
Chiral alkynylcarbinols from marine sponges: asymmetric synthesis and biological relevance†
Dymytrii
Listunov
abcd,
Valérie
Maraval
bc,
Remi
Chauvin
*bc and
Yves
Génisson
*ac
aUMR CNRS 5068, LSPCMIB, Université Paul Sabatier, 118 route de Narbonne, 31062 Toulouse Cedex 9, France. E-mail: genisson@chimie.ups-tlse.fr; Fax: +33-561556011
bUPR CNRS 8241, LCC (Laboratoire de Chimie de Coordination), 205 route de Narbonne, F-31077 Toulouse, France. E-mail: chauvin@lcc-toulouse.fr; Fax: +33 561553003
cUniversité de Toulouse, UPS, INPT, F-31077 Toulouse, France
dKiev National Taras Shevchenko University, 60 Volodymyrska St., 01033 Kiev, Ukraine
First published on 2nd October 2014
Abstract
Covering: up to March 2014. Previous review on the topic: B. W. Gung, C. R. Chim., 2009, 12, 489–505
Chiral α-functional lipidic propargylic alcohols extracted from marine sponges, in particular of the pacific genus Petrosia, constitute a class of acetylenic natural products exhibiting remarkable in vitro biological activities, especially anti-tumoral cytotoxicity. These properties, associated to functionalities that are uncommon among natural products, have prompted recent projects on asymmetric total synthesis. On the basis of a three-sector structural typology, three main sub-types of secondary alkynylcarbinols (with either alkyl, alkenyl, or alkynyl as the second substituent) can be identified as the minimal pharmacophoric units. Selected natural products containing these functionalities have been targeted using previously known or on purpose-designed procedures, where the stereo-determining step can be: (i) a C–C bond forming reaction (e.g. the Zn-mediated addition of alkynyl nucleophiles to aldehydes in the presence of chiral aminoalcohols), (ii) a functional layout (e.g. the asymmetric organo- or metallo-catalytic reduction of ynones), or (iii) an enantiomeric resolution (e.g. a lipase-mediated kinetic resolution via acetylation). The promising medicinal importance of these targets is finally surveyed, and future investigation prospects are proposed, such as: (i) further total synthesis of known or future extraction products; (ii) the synthesis of non-natural analogues, with simpler lipophilic environments of the alkynylcarbinol-based pharmacophoric units; (iii) the variation and optimization of both the pharmacophoric units and their lipophilic environment; and (iv) investigations into the biological mode of action of these unique structures.
 Dymytrii Listunov | Dymytrii Listunov was born in Ovroutch, Ukraine. In 2012, he received a Master’s degree in organic chemistry at the Taras Shevchenko National University of Kyiv. In the same year, he was awarded a Postgraduate Fellowship from the French government to pursue doctoral studies at the Paul Sabatier University, Toulouse, France, at two institutes: the Laboratory of Synthesis and Physical Chemistry of Biologically Active Substances and the Laboratory of Coordination Chemistry, under the supervision of Dr Yves Génisson, Dr Valérie Maraval, and Prof. Remi Chauvin. He is currently working on the synthesis and biological evaluation of novel alkynylcarbinol pharmacophores with anti-tumoral cytotoxicity. |
 Valérie Maraval | Valérie Maraval obtained her Ph.D. in 2000, in the field of phosphorus dendrimer chemistry, under the supervision of Jean-Pierre Majoral in Toulouse, France. In 2001, she worked as a postdoctoral fellow with Bernard Meunier in collaboration with the Aventis Company on the oxidation of terpenes. She then came back to Jean-Pierre Majoral's team as a CNRS Engineer for two years. In 2005, she joined the group of Remi Chauvin at the Laboratoire de Chimie de Coordination (LCC) in Toulouse. Her research activities mainly focus on the synthesis and properties (essentially in optics and electronics) of carbo-meric molecules. |
 Remi Chauvin | Remi Chauvin obtained his Ph.D. in 1988, in the field of asymmetric catalysis under the supervision of H. B. Kagan in Orsay (France). He pursued his career as a post-doctoral fellow with K. B. Sharpless at MIT (USA), then with A. Vasella in Zürich (Switzerland). Returning to France, he worked at the Roussel-Uclaf company in Romainville, and in November 1993 joined the academic researchers at the CNRS in Toulouse. Appointed as a Full Professor at the Paul Sabatier University in 1998, his interests relate to theoretical chemistry, main group-transition metal chemistry, organic chemistry of carbo-mers (for physics) and bio-inspired acetylenics (for biology). |
 Yves Génisson | Yves Génisson was born in Boulogne sur Seine, France, in 1963. He completed his Ph.D. in 1992 at the ICSN in Gif sur Yvette under the supervision of the late Dr C. Marazano. After working for two years as a collaborator of Dr Robert Young at the Merck Frosst Centre of Therapeutic Research in Montréal, Canada, he joined the CNRS as a researcher in the group of Dr A. E. Greene in Grenoble, France. In 1998, he moved to the Université Paul Sabatier of Toulouse where he completed his Habilitation in 2003. He is currently working both in the field of chiral ionic liquids and asymmetric synthesis of bioactive compounds. |
1 Introduction
Among the biologically active acetylenic molecules, those where a triple bond is in the geminal position of a hydroxyl group, i.e. belonging to a propargylic alcohol function, are particularly well represented (Fig. 1). This is the case for almost all the marketed acetylenic drugs, including the steroidal progestins associated to ethynylestradiol in oral contraceptives, and the mifepristone abortifacient (RU-486). In more recently identified representatives, proving highly potent for various pharmaceutical applications, the alkynylcarbinol center is further functionalized by a second geminal unsaturation, like a double bond: the corresponding alkenyl-alkynylcarbinol (AAC) C5 motif indeed occurs in gestodene, a third-generation progestin,1 and in efavirenz, a HIV-1 reverse transcriptase inhibitor.2 Two AAC motifs are also found in the extremely potent anticancer natural antibiotics calicheamicin and esperamicin (extracted from soil bacteria), where the triple bonds also belong to the key enediyne pharmacophore, inducing DNA cleavage upon Bergman cyclization.3 | ||
| Fig. 1 Occurrence of chiral alkynylcarbinol units in marketed drugs (top), and examples of lipidic representatives extracted from marine sponges (box, bottom). The particular AAC and dialkynylcarbinol (DAC) motifs are boxed in blue. | ||
In contrast to the few examples above, most of the natural alkynylcarbinols are found to be associated to their lipidic environment. Though identified on several occasions over the last four decades, chiral lipidic alkynylcarbinols have recently emerged as key structural fragments of natural products extracted from marine organisms, which exhibit potent biological activities, thus confirming and strengthening the pharmacophore status of substituted propargylic alcohol units.4 Selected examples isolated from marine sponges are shown in Fig. 1, where the minimal, but highly functional, three-carbon unit is highlighted. All the representatives contain more or less unsaturated fatty linear backbones.
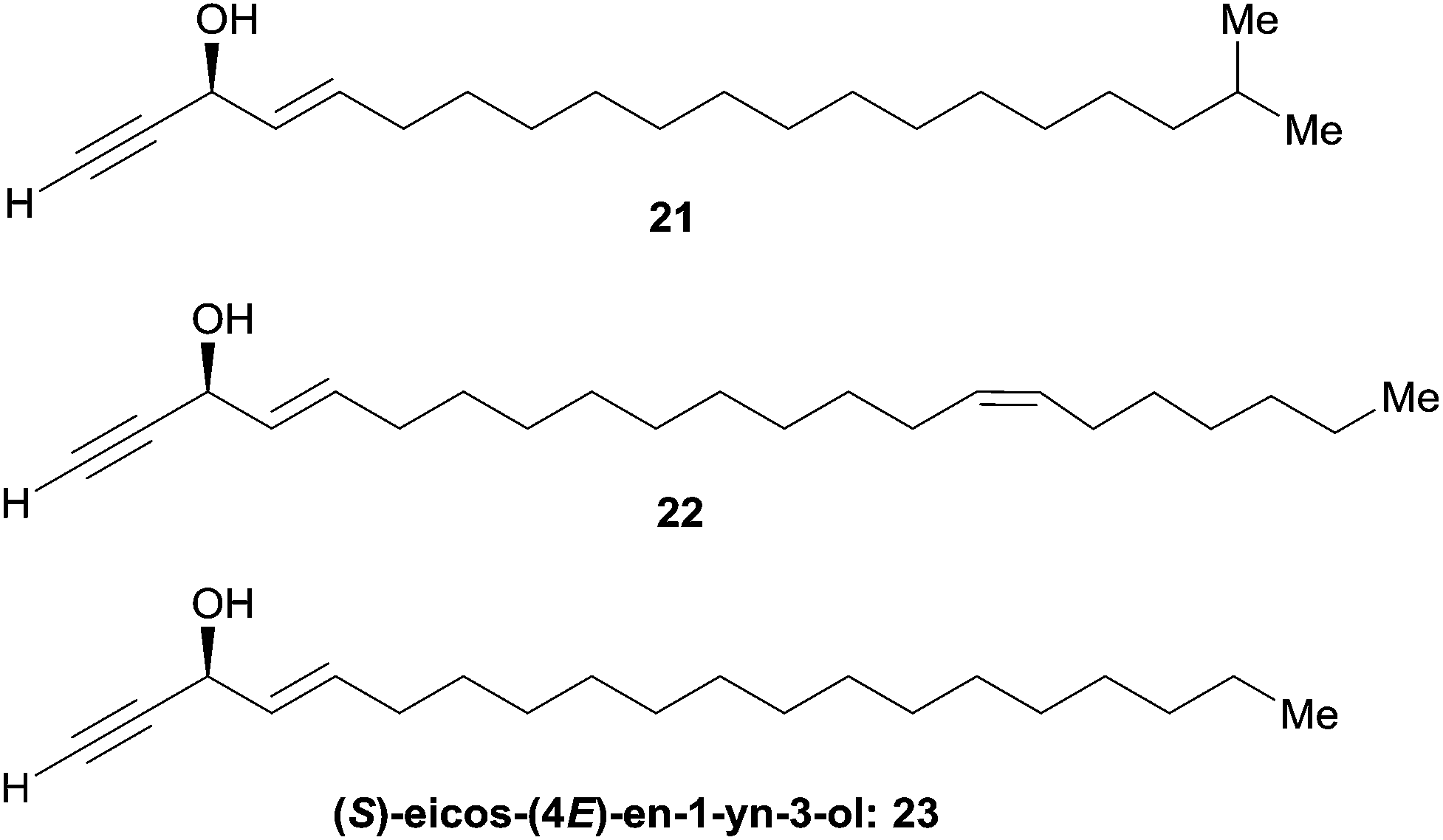
As can be seen from the structures of lembehyne A, strongylodiol A, petrosynol and petrocortyne A, the alkynylcarbinol unit can occur in various local environments along unbranched skeletons, typically made of 20 to more than 40 carbon atoms (Fig. 1). Aiming at a systematic description, three structural segments can be distinguished (Fig. 2): sector A, corresponding to the alkyne substituent R1; sector B, corresponding to the second carbinol substituent R2; and sector C, corresponding to the third substituent R3 (mainly H however) and the associated configuration of the carbinol center, that is typically secondary in nature. In contrast, all the non-lipidic biologically active alkynylcarbinols, shown in Fig. 1 (top), are tertiary carbinols. In sector A, the triple bond of the alkynylcarbinol unit can thus be either terminal (R1 = H) or internal (R1 ≠ H), and in the latter case either isolated (as in petrocortynes, Fig. 1) or conjugated with another triple bond (as in strongylodiols, Fig. 1). In sector B, the propargylic carbinol unit can be simply α-alkylated (as in lembehyne A, Fig. 1) or α-functionalised, either allylic (as in petrosynol and petrocortynes, Fig. 1) or propargylic (as in petrocortyne, Fig. 1, and in several other related compounds such as osirinynes A–F,5 haliclonyne6 and fulvyne,7 not shown). The chiral stereochemistry defined by sector C (typically R1 = H) deserves a specific comment: the present class of natural polyacetylenes includes rare examples of compounds produced in nature in both enantiomeric forms (e.g. durynes, vide infra), if not as scalemic mixtures in a given organism (e.g. strongylodiols and eicos-(4E)-en-1-yn-3-ol, vide infra), therefore making sector C particularly relevant with respect to delineating structure–activity relationships.
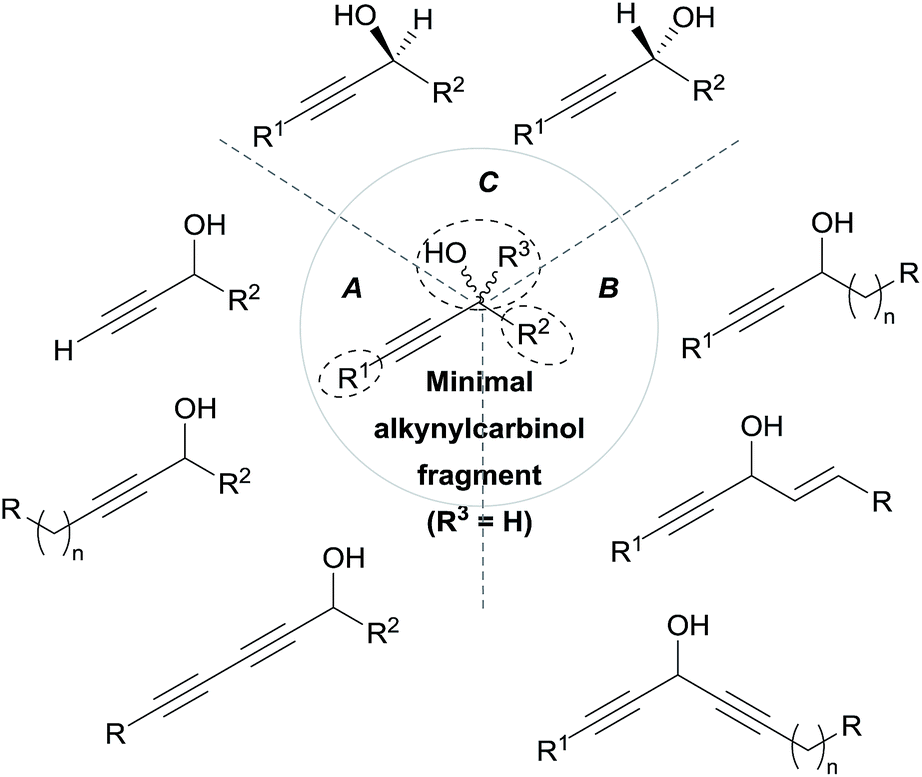 | ||
| Fig. 2 The three structural sectors A, B and C of the alkynylcarbinol motif found in marine products. | ||
The following review aims at highlighting, from the standpoint of total synthesis, the array of marine natural products extracted from sponges, embedding one or more chiral alkynylcarbinol units.8 Based on the structural typology and the sectors A, B, C (see above, Fig. 2), a comprehensive overview of the methodological approaches, implemented for the asymmetric synthesis of these natural products and analogues, is presented. Finally, the biological relevance and the pharmacophoric potential of this minimal structural unit is discussed vs. the nature of the substituents, thus providing a basis to study structure–activity relationships.
Beyond the scope of this review, natural products embedding lipidic alkynylcarbinol motifs, isolated from non-marine sources,4 such as twigs of Ochanostachys amentacea,9 roots of Panax ginseng,10 or flowers of Pratia nummularia, should also be mentioned.11 Whatever their origin is (animals, plants, fungi, bacteria, terrestrial or marine), natural polyacetylenes exhibit common features regarding their biosynthesis (sharing fatty acids and polyketide precursors with mycolic acids) and metabolism.12
2 Synthesis of chiral alkynylcarbinols
Among the two main approaches towards enantio-enriched chiral secondary alkynylcarbinols, the asymmetric reduction of the corresponding ynones (approach (a), Scheme 1) presents the advantage of only exploiting well-established procedures, but requires additional synthetic steps to first secure the carbonyl substrate. The second main approach is based on the asymmetric nucleophilic addition to aldehydes, and corresponds to two possible disconnections (approach (b), Scheme 1). Though more straightforward, the terminal alkyne oxypropargylation route (b) has been limited until recently by a lack of general and reliable protocols.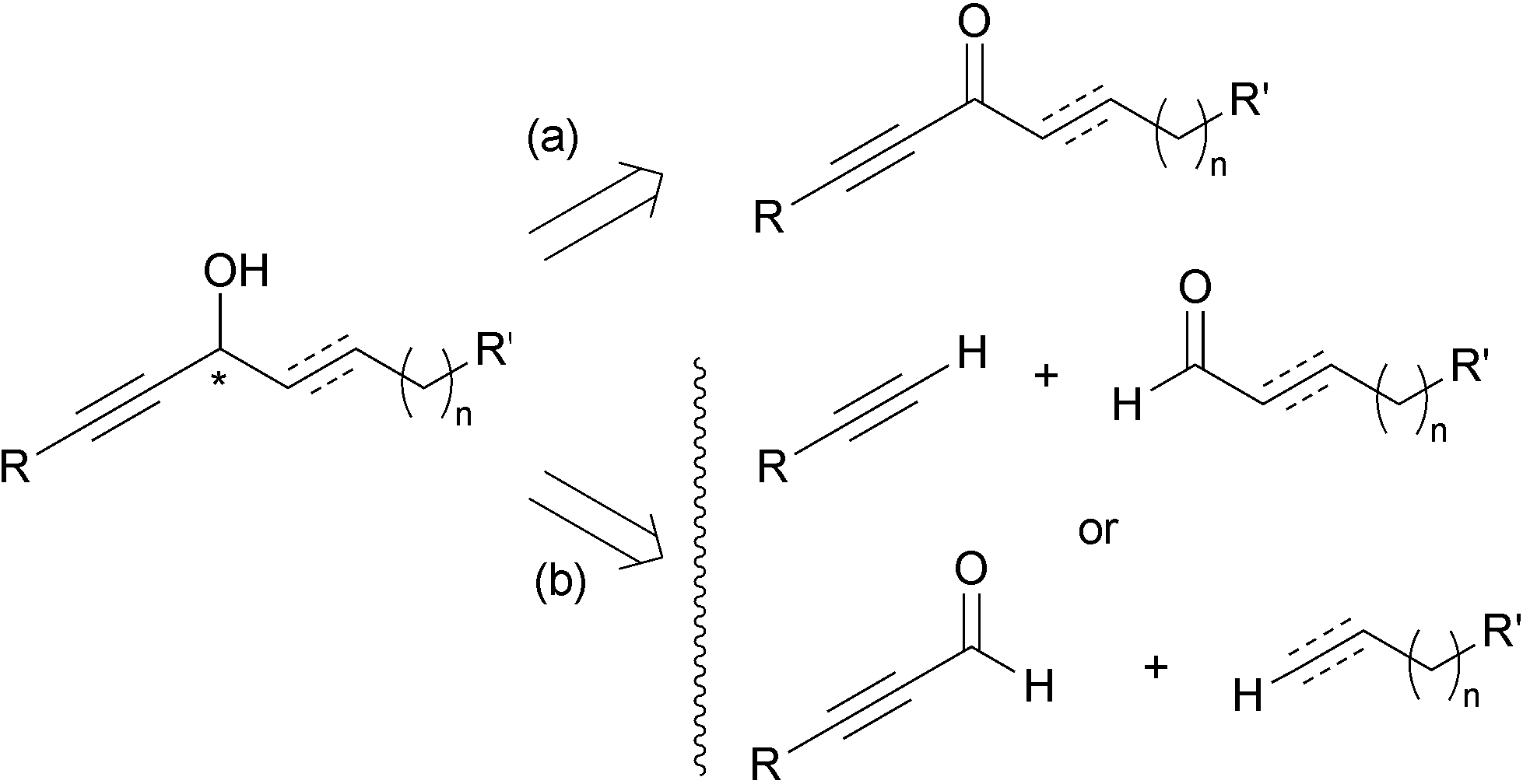 | ||
| Scheme 1 The two main retrosynthetic analyses of chiral secondary alkynylcarbinols. | ||
This section illustrates the use of the two approaches (a) and (b) for the asymmetric synthesis of various types of marine alkynylcarbinols, going from alkyl- and alkenyl-alkynylcarbinols (AACs) to dialkynylcarbinols (DACs).
2.1 α-Alkylated alkynylcarbinols
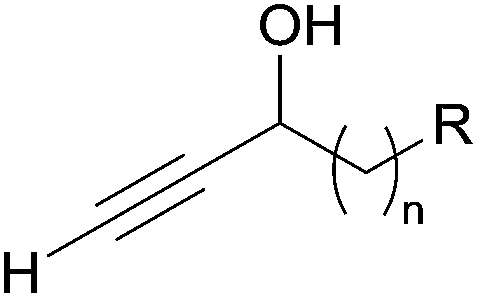 | ||
| Fig. 3 The α-alkylated secondary ethynylcarbinol fragment. | ||
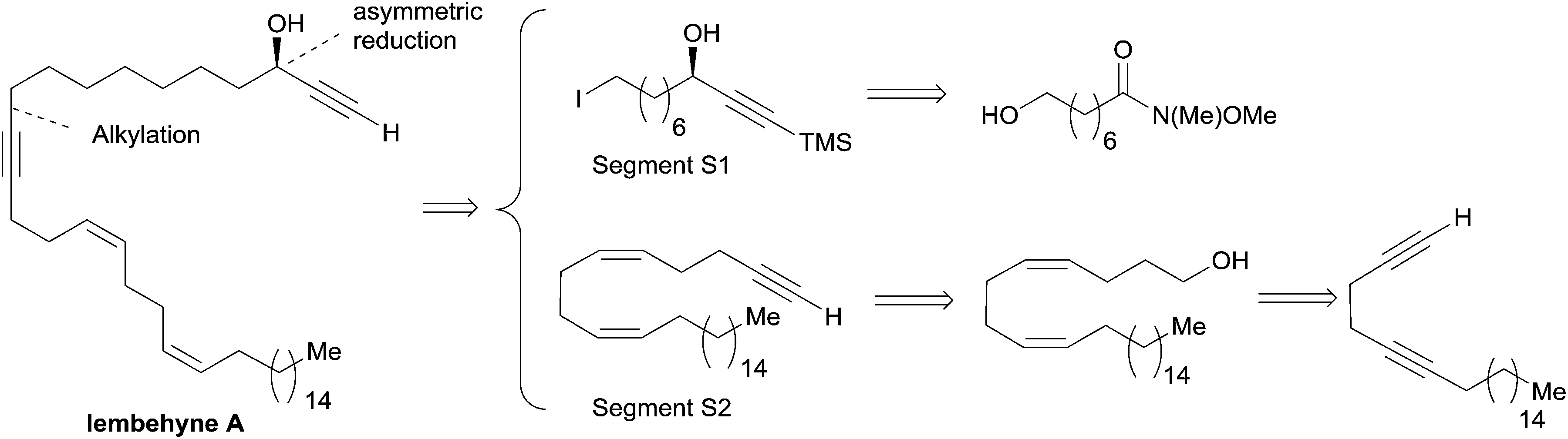
The preparation of the chiral segment S1 is illustrated in Scheme 3. The ynone precursor 1 was obtained from the corresponding Weinreb amide 2 (accessed in two steps from cyclooctanone) by addition of lithium trimethylsilylacetylide, followed by iodination of the primary alcohol. The (R)-carbinol center was generated in 82% yield and 94% ee from 1 using the Alpine-borane reagent reported by H. C. Brown et al.17 The availability of both pinene enantiomers indeed makes Alpine-boranes particularly attractive for asymmetric reductions. Terminal acetylenic ketones are thus quantitatively reduced after 8 h at room temperature in high enantiomeric excesses. The enantioselectivity of this reaction was explained by M. M. Midland considering a simple transition-state model (Scheme 3).17b The boat-like six-membered ring structure of the proposed transition-state should indeed favor the formation of the product, where the larger group (which is generally not the acetylenic substituent) is in the equatorial-like position, thus controlling the corresponding configuration of the carbinol center.
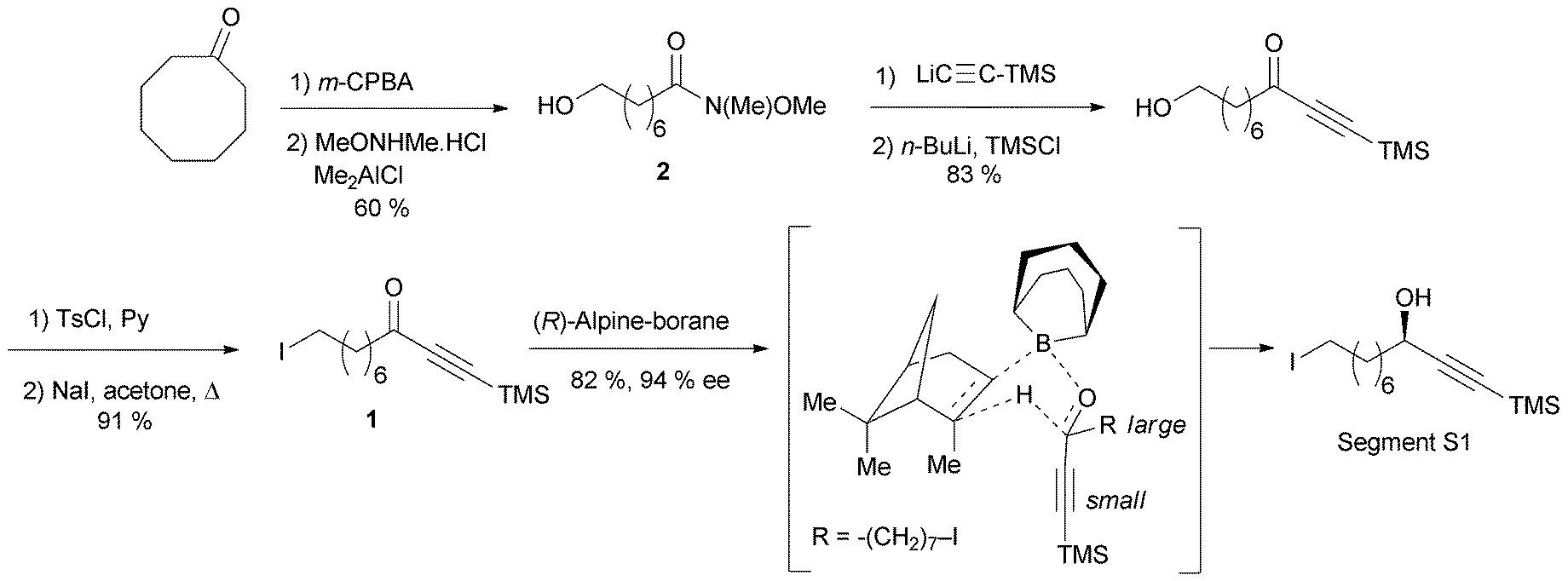 | ||
| Scheme 3 Murakami's synthesis of the segment S1 of lembehyne A (retrosynthesis in Scheme 2). | ||
The segment S2 of lembehyne A (Scheme 2) was prepared from 4-pentyn-1-ol and 1-iodohexadecane (Scheme 4). A one-carbon elongation process using dimethyl-1-diazo-2-oxopropylphosphonate afforded the 1,5-diyne 3 through an aldehyde intermediate. The product 3 was then converted to the 1,5-diene alcohol 4, from which the segment S2 was obtained by means of another one-carbon elongation via the corresponding aldehyde. The final assembly to lembehyne A was accomplished by substitution on the iodinated segment S1 by the terminal alkyne S2 (Scheme 4).
