
Antiviral drug discovery: broad-spectrum drugs from nature
J. P.
Martinez
a,
F.
Sasse
b,
M.
Brönstrup
b,
J.
Diez
c and
A.
Meyerhans
*ad
aInfection Biology Group, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona, Spain. E-mail: andreas.meyerhans@upf.edu
bDepartment of Chemical Biology, Helmholtz Centre for Infection Research, Braunschweig, Germany
cMolecular Virology Group, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona, Spain
dInstitució Catalana de Recerca i Estudis Avançats (ICREA), Barcelona, Spain
First published on 15th October 2014
Abstract
Covering: up to April 2014
The development of drugs with broad-spectrum antiviral activities is a long pursued goal in drug discovery. It has been shown that blocking co-opted host-factors abrogates the replication of many viruses, yet the development of such host-targeting drugs has been met with scepticism mainly due to toxicity issues and poor translation to in vivo models. With the advent of new and more powerful screening assays and prediction tools, the idea of a drug that can efficiently treat a wide range of viral infections by blocking specific host functions has re-bloomed. Here we critically review the state-of-the-art in broad-spectrum antiviral drug discovery. We discuss putative targets and treatment strategies, with particular focus on natural products as promising starting points for antiviral lead development.
 J. P. Martinez | Javier Martinez studied biology at the National University of La Plata, Buenos Aires, Argentina, and obtained his PhD in Virology from the University of the Saarland, Germany, in 2010. After a one-year post-doc position at the Helmholtz Centre for Infection Research (HZI) in Braunschweig, Germany, where he established two antiviral screening assays, he moved to Barcelona, Spain, to join the Infection Biology Laboratory at the University Pompeu Fabra as a Visiting Professor. |
 F. Sasse | Florenz Sasse studied biology and chemistry and obtained his PhD from University of Münster, Germany, in 1977. After a post-doc position he joined the Gesellschaft für Biologische Forschung (GBF) in Braunschweig, and later the Helmholtz Centre for Infection Research (HZI) in 1978, dealing with secondary metabolites of plant cell cultures. After a research sabbatical at the University of Guelph, Canada, in 1983, he joined the group of H. Reichenbach at GBF, dealing with natural products from myxobacteria. In 2005 he moved to the Chemical Biology Department at HZI and became acting head of the department in 2010. F.S. retired at the end of 2013. |
 M. Brönstrup | Mark Brönstrup studied chemistry and obtained his PhD from the TU Berlin in 1999. He joined Aventis in 2000 as a lab head for mass spectrometry and spent a research sabbatical with S. P. Gygi at Harvard Medical School in 2003. Between 2005 and 2010, he was leading the Natural Products Science section at Sanofi Aventis in Frankfurt. Between 2010 and 2013, he was managing sections dealing with biomarkers, bioimaging & biological assays. Since December 2013, he has been head of the Chemical Biology Department at the Helmholtz Centre for Infection Research in Braunschweig and W3 Professor at the University of Hannover. |
 J. Diez | Juana Díez studied biology and obtained her PhD from the University Autónoma de Madrid, Spain. For her post-doc she moved to the Institute of Virology (Madison, USA) where she studied host factors restricting viral replication under the supervision of Dr Paul Ahlquist. In 2001 she obtained a professorship at the Department of Experimental and Health Sciences at the University Pompeu Fabra (Barcelona, Spain). The main focus of her group is to decipher key molecular interactions of viruses with their host cells and to use this knowledge to develop broad-spectrum antivirals. |
 A. Meyerhans | Andreas Meyerhans studied chemistry and obtained his PhD from the University of Hamburg, Germany, in 1987. After a post-doc position in the Molecular Retrovirology Laboratory of Simon Wain Hobson at the Institute Pasteur in Paris, he was appointed Assistant Professor at the University of Freiburg in 1991 and C3 Professor at the University of the Saarland in 1998. His research interests included virus variation and antiviral T cell responses. Since 2010 he has been an ICREA Research Professor and head of the Infection Biology Laboratory at the University Pompeu Fabra in Barcelona. |
1. Introduction and background
This review centres on the concept of host-acting antiviral drugs (HAAs) with broad-spectrum activities as opposed to directly acting antivirals (DAAs) with high virus selectivity. Promising natural products that show diverse antiviral activity ranges are summarized. The focus is on such compounds that are active against different virus families or genera rather than virus serotypes or strains of a given genus.1.1 Viral infections: a global threat
Viruses continue to threaten global health. Of particular significance are the hepatitis B (HBV) and C (HCV) viruses, and the human immunodeficiency virus (HIV) that still are causing a worldwide death toll of approximately 0.6 million, 0.5 million and 2 million individuals per year, respectively1–3 (Table 1). Influenza springs to mind as being amongst the biggest viral killers of all time. It appears as pandemics caused by newly assorted viral strains. Its rapid expansion worldwide is caused by a fast expansion within infected humans with nasal peak virus titers at just 2 days post infection and efficient air-born transmissibility.4 The measles virus, despite the existence of an efficient vaccine and global vaccine coverage of 84% of the world's children, has nonetheless caused around 120 thousand deaths in 2012.5 Then there is the large group of emerging viruses for which no efficient vaccine or specific therapy is available today. They originate in most cases from infected animals and have an RNA genome that guarantees genetic variability with rapid adaptability.6,7 In terms of global spread, the dengue virus with its estimated 100 million apparent and 300 million unapparent infections in the year 2010 is alarming.8 While most infections remain asymptomatic, the number of cases of dengue fever and dengue haemorrhagic fever has significantly increased over the last decades.9 Other viral outbreaks like those of the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2003,10 the Middle East respiratory coronavirus (MERS-CoV) in 2012,11 and the very recent Ebola virus (EBOV) outbreak in West Africa in 201412 were comparatively tiny in numbers. Nonetheless they received large media coverage due to their epidemic potential and high mortality rates of 10%, 30% and up to 90% respectively.| Virus | Infecteda | Newly infecteda | Antiviral treatmentb | Ref. |
|---|---|---|---|---|
| a In millions. b Standard of care. c Cases per year. d ART, antiretroviral therapy, different formulations. e TBCT, tuberculosis combination therapy (rifampin, isoniazid, ethambutol, and pyrazinamide). f Seasonal epidemics. g Cases with severe illness or death. h Prevalence is exacerbated in risk groups, i.e. 80% of drug-injection HIV-infected users are co-infected with HCV and 20% are infected with HBV in endemic areas. N.a.: not assessed. HIV (human immunodeficiency virus), HCV (hepatitis C virus), HBV (hepatitis B virus), DENV (dengue virus), TB (tuberculosis). | ||||
| Mono-infections | ||||
| HIV | 35 | 2.3 | ARTd | 13 |
| HCV | 150 | 3–4 | Peg-IFNα/ribavirin + boceprevir/telaprevir | 2 |
| HBV | 240 | 0.6g | Tenofovir/emtricitabine | 1 and 14 |
| DENV | 50–100c | 0.5g | None | 15 |
| Influenza | 10% adults; 30% childrenf | 3–5g | Oseltamivir; zanamivir | 16 and 17 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Co-infections | ||||
| HIV/HCV | 4h | N.a. | Boceprevir/telaprevir + Peg IFNα/ribavirin + ARTd | 18 |
| HIV/HBV | 3.5h | N.a. | ARTd + tenofovir/emtricitabine | 19 |
| HIV/TB | 11 | 1.1 | ARTd + TBCTe | 20 |
Besides those in our natural surroundings, there are deadly viruses in research laboratories. The smallpox-causing variola viruses have killed an estimated 300–500 million individuals during the 20th century alone.21 Due to tremendous global efforts and the existence of an efficient virus vaccine, smallpox was eradicated in 1979,22 however variola viruses still exist in diverse laboratories while vaccination campaigns have ended. Recent gain-of-function experiments with influenza viruses have artificially generated highly virulent and transmissible new virus strains that have never existed before.23,24 Any release of either of these viruses, be it deliberate or by mistake, could have devastating consequences as they would enter a non-vaccinated, fully susceptible human population.25,26 Thus, considering the continuous spread of major viral pathogens as well as unpredictable viral outbreaks of old or novel virus strains, it seems advisable to have an arsenal of countermeasures ready for the prevention of global health crises.
1.2 Current challenges in antiviral treatment
The arsenal of antivirals is complex and consists of (i) directly acting antivirals (DAAs), i.e. drugs that directly affect virus-derived components including viral proteins and viral genomes, and (ii) host-acting antivirals (HAAs), i.e. modifiers of host factors or host pathways that affect virus life cycles as well as immune response components or immune response modifiers including antibodies, interferons and vaccines. As of March 2014, there were 50 specific DAAs approved by the American Food and Drug Association (FDA). 26 of these are directed against HIV. The other main targets are the hepatitis B and C viruses, different herpes viruses and influenza viruses (data provided by Antiviral InteliStrat, http://www.antiviralintelistrat.com). In most cases, DAAs act on viral polymerases and proteases, however virus entry and exit steps or chromosomal integration of retroviruses are also common points of interference. The group of host-acting antivirals (HAAs) is very heterogeneous. Vaccines, the classical, preventive weapons against viral infections, as well as antibodies or interferons, will not be covered in this review. Instead, we will highlight compounds that transmit their antiviral activity via acting on cellular components or pathways that viruses use for their expansion. Such HAAs are not yet approved by the FDA as antivirals, but are the subject of multiple, promising current research and development projects.The target spectrum of the approved DAAs is a reflection of (1) the need to control particularly persistent infections for which no vaccine is available and (2) the tremendous efforts that were put into HIV research. Indeed it was the HIV epidemic that became apparent in the 1980s plus the publicity generated by AIDS activists that massively boosted antiviral research. With the launch of the first HIV protease inhibitors in 1995, the potency of antiviral therapy to convert a nursing case into a healthy virus carrier became evident. This was the starting point to shift the slowly progressing, fatal HIV-induced immunodeficiency into a controllable chronic infection. At the same time, the rapid dynamics of infecting viruses were recognized together with the error-prone nature of retrovirus and RNA virus replication as the underlying mechanisms for the rapid selection of drug-resistance during antiviral monotherapy. This hurdle can be overcome by antiviral combination therapy, providing that antivirals with non-overlapping resistance profiles are available. The impressive progress in HIV and HCV therapeutics clearly demonstrates that this can be the case. To progress beyond this, there are important challenges ahead. How can we get access to the latent reservoir of infections with HIV and HBV? Can one completely cure HIV or HBV infections? How can we best manage viral co-infections like those of HIV and HCV that require complex drug regimens with drug–drug interactions and overlapping drug toxicities? How can we protect individuals during outbreaks of highly pathogenic viral infections? Possible answers to these and related questions may derive from joining the knowledge of antiviral drug development with the rapidly growing field of systems virology.
2. Strategies for antiviral drug development
Viruses are intracellular parasites with a limited set of encoded genes. Their life cycles are completely dependent on cellular factors and pathways. These features are the underlying principles of two fundamentally different antiviral drug development strategies, the “many for one” and the “one for many” strategy.2.1 Many for one: many drugs, one target
The aim of this antiviral drug development strategy is to find compounds that inhibit a particular viral target of a particular virus. As any virus has a specific set of viral genes, this strategy leads to drugs that are mainly virus-selective with no or little activity against other viruses or even different genotypes of the same virus.27,28 Direct-acting antivirals (DAAs) derived from such a strategy have been proven successful in either curing an infection or maintaining it as asymptomatic. Most notable are the currently developed anti-HCV protease and polymerase inhibitors that in combination have the potential to cure close to all chronic HCV infections.29 However, DAAs are still challenged with unsolved issues such as elevated costs, both for development and implementation,30 the emergence of pathogen resistance,31 poor treatment responses in selected patient groups32–35 or drug–drug interactions leading to toxicity.36–40 These problems become even more relevant in co-infections such as those of HIV and HCV, for which combination treatment can be a clinical challenge.38,41,42 For example, inhibitors of viral proteases are subject to degradation by cytochrome 3A4 (CYP3A4) and the co-administration of so-called booster drugs like ritonavir or cobicistat that inhibit CYP3A4 is often required. As other CYP isoforms are also (partially) inhibited by the boosters, the treatment of co-morbidities with additional drugs may require individual modifications, and the overall assessment of all of the potential drug–drug-interactions is highly complex.39,43–45 Furthermore, these issues come hand in hand with another relevant problem: when there are many drugs against a single viral target, virus variation may generate cross-resistant mutants that would reduce therapy efficiency.46,47 Thus there is a need to develop antiviral drugs that could be individually effective against different viral pathogens. Such broad-spectrum antivirals (BSAs) could in principle alleviate some of the burdens of the current DAAs and expand the application spectrum.2.2 One for many: the broad-spectrum alternative
This antiviral drug development strategy aims at designing drugs with broad-spectrum antiviral activities. The first caveat in the development of BSAs is that viruses are highly diverse, both in structure and in replication strategies. Hence, the development of a DAA with broad-spectrum activity is a difficult task. However, as viruses are bound to utilize the host cellular machinery to propagate, they are critically dependent on cellular factors that are up- or down-regulated as needed. Examples are the down-regulation of membrane receptors,48–53 the up-regulation of the lipid metabolism,54 the use of the mRNA processing machinery55 or the hijacking of components of the endosomal-sorting complex (ESCRT) required for virus export from infected cells56–63 (see 2.4 for details). Moreover, as the life cycle of different viruses share common cellular factors and pathways, it is feasible that these could be used as targets for the design of broad-spectrum antivirals. Indeed, there are a number of chemical compounds that target common host factors and are at various stages of antiviral drug development (see section 3.2). In addition, with the advent of better screening technologies, we now know that the number of host factors associated with viral replication is strikingly large.64–67 This provides a vast space to explore further antiviral targets.68 Nonetheless, the development of such host-acting and broad-spectrum antivirals has its own challenges to meet.2.3 Pros and cons of broad-spectrum antivirals (BSAs)
The putative advantages and disadvantages of BSAs are listed in Fig. 1. The main advantage is that host-acting BSAs can cover multiple viruses and genotypes while reducing at the same time the likelihood of resistance development.69,70 In the clinical setting, applications of BSAs might range from rapid management of new or DAA-resistant viral strains71 and of viral outbreaks12 to reducing therapy complexity of viral co-infections.72–75 In addition, BSAs would be ideal as a first-line treatment or the prophylaxis of acute virus infections such as respiratory tract or sexually transmitted infections.76,77 So far, a main disadvantage associated with host-acting BSAs is the apparent poor translation of in vitro results to in vivo therapy. Thus, excellent antiviral profiles from cell-line-based assays might not be reflected in vivo because systemic mechanisms may compensate the blocked target effect. On the other hand, the identification of host factor targets that are essential for viral replication but redundant for the cell is critical for reducing putative toxicities associated with blocking cellular pathways.69 In the end however, the level of toxicity that can be tolerated will critically depend on the viral threat and the required time of treatment.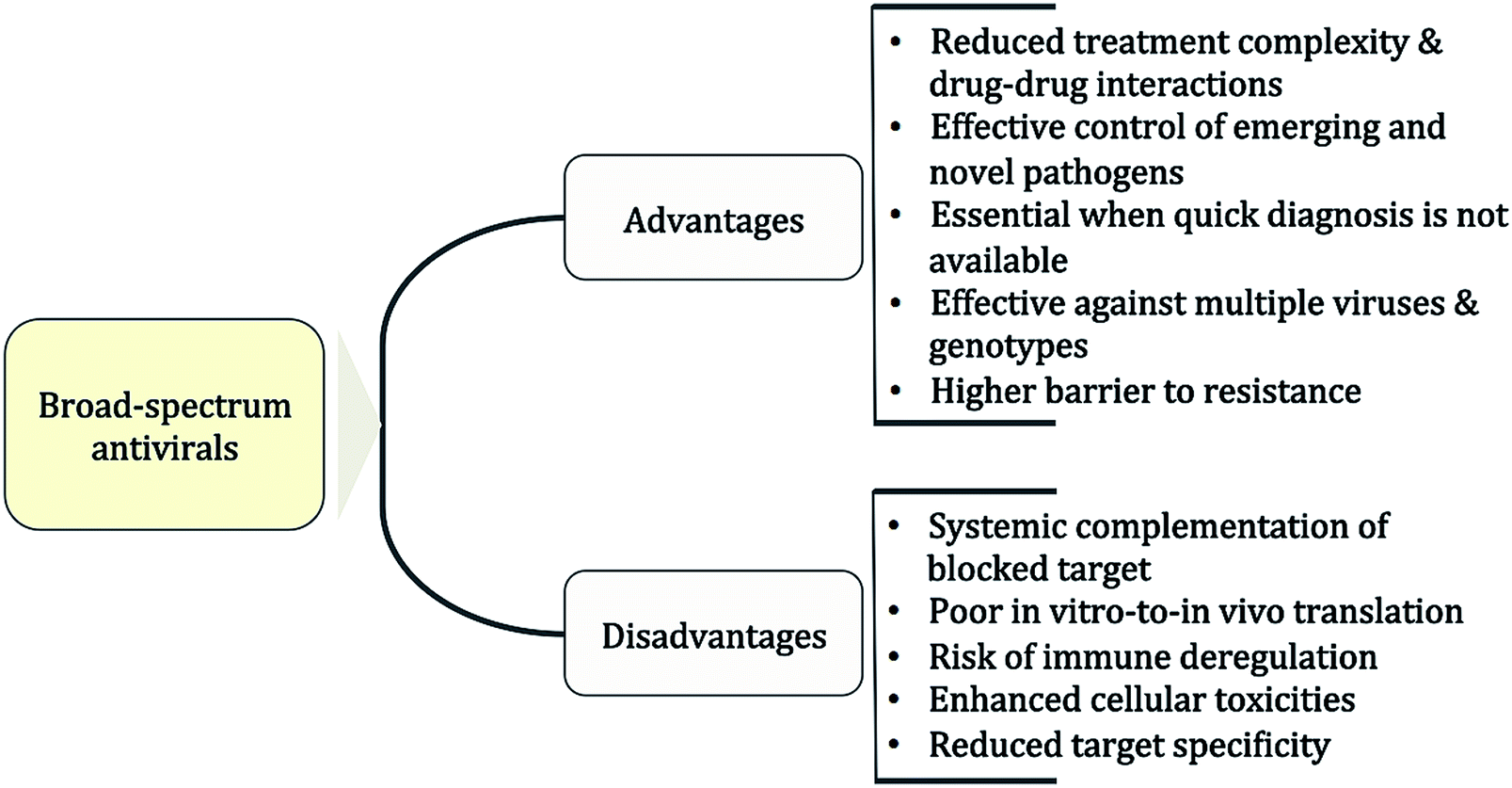 | ||
| Fig. 1 Putative advantages and disadvantages of broad-spectrum antiviral drugs. A drug that targets a common host factor might be effective against different viruses and decrease the likelihood of drug resistance development. However, it may result in a narrower therapeutic window as expected from in vitro studies. See text for details. | ||
2.4 Approaches for BSA design
Current strategies for broad-spectrum antiviral drug development are focused on (i) targeting host factors used for viral replication and, (ii) targeting host factors that are naturally involved in viral restriction.The targeting of host factors associated with viral replication complexes (VRCs) such as ADP-ribosylation factor 1 (ARF1), guanine nucleotide exchange factor 1 (GBF1) and phosphatidylinositol kinase 4III (PI4IIIKα/β) has also been shown to inhibit the replication of HCV, several enteroviruses such as picornavirus (PV), Aichi virus (AiV) and Coxsackie virus B3 (CVB3), as well as rhinovirus, mouse hepatitis coronavirus (MHV) and HIV-1.81,93–101 Some viruses such as DENV and HCV are known to induce the up-regulation of lipid synthesis for their replication.54 Lipid rafts are reported to be involved in the entry, assembly and/or budding of influenza virus, HCV, VSV, HIV-1, Epstein Barr virus (EBV), Ebola virus (EBOV), Marburg virus (MARV), DENV, West Nile virus (WNV) and Herpes Simplex virus (HSV) (Table 2). The down-regulation of lipid metabolism by siRNA or by licensed drugs such as statins has been shown to inhibit the replication of many viruses (see below).
