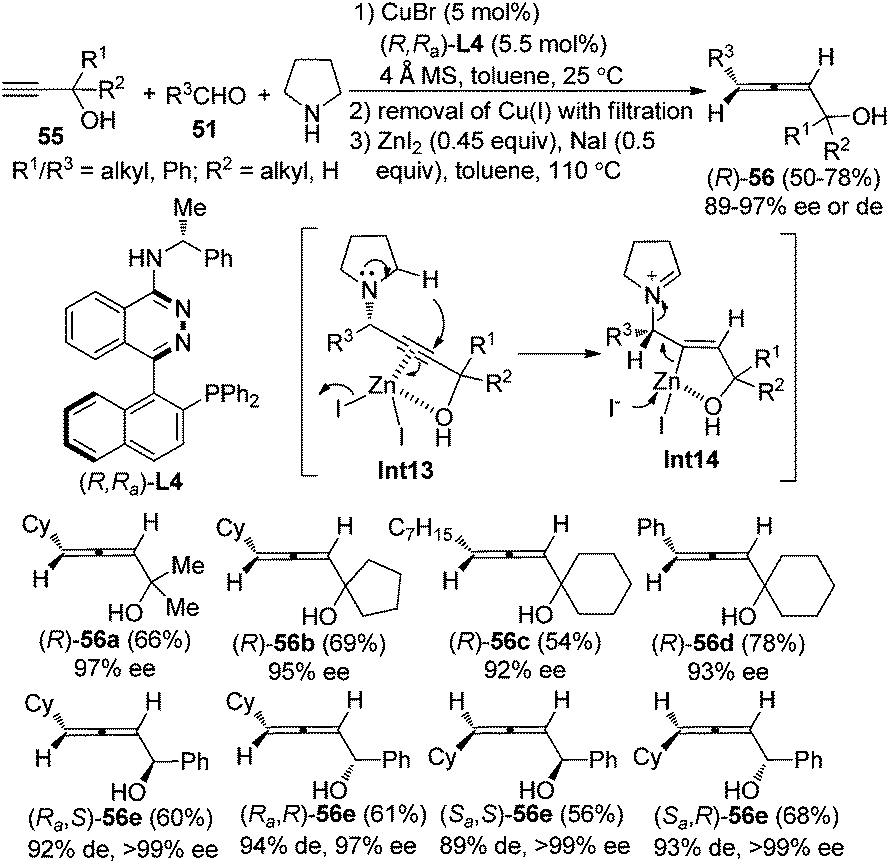
Furthermore, a “chiral ligand” approach was developed to give axially chiral allenes, especially highly useful α-allenols, in decent yields with excellent enantio- or diastereoselectivity from terminal alkynes, aldehydes, and pyrrolidine by applying the chiral ligand (R,Ra)-PINAP L4 (Scheme 19).25 Again, by simply changing the central chirality of secondary propargylic alcohols 55 and axial chirality of the chiral ligand, all of the four diastereoisomers of the secondary α-allenols 56e could be highly stereoselectively prepared. The free hydroxyl group in the terminal alkynes 55 proved to be crucial both for the yield and enantioselectivity of the reaction, which may be explained by its coordination with Cu+ and/or Zn2+ in the first and the second step of this transformation. However, primary propargylic alcohol is not compatible with this procedure, probably resulting from the lack of effective coordination of the hydroxyl group with Zn2+. Nevertheless, the corresponding primary α-allenols may be easily prepared through the “chiral amine” approach (Scheme 18). When ZnI2 was replaced with KAuCl424a or AgNO3,24b only trace amounts of allenol products were detected, suggesting that matching reactivity between substrates and metal salts is crucial for the transformation of propargylic amine to the functionalized allene. Further efforts to design a more practical “one-pot” procedure are in progress in this group.
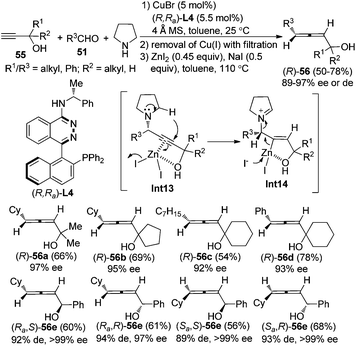 |
| | Scheme 19 Synthesis of axially chiral secondary and tertiary α-allenols via the chiral ligand approach. | |
4. From conjugated enynes
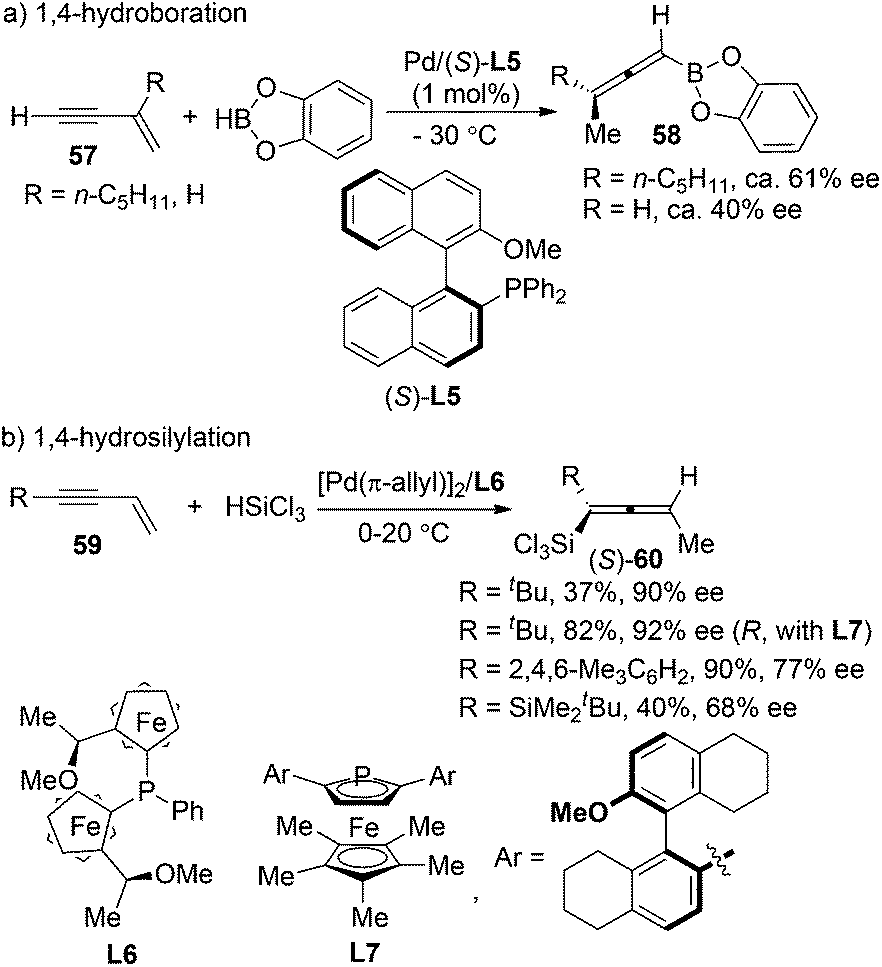
Pd-catalyzed asymmetric 1,4-addition of hydroborane28a or hydrosilane28b,c to conjugated enynes has been established by Hayashi et al. as a very promising strategy for the synthesis of axially chiral allenylboranes or allenylsilanes, which are valuable propynylating reagents in asymmetric synthesis. However, the enantioselectivities were moderate in most cases; the substrate scope also proved to be very limited (Scheme 20).
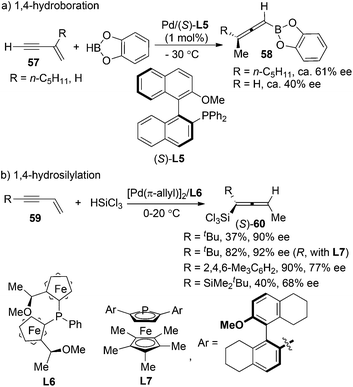 |
| | Scheme 20 Pd-catalyzed 1,4-hydroboration of conjugated enynes. | |
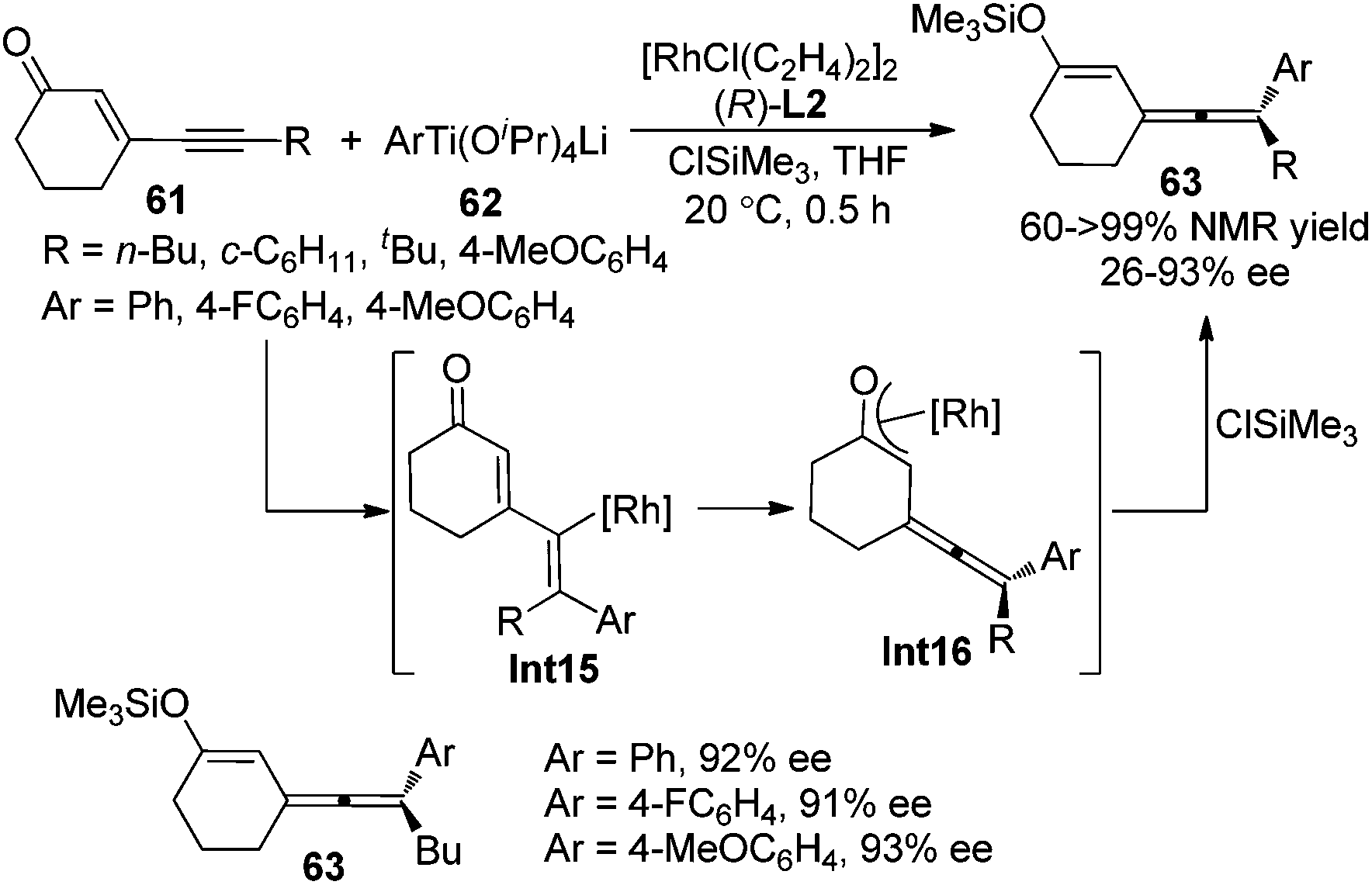
Following these previous studies, the same group developed a Rh-catalyzed asymmetric 1,6-addition of aryltitanates 62 to conjugated enynones 61 to give axially chiral allenylalkenyl silyl enol ethers 63 in the presence of chiral ligand (R)-SEGPHOS L2 and chlorotrimethylsilane (Scheme 21).28d While >90% ee of the products were observed when the terminal substituent (R) is an n-butyl group, only moderate enantioselectivity was obtained in other cases. A mechanism involving carborhodation, enantioselective isomerization, silylation and transmetallation was proposed for the reaction.