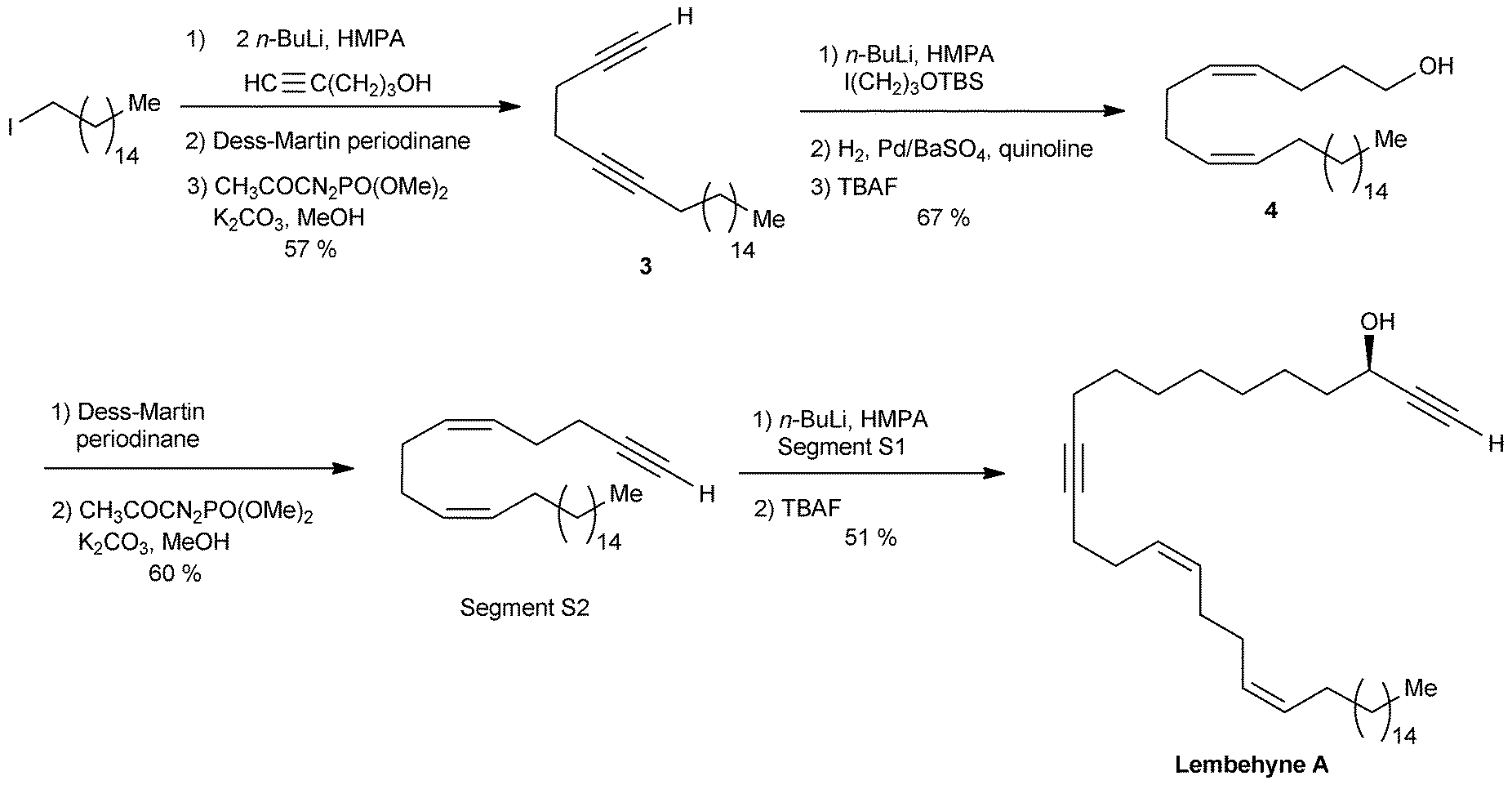 | ||
| Scheme 4 The final steps of Murakami's synthesis of lembehyne A (retrosynthesis in Scheme 2). | ||
Simplified analogues of the natural lembehynes A–C, including a cytotoxic substance named LB-18,18 were synthesized by the same approach using the alternative Corey–Bakshi–Shibata (CBS) asymmetric reduction methodology (Scheme 5).14a As proposed by Parker et al. for the CBS reduction of prochiral ynones,14b the R-configuration of alkynylcarbinol centers, obtained using the (R)-proline-derived oxazaborolidine reagent, can be explained by a careful application of Corey's general transition-state model.14c
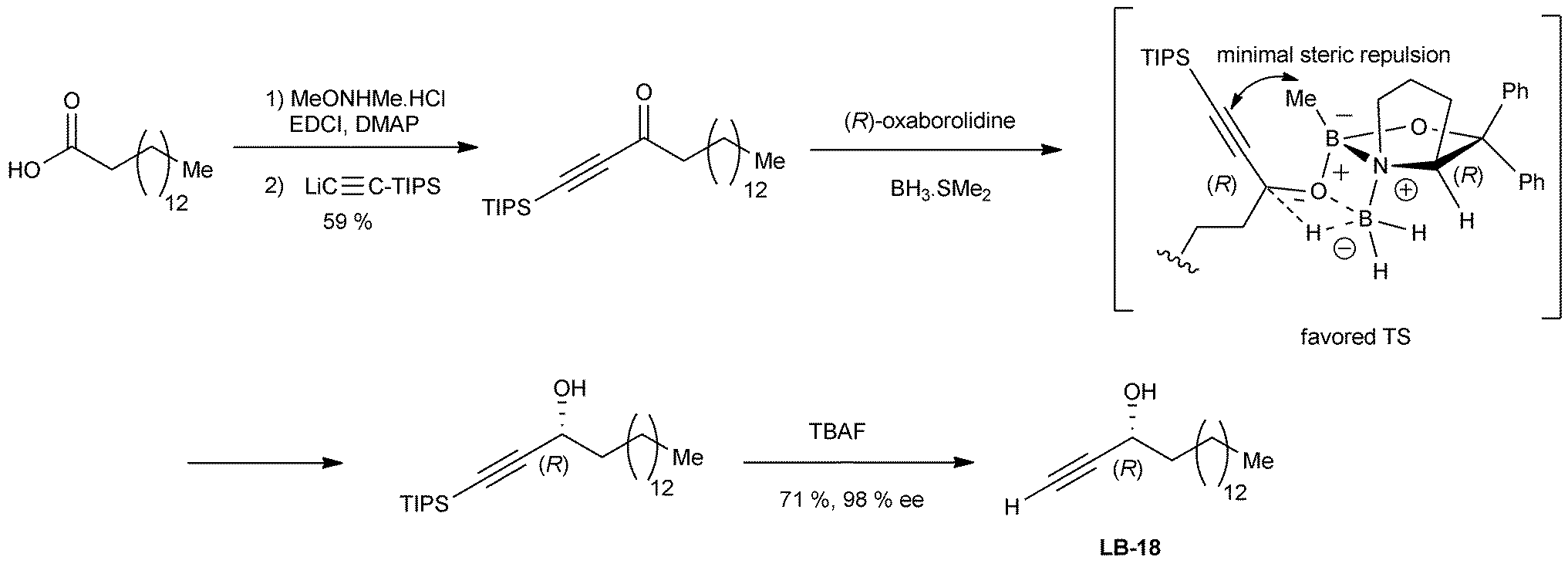 | ||
| Scheme 5 Application of the CBS asymmetric reduction method for the synthesis of LB-18. | ||
The asymmetric transfer hydrogenation method reported by Noyori et al. is also relevant for the preparation of chiral propargylic alcohols.19 Treatment of ynones with 1 mol% of a chiral Ru(II) catalyst, in 2-propanol as the hydrogen donor, delivers the corresponding alkynylcarbinols in high yields and high ee’s (Scheme 6). Baldwin applied this methodology to the synthesis of strongylodiols (vide infra, Section 2.2.2, Schemes 15 and 16).
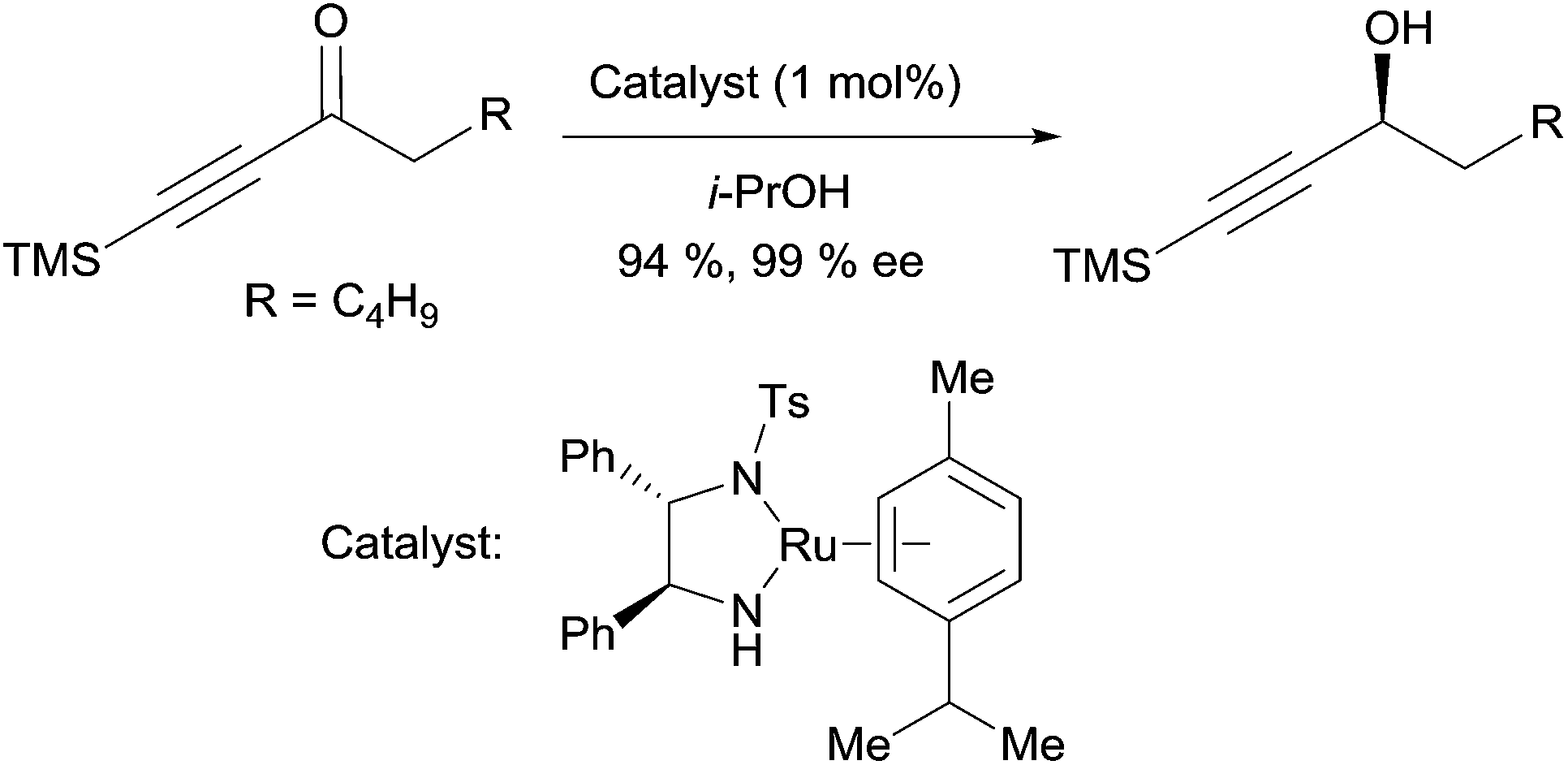 | ||
| Scheme 6 Noyori's asymmetric transfer hydrogenation of propargylic ketones. | ||
2.2 α-Saturated alka-1,3-diynylcarbinols
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 to 97:3 depending on the product).
:16 to 97:3 depending on the product).
 | ||
| Fig. 4 The α-saturated alka-1,3-diynylcarbinol unit of strongylodiols. | ||
2.2.2.1 Elimination reaction from an activated chiral epoxide. In 1990, Yadav et al. proposed a general strategy for the preparation of enantio-enriched chiral propargylic alcohols based on a double elimination from chiral 2,3-epoxychlorides upon treatment with one equivalent of a strong base (Scheme 7).22 This stereospecific transformation leads to the secondary alcohol product whose configuration corresponds to that of the 3-position in the starting epoxide, the latter being readily produced by means of Sharpless' asymmetric epoxidation. The transformation proved to be flexible, also giving access to DACs. A variation of this method, making use of a 4-(chloromethyl)-1,3-dioxolane prepared using Sharpless' asymmetric dihydroxylation, was reported later on.23
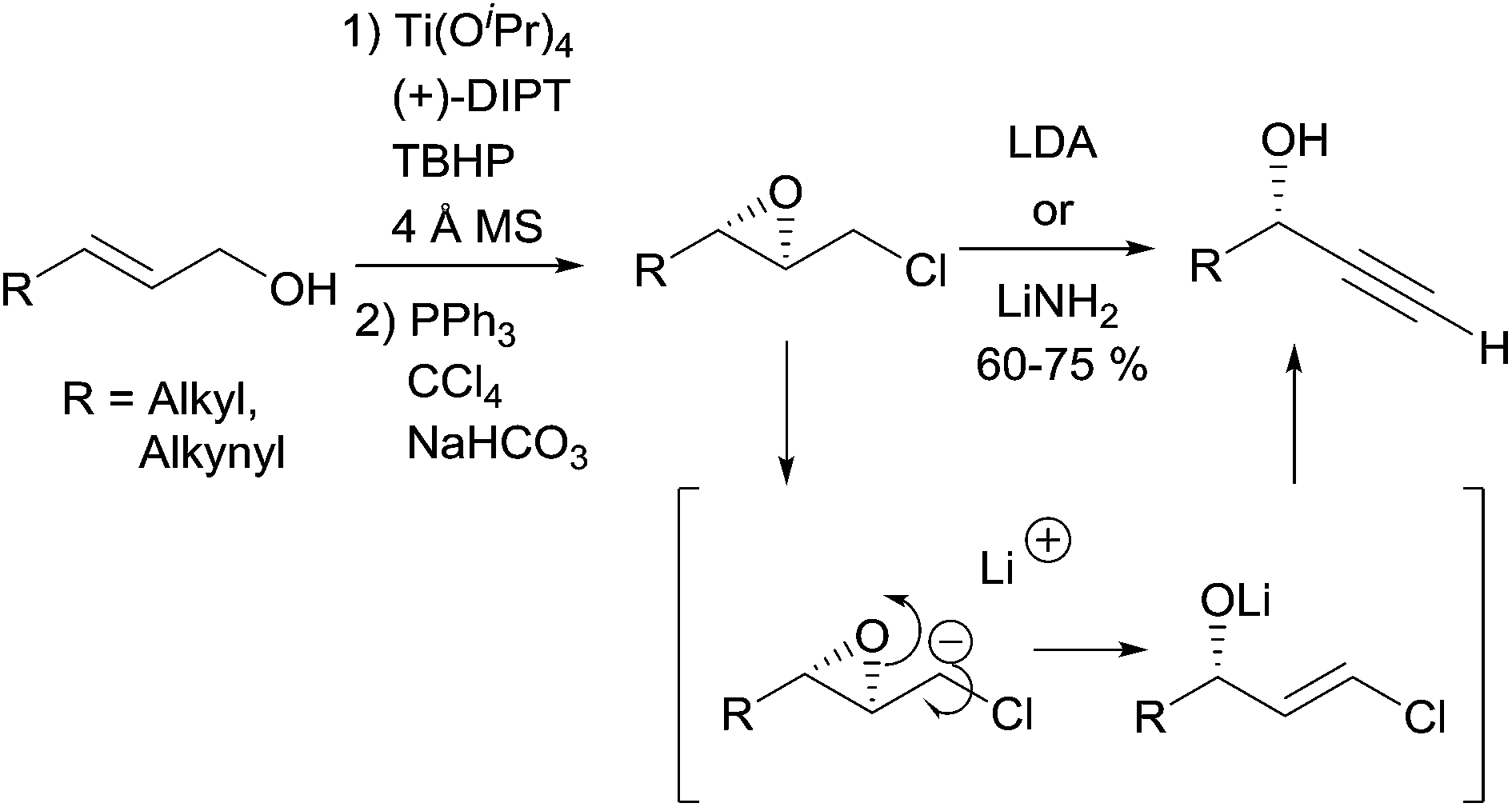 | ||
| Scheme 7 Yadav's approach to chiral alkynylcarbinols. | ||
Although this approach was not, strictly speaking, employed in the preparation of the alka-1,3-diynylcarbinol unit (Fig. 4), it was exploited by Yadav et al. in 2002 in an asymmetric synthesis of strongylodiol A.24 The retrosynthesis was based on a late Cadiot–Chodkiewicz coupling involving a terminal saturated alkynylcarbinol, 5, prepared by the above-mentioned double elimination method (Scheme 8).
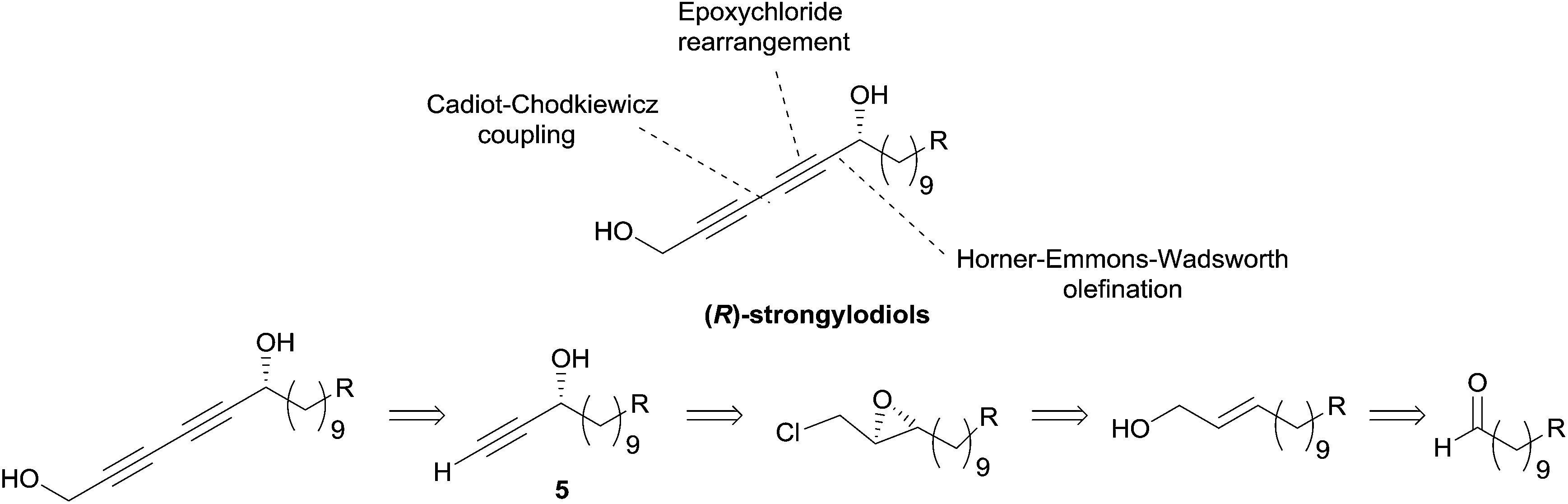 | ||
| Scheme 8 Yadav's retrosynthesis of strongylodiols (synthesis in Scheme 9). | ||
The allylic substrate 6 was obtained from 1,10-decanediol in a six-step sequence, including a stereoselective Wittig olefination step to establish the natural Z-alkene fragment (Scheme 9). Epoxidation of 6 under Sharpless' conditions, followed by chlorination of the primary alcohol, gave the epoxychloride 7. The key double elimination from 7 was efficiently performed using LiNH2 in liquid NH3. The final Cu(I)-catalyzed sp–sp eliminative cross-coupling between 8 and 3-bromopropynol delivered the targeted (R)-strongylodiol A in 85% yield.
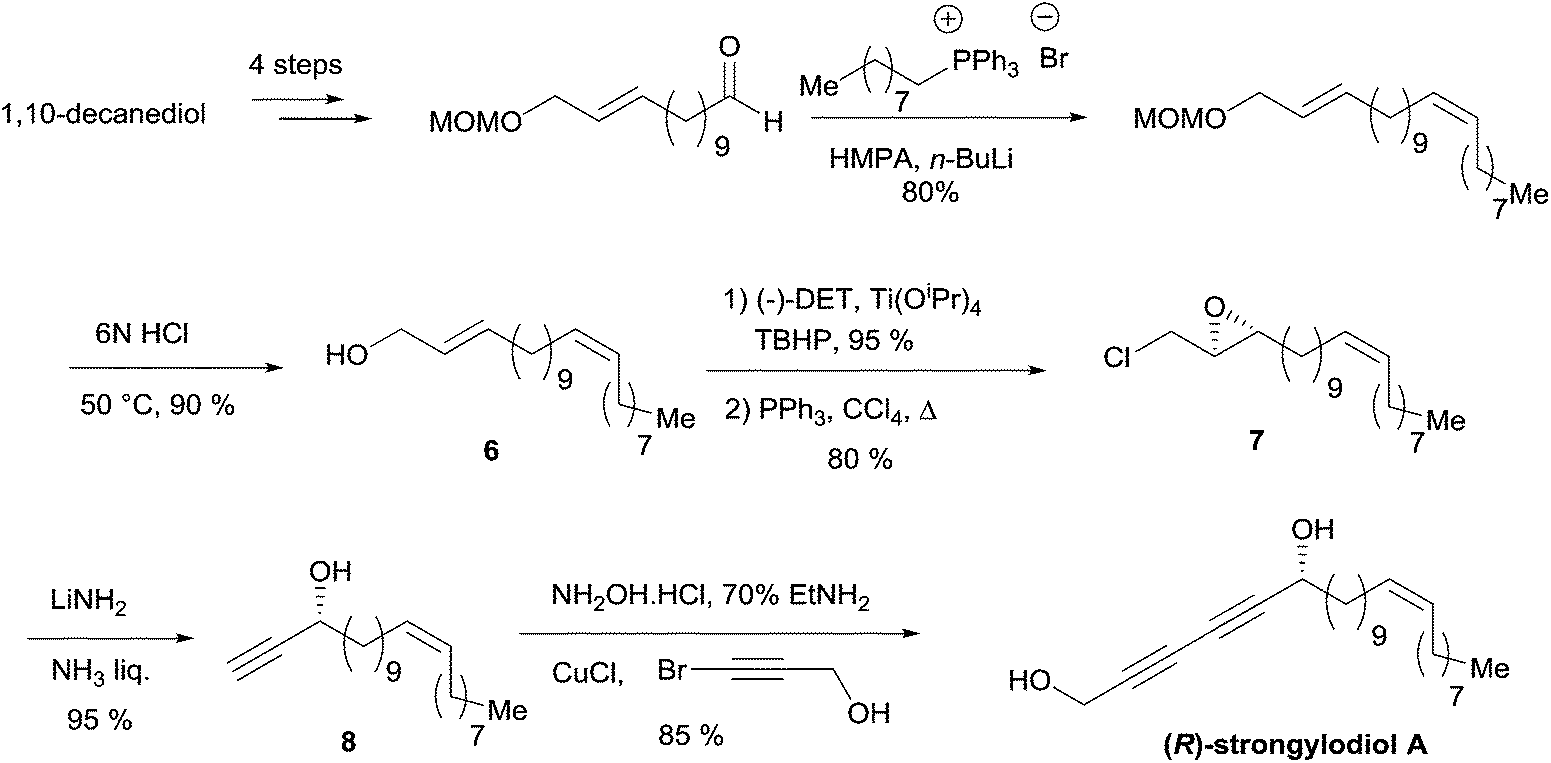 | ||
| Scheme 9 Yadav's enantioselective synthesis of (R)-strongylodiol A (retrosynthesis in Scheme 8). | ||
2.2.2.2 Asymmetric addition of a terminal α-diyne to an aldehyde. The most appealing access to specific chiral secondary propargylic alcohol targets relies on pioneering advances in the Zn-mediated asymmetric addition of terminal alkynes to aldehydes.25 In 2000, indeed, Carreira et al. described a practical method using N-methylephedrine (NME) as the chiral ligand of alkynylzinc(II) centers (Scheme 10).26 The main features, making the procedure user-friendly, are the following: (i) the Zn-acetylide reactant is generated in situ under mild conditions from the stable salt Zn(OTf)2 and Et3N; (ii) the reaction is insensitive to moisture and oxygen; (iii) the NME chiral auxiliary is available in either enantiomeric form; (iv) the stereoselectivity is predictable using Noyori's model;27 (v) the allowed types of substrates are aliphatic and aromatic aldehydes. However, there are several records of unsuccessful attempts applying Carreira's procedure to aliphatic aldehydes28 and α,β-unsaturated aldehydes (enals or ynals). Four examples of such unproductive attempts for the synthesis of acetylenic natural products were reported by Baldwin et al. for the strongylodiols,28 by Gung et al. for eicos-(4E)-en-1-yn-3-ol29 and adociacetylene B,30 and by Curran’s group for petrocortyne A.31 Because of these limitations, further investigations were required before general efficient procedures for the asymmetric addition of alkynes to aldehydes became available. Common features of recently disclosed methodologies are: (i) in situ generation of a zinc acetylide from a highly reactive dialkylzinc reagent (instead of Zn(OTf)2 in Carreira's method), and (ii) subsequent activation via zinc-coordination with a chiral ligand (or alternatively activation of the aldehyde using a chiral Lewis acid), ensuring stereocontrol. Three of these most recent achievements have found applications in the asymmetric total synthesis of chiral marine polyacetylenes: Pu's system based on a BINOL/Ti(Oi-Pr)4 combination,32 Trost's method relying on a ProPhenol-based dinuclear complex,33 and Zhong and Guo's approach using a cyclopropane scaffold-based 1,4-aminoalcohol ligand.34 Whereas Pu's system has been used by Curran et al. for the synthesis of petrocortyne A (vide infra, Section 2.4.2, Schemes 35 and 37), the three other methods – including Carreira's original procedure – were applied to the synthesis of strongylodiols (Scheme 10).
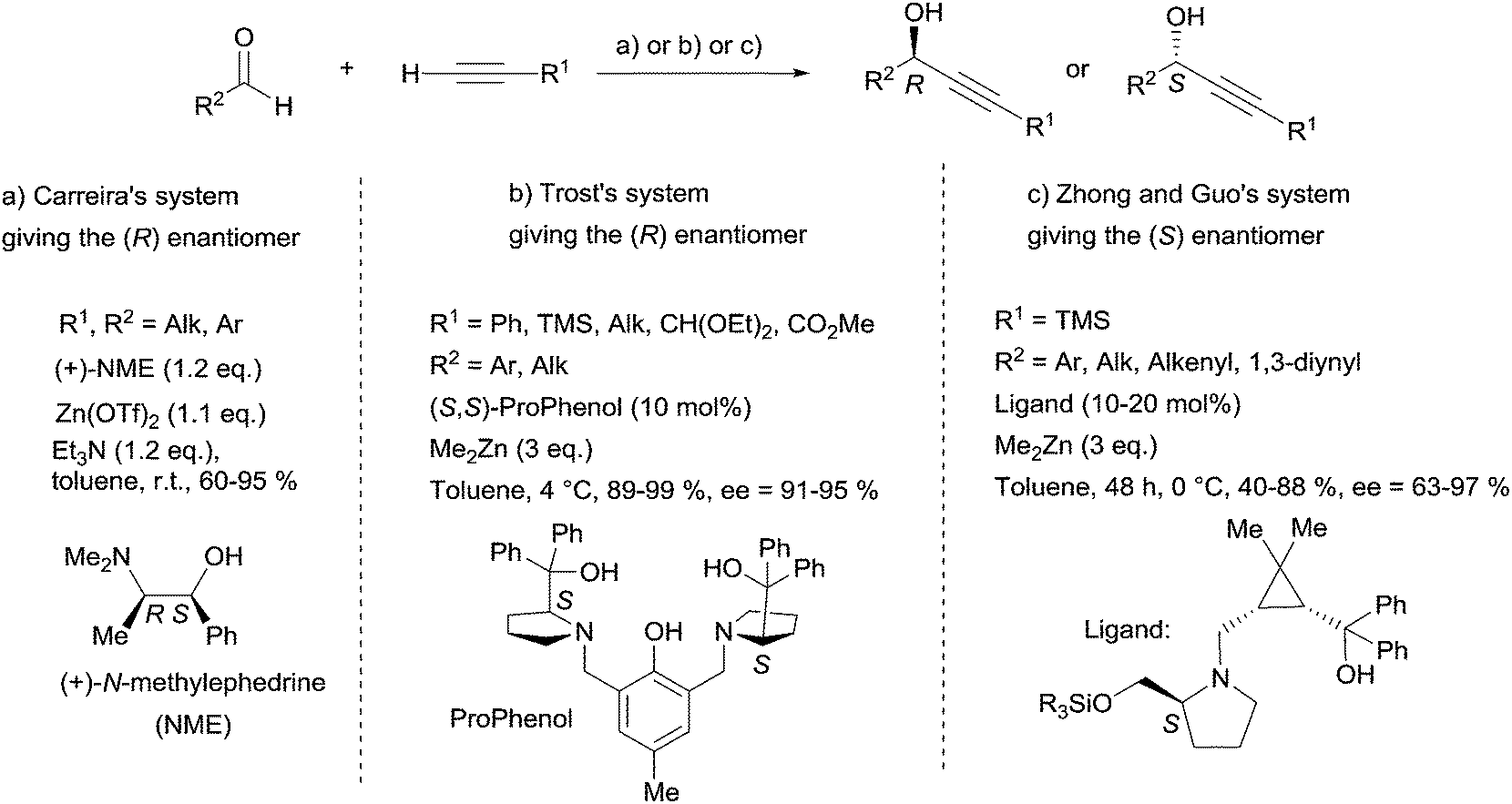 | ||
| Scheme 10 Carreira's, Trost's and Zhong and Guo's systems for the enantioselective addition of terminal alkynes to aldehydes, used in the synthesis of strongylodiols. | ||
The structure of the strongylodiols indeed suggests a general retrosynthetic analysis based on the asymmetric addition of a terminal 1,3-diyne to an aldehyde, which was introduced independently by the respective authors (Scheme 11).