| Compound name | Antiviral against | Available inhibitory data | Ref. |
|---|---|---|---|
| Cyclosporine | HIV | 90% inhibition with 10 μM in HaCaT cells | 105 |
| HPV | 0.07 to 4.7 μM effective concentration 50 (EC50) in TZM-bl assay | 109 | |
| HBV | 70% inhibition with up to 20 μg mL−1 in Huh7 cells | 107 | |
| Influenza | 90% inhibition with 10 μg mL−1 in MDCK cells | 112 | |
| SARS-CoV | 90% inhibition with 16 μM in Vero and Huh7 cells | 108 and 113 | |
| HSV-1 | 90% inhibition with 25 μM in monkey kidney cells | 155 | |
| VV | 97% inhibition with 16 to 40 μM in culture cells | 153 | |
| VSV | 90% inhibition with 26 μM in BHK cells | 106 | |
| HCMV | Virus production delayed by 6 days under 0.5 μM in mice | 111 | |
| HCV | 1 μg mL−1: 80% less viral RNA from MH-14 cells; 45 nM EC50 for alisporivir | 69 and 226 | |
| Statins (several) | HCV | Lovastatin EC50 = 0.9–2.16 μM in OR6 cells | 171 |
| HBV | Selectivity index (SI) = 3.44 in infected HepG2.2.15 cells with fluvastatin | 162 | |
| Influenza | SI = 21 in influenza infection in vitro assays with fluvastatin | 164 and 168 | |
| HIV | 50% less p24 production from isolates in PBMCs under 50 μM lovastatin | 165 and 227 | |
| DENV | Lovastatin SI = 1.4 in Vero cells and 4.5 in HMEC-1 cells | 167 | |
| HCMV | 50% less IE1 protein expression in U373-MG cells with 10 μM simvastatin | 169 | |
| Mycophenolic acid | DENV | EC50 = 1.9 μM in human hepatoma cells | 179 |
| WNV | EC50 = 10 μg mL−1 in primary glial cells | 179 | |
| YFV | EC50 = 0.4 μg mL−1 in Hep3B cells | 179 | |
| HCV | 75% inhibition with 1.0–6.0 μg mL−1 MPA using Luc-viruses | 181 | |
| HIV | 4 μM causes complete suppression of virus replication in CD4 T-cell cultures | 177 | |
| VV | 50% inhibition in plaque reduction assays with 0.2–3 μM in Vero cells | 228 | |
| Castanospermine (CST) & deoxynojirimycin (DNJ) | HCMV | 0.8 mM (CST) and 1 mM (DNJ) plaque reduction assay in HEF cells | 189 |
| HSV-2 | EC50 < 4 μM in plaque assay | 190 and 191 | |
| HIV | 100 μg mL−1 causes 100% syncitia inhibition in H9 and CD4-Jurkat cells | 188, 199, 229 and 230 | |
| BVDV | Celgosivir: 16 μM EC50 in plaque assay; CST: 110 μM | 193 | |
| HCV | CST: low effect; DNJ EC50 > 100 μM; DNJ derivatives: EC50 ≥ 4 μM in Huh7 cells | 118, 194 and 231 | |
| DENV | EC50 = 6 μM in BHK cells | 119, 192, 195 and 232 | |
| WNV | DNJ derivative >90% inhibition under 15 μM in MDBK cells | 102 and 117 | |
| EBOV | DNJ derivative >90% inhibition under 15 μM in MDBK cells | 102 and 117 | |
| LFV | DNJ derivative >90% inhibition under 15 μM in MDBK cells | 102 and 117 | |
| VSV | DNJ derivative >90% inhibition under 15 μM in MDBK cells | 102 and 117 | |
| Influenza | 10 pg mL−1: 90% of the viral glycopeptides endoglucosaminidase H | 187 and 233 | |
| Chebulagic acid & punicalagin | HCMV | SI = 12/17 (chebulagic/punicalagin) in HEL cells | 205 |
| HCV | SI = 19/13 in Huh7.5 cells | 205 | |
| DENV | SI = 12/19 in Vero cells | 205 | |
| MV | SI = 10/11 in CHO cells | 205 | |
| RSV | SI = 642/490 in Hep-2 cells | 205 | |
| HSV | SI = 18.62/14.5 in A549 cells | 207 | |
| AdV | SI = 1.60/1.62 in A549 cells | 205 | |
| Cyanovirin-N | EBOV | EC50 = 100 nM; virus CPE in Vero cells 7 days post infection (d.p.i.) | 216 |
| HCV | EC50 = 1.6 nM in Huh7 cells infected with HCVpp | 217 | |
| Parainfluenza | SI > 1.9 in HEp1 cells | 218 | |
| Influenza A | SI > 228 in MDCK cells | 218 | |
| Influenza B | SI > 20 in MDCK cells | 218 | |
| HIV | EC50 = 0.1–17 nM in PBMC (by reverse transcriptase activity assay of supernatants) | 215 | |
| SIV | EC50 = 11 nM | 215 | |
| HSV-1 | SI = 158 in Vero cells | 219 | |
| EBV | SI = 4.3 in P3Hr1 cells | 218 | |
| HHV-6 | SI = 4.4 in HSB-2 cells | 218 | |
| BVDV | SI = 13 in MDBK cells | 218 | |
| Labyrinthopeptin | HIV | EC50 = 0.70–3.3 μM | 234 and 235 |
| HSV (various) | EC50 = 0.29–2.8 μM | 234 and 235 | |
| Apicularen | HPV | SI = 3–6 in Hela cells | 236 |
| HIV | Z-score = −1.9 in primary screen in TZM-bl cells under 2.5 μM | 237 | |
| HCV | 75% inhibition (replication), 99.5% inhibition (whole life cycle) in Huh7 cells | 238 | |
| Crocapeptin | HCV | Z-score = −1.8 in primary screen in TZM-bl cells under 2.5 μM | 237 |
| HIV | Z-score = −8.6 in primary screen in Huh7 cells under 2.3 μM | 238 | |
| Noricumazole | HIV | Z-score = −1.01 in primary screen in TZM-bl cells under 2.5 μM | 237 |
| HCV | Z-score = −6.3 in primary screen in Huh7 cells under 2.3 μM | 238 | |
| Disorazole | HIV | Z-score = −1.31 to −1.78 in primary screen in TZM-bl cells under 2.5 μM | 237 |
| HCV | Z-score = −6.9 in primary screen in Huh7 cells under 2.3 μM | 238 | |
| Epothilone | HIV | Z-score = −2.38 in primary screen in TZM-bl cells under 2.5 μM | 237 |
| HCV | 95% (replication), around 99% (whole life cycle) inhibition in Huh7 cells under 2.3 μM | 238 | |
| Tubulysin | HIV | Z-score = −1.34 to −2.47 in primary screen in TZM-bl cells under 2.5 μM | 237 |
| HCV | 95% (replication), around 99% (whole life cycle) inhibition in Huh7 cells under 2.3 μM | 238 | |
| Archazolid | HIV | Z-score = −1.4 in primary screen in TZM-bl cells under 2.5 μM | 237 |
| HCV | 77% (replication), 99% (whole life cycle) inhibition in Huh7 cells under 2.3 μM | 238 | |
| Mycalamide | Polio virus | MIC 5 ng per disc (assay not described, n.d.) | 239 |
| HSV-1 | MIC 5 ng perdisc (assay n.d.) | 240 | |
| Influenza | 32 μM of a mycalamide analog; 60 to 90% plaque reduction in MDCK cells | 241 | |
| CoV-A59 | Mice survival 14 days after A59 CoV infection under 0.1 mg kg−1 of 2% mycalamide A | 240 | |
| Dragmacidin & Manzanine | HSV-2 | EC50 = 96 μM HSV in colorimetric plaque-reduction assay; 0.9 μM HIV in MT4 cells | 242–244 |
| HIV | HIV EC50 = 4.2 μM (assay n.d.); anti-HSV MIC = 0.05 μg mL−1 (assay n.d.) | 242–244 | |
| Griffithsin | HIV | SI = 2000 against HIVlai in MT4 cells; SI > 20000 against HIVbal in human PBMCs |
245 |
| HCV | EC50 = 14 nM against JFH1 HCVcc in Huh-7 cells | 246 | |
| SARS-CoV | EC50 = 14 nM in Vero 76 cells | 247 | |
| JEV | EC50 = 20 nM BHK-21 cells | 248 | |
| SIV | SI = 500 against SIVmac in CEMx174 cells | 245 | |
| Squalamine | DENV | 100 μg mL−1 causes 100% inhibition in human endothelial cells | 85 |
| HBV | 20 μg mL−1 causes 80% inhibition in human hepatocytes | 85 | |
| HDV | 20 μg mL−1 causes 80% inhibition in human hepatocytes | 85 | |
| YFV | Hamster 15 mg kg−1 daily dose, 100% survival after 8 days compared to control animals | 85 | |
| MCMV | BALB/c mice, 10 mg kg−1 daily dose intraperitoneal, no virus detected 14 d.p.i. | 85 |
Viruses also hijack host factors involved in protein folding such as cyclophilin A and endoplasmic reticulum (ER)-associated α-glucosidases.102 Cyclophilin A (CypA) belongs to the family of peptidyl-prolyl-cis–trans isomerases (PPIases) and is involved in protein folding, trafficking, formation of multiprotein complexes (MPC) and other cellular functions.103,104 CypA interacts with viral proteins supporting viral replication.69 CypA inhibitors such as cyclosporine A (CsA) have been shown to inhibit the replication of HIV, HCV, influenza virus, CoV, HBV, HSV, human cytomegalovirus (HCMV), VSV, vaccinia virus (VV) and human papillomavirus (HPV).105–114 Alisporivir (Debio-025) and SCY-635, both CsA analogues, have shown antiviral activity against HCV in vivo and are currently in combination with other anti-HCV compounds in various clinical trials.115,116 ER α-glucosidases I and II play a critical role in glycosylation of viral proteins.102 The inhibition of ER α-glucosidases has been shown to affect viral particle assembly and/or secretion of HBV, HIV, HSV-1, influenza virus, parainfluenza virus, measles virus (MV), MARV, EBOV, HCV and other members of the Flaviviridae family, such as bovine viral diarrhea virus (BVDB), DENV, WNV, and Japanese encephalitis virus (JEV).117 Celgosivir has proven effective against HCV and DENV infections in vitro and in vivo.118,119 A description of the antiviral effect of these compounds is provided in Section 3 of this review.
Another putative target for BSA is the endosomal-sorting complex required for transport (ESCRT) that is involved in trafficking viral proteins to the cell surface or into multivesicular bodies.57,60,63 siRNA downregulation of components of the ESCRT and associated factors such as ALIX has been shown to block the cell entry of VSV, LFV, and LCMV, and the cell exit of HIV and hepatitis A virus (HAV).56,58,61,62,87 However, a non-toxic chemical compound has not yet been released for clinical use.