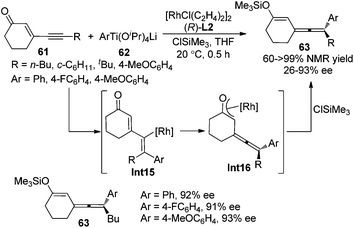 |
| | Scheme 21 Rh-catalyzed asymmetric 1,6-addition of aryltitanates to conjugated enynones. | |
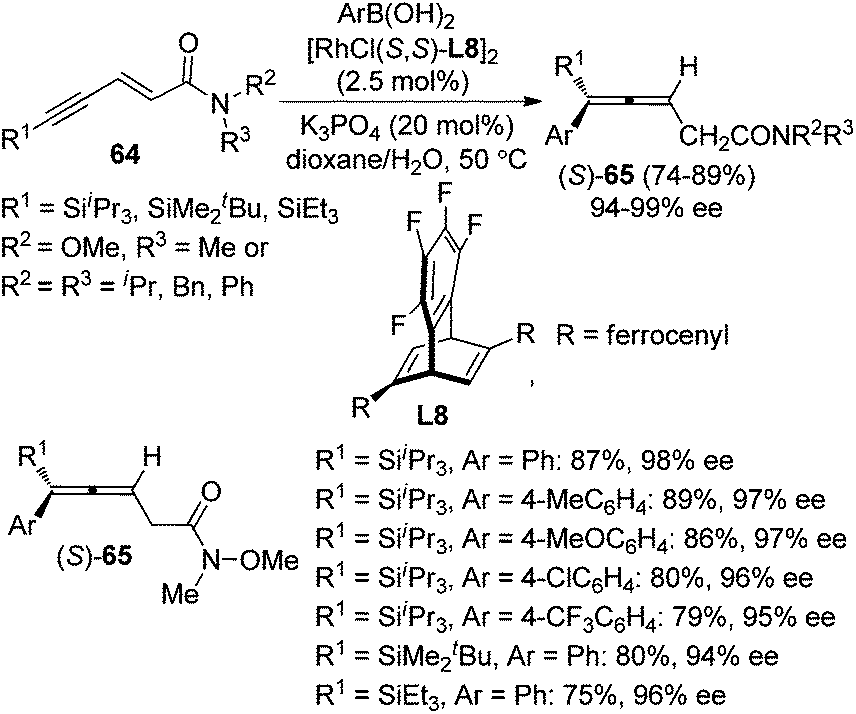
Recently, Hayashi and co-workers further developed a Rh/chiral diene complex-catalyzed enantioselective approach for the synthesis of allenylsilanes 65 with 94–99% ee via the 1,6-addition of arylboronic acids to enynamides 64 (Scheme 22).28e Both the ferrocenyl group of the chiral diene ligand L8 and bulky silyl substituent of the alkyne were found to be critical for achieving high regio- and enantioselectivity.
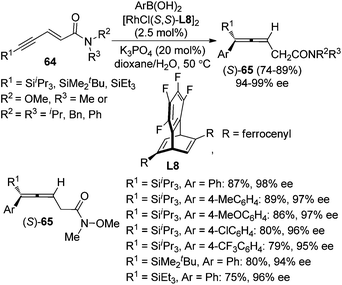 |
| | Scheme 22 Rh-catalyzed asymmetric 1,6-addition of arylboronic acids to enynamides. | |
5. From ketenes
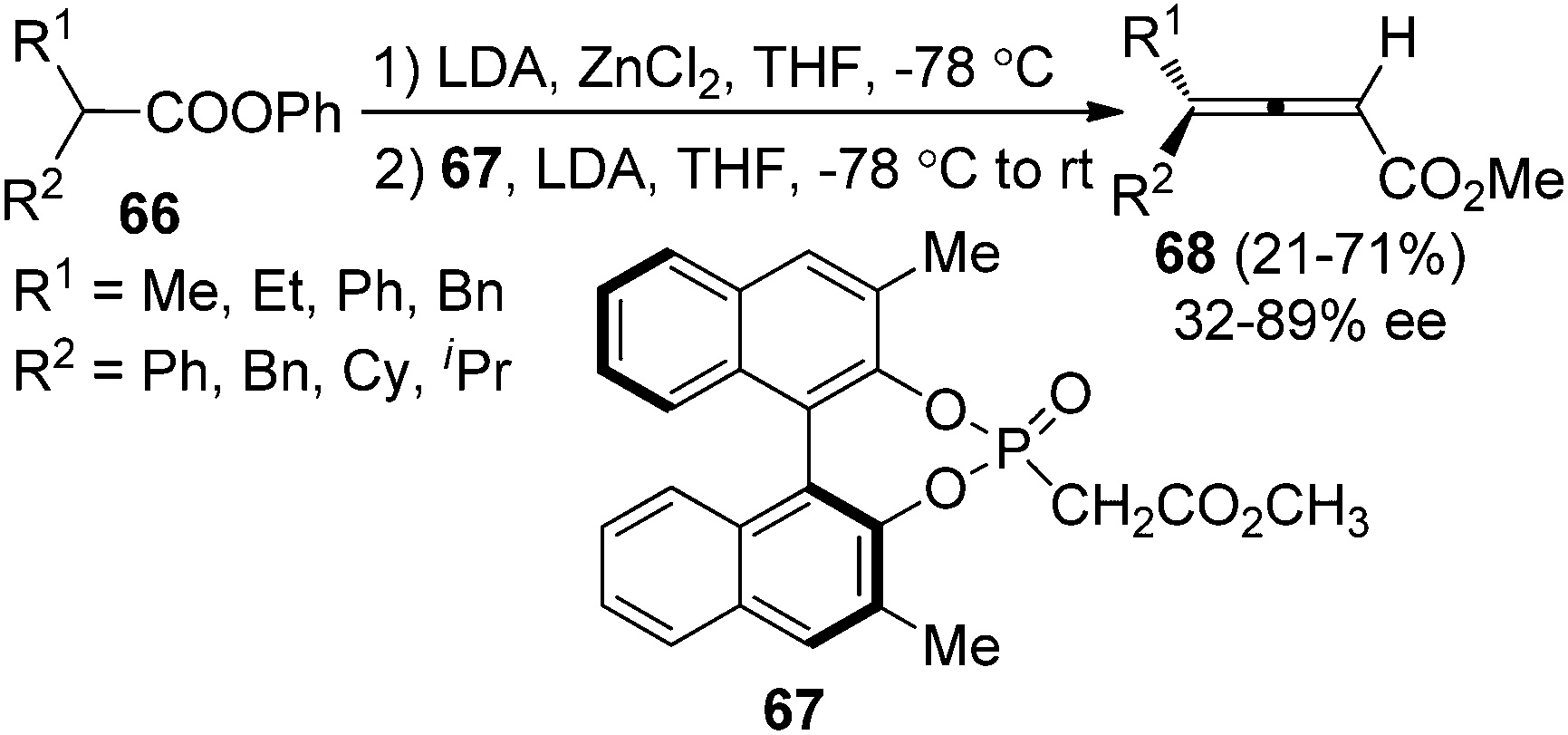
Olefination of ketenes with ylides is also an efficient approach to synthesize allenes.4 Thus, in the presence of a chiral ylide, axially chiral allenes may be accessible. Bestmann29a and Musierowicz29bet al. made pioneering contributions in this area, although the 2,3-allenoates thus obtained exhibited a low optical activity. Tanaka and co-workers showed that chiral ylide derived from BINOL-based phosphinate ester 67 reacted with the in situ generated ketenes to afford 4,4-disubstituted 2,3-allenoates 68 in 21–71% yield with 32–89% ee (Scheme 23).30
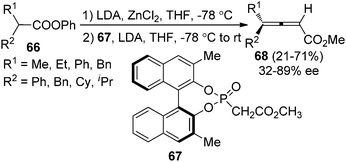 |
| | Scheme 23 Olefination of ketenes with BINOL-based chiral ylide. | |
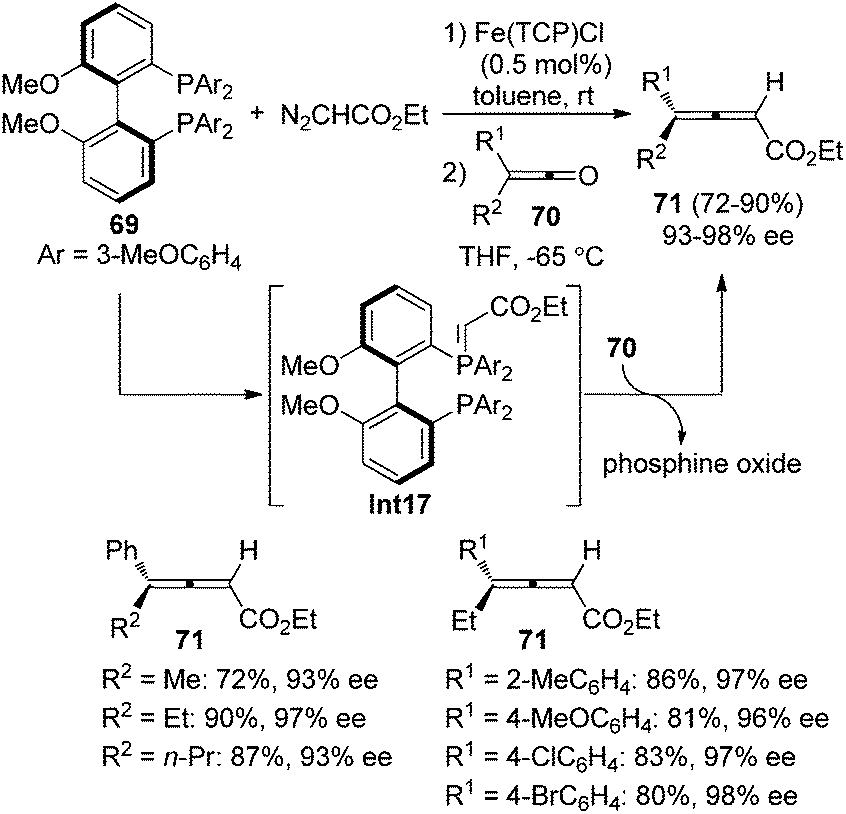
Tang and co-workers reported an iron-porphyrin complex-catalyzed olefination of ketenes 70 with diazoacetate, furnishing 4,4-disubstituted 2,3-allenoates 71 in good yields with excellent enantioselectivity, albeit the substrate scope was somewhat limited (Scheme 24).31a It is worth noting that the chiral phosphine oxide by-product could be easily recycled via reduction. Although diphosphine 69 was utilized, control experiment revealed that the reaction proceeded via monoylide Int17.