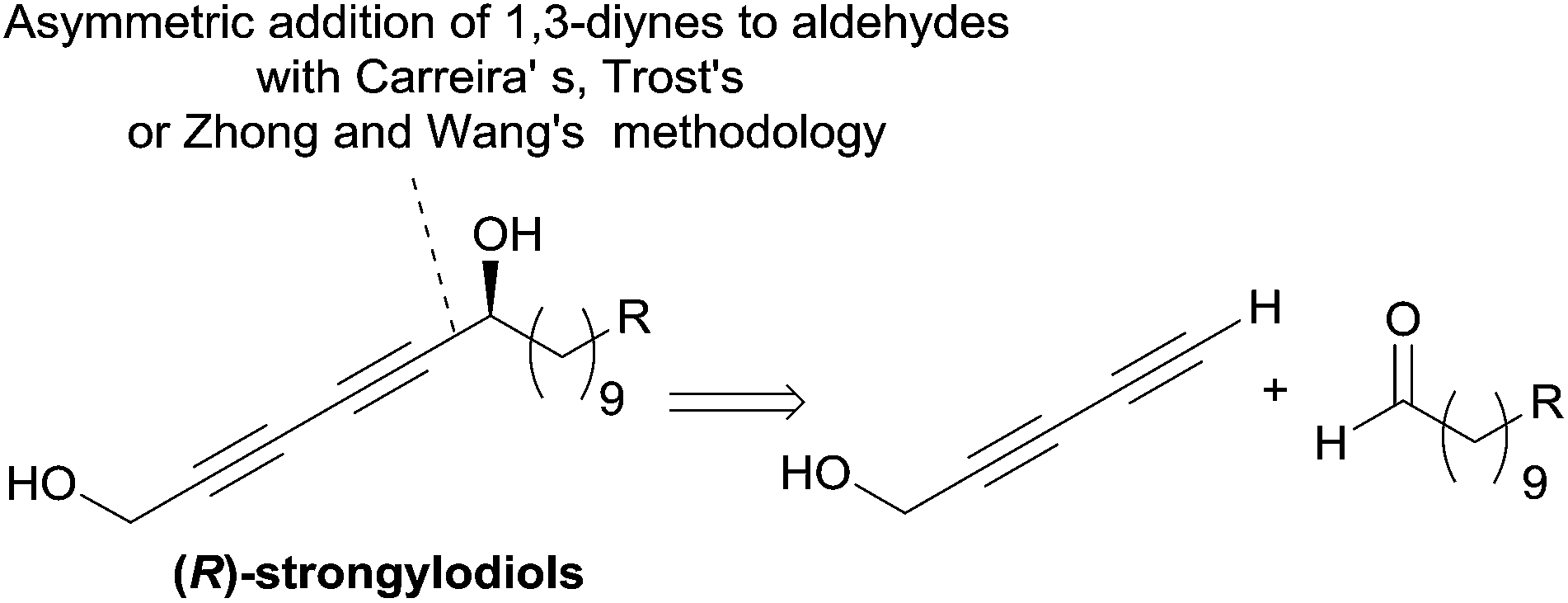 | ||
| Scheme 11 Carreira's, Trost's and Zhong and Wang's retrosynthesis of strongylodiols (syntheses in Schemes 13 and 18). | ||
In 2003, Carreira's group applied its own protocol to synthesis of the strongylodiols A and B.35 The O-protected 1,3-alkadiyne nucleophile 9 was thus first secured in two steps from 1,4-dichlorobutyne via in situ hydroxymethylation of an elusive sodium acetylide (the second “carbo-mer” of NaH,36Scheme 12).
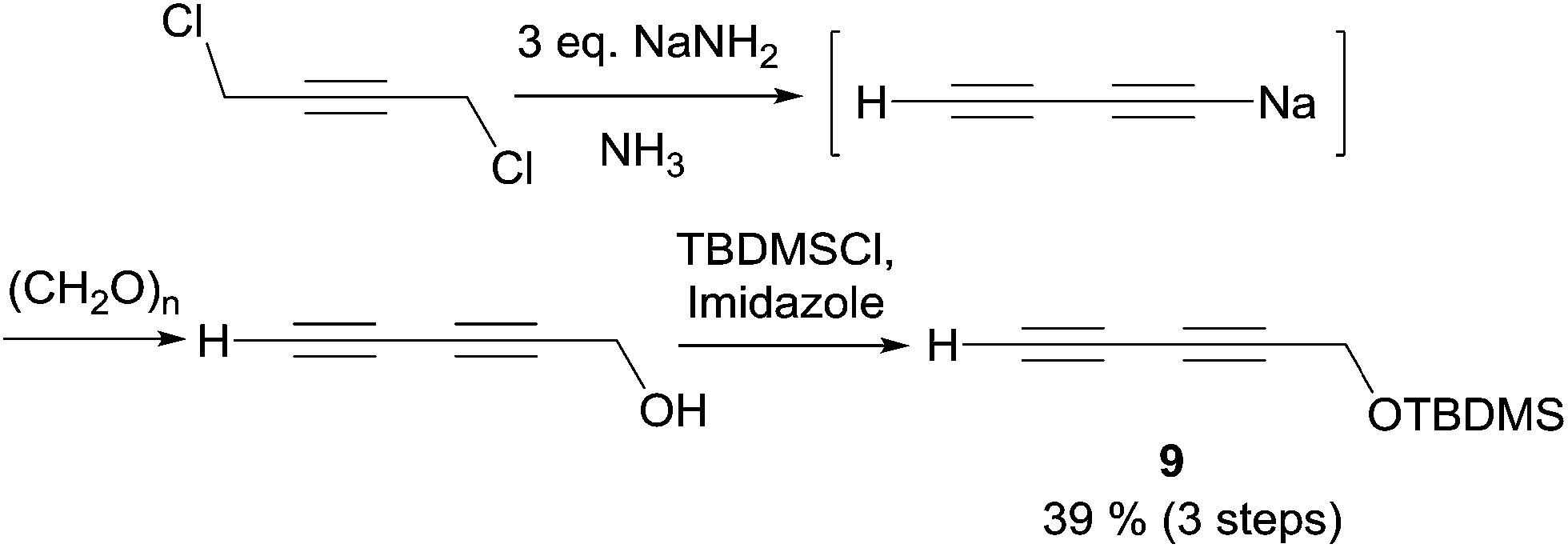 | ||
| Scheme 12 Carreira's synthesis of the 1,3-diynyl fragment of strongylodiols. | ||
The aldehydes 10 and 11 were prepared from the common C20 lipidic ω-alkynol 12, itself generated by monobromination of 1,10-decanediol and subsequent substitution using in situ generated 1-decynyllithium (Scheme 13). Prior to a Swern oxidation step, preparation of aldehyde 11, a precursor of strongylodiol A, required a Z-selective hydrogenation of the triple bond of 12. After optimization, it was found that the use of four equivalents of the auxiliaries (Zn(OTf)2 and Et3N, vs.9), combined with a slow addition modus operandi of the electrophiles 10 or 11, afforded the alkynylcarbinols 13 and 14, precursors of strongylodiols B and A, in 62–68% yield and 80–82% ee, respectively. The application of Carreira's procedure in the enantioselective addition of terminal di- and triynes to (branched) aldehydes has been studied in detail, lately.37
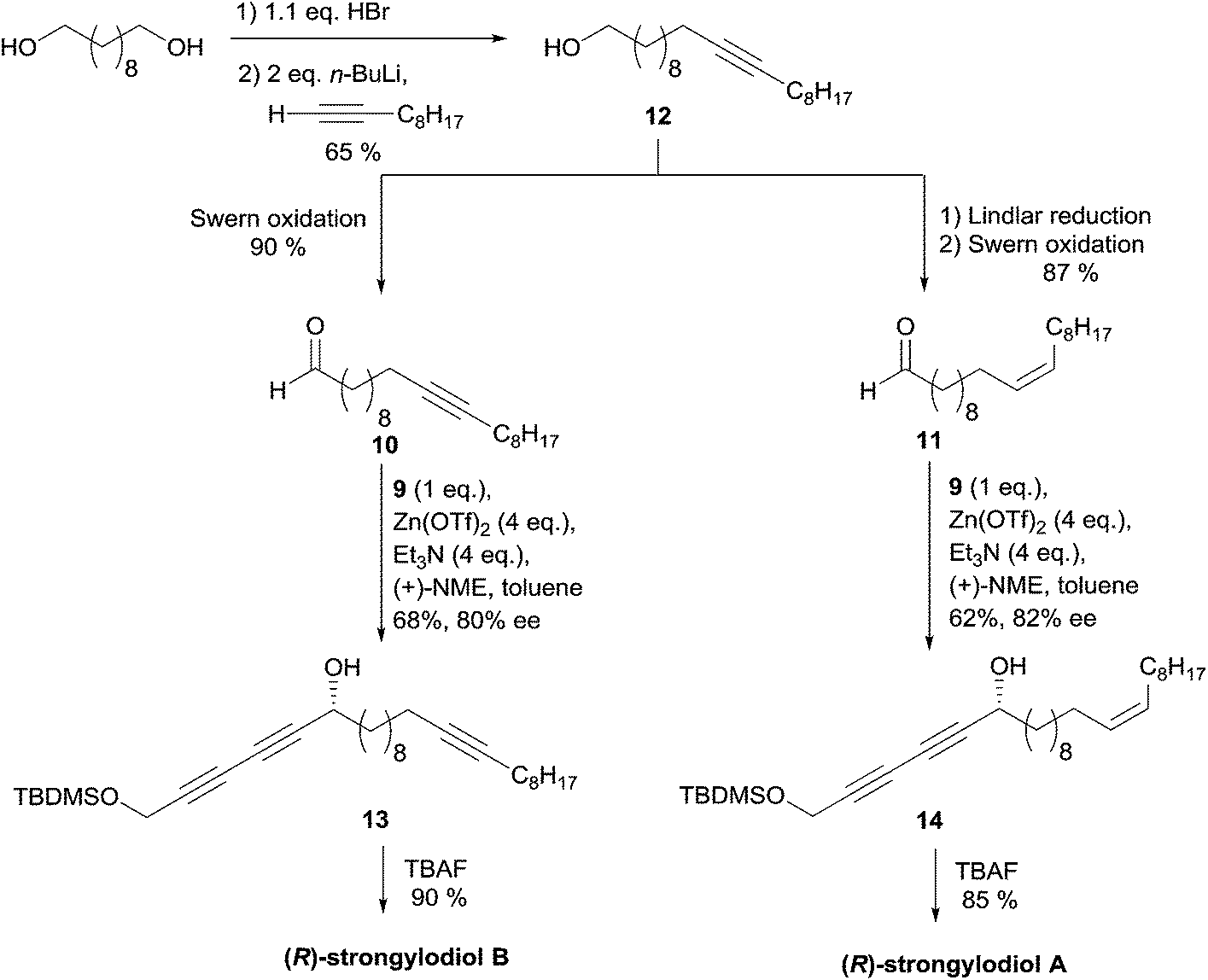 | ||
| Scheme 13 Carreira's late-divergent route to strongylodiols A and B (retrosynthesis in Scheme 11). | ||
A few years later, Baldwin et al. reported on the synthesis of (R)-strongylodiols A and B,38 while discussing in detail the failure of Carreira's procedure in accomplishing the model addition of TMS-acetylene to dodecanal,28 using either the original NME or Jiang's ligand39 derived from the chloramphenicol unit (Scheme 14).
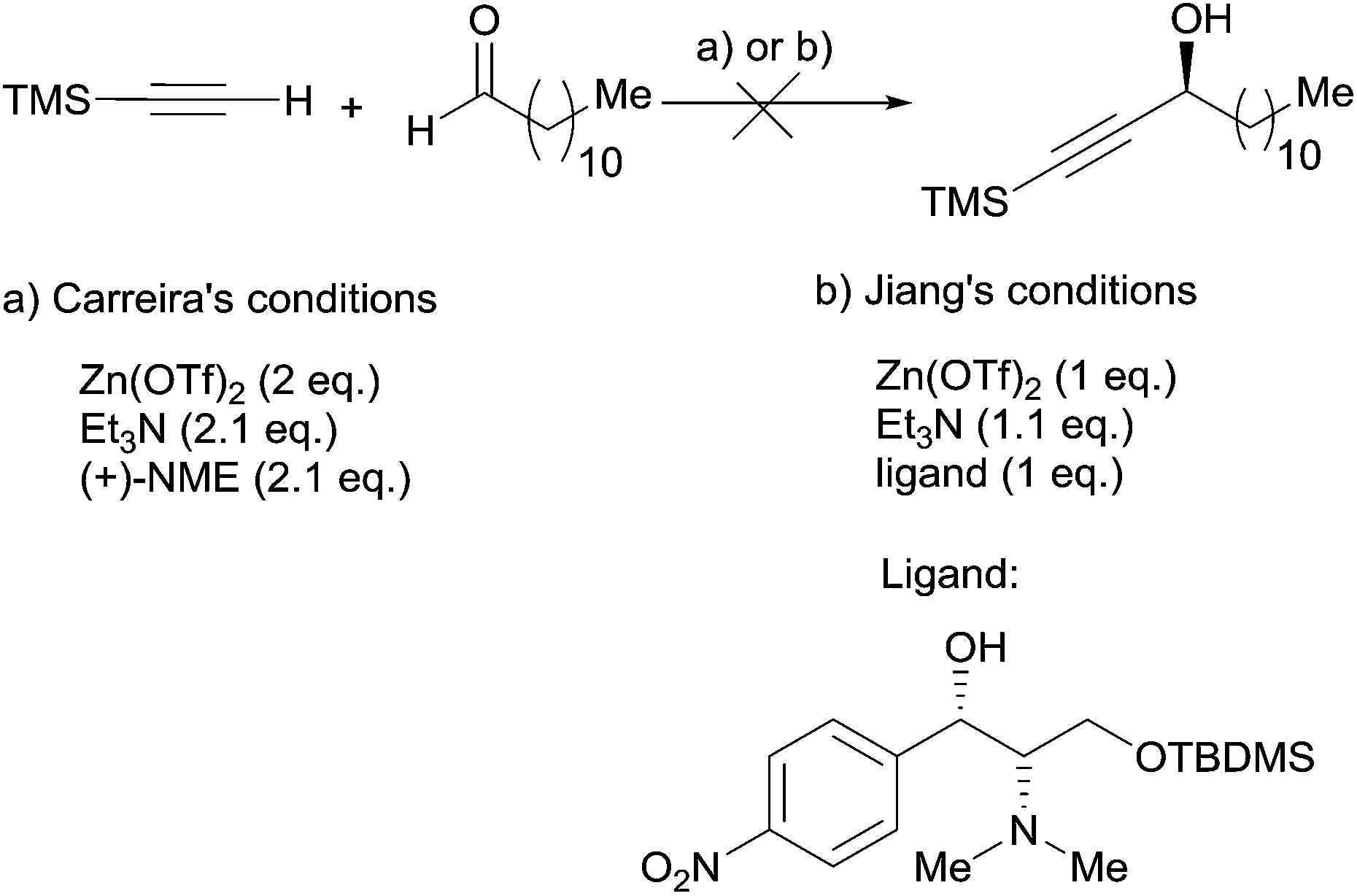 | ||
| Scheme 14 Baldwin's attempts at using Carreira's and Jiang's enantioselective addition procedures. | ||
Subsequently, Baldwin revisited his strategy using Noyori's asymmetric ynone reduction methodology (vide supra, Section 2.1.1, Scheme 6) and a late Cadiot–Chodkiewicz coupling (Scheme 15),28 thus paralleling Yadav's stepwise synthetic approach toward lembehynes (Section 2.2.2.1, Schemes 8 and 9).24
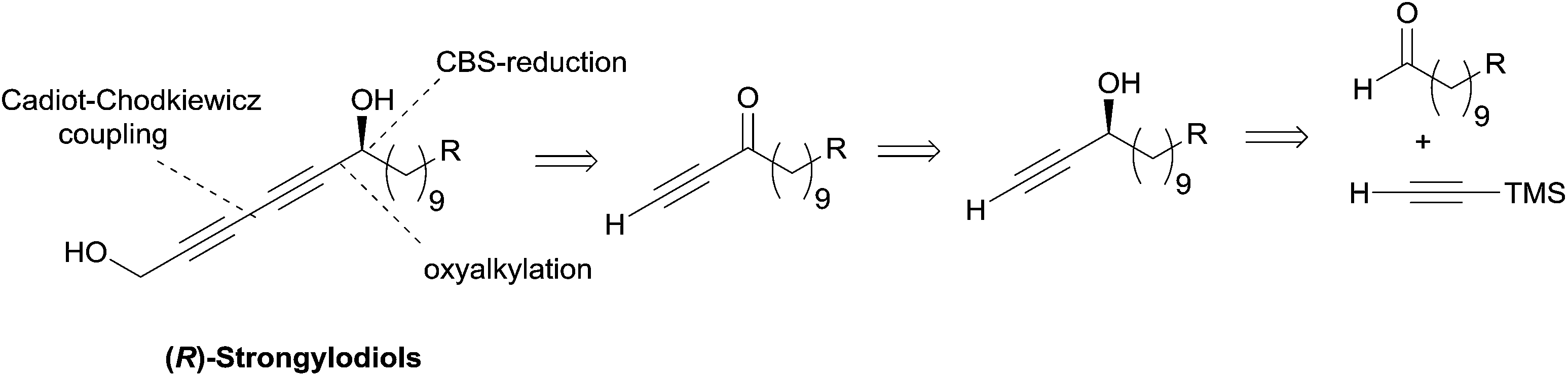 | ||
| Scheme 15 Baldwin's retrosynthesis of strongylodiols (synthesis in Scheme 16). | ||
Baldwin's total synthesis of the (R)-strongylodiols A and B is detailed in Scheme 16. The preparation of the ynone substrates 15 and 16 was accomplished using an addition/oxidation sequence starting from TMS-acetylene and the lipidic aldehydes 10 and 11, respectively, both obtained from alcohol 12 (the formation of 11 requiring an additional step of partial hydrogenation of the triple bond of 12: Scheme 13). The use of the preformed Ru(II) pre-catalyst by Noyori in isopropanol (as the hydrogen donor) afforded the propargylic alcohols 17 and 18 in high ee's. A two-step sequence was then applied to assemble the 1,3-diyne moiety and access the natural products.
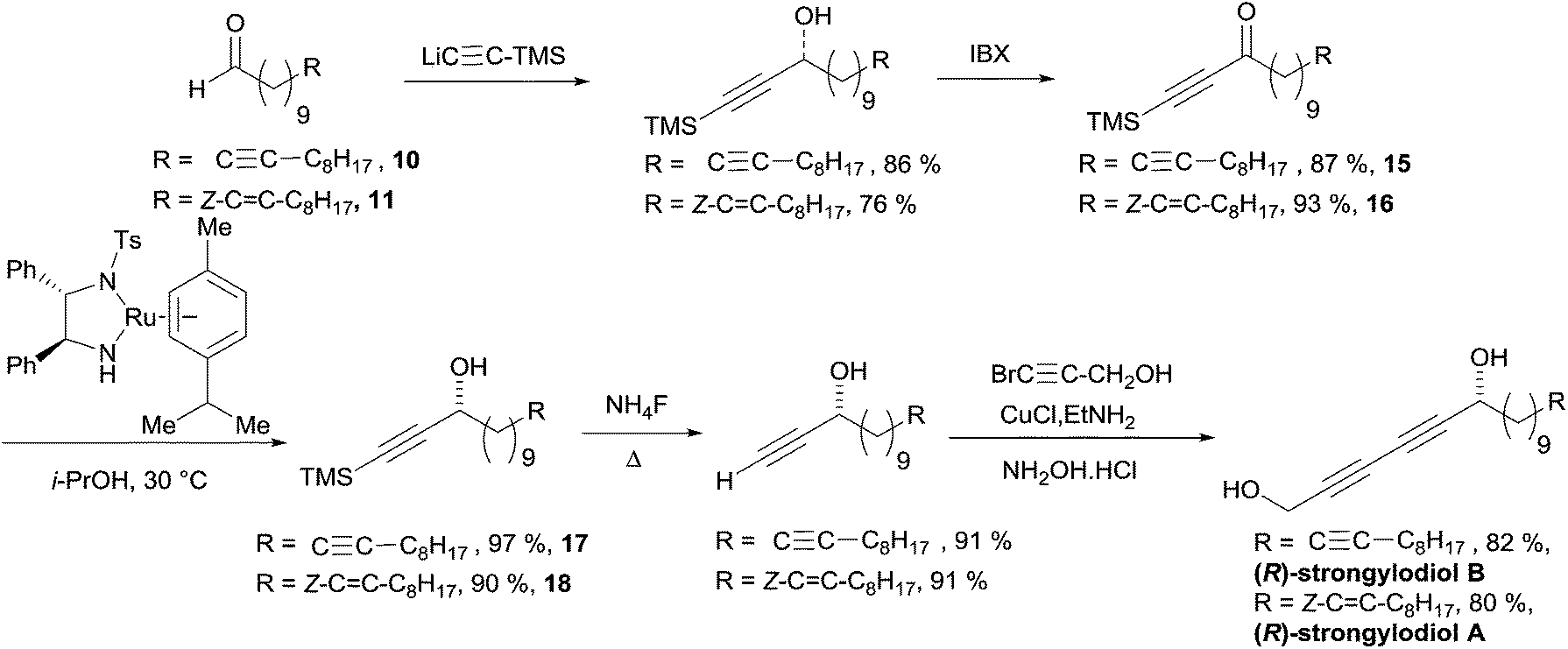 | ||
| Scheme 16 Baldwin's total synthesis of the (R)-strongylodiols A and B (retrosynthesis in Scheme 15). | ||
In 2006 Trost et al. described an efficient method for the asymmetric addition of terminal alkynes to aldehydes using a ProPhenol-derived dinuclear catalytic system (see Scheme 10).33 The procedure, requiring 10 mol% of the ligand and three equivalents of pyrrophoric Me2Zn, proved efficient for a wide range of alkynes, including phenylacetylene, TMS-acetylene and methyl propiolate. Aromatic and α,β-unsaturated aldehydes also proved to be suitable electrophiles. In order to reduce the zinc acetylide stoichiometry, several adaptations were proposed, such as the addition of triphenylphosphine oxide or a pre-mixing step with the alkyne.40 These optimized protocols allowed the reaction of elaborate alkyne reagents and enolizable aliphatic aldehydes. A possible catalytic cycle was proposed by the authors (Scheme 17).40 The reaction of the ProPhenol ligand with two equivalents of Me2Zn would thus afford a dinuclear complex acting as a Brønsted basic and Lewis acidic bifunctional catalyst. The subsequent formation of a zinc acetylide, followed by coordination of the aldehyde to the less sterically demanding site, would then lead to intermediate 19 (Scheme 17). According to the authors, the alkynyl–zinc nucleophile would then “undergo nucleophilic addition to the Si face of an aldehyde”, and an explicit steric interpretation is proposed in Scheme 17 and its legend. The transmetalation to another zinc acetylide would finally release the alkoxide product and regenerate the catalytic species.
