
Gold(I)- and platinum(IV)-catalyzed intramolecular annulations of allenes towards furans†‡
Cheng-Hang
Liu
and
Zhi-Xiang
Yu
*
Beijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry, Peking University, Beijing, 100871, China. E-mail: yuzx@pku.edu.cn; Web: http://www.chem.pku.edu.cn/zxyu/index.htm
First published on 20th October 2014
Abstract
Intramolecular annulations of allenes towards furans to give six-membered ring fused furans with gold and platinum catalysts have been developed. It was found that allenes can be activated by gold catalysts and attacked by the furan rings to generate six-membered rings. When PtCl4 was used as a catalyst, the annulation also gives six-membered ring fused furans, but the carbon–carbon double bond in the products shifted to form conjugation with the furan ring with a high Z/E ratio.
Six-membered ring fused furans are widely found in natural products with biological properties. Fig. 1 shows some selected examples of these natural products.1 These furanosesquiterpenes and furanoditerpenes are ubiquitous metabolites found in varieties of marine invertebrates. For example, (+)-pallescensin A is suggested to be involved in the defensive mechanisms employed by opisthobranch molluscs1b and (−)-furodysinin shows significant activity against parasitic stages of Nippostrongylus brasiliensis.1d Therefore, there is a high demand to develop new methods and strategies for the synthesis of six-membered ring fused furans, which could further be used in target- and function-oriented synthesis of biologically and pharmaceutically interesting natural products with the six-membered ring fused furan skeletons.
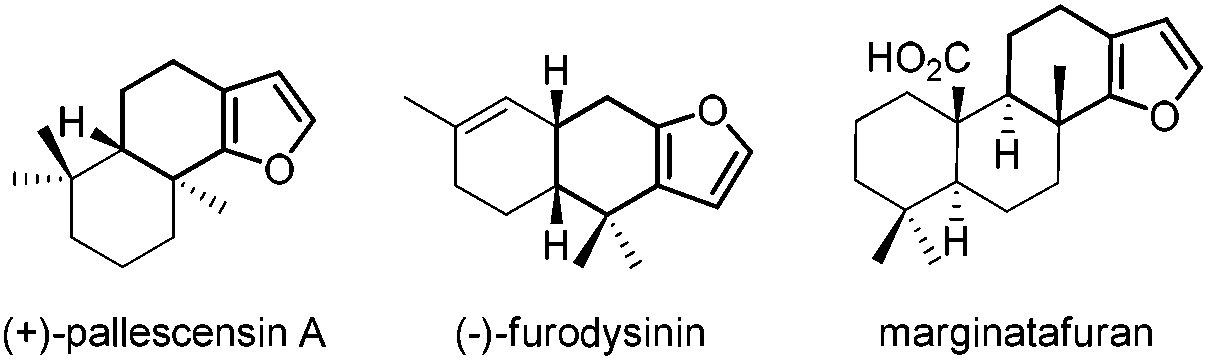 | ||
| Fig. 1 Natural products with six-membered ring fused furans. | ||
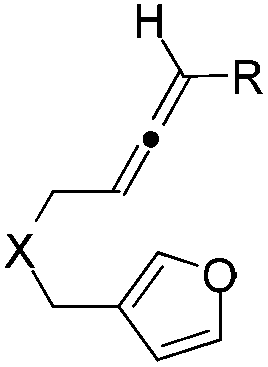
Recently we developed an efficient gold-catalyzed endo-selective intramolecular α-alkenylation of furans with internal alkynes to synthesize the challenging seven-membered ring fused furans (Scheme 1a).2 This strategy was based on the higher reactivity of the α-position over the β-position of furans3 and consequently alkenylation can be realized. We found that six-membered ring fused furans can also be obtained through the intramolecular α-alkenylation strategy via exo-selective cyclization of furans with terminal alkynes or alkynes terminated by electron-withdrawing groups. It was reported that allenes can also be activated and then react with aromatic rings,4 such as electron-rich benzenes,5 indoles,6 and pyrroles,7 to give Friedel–Crafts (F–C) products. We wondered whether the alkynes in our previous substrates can be replaced by allenes so that either six- or seven-membered ring fused furans could be accessed using a gold or a platinum catalyst (Scheme 1c). Herein we report our experimental results of achieving the synthesis of α-alkylated furans from β-allenyl-furans through the gold-catalyzed annulation of allenes towards furans (or hydroarylation of