3. Natural products as a source for broad-spectrum antiviral drugs
3.1 Why natural products?
Natural products have been – and continue to be – a rich source of drugs.136,137 To base the search for treatments of a given medical condition on natural products has a variety of advantages: natural products exhibit a large structural diversity and complexity that remains unmatched by other drug formats.138,139 The diversity of chemical matter is an important prerequisite to address the diversity of a biological target space, in particular in the context of phenotypic screenings that capture full biological pathways rather than single protein domains.140–142 The structural complexity of natural products, often regarded as a drawback with respect to synthetic accessibility, has been successfully mastered due to vastly improved methods of organic synthesis and/or genetic engineering.143 A key advantage of natural products lies in the evolutionary pre-selection and optimization of chemical matter towards biological significance. As natural product biosynthesis is associated with considerable metabolic costs, compounds without significant advantages for their producers would have probably been eliminated in the course of evolution. In fact, numerous studies of natural products have disclosed biological functions that were advantageous for the microbial or eukaryotic producers in their (non-human) environment, but at the same time possessed high relevance for the treatment of human diseases.142 While natural products have been the source of drugs for almost every indication, their importance is most pronounced in the field of infectious diseases where they have provided the chemical template for the majority of antibacterial and antifungal drugs.144 Their role in antiviral drug development is less obvious, though, as nearly all marketed antiviral drugs today are produced by chemical synthesis. However, natural products have made significant contributions to antiviral drug discovery. Nucleoside analogs (containing sugars other than ribose or deoxyribose) represent by far the most important class of antiviral drugs. It should be noted that two early prototypes of nucleotide analogs, named spongouridine (1) and spongothymidine (2), have been discovered in the 1950's from marine sources.145–147 Also the DNA polymerase inhibitor arabinosyladenine (3), marketed as Vidarabin, has been discovered from natural sources – but it happened years after its chemical synthesis.148 Finally, the close similarity between ribavirin (4), a cornerstone in multiple antiviral treatment regimens, and natural products like pyrazomycin (5) or showdomycin is remarkable.149,150 Thus, while nucleotide analoging as a central principle of defeating viruses was addressed by nature, it was independently explored and developed for medical use by means of chemical synthesis.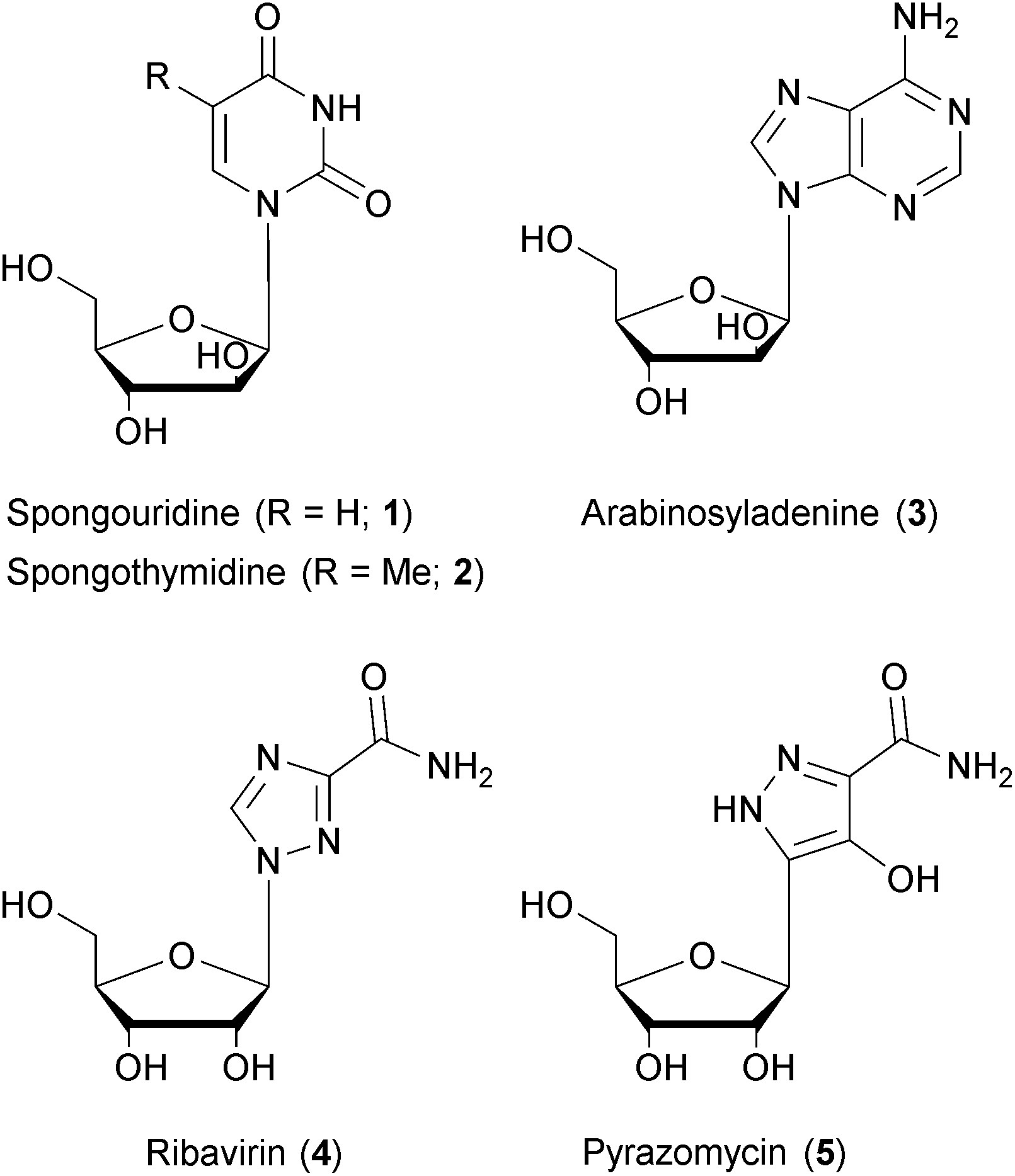
3.2 Natural products with broad-spectrum antiviral activities: source and mode-of-action
Many substances from diverse natural sources such as bacteria, fungi, plants and animals have been described to have antiviral properties. A selection of those exerting BSA activities are shown in Fig. 2 and Table 2 and described below.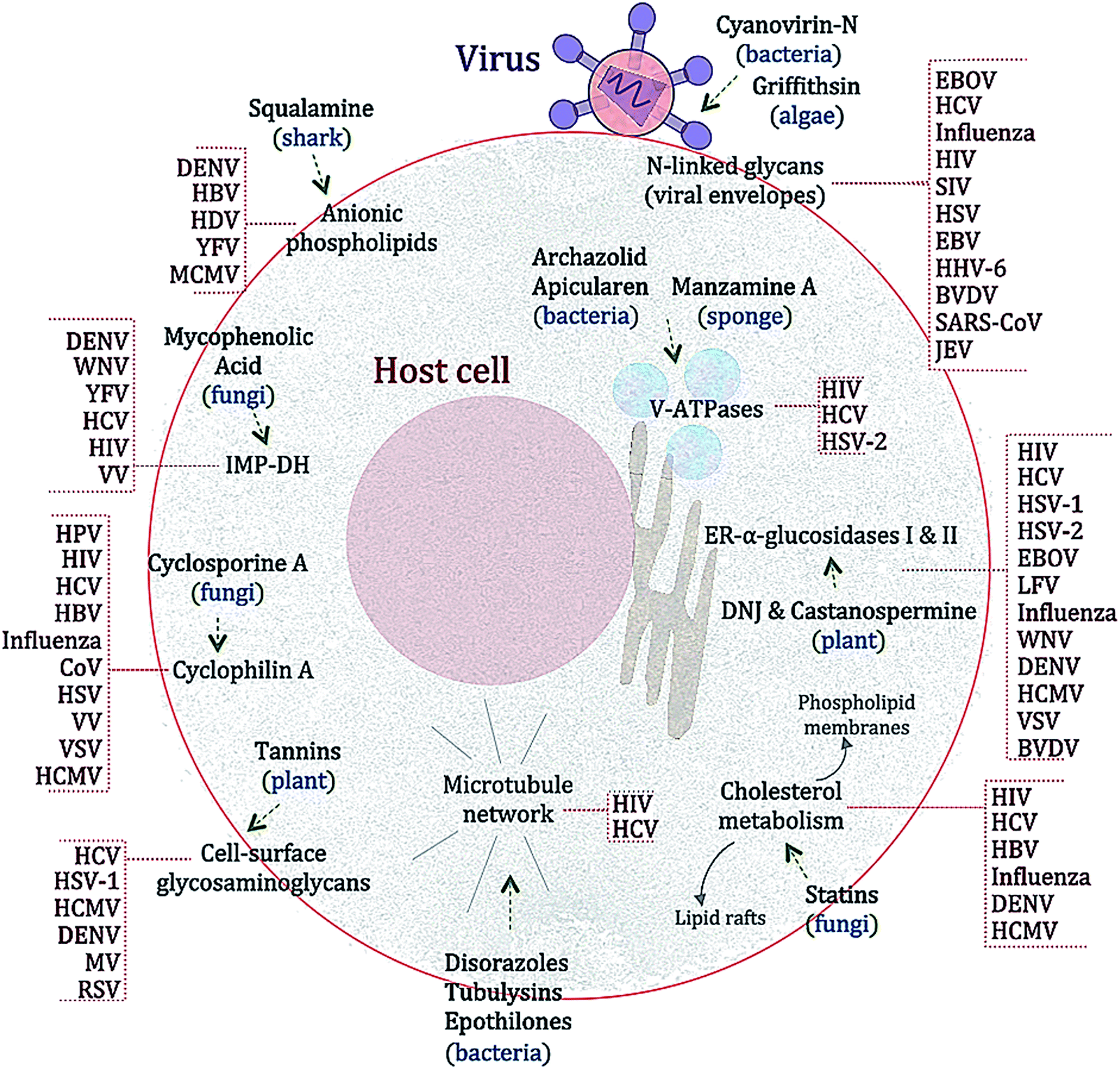 | ||
| Fig. 2 Schematic representation linking selected natural products with their principal targets and their antiviral activity spectra. The sources of the compounds are given in brackets. The compounds are a selection made from an extensive literature search up to April 2014. Abbreviations (in alphabetical order): AdV (adenovirus), BVDV (bovine viral diarrhea virus), CoV-A59 (coronavirus A59), DENV (dengue virus), DNJ (1-deoxynojirimycin), EBOV (Ebola virus), EBV (Epstein Barr virus), HBV (hepatitis B virus), HCMV (human cytomegalovirus), HCV (hepatitis C virus), HDV (hepatitis D virus), HHV-6 (human herpesvirus 6), HIV (human immunodeficiency virus), HPV (human papillomavirus), HSV (herpes simplex virus, various), IMP-DH (inosine monophosphate dehydrogenase), Influenza (Influenza virus), JEV (Japanese encephalitis virus), LFV (Lassa fever virus), MCMV (mouse cytomegalovirus), MV (Measles virus), Parainfluenza (parainfluenza virus), RSV (Rous Sarcoma virus), SARS-CoV (severe acute respiratory syndrome coronavirus), SIV (simian immunodeficiency virus), VSV (vesicular stomatitis virus), VV (vaccinia virus), WNV (West Nile virus), YFV (yellow fever virus). | ||
Statins are HMG-CoA reductase inhibitors that were first isolated from the fungus Penicillium citrinum in the early 1970s.157 The main effect of statin treatment is the decrease in total and low-density lipoprotein (LDL) cholesterol both in vitro and in vivo.158 Statins have also been described to have immune-modulatory properties.159 The antiviral effect of statins was first recognized for HCV.160 In that study, Ye and colleagues demonstrated that treatment with lovastatin (9), which efficiently impaired the replication of HCV sub-genomic replicons in cell cultures by disrupting the membrane components of viral replication complexes. The addition of geranylgeraniol, which is involved in protein trafficking into membrane compartments, restored viral replication, thus suggesting a non-direct antiviral activity of statins. The antiviral effects of statins have also been described for HBV, HIV, influenza virus, DENV, HCMV and norovirus.161–169 However the antiviral efficacy of statins in vivo was only marginal and drug–drug interactions with DAAs have been reported.170,171 While this terminated the further use of statins as antiviral drugs, the data taken together underline the important role of the host's lipid metabolism in viral replication.78
Mycophenolic acid (MPA) (10), an inhibitor of eukaryotic inosine monophosphate dehydrogenase (IMP-DH), was first isolated from the fungus Penicillium stoloniferum.172 Similar to ribavirin, MPA blocks nucleic acid synthesis by interfering with de novo purine biosynthesis.173 However, in contrast to ribavirin, MPA has not been shown to have mutagenic properties. Mycophenolate mofetil, a MPA prodrug, has been described to have immunosuppressive properties.174 MPA was first observed to limit the cytopathic effects of VV, HSV and MV in cell cultures.175 Since then, the drug has been reported to inhibit the replication of Hantaan River virus, DENV, WNV, HBV, HEV, HCV, HIV and poxviruses by affecting viral nucleic acid synthesis.176–182In vivo MPA alone is not effective against HSV, but it has been shown to enhance the anti-HSV activities of acyclovir, gancyclovir and pencyclovir in experimental animal studies.183 Interestingly, MPA has also been shown to synergize with antiretroviral drugs184,185 as well as with cyclosporin A and IFN-a in HCV inhibition.181 However, due to its mode of action, the long-term use of MPA might lead to the development of drug resistance, as has been previously shown for the Sindbis virus.186
This addition is an intermediate step that enhances the efficiency of protein folding in the glycoprotein maturation process.198 The BSA mechanism of CST and DNJ is thus suggested to be the disruption of the folding of some viral glycoproteins leading to the poor expression of mature envelopes and reduced infectivity.189,199,200 Given the broad range antiviral effects of blocking the host glycoprotein processing machinery, new screening campaigns with different glucosidase inhibitors might identify compounds with enhanced BSA effects. Indeed, many other glucosidase inhibitors isolated from natural sources exist (see ref. 201 for an extensive overview) that may be considered for testing as BSAs.
Tannins are antimicrobial secondary metabolites commonly found in plants.202 Hydrolysable tannins have been described to exert inhibitory effects against viruses, bacteria and eukaryotic microorganisms.203 Chebulagic acid (CHLA) (13) and punicalagin (PUG) (14) are two hydrolysable tannins isolated from the tree Terminalia chebula that were initially found to inhibit HIV.204 CHLA and PUG also have antiviral activities against HCMV, HCV, DENV, MV and RSV.205 PUG treatment also protected mice challenged with an otherwise lethal dose of enterovirus.206 While the exact mechanism of action of CHLA and PUG has not been entirely elucidated, it is suggested that these compounds inhibit the interaction between viral glycoproteins and cellular glycosaminoglycans (GAGs).207 GAGs are carbohydrates present on the surface and in the extracellular matrix of cells that have been shown to be required for the infection of several viruses.208–213 Of note, two other hydrolysable tannins isolated from mango (Mangifera indica) were shown to inhibit influenza virus and Coxsackie virus in vitro.214 Although more studies are needed to clarify the antiviral mechanism of action exerted by hydrolysable tannins, these plant-derived substances might be a good starting point for BSA development.

Nevertheless, antiviral studies with CV-N in vivo against neuroaminidase-inhibitor resistant influenza, Zaire strain of EBOV in mice, and studies performed with transmission models of HIV and SIV suggest that CV-N has potential for use as a prophylaxis and early post-exposure treatment.216,219,223–225 Still, whether CV-N has a binding partner in the host has not been determined yet and further studies are needed to assess the safety of CV-N as a therapeutic drug.
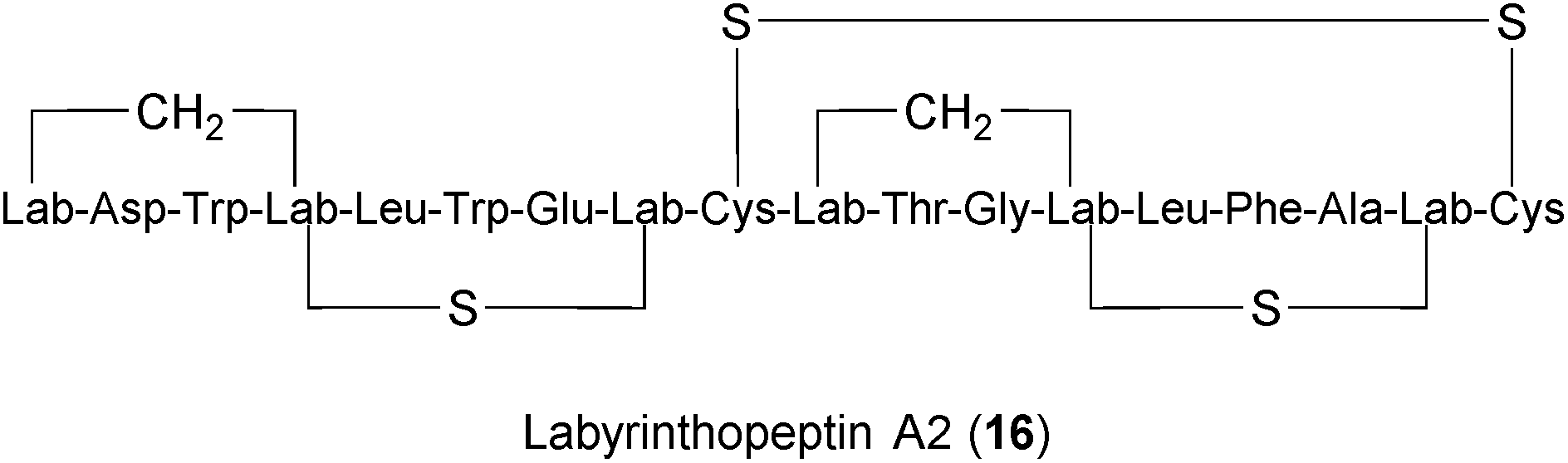
Lantibiotics are peptides with unusual amino acids produced by several Gram-positive bacteria.249,250 Labyrinthopeptin A1 (LabyA1) (16) belongs to a novel class of carbacyclic lantibiotics,251,252 that was isolated from the actinomycete Actinomadura namibiensis. LabyA1 was recently shown to inhibit both HIV and HSV at sub-micromolar concentrations in vitro.234,235 The compound is suggested to block viral entry by interacting with viral envelopes and to prevent cell-to-cell transmission. What makes LabyA1 appealing is its effectiveness against resistant HIV and HSV viruses, its synergistic effects with standard antiretroviral drugs, and the absence of an inflammatory response in peripheral blood mononuclear cells (PBMCs). LabyA1 was nontoxic to vaginal lactobacilli,234 thus making it an excellent candidate microbicide for the prevention of sexually transmitted virus infections. Although HIV and HSV are non-related viruses, the question remains whether LabyA1 might be effective against a broader range of viral pathogens.