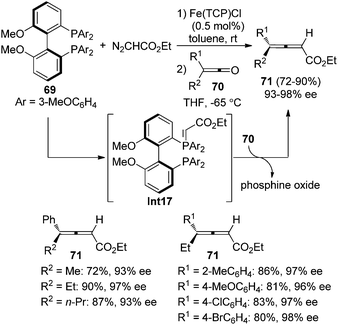 |
| | Scheme 24 Fe-catalyzed olefination of ketenes with diazoacetate. | |
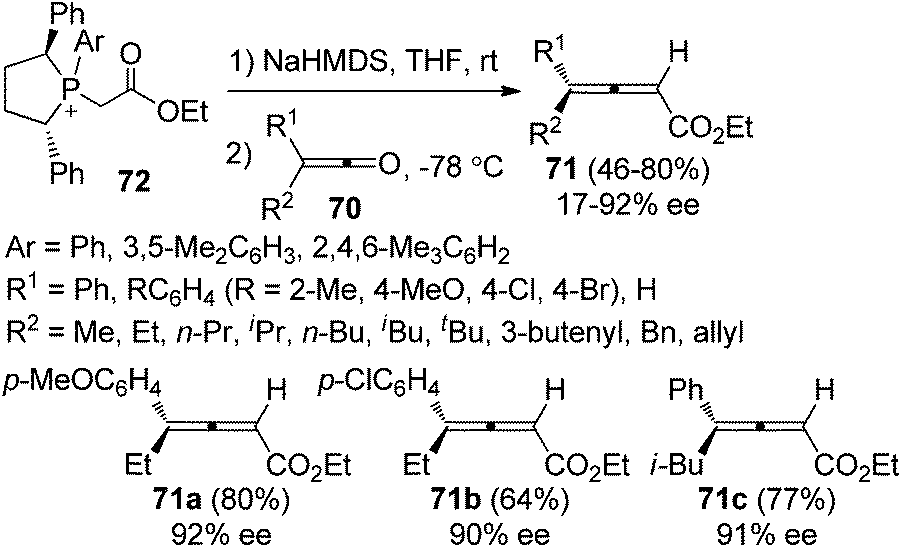
Tang et al. further developed pseudo-C2-symmetric monoylides 72 for the synthesis of optically active 2,3-allenoates. The scope of ketenes was expanded; however, the enantioselectivities were generally moderate with only three products 71a–c being obtained with ≥90% ee (Scheme 25).31b,c
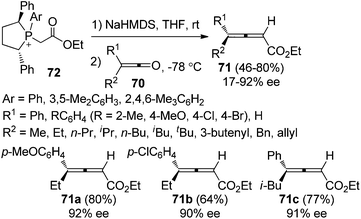 |
| | Scheme 25 Olefination of ketenes with pseudo-C2-symmetric ylides. | |
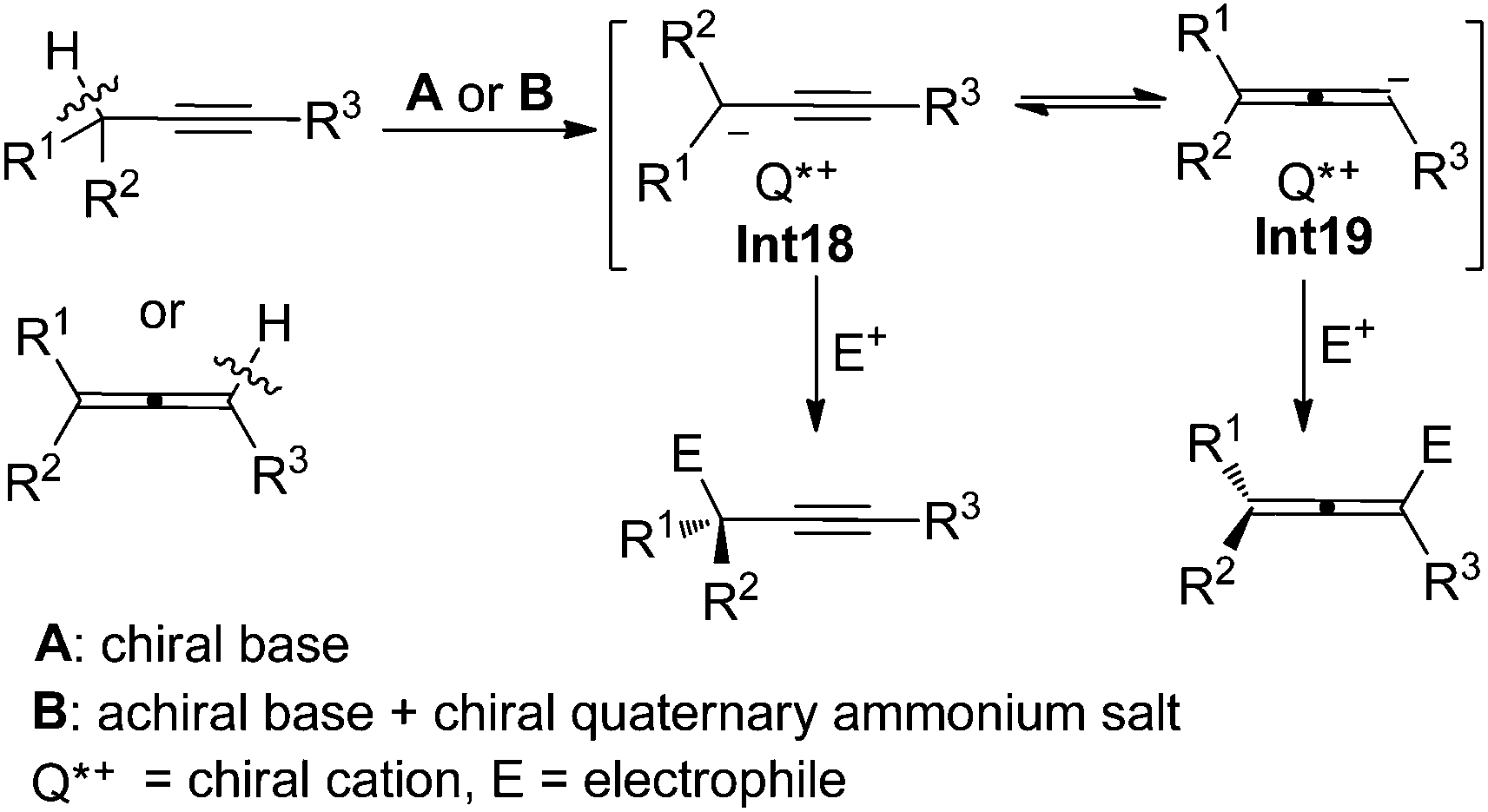
6. From propargyl/1,2-allenyl anion intermediates
In the presence of a chiral base or an achiral base combined with a chiral quaternary ammonium salt, properly substituted propargylic compounds or allenes would be deprotonated to afford propargyl/1,2-allenyl anion intermediates Int18/Int19 with a chiral cation, which may react with electrophiles enantioselectively to furnish propargylic compounds with a central chirality and/or allenes with an axial chirality depending on the substituents of substrates and the type of electrophiles (Scheme 26).
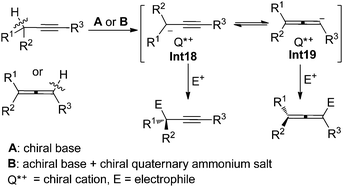 |
| | Scheme 26 Two strategies for the generation of propargyl/1,2-allenyl anion intermediates with a chiral cation. | |
6.1 Alkyne isomerization
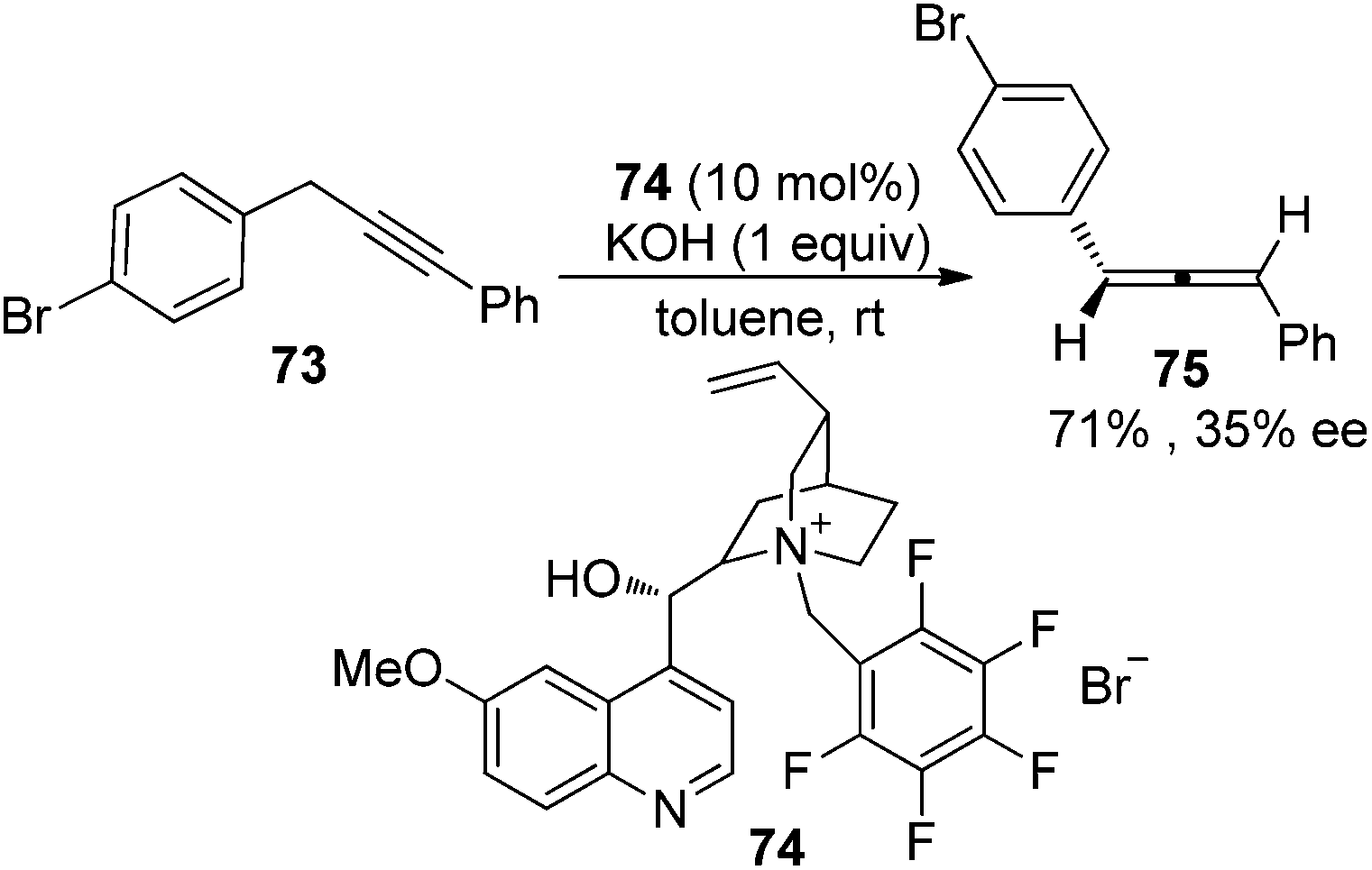
In the first place, base-promoted isomerization of alkynes has long been recognized as another straightforward protocol to allenes;4 however, catalytic asymmetric approaches have rarely been reported.32 In 2000, Arai and Shioiri et al. reported a single example of asymmetric isomerization of 1,3-diaryalkyne 73 to axially chiral allene 75 in the presence of phase-transfer catalyst 74 with a low enantioselectivity (Scheme 27).33
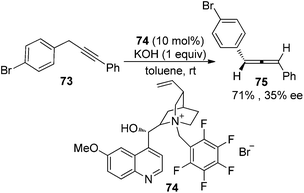 |
| | Scheme 27 Alkyne isomerization in the presence of a chiral phase-transfer catalyst. | |
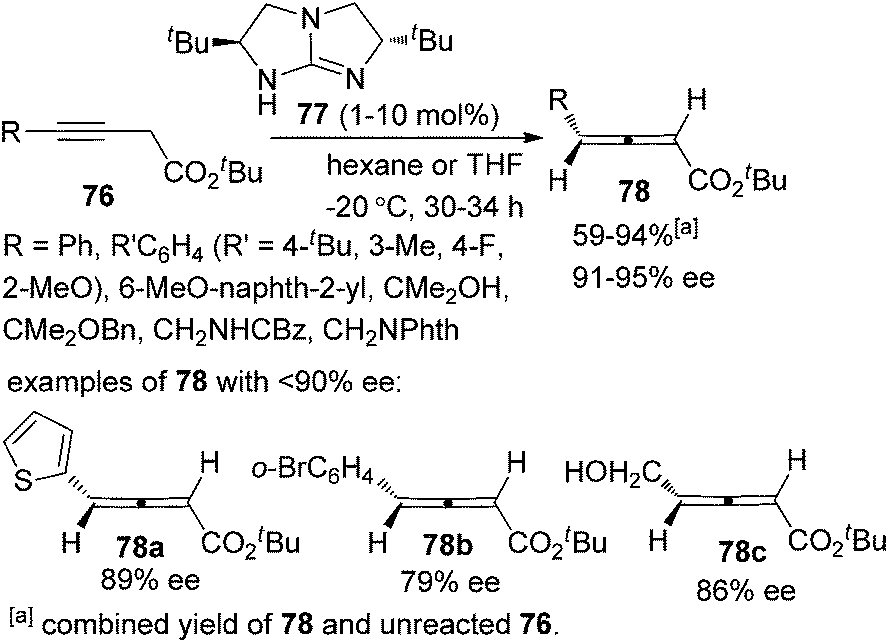
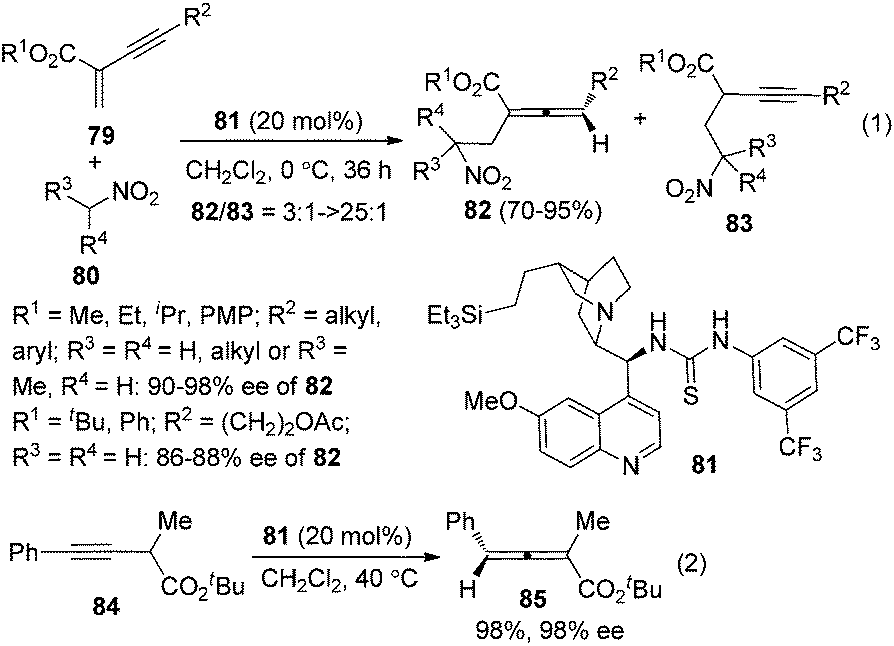
In 2009, Huang and Tan et al. demonstrated that guanidine 77 was a highly enantioselective catalyst for the isomerization of 3-alkynoates 76 to disubstituted 2,3-allenoates 78 (Scheme 28).34 The bulky tert-butyl in the ester group is crucial for the reaction to proceed in high enantioselectivity. While 91–95% ee was obtained for most of the substrates, relatively lower enantioselectivities were observed when the terminal substituent (R) is a 2-thienyl, o-BrC6H4, or CH2OH group. A flaw in this procedure is that the reaction cannot reach full conversion even after extending the reaction time, which makes the isolation of pure 2,3-allenoates 78 problematic due to the similar polarity of the unreacted 3-alkynoates. Besides, the reactivity of 2-substituted 3-alkynoate has not been demonstrated, although it might be useful for the synthesis of trisubstituted 2,3-allenoates. This limitation has been nicely addressed very recently by Zhang and Sun,35 who developed a tandem conjugate addition/isomerization sequence from activated enynes 79 and nitroalkanes 80 using the newly developed cinchona-based thiourea catalyst 81, resulting in trisubstituted 2,3-allenoates 82 in high yields with excellent enantioselectivity, albeit in some cases contaminated with a minor amount of alkynoate intermediates 83, which can be further isomerized to the corresponding 2,3-allenoates under the same reaction conditions (eqn (1), Scheme 29). To further demonstrate the generality of thiourea 81 as an isomerization catalyst, the authors also realized the highly enantioselective isomerization of racemic alkynoate 84 to trisubstituted 2,3-allenoate 85 in 98% yield with 98% ee (eqn (2), Scheme 29).