allenes with furans). The method is of high stereoselectivity and only the E configuration products were obtained. We also noticed that the carbon–carbon double bond can shift to the position conjugated with the furan ring with a high Z/E ratio when PtCl4 was used. This reaction is different from previous reports from Mascareñas8 and Toste,9 who independently showed that α-substituted allenyl-furans under gold and platinum catalysis gave [4 + 2] and [4 + 3] type products,10 not the alkylation products (Scheme 1b).11
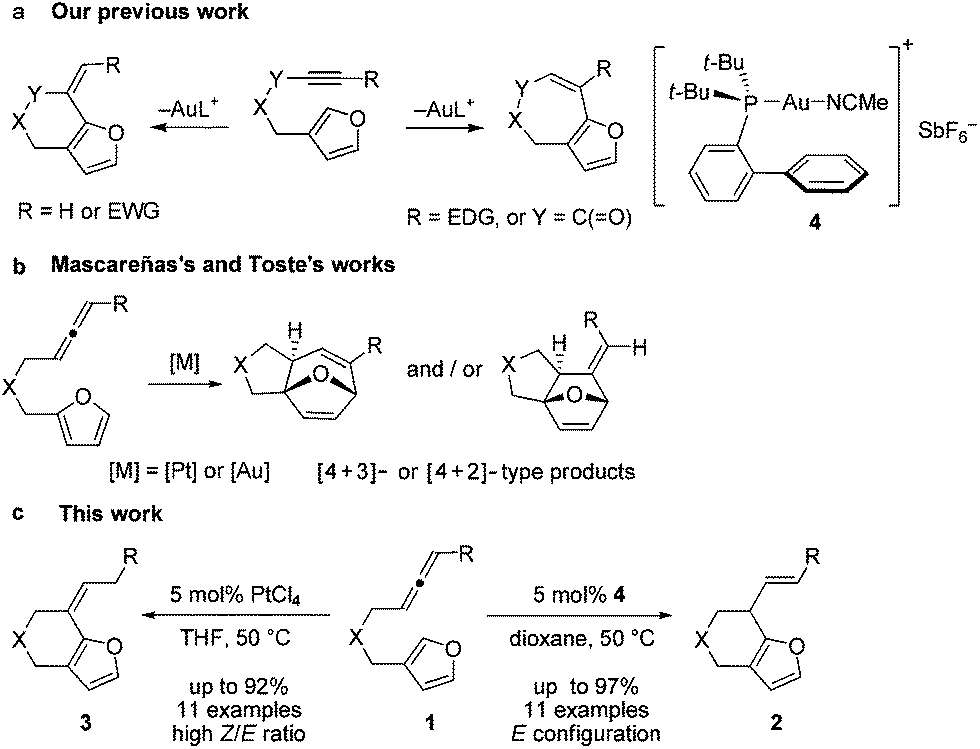 | ||
| Scheme 1 (a) Our previous work about the α-alkenylation of β-yne-furans. (b) [4 + 2] and [4 + 3] type reactions of α-allenyl-furans. (c) The annulation of β-allenyl-furans. | ||
We commenced our investigation by subjecting compound 1a to the standard conditions for α-alkenylation of β-yne-furans using catalyst 4 (Echavarren's catalyst), finding that the desired product 2a can be isolated in a modest yield (65%) and a very long reaction time (60 h) was needed (Table 1, entry 1). This transformation was highly stereoselective because only the annulation product with the E configuration was observed. The reaction yield can be further increased to 86% and the reaction time can be shortened to 7 h when the reaction was carried out at 50 °C (Table 1, entry 2). Then we screened various gold complexes as catalysts (Table 1, entries 3–7). No desired product was obtained when using Au(JohnPhos)Cl as the catalyst, indicating that a ‘naked’ gold cation was required. When using cationic gold catalyst generated from Au(IPr)Cl and AgSbF6 for the annulation reaction, we found that substrate 1a was consumed after 4 h, but the reaction yield was low. Other gold catalysts including the simple AuCl3 and AuCl have also been tested, showing that only a trace amount of 2a was detected by TLC (Table S1,† entries 9 and 10). Several widely used Brønsted acids and Lewis acids such as TfOH, AlCl3, BF3·OEt2 and InCl3 were proved to be ineffective for the target transformation (Table S1,† entries 12–15). We then investigated how solvents affect the reaction outcome, finding that dioxane turned out to be the best solvent and the product 2a can be isolated in 91% yield, whereas no product was detected in MeCN (Table 1, entries 8–12). A catalytic amount of water is required for this transformation (no desired product was observed when 4 Å MS was added to the reaction system), which is similar to our previous gold-catalyzed α-alkenylation reaction of β-yne-furans (Table S1,† entry 23).2
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solventa | Catalyst | Temp. | Time [h] | Product | Yieldb [%] |
a Concentration: 0.05 M.
b Isolated yield, the Z/E ratio of 3a was above 20/1.
c Gold and silver salts were added at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
d 27% 1a was recovered.
e 28% 1a was recovered.
f Yield based on NMR. :1.
d 27% 1a was recovered.
e 28% 1a was recovered.
f Yield based on NMR.