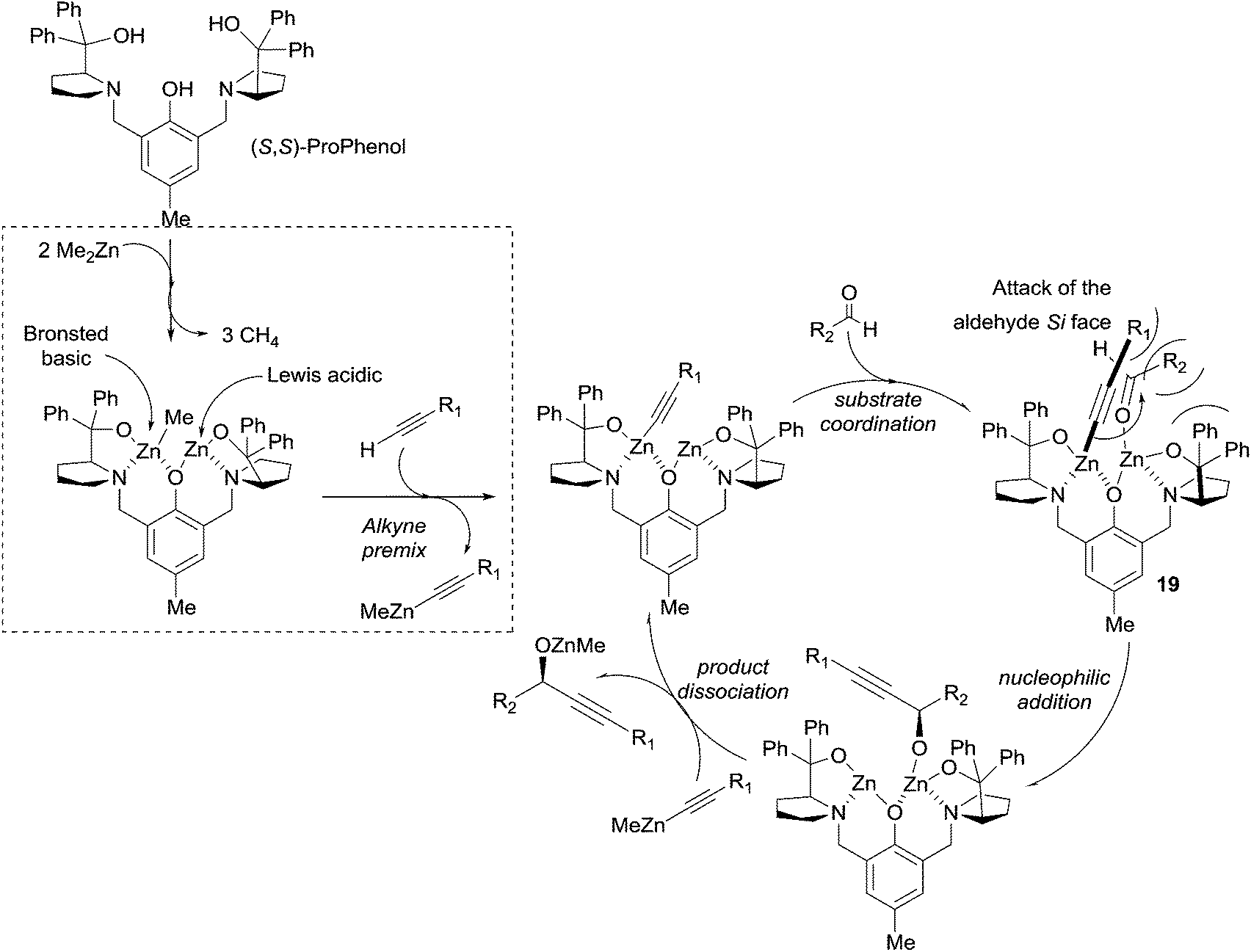 | ||
| Scheme 17 Proposed catalytic cycle for Trost's enantioselective addition of terminal alkynes to aldehydes, and proposed origin of the enantioselectivity (repulsion of the R2 substituent by both the attacking alkynyl group and the CPh2 group of (S)-diphenylprolinol). | ||
Trost et al. then extended the methodology to terminal α-diyne nucleophiles. An optimized protocol, relying on the combined use of TPPO (20 mol%) and ProPhenol (10 mol%) with penta-2,4-diynyl acetate as the nucleophile, was applied in the convergent syntheses of (R)-strongylodiols A and B (Scheme 18).41
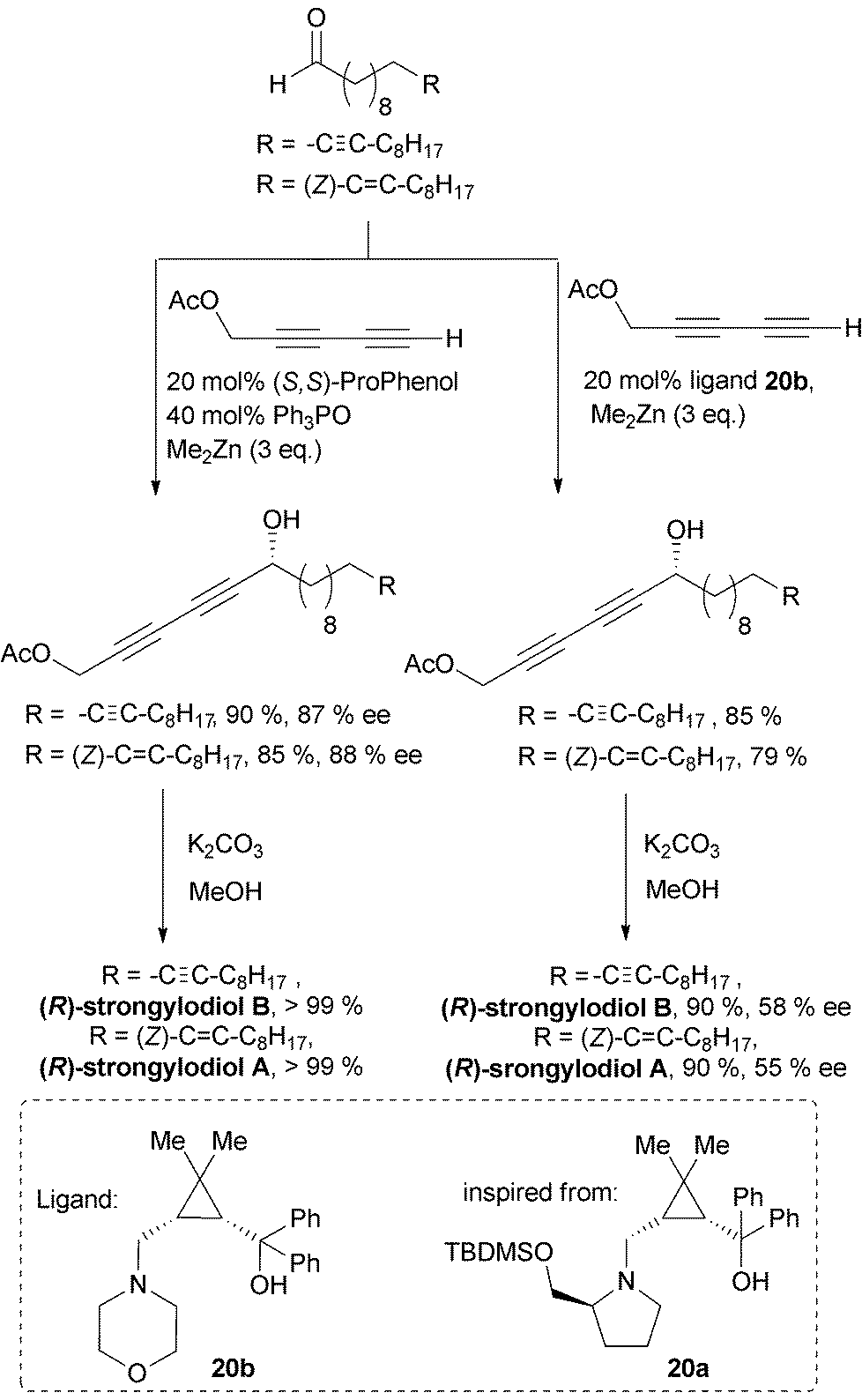 | ||
| Scheme 18 Trost's (left) and Zhong and Wang's (right) syntheses of strongylodiols A and B. | ||
In 2011, Zhong and Guo described the 1,4-aminoalcohol 20a, based on a cis-prolino-chrysanthemic scaffold (Scheme 18), that proved effective in the enantioselective catalysis of the Me2Zn-mediated addition of TMS-acetylene to various types of aldehydes (see Scheme 10).34 The procedure was applied to the synthesis of (S)-eicos-(4E)-en-1-yn-3-ol (see Section 2.3.2.2, Scheme 24), and one year later, Zhong and Wang further screened related ligands for the reaction of terminal α-diyne nucleophiles.42 The use of 10 mol% of the 1,4-aminoalcohol 20b, containing a morpholine moiety, in the presence of an excess of Me2Zn, proved highly efficient in promoting the enantioselective addition of 1,3-diynes to various acceptors, including aromatic, α,β-unsaturated and branched aliphatic aldehydes. The flexibility of this procedure was also challenged in an asymmetric synthesis of (R)-strongylodiols A and B using penta-2,4-diynyl acetate as the nucleophile (Scheme 18).
2.3 Alkenyl alkynylcarbinols
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH(OH)(C
CHCH(OH)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) and R2 = (CH2)9CHCHCH(OH)(CCH), both arms embedding a homochiral (S)-4-en-1-yn-3-ol unit.46
CH) and R2 = (CH2)9CHCHCH(OH)(CCH), both arms embedding a homochiral (S)-4-en-1-yn-3-ol unit.46
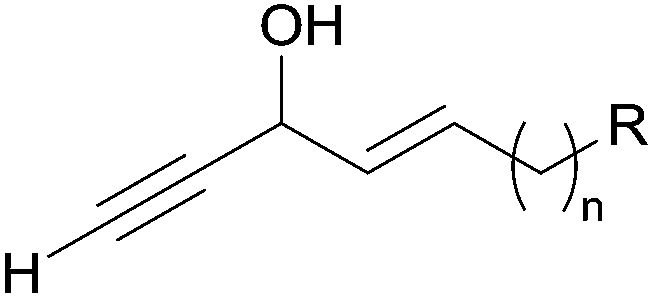 | ||
| Fig. 5 The C5 AAC fragment found in many marine natural products. | ||
As a typology extension, the central motif of petrosynol can be seen as an internal cis-fused bis-AAC (Fig. 6) where R1 = alkyl and R2 = alkenyl in Fig. 2. Synthetic methodologies for such a substructure have not been reported so far.
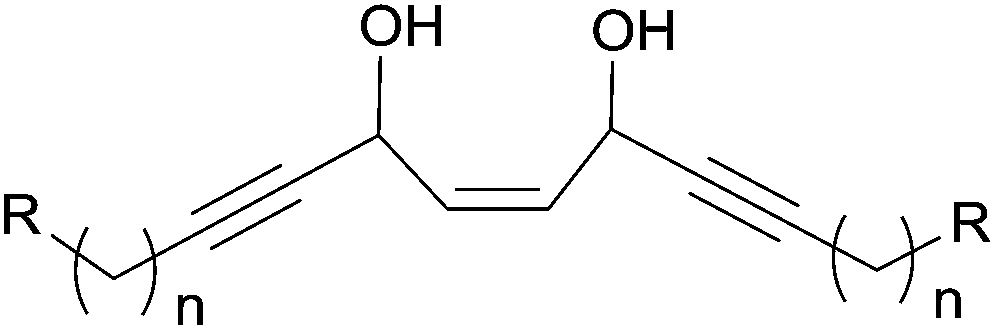 | ||
| Fig. 6 The cis-fused bis-AAC motif of petrosynol (n = 6, R = (E)-CHCH–CH(OH)–CCH, see Fig. 1). | ||
 | ||
| Fig. 7 Cytotoxic marine AAC products. | ||
2.3.2.1 Enzymatic kinetic resolution of racemic propargylic alcohols. The use of various lipases (Candida rugosa, Pseudomonas, Candida antarctica) proved highly efficient in the preparation of enantio-enriched AACs using the kinetic enzymatic resolution of racemic mixtures. Upon treatment with a selected enzyme, one enantiomer of the carbinol is selectively trans-acylated, allowing the subsequent separation of a scalemic ester from the unreacted propargylic alcohol of the complementary scalemic composition.
In a pioneering report back in 1998, Chattopadhyay et al. described a chemoenzymatic synthesis of (S)-eicos-(4E)-en-1-yn-3-ol (23) via an enantio-enriched C5 DAC key building block (Scheme 19).49
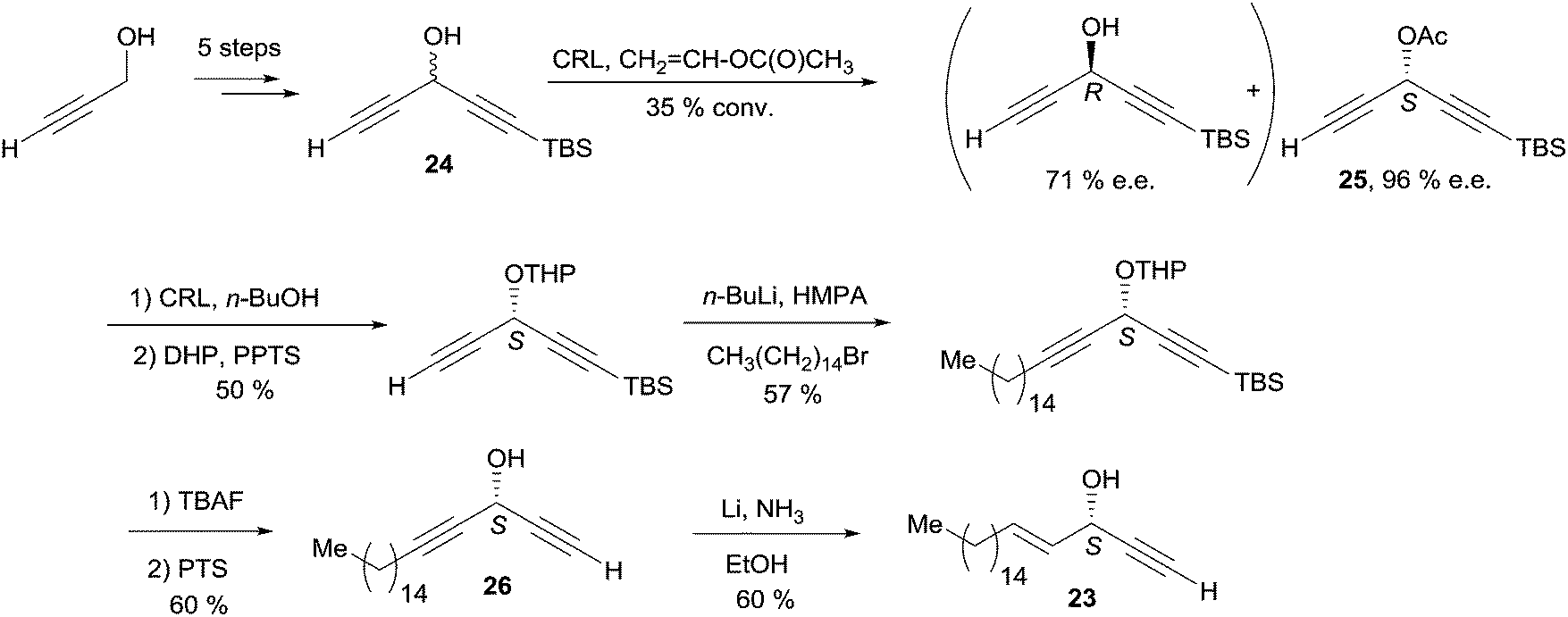 | ||
| Scheme 19 Chattopadhyay's chemoenzymatic synthesis of (S)-eicos-(4E)-en-1-yn-3-ol (23). | ||
Propynol was first transformed in five steps to racemic DAC 24 (Scheme 19), which was selectively acylated upon addition of the Candida Rugosa lipase (CRL) and vinyl acetate to deliver the (S)-acetate 25 in 96% ee.50 Treatment of 25 with the same enzyme in the presence of n-BuOH induced the reverse trans-acylation. After protection of the resulting alcohol as a THP acetal, C-alkylation with 1-bromopentadecane followed by hydrolytic C-desilylation afforded the mono-terminal dialkynylcarbinol 26 in 34% yield over two steps. The AAC target 23 was finally obtained by the regio- and stereoselective reduction of the internal (and most hindered) triple bond of 26 under Birch-type conditions (Scheme 19). It is noteworthy that the primary value of this route is an access to non-racemic DACs, such as the intermediate 26 (vide infra). One year later, Naoshima et al. reported on a similar approach for the synthesis of a close analogue of 23, (S)-(+)-docosa-(4E,15Z)-dien-1-yn-3-ol (27), thus allowing the confirmation of the 3S/15Z configuration of the natural product (Scheme 20).51
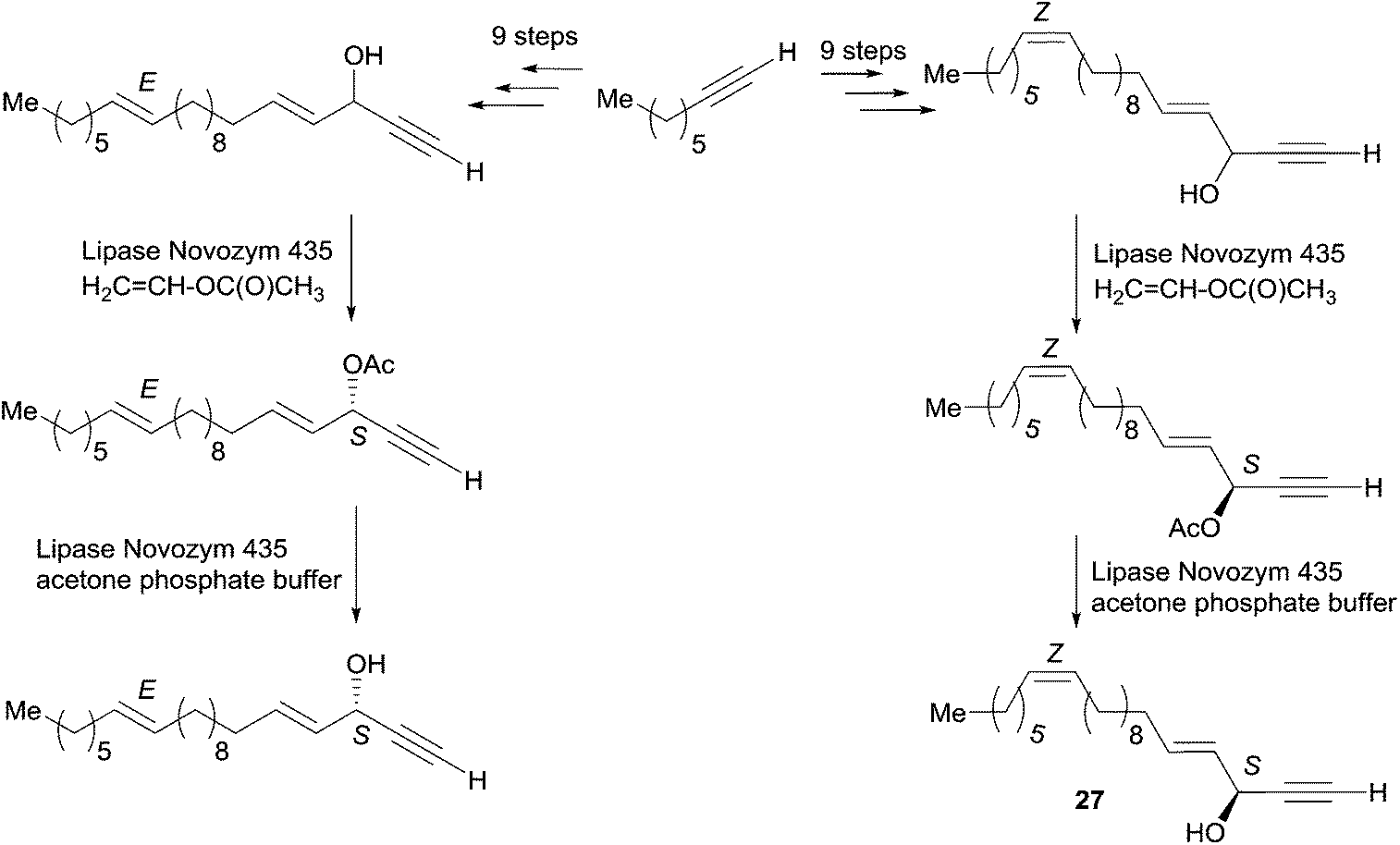 | ||
| Scheme 20 Naoshima's chemoenzymatic synthesis of (S)-(+)-docosa-(4E,15Z)-dien-1-yn-3-ol (27) and the (E,E)-isomer. | ||
The synthesis of (S)-(+)-eicos-(4E)-en-1-yn-3-ol (23) was also performed by Gung et al. using a more concise strategy (Scheme 21).29 Due to a failure of Carreira's method in this case, the kinetic resolution of a racemic sample of 23 with the lipase AK from Amano was used to secure the (S)-acetate in an optimal 98% ee and 25% conversion. Subsequent hydrolysis of the acetate gave the dextrorotatory natural compound.
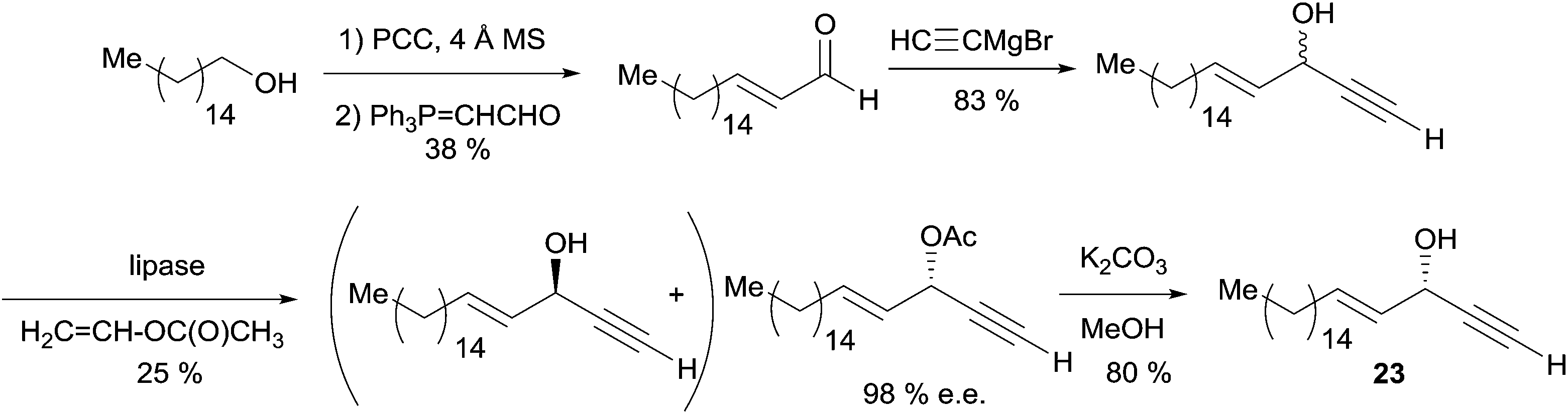 | ||
| Scheme 21 Gung's chemoenzymatic synthesis of (S)-(+)-eicos-(4E)-en-1-yn-3-ol (23). | ||
Lately, a similar lipase-based approach was used by Mori et al. to synthesize the different stereoisomers of miyakosyne A, allowing the determination of the absolute configuration of its major component.46
2.3.2.2 Non-enzymatic routes to natural alkenyl alkynylcarbinols. The asymmetric synthesis of eicos-(4E)-en-1-yn-3-ol (23) and its analogues was also tackled using chemical methodologies described above.
Enantioselective reduction of ynones. In 1999, Garcia et al. described the synthesis of (3R,4E)-19-methylicos-4-en-1-yn-3-ol (21), using the enantioselective reduction of ynone 28 in a key step (Scheme 22).52 The use of the CBS technology with one equivalent of (R)-oxazaborolidine provided the (R)-alcohol target 21 in a few minutes, 72% yield and 95% ee. The use of Alpine-borane gave comparable results after longer reaction times.
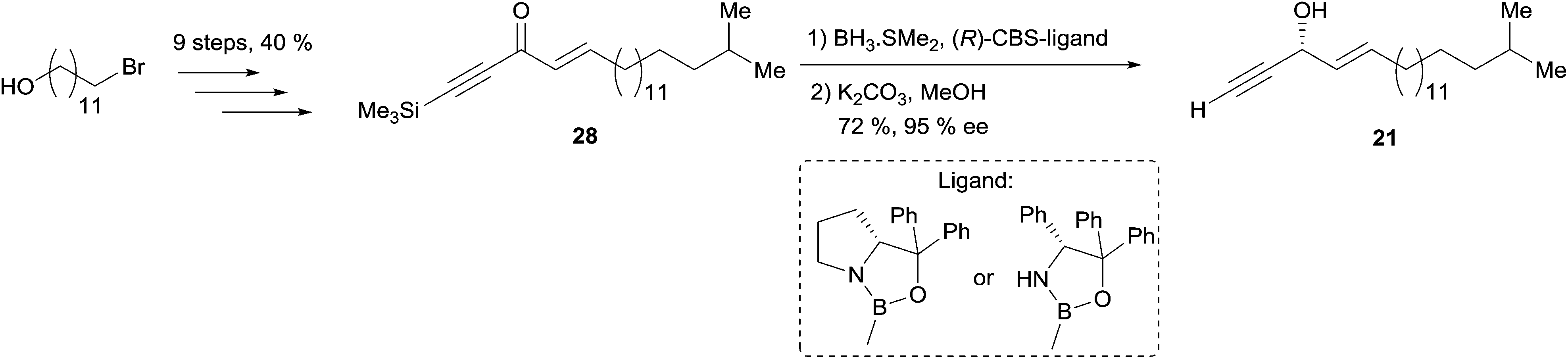 | ||
| Scheme 22 Garcia's synthesis of (3R,4E)-19-methylicos-4-en-1-yn-3-ol (21) by the enantioselective reduction of an ynone. | ||
Double elimination reaction. In 1999, using Yadav's double elimination methodology,22 Cai et al. reported the first co-preparation of both the (S) and (R)-enantiomers of eicos-(4E)-en-1-yn-3-ol (23), thus providing the confirmation of the absolute configuration of the natural enantiomer. Each optical isomer was obtained from either key β-alkoxy chloride 29 or 30 (Scheme 23).53 Later on, Bittman et al. applied a similar principle in the preparation of the Z-isomer from a β-alkoxy chloride, obtained by means of Sharpless' asymmetric dihydroxylation.23
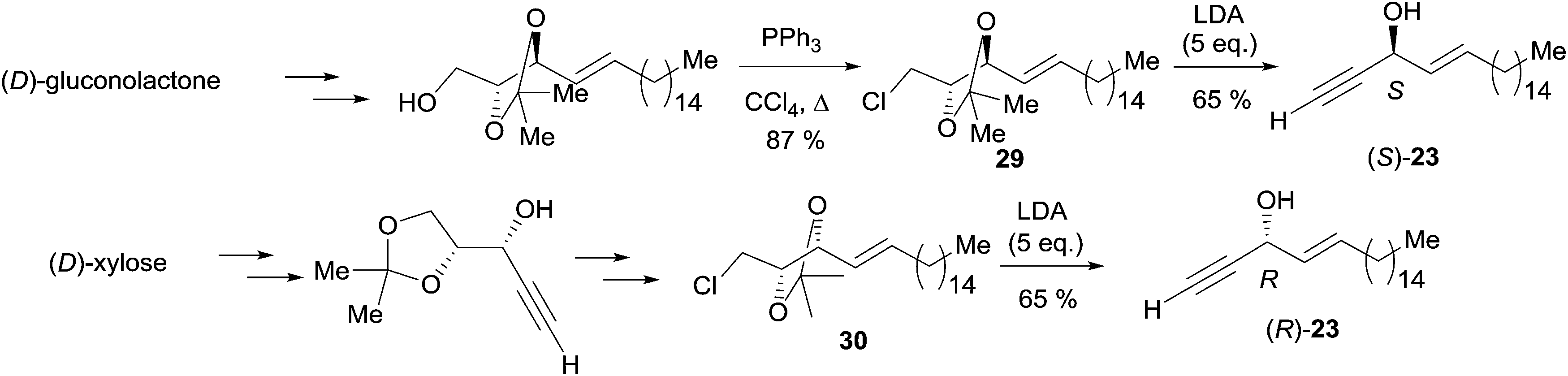 | ||
| Scheme 23 Cai's synthesis of the (R)- and (S)-eicos-(4E)-en-1-yn-3-ol (23) using Yadav's double elimination reaction. | ||
Asymmetric addition of terminal alkynes to aldehydes. Zhong and Guo et al. applied their Me2Zn-mediated asymmetric addition procedure (resorting to the prolino-chrysanthemic ligand 20a, see Scheme 18) to the synthesis of (S)-eicos-(4E)-en-1-yn-3-ol (23) (Scheme 24).34 Addition of TMS-acetylene to (E)-octadec-2-enal thus furnished the C-silylated AAC 31 in 80% ee, that was upgraded to 97% ee by recrystallization of the desilylated target 23.
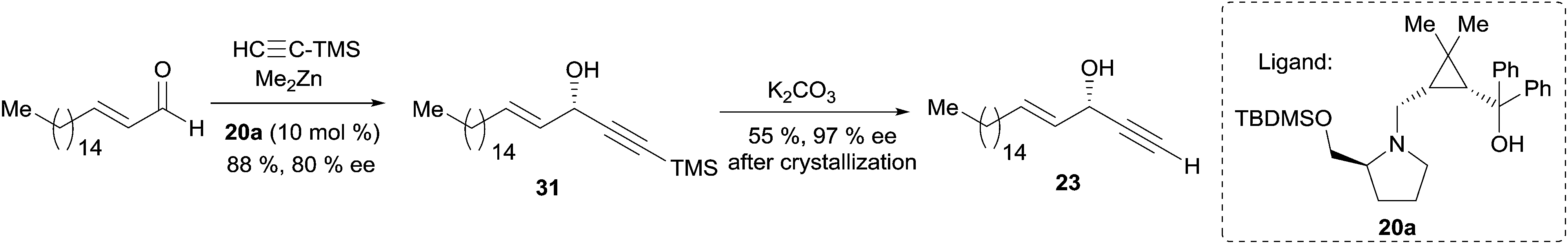 | ||
| Scheme 24 Zhong and Guo's synthesis of (S)-eicos-(4E)-en-1-yn-3-ol (23) by the enantioselective addition of a terminal alkyne to an aldehyde. | ||
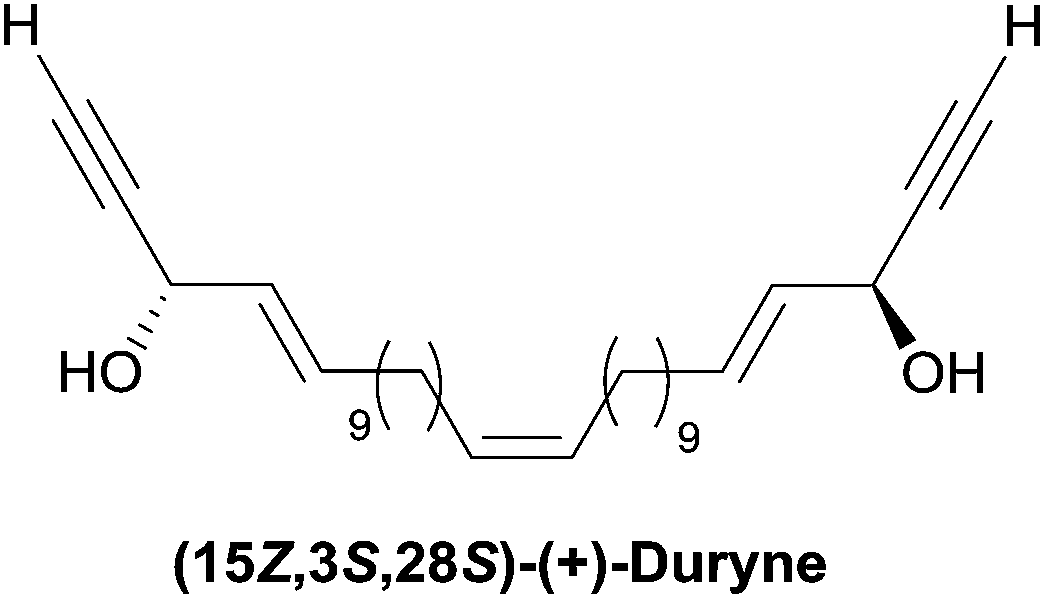 | ||
| Fig. 8 The (S,S)-duryne structure. | ||
2.3.3.1 Asymmetric organo-catalyzed aldehyde α-hydroxylation/Ohira–Bestmann reaction. Within the context of the total synthesis of (+)-duryne, an efficient method was devised by Kumaraswamy et al. for the stepwise access to (4E)-en-1-yn-3-ol motifs (Scheme 25).54 The asymmetric carbinol center was here first elaborated in the key chiral allylic alcohol 32 using the proline-catalyzed α-hydroxylation of an aldehyde (Scheme 26), both the alkene and alkyne C2 units being then installed in an Ohira–Bestmann ethynylation step. In the retrosynthetic scheme toward duryne, the central olefinic core is assembled using a Wittig-type olefination (Scheme 25). Both partners of this reaction are derived from a common C14 precursor (intermediate 37 in Scheme 27), containing the (S)-AAC unit and obtained by alkene cross-metathesis between 32 and the lipidic ethylenic alcohol 33.
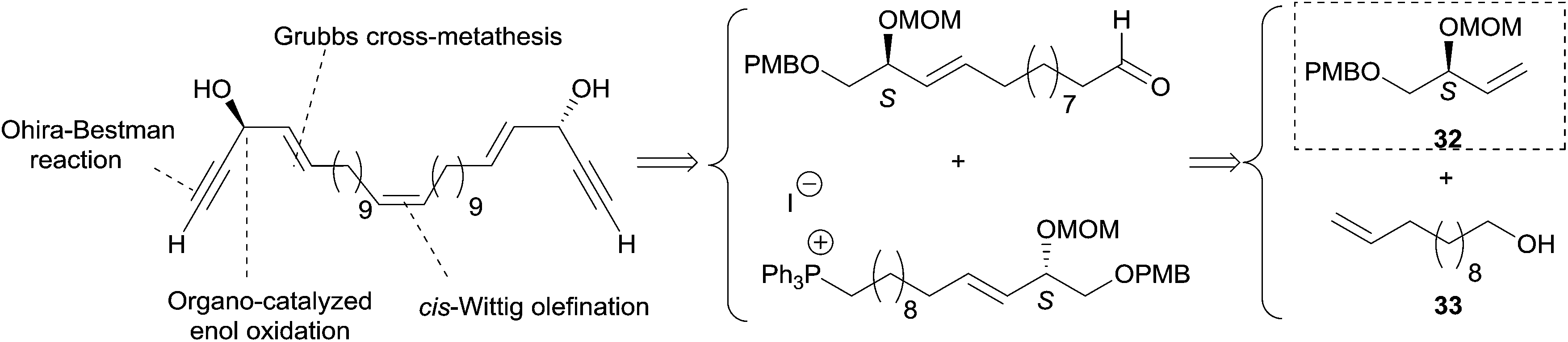 | ||
| Scheme 25 Kumaraswamy's retrosynthesis of (+)-duryne (synthesis Scheme 27). | ||
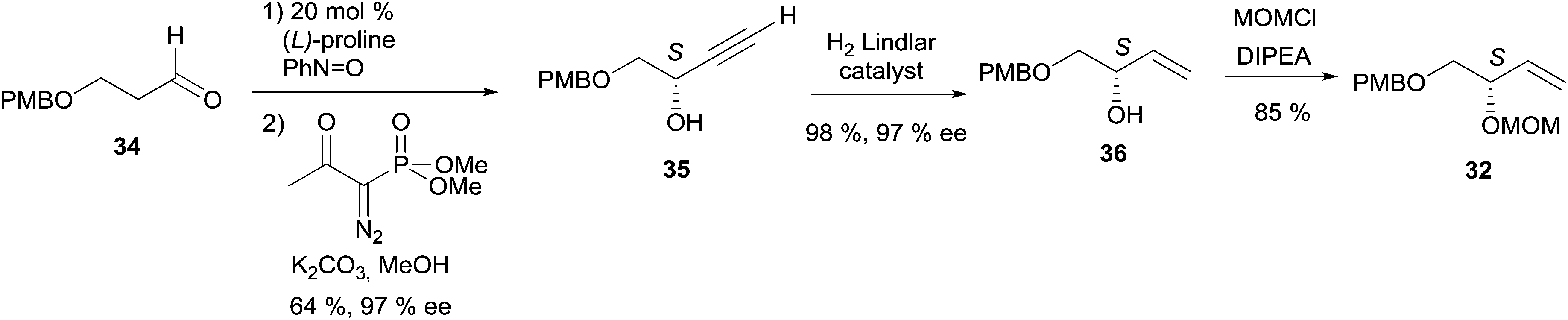 | ||
| Scheme 26 Kumaraswamy's synthesis of the key chiral C4 precursor 32 of (+)-duryne. | ||
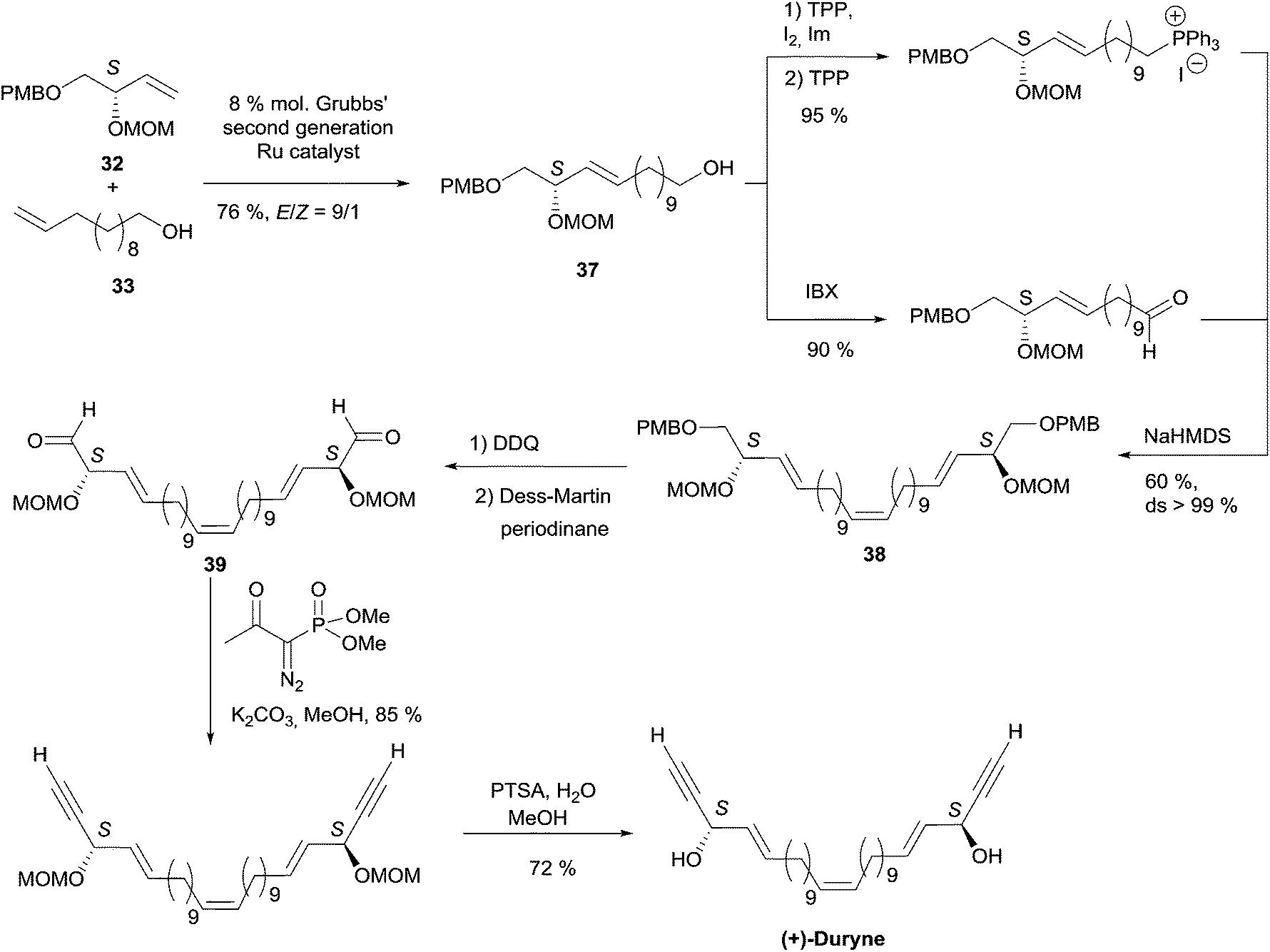 | ||
| Scheme 27 Kumaraswamy's synthesis of (+)-duryne (retrosynthesis Scheme 25). | ||
The key building block (S)-32 was thus generated in 97% ee via proline-catalyzed α-aminoxylation of the enolizable aldehyde 34, through propargylic alcohol 35 (after in situ methanol-induced N–O bond cleavage of the labile aminoxylated aldehyde and trapping with the Ohira–Bestmann diazo-phosphonate) and via the allylic alcohol 36 (obtained using Lindlar hydrogenation, Scheme 26).
Using the Grubbs-II (second generation) catalyst, the cross-metathesis of 32 and 33 afforded the C14 intermediate 37, which was then converted to either of the two precursors of the Wittig reaction to produce the central cis-double bond of the C28 intermediate 38 (Scheme 27). After deprotection–oxidation of the two primary carbinol centers, double Ohira–Bestmann homologation of the resulting dialdehyde 39 afforded the C30 targeted (+)-duryne.
2.3.3.2 Enzymatic kinetic resolution. Access to the AAC fragments of duryne using enzymatic kinetic resolution has been addressed by the groups of Chattopadhyay and Gung. Chattopadhyay's approach to (S)-eicos-(4E)-en-1-yn-3-ol (23) (Section 2.3.2, Scheme 19) was introduced to access the non-natural (15E,R,R)-isomer of duryne (Scheme 28).50 Treatment of the racemic DAC, (rac)-40, with vinyl acetate and CRL afforded (R)-40 in 71% ee and 35% conversion. Further optical enrichment to 93% ee of the slow-reacting enantiomer was accomplished by partial re-acylation of the mixture with 30% conversion. Assembly of the full C30 skeleton was performed via double substitution of a linear 1,20-dibromoalkyne by the O-protected enantio-enriched carbinol 40. Partial reduction under Birch conditions of the resulting pentayne 42 (after full deprotection) finally established, in a single step, the three E-alkene units of the targeted (15E,R,R)-duryne.
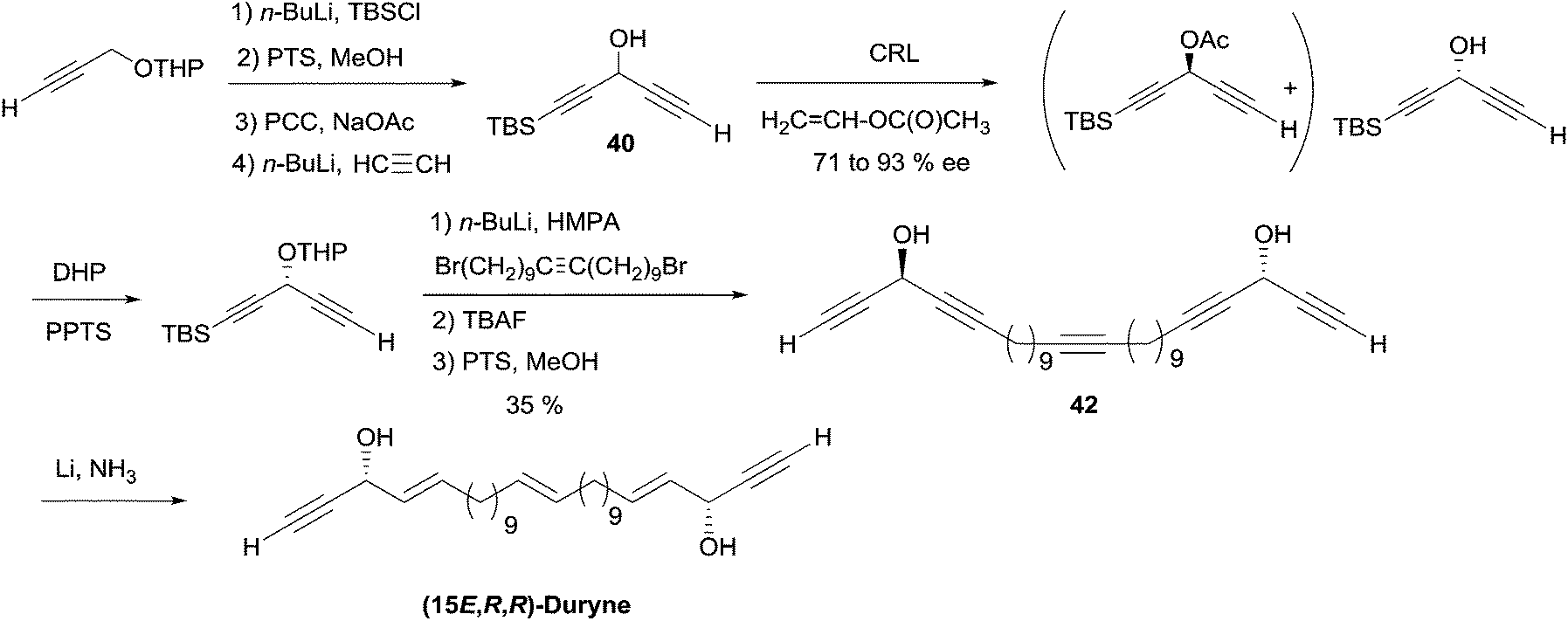 | ||
| Scheme 28 Chattopadhyay's synthesis of the non-natural (15E,R,R)-duryne. | ||
More recently, Gung et al. reported on the kinetic resolution of a mixture of the three stereoisomers (R,R/S,S/R,S) of duryne with a fixed Z-configuration at the central double bond (Scheme 29).55 The stereoisomeric mixture was readily prepared from the C22 diol (Z)-43. Treatment of this mixture with vinyl acetate and the lipase AK resulted in a faster acetylation of the S-configured carbinol units. The bis-acetylated (S,S)-isomer was thus isolated in 18% yield, the unreacted (R,R)-diol was recovered in 21% yield, while the monoacetylated meso (R,S)-isomer was obtained in 45% yield. Smooth saponification of the bis-acetylated product gave (S,S)-(Z)-duryne. The (S,S)-(E)-isomer was obtained via the same route, and the comparison of the optical rotations of the two isomers allowed confirmation of the Z-geometry of the central double bond of the natural product.
 | ||
| Scheme 29 Gung's kinetic enzymatic resolution of the three stereoisomers of duryne. | ||
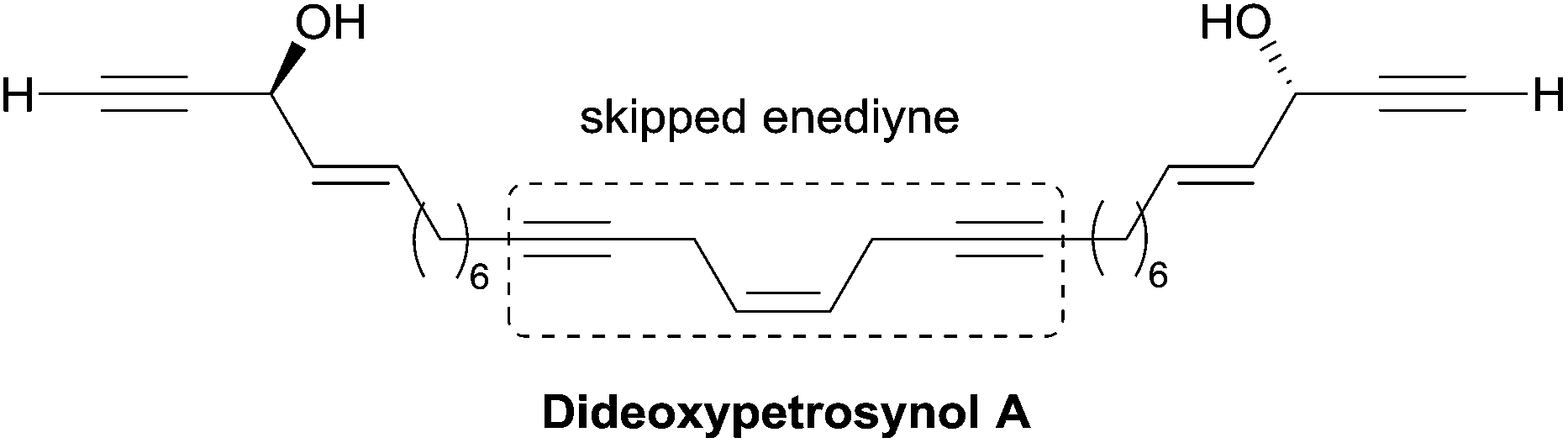 | ||
| Fig. 9 The structure of dideoxypetrosynol A. | ||
A single total asymmetric synthesis of dideoxypetrosynol A has been described to date. As in the synthesis of duryne (see Section 2.3.3.2, Scheme 29), Gung et al. made use of a late enzymatic kinetic resolution using lipase AK from Pseudomonias sp., favoring the acetylation of the (S,S)-diastereoisomer (Scheme 30).59 The key principle of this route is the unconventional assembly of the skipped enediyne motif using an oxidative Wittig-type coupling of a homopropargylic phosphonium ylide. The dextrorotatory product was assigned to the S,S-configuration using Burgess' Pseudomonas sp. active site model, and by comparison with the positive optical rotations of related polyacetylene products with an S-configuration.
 | ||
| Scheme 30 Gung's kinetic chemoenzymatic resolution of dideoxypetrosynol A. | ||
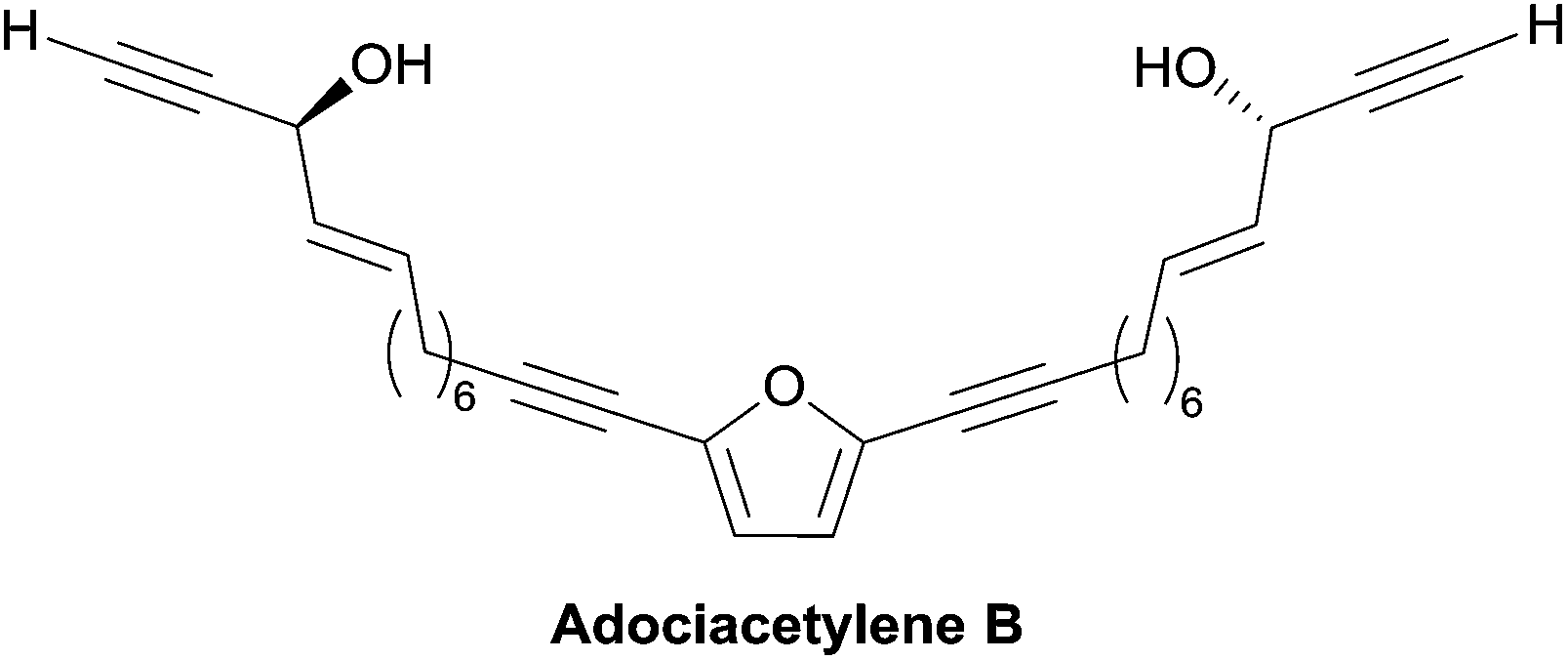 | ||
| Fig. 10 Structure of the adociacetylene B (or petrofuran). | ||
2.3.5.1 Asymmetric reduction of ynones. In 1999, Garcia et al. devised a bidirectional approach to adociacetylene B (petrofuran), based on the asymmetric reduction of a bis-ynone precursor generated from 2,5-dibromofuran (Scheme 31).62 The choice of the CBS reduction methodology was guided by preliminary experiments performed on a model substrate, which indicated the phenylglycine-derived oxazaborolidine as an optimal reagent in combination with BH3·SMe2 (other reagents, like Alpine-borane or DIP-Cl, were found to give lower yields or enantioselectivities).
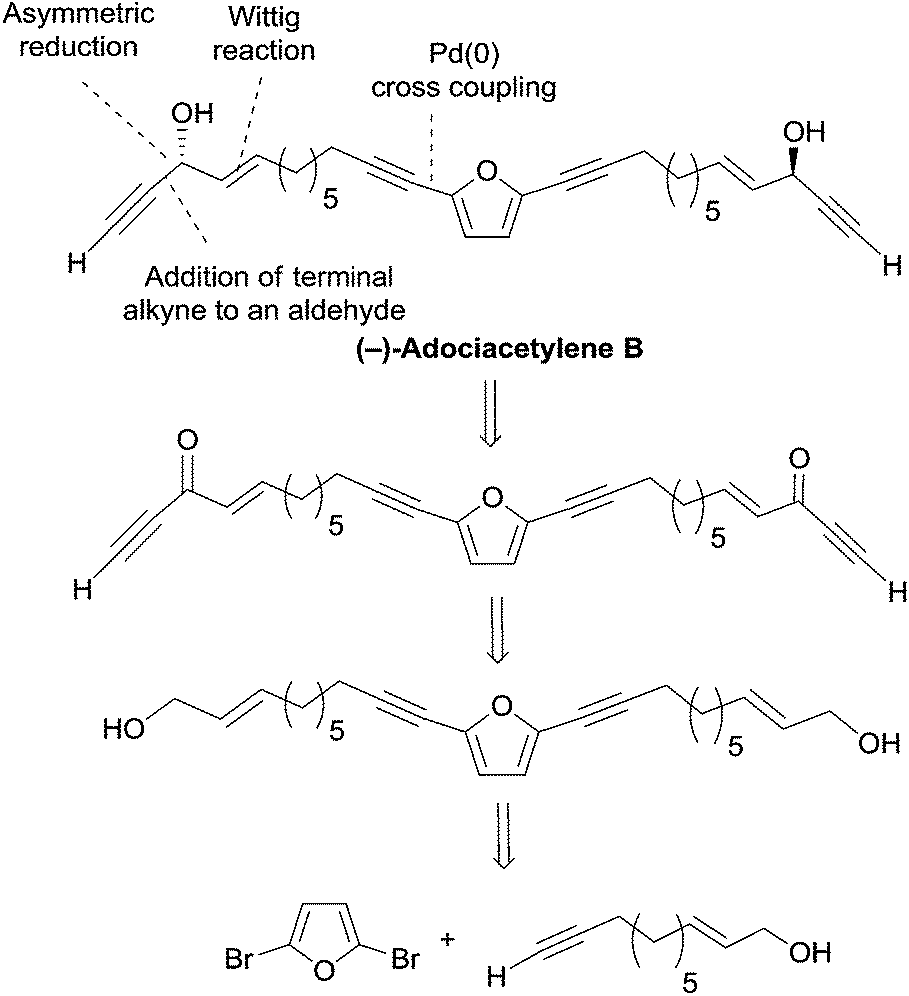 | ||
| Scheme 31 Garcia's retrosynthetic approach to (−)-adociacetylene B (synthesis in Scheme 32). | ||
The C26 central core of the adociacetylene skeleton was built in a single step by means of a double Sonogashira-type coupling reaction (Scheme 32). In this event, the use of Pd(PPh3)4 as the catalyst in refluxing pyrrolidine and in the absence of copper salt, gave optimal results. Oxidation of the allylic primary carbinol centers, and subsequent addition of trimethylsilylacetylene to the resulting aldehyde groups, followed by oxidation of the propargylic secondary carbinol centers, afforded bis-ynone 44. Final CBS reduction and C-desilylation afforded (S,S)-45, i.e. (+)-adociacetylene B, in 98% ee.
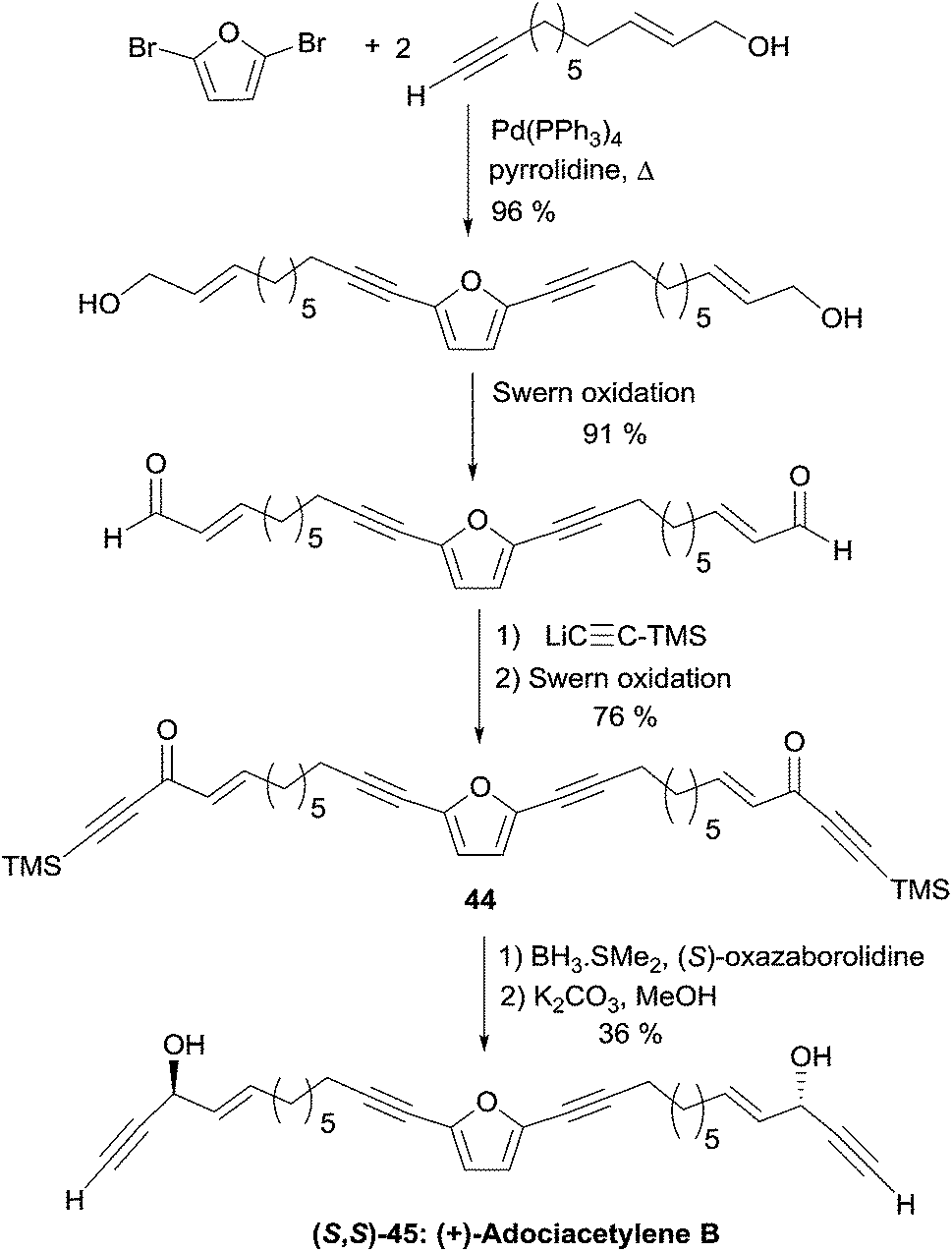 | ||
| Scheme 32 Garcia's synthesis of adociacetylene B (retrosynthesis in Scheme 31). | ||
2.3.5.2 Enzymatic kinetic resolution. In 2001, as Carreira's procedure failed to produce the targeted diol, Gung et al. devised an enzymatic kinetic resolution method for the preparation of adociacetylene B, which was later on applied to the synthesis of duryne (Section 2.3.3.2, Scheme 29) and dideoxypetrosynol (Section 2.3.4, Scheme 30).30 The substrate is a mixture of the C2-symmetrical bis-propargylic alcohols DL-45 and meso-45, prepared via a six-step sequence starting with a double Pd-catalyzed sp–sp2 Negishi coupling of 2,5-dibromofuran with the terminal ω-hydroxy-alkyne 46 (Scheme 33).
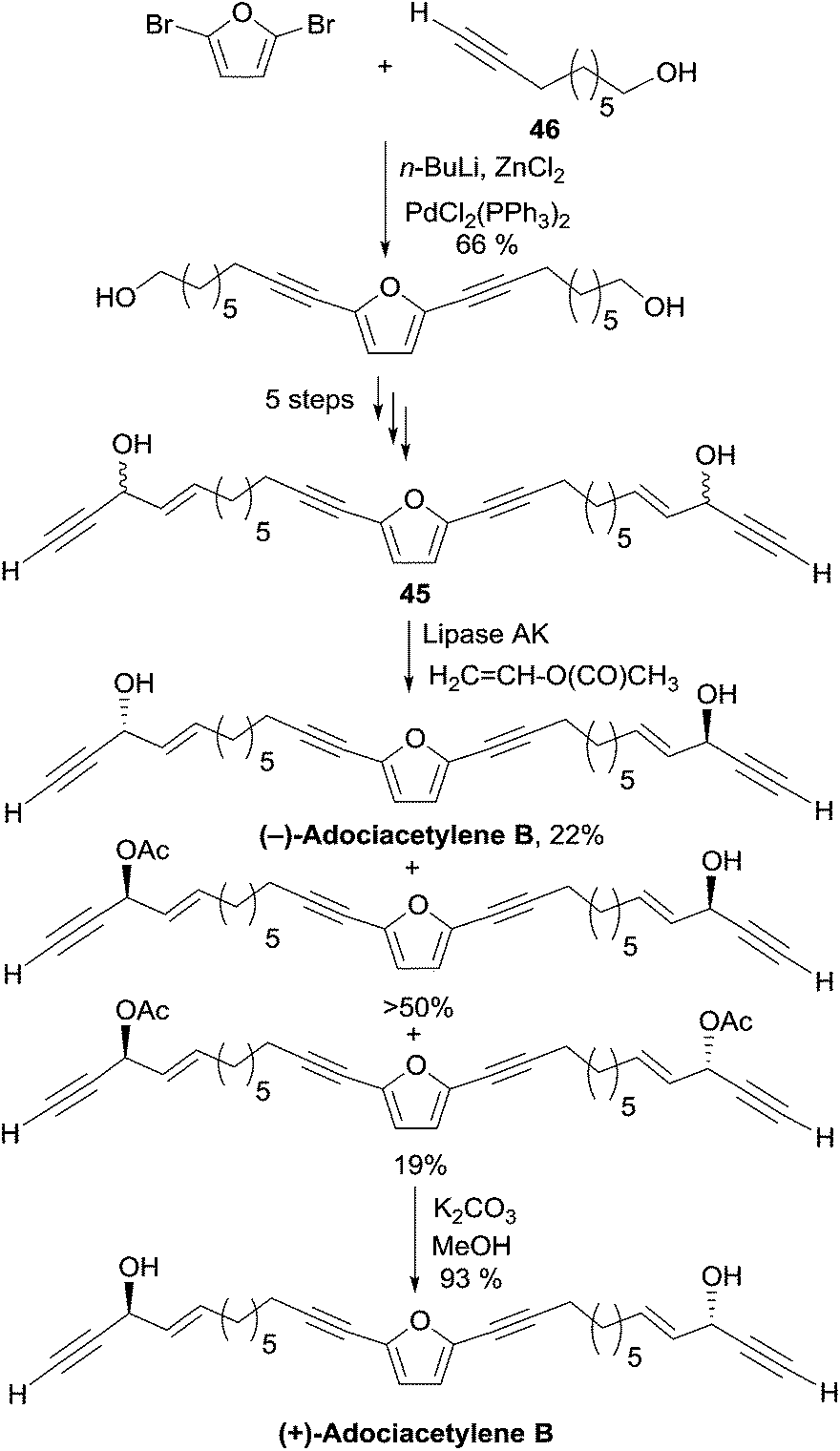 | ||
| Scheme 33 Gung's enzymatic kinetic resolution of adociacetylene B. | ||
Upon treatment of this mixture of isomers with vinyl acetate in the presence of the lipase AK, a faster acylation of the natural (S,S)-enantiomer was observed leaving the (R,R)-enantiomer unreacted, while the meso stereoisomer was mono-acetylated at its (S)-carbinol center. The formal discrepancy between the experimental result and Burgess’ model for the enantioselectivity prediction is due to the fact that the smallest carbinol substituent (the alkynyl group) has also the highest CIP priority.63 Smooth saponification of the diacetylated product led to the corresponding diol, whose optical rotation of [α]D = +21° was consistent with that of a natural sample of (+)-adociacetylene B. An enantiomeric excess of 90% and the confirmation of the S,S-configuration were provided by a 1H NMR study of the corresponding O-methyl mandelate ester.
2.3.5.3 Asymmetric addition of terminal alkynes to aldehydes. In 2006, Trost et al. applied the Me2Zn-ProPhenol procedure for the asymmetric addition of terminal alkynes to aldehydes in a total synthesis of adociacetylene B, using a bidirectional strategy (Scheme 34).64 The bis-enal 47 was first obtained via Ru-catalyzed redox isomerization of the corresponding bis-propargylic alcohol, which was assembled from 2,5-dibromofuran by means of a copper-free Sonogashira cross-coupling. Double addition of TMS-acetylene to the dialdehyde 47 in the presence of six equivalents of Me2Zn and 20 mol% of the (S,S)-ProPhenol catalyst afforded the chiral direct precursor of adociacetylene B as the major component in a 9
:1 mixture of dl/meso stereoisomers. After C-desilylation, (R,R)-adociacetylene B was isolated in high ee (>99% according to chiral HPLC analysis). Significant improvement was gained by running the alkynylation process with methyl propiolate instead of TMS-acetylene. A higher yield and dl/meso ratio (17:1) were obtained, while retaining a 99% ee. In this case, (R,R)-(−)-adociacetylene B was generated using saponification and CuCl-promoted C-decarboxylation (Scheme 34).
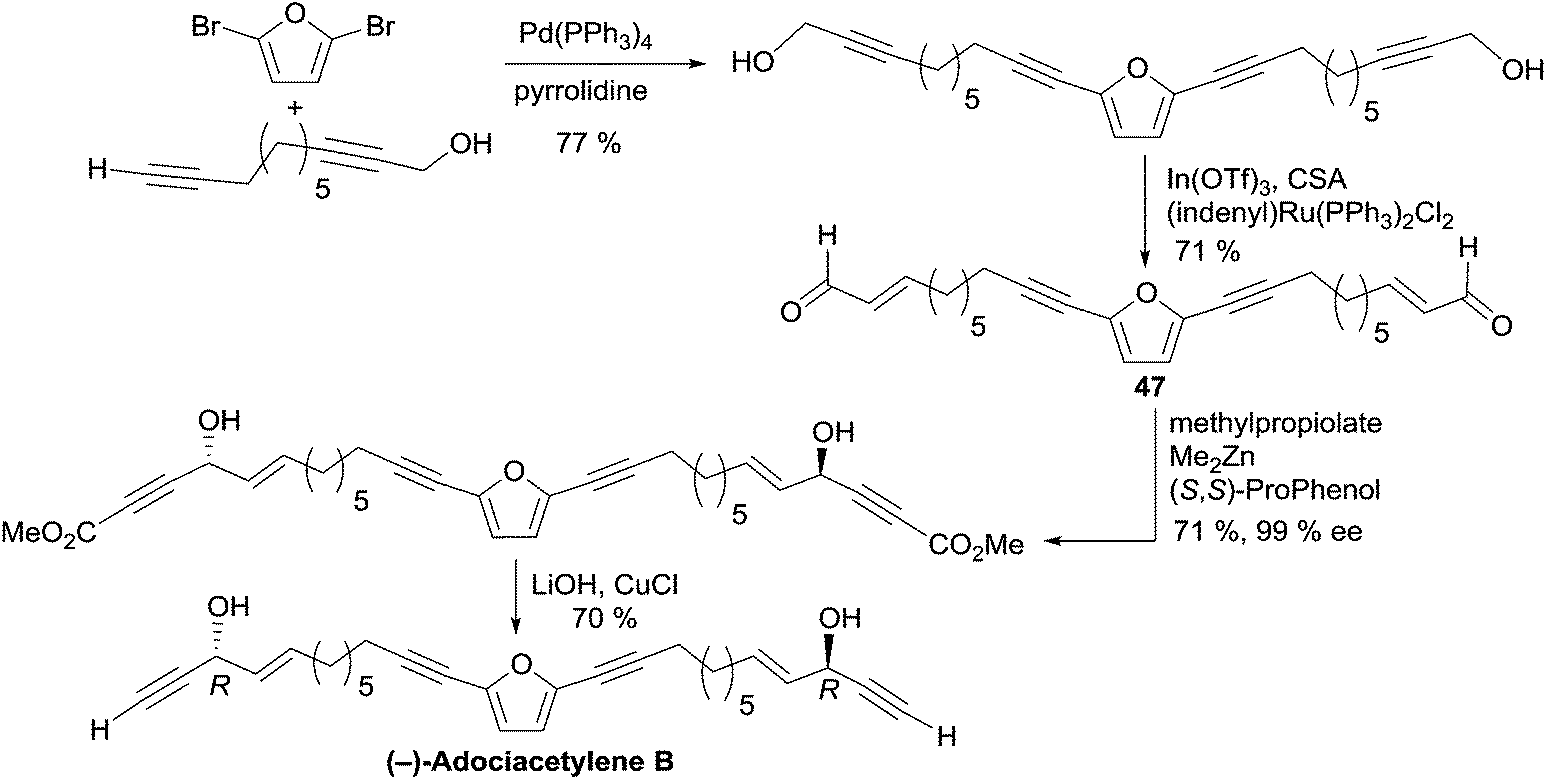 | ||
| Scheme 34 Trost's catalytic enantioselective synthesis of (−)-adociacetylene B. | ||
2.4 Dialkynylcarbinols
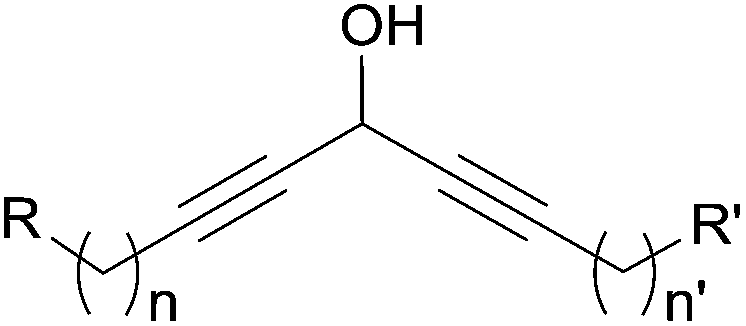 | ||
| Fig. 11 The DAC (“carbinol-skipped diyne”) fragment found in a few marine natural products. | ||
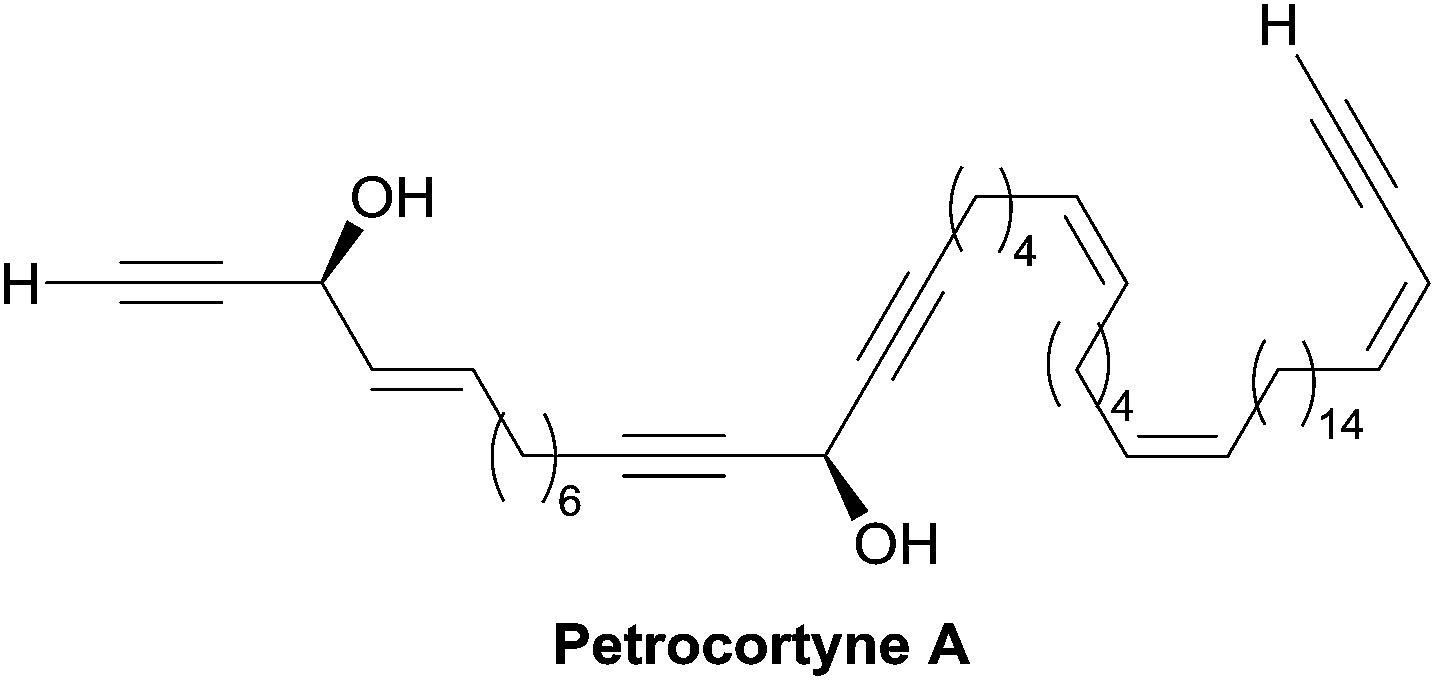 | ||
| Fig. 12 Structure of petrocortyne A. | ||
Curran's approach aimed at the four possible epimers at C-3 and C-14 of petrocortyne A using the sophisticated fluorous mixture synthesis. According to this “split and mix” strategy, the four individual stereoisomers were targeted through a late demixing, by fluorous HPLC of so-called “quasiisomers”, i.e. stereoisomers encoded by different fluorous tags. Curran's synthetic route made use of the CBS reduction described earlier for the preparation of the (R)- and (S)-(4E)-en-1-yn-3-ol motif at C-3. On the other hand, after an unproductive attempt using Carreira's procedure,31 the efficient general procedure developed by Pu for the asymmetric addition of terminal alkynes to aldehydes was applied to the enantioselective synthesis of the C-14 DAC unit of petrocortyne A.32,75 In Pu's method, a zinc acetylide species is first generated using dialkylzinc, as in Trost's and Zhong and Guo's methods (see Section 2.2.2, Scheme 10), but, in this case, the aldehyde is then reacted in the presence of a combination of enantiopure BINOL and Ti(Oi-Pr)4, with the use of (S)-BINOL yielding (R)-propargylic alcohols (Scheme 35). This system proved flexible, working equally well for the addition of phenylacetylene (or trialkylsilylacetylene) to aromatic, α,β-unsaturated, or aliphatic aldehydes. This procedure was also applied in the enantioselective preparation of γ-hydroxy-α,β-acetylenic esters with methyl propiolate,76 and, in addition, several ligand optimizations were reported.77
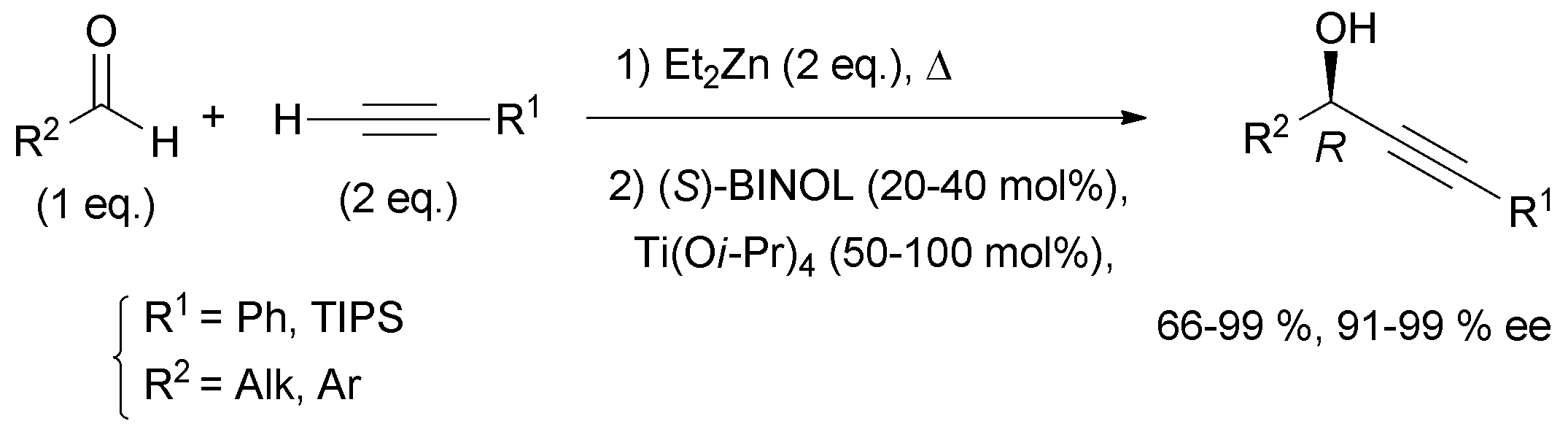 | ||
| Scheme 35 Pu's method for asymmetric addition of alkynes to aldehydes. | ||
Curran's disconnection led to three key segments (Scheme 36, where only the (3S,14S)-isomer is shown): a western C-1–C-11 segment, 48, embedding the terminal (4E)-en-1-yn-3-ol motif, a C-12–C-21 segment, 49, bearing the DAC unit, and an eastern C-22–C-46 segment, 50.
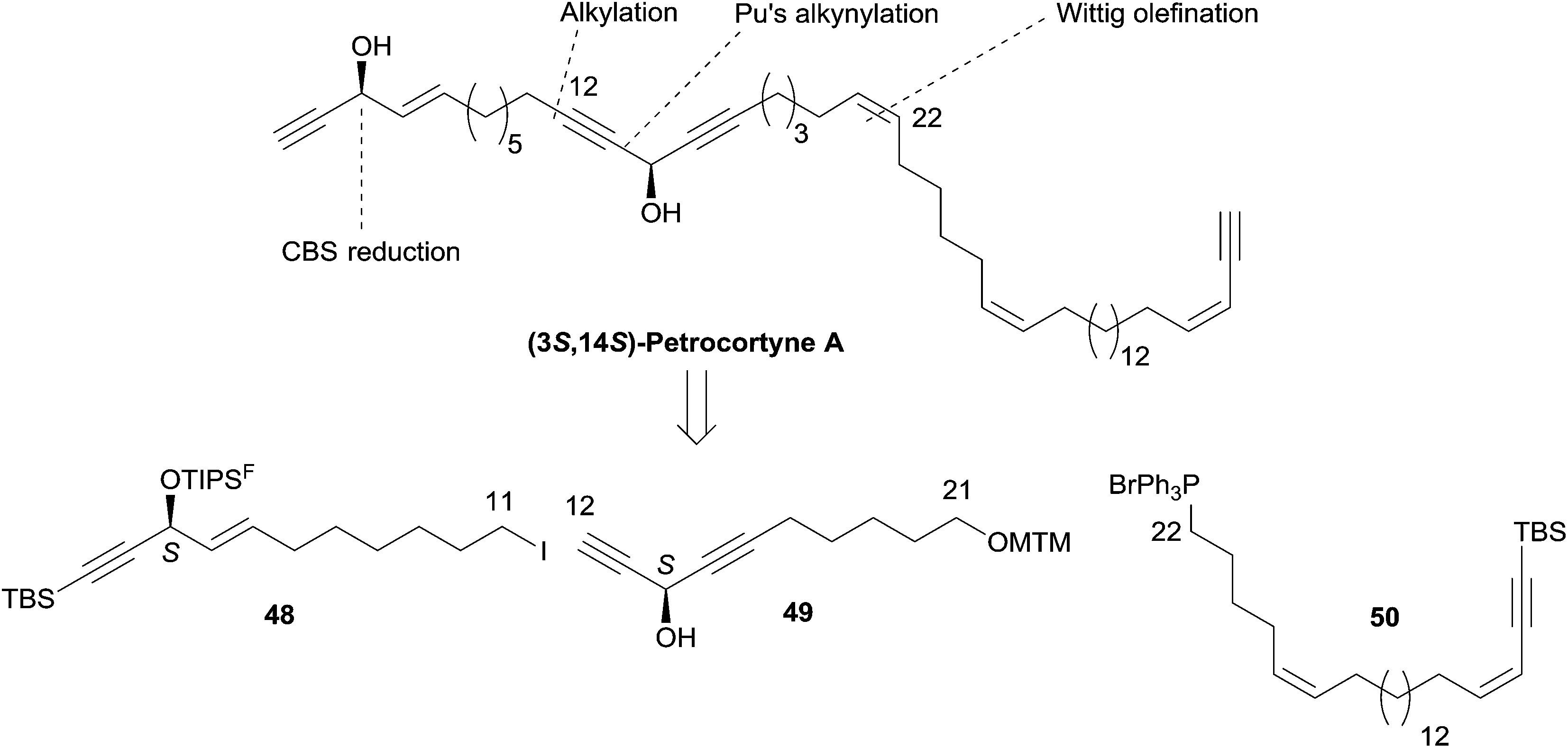 | ||
| Scheme 36 Curran's retrosynthesis of petrocortyne A. | ||
The C-12–C-21 segment was targeted using Pu's methodology (Scheme 37).74b For the sake of a preliminary NMR study, the DACs 52a (R = PMB) were first secured in 83% ee by addition of dimethylphenylacetylene to the respective aldehydes 51a. This procedure was also applied to several other alkynes which were reacted with oct-2-ynal, thus delivering the expected DACs in ee’s ranging from 78 to 90%. Although chiral preparative HPLC resolution of racemic 52b (R = MTM) was ultimately preferred to produce highly enantio-enriched samples of (S)- and (R)-49 (Scheme 37), these results demonstrated the unique relevance of Pu's procedure for the preparation of chiral DACs.
The preparation of the western C-1–C-11 segment 48 is depicted in Scheme 38. The individual asymmetric synthesis and tagging of each enantiomeric AAC unit with a distinct fluorinated trialkylsilyl group led to two quasienantiomers which were then mixed to generate the quasiracemate qrac-48.
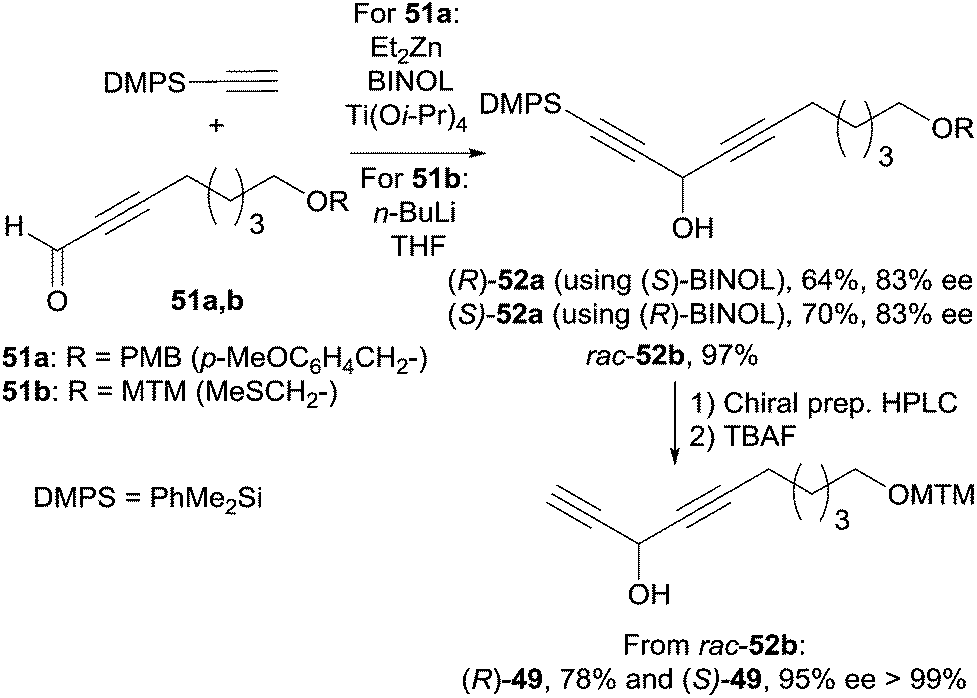 | ||
| Scheme 37 Model study using Pu's asymmetric addition of alkynes to aldehydes for the synthesis the C-12–C-21 segment of petrocortyne A. | ||
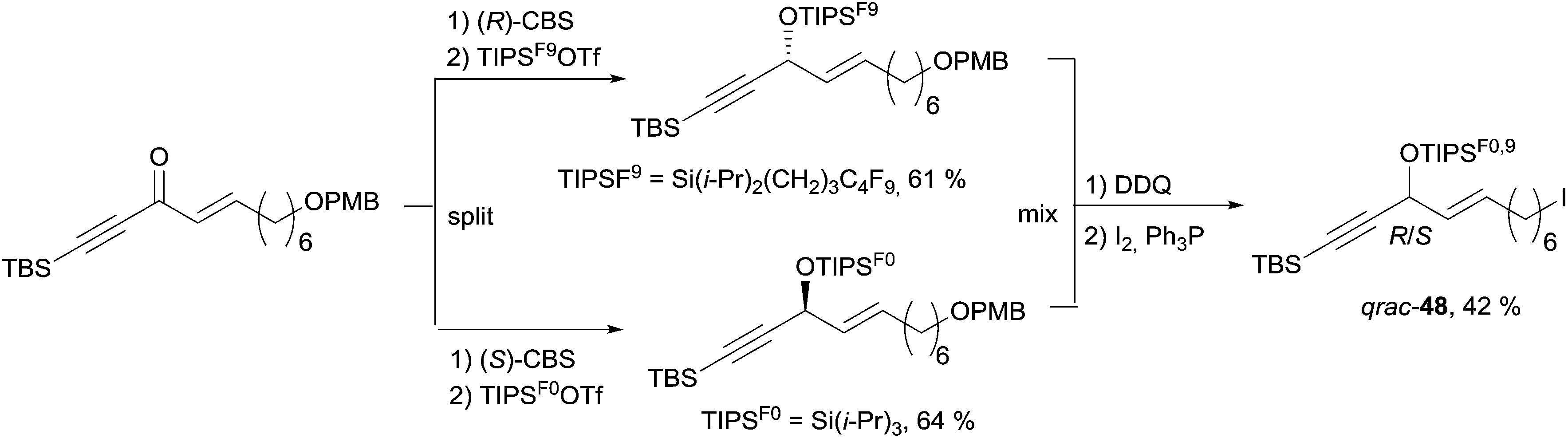 | ||
| Scheme 38 Curran's preparation of the C-1–C-11 segment of petrocortyne A. | ||
After treatment with two equivalents of n-BuLi, each enantiomer of DAC 49 was individually alkylated with the mixture qrac-48 (Scheme 39). The resulting intermediates were differentially tagged by silylation of the secondary carbinol centers with two differently fluorinated TIPS residues. Subsequent MTM cleavage and oxidation of the mixed quasistereoisomeric alcohols then led to a quasidiastereoisomeric mixture of the aldehydes qdim-53.
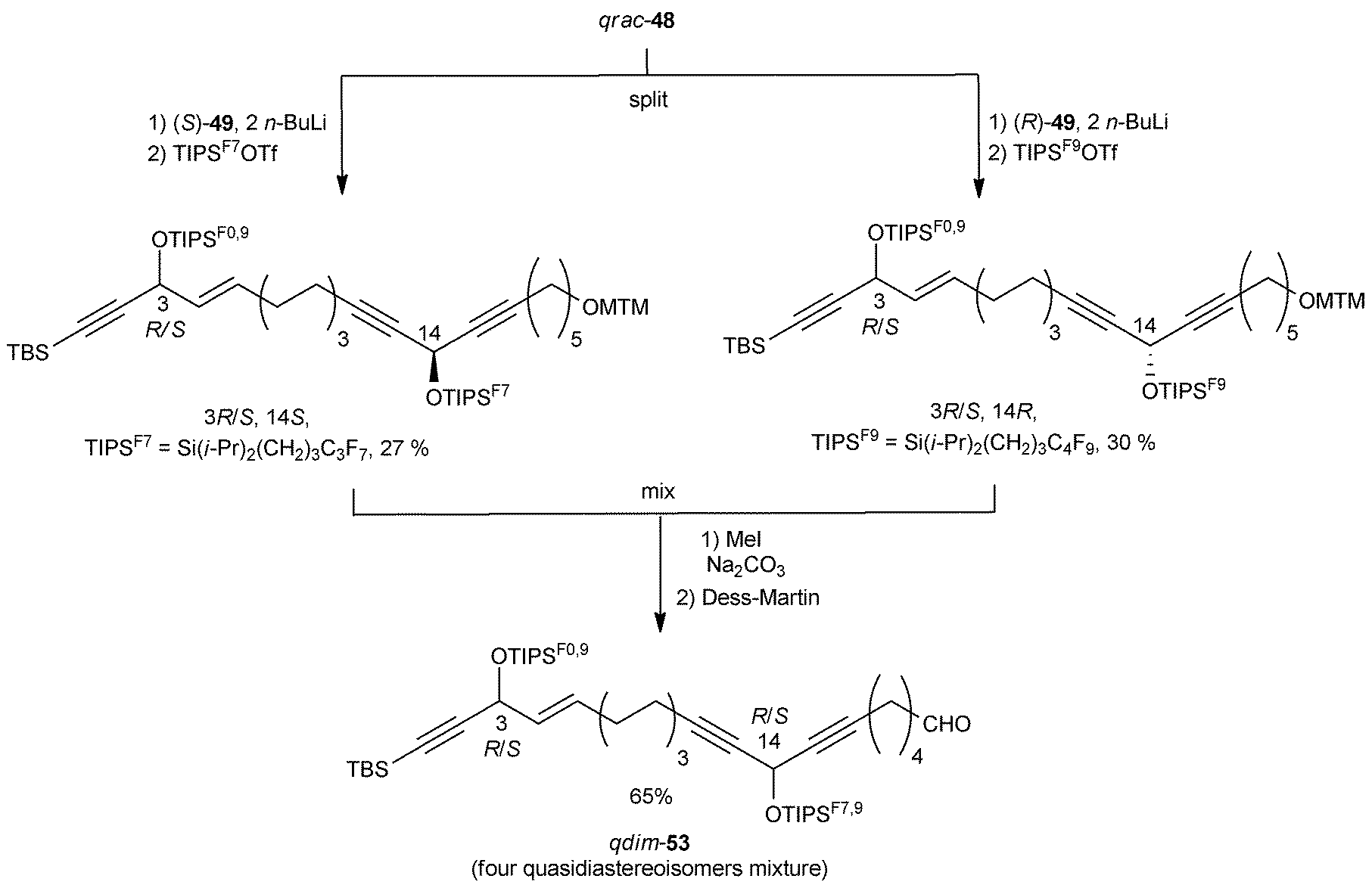 | ||
| Scheme 39 Curran's preparation of the C-1–C-21 fragment of petrocortyne A. | ||
Final assembly of the petrocortyne A skeleton was accomplished by Wittig olefination of the mixture of aldehydes, qdim-53, with phosphonium 50, thus delivering qdim-54 which was demixed by semipreparative fluorous HPLC (Scheme 40). Subsequent individual deprotection with TBAF led to the four pure stereoisomers of petrocortyne A.
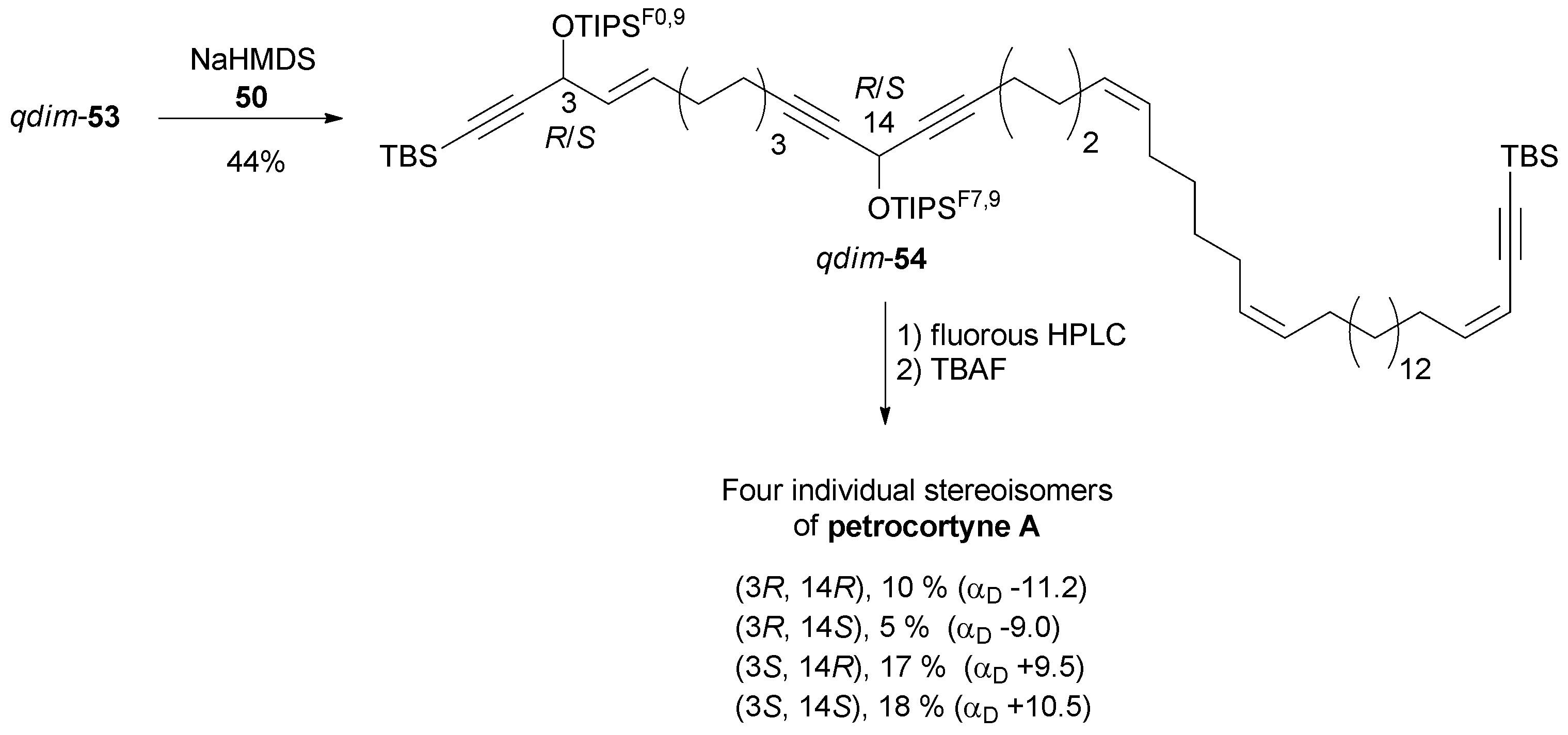 | ||
| Scheme 40 Curran's final assembly of the four diastereoisomers of petrocortyne A. | ||
NMR spectra did not allow these four isomers to be distinguished. The value of αD could be used to assign the configuration at C-3 (the 3R,14R/3R,14S pair being dextrorotatory, and the 3S,14R/3S,14S pair laevorotatory), the absolute configuration of the remote stereocenter at C-14 having a negligible contribution to the global optical rotation of 54 (Scheme 40). Curran based the final structure assignment on the NMR analysis of the four individual Mosher derivatives. Not only did the advanced Mosher rule prove applicable to the assignment of the C-14 configuration, but also a single set of bis-Mosher ester NMR spectra was found to match those reported by both Shin and Jung. These observations unambiguously demonstrated that the configuration of the natural product isolated by Shin's and Jung's groups was (3S,14S).
3 Biological relevance of marine polyacetylenes
Marine polyacetylenic polyols were reported to display a wide range of biological activities. Most of these properties were reported along with the isolation of the natural product itself. For example, petrocortynes were shown to be cytotoxic against human solid tumor66b,c,d and leukemia cell lines,66a and to inhibit DNA replication66c,d and phospholipase A2 activity.66a Petrocortyne A was also the subject of further studies on DNA replication inhibition,78 and anti-inflammatory and pro-aggregative effects.79 The pro-apoptotic behavior of the moderately cytotoxic petrotetrayndiol A, isolated along with petrocortyne A,65 was also demonstrated.80 A moderate cytotoxicity and inhibitory potency against ATPase and reverse transcriptase was recorded for the related osirisynes.5 On the other hand, fulvyne was described as an antimicrobial agent against a chloramphenicol-resistant strain of Bacillus subtilis.7Among the C2-symmetrical derivatives, adociacetylenes and petrosynol exhibit moderate inhibition of the neutrophil leukocyte adhesion to tumor necrosis factor-α endothelial cells and of the HIV reverse transcriptase.60,81 In contrast, a strong cytotoxicity against several tumor cell lines was found for the related C46 unsaturated derivative (3S,44S)-(−)-fulvinol44 (Fig. 13), and both enantiomers of durynes display cytotoxic activities with IC50 values in the submicromolar range.45
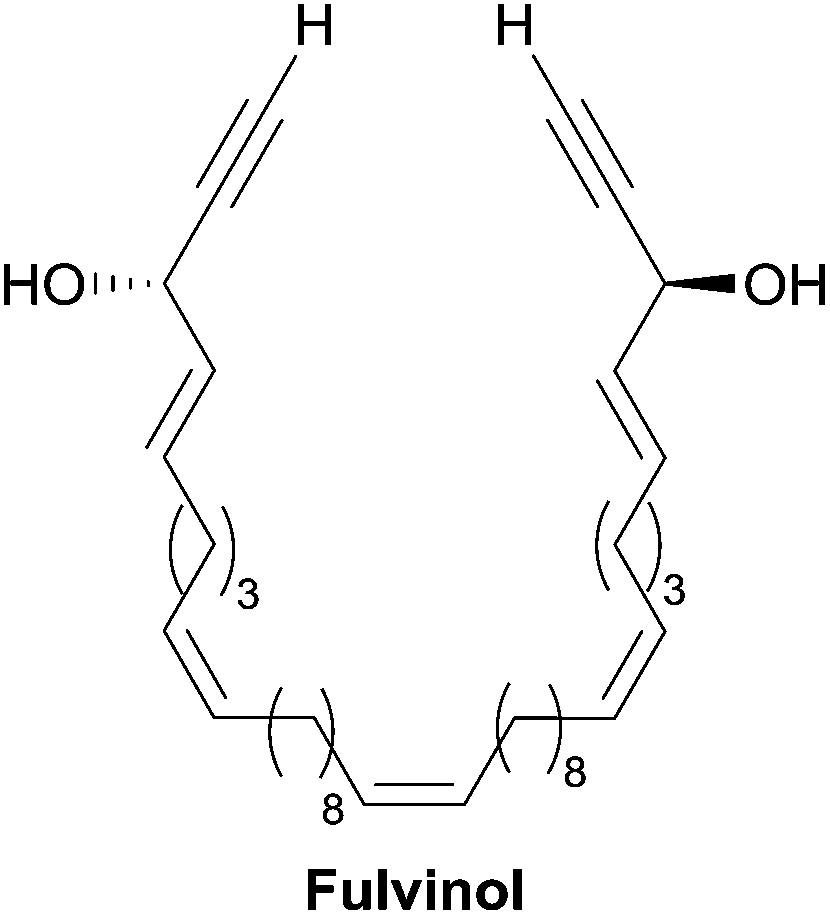 | ||
| Fig. 13 Structure of fulvinol. | ||
Dideoxypetrosynols are the most potent, and also the most studied, petrosiacetylenes, likely also because of their “simple” C2-symmetrical structure. In particular, dideoxypetrosynol A was reported to exhibit a significant brine shrimp toxicity,56 RNA cleaving activity56 and a potent cytotoxicity against human cancer cell lines.15 Extensive studies on the pro-apoptotic activity of dideoxypetrosynol A revealed various antiproliferative mechanisms,82 such as the inhibition of DNA replication,78 inhibition of cyclooxygenase-2 and telomerase,83 induction of the cyclin-dependant kinase inhibitor p16, and the down-regulation of the retinoblastoma protein phosphorylation.84 Rational attempts to bring out structure–activity relationships for these polacetylenes remain scarce, mainly because of the lack of systematic studies leading to homogenous series of data. On a qualitative level, however, the bioactivity of such metabolites does not appear directly related to their structural complexity. For example, “simpler” C2-symmetrical representatives display a larger panel of effects, including potent cytotoxicity, than the petrocortynes. For example, a 70 times higher cytotoxicity against SK-MEL-2 cells was reported by Jung and colleagues for dideoxypetrosynol A15 compared to petrocortyne A.65 Furthermore, within the C2-symmetrical C30 series, the gradual simplification of the central core, from those of adociacetylene (tetrahydrofuran, Fig. 10) or petrosynol (carbinol-skipped vic-ene-diyne, Fig. 6) to those of dideoxypetrosynol (methylene-skipped vic-ene-diyne, Fig. 9) and duryne (simple alkene, Fig. 8), is also associated with an increase of cytotoxicity. More generally, this trend raises the question of the influence of the central structural unit on cytotoxicity. A C20 analogue of the durynes with a saturated central region was isolated from the marine sponge Callyspongia pseudoreticulata,85 and its cytotoxicity against TR-LE cells was very recently described in a detailed study allowing identification of a trend in the structure–activity relationship (Fig. 14).86
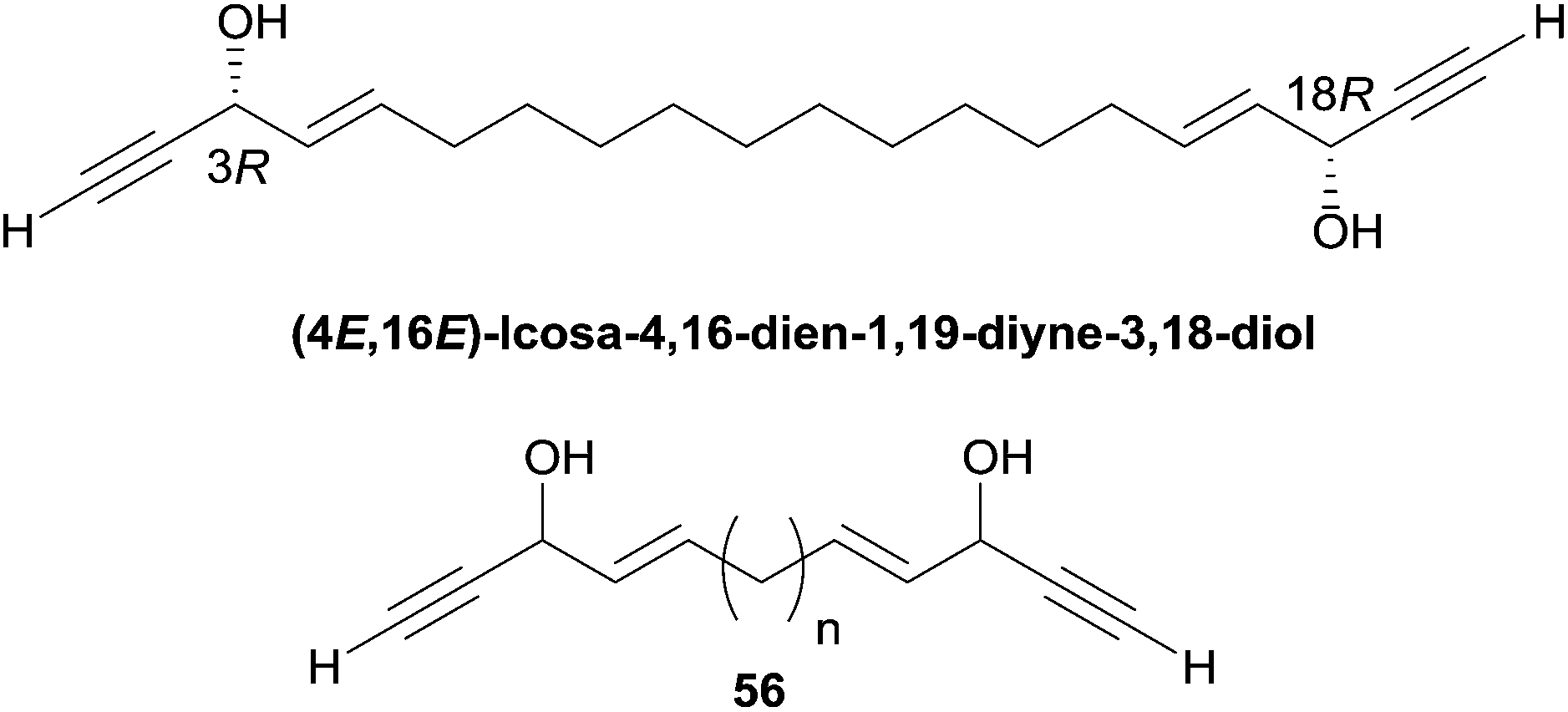 | ||
| Fig. 14 Structures of (4E,16E)-icosa-4,16-dien-1,19-diyne-3,18-diol and synthetic analogues. | ||
Both enantiomers displayed a submicromolar cytotoxicity in the same range as (+)- or (−)-duryne. Furthermore, in the racemic series, a direct influence of the aliphatic spacer between the two AAC moieties was observed. While the natural C20 product (56, n = 10, Fig. 14) displayed an IC50 of 0.6 μM, the shorter C16 analogue (56, n = 6, Fig. 14) was 16 times less potent (IC50 9.4 μM) and the longer C24 homologue (56, n = 14, Fig. 14) proved 3 times more active (IC50 0.17 μM). These observations suggest that the nature of the central fragment of the C2-symmetrical double-headed polyacetylenic polyols has a moderate influence on their cytotoxicity, while the overall length of their backbone has a more sizable effect.
A related issue is the identification of a “minimal” pharmacophoric unit responsible for the cytotoxicity in the structure of polyacetylenic polyols. The potent cytotoxicity of the plain chain eicos-(4E)-en-1-yn-3-ol and of its homologues provides some indication in this regard.43,87 Indeed, the chain-terminal AAC fragment is ubiquitous not only in all the cytotoxic C2-symmetrical marine polyacetylenic polyols, but also in other bio-active natural products such as the pelynols (Fig. 15).88 As noticed by Molinski et al.,89 the lack of potency of the corresponding enynones, named halicynones, indicates the crucial role of the (4E)-en-1-yn-3-ol terminal fragment. A parallel behavior was recently recorded in the course of a study on the naturally occurring C20 saturated analogue of duryne (Fig. 14), where the replacement of one or both of the (4E)-en-1-yn-3-ol units by their corresponding enynones was correlated with a drop in activity.86
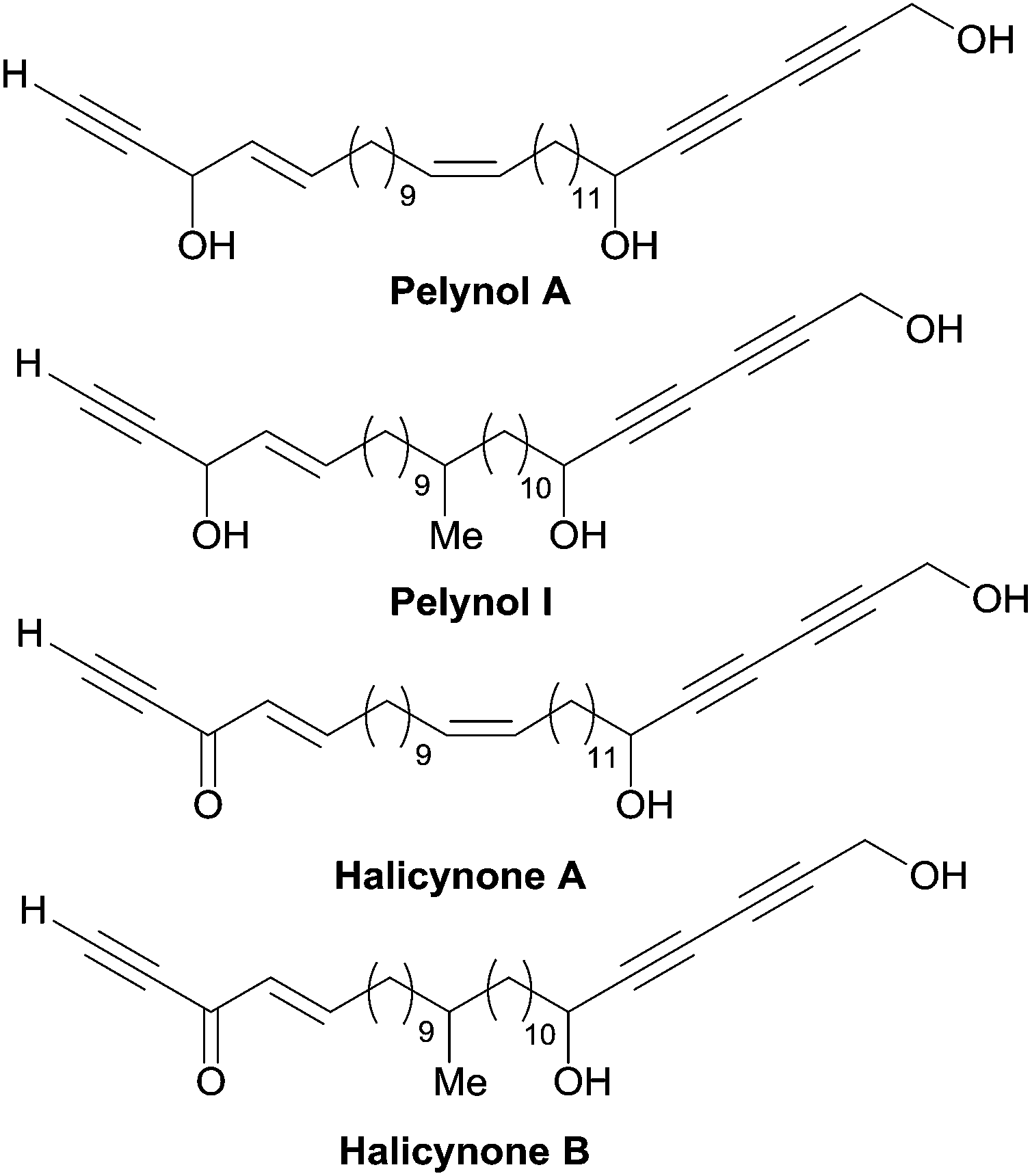 | ||
| Fig. 15 Structures of pelynols and halicynones. | ||
Several simple alkynylcarbinols (substituted propargylic alcohols without an adjacent alkene fragment) were shown to possess comparable or higher cytotoxicity than their AAC analogues, thus raising the question of the requirement of the alkenyl substituent for anti-proliferative activity.90 In this regard, lembehynes, embedding a characteristic α-alkylated ethynylcarbinol fragment (Section 2.1.2, Scheme 2), were shown to induce a bipolar neuritogenesis of Neuro2A cells (by specific blocking of the cell cycle at the G1 phase), and to exhibit a pronounced influence of the carbinol configuration on the cell differentiation activity (the (R)-enantiomer being the most active).14 LB-18, a synthetic analogue of lembehyne A, was finally shown to induce cell death, but, unlike dideoxypetrosynol A, following a non-apoptotic caspase-independent pathway (Section 2.1.2, Scheme 5).18 These data would thus support the relevance of the (4E)-en-1-yn-3-ol AAC motif as a minimal pharmacophoric unit for pro-apoptotic properties.
4 Conclusions
The long-reported pharmacological relevance of natural polyacetylenic polyols, most of them extracted from marine sponges,4 has only recently evoked total synthesis efforts, calling for the optimization of the enantioselective synthesis tools. The chemical complexity of these natural products, even within the sub-family of C2-symmetrical derivatives, remains, however, challenging for future medicinal developments. Beyond the identification of “minimal pharmacophoric units”, key issues remain unexplored, such as (i) the determination of the eutomeric configuration of the carbinol center and (ii) the optimization of the length and functionality of the lipidic backbone. Advances in this direction have just been achieved through a systematic four-parameter pharmacophoric study (Scheme 41, top), based on a specific structural analysis of the sectors A, B and C (Fig. 2, Scheme 41).91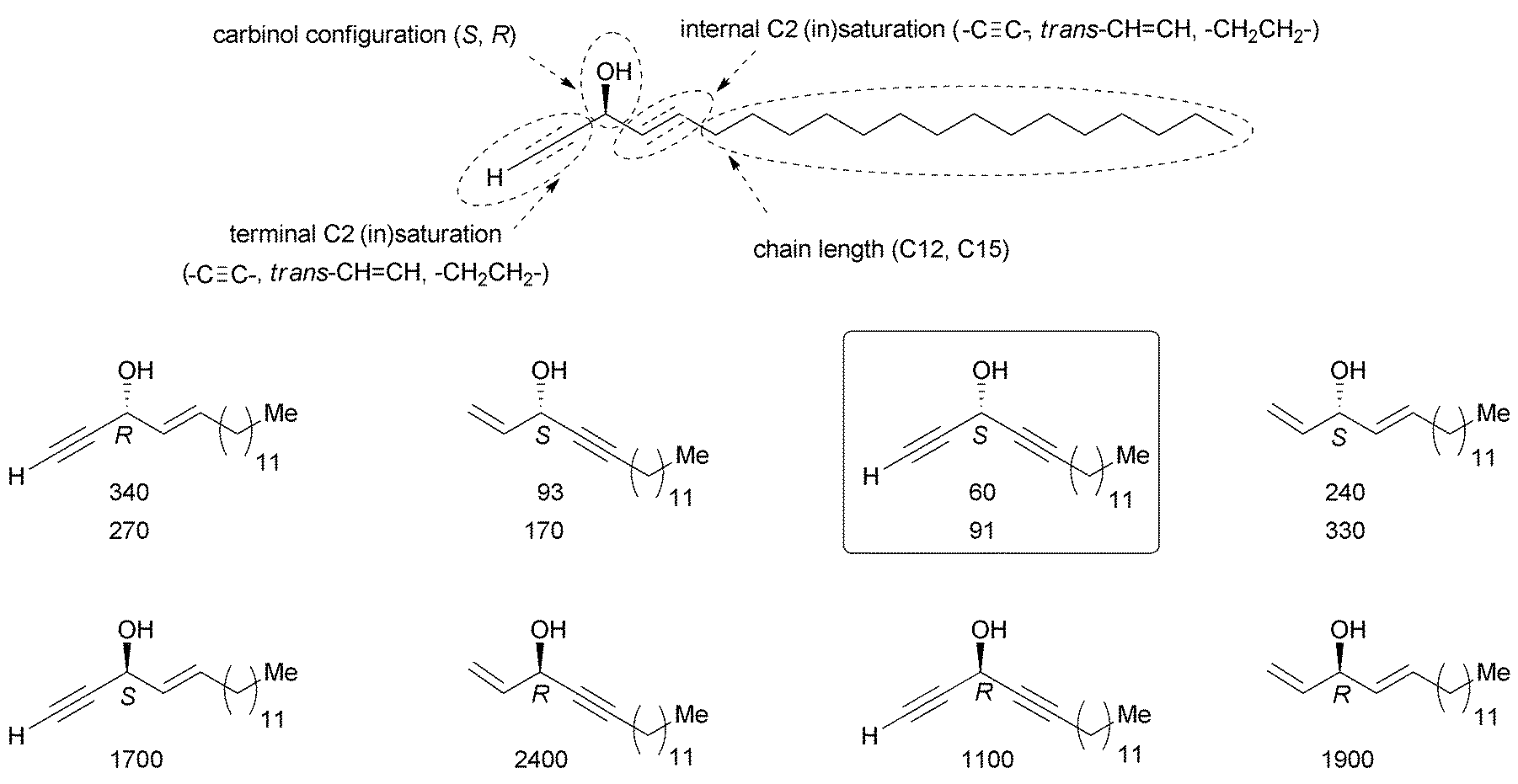 | ||
| Scheme 41 Systematic four-parameter pharmacophore analysis based on a variation of the structure of (S)-eicos-(4E)-en-1-yn-3-ol (23) (top), and selected data for the cytotoxicity towards HCT 116 cancer cells highlighting two structure–activity relationships (middle & bottom): (i) the influence of the carbinol CIP configuration (vertical entries), and (ii) the internal/terminal unsaturation levels (horizontal entries). IC50 values in nM; for eutomers (middle), two sets of data, obtained from different samples of HCT 116 and with different treatment times, are given. | ||
In line with the natural (S)-eicos-(4E)-en-1-yn-3-ol (23) (Section 3.3.2, Fig. 7, Schemes 19, 21, 23 and 24), the results show that simple synthetic molecules, containing a single AAC or DAC pharmacophoric unit and a fully saturated linear lipidic substituent, exhibit high cytotoxicity on cancer cell lines, like HCT116 colorectal adenocarcinoma cells, with IC50 values as low as 60 nM (Scheme 41, bottom). It was also demonstrated that two unsaturations adjacent to the carbinol center are required for considerable activity, and that the natural chiral configuration (terminal triple bond + internal double bond corresponding to that of (S)-eicos-(4E)-en-1-yn-3-ol (23)) is less potent that the opposite one (terminal double bond + internal triple bond). Moreover, the most potent pharmacophore is shown to be a terminal DAC unit (terminal and internal triple bonds), which has not been exemplified among natural products hitherto. Finally, a constant effect, by ca. one order of magnitude, of the carbinol stereochemical configuration was evidenced (“biological chiral effect”): the eutomer, in terms of cytotoxicity, is systematically assigned to the “pseudo-S” configuration, where the priority is formally given to the lipidic chain over the C2 substituent, whatever the actual unsaturations are. This chiral effect is also consistent with several reports on other cytotoxic AACs.90
These results thus pave the way for further synthetic chemistry-driven investigations into alkynylcarbinol derivatives as potential anti-tumor compounds. The optimization of the cytotoxic activity of chiral DAC or AAC derivatives may in particular be achieved by fine-tuning of their lipophilic environment (i.e. lipidic chain length, branching, unsaturation level, position(s), and configurations (Z vs. E)) and by comparison between single- and double-headed representatives (aiming at optimal cell uptake and intracellular localization). Efficient synthetic tools to create large molecular libraries should also facilitate the search for optimal bio-disponibility (“chemical galenic”): along this line, the recent four-parameter study (Scheme 41) has evidenced a dramatic effect of the chain length (the elongation of the lipidic chain from C12 to C15 indeed induces a decrease in cyctotoxicity on HCT 116 cells by ca. one order of magnitude while retaining a “pseudo-S” eutomeric configuration).91 Likewise, the properties of tertiary alkynylcarbinol derivatives (R3 = alkyl, aryl, alkynyl in sector C, Fig. 2) should also be examined.