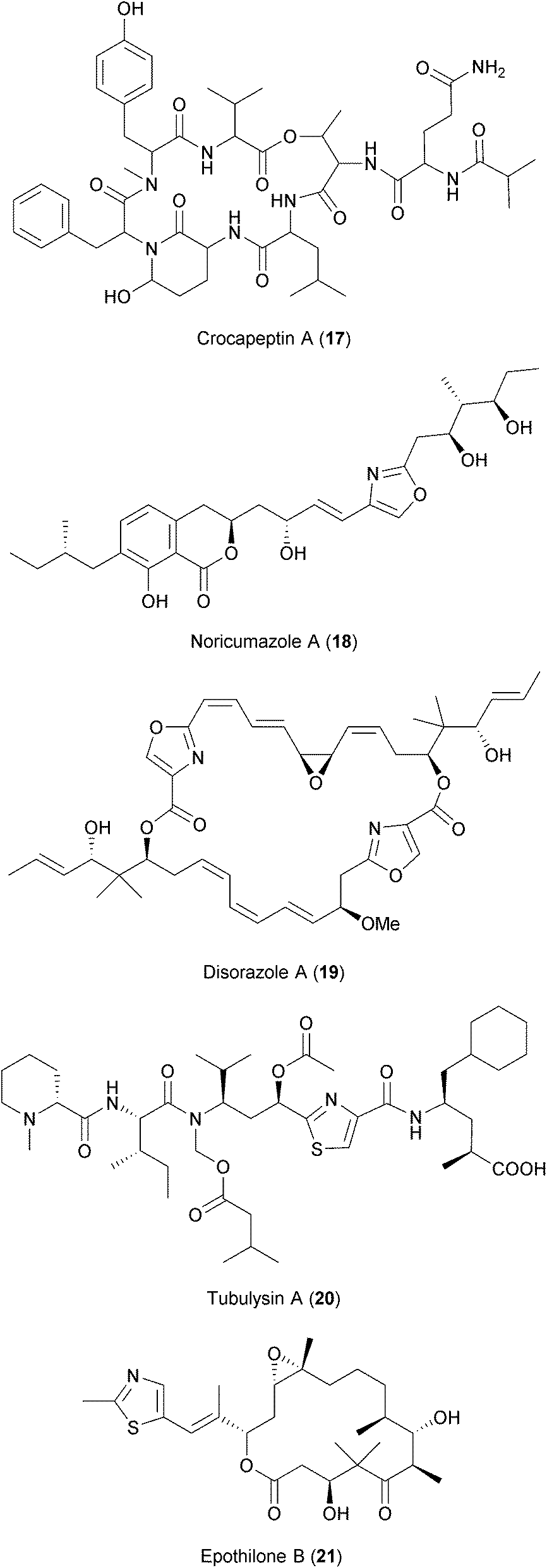
Myxobacteria are soil bacteria known to be producers of highly bioactive secondary metabolites.253 These have been shown to exhibit a wide range of activities such as antifungal and antibacterial properties (see ref. 254 and 255 for further details). Two recent antiviral screens using a library of compounds derived from the secondary metabolism of myxobacteria have identified several compounds with overlapping activities against HIV and HCV237,238 (Table 2). Among these are crocapeptin B (17), a cyclic depsipeptide isolated from the myxobacterium Chondromyces crocatus described to have inhibitory activity against serine proteases256 and noricumazole A (18), an oxazole- and isochromanone-containing metabolite isolated from Sorangium cellulosum shown to block ion channels.257,258
Other anti-HIV and anti-HCV hits were compounds known to inhibit tubulin polymerization, namely disorazoles (19), polyketides isolated from the myxobacterium Sorangium cellulosum, and tubulysins (20), unusual peptides derived from the myxobacterium Archangium gephyra (ref. 237 and references therein). Epothilones (21), a group of macrolides that enhance tubulin polymerization and that are approved for cancer treatment, were also found to be inhibiting HIV and HCV. Modulation of the host's microtubule network is known to influence the replication of many diverse viruses.259,260 However, the chemical blockade of microtubules is associated with toxicities that, so far, hamper the development of these compounds as antiviral drugs.
Both antiviral screens also identified two highly specific V-ATPase inhibitors, apicularen (22) and archazolid (23), as anti-HIV and anti-HCV hits.237,238 Recently, Müller et al. also described apicularen as an inhibitor of HPV replication.236 Evidence from genome-wide siRNA screens and other studies highlighted the dependency on the host's V-ATPases for the replication of diverse viruses like HCV, DENV, WNV, influenza virus, and HIV (Table 2 and references therein).
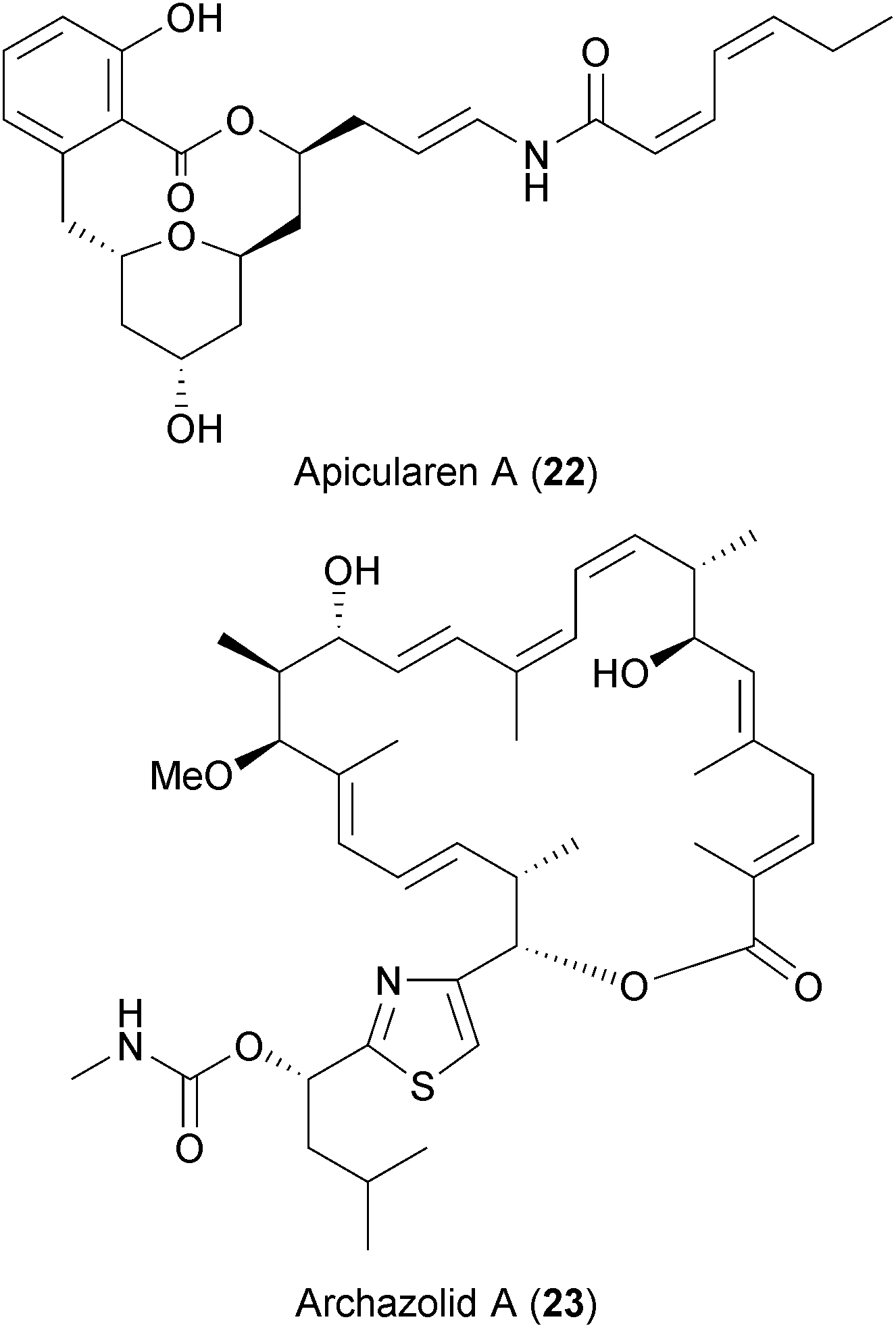
V-ATPases translocate protons from the cytoplasm into intracellular compartments and through the plasma membrane. This activity is important for the function and trafficking of internal organelles such as vacuoles, endosomes, or lysosomes, which are in turn used by viruses for entry, translation, assembly or budding.261,262 However, blocking these proton pumps also leads to other physiological changes in the host cell (ref. 263 and references therein), and thus the benefit-risk ratio of such compounds as BSAs in vivo remains to be determined.
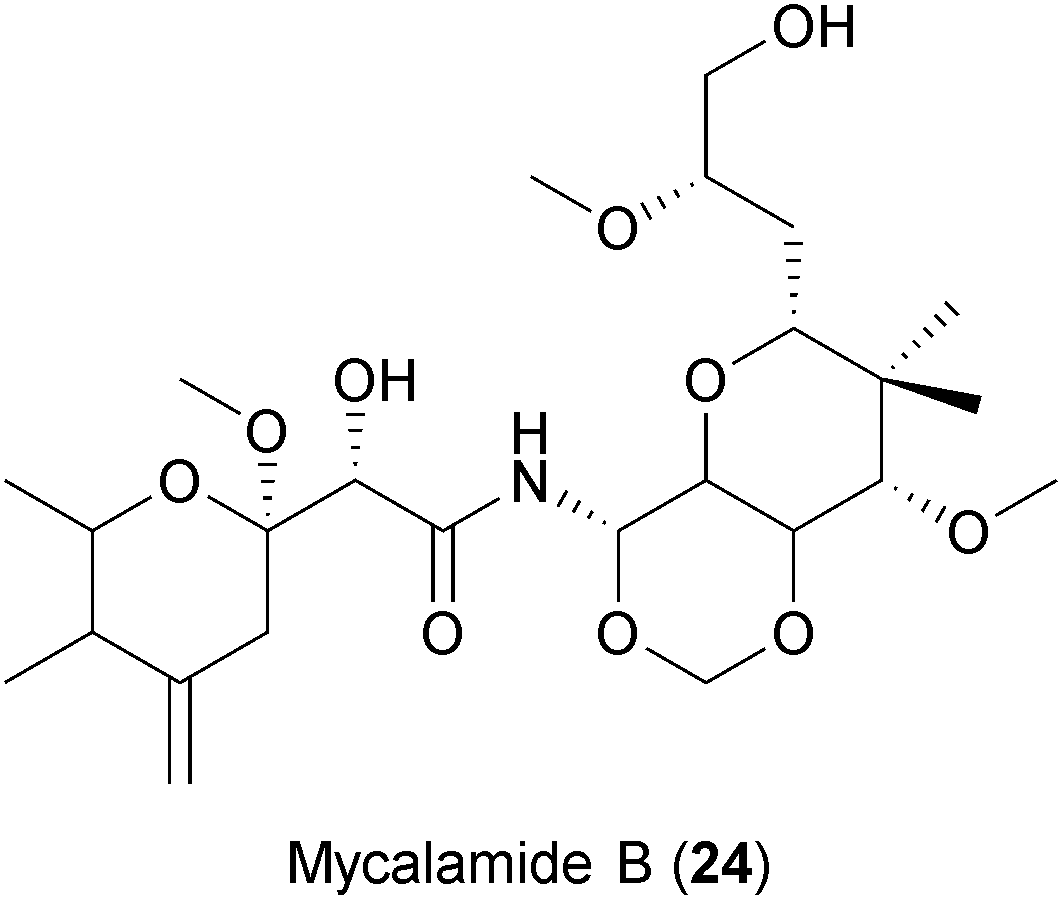
Similar to myxobacteria, marine sponges produce an ample number of secondary metabolites with diverse biological activities.267 Of note, the first antiviral drug approved by the FDA, the nucleoside Ara-A (Vidarabine), was isolated from a marine sponge.239 Mycalamide A and B (24), two natural products isolated from Mycale sponges, have shown antiviral activities against coronaviruses,240 HSV and Polio virus.268 It was suggested that the compounds inhibit viral protein synthesis by direct binding to ribosomes, a well-described host cell target for the compound class.269 However, some analogues of mycalamides are described to inhibit influenza virus in vitro by binding to the viral nucleoprotein (NP), thereby impeding its association with viral RNA.241 Thus, whether mycalamides exert their antiviral action by targeting host factors, viral components or both is still not clear.
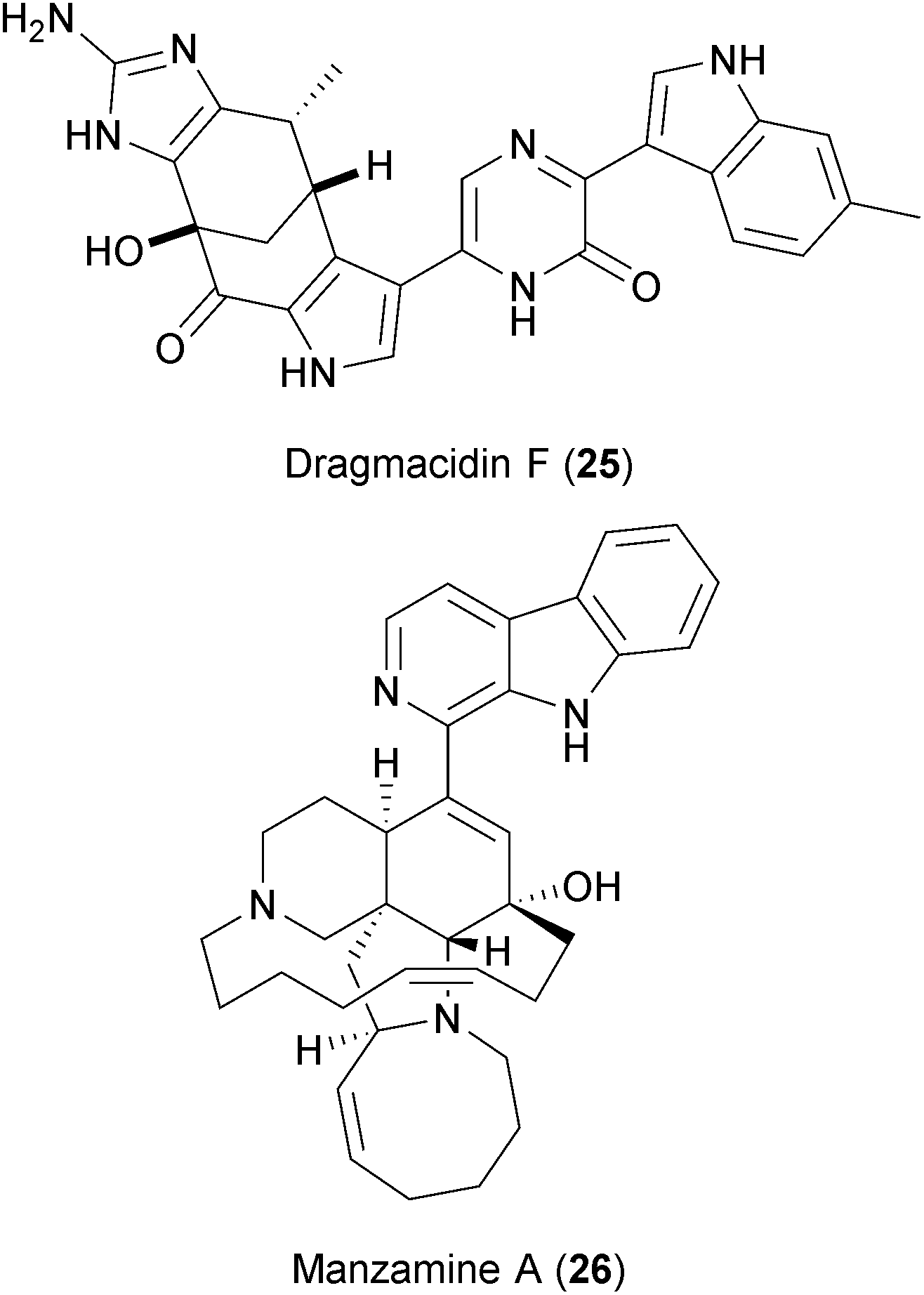
Two alkaloids isolated from Halicortex sponges, dragmacidin F (25) and manzamine A (26), have also been reported to inhibit HSV and HIV.270 Although the exact mechanism of viral inhibition is not clear, dragmacidin F is a serine-threonine protein phosphatase inhibitor271 and manzamine A targets V-ATPases,272 thus providing clues of their BSA action. The viral dependency on the host's V-ATPases was described above, and serine-threonine phosphatases are known to play several roles in viral replication.273,274 Whether dragmacidin targets host and virus-encoded serine-threonine phosphatases275 is not known.