|
||||||
| 1 | DME | [Au(NCMe)JohnPhos]SbF6 | rt | 60 | 2a | 65 |
| 2 | DME | [Au(NCMe)JohnPhos]SbF6 | 50 °C | 7 | 2a | 86 |
| 3 | DME | Au(JohnPhos)Cl | 50 °C | 12 | — | No reaction |
| 4 | DME | Au(PPh3)Cl/AgSbF6c |
50 °C | 24 | 2a | 49d |
| 5 | DME | Au(IPr)Cl/AgSbF6c |
50 °C | 4 | 2a | 83 |
| 6 | DME | Au[P(OPh-2,4-t-Bu)3]Cl/AgSbF6c |
50 °C | 15 | 2a | 71 |
| 7 | DME | Au(XPhos)NTf2 | 50 °C | 48 | 2a | 48e |
| 8 | MeCN | [Au(NCMe)JohnPhos]SbF6 | 50 °C | 7 | — | No reaction |
| 9 | DCE | [Au(NCMe)JohnPhos]SbF6 | 50 °C | 42 | 2a | 76f |
| 10 | Toluene | [Au(NCMe)JohnPhos]SbF6 | 50 °C | 13 | 2a | 88 |
| 11 | THF | [Au(NCMe)JohnPhos]SbF6 | 50 °C | 9 | 2a | 36 |
| 12 | Dioxane | [Au(NCMe)JohnPhos]SbF6 | 50 °C | 6 | 2a | 91 |
| 13 | DME | PtCl2 | 50 °C | 17 | 3a | 73 |
| 14 | DME | PtCl4 | 50 °C | 2 | 3a | 74 |
| 15 | Dioxane | PtCl4 | 50 °C | 2 | 3a | 76 |
| 16 | Toluene | PtCl4 | 50 °C | 4 | 3a | 40f |
| 17 | THF | PtCl4 | 50 °C | 2 | 3a | 92 |
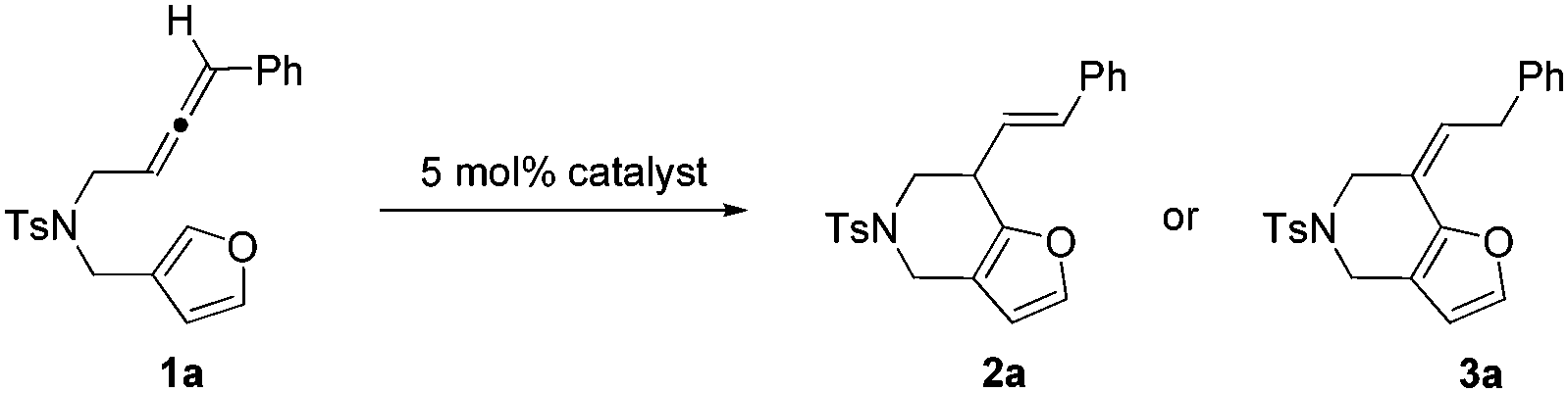
Interestingly, a new product 3a, which is the isomer of 2a and has the double bond in conjugation with furan, was isolated when we used PtCl2 as the catalyst (Table 1, entry 13). Compound 3a has a high Z/E ratio (>20:1). We also found that the reaction time can be dramatically shortened to 2 h without the loss of reaction yield when using PtCl4 instead of PtCl2 as the catalyst (Table 1, entry 14). We screened several common solvents using PtCl4 as the catalyst (Table 1, entries 14–17), and found that product 3a can be obtained in 92% yield in THF. Consequently, THF was chosen as the optimal solvent for the annulation reaction under the catalysis of PtCl4.
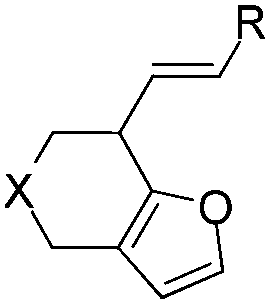
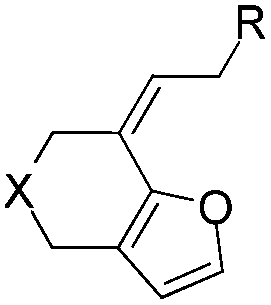
With the optimized reaction conditions in hand, the scope of this intramolecular annulation reaction of β-allenyl-furans was studied. At first, we investigated the influence of the substitutions in the allene moiety, finding that other aryl groups such as para-bromo phenyl, para-methoxy phenyl could also be used in the substrates for both gold- and platinum-catalyzed reactions, giving their corresponding products 2 and 3, respectively (Table 