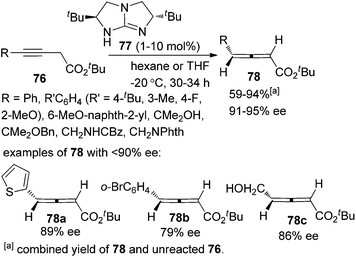 |
| | Scheme 28 Guanidine-catalyzed isomerization of 3-alkynoates to disubstituted 2,3-allenoates. | |
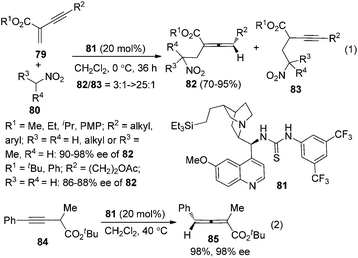 |
| | Scheme 29 Cinchona-based thiourea-catalyzed asymmetric synthesis of trisubstituted 2,3-allenoates. | |
6.2 Electrophilic addition and substitution of 1-alkylallene-1,3-dicarboxylates
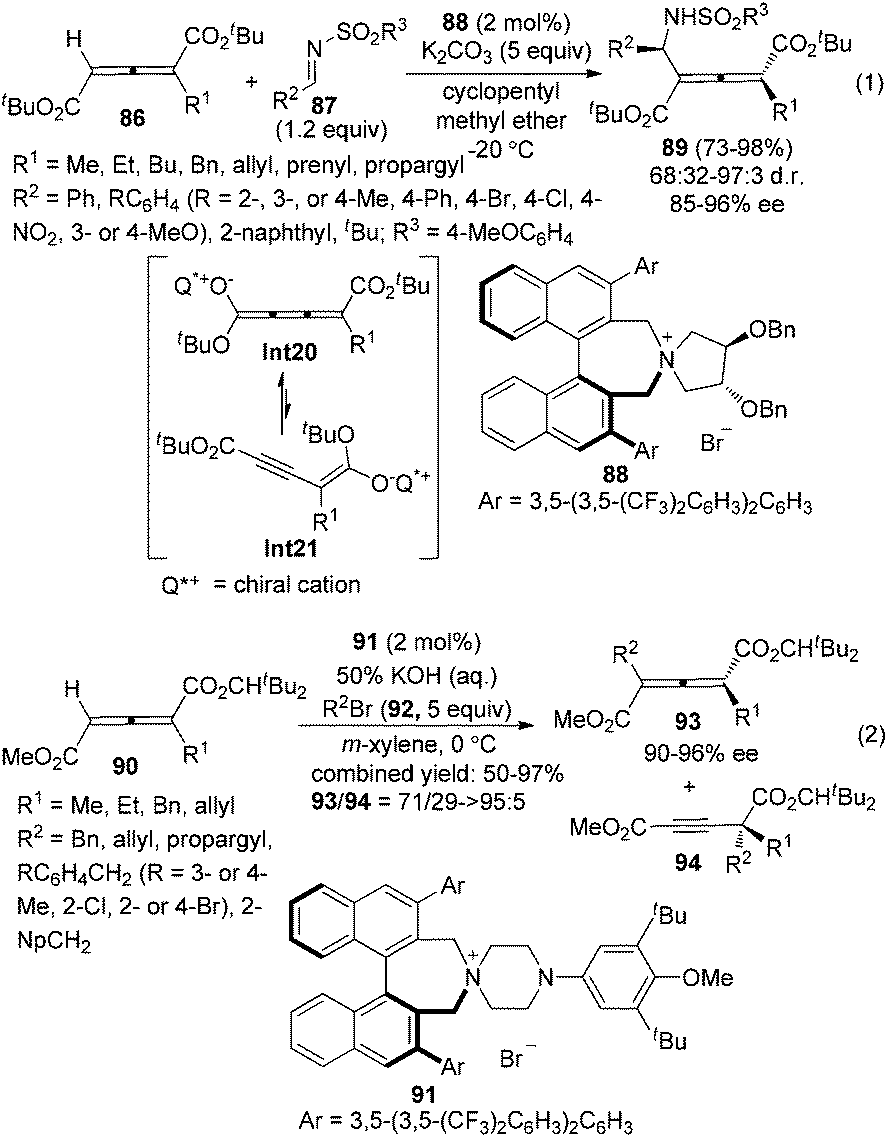
Although considerable progress has been made in the enantioselective synthesis of di- or trisubstituted allenes in the past few decades, such a synthesis of tetrasubstituted allenes has rarely been explored.4 Recently, Maruoka and co-workers reported a phase-transfer-catalyzed approach for the generation of axially chiral tetrasubstituted allenes from 1-alkylallene-1,3-dicarboxylates 86 or 90 and N-arylsulfonyl imines 87 or alkyl bromides 92 (Scheme 30).36 Chiral cumulenolate Int20 and α-alkynyl enolate Int21 generated in situ from 1-alkylallene-1,3-dicarboxylates in the presence of a chiral quaternary ammonium salt under basic conditions were acting as the nucleophiles. Interestingly, when N-arylsulfonyl imines 87 were utilized as the electrophile, tetrasubstituted allenes 89 were obtained as a single regioisomer with 68/32–97/3 dr and 85–96% ee in the presence of phase-transfer catalyst 88 (eqn (1), Scheme 30). In contrast, with alkyl bromides 92 as the electrophile under the catalysis of chiral quaternary ammonium salt 91, poorer regioselectivities were observed: both tetrasubstituted allenes 93 (90–96% ee) and alkynes 94 were obtained with the ratio of 93/94 ranging from 71/29 to >95/5 (eqn (2), Scheme 30).
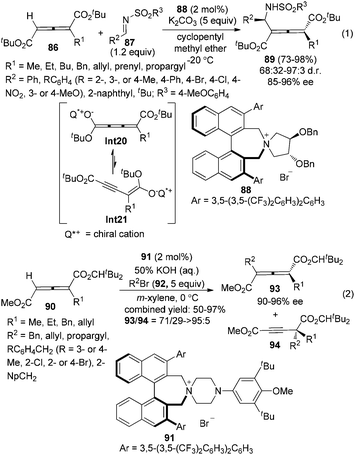 |
| | Scheme 30 Electrophilic addition and substitution reactions of 1-alkylallene-1,3-dicarboxylates. | |