Griffithsin (GRFT) (27), a 13 kDa lectin isolated from the red alga Griffithsia sp.,276 was first shown to bind to oligosaccharides on the surface of the HIV envelope glycoprotein gp120 and block viral entry.276 Similar to CV-N, it interacts with terminal mannose residues found in N-linked glycans of the viral envelope.277 By a similar mechanism, GRFT inhibits SIV, HCV, SARS-CoV, HSV and JEV,245–248,278,279 thus exhibiting broad-spectrum antiviral activities. When applied in combination with antiretroviral therapy, GRFT shows a synergistic inhibitory effect.280 Interestingly, unlike other lectins, GRFT does not induce the production of pro-inflammatory cytokines in treated human peripheral blood mononuclear cells.278 It has been shown to safely protect mice from genital HSV279 and monkeys from vaginal SIV infection,245 thus having promising properties for preventing viral infections. The selection of GRFT-resistant HIV variants has been observed. However, as this requires an extensive loss of glycans and multiple amino acid sequence changes,281 GRFT represents an interesting candidate natural product to be developed into a broad-spectrum antiviral drug.

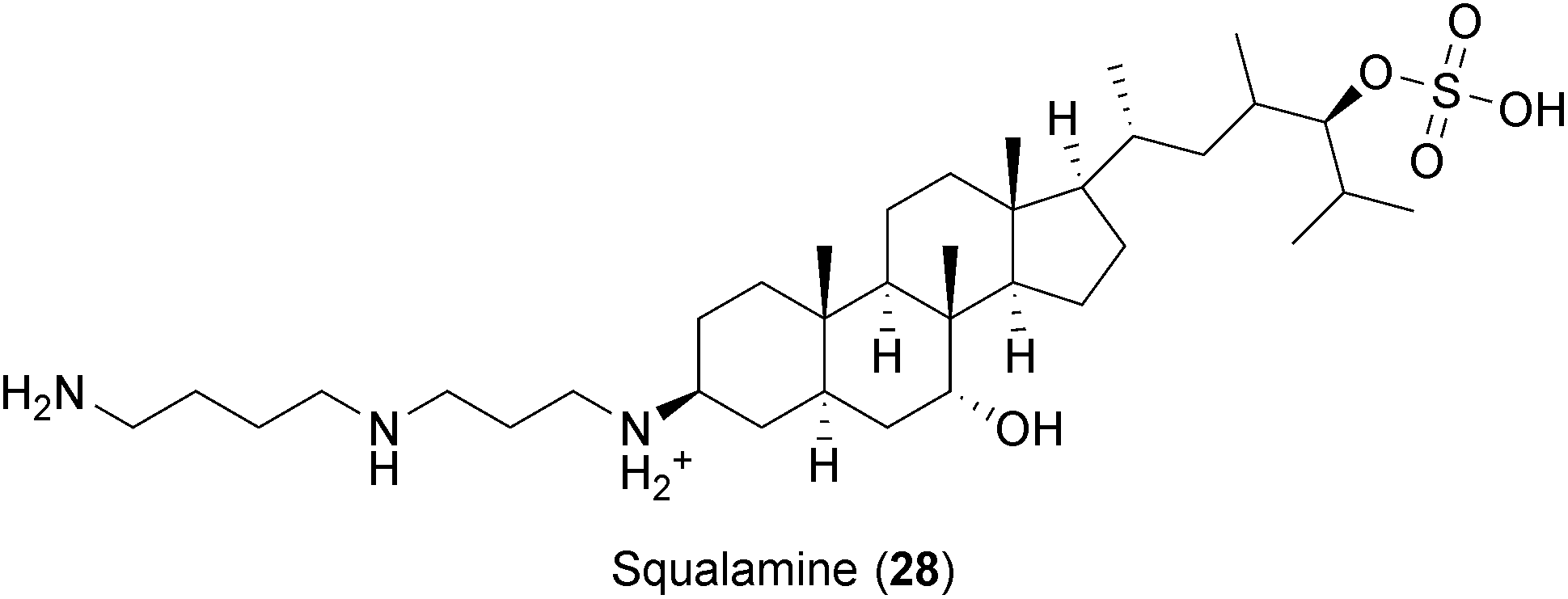
Squalamine (28), an amphipathic sterol isolated from the tissue of the dogfish shark Squalus acanthias,282 has been recently described to inhibit infections by DENV, HBV, HDV, YFV and mouse cytomegalovirus (MCMV).85 Squalamine has a high affinity for anionic phospholipids and is able to neutralize the negative charge of its membrane-associated targets without affecting cell membrane composition.283 Thus, it was suggested that squalamine might disturb the electrostatic associations of host and viral proteins in membrane compartments, rendering the cells unable to support viral replication.85 More studies are needed to determine whether protein displacement by squalamine affects the replication of other relevant viruses such as influenza and HIV.
4. A brief note on BSAs from other sources
Plants and animals also produce peptidic BSAs as part of their defenses against viral pathogens. Likewise, some BSAs are generated de novo by synthetic chemistry. While both groups of compounds are not the main topic of this review, we will briefly mention some for completeness.4.1 Host defense factors
Antiviral defense factors other than ISGs have been described. Defensins and cathelicidins are antimicrobial polypeptides that can be constitutively produced by the host or induced after innate immune recognition of pathogens.284 Two excellent recent reviews refer to the BSA activities of these host defense factors.285,286 Wilson et al. have recently summarized the BSA effect of α- and β-defensins against various viruses including HIV, VSV, AAV, VV, RSV, HSV, and HPV.286 Their proposed mechanisms of action are: interacting with lipid bilayers, binding to glycoproteins and blocking protein–protein or protein–DNA interactions. Thus, defensins can potentially block different steps in viral life cycles.286 Interestingly, defensins seem to be conserved among different organisms. Indeed, mastoparan, a defense peptide found in the venom of wasps, has been described to have BSA activities against VSV, WNV, DENV, HSV, RSV, influenza virus and AdV mainly by disrupting the viral envelope structure.287 Cathelicidins are another type of defense peptides that carry a conserved cathelin-like domain.288 Barlow and colleagues have recently reviewed the BSA effect of cathelicidins.285 In particular, the human cathelicidin LL-37 has been shown to affect several viruses including VV, RSV, influenza virus, HIV, HSV, DENV and AdV by distinct mechanisms such as envelope disruption and polymerase or protease inhibition.285 However, the mere co-existence of cellular defense mechanisms and viruses already implies that there is an ongoing evolutionary race between the arming of cells with new antiviral weapons and the arming of viruses with new anti-host defense mechanisms to stay in place. Recent elegant studies emphasize this “arms race” as part of the evolution of the human innate immune system.289 The extent to which this holds true for the interrelationships of defensins and cathelicidins with viruses has yet to be determined.4.2 Non-natural synthetic ligands
Drug-like small synthetic compounds have also been reported to exhibit BSA activities. Still, most of these chemicals are only able to efficiently block the replication of certain viral groups such as RNA viruses. Examples are arbidol, an indole derivative (reviewed in ref. 290), T-705 (Favipiravir) (reviewed in ref. 291) and more recently BCX4430, a novel nucleoside analog shown to have strong antiviral effects against filoviruses, and mild effects against bunyaviruses, arenaviruses, paramyxoviruses, coronaviruses and flaviviruses.292 Highly encouraging results in vitro and in cynomolgus macaques have yet to be confirmed by clinical studies.5. Conclusions and perspectives
While humans depend on their immune system to repel microbial threats, simpler organisms like plants, bacteria and fungi produce a variety of metabolites for this purpose. These compounds often target general cellular pathways and regulatory elements that are also exploited by viruses for their propagation. Given the intriguing conservation of some of these elements between species and their indiscriminate use by divergent viruses, it is not too surprising to find a rich arsenal of antivirus-acting natural products when analyzing simpler organisms. Indeed, the cyclophilin inhibitors mentioned above are good examples. More recent examples are the described overlapping hits from a myxobacterial library that inhibit both HIV and HCV as well as host factors involved in the processing machinery of cellular and viral RNA.55,237,238,263Today, DAAs are the dominant class of antiviral drugs in use. They are highly successful against clinically important infections like HIV, HCV and several others. Nonetheless, obstacles like drug resistant viruses or emerging viruses with no available selective antivirals in the market call for the utilization of the armament of simpler organisms that target host factors and show broad-spectrum antiviral activities. The observation that some host factors like the HIV co-receptor CCR5 can be targeted without major toxicities is encouraging for the overall concept of targeting the host to inhibit a virus. Compounds like alisporivir with a potentially broader antiviral application range than CCR5 inhibitors are in clinical trials. With (i) the rapidly increasing knowledge of virus–host factor interactions via system-wide screening campaigns, (ii) the highly advanced techniques of natural product isolation, characterization and modification either via chemistry or genetic modifications of producer strains, and (iii) the advances of in silico tools for dynamic molecule simulations and toxicity predictions, the usable antiviral drug space will significantly increase in the coming years. Thus, despite perpetual worrisome news of novel or re-emerging viral threats, there is a rich source of weaponry in nature to appease these threats.
Acknowledgements
We thank Dr Jocelyn Yelle of Antiviral InteliStrat Inc. for providing the referred-to list of FDA-approved antivirals and Sara Ursula Abad for an initial literature search. The authors are supported by grants SAF2013-46077-R (to AM and JM) and BFU2013-44629-R (to JD), both from the Spanish Ministry of Economy and Competitivity, and funds from the German Centre for Infection Research (DZIF, to MB and FS). We thank Dr Heng-Chang Chen, CRG, Barcelona, for the artwork of the graphical abstract.References
- WHO, 2012, http://www.who.int/mediacentre/factsheets/fs204/en/.
- WHO, 2014, http://www.who.int/mediacentre/factsheets/fs164/en/.
- WHO, 2014, http://www.who.int/gho/hiv/en/.
- P. Baccam, C. Beauchemin, C. A. Macken, F. G. Hayden and A. S. Perelson, J. Virol., 2006, 80, 7590–7599 CrossRef CAS PubMed.
- WHO, 2014, http://www.who.int/mediacentre/factsheets/fs286/en/.
- K. E. Jones, N. G. Patel, M. A. Levy, A. Storeygard, D. Balk, J. L. Gittleman and P. Daszak, Nature, 2008, 451, 990–993 CrossRef CAS PubMed.
- L. H. Taylor, S. M. Latham and M. E. Woolhouse, Philos. Trans. R. Soc., B, 2001, 356, 983–989 CrossRef CAS PubMed.
- S. Bhatt, P. W. Gething, O. J. Brady, J. P. Messina, A. W. Farlow, C. L. Moyes, J. M. Drake, J. S. Brownstein, A. G. Hoen, O. Sankoh, M. F. Myers, D. B. George, T. Janeisch, G. R. Wint, C. P. Simmons, T. W. Scott, J. J. Farrar and S. I. Hay, Nature, 2013, 496, 504–507 CrossRef CAS PubMed.
- D. J. Gubler, Trends Microbiol., 2002, 10, 100–103 CrossRef CAS PubMed.
- K. Stadler, V. Masignani, M. Eickmann, S. Becker, S. Abrignani, H.-D. Klenk and R. Rappuoli, Nat. Rev. Microbiol., 2003, 1, 209–218 CrossRef CAS PubMed.
- V. S. Raj, A. D. Osterhaus, R. A. Fouchier and B. L. Haagmans, Curr. Opin. Virol., 2014, 5, 58–62 CrossRef PubMed.
- S. Baize, D. Pannetier, L. Oestereich, T. Rieger, L. Koivogui, N. F. Magassouba, B. Soropogui, M. S. Sow, S. Keïta and H. De Clerck, N. Engl. J. Med., 2014, 371, 1418–1425 CrossRef CAS PubMed.
- WHO, 2014, http://www.who.int/hiv/en/.
- J. Sheldon, B. Ramos, C. Toro, P. Ríos, J. Martínez-Alarcón and M. Romero, Antiviral Ther., 2008, 13, 97 CAS.
- WHO, 2014, http://www.who.int/mediacentre/factsheets/fs117/en/.
- T. M. Uyeki, N. Engl. J. Med., 2014, 370, 3 CrossRef PubMed.
- WHO, 2014, http://www.who.int/mediacentre/factsheets/fs211/en/.
- P. Barreiro, J. V. Fernandez-Montero, C. de Mendoza, P. Labarga and V. Soriano, Antiviral Res., 2014, 105, 1–7 CrossRef CAS PubMed.
- A. P. Kourtis, M. Bulterys, D. J. Hu and D. J. Jamieson, N. Engl. J. Med., 2012, 366, 1749–1752 CrossRef CAS PubMed.
- S. S. Abdool Karim, K. Naidoo, A. Grobler, N. Padayatchi, C. Baxter, A. L. Gray, T. Gengiah, S. Gengiah, A. Naidoo and N. Jithoo, N. Engl. J. Med., 2011, 365, 1492–1501 CrossRef CAS PubMed.
- D. A. Koplow, Smallpox: the fight to eradicate a global scourge, University of California Press, 2004 Search PubMed.
- J. G. Breman and I. Arita, N. Engl. J. Med., 1980, 303, 1263–1273 CrossRef CAS PubMed.
- M. Imai, T. Watanabe, M. Hatta, S. C. Das, M. Ozawa, K. Shinya, G. Zhong, A. Hanson, H. Katsura, S. Watanabe, C. Li, E. Kawakami, S. Yamada, M. Kiso, Y. Suzuki, E. A. Maher, G. Neumann and Y. Kawaoka, Nature, 2012, 486, 420–428 CAS.
- S. Herfst, E. J. Schrauwen, M. Linster, S. Chutinimitkul, E. de Wit, V. J. Munster, E. M. Sorrell, T. M. Bestebroer, D. F. Burke and D. J. Smith, Science, 2012, 336, 1534–1541 CrossRef CAS PubMed.
- S. Merler, M. Ajelli, L. Fumanelli and A. Vespignani, BMC Med., 2013, 11, 252 CrossRef PubMed.
- F. Rey, O. Schwartz and S. Wain-Hobson, Science, 2013, 342, 311 CrossRef CAS PubMed.
- B. R. Bacon, S. C. Gordon, E. Lawitz, P. Marcellin, J. M. Vierling, S. Zeuzem, F. Poordad, Z. D. Goodman, H. L. Sings and N. Boparai, N. Engl. J. Med., 2011, 364, 1207–1217 CrossRef CAS PubMed.
- A. S. Lok, D. F. Gardiner, E. Lawitz, C. Martorell, G. T. Everson, R. Ghalib, R. Reindollar, V. Rustgi, F. McPhee and M. Wind-Rotolo, N. Engl. J. Med., 2012, 366, 216–224 CrossRef CAS PubMed.
- M. P. Manns and T. von Hahn, Nat. Rev. Drug Discovery, 2013, 12(8), 595–610 CrossRef CAS PubMed.
- J. H. Hoofnagle and A. H. Sherker, N. Engl. J. Med., 2014, 370, 1552–1553 CrossRef CAS PubMed.
- E. De Clercq, Med. Res. Rev., 2013 Search PubMed.
- S. Beinhardt, K. Rutter, A. F. Stättermayer and P. Ferenci, Clin. Infect. Dis., 2013, 56, 118–122 CrossRef PubMed.
- J. Berenguer, M. A. von Wichmann, C. Quereda, P. Miralles, J. Mallolas, J. Lopez-Aldeguer, J. Alvarez-Pellicer, J. De Miguel, M. Crespo, J. M. Guardiola, M. J. Tellez, M. J. Galindo, S. Arponen, E. Barquilla, J. M. Bellon and J. Gonzalez-Garcia, J. Antimicrob. Chemother., 2011, 66, 2843–2849 CrossRef CAS PubMed.
- F.-X. Blanc, T. Sok, D. Laureillard, L. Borand, C. Rekacewicz, E. Nerrienet, Y. Madec, O. Marcy, S. Chan and N. Prak, N. Engl. J. Med., 2011, 365, 1471–1481 CrossRef CAS PubMed.
- R. J. Galán, E. C. Cidoncha, M. F. Martin, C. C. Rodriguez, C. Almeida and R. M. Verdugo, J. Manag. Care Pharm., 2013, 19, 448–453 Search PubMed.
- D. Back and L. Else, Dig. Liver Dis., 2013, 45(suppl. 5), S343–S348 CrossRef CAS PubMed.