2, entries 2 and 3). Treating compound 1c with PtCl4, a mixture of the alkene shifted product 3c and unshifted product 2c (the ratio of 3c/2c is 4.3:1) was obtained in a combined isolated yield of 58%. The allene moiety of the substrate can be substituted by simple aliphatic groups such as methyl and n-propyl groups, finding that the desired products in high yields were generated under both gold and platinum catalysis (Table 2, entries 4 and 5). Unfortunately, the monosubstituted substrate 1f decomposed under the standard reaction conditions (Table 2, entry 6). For trisubstituted allene substrate 1g, the annulation product 2g could also be formed in an excellent yield using the gold catalyst 4. But a mixture of 2g and 3g was obtained using PtCl4 as the catalyst (Table 2, entry 7).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product 2 yielda [%], time | Product 3 yielda [%], time |
| a Isolated yield. The Z/E ratio of 3 was above 20/1 if not mentioned. b 3c/2c = 4.3/1 based on NMR. c 3g/2g = 1/2.4 based on NMR. d Carried out at 80 °C using a 10 mol% catalyst. e Carried out at 80 °C in dioxane instead of THF using a 10 mol% catalyst. f Z/E = 10/1 based on NMR. g Z/E = 1.3/1 based on NMR. h Z/E = 8/1 based on NMR. | |||
|
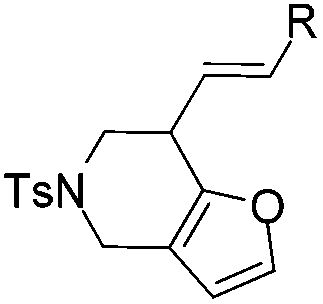
|
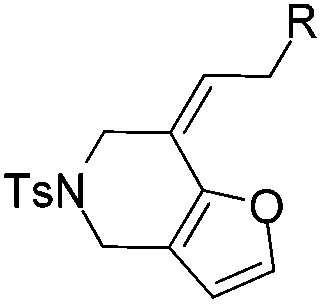
|
|
| 1 | 1a, R = Ph | 2a, 91, 6 h | 3a, 92, 2 h |
| 2 | 1b, R = p-BrPh | 2b, 78, 7 h | 3b, 79, 2.5 h |
| 3 | 1c, R = p-OMePh | 2c, 67, 3 h | 3c, 58b, 2 h |
| 4 | 1d, R = Me | 2d, 79, 5 h | 3d, 80, 1 h |
| 5 | 1e, R = n-Pr | 2e, 91, 4 h | 3e, 80, 2 h |
| 6 | 1f, R = H | Decomposed | Decomposed |
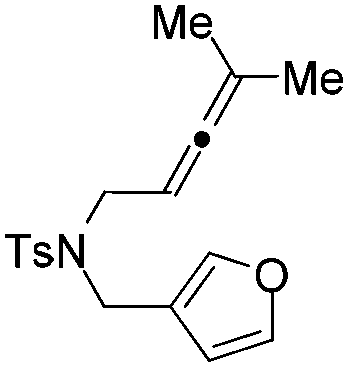
|
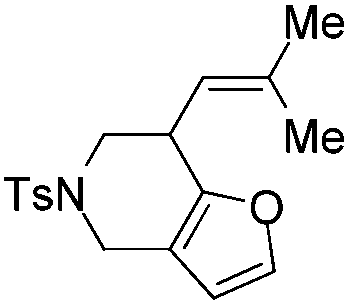
|
|
|
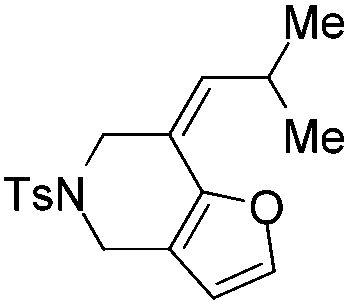
| 7 | 1g | 2g, 97, 4 h | 3g, 88c,4 h |
|
|
|
|
| 8 | 1h, X = NBs, R = p-BrPh | 2h, 93, 8 h | 3h, 84, 2 h |
| 9 | 1i, X = O, R = Ph | 2i , 83, 8 h | 3i , 67, 4 h |
| 10 | 1j, X = C(CO2Me)2, R = Ph | 2j, 86, 4 h | 3j, 89f, 2 h |
| 11 | 1k, X = C(CO2Me)2, R = H | 2k , 63, 3 h | 3k , 72g, 2 h |
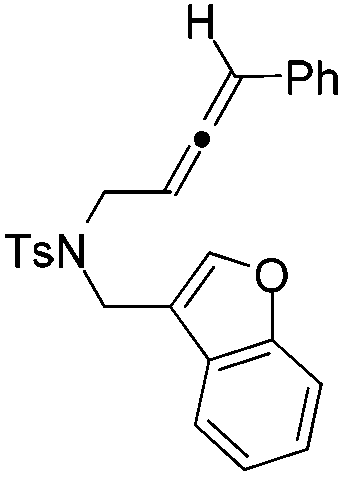
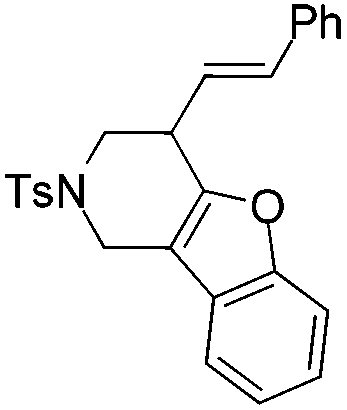
|
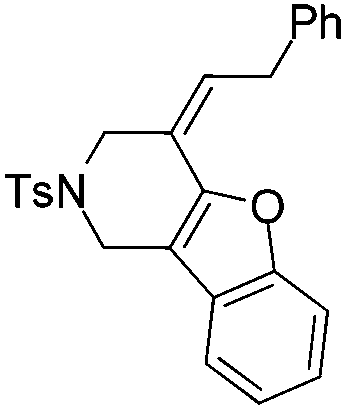
|