In parallel, continuing the collection of new natural products,7 possibly from novel sponge genera and/or species, will extend the family of the lipidic α-functional alkynylcarbinols for further investigations of their biological properties. The mode of action of these molecules, in particular the mechanism of their cytotoxicity, remains poorly explored, and related investigations are also desired. Reported data somehow converge towards nucleic acid-related activities such as inhibition of the Na+/K+ ATPase,5,56 DNA replication,66,78 telomerase83 and of the reverse transcriptase,5 or RNA cleaving ability.56 Although an in vitro enzymatic inhibitory effect on DNA polymerases has been argued,78 direct chemical-damaging interactions with DNA/RNA nucleotides can also be envisaged on the basis of the structural analogy between the α-unsaturated alkynylcarbinols and the so-called “ene–diyne antibiotics” (Scheme 42).3 The AAC motif can indeed be identified (in duplicate) in the strained cyclic pharmacophore of calicheamicin, and the same motif has been theoretically shown to be prone to undergo Schreiner cyclization, namely the Bergman-like diradical cyclo-aromatization.92 The value of the pharmacophore concept, which has been deeply rationalized in recent years,93 is further illustrated by the examples of the AAC and DAC units.92 Similar systematic studies based on the same approach can be anticipated in the near future.
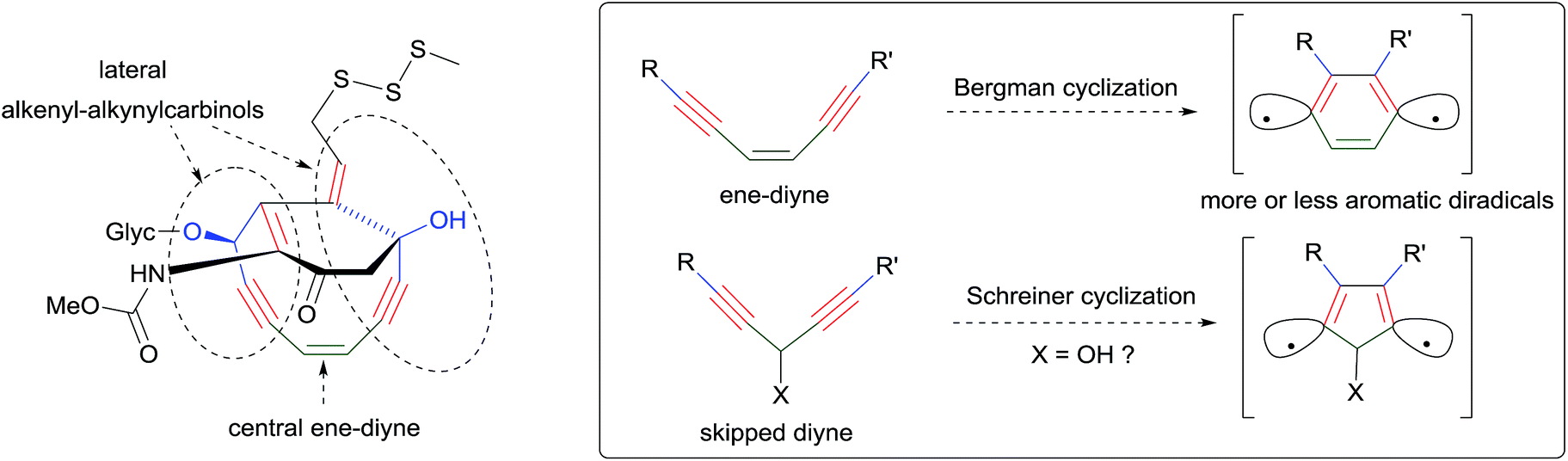 | ||
| Scheme 42 Relations of α-functional carbinols with ene–diyne anticancer molecules: identification of AAC pharmacophoric units in calicheamicin (left), and theoretical parallel between Bergman's and Schreiner's cycloaromatization processes (right). | ||
5 Acknowledgements
D. L. was supported by the French Embassy in Kiev, Ukraine. The investigations having been performed within the framework of the GDRI (Groupement Franco-Ukrainien en Chimie Moléculaire) funded by the CNRS. The French Embassy in Kiev, and the CNRS are thus sincerely acknowledged.6 Notes and references
- H. Hofmeister, K. Annen, H. Laurent, K. Petzoldt and R. Wiechert, Arzneim. Forsch., 1986, 36, 781–783 CAS.
- A. S. Thompson, E. G. Corley, M. F. Huntington and E. J. J. Grabowski, Tetrahedron Lett., 1995, 36, 8937–8940 CrossRef.
- (a) K. C. Nicolaou and W. M. Dai, Angew. Chem., Int. Ed., 1991, 30, 1387–1416 CrossRef; (b) G. A. Ellestad, Chirality, 2011, 23, 660–671 CrossRef CAS PubMed.
- (a) V. M. Dembitsky, Lipids, 2006, 41, 883–924 CrossRef CAS PubMed; (b) A. Siddiq and V. Dembitsky, Anticancer Agents Med. Chem., 2008, 8, 132–170 CrossRef CAS.
- (a) J. H. Shin, Y. W. Seo, K. W. Cho, J. R. Rho and V. J. Paul, Tetrahedron, 1998, 54, 8711–8720 CrossRef CAS; (b) J. Shin, Y. See, K. W. Cho, J. R. Rho and V. J. Paul, Tetrahedron, 1998, 54, 14636 CrossRef CAS.
- L. Chill, A. Miroz and Y. Kashman, J. Nat. Prod., 2000, 63, 523–526 CrossRef CAS PubMed.
- G. Nuzzo, M. L. Ciavatta, G. Villani, E. Manzo, A. Zanfardino, M. Varcamonti and M. Gavagnin, Tetrahedron, 2012, 68, 754–760 CrossRef CAS PubMed.
- For a precedent review on total synthesis of polyyne natural products in general, see: B. W. Gung, C. R. Chim., 2009, 12, 489–505 CrossRef CAS PubMed.
- A. Ito, B. L. Cui, D. Chavez, H. B. Chai, Y. G. Shin, K. Kawanishi, L. B. S. Kardono, S. Riswan, N. R. Farnsworth, G. A. Cordell, J. M. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 2001, 64, 246–248 CrossRef CAS.
- M. K. Gurjar, V. S. Kumar and B. V. Rao, Tetrahedron, 1999, 55, 12563–12576 CrossRef CAS.
- K. Ishimaru, M. Osabe, L. Yan, T. Fujioka, K. Mihashi and N. Tanaka, Phytochemistry, 2003, 62, 643–646 CrossRef CAS.
- R. E. Minto and B. J. Blacklock, Prog. Lipid Res., 2008, 47, 233–306 CrossRef CAS PubMed.
- S. Aoki, K. Matsui, K. Tanaka, R. Satari and M. Kobayashi, Tetrahedron, 2000, 56, 9945–9948 CrossRef CAS.
- (a) S. Aoki, K. Matsui, H. Wei, N. Murakami and M. Kobayashi, Tetrahedron, 2002, 58, 5417–5422 CrossRef CAS; (b) K. A. Parker and M. W. Ledeboer, J. Org. Chem., 1996, 61, 3214–3217 CrossRef CAS; (c) E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925–7926 CrossRef CAS.
- J. S. Kim, K. S. Im, J. H. Jung, Y. L. Kim, J. Kim, C. J. Shim and C. O. Lee, Tetrahedron, 1998, 54, 3151–3158 CrossRef CAS.
- N. Murakami, T. Nakajima and M. Kobayashi, Tetrahedron Lett., 2001, 42, 1941–1943 CrossRef CAS.
- (a) H. C. Brown and G. G. Pai, J. Org. Chem., 1985, 50, 1384–1394 CrossRef CAS; (b) M. M. Midland, Chem. Rev., 1989, 89, 1553–1561 CrossRef CAS.
- M. Izumi, S. Yogosawa, S. Aoki, H. Watanabe, J. Kamiyama, Y. Takahara, Y. Sowa, M. Kobayashi, H. Hosoi, T. Sugimoto and T. Sakai, Int. J. Oncol., 2006, 29, 169–173 CAS.
- (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (b) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS; (c) K. J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1997, 36, 285–288 CrossRef CAS.
- K. Watanabe, Y. Tsuda, Y. Yamane, H. Takahashi, K. Iguchi, H. Naoki, T. Fujita and R. W. M. Van Soest, Tetrahedron Lett., 2000, 41, 9271–9276 CrossRef CAS.
- A. L. K. S. Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef CAS PubMed.
- J. S. Yadav, P. K. Deshpande and G. V. M. Sharma, Tetrahedron, 1990, 46, 7033–7046 CrossRef CAS.
- J. Chun, H. S. Byun and R. Bittman, Tetrahedron Lett., 2002, 43, 8043–8045 CrossRef CAS.
- J. S. Yadav and R. K. Mishra, Tetrahedron Lett., 2002, 43, 1739–1741 CrossRef CAS.
- For major publications, see: (a) D. Boyall, F. Lopez, H. Sasaki, D. Frantz and E. M. Carreira, Org. Lett., 2000, 2, 4233–4236 CrossRef CAS PubMed; (b) N. K. Anand and E. M. Carreira, J. Am. Chem. Soc., 2001, 123, 9687–9688 CrossRef CAS; (c) D. Boyall, D. E. Frantz and E. M. Carreira, Org. Lett., 2002, 4, 2605–2606 CrossRef CAS PubMed.
- For the initial reports, see: (a) D. E. Frantz, R. Fassler and E. M. Carreira, J. Am. Chem. Soc., 2000, 122, 1806–1807 CrossRef CAS; (b) D. E. Frantz, R. Fassler, C. S. Tomooka and E. M. Carreira, Acc. Chem. Res., 2000, 33, 373–381 CrossRef CAS PubMed.
- R. Noyori and M. Kitamura, Angew. Chem., Int. Ed., 1991, 30, 49–69 CrossRef.
- For a detailed discussion on the substrate sensitivity of Carreira's method, see: J. E. D. Kirkham, T. D. L. Courtney, V. Lee and J. E. Baldwin, Tetrahedron, 2005, 61, 7219–7232 CrossRef CAS PubMed and references cited therein.
- B. W. Gung, H. D. Dickson, S. Seggerson and K. Bluhm, Synth. Commun., 2002, 32, 2733–2740 CrossRef CAS PubMed.
- B. W. Gung, H. Dickson and S. Shockley, Tetrahedron Lett., 2001, 42, 4761–4763 CrossRef CAS.
- B. Sui, Fluorous mixture synthesis and structure assignment of petrocortyne A and its stereoisomers, Ph.D. thesis, University of Pittsburg, 2009.
- (a) D. Moore and L. Pu, Org. Lett., 2002, 4, 1855–1857 CrossRef CAS PubMed; (b) G. Gao, D. Moore, R. G. Xie and L. Pu, Org. Lett., 2002, 4, 4143–4146 CrossRef CAS PubMed.
- B. M. Trost, A. H. Weiss and A. Jacobi von Wangelin, J. Am. Chem. Soc., 2006, 128, 8–9 CrossRef CAS PubMed.
- Z. Y. Li, M. Wang, Q. H. Bian, B. Zheng, J. Y. Mao, S. N. Li, S. Z. Liu, M. A. Wang, J. C. Zhong and H. C. Guo, Chem.–Eur. J., 2011, 17, 5782–5786 CrossRef CAS PubMed.
- S. Reber, T. F. Knopfel and E. M. Carreira, Tetrahedron, 2003, 59, 6813–6817 CrossRef CAS.
- V. Maraval and R. Chauvin, New J. Chem., 2007, 31, 1853–1873 RSC.
- E. R. Graham and R. R. Tykwinski, J. Org. Chem., 2011, 76, 6574–6583 CrossRef CAS PubMed.
- J. E. D. Kirkham, T. D. L. Courtney, V. Lee and J. E. Baldwin, Tetrahedron Lett., 2004, 45, 5645–5647 CrossRef CAS PubMed.
- B. Jiang, Z. L. Chen and W. N. Xiong, Chem. Commun., 2002, 1524–1525 RSC.
- B. M. Trost, M. J. Bartlett, A. H. Weiss, A. Jacobi von Wangelin and V. S. Chan, Chem.–Eur. J., 2012, 18, 16498–16509 CrossRef CAS PubMed.
- B. M. Trost, V. S. Chan and D. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5186–5192 CrossRef CAS PubMed.
- B. Zheng, S. N. Li, J. Y. Mao, B. Wang, Q. H. Bian, S. Z. Liu, J. C. Zhong, H. C. Guo and M. Wang, Chem.–Eur. J., 2012, 18, 9208–9211 CrossRef CAS PubMed.
- Y. F. Hallock, J. H. Cardellina, M. S. Balaschak, M. R. Alexander, T. R. Prather, R. H. Shoemaker and M. R. Boyd, J. Nat. Prod., 1995, 58, 1801–1807 CrossRef CAS.
- M. J. Ortega, E. Zubia, J. L. Carballo and J. Salva, J. Nat. Prod., 1996, 59, 1069–1071 CrossRef CAS.
- (a) A. E. Wright, O. J. Mcconnell, S. Kohmoto, M. S. Lui, W. Thompson and K. M. Snader, Tetrahedron Lett., 1987, 28, 1377–1379 CrossRef CAS; (b) Y. Hitora, K. Takada, S. Okada, Y. Ise and S. Matsunaga, J. Nat. Prod., 2011, 74, 1262–1267 CrossRef CAS PubMed.
- (a) Y. Hitora, K. Takada, S. Okada and S. Matsunaga, Tetrahedron, 2011, 67, 4530–4534 CrossRef CAS PubMed; (b) Y. Hitora, K. Takada and S. Matsunaga, Tetrahedron, 2013, 69, 11070–11073 CrossRef CAS PubMed; (c) K. Mori, K. Akasaka and S. Matsunaga, Tetrahedron, 2014, 70, 392–401 CrossRef CAS PubMed.
- S. P. Gunasekera and G. T. Faircloth, J. Org. Chem., 1990, 55, 6223–6225 CrossRef CAS.
- A. Aiello, E. Fattorusso, M. Menna and M. Pansini, J. Nat. Prod., 1992, 55, 1275–1280 CrossRef CAS.
- A. Sharma and S. Chattopadhyay, Tetrahedron: Asymmetry, 1998, 9, 2635–2639 CrossRef CAS.
- A. Sharma and S. Chattopadhyay, J. Org. Chem., 1998, 63, 6128–6131 CrossRef CAS PubMed.
- K. Morishita, M. Kamezawa, T. Ohtani, H. Tachibana, M. Kawase, M. Kishimoto and Y. Naoshima, J. Chem. Soc., Perkin Trans. 1, 1999, 513–518 RSC.
- J. Garcia, M. Lopez and J. Romeu, Tetrahedron: Asymmetry, 1999, 10, 2617–2626 CrossRef CAS.
- W. Lu, G. R. Zheng and J. C. Cai, Tetrahedron, 1999, 55, 4649–4654 CrossRef CAS.
- G. Kumaraswamy, K. Sadaiah and N. Raghu, Tetrahedron: Asymmetry, 2012, 23, 587–593 CrossRef CAS PubMed.
- B. W. Gung and A. O. Omollo, J. Org. Chem., 2008, 73, 1067–1070 CrossRef CAS PubMed.
- For isolation of both petrosyacetylene (didepoxypetrosynol) and petrocortyne A, see: Y. Seo, K. W. Cho, J. R. Rho, J. Shin and C. J. Sim, Tetrahedron, 1998, 54, 447–462 CrossRef CAS.
- (a) N. Fusetani, Y. Kato, S. Matsunaga and K. Hashimoto, Tetrahedron Lett., 1983, 24, 2771–2774 CrossRef CAS; (b) N. Fusetani, T. Shiragaki, S. Matsunaga and K. Hashimoto, Tetrahedron Lett., 1987, 28, 4313–4314 CrossRef CAS.
- R. Gleiter and R. Merger, Tetrahedron Lett., 1990, 31, 1845–1848 CrossRef CAS.
- B. W. Gung and A. O. Omallo, Eur. J. Org. Chem., 2008, 4790–4795 CrossRef CAS PubMed.
- M. Kobayashi, T. Mahmud, H. Tajima, W. Q. Wang, S. Aoki, S. Nakagawa, T. Mayumi and I. Kitagawa, Chem. Pharm. Bull., 1996, 44, 720–724 CrossRef CAS.
- T. Higa, J. Tanaka, A. Kitamura, T. Koyama, M. Takahashi and T. Uchida, Pure Appl. Chem., 1994, 66, 2227–2230 CrossRef CAS.
- J. Garcia, M. Lopez and J. Romeu, Synlett, 1999, 429–431 CrossRef CAS PubMed.
- K. Burgess and L. D. Jennings, J. Am. Chem. Soc., 1990, 112, 7434–7436 CrossRef CAS.
- B. M. Trost and A. H. Weiss, Org. Lett., 2006, 8, 4461–4464 CrossRef CAS PubMed.
- For isolation of petrocortyne A, see: J. S. Kim, Y. J. Lim, K. S. Im, J. H. Jung, C. J. Shim, C. O. Lee, J. Hong and H. Lee, J. Nat. Prod., 1999, 62, 554–559 CrossRef CAS PubMed.
- For isolation of other petrocortynes or related products such as petrotetrayndiol or norpetrocortyne, see: (a) J. Shin, Y. Seo and K. W. Cho, J. Nat. Prod., 1998, 61, 1268–1273 CrossRef CAS PubMed; (b) Y. J. Lim, J. S. Kim, K. S. Im, J. H. Jung, C. O. Lee, J. Hong and D. Kim, J. Nat. Prod., 1999, 62, 1215–1217 CrossRef CAS PubMed; (c) Y. J. Lim, H. S. Park, K. S. Im, C. O. Lee, J. Hong, M. Y. Lee, D. K. Kim and J. H. Jung, J. Nat. Prod., 2001, 64, 46–53 CrossRef CAS PubMed; (d) Y. J. Lim, C. O. Lee, J. Hong, D. K. Kim, K. S. Im and J. H. Jung, J. Nat. Prod., 2001, 64, 1565–1567 CrossRef CAS PubMed.
- C. Tedeschi, C. Saccavini, L. Maurette, M. Soleilhavoup and R. Chauvin, J. Organomet. Chem., 2003, 670, 151–169 CrossRef CAS.
- (a) L. Maurette, C. Tedeschi, E. Sermot, M. Soleilhavoup, F. Hussain, B. Donnadieu and R. Chauvin, Tetrahedron, 2004, 60, 10077–10098 CrossRef CAS PubMed; (b) C. Saccavini, C. Tedeschi, L. Maurette, C. Sui-Seng, C. Zou, M. Soleilhavoup, L. Vendier and R. Chauvin, Chem.–Eur. J., 2007, 13, 4895–4913 CrossRef CAS PubMed; (c) C. H. Zou, C. Duhayon, V. Maraval and R. Chauvin, Angew. Chem., Int. Ed., 2007, 46, 4337–4341 CrossRef CAS PubMed.
- A. Rives, V. Maraval, N. Saffon-Merceron and R. Chauvin, Chem.–Eur. J., 2012, 18, 14702–14707 CrossRef CAS PubMed; A. Rives, V. Maraval, N. Saffon-Merceron and R. Chauvin, Chem.–Eur. J., 2014, 20, 483–492 CrossRef PubMed.
- (a) V. Maraval and R. Chauvin, Chem. Rev., 2006, 106, 5317–5343 CrossRef CAS PubMed; (b) L. Leroyer, V. Maraval and R. Chauvin, Chem. Rev., 2012, 112, 1310–1343 CrossRef CAS PubMed.
- M. Oukessou, Y. Genisson, D. El Arfaoui, A. Ben-Tama, E. El Hadrami and R. Chauvin, Tetrahedron Lett., 2013, 54, 4362–4364 CrossRef CAS PubMed.
- V. Convertino, P. Manini, W. B. Schweizer and F. Diederich, Org. Biomol. Chem., 2006, 4, 1206–1208 CAS.
- M. Nechab and N. Vanthuyne, Org. Lett., 2012, 14, 3974–3977 CrossRef CAS PubMed.
- (a) D. P. Curran and B. Sui, J. Am. Chem. Soc., 2009, 131, 5411–5413 CrossRef CAS PubMed; (b) B. Sui, E. A. H. Yeh and D. P. Curran, J. Org. Chem., 2010, 75, 2942–2954 CrossRef CAS PubMed.
- For a recent review of the synthetic utility of this transformation, see: Q. Wang and L. Pu, Synlett, 2013, 24, 1340–1363 CrossRef CAS PubMed.
- G. Gao, Q. Wang, X. Q. Yu, R. G. Xie and L. Pu, Angew. Chem., Int. Ed., 2006, 45, 122–125 CrossRef CAS PubMed.
- (a) M. H. Xu and L. Pu, Org. Lett., 2002, 4, 4555–4557 CrossRef CAS PubMed; (b) M. Turlington, A. M. DeBerardinis and L. Pu, Org. Lett., 2009, 11, 2441–2444 CrossRef CAS PubMed.
- D. K. Kim, M. Y. Lee, H. S. Lee, D. S. Lee, J. R. Lee, B. J. Lee and J. H. Jung, Cancer Lett., 2002, 185, 95–101 CrossRef CAS.
- S. Y. Hong, S. H. Kim, M. H. Rhee, A. R. Kim, J. H. Jung, T. Chun, E. S. Yoo and J. Y. Cho, Naunyn-Schmiedeberg's Arch. Pharmacol., 2003, 368, 448–456 CrossRef CAS PubMed.
- H. J. Choi, S. B. Yee, S. E. Park, E. Im, J. H. Jung, H. Y. Chung, Y. H. Choi and N. D. Kim, Cancer Lett., 2006, 232, 214–225 CrossRef CAS PubMed.
- S. Isaacs, Y. Kashman, S. Loya, A. Hizi and Y. Loya, Tetrahedron, 1993, 49, 10435–10438 CrossRef CAS.
- H. J. Choi, S. J. Bae, N. D. Kim, J. H. Jung and Y. H. Choi, Int. J. Mol. Med., 2004, 14, 1091–1096 CAS.
- C. Park, J. H. Jung, N. D. Kim and Y. H. Choi, Int. J. Oncol., 2007, 30, 291–298 CAS.
- C. Park, G. Y. Kim, G. D. Kim, W. H. Lee, J. H. Cheong, N. D. Kim, S. J. Bae, J. H. Jung and Y. H. Choi, Oncol. Rep., 2006, 16, 171–176 CAS.
- J. C. Braekman, D. Daloze, C. Devijver, D. Dubut and R. W. M. van Soest, J. Nat. Prod., 2003, 66, 871–872 CrossRef CAS PubMed.
- T. Shirouzu, K. Watari, M. Ono, K. Koizumi, I. Saiki, C. Tanaka, R. W. M. van Soest and T. Miyamoto, J. Nat. Prod., 2013, 76, 1337–1342 CrossRef CAS PubMed.
- H. S. Lee, J. H. Lee, H. Won, S. K. Park, H. M. Kim, H. J. Shin, H. S. Park, C. J. Sim and H. K. Kim, Lipids, 2009, 44, 71–75 CrossRef CAS PubMed.
- (a) X. Fu, S. A. Abbas, F. J. Schmitz, I. Vidavsky, M. L. Gross, M. Laney, R. C. Schatzman and R. D. Cabuslay, Tetrahedron, 1997, 53, 799–814 CrossRef CAS; (b) M. A. Rashid, K. R. Gustafson and M. R. Boyd, Nat. Prod. Lett., 2000, 14, 387–392 CrossRef CAS.
- G.-X. Zhou and T. F. Molinski, Mar. Drugs, 2003, 1, 46–53 CrossRef CAS PubMed.
- (a) Regarding panaxytriol, see: T. C. Chou, H. J. Dong, X. G. Zhang, X. G. Lei, J. Hartung, Y. D. Zhang, J. H. Lee, R. M. Wilson and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14336–14341 CrossRef CAS PubMed; (b) Regarding a C2-symmetrical C20 derivative, see ref. 86.
- D. El Arfaoui, D. Listunov, I. Fabing, M. Oukessou, C. Frongia, V. Lobjois, A. Samson, F. Ausseil, A. Ben-Tama, E. El Hadrami, R. Chauvin and Y. Génisson, ChemMedChem, 2013, 8, 1779–1786 CrossRef CAS PubMed.
- S. P. Kawatkar and P. R. Schreiner, Org. Lett., 2002, 4, 3643–3646 CrossRef CAS PubMed.
- See for example: (a) Pharmacophores and Pharmacophore Searches, ed. T. Langer and R. D. Hoffmann, Wiley, Weinheim, 2006 Search PubMed; (b) B. Meunier, Acc. Chem. Res., 2008, 41, 69–77 CrossRef CAS PubMed; (c) S.-Y. Yang, Drug Discovery Today, 2010, 15, 444–450 CrossRef CAS PubMed.
Footnote |
| † This article is dedicated to Dr Bernard Meunier, especially in honor and recognition of his decisive achievements in the field of pharmacophore analysis. |
| This journal is © The Royal Society of Chemistry 2015 |