- D. Burger and D. Back, Antiviral Ther., 2013, 18, 649–650 CrossRef CAS PubMed.
- J. J. Kiser, J. R. Burton Jr and G. T. Everson, Nat. Rev. Gastroenterol. Hepatol., 2013, 10, 596–606 CrossRef CAS PubMed.
- B. Maasoumy, K. Port, B. Calle Serrano, A. A. Markova, L. Sollik, M. P. Manns, M. Cornberg and H. Wedemeyer, Aliment. Pharmacol. Ther., 2013, 38(11–12), 1365–1372 CrossRef CAS PubMed.
- A. Tseng and M. Foisy, Curr. Infect. Dis. Rep., 2012, 14, 67–82 CrossRef PubMed.
- L. Hamzaoui, S. El Bouchtili, K. Siai, M. Mahmoudi and M. M. Azzouz, Clin. Res. Hepatol. Gastroenterol., 2013, 37, e16–e20 CrossRef PubMed.
- E. A. Operskalski and A. Kovacs, Curr. HIV/AIDS Rep., 2011, 8, 12–22 CrossRef PubMed.
- P. Cahn and O. Sued, J. Infect. Dis., 2013, 208, 4–6 CrossRef PubMed.
- R. P. van Heeswijk, M. Beumont, R. S. Kauffman and V. Garg, Antiviral Ther., 2013, 18, 553–560 CrossRef CAS PubMed.
- F. Müller and M. F. Fromm, Pharmacogenomics, 2011, 12, 1017–1037 CrossRef PubMed.
- G. L. Melikian, S.-Y. Rhee, V. Varghese, D. Porter, K. White, J. Taylor, W. Towner, P. Troia, J. Burack and E. DeJesus, J. Antimicrob. Chemother., 2014, 69, 12–20 CrossRef CAS PubMed.
- D. L. Wyles, J. Infect. Dis., 2013, 207, S33–S39 CrossRef CAS PubMed.
- L. R. Gray, D. Gabuzda, D. Cowley, A. Ellett, L. Chiavaroli, S. L. Wesselingh, M. J. Churchill and P. R. Gorry, J. NeuroVirol., 2011, 17, 82–91 CrossRef CAS PubMed.
- J. Larrubia, M. Lokhande, S. García-Garzón, J. Miquel, A. González-Praetorius, T. Parra-Cid and E. Sanz-de-Villalobos, J. Viral Hepat., 2013, 20, 85–94 CrossRef CAS PubMed.
- D. N. Martin and S. L. Uprichard, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10777–10782 CrossRef CAS PubMed.
- J. L. Nieva, V. Madan and L. Carrasco, Nat. Rev. Microbiol., 2012, 10, 563–574 CrossRef CAS PubMed.
- D. van Riel, L. M. Leijten, G. Kochs, A. D. Osterhaus and T. Kuiken, Am. J. Pathol., 2013, 183, 1382–1389 CrossRef CAS PubMed.
- H. Xu, X. Wang, A. A. Lackner and R. S. Veazey, J. Leukocyte Biol., 2013, 93, 943–950 CrossRef CAS PubMed.
- N. S. Heaton, R. Perera, K. L. Berger, S. Khadka, D. J. LaCount, R. J. Kuhn and G. Randall, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17345–17350 CrossRef CAS PubMed.
- N. Scheller, L. B. Mina, R. P. Galao, A. Chari, M. Gimenez-Barcons, A. Noueiry, U. Fischer, A. Meyerhans and J. Diez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13517–13522 CrossRef CAS PubMed.
- V. Baumgärtel, S. Ivanchenko, A. Dupont, M. Sergeev, P. W. Wiseman, H.-G. Kräusslich, C. Bräuchle, B. Müller and D. C. Lamb, Nat. Cell Biol., 2011, 13, 469–474 CrossRef PubMed.
- J. G. Carlton and J. Martin-Serrano, Biochem. Soc. Trans., 2009, 37, 195–199 CrossRef CAS PubMed.
- Z. Feng, L. Hensley, K. L. McKnight, F. Hu, V. Madden, L. Ping, S. H. Jeong, C. Walker, R. E. Lanford and S. M. Lemon, Nature, 2013, 496, 367–371 CrossRef CAS PubMed.
- W. M. Henne, N. J. Buchkovich and S. D. Emr, Dev. Cell, 2011, 21, 77–91 CrossRef CAS PubMed.
- B. McDonald and J. Martin-Serrano, J. Cell Sci., 2009, 122, 2167–2177 CrossRef CAS PubMed.
- G. Pasqual, J. M. Rojek, M. Masin, J. Y. Chatton and S. Kunz, PLoS Pathog., 2011, 7, e1002232 CAS.
- D. Silva-Ayala, T. Lopez, M. Gutierrez, N. Perrimon, S. Lopez and C. F. Arias, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(25), 10270–10275 CrossRef CAS PubMed.
- J. Votteler and W. I. Sundquist, Cell Host Microbe, 2013, 14, 232–241 CAS.
- Y. Ma-Lauer, J. Lei, R. Hilgenfeld and A. von Brunn, Curr. Opin. Virol., 2012, 2, 614–621 CrossRef CAS PubMed.
- M. L. Shaw, Rev. Med. Virol., 2011, 21, 358–369 CrossRef CAS PubMed.
- O. M. Sessions, N. J. Barrows, J. A. Souza-Neto, T. J. Robinson, C. L. Hershey, M. A. Rodgers, J. L. Ramirez, G. Dimopoulos, P. L. Yang, J. L. Pearson and M. A. Garcia-Blanco, Nature, 2009, 458, 1047–1050 CrossRef CAS PubMed.
- H. Zhou, M. Xu, Q. Huang, A. T. Gates, X. D. Zhang, J. C. Castle, E. Stec, M. Ferrer, B. Strulovici, D. J. Hazuda and A. S. Espeseth, Cell Host Microbe, 2008, 4, 495–504 CAS.
- B. de Chassey, L. Meyniel-Schicklin, A. Aublin-Gex, P. Andre and V. Lotteau, Curr. Opin. Virol., 2012, 2, 606–613 CrossRef CAS PubMed.
- K. Lin and P. Gallay, Antiviral Res., 2013, 99, 68–77 CrossRef CAS PubMed.
- M. B. Zeisel, J. Lupberger, I. Fofana and T. F. Baumert, J. Hepatol., 2013, 58, 375–384 CrossRef PubMed.
- E. De Clercq, Nat. Rev. Drug Discovery, 2002, 1, 13–25 CrossRef CAS PubMed.
- C. C. Chang, M. Crane, J. Zhou, M. MIna, J. J. Post, B. A. Cameron, A. R. Lloyd, A. Jaworowski, M. A. French and S. R. Lewin, Immunol. Rev., 2013, 254(1), 114–142 CrossRef PubMed.
- C. L. Clouser, S. E. Patterson and L. M. Mansky, J. Virol., 2010, 84, 9301–9309 CrossRef CAS PubMed.
- J. J. Feld and J. H. Hoofnagle, Nature, 2005, 436, 967–972 CrossRef CAS PubMed.
- B. B. Gowen, M. H. Wong, K. H. Jung, A. B. Sanders, M. Mendenhall, K. W. Bailey, Y. Furuta and R. W. Sidwell, Antimicrob. Agents Chemother., 2007, 51, 3168–3176 CrossRef CAS PubMed.
- A. Garcia-Sastre, Nature, 2013, 494, 181–182 CrossRef CAS PubMed.
- B. X. Wang and E. N. Fish, Trends Immunol., 2012, 33, 190–197 CrossRef CAS PubMed.
- N. S. Heaton and G. Randall, Trends Microbiol., 2011, 19, 368–375 CrossRef CAS PubMed.
- P. D. Nagy and J. Pogany, Nat. Rev. Microbiol., 2012, 10, 137–149 CAS.
- F. S. Vieira, G. Correa, M. Einicker-Lamas and R. Coutinho-Silva, Biol. Cell, 2010, 102, 391–407 CrossRef CAS PubMed.
- L. Delang, J. Paeshuyse and J. Neyts, Biochem. Pharmacol., 2012, 84, 1400–1408 CrossRef CAS PubMed.
- L. Rajendran, H. J. Knolker and K. Simons, Nat. Rev. Drug Discovery, 2010, 9, 29–42 CrossRef CAS PubMed.
- S. Reiss, C. Harak, I. Romero-Brey, D. Radujkovic, R. Klein, A. Ruggieri, I. Rebhan, R. Bartenschlager and V. Lohmann, PLoS Pathog., 2013, 9, e1003359 CAS.
- V. Chukkapalli, I. B. Hogue, V. Boyko, W.-S. Hu and A. Ono, J. Virol., 2008, 82, 2405–2417 CrossRef CAS PubMed.
- M. Zasloff, A. P. Adams, B. Beckerman, A. Campbell, Z. Han, E. Luijten, I. Meza, J. Julander, A. Mishra and W. Qu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15978–15983 CrossRef CAS PubMed.
- E. Ikonen, Nat. Rev. Mol. Cell Biol., 2008, 9, 125–138 CrossRef CAS PubMed.
- I. Le Blanc, P. P. Luyet, V. Pons, C. Ferguson, N. Emans, A. Petiot, N. Mayran, N. Demaurex, J. Faure, R. Sadoul, R. G. Parton and J. Gruenberg, Nat. Cell Biol., 2005, 7, 653–664 CrossRef CAS PubMed.
- E. Sun, J. He and X. Zhuang, J. Virol., 2013, 87, 2673–2685 CrossRef CAS PubMed.
- C. Bissig, M. Lenoir, M.-C. Velluz, I. Kufareva, R. Abagyan, M. Overduin and J. Gruenberg, Dev. Cell, 2013, 25, 364–373 CrossRef CAS PubMed.
- N. Arhel and F. Kirchhoff, Biochim. Biophys. Acta, 2010, 1802, 313–321 CrossRef CAS PubMed.
- D. K. Cureton, R. Burdeinick-Kerr and S. P. Whelan, J. Virol., 2012, 86, 655–666 CrossRef CAS PubMed.
- R. Konig, S. Stertz, Y. Zhou, A. Inoue, H. H. Hoffmann, S. Bhattacharyya, J. G. Alamares, D. M. Tscherne, M. B. Ortigoza, Y. Liang, Q. Gao, S. E. Andrews, S. Bandyopadhyay, P. De Jesus, B. P. Tu, L. Pache, C. Shih, A. Orth, G. Bonamy, L. Miraglia, T. Ideker, A. Garcia-Sastre, J. A. Young, P. Palese, M. L. Shaw and S. K. Chanda, Nature, 2010, 463, 813–817 CrossRef PubMed.
- P. Ahlquist, Nat. Rev. Microbiol., 2006, 4, 371–382 CrossRef CAS PubMed.
- G. A. Belov, N. Altan-Bonnet, G. Kovtunovych, C. L. Jackson, J. Lippincott-Schwartz and E. Ehrenfeld, J. Virol., 2007, 81, 558–567 CrossRef CAS PubMed.
- J. A. den Boon, A. Diaz and P. Ahlquist, Cell Host Microbe, 2010, 8, 77–85 CAS.
- N. Y. Hsu, O. Ilnytska, G. Belov, M. Santiana, Y. H. Chen, P. M. Takvorian, C. Pau, H. van der Schaar, N. Kaushik-Basu, T. Balla, C. E. Cameron, E. Ehrenfeld, F. J. van Kuppeveld and N. Altan-Bonnet, Cell, 2010, 141, 799–811 CrossRef CAS PubMed.
- A. Joshi, H. Garg, K. Nagashima, J. S. Bonifacino and E. O. Freed, Mol. Cell, 2008, 30, 227–238 CrossRef CAS PubMed.
- K. H. Lanke, H. M. van der Schaar, G. A. Belov, Q. Feng, D. Duijsings, C. L. Jackson, E. Ehrenfeld and F. J. van Kuppeveld, J. Virol., 2009, 83, 11940–11949 CrossRef CAS PubMed.
- J. Sasaki, K. Ishikawa, M. Arita and K. Taniguchi, EMBO J., 2012, 31, 754–766 CrossRef CAS PubMed.
- M. H. Verheije, M. Raaben, M. Mari, E. G. Te Lintelo, F. Reggiori, F. J. van Kuppeveld, P. J. Rottier and C. A. de Haan, PLoS Pathog., 2008, 4, e1000088 Search PubMed.
- H. M. van der Schaar, L. van der Linden, K. H. Lanke, J. R. Strating, G. Pürstinger, E. de Vries, C. A. de Haan, J. Neyts and F. J. van Kuppeveld, Cell Res., 2012, 22, 1576–1592 CrossRef CAS PubMed.
- J. Chang, T. M. Block and J.-T. Guo, Antiviral Res., 2013, 99, 251–260 CrossRef CAS PubMed.
- T. L. Davis, J. R. Walker, V. Campagna-Slater, P. J. Finerty Jr, R. Paramanathan, G. Bernstein, F. MacKenzie, W. Tempel, H. Ouyang and W. H. Lee, PLoS Biol., 2010, 8, e1000439 Search PubMed.
- P. Nigro, G. Pompilio and M. Capogrossi, Cell Death Dis., 2013, 4, e888 CrossRef CAS PubMed.
- M. Bienkowska-Haba, C. Williams, S. M. Kim, R. L. Garcea and M. Sapp, J. Virol., 2012, 86, 9875–9887 CrossRef CAS PubMed.
- S. Bose, M. Mathur, P. Bates, N. Joshi and A. K. Banerjee, J. Gen. Virol., 2003, 84, 1687–1699 CrossRef CAS PubMed.
- S. Chokshi, S. Phillips, A. Riva and N. Naoumov, Gut, 2012, 61, A11 CrossRef.
- A. H. de Wilde, J. C. Zevenhoven-Dobbe, Y. van der Meer, V. Thiel, K. Narayanan, S. Makino, E. J. Snijder and M. J. van Hemert, J. Gen. Virol., 2011, 92, 2542–2548 CrossRef CAS PubMed.
- P. A. Gallay, R. G. Ptak, M. D. Bobardt, J. M. Dumont, G. Vuagniaux and B. Rosenwirth, Viruses, 2013, 5, 981–997 CrossRef CAS PubMed.
- J. A. Garcia-Rivera, M. Bobardt, U. Chatterji, S. Hopkins, M. A. Gregory, B. Wilkinson, K. Lin and P. A. Gallay, Antimicrob. Agents Chemother., 2012, 56, 5113–5121 CrossRef CAS PubMed.
- L. R. Keyes, M. G. Bego, M. Soland and S. St Jeor, J. Gen. Virol., 2012, 93, 722–732 CrossRef CAS PubMed.
- X. Liu, Z. Zhao, Z. Li, C. Xu, L. Sun, J. Chen and W. Liu, PLoS One, 2012, 7, e37277 CAS.
- S. Pfefferle, J. Schopf, M. Kogl, C. C. Friedel, M. A. Muller, J. Carbajo-Lozoya, T. Stellberger, E. von Dall'Armi, P. Herzog, S. Kallies, D. Niemeyer, V. Ditt, T. Kuri, R. Zust, K. Pumpor, R. Hilgenfeld, F. Schwarz, R. Zimmer, I. Steffen, F. Weber, V. Thiel, G. Herrler, H. J. Thiel, C. Schwegmann-Wessels, S. Pohlmann, J. Haas, C. Drosten and A. von Brunn, PLoS Pathog., 2011, 7, e1002331 CAS.
- M. W. Yap, M. P. Dodding and J. P. Stoye, J. Virol., 2006, 80, 4061–4067 CrossRef CAS PubMed.
- F. E. Membreno, J. C. Espinales and E. J. Lawitz, Clin. Liver Dis., 2013, 17, 129–139 CrossRef PubMed.
- M. Peel and A. Scribner, Bioorg. Med. Chem. Lett., 2013, 23, 4485–4492 CrossRef CAS PubMed.
- J. Chang, T. K. Warren, X. Zhao, T. Gill, F. Guo, L. Wang, M. A. Comunale, Y. Du, D. S. Alonzi, W. Yu, H. Ye, F. Liu, J. T. Guo, A. Mehta, A. Cuconati, T. D. Butters, S. Bavari, X. Xu and T. M. Block, Antiviral Res., 2013, 98, 432–440 CrossRef CAS PubMed.
- D. Durantel, Curr. Opin. Invest. Drugs, 2009, 10, 860–870 CAS.
- A. P. Rathore, P. N. Paradkar, S. Watanabe, K. H. Tan, C. Sung, J. E. Connolly, J. Low, E. E. Ooi and S. G. Vasudevan, Antiviral Res., 2011, 92, 453–460 CrossRef CAS PubMed.