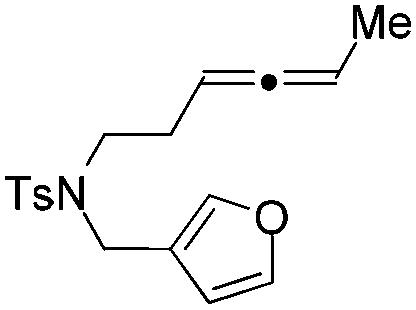
|
|
| 12 | 1l | 2l , 85, 5 h | 3l , 83h, 1 h |
|
|||
| 13 | 1m | No reaction | No reaction |
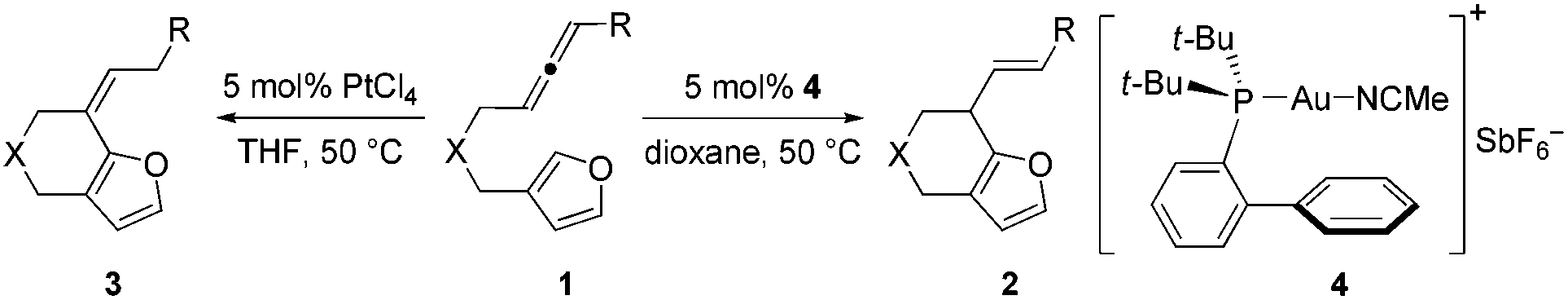
The tether in β-allenyl-furans can be NBs, O and C(CO2Me)2 for the annulation reactions using gold and platinum catalysts (Table 2, entries 8–11). The Z configuration of the double bond in 3 was further confirmed by single X-ray crystallography analysis of product 3h (Fig. 2). For substrates 1i and 1k, the desired annulation reactions had to be carried out at 80 °C in dioxane, considering that their reactions were too slow upon using the above-mentioned standard optimized reaction conditions (Table 2, entries 9 and 11). We observed that the Z/E ratios of the final product were not satisfying for the C-tethered substrate when PtCl4 was used as the catalyst (Table 2, entries 10 and 11). The furan moiety could also be replaced by benzofuran, but the corresponding annulation reaction for β-allenyl-benzofuran substrate 1l had to be carried out at 80 °C in order to obtain a reasonable reaction yield (Table 2, entry 12). Finally, we tested the influence of the tether length, showing that no seven-membered ring fused furans were observed for substrate 1m, even though the reactions were performed at 80 °C (Table 2, entry 13). This further demonstrated that synthesis of the seven-membered ring fused furan was not trivial.
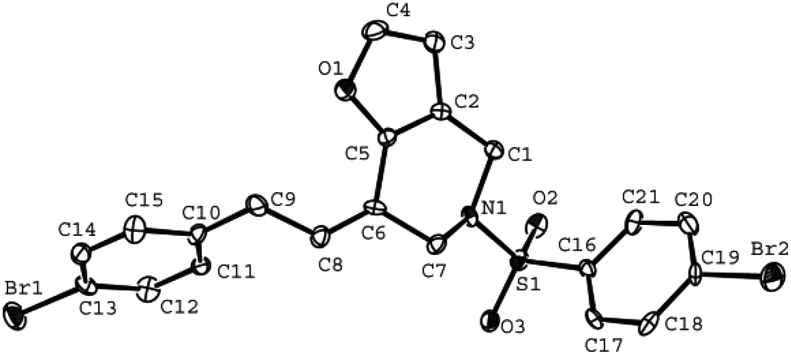 | ||
| Fig. 2 ORTEP representation of crystallized compound 3h (hydrogen atoms are omitted). | ||
We hypothesized that the annulation reaction using gold and platinum catalysts gave the same product 2, but 2 could further isomerize to 3 under platinum catalysis. When we treated 2h with PtCl4, no alkene shifted product 3h was detected (Scheme 2a), showing that compound 3 was not generated through olefin isomerization from 2. To obtain more information on the platinum catalysis, we tried to add additive D2O to the platinum-catalyzed reaction system (Scheme 2b). We found that 3h-d with 15% D incorporation at the C-2 position and 40% D incorporation at the C-1 position was obtained, which indicated that D2O is involved in the olefin isomerization process.
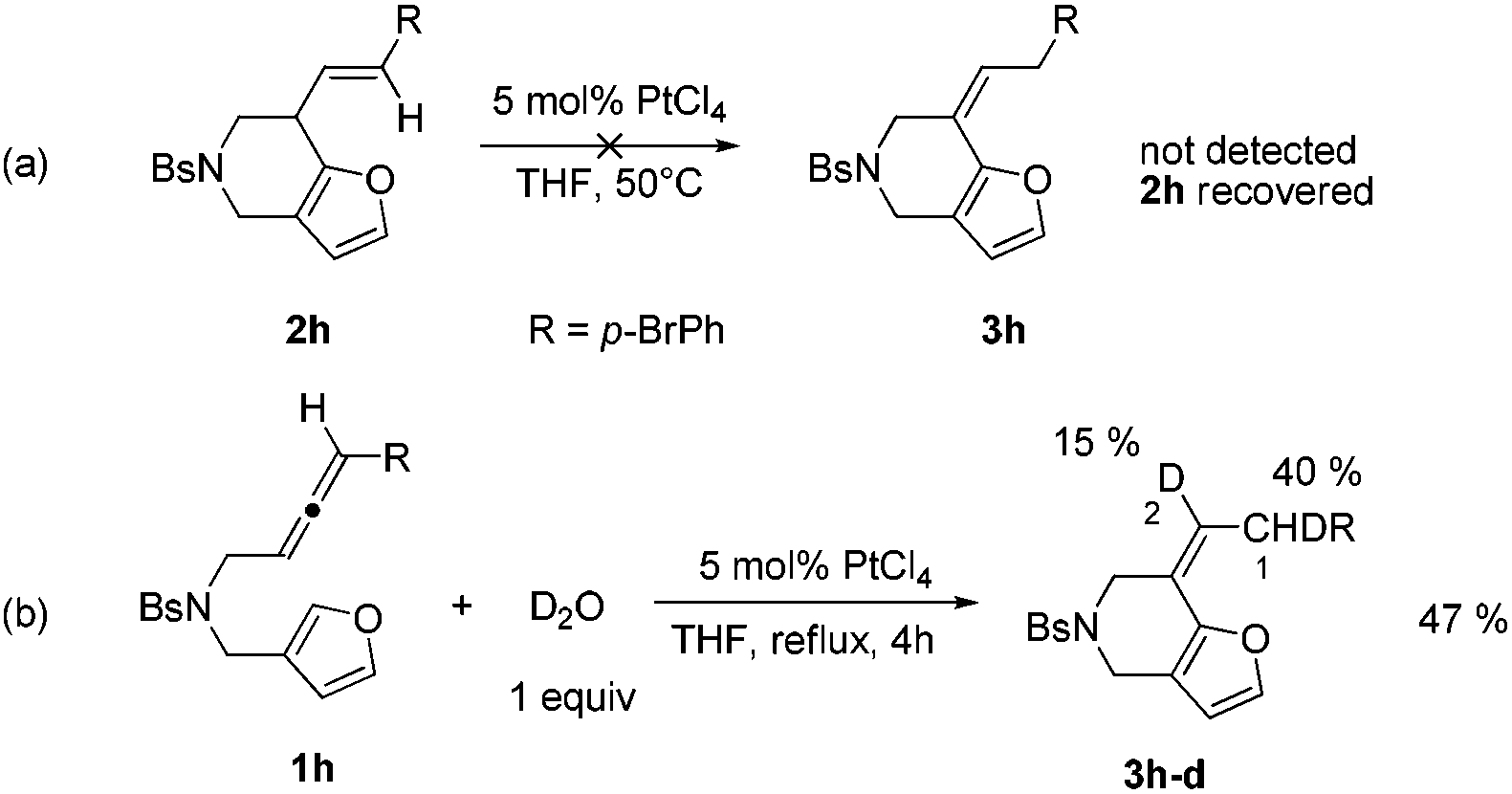 | ||
| Scheme 2 A preliminary mechanistic study. | ||
Based on the experiments, we proposed the following mechanisms to account for the annulation reactions of allenes towards furans using gold and platinum catalysts (Scheme 3). The annulation reaction begins with the coordination of the allene part of the substrate to the metal catalyst, generating a metal–allene complex, intermediate INT I. Then nucleophilic anti-attack of the α-position in the furan rings on the metal-complexed allene of intermediate INT I gives rise to the intermediate INT II.12 When the catalyst used is the cationic gold complex, intermediate INT II will undergo a protodeauration process to form six-membered ring fused furan product 2 and release the gold catalyst for the next catalytic cycle. When the catalyst is a platinum complex, we propose that there is a water-assisted proton transfer process via the platinum carbene intermediate INT III.13 Finally, the alkene shifted product 3 can be afforded with a high Z/E ratio after the protodeplatination process from INT IV. However, the reason why different products can be achieved using diverse catalysts is still not known.
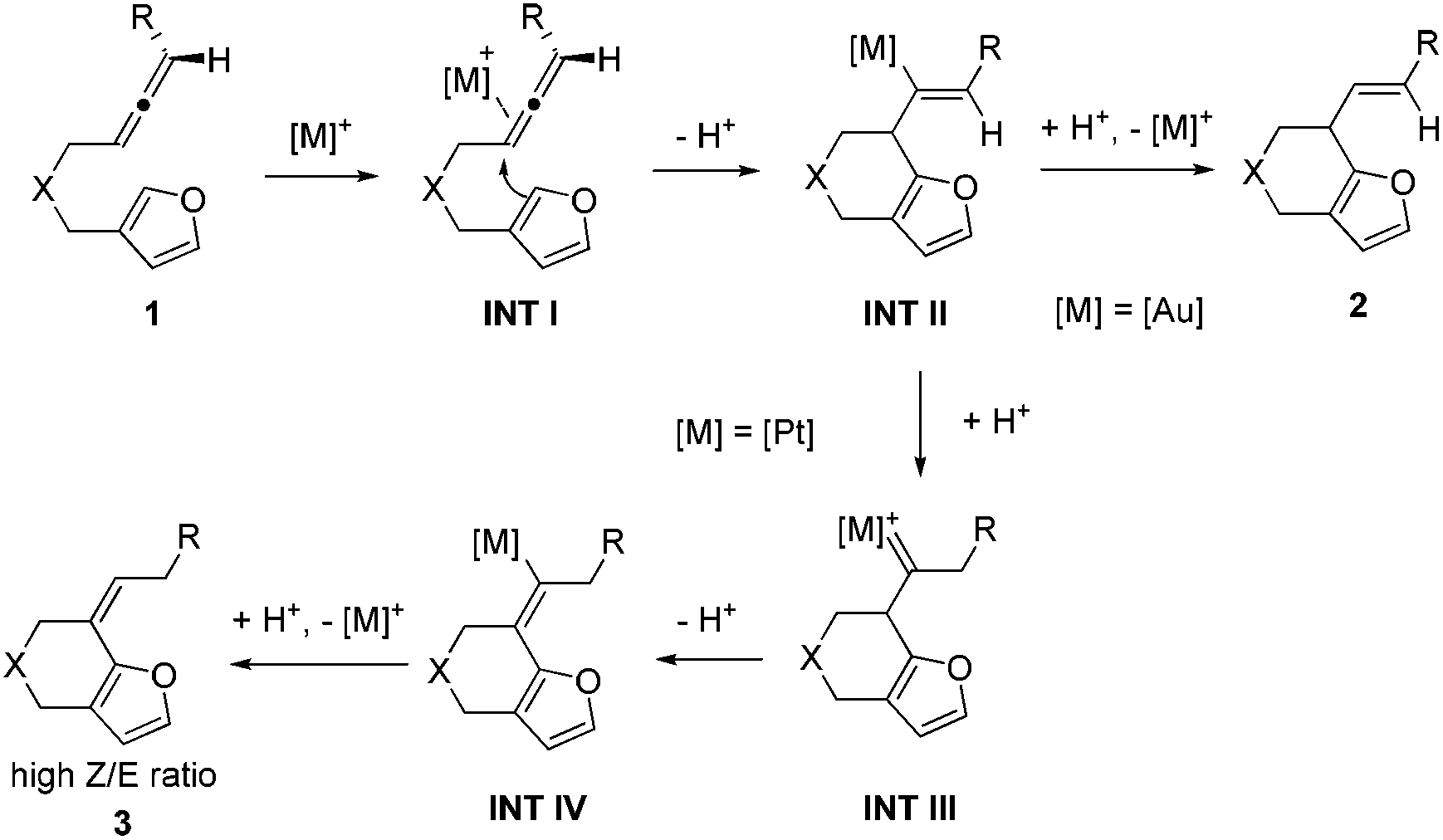 | ||
| Scheme 3 The proposed mechanism. | ||
In conclusion, an efficient intramolecular annulation of allenes towards furans has been developed to synthesize six-membered ring fused furans in good yields and high stereoselectivities. To the best of our knowledge, this is the first report of the intramolecular Friedel–Crafts-type annulation of allenes with furans. Two different products can be generated under gold and platinum catalysis. Under gold catalysis, a nucleophilic anti-attack mechanism was involved and the products with E configuration were generated. While using PtCl4 as the catalyst, alkene shifted products 3 were isolated with a high Z/E ratio, especially for N- and O-tethered substrates.
Experimental section
General procedure
A solution of the β-allenyl-furan substrate and catalyst (5 mol% or 10 mol%, [Au(NCMe)JohnPhos]SbF6 or PtCl4) in an anhydrous solvent (dioxane or THF, 0.05 M) was stirred under an argon atmosphere at 50 °C (or 80 °C). When TLC indicated the disappearance of the starting material, the reaction mixture was cooled to room temperature and concentrated. The crude mixture was submitted to flash column chromatography on silica gel to afford the corresponding product.Acknowledgements
We thank the National Basic Research Program of China-973 Program (2011CB808600) and the Natural Science Foundation of China (21232001) for financial support. We also thank Dr Wen-Xiong Zhang and Dr Neng-Dong Wang for X-ray crystal analysis.Notes and references
- (a) G. Cimino, S. De Stefano, A. Guerriero and L. Minale, Tetrahedron Lett., 1975, 1425 CrossRef CAS; (b) A. J. Allen, V. Vaillancourt and K. F. Albizati, Org. Prep. Proced. Int., 1994, 26, 1 CrossRef CAS; (c) R. Kazlauskas, P. T. Murphy and R. J. Wells, Tetrahedron Lett., 1978, 4951 CrossRef CAS; (d) P. Horton, W. Inman and P. Crews, J. Nat. Prod., 1990, 53, 143 CrossRef CAS; (e) K. Gustafson and R. J. Andersen, Tetrahedron Lett., 1985, 2521 CrossRef CAS.
- Z. Dong, C.-H. Liu, Y. Wang, M. Lin and Z.-X. Yu, Angew. Chem., Int. Ed., 2013, 52, 14157 CrossRef CAS PubMed.
- L. A. Paquette, in Principles of Modern Heterocyclic Chemistry, Benjamin, New York, 1968 Search PubMed.
- N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed.
- (a) N. Marion, S. Díez-González, P. de Frémont, A. R. Noble and S. P. Nolan, Angew. Chem., Int. Ed., 2006, 45, 3647 CrossRef CAS PubMed; (b) J. Mo and P. H. Lee, Org. Lett., 2010, 12, 2570 CrossRef CAS PubMed; (c) C. Park and P. H. Lee, Org. Lett., 2008, 10, 3359 CrossRef CAS PubMed; (d) W. Kong, C. Fu and S. Ma, Eur. J. Org. Chem., 2010, 6545 CrossRef CAS; (e) M. A. Tarselli and M. R. Gagné, J. Org. Chem., 2008, 73, 2439 CrossRef CAS PubMed; (f) M. A. Tarselli, A. Liu and M. R. Gagné, Tetrahedron, 2009, 65, 1785 CrossRef CAS PubMed; (g) D. Weber, M. A. Tarselli and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 5733 CrossRef CAS PubMed; (h) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS PubMed; (i) D. Weber and M. R. Gagné, Chem. Sci., 2013, 4, 335 RSC; (j) R. Skouta and C.-J. Li, Can. J. Chem., 2008, 86, 616 CrossRef CAS; (k) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2007, 9, 4821 CrossRef CAS PubMed; (l) D. Pflästerer, P. Dolbundalchok, S. Rafique, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2013, 355, 1383 CrossRef.
- (a) Z. Zhang, C. Liu, R. E. Kinder, X. Han, H. Qian and R. A. Widenhoefer, J. Am. Chem. Soc., 2006, 128, 9066 CrossRef CAS PubMed; (b) C. Liu and R. A. Widenhoefer, Org. Lett., 2007, 9, 1935 CrossRef CAS PubMed; (c) K. L. Toups, G. T. Liu and R. A. Widenhoefer, J. Organomet. Chem., 2009, 694, 571 CrossRef CAS PubMed; (d) M.-Z. Wang, C.-Y. Zhou, Z. Guo, E. L.-M. Wong, M.-K. Wong and C.-M. Che, Chem. – Asian J., 2011, 6, 812 CrossRef CAS PubMed; (e) M. P. Muñoz, M. C. de la Torre and M. A. Sierra, Chem. – Eur. J., 2012, 18, 4499 CrossRef PubMed; (f) W. Kong, C. Fu and S. Ma, Chem. Commun., 2009, 4572 RSC; (g) W. Kong, C. Fu and S. Ma, Org. Biomol. Chem., 2012, 10, 2164 RSC; (h) W. Kong, Y. Qiu, X. Zhang, C. Fu and S. Ma, Adv. Synth. Catal., 2012, 354, 2339 CrossRef CAS; (i) B. Alcaide, P. Almendros, J. M. Alonso, M. T. Quirós and P. Gadziński, Adv. Synth. Catal., 2011, 353, 1871 CrossRef CAS; (j) Y. Qiu, D. Ma, C. Fu and S. Ma, Org. Biomol. Chem., 2013, 11, 1666 RSC; (k) W. Kong, C. Fu and S. Ma, Chem. – Eur. J., 2011, 17, 13134 CrossRef CAS PubMed; (l) J. Barluenga, M. Piedrafita, A. Ballesteros, A. L. Suárez-Sobrino and J. M. González, Chem. – Eur. J., 2010, 16, 11827 CrossRef CAS PubMed; (m) R. M. Zeldin and F. D. Toste, Chem. Sci., 2011, 2, 1706 RSC; (n) B. Chen, W. Fan, G. Chai and S. Ma, Org. Lett., 2012, 14, 3616 CrossRef CAS PubMed; (o) E. Álvarez, P. García-García, M. A. Fernández-Rodríguez and R. Sanz, J. Org. Chem., 2013, 78, 9758 CrossRef PubMed.
- See ref. 5e, and also: (a) V. Mamane, P. Hannen and A. Fürstner, Chem. – Eur. J., 2004, 10, 4556 CrossRef CAS PubMed; (b) E. Soriano and J. Marco-Contelles, Organometallics, 2006, 25, 4542 CrossRef CAS; (c) Z. Liu, A. S. Wasmuth and S. G. Nelson, J. Am. Chem. Soc., 2006, 128, 10352 CrossRef CAS PubMed.
- (a) B. Trillo, F. López, M. Gulías, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2008, 47, 951 CrossRef CAS PubMed; (b) B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, Chem. – Eur. J., 2009, 15, 3336 CrossRef CAS PubMed; (c) I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020 CrossRef CAS PubMed.
- P. Mauleón, R. M. Zeldin, A. Z. González and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6348 CrossRef PubMed.
- Another example about the [4 + 3] type reaction between allenes and furans: B. W. Gung, D. T. Craft, L. N. Bailey and K. Kirschbaum, Chem. – Eur. J., 2010, 16, 639 CrossRef CAS PubMed.
- Intermolecular hydroarylation of allenes with furans was reported by Hashmi but the yields were low and the alkylation products were afforded as byproducts: A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS.
- For capture of the organometallic intermediate in the reactions involving allenes, see: (a) L.-P. Liu, B. Xu, M. S. Mashuta and G. B. Hammond, J. Am. Chem. Soc., 2008, 130, 17642 CrossRef CAS PubMed; (b) L.-P. Liu and G. B. Hammond, Chem. – Asian J., 2009, 4, 1230 CrossRef CAS PubMed; (c) D. Weber, M. A. Tarselli and M. R. Gagné, Angew. Chem., Int. Ed., 2009, 48, 5733 CrossRef CAS PubMed; (d) T. J. Brown, D. Weber, M. R. Gagné and R. A. Widenhoefer, J. Am. Chem. Soc., 2012, 134, 9134 CrossRef CAS PubMed.
- For selected examples involving the platinum carbene intermediate, see: (a) H. Funami, H. Kusama and N. Iwasawa, Angew. Chem., Int. Ed., 2007, 46, 909 CrossRef CAS PubMed; (b) H. Kusama, Y. Miyashita, J. Takaya and N. Iwasawa, Org. Lett., 2006, 8, 289 CrossRef CAS PubMed; (c) G. Li, X. Huang and L. Zhang, Angew. Chem., Int. Ed., 2008, 47, 346 CrossRef CAS PubMed; (d) K. Saito, H. Sogou, T. Suga, H. Kusama and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 689 CrossRef CAS PubMed; (e) H. Kusama, H. Sogo, K. Saito, T. Suga and N. Iwasawa, Synlett, 2013, 1364 CAS; (f) W. Kong, Y. Qiu, X. Zhang, C. Fu and S. Ma, Adv. Synth. Catal., 2012, 354, 2339 CrossRef CAS; (g) Y. Qiu, D. Ma, W. Kong, C. Fu and S. Ma, Org. Chem. Front., 2014, 1, 62 RSC.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and analytical data; CIF file for single crystal X-ray structural analysis. CCDC 1018190. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00223g |
| This journal is © the Partner Organisations 2014 |