7. From racemic allenes
7.1 Kinetic resolution or desymmetrization
7.1.1 Kinetic resolution or desymmetrization using enzyme.
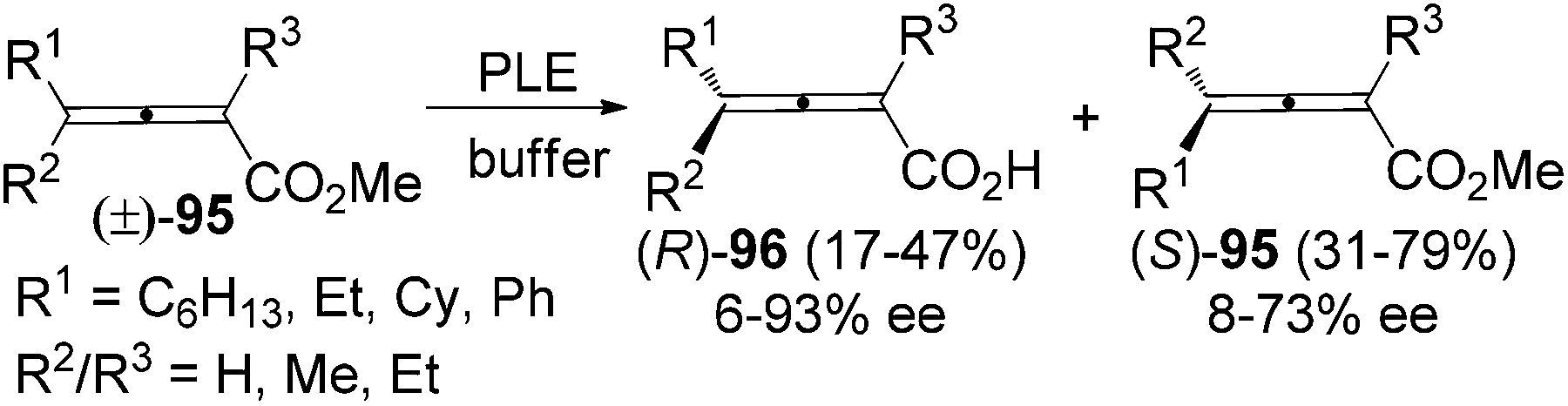
Resolution of racemic allenes with inexpensive enzyme is another approach to obtain axially chiral allenes. However, a lack of generality is the common problem of this method. Jones et al. found that optically active 2,3-allenoic acids 96 could be accessed via hydrolysis of racemic 2,3-allenoates 95 in the presence of PLE. However, only two substrates (R1 = Ph, R2 = R3 = Me or Et) afforded the corresponding 2,3-allenoic acids with >90% ee (Scheme 31).37
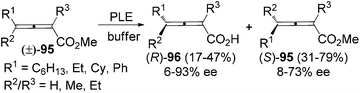 |
| | Scheme 31 PLE-catalyzed hydrolysis of 2,3-allenoates. PLE = pig liver esterase. | |
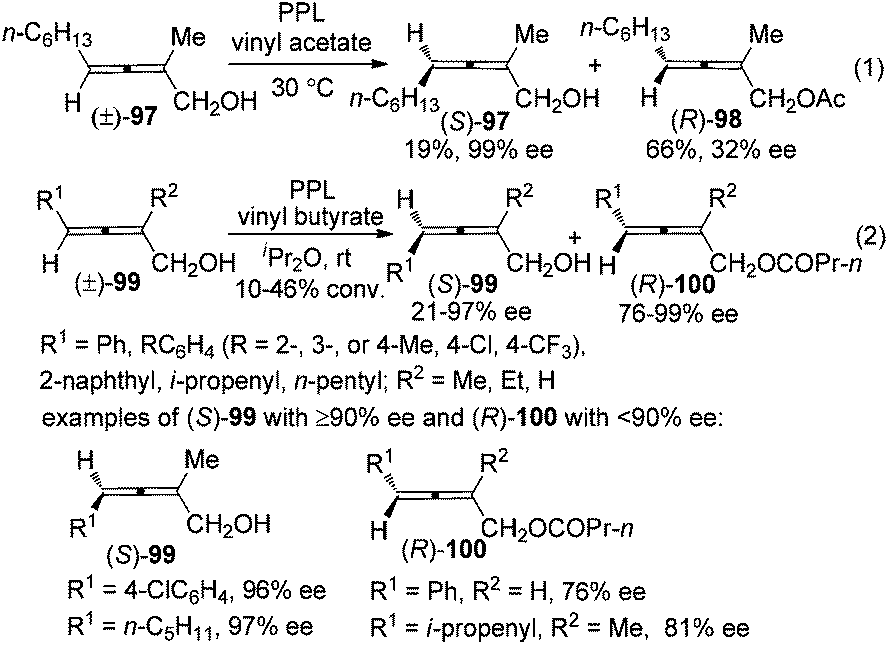
For 2-substituted axially chiral 2,3-allenols, this group found that PPL is an optimal enzyme for the resolution of this type of substrates; for example, trisubstituted 2,3-allenol (S)-97 was obtained with 99% ee, albeit in low yield (eqn (1), Scheme 32).38 Bäckvall et al. studied the kinetic resolution of this type of allenols using vinyl butyrate as the acyl donor and found that 4-aryl substituted allenols were resolved more efficiently than 4-alkyl substituted ones, affording the corresponding allenyl esters (R)-100 with excellent enantioselectivity. A substituent at 2-position is required as when R2 is hydrogen, a low reaction rate and selectivity were observed (eqn (2), Scheme 32).39a
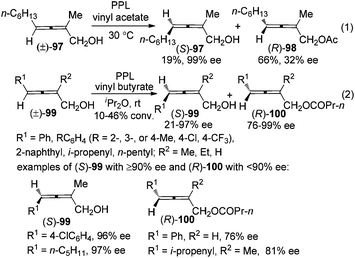 |
| | Scheme 32 Resolution of 2-substituted 2,3-allenols with PPL. PPL = porcine pancreatic lipase. | |
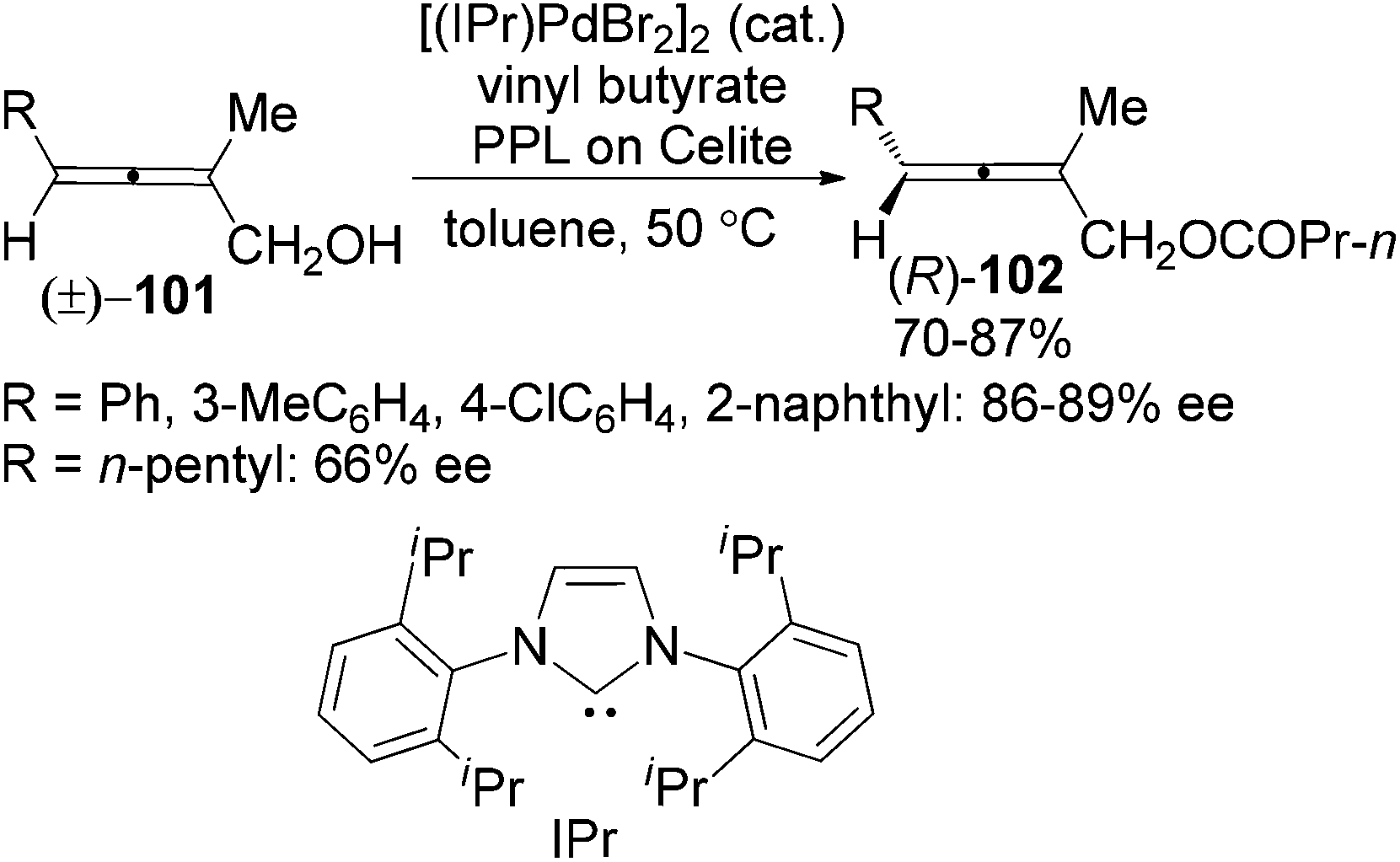
Following this study, the same group developed a dynamic kinetic resolution (DKR) of trisubstituted 2,3-allenols 101 by combining enzymatic dynamic kinetic resolution with Pd-catalyzed in situ racemization of allenes, providing allenyl butyrates 102 in 70–87% yields with 86–89% ee for aryl substrates and 66% ee when R is an n-pentyl group (Scheme 33).39b However, the substrate scope was very limited as only 2-methyl substituted substrates are applicable. The N-heterocyclic carbene (IPr) was found to be the optimal ligand for ensuring a faster racemization of the chiral allene moiety in the allenol than the allenyl ester, which is of critical importance for the DKR process.
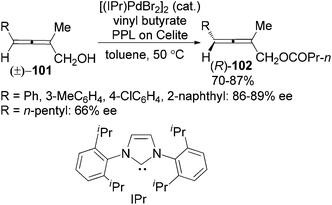 |
| | Scheme 33 Dynamic kinetic resolution of trisubstituted 2,3-allenols. | |
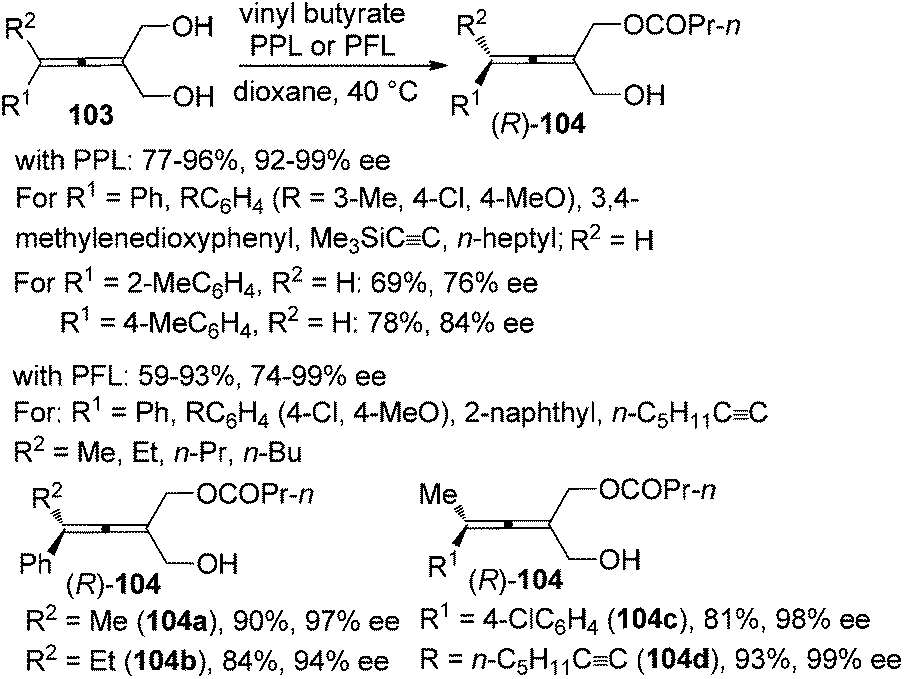
Enzymatic desymmetrization of prochiral allenes is another way for the preparation of axially chiral allenes, which may also overcome the intrinsic yield limitation (≤50%) encountered in the traditional kinetic resolution. Deska and co-workers demonstrated that porcine pancreatic lipase (PPL) is an excellent biocatalyst for the desymmetrization of trisubstituted prochiral allenic diols 103, producing 2,3-allenols with an extra ester group 104 in moderate to high yields with excellent enantioselectivity except when R1 is a 2-MeC6H4 or 4-MeC6H4 group (Scheme 34).40a For the tetrasubstituted substrates, the authors revealed that the lipase from Pseudomonas fluorescens (PFL) is a more effective catalyst in terms of reaction rate and selectivity and tetrasubstituted 2,3-allenols 104a–d were obtained with >90% ee.40b
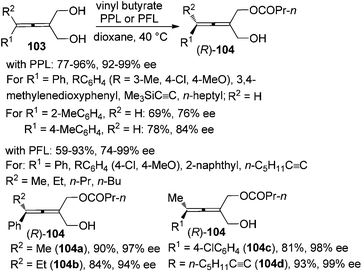 |
| | Scheme 34 Enzymatic desymmetrization of prochiral allenic diols. PFL = Pseudomonas fluorescens lipase. | |