- R. E. Hancock, A. Nijnik and D. J. Philpott, Nat. Rev. Microbiol., 2012, 10, 243–254 CrossRef CAS PubMed.
- N. Yan and Z. J. Chen, Nat. Immunol., 2012, 13, 214–222 CrossRef CAS PubMed.
- J. W. Schoggins, S. J. Wilson, M. Panis, M. Y. Murphy, C. T. Jones, P. Bieniasz and C. M. Rice, Nature, 2011, 472, 481–485 CrossRef CAS PubMed.
- R. Baccala, K. Hoebe, D. H. Kono, B. Beutler and A. N. Theofilopoulos, Nat. Med., 2007, 13, 543–551 CrossRef CAS PubMed.
- M. Colonna, Eur. J. Immunol., 2007, 37, 306–309 CrossRef CAS PubMed.
- O. Haller, Cell Host Microbe, 2013, 14, 371–373 CAS.
- O. Haller and G. Kochs, J. Interferon Cytokine Res., 2011, 31, 79–87 CrossRef CAS PubMed.
- M. S. Diamond and M. Farzan, Nat. Rev. Immunol., 2013, 13, 46–57 CrossRef CAS PubMed.
- J. Rehwinkel and C. Reis e Sousa, Science, 2010, 327, 284–286 CrossRef CAS PubMed.
- E. J. Hennessy, A. E. Parker and L. A. O'Neill, Nat. Rev. Drug Discovery, 2010, 9, 293–307 CrossRef CAS PubMed.
- M. Carroll, S. Yerkovich and J. Upham, Am. J. Respir. Crit. Care Med., 2010, 181, A4230 Search PubMed.
- H. Hemmi, T. Kaisho, O. Takeuchi, S. Sato, H. Sanjo, K. Hoshino, T. Horiuchi, H. Tomizawa, K. Takeda and S. Akira, Nat. Immunol., 2002, 3, 196–200 CrossRef CAS PubMed.
- A. Terlou, M. v. Seters, A. Kleinjan, C. Heijmans-Antonissen, L. A. Santegoets, I. Beckmann, M. v. Beurden, T. J. Helmerhorst and L. J. Blok, Int. J. Cancer, 2010, 127, 2831–2840 CrossRef CAS PubMed.
- G. M. Bahr, J. Antimicrob. Chemother., 2003, 51, 5–8 CrossRef CAS PubMed.
- D. T. O'Hagan and E. De Gregorio, Drug Discovery Today, 2009, 14, 541–551 CrossRef PubMed.
- M. Hedayat, M. G. Netea and N. Rezaei, Lancet Infect. Dis., 2011, 11, 702–712 CrossRef CAS PubMed.
- M. S. Butler, Nat. Prod. Rep., 2008, 25, 475–516 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- J. Rosen, J. Gottfries, S. Muresan, A. Backlund and T. I. Oprea, J. Med. Chem., 2009, 52, 1953–1962 CrossRef CAS PubMed.
- J. Clardy and C. Walsh, Nature, 2004, 432, 829–837 CrossRef CAS PubMed.
- N. Dixon, L. S. Wong, T. H. Geerlings and J. Micklefield, Nat. Prod. Rep., 2007, 24, 1288–1310 RSC.
- J. J. La Clair, Nat. Prod. Rep., 2010, 27, 969–995 RSC.
- E. K. Schmitt, C. M. Moore, P. Krastel and F. Petersen, Curr. Opin. Chem. Biol., 2011, 15, 497–504 CrossRef CAS PubMed.
- A. Bauer and M. Brönstrup, Nat. Prod. Rep., 2014, 31, 35–60 RSC.
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2013, 66, 571–591 CrossRef CAS PubMed.
- W. Bergmann and D. C. Burke, J. Org. Chem., 1956, 21, 226–228 CrossRef CAS.
- W. Bergmann and R. J. Feeney, J. Am. Chem. Soc., 1950, 72, 2809–2810 CrossRef CAS.
- W. Bergmann and R. J. Feeney, J. Org. Chem., 1951, 16, 981–987 CrossRef CAS.
- D. J. Newman, G. M. Cragg and K. M. Snader, Nat. Prod. Rep., 2000, 17, 215–234 RSC.
- K. R. Darnall, L. B. Townsend and R. K. Robins, Proc. Natl. Acad. Sci. U. S. A., 1967, 57, 548–553 CrossRef CAS.
- H. Nishimura, M. Mayama, Y. Komatsu, H. Kato, N. Shimaoka and Y. Tanaka, J. Antibiot., 1964, 17, 148–155 CAS.
- S. Matsuda and S. Koyasu, Immunopharmacology, 2000, 47, 119–125 CrossRef CAS PubMed.
- J. Luban, K. L. Bossolt, E. K. Franke, G. V. Kalpana and S. P. Goff, Cell, 1993, 73, 1067–1078 CrossRef CAS PubMed.
- C. R. Damaso and S. Keller, Arch. Virol., 1994, 134, 303–319 CrossRef CAS PubMed.
- C. Damaso and N. Moussatch, J. Gen. Virol., 1998, 79, 339–346 CrossRef CAS PubMed.
- A. Vahlne, P.-A. Larsson, P. Horal, J. Ahlmen, B. Svennerholm, J. Gronowitz and S. Olofsson, Arch. Virol., 1992, 122, 61–75 CrossRef CAS PubMed.
- U. Chatterji, J. A. Garcia-Rivera, J. Baugh, K. Gawlik, K. A. Wong, W. Zhong, C. A. Brass, N. V. Naoumov and P. A. Gallay, Antimicrob. Agents Chemother., 2014, 58(6), 3327–3334 CrossRef PubMed.
- A. Endo, International Congress Series, 2004 Search PubMed.
- J. de Lemos, E. Braunwald, M. Blazing, S. Murphy, J. Downs, A. Gotto, M. Clearfield, H. Holdaas, D. Gordon and B. Davis, Lancet, 2010, 376, 1670–1681 CrossRef.
- B. Kwak, F. Mulhaupt, S. Myit and F. Mach, Nat. Med., 2000, 6, 1399–1402 CrossRef CAS PubMed.
- J. Ye, C. Wang, R. Sumpter, M. S. Brown, J. L. Goldstein and M. Gale, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15865–15870 CrossRef CAS PubMed.
- T. Amet, M. Nonaka, M. Z. Dewan, Y. Saitoh, X. Qi, S. Ichinose, N. Yamamoto and S. Yamaoka, Microbes Infect., 2008, 10, 471–480 CrossRef CAS PubMed.
- T. Bader and B. Korba, Antiviral Res., 2010, 86, 241–245 CrossRef CAS PubMed.
- G. del Real, S. Jiménez-Baranda, E. Mira, R. A. Lacalle, P. Lucas, C. Gómez-Moutón, M. Alegret, J. M. Peña, M. Rodríguez-Zapata and M. Alvarez-Mon, J. Exp. Med., 2004, 200, 541–547 CrossRef CAS PubMed.
- D. S. Fedson, Antiviral Res., 2013, 99, 417–435 CrossRef CAS PubMed.
- J.-F. Giguère and M. J. Tremblay, J. Virol., 2004, 78, 12062–12065 CrossRef PubMed.
- K. Jung, Q. Wang, Y. Kim, K. Scheuer, Z. Zhang, Q. Shen, K.-O. Chang and L. J. Saif, PLoS One, 2012, 7, e41619 CAS.
- M. Martínez-Gutierrez, J. E. Castellanos and J. C. Gallego-Gómez, Intervirology, 2011, 54, 202–216 CrossRef PubMed.
- J. Peng, D. Zhang, Y. Ma, G. Wang, Z. Guo and J. Lu, Mol. Med. Rep., 2014, 9, 2221–2226 CAS.
- H. Sadanari, T. Murayama, X. Zheng, R. Yamada, K. Matsubara, H. Yoshida and T. Takahashi, J. Exp. Clin. Med., 2013, 5, 187–193 CrossRef CAS.
- B. Chauvin, S. Drouot, A. Barrail-Tran and A.-M. Taburet, Clin. Pharmacokinet., 2013, 1–17 Search PubMed.
- M. Ikeda, K. Abe, M. Yamada, H. Dansako, K. Naka and N. Kato, Hepatology, 2006, 44, 117–125 CrossRef CAS PubMed.
- T. Franklin and J. M. Cook, Biochem. J., 1969, 113, 515–524 CrossRef CAS PubMed.
- J. E. Silverman Kitchin, M. K. Pomeranz, G. Pak, K. Washenik and J. L. Shupack, J. Am. Acad. Dermatol., 1997, 37, 445–449 CrossRef.
- A. C. Allison and E. M. Eugui, Immunopharmacology, 2000, 47, 85–118 CrossRef CAS PubMed.
- J. Cline, J. D. Nelson, K. Gerzon, R. Williams and D. Delong, Appl. Microbiol., 1969, 18, 14–20 CAS.
- Y. Wang, X. Zhou, Y. Debing, K. Chen, L. J. Van Der Laan, J. Neyts, H. L. Janssen, H. J. Metselaar, M. P. Peppelenbosch and Q. Pan, Gastroenterology, 2014, 146, 1775–1783 CrossRef CAS PubMed.
- A. G. Chapuis, G. P. Rizzardi, C. D'Agostino, A. Attinger, C. Knabenhans, S. Fleury, H. Acha-Orbea and G. Pantaleo, Nat. Med., 2000, 6, 762–768 CrossRef CAS PubMed.
- D. H. Chung, Y. Sun, W. B. Parker, J. B. Arterburn, A. Bartolucci and C. B. Jonsson, J. Virol., 2007, 81, 11722–11729 CrossRef CAS PubMed.
- M. S. Diamond, M. Zachariah and E. Harris, Virology, 2002, 304, 211–221 CrossRef CAS PubMed.
- Z. Gong, S. De Meyer, C. Clarysse, C. Verslype, J. Neyts, E. De Clercq and S. H. Yap, J. Viral Hepat., 1999, 6, 229–236 CrossRef CAS PubMed.
- S. D. Henry, H. J. Metselaar, R. C. Lonsdale, A. Kok, B. L. Haagmans, H. W. Tilanus and L. J. Van der Laan, Gastroenterology, 2006, 131, 1452–1462 CrossRef CAS PubMed.
- J. D. Morrey, D. F. Smee, R. W. Sidwell and C. Tseng, Antiviral Res., 2002, 55, 107–116 CrossRef CAS PubMed.
- J. Neyts, G. Andrei and E. De Clercq, Antimicrob. Agents Chemother., 1998, 42, 216–222 CAS.
- M. M. Hossain, J. J. Coull, G. L. Drusano and D. M. Margolis, Antiviral Res., 2002, 55, 41–52 CrossRef CAS PubMed.
- D. Margolis, A. Heredia, J. Gaywee, D. Oldach, G. Drusano and R. Redfield, J. Acquired Immune Defic. Syndr., 1999, 21, 362–370 CrossRef CAS PubMed.
- L. M. Scheidel and V. Stollar, Virology, 1991, 181, 490–499 CrossRef CAS PubMed.
- Y. Pan, H. Hori, R. Saul, B. A. Sanford, R. J. Molyneux and A. D. Elbein, Biochemistry, 1983, 22, 3975–3984 CrossRef CAS PubMed.
- P. S. Sunkara, T. L. Bowlin, P. S. Liu and A. Sjoerdsma, Biochem. Biophys. Res. Commun., 1987, 148, 206–210 CrossRef CAS PubMed.
- D. Taylor, L. Fellows, G. Farrar, R. Nash, D. Taylor-Robinson, M. Mobberley, T. Ryder, D. Jeffries and A. Tyms, Antiviral Res., 1988, 10, 11–26 CrossRef CAS PubMed.
- S. P. Ahmed, R. J. Nash, C. G. Bridges, D. L. Taylor, M. S. Kang, E. A. Porter and A. S. Tyms, Biochem. Biophys. Res. Commun., 1995, 208, 267–273 CrossRef CAS PubMed.
- C. G. Bridges, S. P. Ahmed, M. S. Kang, R. J. Nash, E. A. Porter and A. S. Tyms, Glycobiology, 1995, 5, 249–253 CrossRef CAS PubMed.
- S.-F. Wu, C.-J. Lee, C.-L. Liao, R. A. Dwek, N. Zitzmann and Y.-L. Lin, J. Virol., 2002, 76, 3596–3604 CrossRef CAS PubMed.
- K. Whitby, D. Taylor, D. Patel, P. Ahmed and A. S. Tyms, Antiviral Chem. Chemother., 2004, 15, 141–151 CrossRef CAS.
- L. Sorbera, J. Castaner and L. Garcia-Capdevila, Drugs Future, 2005, 30, 545–552 CrossRef CAS.
- K. Whitby, T. C. Pierson, B. Geiss, K. Lane, M. Engle, Y. Zhou, R. W. Doms and M. S. Diamond, J. Virol., 2005, 79, 8698–8706 CrossRef CAS PubMed.
- Y. Tanaka, J. Kato, M. Kohara and M. S. Galinski, Antiviral Res., 2006, 72, 1–9 CrossRef CAS PubMed.
- B. Gu, P. Mason, L. Wang, P. Norton, N. Bourne, R. Moriarty, A. Mehta, M. Despande, R. Shah and T. Block, Antiviral Chem. Chemother., 2007, 18, 49 CrossRef CAS.
- A. Helenius and M. Aebi, Annu. Rev. Biochem., 2004, 73, 1019–1049 CrossRef CAS PubMed.
- P. S. Sunkara, M. S. Kang, T. L. Bowlin, P. S. Liu, A. S. Tyms and A. Sjoerdsma, Ann. N. Y. Acad. Sci., 1990, 616, 90–96 CrossRef CAS PubMed.
- V. C. Chu and G. R. Whittaker, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18153–18158 CrossRef CAS PubMed.
- D. J. Wardrop and S. L. Waidyarachchi, Nat. Prod. Rep., 2010, 27, 1431–1468 RSC.
- A. Scalbert, Phytochemistry, 1991, 30, 3875–3883 CrossRef CAS.
- P. Buzzini, P. Arapitsas, M. Goretti, E. Branda, B. Turchetti, P. Pinelli, F. Ieri and A. Romani, Mini-Rev. Med. Chem., 2008, 8, 1179–1187 CrossRef CAS PubMed.
- H. Nakashima, T. Murakami, N. Yamamoto, H. Sakagami, S.-i. Tanuma, T. Hatano, T. Yoshida and T. Okuda, Antiviral Res., 1992, 18, 91–103 CrossRef CAS PubMed.
- L.-T. Lin, T.-Y. Chen, S.-C. Lin, C.-Y. Chung, T.-C. Lin, G.-H. Wang, R. Anderson, C.-C. Lin and C. D. Richardson, BMC Microbiol., 2013, 13, 187 CrossRef CAS PubMed.
- Y. Yang, J. Xiu, L. Zhang, C. Qin and J. Liu, Phytomedicine, 2012, 20, 67–70 CrossRef CAS PubMed.
- L.-T. Lin, T.-Y. Chen, C.-Y. Chung, R. S. Noyce, T. B. Grindley, C. McCormick, T.-C. Lin, G.-H. Wang, C.-C. Lin and C. D. Richardson, J. Virol., 2011, 85, 4386–4398 CrossRef CAS PubMed.
- E. Kamhi, E. J. Joo, J. S. Dordick and R. J. Linhardt, Biol. Rev. Cambridge Philos. Soc., 2013, 88, 928–943 CrossRef PubMed.
- P. M. Matos, D. Andreu, N. C. Santos and R. Gutiérrez-Gallego, Arch. Virol., 2013, 1–6 Search PubMed.
- I. Montanuy, A. Alejo and A. Alcami, FASEB J., 2011, 25, 1960–1971 CrossRef CAS PubMed.
- B. Salvador, N. R. Sexton, R. Carrion, J. Nunneley, J. L. Patterson, I. Steffen, K. Lu, M. O. Muench, D. Lembo and G. Simmons, J. Virol., 2013, 87, 3295–3304 CrossRef CAS PubMed.
- R. M. Schowalter, D. V. Pastrana and C. B. Buck, PLoS Pathog., 2011, 7, e1002161 CAS.
- D. Watterson, B. Kobe and P. R. Young, J. Gen. Virol., 2012, 93, 72–82 CrossRef CAS PubMed.
- W. M. Abdel-Mageed, S. A. Bayoumi, C. Chen, C. J. Vavricka, L. Li, A. Malik, H. Dai, F. Song, L. Wang and J. Zhang, Bioorg. Med. Chem., 2014, 22(7), 2236–2243 CrossRef CAS PubMed.