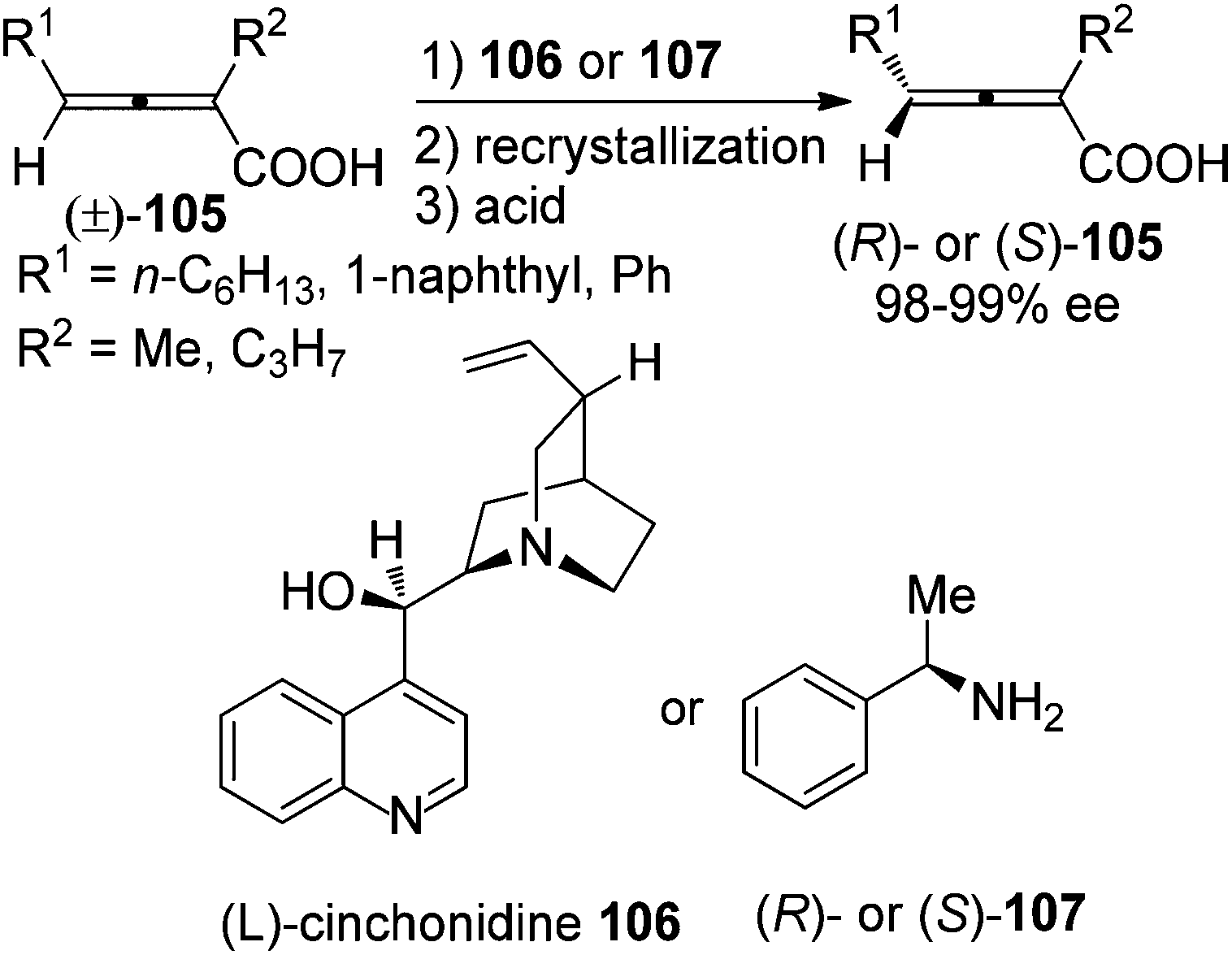
7.1.2 Kinetic resolution using other chiral reagents.
In the presence of a readily available and inexpensive chiral amine such as (L)-cinchonidine 106 or methylbenzylamine 107, racemic 2,3-allenoic acids 105 were classically resolved to afford the corresponding axially chiral ones with 98–99% ee after recrystallization and acidification of the acid–base salts with very low efficiency and limited scope (Scheme 35).41
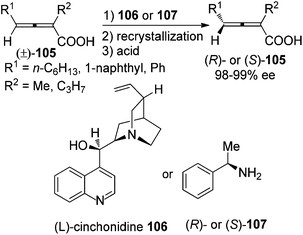 |
| | Scheme 35 Resolution of 2,3-allenoic acids with chiral amines. | |
Besides enzyme and chiral amine, organocatalysts such as bisphosphoric acid 109 can also be applied to the kinetic resolution of racemic 2,3-allenoates in the presence of aldehyde 110 and amine 111: optically active 2,3-allenoates 108 and 1,3-dipolar cycloaddition products 3-methylenepyrrolidine derivatives 112 were obtained in 35–48% yield with 85–99% ee and in 39–57% yield with 64–94% ee, respectively (Scheme 36).42 However, this reaction may not be applied to aryl-substituted substrates.
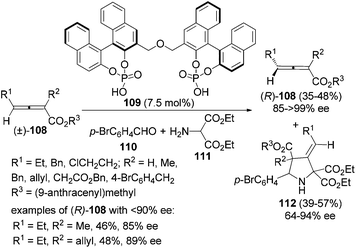 |
| | Scheme 36 Kinetic resolution of racemic 2,3-allenoates via organocatalysis. | |
7.2 Pd-catalyzed nucleophilic allenylation reactions
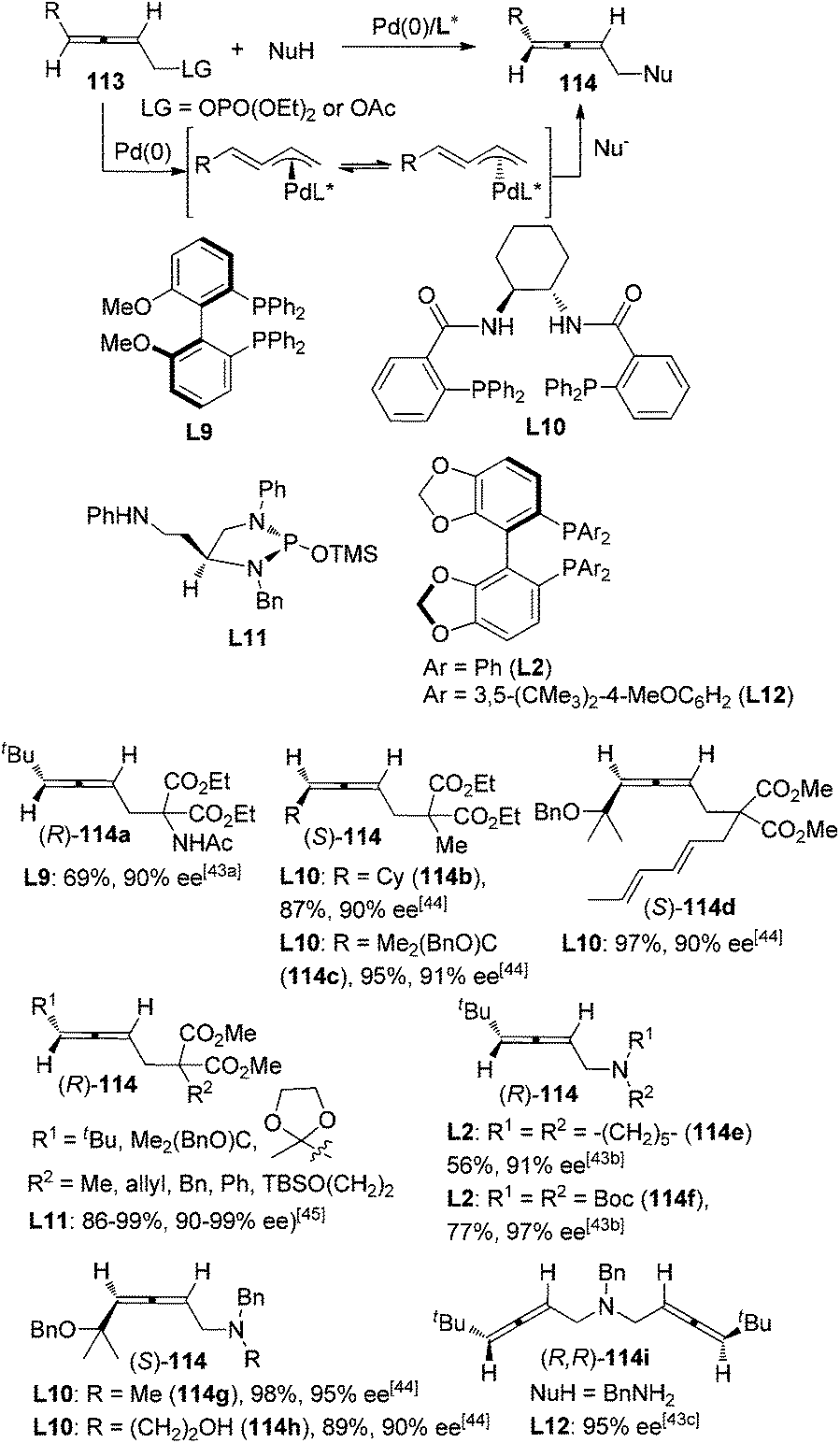
Pd-catalyzed asymmetric synthesis of axially chiral allenes with a nucleophilic functionality from 2,3-allenyl phosphates or carboxylates 113 in the presence of a chiral ligand and a nucleophile such as amine or malonate derivative has been extensively studied by Imada,43 Murahashi,43a,b Naota,43b,c Trost,44 and Hamada45et al. since 2002, leading to functionalized allenes 114 in practical yield with moderate to good enantiopurity. Allene products with high enantiopurity (≥90% ee) are shown in Scheme 37. This part of the work has been well summarized by Ogasawara in 20094e and thus will not be discussed in detail here. However, it should be noted that a sterically bulky substituent on the allene moiety is generally required for achieving a high level of enantioselectivity. In addition, the scope of nucleophiles also awaits further expansion.
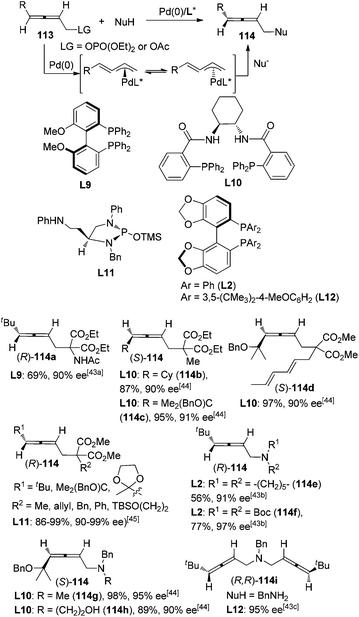 |
| | Scheme 37 Pd-catalyzed asymmetric synthesis of axially chiral allenes from 2,3-allenyl phosphates or carboxylates. | |
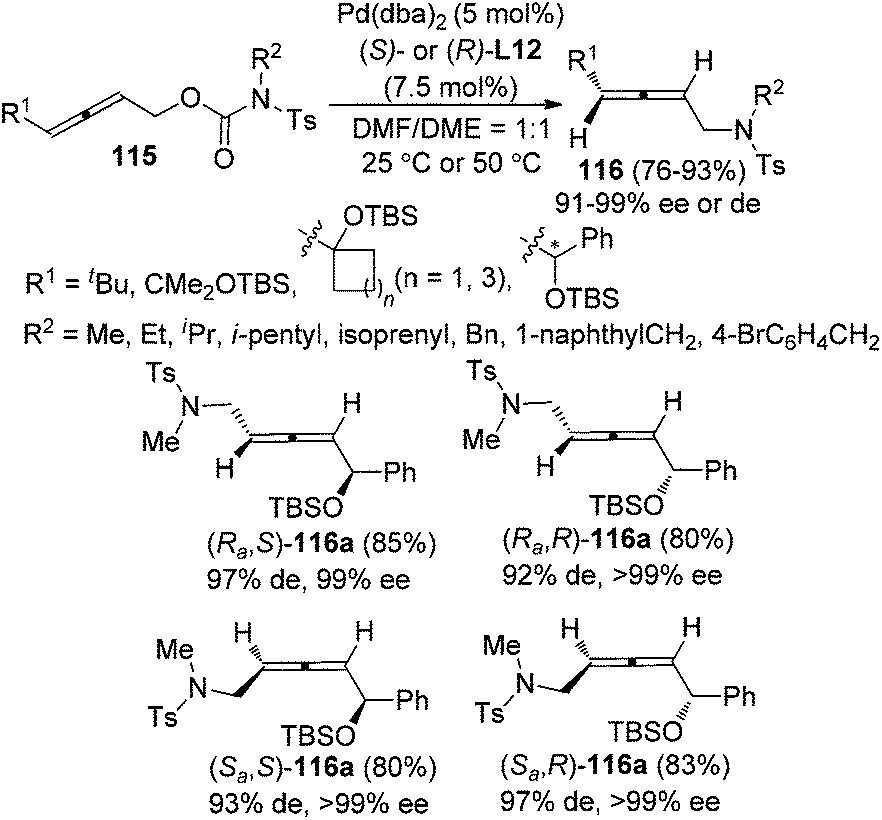
While previous studies have focused on intramolecular reactions, an enantioselective synthesis of axially chiral allenyl amines 116via “intramolecular” decarboxylative amination of allenyl N-tosylcarbamates 115 was recently developed by our group (Scheme 38).46 The chiral ligand (S)- or (R)-L12 imparted remarkable enantio- and diastereoselectivity under Pd catalysis. However, as observed in previous studies, a sterically bulky substituent on the allene moiety (R1) is required in order to achieve high enantioselectivity.
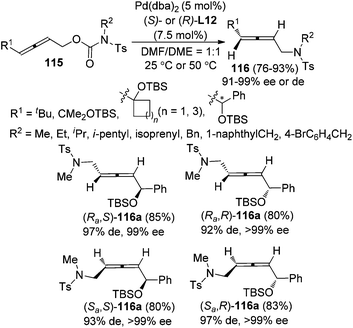 |
| | Scheme 38 Pd-catalyzed synthesis of axially chiral allenyl amines via decarboxylative amination. | |
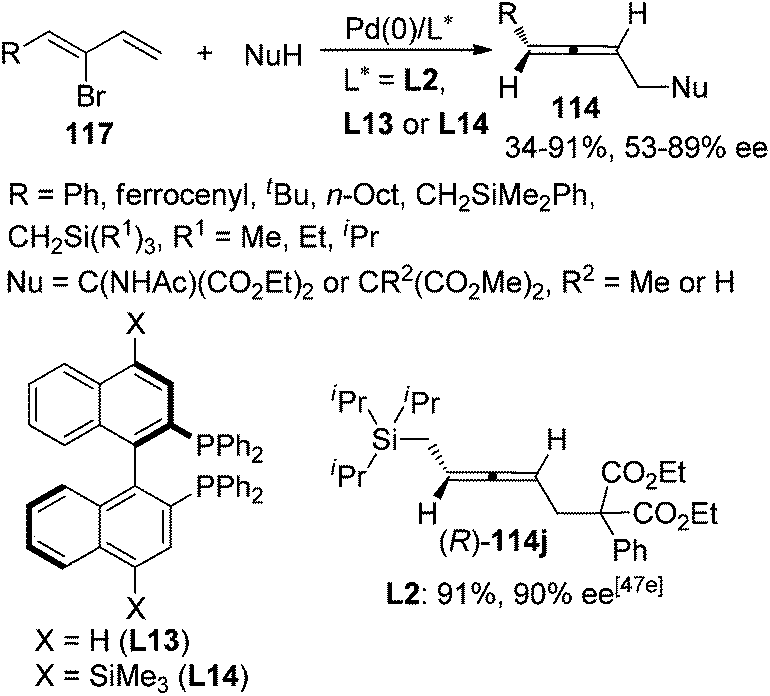
Besides 2,3-allenyl phosphates or carboxylates 113, 2-bromo-1,3-dienes 117 have also been utilized by Hayashi,47a,c Takahashi,47d,e and Ogasawara47et al. to synthesize functionalized axially chiral allenes in a similar manner, although the enantioselectivities were generally lower than using 2,3-allenyl phosphates or carboxylates 113 as the starting material and only one product (R)-114j was obtained with high enantiopurity (≥90% ee) (Scheme 39).
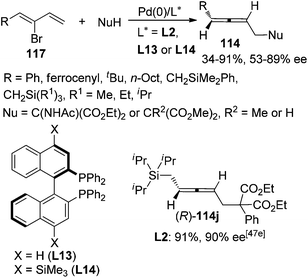 |
| | Scheme 39 Pd-catalyzed asymmetric synthesis of axially chiral allenes from 2-bromo-1,3-dienes. | |
8. From enol triflates via β-hydride elimination
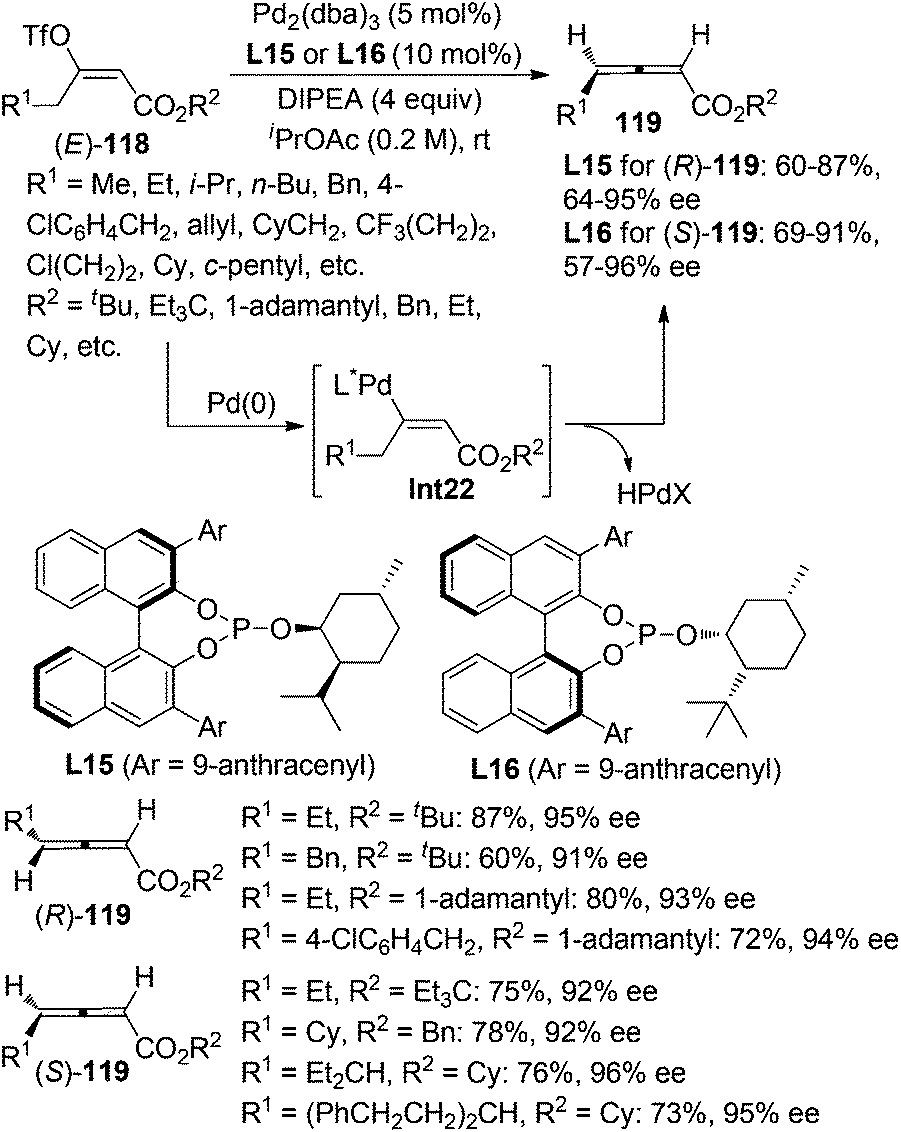
While β-hydride elimination of alkyl palladium species is commonly observed in the traditional Heck reaction,48a β-hydride elimination of vinyl palladium species has rarely been observed49 as it was considered to be an energetically unfavorable process,48 albeit formation of allene intermediate via β-hydride elimination has been proposed in several transformations.50 However, Miura and co-workers reported that trisubstituted allenes could be prepared via the Pd-catalyzed coupling reactions of dialkylacetylenes with aryl bromides, which was believed to proceed via β-hydride elimination of vinyl palladium intermediate under a very high temperature (130 °C).49a Very recently, Frantz and co-workers realized such a concept by developing a Pd-catalyzed asymmetric β-hydride elimination for the synthesis of axially chiral 2,3-allenoates 119 from (E)-enol triflates 118 in the presence of the newly developed chiral phosphite ligand L15 or L16 (Scheme 40).51a In sharp contrast with the aforementioned work of Miura et al.,49a the current reaction may be carried out at room temperature, which may explain why further hydropalladation of the allene products delivering 1,3-dienes in the authors’ previous study51b was inhibited in this catalytic system. In addition, low temperature is required to avoid in situ racemization of the electron-deficient chiral allene products. A limitation is that aryl-substituted 2,3-allenoate may not be accessible via this procedure. Besides, trisubstituted 2,3-allenoates were obtained with low conversion and enantioselectivity from the corresponding fully substituted (E)-enol triflates.
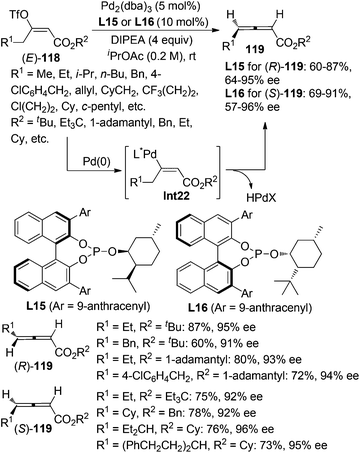 |
| | Scheme 40 Pd-catalyzed asymmetric synthesis of 2,3-allenoates from (E)-enol triflates. | |
9. Conclusions and perspectives
Despite the substantial advances that have been made so far, enantioselective approaches to obtain axially chiral allenes are still just at the very early stage and thus of high current interest, especially in the area of asymmetric catalysis: developing chiral ligands of new skeletons or modifying known privileged ligands for catalytic enantioselective synthesis of allenes would undoubtedly be one of the most active areas as many reactions that were once considered as impossible or formidable are now realized with the aid of novel chiral ligands.
Secondly, although the first synthesized axially chiral allene 3 was prepared via organocatalysis (Scheme 1), this area remains largely underdeveloped and may be worth exploring as many types of chiral organocatalysts are readily available nowadays. The chiral base-catalyzed isomerization reactions (section 6.1), for example, still suffer from limited substrate scope and more general approaches are highly sought after. In addition, propargylic compounds have only been used in isomerization reactions to date; a more desirable and challenging goal is to access tri- or tetrasubstituted allenes in the presence of an external electrophile (Scheme 26).
Thirdly, as β-hydride elimination of vinyl palladium species has been proven to be feasible for the synthesis of both racemic and enantioenriched allenes, further efforts to establish more protocols with broader substrate scope and better reactivity and enantioselectivity should be a promising area. In addition to palladium, other transition metals such as rhodium and iridium may also be considered for such a β-hydride elimination strategy. Other types of β-elimination will also be applied.
Last but not least, completely new approaches to obtain allenes will be highly expected. Overall, given the rapid development of allene chemistry and the widespread utility of axially chiral allenes, chemists around the world will surely spare no efforts to overcome the challenges in the next few decades. The efforts to obtain axially chiral allenes have already been initiated.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21232006) and the National Basic Research Program of China (2011CB808700) for our own research in this area is greatly appreciated.
Notes and references
-
Stereochemistry of Organic Compounds, ed. E. L. Eliel, S. H. Wilen and L. N. Mander, John Wiley & Sons, New York, 1994 Search PubMed.
-
(a) J. H. van't Hoff, Arch. Neerl. Sci. Exactes Nat., 1874, 9, 445 Search PubMed;
(b) J. A. Le Bel, Bull. Soc. Chim. Fr., 1874, 22, 337 Search PubMed.
- For selected reviews on the chemistry of allenes, see:
(a) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef PubMed;
(b) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed;
(c) M. Brasholz, H. U. Reissig and R. Zimmer, Acc. Chem. Res., 2009, 42, 45 CrossRef CAS PubMed;
(d) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS PubMed;
(e) B. Alcaide, P. Almendros and T. Martínez del Campo, Chem. – Eur. J., 2010, 16, 5836 CrossRef CAS PubMed;
(f) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed;
(g) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed;
(h) F. López and J. L. Mascareñas, Chem. – Eur. J., 2011, 17, 418 CrossRef PubMed;
(i) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed;
(j) J. Te and S. Ma, Acc. Chem. Res., 2014, 47, 989 CrossRef PubMed.
- For selected reviews on the synthesis of allenes, see:
(a) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2002, 41, 2933 CrossRef;
(b)
Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, Germany, 2004, vol. 1 and 2 Search PubMed;
(c) N. Krause and A. Hoffmann-Röder, Tetrahedron, 2004, 60, 11671 CrossRef CAS PubMed;
(d) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795 CrossRef CAS PubMed;
(e) M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259 CrossRef CAS PubMed;
(f) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC;
(g) R. K. Neff and D. E. Frantz, ACS Catal., 2014, 4, 519 CrossRef CAS.
-
(a) P. Maitland and W. H. Mills, Nature, 1935, 135, 994 CrossRef CAS;
(b) P. Maitland and W. H. Mills, J. Chem. Soc., 1936, 987 RSC.
-
J. H. van't Hoff, La Chimie dans L'Espace, Bazemdijk, Rotterdam, 1875 Search PubMed.
-
(a) P. Rona and P. Crabbé, J. Am. Chem. Soc., 1968, 90, 4733 CrossRef CAS;
(b) P. Rona and P. Crabbé, J. Am. Chem. Soc., 1969, 91, 3289 CrossRef CAS.
- For early leading references, see:
(a) J.-L. Luche, E. Barreiro, J.-M. Dollat and P. Crabbé, Tetrahedron Lett., 1975, 16, 4615 CrossRef;
(b) J. M. Dollat, J. L. Luche and P. Crabbé, J. Chem. Soc., Chem. Commun., 1977, 761 RSC;
(c) A. Claesson and L.-I. Olsson, Acta Chem., Scand., 1979, B33, 679 Search PubMed;
(d) C. J. Elsevier, P. Vermeer, A. Gedanken and W. Runge, J. Org. Chem., 1985, 50, 364 CrossRef CAS;
(e) C. J. Elsevier and P. Vermeer, J. Org. Chem., 1989, 54, 3726 CrossRef CAS;
(f) I. Marek, P. Mangeney, A. Alexakis and J. F. Normant, Tetrahedron Lett., 1986, 27, 5499 CrossRef CAS;
(g) A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, J.
Am. Chem. Soc., 1990, 112, 8042 CAS;
(h) A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, Tetrahedron, 1991, 47, 1677 CrossRef CAS;
(i) O. W. Gooding, C. C. Beard, D. Y. Jackson, D. L. Wren and G. F. Cooper, J. Org. Chem., 1991, 56, 1083 CrossRef CAS.
- Racemization of chiral allenes by organocuprate reagents:
(a) A. Claesson and L.-I. Olsson, J. Chem. Soc., Chem. Commun., 1979, 524 RSC;
(b) H. Westmijze, I. Nap, J. Meijer, H. Kleijn and P. Vermeer, Recl. Trav. Chim. Pays-Bas, 1983, 102, 154 CrossRef CAS.
-
(a) H. Ohmiya, U. Yokobori, Y. Makida and M. Sawamura, Org. Lett., 2011, 13, 6312 CrossRef CAS PubMed;
(b) M. Yang, N. Yokokawa, H. Ohmiya and M. Sawamura, Org. Lett., 2012, 14, 816 CrossRef CAS PubMed.
- M. R. Uehling, S. T. Marionni and G. Lalic, Org. Lett., 2012, 14, 362 CrossRef CAS PubMed.
- For a review, see: Y. Deng, Z. Gu and S. Ma, Chin. J. Org. Chem., 2006, 26, 1468 CAS.
- K. Kobayashi, H. Naka, A. E. H. Wheatley and Y. Kondo, Org. Lett., 2008, 10, 3375 CrossRef CAS PubMed.
- H. Li, D. Müller, L. Guénée and A. Alexakis, Org. Lett., 2012, 14, 5880 CrossRef CAS PubMed.
-
(a) P. H. Dixneuf, T. Guyot, M. D. Ness and S. M. Roberts, Chem. Commun., 1997, 2083 RSC;
(b) R. Riveiros, D. Rodríguez, J. Sestelo and L. Sarandeses, Org. Lett., 2006, 8, 1403 CrossRef CAS PubMed;
(c) M. Yoshida, T. Okada and K. Shishido, Tetrahedron, 2007, 63, 6996 CrossRef CAS PubMed.
- J. A. Marshall, M. A. Wolf and E. M. Wallace, J. Org. Chem., 1997, 62, 367 CrossRef CAS PubMed.
-
(a) Y. Wang and S. Ma, Adv. Synth. Catal., 2013, 355, 741 CrossRef CAS;
(b) Y. Wang, W. Zhang and S. Ma, Org. Chem. Front., 2014, 1, 807 RSC.
- Y. Wang, W. Zhang and S. Ma, J. Am. Chem. Soc., 2013, 135, 11517 CrossRef CAS PubMed.
- K. Mikami and A. Yoshida, Angew. Chem., Int. Ed. Engl., 1997, 36, 858 CrossRef CAS.
- A. G. Myers and B. Zheng, J. Am. Chem. Soc., 1996, 118, 4492 CrossRef CAS.
- X. Pu and J. M. Ready, J. Am. Chem. Soc., 2008, 130, 10874 CrossRef CAS PubMed.
- P. Crabbé, H. Fillion, D. André and J.-L. Luche, J. Chem. Soc., Chem. Commun., 1979, 859 RSC.
-
(a) J. Kuang and S. Ma, J. Org. Chem., 2009, 74, 1763 CrossRef CAS PubMed;
(b) J. Kuang and S. Ma, J. Am. Chem. Soc., 2010, 132, 1786 CrossRef CAS PubMed;
(c) X. Tang, C. Zhu, T. Cao, J. Kuang, W. Lin, S. Ni, J. Zhang and S. Ma, Nat. Commun., 2013, 4 DOI:10.1038/ncomms3450.
-
(a) V. K.-Y. Lo, M.-K. Wong and C.-M. Che, Org. Lett., 2008, 10, 517 CrossRef CAS PubMed;
(b) V. K.-Y. Lo, C.-Y. Zhou, M.-K. Wong and C.-M. Che, Chem. Commun., 2010, 46, 213 RSC;
(c) V. K.-Y. Lo, Y. Liu, M.-K. Wong and C.-M. Che, Org. Lett., 2006, 8, 1529 CrossRef CAS PubMed.
- J. Ye, S. Li, B. Chen, W. Fan, J. Kuang, J. Liu, Y. Liu, B. Miao, B. Wan, Y. Wang, X. Xie, Q. Yu, W. Yuan and S. Ma, Org. Lett., 2012, 14, 1346 CrossRef CAS PubMed.
-
(a) J. Ye, R. Lü, W. Fan and S. Ma, Tetrahedron, 2013, 69, 8959 CrossRef CAS PubMed;
(b) R. Lü, J. Ye, T. Cao, B. Chen, W. Fan, W. Lin, J. Liu, H. Luo, B. Miao, S. Ni, X. Tang, N. Wang, Y. Wang, X. Xie, Q. Yu, W. Yuan, W. Zhang, C. Zhu and S. Ma, Org. Lett., 2013, 15, 2254 CrossRef PubMed;
(c) J. Ye, W. Fan and S. Ma, Chem. – Eur. J., 2013, 19, 716 CrossRef CAS PubMed.
-
(a) M. Periasamy, N. Sanjeevakumar, M. Dalai, R. Gurubrahamam and P. O. Reddy, Org. Lett., 2012, 14, 2932 CrossRef CAS PubMed. For reports on other optically active amines, see:
(b) R. Gurubrahamam and M. Periasamy, J. Org. Chem., 2013, 78, 1463 CrossRef CAS PubMed;
(c) M. Periasamy, P. O. Reddy and N. Sanjeevakumar, Eur. J. Org. Chem., 2013, 3866 CrossRef CAS.
-
(a) Y. Matsumoto, M. Naito, Y. Uozumi and T. Hayashi, J. Chem. Soc., Chem. Commun., 1993, 1468 RSC;
(b) J. W. Han, N. Tokunaga and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 12915 CrossRef CAS;
(c) M. Ogasawara, A. Ito, K. Yoshida and T. Hayashi, Organometallics, 2006, 25, 2715 CrossRef CAS;
(d) T. Hayashi, N. Tokunaga and K. Inoue, Org. Lett., 2004, 6, 305 CrossRef CAS PubMed;
(e) T. Nishimura, H. Makino, M. Nagaosa and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 12865 CrossRef CAS PubMed.
-
(a) I. Tömösközi and H. J. Bestmann, Tetrahedron Lett., 1964, 5, 1293 CrossRef;
(b) S. Musierowicz, A. E. Wróblewski and H. Krawczyk, Tetrahedron Lett., 1975, 16, 437 CrossRef.
- J. Yamazaki, T. Watanabe and K. Tanaka, Tetrahedron: Asymmetry, 2001, 12, 669 CrossRef CAS.
-
(a) C. Li, X. Wang, X. Sun, Y. Tang, J. Zheng, Z. Xu, Y. Zhou and L. Dai, J. Am. Chem. Soc., 2007, 129, 1494 CrossRef CAS PubMed;
(b) C. Li, X. Sun, Q. Jing and Y. Tang, Chem. Commun., 2006, 2980 RSC;
(c) C. Li, B. Zhu, L. Ye, Q. Jing, X. Sun, Y. Tang and Q. Shen, Tetrahedron, 2007, 63, 8046 CrossRef CAS PubMed.
- For two earlier examples affording allenes with low optical activity, see:
(a) T. L. Jacobs and D. Dankner, J. Org. Chem., 1957, 22, 1424 CrossRef CAS;
(b) U. Mödlhammer and H. Hopf, Angew. Chem., Int. Ed. Engl., 1975, 14, 501 CrossRef.
- M. Oku, S. Arai, K. Katayama and T. Shioiri, Synlett, 2000, 493 CAS.
- H. Liu, D. Leow, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2009, 131, 7212 CrossRef CAS PubMed.
- H. Qian, X. Yu, J. Zhang and J. Sun, J. Am. Chem. Soc., 2013, 135, 18020 CrossRef CAS PubMed.
- T. Hashimoto, K. Sakata, F. Tamakuni, M. J. Dutton and K. Maruoka, Nat. Chem., 2013, 5, 240 CrossRef CAS PubMed.
- S. Ramaswamy, R. Hui and J. B. Jones, J. Chem. Soc., Chem. Commun., 1986, 1545 RSC.
-
D. Xu, Enzyme-Catalyzed Kinetic Resolution of Allenols and Propargyl Alcohols and Their Synthetic Applications, Doctoral Dissertation of Shanghai Institute of Organic Chemistry, Shanghai, China, 2003, p. 47 Search PubMed.
-
(a) J. Deska and J.-E. Bäckvall, Org. Biomol. Chem., 2009, 7, 3379 RSC;
(b) J. Deska, C. del Pozo Ochoa and J.-E. Bäckvall, Chem. – Eur. J., 2010, 16, 4447 CrossRef CAS PubMed.
-
(a) C. Sapu, J.-E. Bäckvall and J. Deska, Angew. Chem., Int. Ed., 2011, 50, 9731 CrossRef CAS PubMed;
(b) M. Hammel and J. Deska, Synthesis, 2012, 3789 CAS.
- S. Ma and S. Wu, Chem. Commun., 2001, 441 RSC.
- J. Yu, W. Chen and L. Gong, Org. Lett., 2010, 12, 4050 CrossRef CAS PubMed.
-
(a) Y. Imada, K. Ueno, K. Kutsuwa and S.-I. Murahashi, Chem. Lett., 2002, 140 CrossRef CAS;
(b) Y. Imada, M. Nishida, K. Kutsuwa, S.-I. Murahashi and T. Naota, Org. Lett., 2005, 7, 5837 CrossRef CAS PubMed;
(c) Y. Imada, M. Nishida and T. Naota, Tetrahedron Lett., 2008, 49, 4915 CrossRef CAS PubMed.
- B. M. Trost, D. R. Fandrick and D. C. Dinh, J. Am. Chem. Soc., 2005, 127, 14186 CrossRef CAS PubMed.
- T. Nemoto, M. Kanematsu, S. Tamura and Y. Hamada, Adv. Synth. Catal., 2009, 351, 1773 CrossRef CAS.
- B. Wan and S. Ma, Angew. Chem., Int. Ed., 2013, 52, 441 CrossRef CAS PubMed.
-
(a) M. Ogasawara, H. Ikeda, T. Nagano and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 2089 CrossRef CAS;
(b) M. Ogasawara, K. Ueyama, T. Nagano, Y. Mizuhata and T. Hayashi, Org. Lett., 2003, 5, 217 CrossRef CAS PubMed;
(c) M. Ogasawara, T. Nagano and T. Hayashi, J. Org. Chem., 2005, 70, 5764 CrossRef CAS PubMed;
(d) M. Ogasawara, H. L. Ngo, T. Sakamoto, T. Takahashi and W. Lin, Org. Lett., 2005, 7, 2881 CrossRef CAS PubMed;
(e) M. Ogasawara, Y. Ge, A. Okada and T. Takahashi, Eur. J. Org. Chem., 2012, 1656 CrossRef CAS.
-
(a)
J. Tsuji, Palladium Reagents and Catalysts, John Wiley & Sons Ltd, England, 2004 Search PubMed;
(b) E. Negishi, C. Copéret, S. Ma, S. Liou and F. Liu, Chem. Rev., 1996, 96, 365 CrossRef CAS PubMed.
-
(a) S. Pivsa-Art, T. Satoh, M. Miura and M. Nomura, Chem. Lett., 1997, 823 CrossRef CAS;
(b) M. Catellani, E. Motti and S. Baratta, Org. Lett., 2001, 3, 3611 CrossRef CAS PubMed.
-
(a) H. Sheng, S. Lin and Y. Huang, Tetrahedron Lett., 1986, 27, 4893 CrossRef CAS;
(b) B. M. Trost and T. Schmidt, J. Am. Chem. Soc., 1988, 110, 2301 CrossRef CAS;
(c) X. Lu, J. Ji, D. Ma and W. Shen, J. Org. Chem., 1991, 56, 5774 CrossRef CAS;
(d) I. Kadota, A. Shibuya, Y. S. Gyoung and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 10262 CrossRef CAS;
(e) I. Kadota, A. Shibuya, L. M. Lutete and Y. Yamamoto, J. Org. Chem., 1999, 64, 4570 CrossRef CAS;
(f) L. M. Lutete, I. Kadota and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 1622 CrossRef CAS PubMed;
(g) C. Fu and S. Ma, Org. Lett., 2005, 7, 1605 CrossRef CAS PubMed.
-
(a) I. T. Crouch, R. K. Neff and D. E. Frantz, J. Am. Chem. Soc., 2013, 135, 4970 CrossRef CAS PubMed;
(b) I. T. Crouch, T. Dreier and D. E. Frantz, Angew. Chem., Int. Ed., 2011, 50, 6128 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. Negishi on the occasion of his 80th birthday. |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.