- M. R. Boyd, K. R. Gustafson, J. B. McMahon, R. H. Shoemaker, B. R. O'Keefe, T. Mori, R. J. Gulakowski, L. Wu, M. I. Rivera and C. M. Laurencot, Antimicrob. Agents Chemother., 1997, 41, 1521–1530 CAS.
- L. G. Barrientos, B. R. O'Keefe, M. Bray, A. Sanchez, A. M. Gronenborn and M. R. Boyd, Antiviral Res., 2003, 58, 47–56 CrossRef CAS PubMed.
- F. Helle, C. Wychowski, N. Vu-Dac, K. R. Gustafson, C. Voisset and J. Dubuisson, J. Biol. Chem., 2006, 281, 25177–25183 CrossRef CAS PubMed.
- B. R. O'Keefe, D. F. Smee, J. A. Turpin, C. J. Saucedo, K. R. Gustafson, T. Mori, D. Blakeslee, R. Buckheit and M. R. Boyd, Antimicrob. Agents Chemother., 2003, 47, 2518–2525 CrossRef.
- H. Yu, Z.-t. Liue, R. Lv and W.-q. Zhang, Virol. Sin., 2010, 25, 432–439 CrossRef CAS PubMed.
- A. J. Bolmstedt, B. R. O'Keefe, S. R. Shenoy, J. B. McMahon and M. R. Boyd, Mol. Pharmacol., 2001, 59, 949–954 CAS.
- Q. Hu, N. Mahmood and R. J. Shattock, Virology, 2007, 368, 145–154 CrossRef CAS PubMed.
- D. F. Smee, M. K. Wandersee, M. B. Checketts, B. R. O'Keefe, C. Saucedoe, M. R. Boyd, V. P. Mishin and L. V. Gubareva, Antiviral Chem. Chemother., 2007, 18, 317–327 CrossRef CAS.
- D. F. Smee, K. W. Bailey, M.-H. Wong, B. R. O'Keefe, K. R. Gustafson, V. P. Mishin and L. V. Gubareva, Antiviral Res., 2008, 80, 266–271 CrossRef CAS PubMed.
- C.-C. Tsai, P. Emau, Y. Jiang, M. B. Agy, R. J. Shattock, A. Schmidt, W. R. Morton, K. R. Gustafson and M. R. Boyd, AIDS Res. Hum. Retroviruses, 2004, 20, 11–18 CrossRef CAS PubMed.
- C.-C. Tsai, P. Emau, Y. Jiang, B. Tian, W. R. Morton, K. R. Gustafson and M. R. Boyd, AIDS Res. Hum. Retroviruses, 2003, 19, 535–541 CrossRef CAS PubMed.
- K. Watashi, M. Hijikata, M. Hosaka, M. Yamaji and K. Shimotohno, Hepatology, 2003, 38, 1282–1288 CrossRef CAS PubMed.
- E. Negredo, B. Clotet, J. Puig, N. Pérez-Álvarez, L. Ruiz, J. Romeu, J. Moltó, C. Rey-Joly and J. Blanco, AIDS, 2006, 20, 619–621 CrossRef CAS PubMed.
- D. F. Smee, M. Bray and J. W. Huggins, Antiviral Chem. Chemother., 2001, 12, 327–336 CrossRef CAS.
- G. Roja and M. Heble, Phytother. Res., 1995, 9, 540–542 CrossRef CAS.
- D. L. Taylor, P. S. Sunkara, P. S. Liu, M. S. Kang, T. L. Bowlin and A. S. Tyms, AIDS, 1991, 5, 693–698 CrossRef CAS PubMed.
- X. Qu, X. Pan, J. Weidner, W. Yu, D. Alonzi, X. Xu, T. Butters, T. Block, J.-T. Guo and J. Chang, Antimicrob. Agents Chemother., 2011, 55, 1036–1044 CrossRef CAS PubMed.
- S. Watanabe, A. P. Rathore, C. Sung, F. Lu, Y. M. Khoo, J. Connolly, J. Low, E. E. Ooi, H. S. Lee and S. G. Vasudevan, Antiviral Res., 2012, 96, 32–35 CrossRef CAS PubMed.
- T. Saito and I. Yamaguchi, J. Vet. Med. Sci., 2000, 62, 575–581 CrossRef CAS PubMed.
- G. Férir, M. I. Petrova, G. Andrei, D. Huskens, B. Hoorelbeke, R. Snoeck, J. Vanderleyden, J. Balzarini, S. Bartoschek and M. Brönstrup, PLoS One, 2013, 8, e64010 Search PubMed.
- G. Férir, M. I. Petrova, G. Andrei, R. Snoeck, M. Brönstrup, R. D. Süssmuth and D. Schols, BMC Infect. Dis., 2014, 14, P79 CrossRef.
- K. H. Müller, G. A. Spoden, K. D. Scheffer, R. Brunnhöfer, J. K. De Brabander, M. E. Maier, L. Florin and C. P. Muller, Antimicrob. Agents Chemother., 2014, 02284–02313 Search PubMed.
- J. P. Martinez, B. Hinkelmann, E. Fleta-Soriano, H. Steinmetz, R. Jansen, J. Diez, R. Frank, F. Sasse and A. Meyerhans, Microb. Cell Fact., 2013, 12, 85 CrossRef PubMed.
- J. Gentzsch, B. Hinkelmann, L. Kaderali, H. Irschik, R. Jansen, F. Sasse, R. Frank and T. Pietschmann, Antiviral Res., 2011, 89, 136–148 CrossRef CAS PubMed.
- S. Sagar, M. Kaur and K. P. Minneman, Mar. Drugs, 2010, 8, 2619–2638 CrossRef CAS PubMed.
- N. B. Perry, J. W. Blunt, M. H. Munro and L. K. Pannell, J. Am. Chem. Soc., 1988, 110, 4850–4851 CrossRef CAS.
- K. Hagiwara, Y. Kondoh, A. Ueda, K. Yamada, H. Goto, T. Watanabe, T. Nakata, H. Osada and Y. Aida, Biochem. Biophys. Res. Commun., 2010, 394, 721–727 CrossRef CAS PubMed.
- T. Ichiba, J. M. Corgiat, P. J. Scheuer and M. Kelly-Borges, J. Nat. Prod., 1994, 57, 168–170 CrossRef CAS.
- C. McLaren, M. Ellis and G. Hunter, Antiviral Res., 1983, 3, 223–234 CrossRef CAS PubMed.
- M. Yousaf, N. L. Hammond, J. Peng, S. Wahyuono, K. A. McIntosh, W. N. Charman, A. M. Mayer and M. T. Hamann, J. Med. Chem., 2004, 47, 3512–3517 CrossRef CAS PubMed.
- P. Emau, B. Tian, B. R. O'Keefe, T. Mori, J. B. McMahon, K. E. Palmer, Y. Jiang, G. Bekele and C. C. Tsai, J. Med. Primatol., 2007, 36, 244–253 CrossRef CAS PubMed.
- P. Meuleman, A. Albecka, S. Belouzard, K. Vercauteren, L. Verhoye, C. Wychowski, G. Leroux-Roels, K. E. Palmer and J. Dubuisson, Antimicrob. Agents Chemother., 2011, 55, 5159–5167 CrossRef CAS PubMed.
- B. R. O'Keefe, B. Giomarelli, D. L. Barnard, S. R. Shenoy, P. K. Chan, J. B. McMahon, K. E. Palmer, B. W. Barnett, D. K. Meyerholz, C. L. Wohlford-Lenane and P. B. McCray Jr, J. Virol., 2010, 84, 2511–2521 CrossRef PubMed.
- H. Z. Ishag, C. Li, L. Huang, M. X. Sun, F. Wang, B. Ni, T. Malik, P. Y. Chen and X. Mao, Arch. Virol., 2013, 158, 349–358 CrossRef CAS PubMed.
- H.-G. Sahl and G. Bierbaum, Annu. Rev. Microbiol., 1998, 52, 41–79 CrossRef CAS PubMed.
- J. M. Willey and W. A. van Der Donk, Annu. Rev. Microbiol., 2007, 61, 477–501 CrossRef CAS PubMed.
- K. Meindl, T. Schmiederer, K. Schneider, A. Reicke, D. Butz, S. Keller, H. Gühring, L. Vértesy, J. Wink and H. Hoffmann, Angew. Chem., Int. Ed., 2010, 49, 1151–1154 CrossRef CAS PubMed.
- J. M. Krawczyk, G. H. Völler, B. Krawczyk, J. Kretz, M. Brönstrup and R. D. Süssmuth, Chem. Biol., 2013, 20, 111–122 CrossRef CAS PubMed.
- J. Diez, J. P. Martinez, J. Mestres, F. Sasse, R. Frank and A. Meyerhans, Microb. Cell Fact., 2012, 11, 1–3 CrossRef PubMed.
- T. F. Schäberle, F. Lohr, A. Schmitz and G. M. König, Nat. Prod. Rep., 2014, 31(7), 953–972 RSC.
- K. J. Weissman and R. Müller, Nat. Prod. Rep., 2010, 27, 1276–1295 RSC.
- K. Viehrig, F. Surup, K. Harmrolfs, R. Jansen, B. Kunze and R. Müller, J. Am. Chem. Soc., 2013, 135, 16885–16894 CrossRef CAS PubMed.
- J. Barbier, R. Jansen, H. Irschik, S. Benson, K. Gerth, B. Böhlendorf, G. Höfle, H. Reichenbach, J. Wegner, C. Zeilinger, A. Kirschning and R. Müller, Angew. Chem., Int. Ed., 2012, 51, 1256–1260 CrossRef CAS PubMed.
- J. Barbier, J. Wegner, S. Benson, J. Gentzsch, T. Pietschmann and A. Kirschning, Chemistry, 2012, 18, 9083–9090 CrossRef CAS PubMed.
- U. F. Greber and M. Way, Cell, 2006, 124, 741–754 CrossRef CAS PubMed.
- K. Radtke, K. Döhner and B. Sodeik, Cell. Microbiol., 2006, 8, 387–400 CrossRef CAS PubMed.
- A. Hinton, S. Bond and M. Forgac, Pflugers Arch., 2009, 457, 589–598 CrossRef CAS PubMed.
- N. Chazal and D. Gerlier, Microbiol. Mol. Biol. Rev., 2003, 67, 226–237 CrossRef CAS PubMed.
- J. P. Martinez, G. Perez-Vilaro, Y. Muthukumar, N. Scheller, T. Hirsch, R. Diestel, H. Steinmetz, R. Jansen, R. Frank, F. Sasse, A. Meyerhans and J. Diez, RNA Biol., 2013, 10, 1661–1669 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31(2), 160–258 RSC.
- R. C. F. Cheung, J. H. Wong, W. L. Pan, Y. S. Chan, C. M. Yin, X. L. Dan, H. X. Wang, E. F. Fang, S. K. Lam and P. H. K. Ngai, Appl. Microbiol. Biotechnol., 2014, 1–20 Search PubMed.
- J. Yasuhara-Bell and Y. Lu, Antiviral Res., 2010, 86, 231–240 CrossRef CAS PubMed.
- A. S. Sarma, T. Daum and W. E. Müller, Secondary metabolites from marine sponges, Ullstein Mosby, 1993 Search PubMed.
- N. B. Perry, J. W. Blunt, M. H. Munro and A. M. Thompson, J. Org. Chem., 1990, 55, 223–227 CrossRef CAS.
- G. Gürel, G. Blaha, T. A. Steitz and P. B. Moore, Antimicrob. Agents Chemother., 2009, 53, 5010–5014 CrossRef PubMed.
- A. Cutignano, G. Bifulco, I. Bruno, A. Casapullo, L. Gomez-Paloma and R. Riccio, Tetrahedron, 2000, 56, 3743–3748 CrossRef CAS.
- R. J. Capon, F. Rooney, L. M. Murray, E. Collins, A. T. Sim, J. A. Rostas, M. S. Butler and A. R. Carroll, J. Nat. Prod., 1998, 61, 660–662 CrossRef CAS PubMed.
- G. Kallifatidis, D. Hoepfner, T. Jaeg, E. A. Guzmán and A. E. Wright, Mar. Drugs, 2013, 11, 3500–3516 CrossRef CAS PubMed.
- D. D. Dunigan and J. C. Madlener, Virology, 1995, 207, 460–466 CrossRef CAS PubMed.
- F. H. Duong, M. Filipowicz, M. Tripodi, N. La Monica and M. H. Heim, Gastroenterology, 2004, 126, 263–277 CrossRef CAS.
- T. Jacob, C. Van den Broeke and H. W. Favoreel, J. Virol., 2011, 85, 1158–1173 CrossRef CAS PubMed.
- T. Mori, B. R. O'Keefe, R. C. Sowder 2nd, S. Bringans, R. Gardella, S. Berg, P. Cochran, J. A. Turpin, R. W. Buckheit Jr, J. B. McMahon and M. R. Boyd, J. Biol. Chem., 2005, 280, 9345–9353 CrossRef CAS PubMed.
- T. Moulaei, S. R. Shenoy, B. Giomarelli, C. Thomas, J. B. McMahon, Z. Dauter, B. R. O'Keefe and A. Wlodawer, Structure, 2010, 18, 1104–1115 CrossRef CAS PubMed.
- C. Barton, J. C. Kouokam, A. B. Lasnik, O. Foreman, A. Cambon, G. Brock, D. C. Montefiori, F. Vojdani, A. A. McCormick, B. R. O'Keefe and K. E. Palmer, Antimicrob. Agents Chemother., 2014, 58, 120–127 CrossRef PubMed.
- B. Nixon, M. Stefanidou, P. M. Mesquita, E. Fakioglu, T. Segarra, L. Rohan, W. Halford, K. E. Palmer and B. C. Herold, J. Virol., 2013, 87, 6257–6269 CrossRef CAS PubMed.
- G. Ferir, K. E. Palmer and D. Schols, Virology, 2011, 417, 253–258 CrossRef CAS PubMed.
- K. B. Alexandre, P. L. Moore, M. Nonyane, E. S. Gray, N. Ranchobe, E. Chakauya, J. B. McMahon, B. R. O'Keefe, R. Chikwamba and L. Morris, Virology, 2013, 446, 66–76 CrossRef CAS PubMed.
- K. S. Moore, S. Wehrli, H. Roder, M. Rogers, J. N. Forrest Jr, D. McCrimmon and M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1354–1358 CrossRef CAS.
- T. Yeung, G. E. Gilbert, J. Shi, J. Silvius, A. Kapus and S. Grinstein, Science, 2008, 319, 210–213 CrossRef CAS PubMed.
- T. Ganz, Nat. Rev. Immunol., 2003, 3, 710–720 CrossRef CAS PubMed.
- P. G. Barlow, E. G. Findlay, S. M. Currie and D. J. Davidson, Future Microbiol., 2014, 9, 55–73 CrossRef CAS PubMed.
- S. S. Wilson, M. E. Wiens and J. G. Smith, J. Mol. Biol., 2013, 425, 4965–4980 CrossRef CAS PubMed.
- C. J. Sample, K. E. Hudak, B. E. Barefoot, M. D. Koci, M. S. Wanyonyi, S. Abraham, H. F. Staats and E. A. Ramsburg, Peptides, 2013, 48C, 96–105 CrossRef PubMed.
- M. Pazgier, B. Ericksen, M. Ling, E. Toth, J. Shi, X. Li, A. Galliher-Beckley, L. Lan, G. Zou, C. Zhan, W. Yuan, E. Pozharski and W. Lu, Biochemistry, 2013, 52, 1547–1558 CrossRef CAS PubMed.
- N. K. Duggal and M. Emerman, Nat. Rev. Immunol., 2012, 12, 687–695 CrossRef CAS PubMed.
- J. Blaising, S. J. Polyak and E.-I. Pécheur, Antiviral Res., 2014, 107, 84–94 CrossRef CAS PubMed.
- Y. Furuta, K. Takahashi, K. Shiraki, K. Sakamoto, D. F. Smee, D. L. Barnard, B. B. Gowen, J. G. Julander and J. D. Morrey, Antiviral Res., 2009, 82, 95–102 CrossRef CAS PubMed.
- T. K. Warren, J. Wells, R. G. Panchal, K. S. Stuthman, N. L. Garza, S. A. Van Tongeren, L. Dong, C. J. Retterer, B. P. Eaton, G. Pegoraro, S. Honnold, S. Bantia, P. Kotian, X. Chen, B. R. Taubenheim, L. S. Welch, D. M. Minning, Y. S. Babu, W. P. Sheridan and S. Bavari, Nature, 2014, 508, 402–405 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |