
Graphitic carbon nitride based nanocomposites: a review
Zaiwang
Zhao
,
Yanjuan
Sun
and
Fan
Dong
*
Chongqing Key Laboratory of Catalysis and Functional Organic Molecules, College of Environmental and Biological Engineering, Chongqing Technology and Business University, Chongqing, 400067, China. E-mail: dfctbu@126.com; Fax: +86-23-62769785-605; Tel: +86-23-62769785-605
First published on 10th September 2014
Abstract
Graphitic carbon nitride (g-C3N4), as an intriguing earth-abundant visible light photocatalyst, possesses a unique two-dimensional structure, excellent chemical stability and tunable electronic structure. Pure g-C3N4 suffers from rapid recombination of photo-generated electron–hole pairs resulting in low photocatalytic activity. Because of the unique electronic structure, the g-C3N4 could act as an eminent candidate for coupling with various functional materials to enhance the performance. According to the discrepancies in the photocatalytic mechanism and process, six primary systems of g-C3N4-based nanocomposites can be classified and summarized: namely, the g-C3N4 based metal-free heterojunction, the g-C3N4/single metal oxide (metal sulfide) heterojunction, g-C3N4/composite oxide, the g-C3N4/halide heterojunction, g-C3N4/noble metal heterostructures, and the g-C3N4 based complex system. Apart from the depiction of the fabrication methods, heterojunction structure and multifunctional application of the g-C3N4-based nanocomposites, we emphasize and elaborate on the underlying mechanisms in the photocatalytic activity enhancement of g-C3N4-based nanocomposites. The unique functions of the p–n junction (semiconductor/semiconductor heterostructures), the Schottky junction (metal/semiconductor heterostructures), the surface plasmon resonance (SPR) effect, photosensitization, superconductivity, etc. are utilized in the photocatalytic processes. Furthermore, the enhanced performance of g-C3N4-based nanocomposites has been widely employed in environmental and energetic applications such as photocatalytic degradation of pollutants, photocatalytic hydrogen generation, carbon dioxide reduction, disinfection, and supercapacitors. This critical review ends with a summary and some perspectives on the challenges and new directions in exploring g-C3N4-based advanced nanomaterials.
 Zaiwang Zhao | Zaiwang Zhao obtained his BS degree from Chongqing Technology and Business University (CTBU) in 2012. He is currently a graduate student under the supervision of Prof. Fan Dong at CTBU. His research interests are focused on the design and synthesis of photocatalytic materials for energy and environmental applications. |
 Yanjuan Sun | Yanjuan Sun received her BS in Environmental Engineering and MS in Chemical Technology from Hubei University of Technology. In 2011, she became an Assistant Professor at Chongqing Technology and Business University. Her research interests include photocatalytic materials for environmental and energetic applications, and separation technology. |
 Fan Dong | Fan Dong received his PhD in Environmental Engineering in 2010 from Zhejiang University. Currently, he is a Professor at Chongqing Key Laboratory of Catalysis and Functional Organic Molecules, College of Environmental and Biological Engineering, Chongqing Technology and Business University. He was a visiting scholar from 2009 to 2010 at Hong Kong Polytechnic University. His research interests include semiconductor and plasmonic photocatalysis, CO2 capture and utilization, and air pollution control. He has coauthored more than 50 papers on the topics of his research and has an H index of 20. |
1. Introduction
Over the past few decades, the growing awareness of environmental protection and energy conservation has stimulated intensive research on solar energy utilization.1 In the domain of pollutant elimination and solar energy conversion, semiconductor photocatalysis has emerged as one of the most fascinating technologies.2–5 Nevertheless, the wide band gap and the low solar-energy utilization efficiency remain the “bottleneck” of the photocatalysts to satisfy the requirements of applications in a practical way. For instance, the traditional TiO2 is limited owing to its poor performances associated with visible light application. As a result, it is necessary to seek efficient visible-light-driven (VLD) photocatalysts. For this objective, various modified TiO2 and TiO2-alternative photocatalysts have been fabricated.6–17 Currently, it is still a challenge to design new photocatalysts that are abundant, stable and facile in fabrication besides showing high visible-light performance.In the search for robust and stable VLD semiconductor photocatalysts, a polymeric semiconductor, graphitic carbon nitride (g-C3N4), has recently attracted tremendous attention. The heptazine ring structure and the high condensation degree enable metal-free g-C3N4 to possess many advantages such as good physicochemical stability, as well as an appealing electronic structure combined with a medium band gap (2.7 eV).18 These unique properties make g-C3N4 a promising candidate for visible light photocatalytic applications utilizing solar energy. In addition, g-C3N4 is abundant and easily-prepared via one-step polymerization of cheap feedstocks like cyanamide,18,19 urea,20–22 thiourea,23,24 melamine25–27 and dicyandiamide.28,29 Nevertheless, pure g-C3N4 suffers from shortcomings such as rapid recombination of photo-generated electron–hole pairs, a small specific surface area and a low visible light utilization efficiency.21–29 Consequently, the exploration of facile and dependable strategies to synthesize the modified g-C3N4-based photocatalysts with improved physicochemical properties and high photocatalytic activities is of increasing requirement. The g-C3N4 has a unique two-dimensional layered structure, which is favorable for hybridizing with other components. Very recently, several approaches have been employed to enhance the visible light photocatalytic performance of g-C3N4, for example, formation of surface coupling hybridization utilizing TaON,30 Bi2WO6,31 or graphene,32 construction of mesoporous structures,33 doping with metal or nonmetal species Fe,34 Ag,35 Au,36 Pd,37 S,38 B39 and P40 and sensitizing with organic dyes.41 Among these approaches, formation of heterostructures demonstrates a great potential to promote the photocatalytic performance of g-C3N4 because the electron–hole pairs can be efficiently separated, and charge carriers could transfer across the interface of the heterostructure to restrain the recombination.
In a coupling process, g-C3N4 based heterostructures not only can be formed by combining with visible light excited photocatalytic semiconductor materials with a narrow band gap (such as CdS,42 Bi2WO3,43 and BiOI44), but also can combine with UV excited photocatalysts with large band gaps (such as TiO2,45 ZnO,46 and ZnWO447), which can largely broaden the application of the g-C3N4 based nanocomposites. Nevertheless, not all materials can couple with g-C3N4 to form a heterostructure. The most important prerequisite condition to form an effective visible light excited g-C3N4 based heterostructure is that the candidates should have an appropriate band structure which is beneficial to create a coupling hybridization. Besides, the difference in chemical potential between the coupling semiconductors A and B generates band bending at the interface of junctions. The band bending induces a built-in field, which impels the photogenerated electrons and holes to transfer in opposite directions, resulting in a spatially efficient separation of the electrons and holes pairs on different sides of the heterojunction.48,49 In addition, the crystal structure in the junction domain of the heterostructure is also important in strengthening the quantum efficiency of the photocatalyst. A distinction in lattice spacing between two semiconductors could probably cause lattice mismatch. The lattice mismatch at the interface may cause defects, which will capture the photo-generated electronic carriers and thus inhibit the diffusion of electrons and holes. Thus, the formation of g-C3N4 based heterostructures is an effective approach to enhance charge separation efficiency for an improved photocatalytic performance.
Recently, numerous research studies have been carried out to couple g-C3N4 with various semiconductors to enhance the photocatalytic activities.36,50,51 For instance, Wang and co-workers firstly reported TiN/g-C3N4 multi-layer hybridization using a dual-facing-target magnetron sputtering method at room temperature and exhibited enhanced properties in photocatalytic applications in 2008.50 This pioneering work has stimulated tremendous interest in the fabrication, modification, and application of g-C3N4-based semiconductor photocatalysts. Yan et al. successfully developed a g-C3N4/TaON organic–inorganic composite photocatalyst with visible-light response by a milling-heat treatment method that demonstrated an enhanced photocatalytic performance for photodegradation of rhodamine B (RhB) in aqueous solutions.51 Di et al. prepared the Au/g-C3N4 nanocomposite photocatalyst by depositing gold nanoparticles on the surface of a g-C3N4 semiconductor to generate metal–semiconductor junctions, which showed well-improved photocatalytic hydrogen evolution with visible light.36
Now that significant advances have been made on g-C3N4-based photocatalysts in recent years, we believe that a comprehensive review on this subject is necessary to accelerate further developments in this exciting research domain. This review article is focused on recent progress in the design, fabrication, mechanistic understanding, and potential applications of these g-C3N4-based nanocomposites in various realms such as photodegradation of nitrogen oxides, organic contaminant photodegradation, photocatalytic hydrogen evolution, conversion of carbon dioxide to methane fuel, oxygen reduction reaction (ORR), and photoelectrochemical determination of Cu2+. Eventually, some concluding remarks and invigorating perspectives on the current situation and further prospects of the g-C3N4-related research studies are presented, which may promote the understanding and large scale application of g-C3N4-based nanocomposites.
2. g-C3N4-based nanocomposites
2.1. Preparation of g-C3N4
Generally, carbon nitride materials fabricated by direct condensation of nitrogen-containing organic precursors (for example, urea, thiourea, melamine, dicyandiamide, cyanamide, and guanidine hydrochloride) are bulk materials with a small surface area, normally below 10 m2 g−1. For practical applications of the materials in domains such as a single catalyst or a support substrate of co-catalysts (such as heterojunctions), the introduction of well-controlled porosity at the nanoscale in the bulk carbon nitride is mandatory to strengthen its utilization to a large scale. It is worth noting that formation of mesoporous structure and augmentation of specific surface area are crucial to fine-tune the physicochemical properties and improve the photocatalytic performance.52 As previously reported, the mesoporous g-C3N4 (mpg-C3N4) was first obtained by nanocasting/replication of mesoporous silica matrices, which were famous for the generation of the corresponding carbon nanostructures.53–55 Inspired by this hard template method, tremendous attempts were made to explore new strategies for g-C3N4 modification, such as the soft template method,56–58 the ultrasonic dispersion method,59 acidic solution impregnation60,61 and chemical functionalization.62–66 As universally applied, the methods mentioned above were good as a proof-of-principle in tailoring the texture and surface chemical properties, as well as the electronic properties.2.2. Design considerations for g-C3N4-based nanocomposites
To develop effective g-C3N4-based nanocomposites working for enhanced performance, several pivotal requirements must be considered. First of all, the semiconductor light harvesting antenna must have a narrow band gap to allow for efficient absorption of the solar spectrum. Second, there must be a driving force boosting charge separation and accelerating the transportation process. Furthermore, the semiconductor should possess adequate redox potential for the desired photochemical reactions. Ultimately, a mechanism should be accompanied to guarantee the photochemical stability of the photocatalyst. It is unlikely that a single material system could satisfy all these requirements, while the nanocomposite photocatalysts may have the potential to accomplish these goals. In general, nanocomposite photocatalysts could offer several potential merits: (1) cocatalyst effect: the integration with a proper cocatalyst can lower the redox overpotential at the respective active sites; (2) strengthened light absorption: for instance, semiconductors with small band gaps possessing a high absorption coefficiency can be utilized to functionalize (or sensitize) semiconductor materials with large band gaps; (3) efficient charge separation and transportation: a p–n junction (semiconductor/semiconductor heterostructures) or the Schottky junction (metal/semiconductor heterostructures) with built-in electrical potential can be constructed in heterogeneous photocatalysts to effectively expedite electron–hole pair separation and transportation; (4) decent stability: if the active sites and functional groups on the semiconductor surface can be protected through appropriate surface passivation, it is possible to improve the stability.In consideration of the diverse mechanisms of different g-C3N4 based nanocomposites in photocatalytic reactions, g-C3N4 based heterojunctions were summarized and classified into six different main combination systems. Below, we review the recent progress on the g-C3N4 based metal-free heterojunction, the g-C3N4/metal oxide (metal sulfide) heterojunction, g-C3N4/inorganic acid salt composites, the g-C3N4/halide heterojunction, g-C3N4/noble metal heterostructures, and the g-C3N4 based multi-component heterojunction for improved photocatalysis.
3. Categories of g-C3N4 based nanocomposites
3.1. g-C3N4 based metal-free layered heterojunctions
To our knowledge, metal-free materials, owing to their various advantages such as low cost and environmentally amicable, have attracted multitudinous attention because of their great potential in solving environmental and energy problems. Hence, it is extremely attractive to construct hybridized materials via coupling two metal-free materials. For example, graphene, a two-dimensional macromolecular sheet of carbon atoms, has preferentially attracted much attention due to its outstanding mechanical, thermal, easily-obtained, and electrical properties. It has been widely used in nanoelectronics, biosensing, capacitors, and catalysis domains.67,68 In particular, it possesses an extremely high specific surface area (∼2600 m2 g−1)69 and marvelous thermal conductivity (∼5000 W m−1 K−1), guaranteeing a superb mobility of charge carriers (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1). Therefore, owing to the merits mentioned above, graphene is the first metal-free candidate, and is preferentially used to construct layered graphene/g-C3N4 nanocomposites. Sun et al. prepared graphene/g-C3N4 nanoheterojunctions by the absorption–calculation method, which demonstrated apparently enhanced conductivity and electrocatalytic performance on oxygen reduction reaction (ORR).70 Xiang et al. also successfully obtained graphene/g-C3N4 nanocomposites by a combined impregnation–chemical reduction strategy, and firstly utilized this graphene/g-C3N4 nanoheterojunction in visible-light photocatalytic H2-production containing Pt as a cocatalyst.32Fig. 1a and 1b show a typical TEM image of graphene oxide and graphene/g-C3N4 nanocomposites. In comparison with graphene oxide, the graphene/g-C3N4 composite possessed a more compact structure due to the fact that g-C3N4 was sandwiched between graphene sheets through polymerization of melamine molecules pre-adsorbed on the GO sheets. In the aforementioned trials, high H2-production activity over the graphene/g-C3N4 nanocomposites under visible-light irradiation can be achieved. The mechanism of the activity enhancement is illustrated in Fig. 1c.
000 cm2 V−1 s−1). Therefore, owing to the merits mentioned above, graphene is the first metal-free candidate, and is preferentially used to construct layered graphene/g-C3N4 nanocomposites. Sun et al. prepared graphene/g-C3N4 nanoheterojunctions by the absorption–calculation method, which demonstrated apparently enhanced conductivity and electrocatalytic performance on oxygen reduction reaction (ORR).70 Xiang et al. also successfully obtained graphene/g-C3N4 nanocomposites by a combined impregnation–chemical reduction strategy, and firstly utilized this graphene/g-C3N4 nanoheterojunction in visible-light photocatalytic H2-production containing Pt as a cocatalyst.32Fig. 1a and 1b show a typical TEM image of graphene oxide and graphene/g-C3N4 nanocomposites. In comparison with graphene oxide, the graphene/g-C3N4 composite possessed a more compact structure due to the fact that g-C3N4 was sandwiched between graphene sheets through polymerization of melamine molecules pre-adsorbed on the GO sheets. In the aforementioned trials, high H2-production activity over the graphene/g-C3N4 nanocomposites under visible-light irradiation can be achieved. The mechanism of the activity enhancement is illustrated in Fig. 1c.
 | ||
| Fig. 1 (a) TEM images of graphene oxide and (b) the graphene/g-C3N4 nanocomposites; (c) proposed mechanism for the enhanced electron transfer in the graphene/g-C3N4 composites. (Reprinted with permission from ref. 32. Copyright (2011) American Chemical Society.) | ||
Under visible-light illumination, electrons (e−) of g-C3N4 are excited from the valence band (VB) to the conduction band (CB), generating holes (h+) in the VB. Generally, these charge carriers quickly recombine and only a section of electrons are injected into the Pt nanoparticles due to a Schottky barrier.71,72 The injected electrons accumulate on the Pt nanoparticles and can effectively reduce H2O (or H+) to produce H2, while holes accumulate at the valence band of g-C3N4 and can react with methanol as a sacrificial reagent. However, when g-C3N4 is immobilized on the surface of graphene sheets to form the layered composites, these photogenerated electrons on the CB of g-C3N4 could tend to transfer to graphene sheets due to their excellent electronic conductivity, restraining the electron–hole pair recombination.68 The transferred electrons will accumulate on the Pt nanoparticles loaded on the graphene sheets via a percolation mechanism73 and then participate in H2 generation. The major reaction steps in this photocatalytic water-splitting mechanism under visible-light illumination are summarized in eqn (1)–(4).
 | (1) |
| graphene (e−) + Pt→ graphene + Pt (e−) | (2) |
| Pt (e−) + 2H+ → Pt + H2 | (3) |
| g-C3N4 (h+) + CH3OH + 6OH− → g-C3N4 + CO2 + 5H2O | (4) |
Cross-linked g-C3N4/rGO (reduced graphene oxide) nanocomposites with tunable band structure were synthesized by Li et al. which demonstrated well-enhanced visible light photocatalytic activity.74 Later, Wang et al. noticed that a new class of ternary g-C3N4/graphene/S nanoheterojunctions was prepared by wrapping reduced graphene oxide and g-C3N4 sheets on crystals of cyclooctasulfur (α-S8) in bacterial inactivation under visible light.75 Except for the aforementioned achievement, a large number of attempts have been made to explore novel materials coupling with g-C3N4 for broadening the applications of g-C3N4-based coupling heterojunctions. In summary, these excellent g-C3N4 based non-metal nanoheterojunctions are MWNTs (multi-walled carbon nanotubes)/g-C3N4,76,77 polypyrrole/C3N4,78 P/C3N4,79 CN/CNS,80 g-C3N4(from thiourea)/g-C3N4(from urea),81 and C60/g-C3N4, as shown in Table 1.82
| Composite photocatalyst | Mass or molar fraction of g-C3N4 | Typical parameters of photocatalytic experiments | Photocatalytic activity | Reference photocatalyst; photocatalytic activity | Enhancement factor over the reference photocatalyst | Reference |
|---|---|---|---|---|---|---|
| Graphene/g-C3N4 | Mass: 99% | Photocatalytic H2 evolution under visible light; co-catalyst: Pt; sacrificial reagent: methanol | RH2: 451 μmol h−1 g−1 | g-C3N4: 147 μmol h−1 g−1 | 3.07 | 32 |
| Graphene: no data | 1.21 | |||||
| Reduced graphene oxide (rGO)/g-C3N4 | Mass: 98.4% | Decomposing rhodamine B (RhB) under visible light | 84% in 75 min | g-C3N4: 80% | 1.05 | 74 |
| 97.5% | 100% in 75 min | rGO: no data | 1.25 | |||
| 94.9% | 52% in 75 min | 0.65 | ||||
| 80.4% | 36% in 75 min | 0.45 | ||||
| Mass: 98.4% | Decomposing 4-nitrophenol under visible light | No data in 150 min | g-C3N4: no data | No data | ||
| 97.5% | No data in 150 min | rGO: no data | No data | |||
| 94.9% | No data in 150 min | |||||
| 80.4% | No data in 150 min | |||||
| Multi-walled carbon nanotubes (MWNTs)/g-C3N4 | Mass: 98% | Photocatalytic H2 evolution under visible light; co-catalyst: Pt; sacrificial reagent: methanol | RH2: 7.58 μmol h−1 | g-C3N4: 2.03 μmol h−1 | 3.7 | 76 |
| MWNTs: no data | No data | |||||
| Multi-walled carbon nanotubes (CNT)/white C3N4 | Mass: no data | Decomposing methyl orange (MO) under visible light | 89.7%, in 3 h | White C3N4: no data | No data | 77 |
| CNT: no data | No data | |||||
| Mass: no data | Decomposing (RhB) under visible light | 85.4%, in 3 h | White C3N4: no data | No data | ||
| CNT: no data | No data | |||||
| Polypyrrole (PPy)/g-C3N4 | Mass: 98.5% | Photocatalytic H2 evolution under visible light; co-catalyst: Pt; sacrificial reagent: no mention | RH2: 15.4 μmol h−1 | g-C3N4: no data | No data | 78 |
| 99.5% | No data | PPy: no data | No data | |||
| 97.5% | No data | |||||
| 96% | No data | |||||
| Red phosphor (r-P)/g-C3N4 | Mass: 5% | Photocatalytic H2 evolution under visible light; co-catalyst: Pt; sacrificial reagent: ascorbic acid | RH2: 310 μmol h−1 g−1 | g-C3N4: 340 μmol h−1 g−1 | 0.91; 14.09 | 79 |
| 30% | 1000 μmol h−1 g−1 | r-P: 22 μmol h−1 g−1 | 2.94; 45.45 | |||
| Red phosphor (r-P)/g-C3N4 | Mass: 30% | Photocatalytic CO2 conversion into valuable hydrocarbon fuel (CH4) in the presence of water vapor | 295 μmol h−1 g−1 | g-C3N4: 107 μmol h−1 g−1 | 2.76 | |
| r-P: 145 μmol h−1 g−1 | 2.03 | |||||
| RGO/g-C3N4/α-S8 | Mass: 30% | Bacterial inactivation (E. coli K-12) under visible-light | No data | g-C3N4: no data | No data | 75 |
| g-C3N4/RGO/α-S8 | RGO: no data | No data | ||||
| α-S8: no data | No data | |||||
| g-C3N4/C60 | Mass: 99.5% | Decomposing (RhB) under visible light | 87% in 60 min | g-C3N4: 54% | 1.61; 1.80; 1.56 | 82 |
| 99% | 97% in 60 min | C60: no data | No data | |||
| 98% | 84% in 60 min | |||||
| CNS/CN-2 | Mass: no data | Photocatalytic H2 evolution under visible light; co-catalyst: Pt; sacrificial reagent: no mention | No data | CN: no data | 11 | 80 |
| CN/CNS-2 | CNS: no data | 2.3 | ||||
| g-C3N4/g-C3N4 | Mass: no data | Decomposing | 47.6% in 30 min | g-C3N4:(T) 27.3% | 1.74 | 81 |
| NO under visible light | g-C3N4:(U) 31.7% | 1.50 | ||||
| Graphene/g-C3N4 | Mass: no data | Conductivity and electrocatalytic performance on oxygen reduction reaction (ORR) | No data | g-C3N4: no data | No data | 70 |
| Graphene: no data | No data |
Significantly, Wang et al. obtained two types of host–guest CN/CNS heterojunctions (Fig. 2) with a facile band alignment by the surface-assisted polymerization: CNS–CN (CN serving as the host) and CN–CNS (CNS serving as the host).80 CN/CNS isotype heterojunctions were obtained based on the band alignment between CN and CNS, owing to the slight difference in their electronic band structures. As an amazing result, these two types of CN/CNS heterojunctions mentioned above demonstrated conspicuous enhancement in the photocatalytic activity and stability for hydrogen evolution.
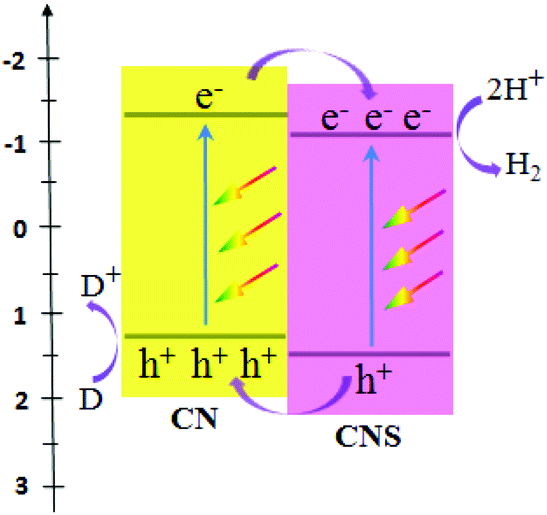 | ||
| Fig. 2 Schematic illustration of organic heterojunctions formed between CN and CNS. (CN refers to g-C3N4, CNS refers to sulfur-mediated CN, and D = donor.) | ||
Inspired by Wang's work, the layered g-C3N4/g-C3N4 metal-free isotype heterojunction (Fig. 3a) was produced by treating the molecular composite precursors of urea and thiourea under the same thermal conditions. The molecular structure of this CN-T/CN-U heterojunction is illustrated in Fig. 3b. This isotype heterojunction was constructed on the basis that the g-C3N4 prepared from separate urea and thiourea have different band structures. Upon visible light irradiation, the photogenerated electrons transfer from g-C3N4 (thiourea) to g-C3N4 (urea) driven by the conduction band offset of 0.10 eV, whereas the photogenerated holes transfer from g-C3N4 (urea) to g-C3N4 (thiourea) driven by the valence band offset of 0.40 eV.81 These two charge transfer processes are beneficial for overcoming the high dissociation barrier of the Frenkel exciton and stabilizing electrons and holes. The redistribution of electrons on the one side of the heterojunction (CN-U) and holes on the opposite side (CN-T) could reduce the electron–hole pair recombination as well as prolonged lifetime of charge carriers, resulting in an exceptionally high photocatalytic capability. These two successful attempts demonstrated that rational design and construction of isotype heterojunctions could open up a new avenue for the development of new efficient visible-light photocatalysts.
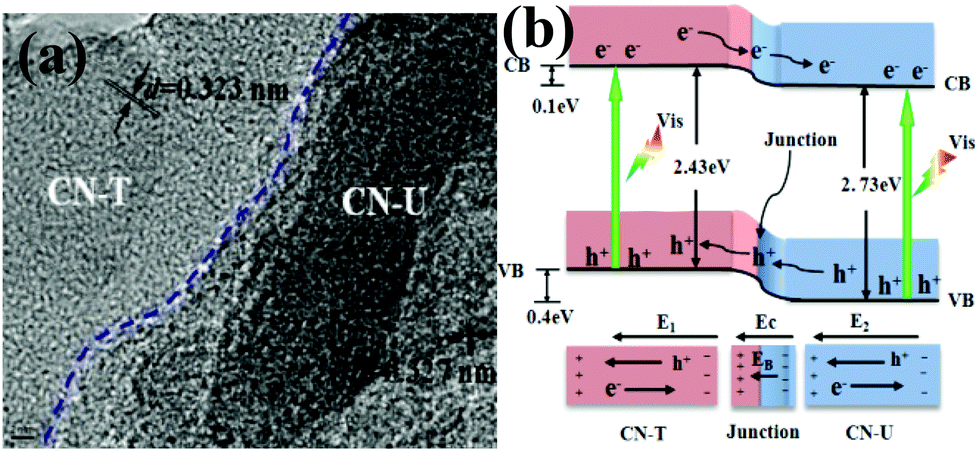 | ||
| Fig. 3 (a) TEM images of CN-TU; (b) schematic illustration of electron–hole separation and transport at the g-C3N4–g-C3N4 heterojunction interface and in both semiconductors: EC is the contact electric field for the two components; EB is the potential barrier in the interfacial depletion layer (EB < EC during the photocatalytic reaction); E1 and E2 are the internal electric fields induced by the redistribution of the spatial charges in CN-T and CN-U, respectively. CN-T refers to g-C3N4 from thiourea and CN-U refers to g-C3N4 from urea. (Reprinted with permission from ref. 81. Copyright (2013) American Chemical Society.) | ||
3.2. g-C3N4/single metal oxide (metal sulfide)
So far, various single metal oxides (metal sulfides) have been coupled with g-C3N4 for enhanced visible light photocatalysis.83–87 Among abundant metal oxides, TiO2 (e.g. 3.2 eV), as a widely used photocatalyst, preferentially acted as a candidate for constructing g-C3N4 based heterojunctions. The work of Zhou et al., for example, demonstrated that a g-C3N4/TiO2 nanotube array heterojunction was successfully achieved by a simple electrochemical method, showing highly effective visible-light performance with respect to bare g-C3N4 and TiO2 nanotubes.45 Zhao et al. obtained g-C3N4/TiO2 hybrid photocatalysts by a facile hydrolysis approach, accompanied by a remarkable enhancement of photocatalytic capability in degradation of phenol both under visible and UV light irradiation.83 Sridharan et al. successfully fabricated a g-C3N4/TiO2 composite with decent photocatalytic performance which was utilized in the treatment of methylene blue (MB) and reduction of hazardous Cr(VI) ions.84 Very recently, direct Z-scheme g-C3N4/TiO2 photocatalysts were successfully prepared via simple one-step calcinations with good photocatalytic response for formaldehyde decomposition in air, reported by Yu et al.85WO3 is another metal oxide that can be used to couple with g-C3N4. The band gap of WO3 is between 2.6 and 2.8 eV.88–90 For instance, Zang et al. obtained environmentally benign g-C3N4/WO3 composites via a facile mixing–heating procedure, which showed superb performance in methyl orange (MO) degradation.88 A WO3/g-C3N4 heterojunction (Fig. 4a and 4b) was prepared by a calcination process accompanied by marvelous visible-light-activity by degradation of MB and 4-chlorophenol, documented by Huang et al.89 Apparently, all of the WO3/g-C3N4 photocatalysts demonstrated higher photocatalytic performance than the pristine WO3, g-C3N4 as well as the mechanically blended WO3 and g-C3N4 sample under visible light irradiation (Fig. 4c). Equally important, Katsumata et al. have associated g-C3N4 with WO3 by a physical mixing method to decontaminate the organic gas pollution acetaldehyde (CH3CHO).90
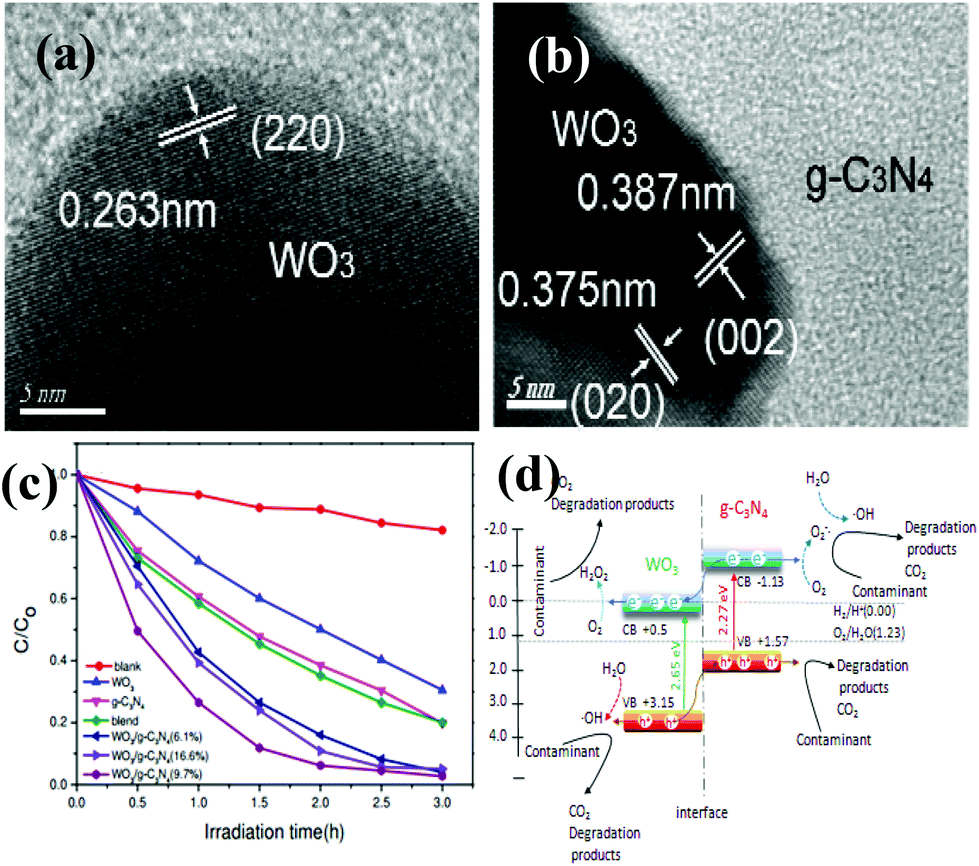 | ||
| Fig. 4 (a, b) HRTEM images of WO3/g-C3N4 (9.7%) composites; (c) photocatalytic degradation efficiency of MB by g-C3N4, WO3, blend and WO3/g-C3N4 samples with different WO3 contents under visible light; (d) proposed mechanism for the photodegradation of contaminants on WO3/g-C3N4 composites. (Reproduced from ref. 89 with permission from The Royal Society of Chemistry.) | ||
The remarkably highly increased performance of WO3/g-C3N4 was mainly ascribed to the synergistic effects of the enhanced optical absorption in the visible region, enlarged specific surface areas and suitable band positions. A conceivable degradation mechanism was put forward in Fig. 4d.
As can be seen in Fig. 4d, the top of the VB and the bottom of the CB of WO3 were calculated to be 3.15 eV and 0.5 eV, respectively.89 In the case of g-C3N4, the top of the VB is 1.57 V and the bottom of the CB is −1.13 V.89 The band gaps of g-C3N4 and WO3 were 2.70 eV and 2.65 eV, respectively. The pristine g-C3N4 and WO3 are two semiconductors which can be both excited to produce photogenerated electron–hole pairs under visible light irradiation. Since the CB potential (−1.13 eV) is lower than the CB edge of WO3 (0.5 eV), the excited-state electrons on g-C3N4 can directly inject into the CB of WO3. Similarly, the VB position of WO3 (+3.15 eV) is higher than the VB of g-C3N4 (+1.57 eV). The photogenerated holes on the VB of WO3 can be directly injected into the VB of g-C3N4. The redistribution of electrons on the one side of the junction (WO3) and holes on the opposite side (g-C3N4) greatly reduces the electron–hole recombination, which is beneficial to promote the photocatalytic performance. As a result, the photocatalytic activity of WO3/g-C3N4 composites was much higher than that of WO3 and the pristine g-C3N4. Therefore, the enhanced photocatalytic activity was mainly associated with the heterojunction structure and suitable band-edge positions of the two semiconductors, except for enhanced optical absorption in the visible region and enlarged specific surface areas of WO3/g-C3N4 nanojunctions.
Besides, cadmium sulfide (CdS) is a fascinating semiconductor with a relatively low band gap of 2.4 eV, which makes it able to absorb solar light up to 520 nm or even longer.86,87 Comparing the energy levels of C3N4 with CdS, it is fortunate to find that their well-matched band-structures are quite suitable to construct heterostructures that would bring an effective separation and transfer of photogenerated charges. To date, novel CdS/g-C3N4 organic–inorganic composites composed of two visible light responsive semiconductors have been fabricated via an “in situ” precipitation–deposition method by Fu et al., exhibiting outstanding visible light photocatalytic activity in 4-aminobenzoic acid removal.42
On account of multiple advantages of combining g-C3N4 with oxide (sulfide), a host of attempts have been devoted to exploit novel candidates for coupling with g-C3N4.88–104 They mainly include g-C3N4/Fe2O3,91,92 g-C3N4/CeO2,93 g-C3N4/MoO3,94 g-C3N4/Fe3O4,95 g-C3N4/Ni(OH)2,96 g-C3N4/Ag2O,97 g-C3N4/MoS2,98 g-C3N4/NiS,99 g-C3N4/TaON,30 g-C3N4/ZnO,100,101 g-C3N4/In2O3,102 g-C3N4/WO3,88,89,103 and SiO2/g-C3N4.104 These g-C3N4 based nanocomposites have become an important family of visible light photocatalysts for contaminant degradation and hydrogen production (Table 2).
| Composite photocatalyst | Mass fraction or molar of g-C3N4 | Typical parameters of photocatalytic experiments | Photocatalytic activity | Reference photocatalyst; photocatalytic activity | Enhancement factor over the reference photocatalyst | Reference |
|---|---|---|---|---|---|---|
| TiO2/g-C3N4 | No data | Decomposing methyl orange (MO) under visible light | 100% degradation in 30 min | g-C3N4: no data | No data | 45 |
| TiO2: no data | 3 | |||||
| TiO2/g-C3N4 | 300 °C | Decomposing methylene blue (MB) under visible light | 93% | g-C3N4: no data | No data | 84 |
| 500 °C | 75% degradation in 120 min | TiO2: no data | No data | |||
| TiO2/g-C3N4 | 300 °C | Photocatalytic reduction of Cr(VI) under visible light | 72% | g-C3N4: no data | No data | |
| 500 °C | 45% in 100 min | TiO2: no data | No data | |||
| TiO2/g-C3N4 | Mass: 20% | Decomposing phenol under UV light | 96.6% degradation in 60 min | g-C3N4: 79.5% | 1.2 | 83 |
| Mass: 20% | Decomposing phenol under visible light | 67.7% degradation in 180 min | TiO2: 65.9% | 1.5 | ||
| TiO2/g-C3N4 | Mass: 16.7% | Decomposing phenol under UV light | 69.1% in 60 min | g-C3N4: 43.8% | 1.5 | |
| 50% | 82.7% in 60 min | TiO2: no data | No data | |||
| 66.7% | 96.6% in 60 min | g-C3N4: no data | No data | |||
| 83.3% | 82.8% in 60 min | TiO2: no data | No data | |||
| TiO2/g-C3N4 (Z-scheme) | Mass ratio: 100% (urea: P25) | Decomposing formaldehyde under UV light | 94% in 60 min apparent rate constant k: 7.36 × 10−2 min−1 | g-C3N4: no data | No data | 85 |
| TiO2: no data | No data | |||||
| g-C3N4: no data | No data | |||||
| P25: 3.53 × 10−2 min−1 | 2.1 | |||||
| WO3/g-C3N4 | Mass ratio: no data | Decomposing (MO) under visible light | No data | g-C3N4: no data | No data | 88 |
| WO3: no data | No data | |||||
| WO3/g-C3N4 | Mass: 80% | Decomposing acetaldehyde (CH3CHO) under visible light | No data | g-C3N4: no data | No data | 90 |
| 60% | No data | WO3: no data | No data | |||
| 40% | No data | |||||
| 20% | No data | |||||
| WO3/g-C3N4 | Mass: 90.3% | Decomposing MB under visible light | 97% degradation in 2 h | g-C3N4: 81%, 3 h | 1.2 | 89 |
| WO3: 73%, 3 h | 1.3 | |||||
| Mass: 90.3% | Decomposing 4-chlorophenol (4-CP) under visible light | 43% degradation in 6 h | g-C3N4: 3%, 6 h | 14.3 | ||
| WO3: no data | ||||||
| a-Fe2O3/g-C3N4 | No data | Electrochemical properties | Supercapacitive performance | g-C3N4: no data | No data | 91 |
| a-Fe2O3: 72 F g−1 | 2.3; 1.9 | |||||
| Precursors: [Bmim]FeCl4 anhydrous FeCl | 167 F g−1 | |||||
| 140 F g−1 | ||||||
| Fe2O3/g-C3N4 | Mass: 97.2% | Decomposing rhodamine B (RhB) under visible light | 100% in 120 min | g-C3N4: 100%, 180 min | No data | 92 |
| 95.1% | 100% in 160 min | Fe2O3: no data | No data | |||
| 93.5% | 95.7% in 180 min | 0.96 | ||||
| 92% | 75.8% in 180 min | 0.76 | ||||
| 88.4% | 52.5% in 180 min | 0.53 | ||||
| ZnO/g-C3N4 | Mass: 95.1% | Decomposing rhodamine B (RhB) under visible light | No data | g-C3N4: no data | No data | 100 |
| 91.6% | No data | ZnO: no data | No data | |||
| 84.4% | 97% in 80 min | |||||
| 41.9% | No data | |||||
| ZnO/g-C3N4 | Mass: 84.4% | Decomposing p-nitrophenol under visible light | No data | g-C3N4: 30%, 5 h | No data | |
| ZnO: no data | No data | |||||
| ZnO/g-C3N4 | Mass: 5.0% | Decomposing (RhB) under visible light photoreduction of Cr6+ under visible light irradiation | 97.4% in 100 min | g-C3N4: no data | No data | 101 |
| ZnO: no data | No data | |||||
| Mass: 5.0% | 75.5% in 100 min | g-C3N4: no data | No data | |||
| ZnO: no data | No data | |||||
| CeO2/g-C3N4 | Mass: 87% | Decomposing MB under visible light | 95% in 120 min | g-C3N4: 75%, 3 h | 1.3 | 93 |
| Mass: 94.1% | 30% in 5 h | CeO2: 28%, 3 h | 3.4 | |||
| 87.0% | decomposing 4-chlorophenol (4-CP) under visible light | 45% in 5 h | g-C3N4: 2.3% | 13.0; 2.0 | ||
| 77.6% | 37% in 5 h | CeO2: 15.1% | 19.6; 3.0 | |||
| 16.1; 2.5 | ||||||
| MoO3/g-C3N4 | Mass: 7.0% | Decomposing MB under visible light | 93% in 3 h | g-C3N4: no data | No data | 94 |
| MoO3: no data | No data | |||||
| Fe3O4/g-C3N4 | Mass: 98% | Decomposing MO under visible light | No data | g-C3N4: no data | No data | 95 |
| Fe3O4: no data | No data | |||||
| Ni(OH)2/g-C3N4 | Molar: 99.5% | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: triethanolamine | RH2: 7.6 mmol h−1 | g-C3N4: no data | No data | 96 |
| 99.9% | RH2: 3.3 mmol h−1 | Ni(OH)2: no data | No data | |||
| MoS2/g-C3N4 | Mass: 99.5% | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: lactic acid | RH2: 20.6 mmol h−1 | g-C3N4: no data | No data | 98 |
| MoS2: no data | No data | |||||
| 0.5 wt% Pt/g-C3N4: 4.8 mmol h−1 | 4.3 | |||||
| CdS/g-C3N4 | Mass: 70% | Decomposing (MO) under visible light | 92% in 20 min | g-C3N4: 16%, in 25 min | No data | 42 |
| CdS: no data | No data | |||||
| 0.3CdS-0.7TiO2: 19%, in 25 min | No data | |||||
| Mass: 70% | Decomposing 4-aminobenzoic acid under visible light | 73% in 60 min | 0.7C3N4-0.3TiO2: 22%, in 25 min | No data | ||
| g-C3N4: 3% | 2.7 | |||||
| CdS: 38% | 41.6 | |||||
| NiS/g-C3N4 | Mass: 99.5% | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: triethanolamine | RH2: 9.2 mmol h−1 | g-C3N4: 0.2 mmol h−1 | 46.0 | 99 |
| 98.75% | RH2: 48.2 mmol h−1 | NiS: no data | 241.0 | |||
| 98% | RH2: 32.1 mmol h−1 | 160.5 | ||||
| 97.5% | RH2: 13.5 mmol h−1 | 67.5 | ||||
| TaON/g-C3N4 | Mass: 60% | Decomposing (RhB) under visible light | 100% in 50 min | g-C3N4: 100%, in 80 min | No data | 30 |
| TaON: 34%, in 80 min | No data | |||||
| In2O3/g-C3N4 | Mass: 90% | CO2 reduction into hydrocarbon fuels | CH4 production yield: 76.7 ppm | g-C3N4: no data | 3.0 | 102 |
| In2O3: no data | 4.0 | |||||
| WO3/g-C3N4 | Mass: 50% | Oxidation of acetaldehyde into CO2 | At least 600 ppm of acetaldehyde had been removed in 48 h | g-C3N4: no data | No data | 103 |
| WO3: no data | No data | |||||
| SiO2/g-C3N4 | No data | Decomposing RhB under visible light | Fully degraded in 60 min | g-C3N4: no data | No data | 104 |
| SiO2: no data | No data |
3.3. g-C3N4/composite oxide
During the past few years, many groups have reported different types of g-C3N4/composite oxide heterojunction photocatalysts. These research studies greatly improve the efficiencies of the photocatalysts and promote their applications in the energy production and environmental remediation.105,106Table 3 summarizes and compares the g-C3N4/composite oxide heterojunction photocatalysts. Among these photocatalytic systems, Bi2WO6/g-C3N4 has been the most widely investigated. Ge et al. prepared g-C3N4/Bi2WO6 heterostructured photocatalysts via mixing and heating methods.31 The g-C3N4/Bi2WO6 photocatalyst had a remarkably enhanced MO photodegradation activity than pure g-C3N4 and Bi2WO6 under visible light irradiation. This enhancement could be attributed to the enhanced visible-light utilization efficiency and accelerated transfer of photogenerated electron–hole pairs at the intimate interface of heterojunctions, which rationally can be ascribed to the well-aligned overlapping band-structures of g-C3N4 and Bi2WO6. Subsequently, Wang et al. prepared Bi2WO6 hybridized with g-C3N4 by facile chemisorptions, which exhibited enhanced photocatalytic performance in MB degradation.105 Tian et al. hydrothermally synthesized a heterojunction by combining g-C3N4 with Bi2WO6 with enhanced visible light photocatalytic capability.106
| Composite photocatalyst | Mass or molar fraction of g-C3N4 | Typical parameters of photocatalytic experiments | Photocatalytic activity | Reference photocatalyst; photocatalytic activity | Enhancement factor over the reference photocatalyst | Reference |
|---|---|---|---|---|---|---|
| a RH2 represents H2 production rate. | ||||||
| BiPO4/g-C3N4 | Mass: 4% | Decomposing methyl blue (MB) under UV light | 90% degradation in 5 min | BiPO4: no data | 2.5 | 117 |
| P25(TiO2): no data | 4.5 | |||||
| Mass: 10% | Decomposing MB under visible light | Apparent rate constant k: 0.31 h−1 | g-C3N4: k = 0.06 h−1 | 5 | ||
| Bi2WO6/g-C3N4 | Mass: 2% | Decomposing (MB) under simulated solar irradiation | Apparent rate constant: k: 1.0291 h−1 | Bi2WO6; k: 0.6060 h−1 | 1.67 | 105 |
| Bi2WO6/g-C3N4 | Mass: 50% | Decomposing methyl orange (MO) under visible light | 93% degradation in 120 min | g-C3N4: 31% | 3 | 106 |
| Bi2WO6: 0.6% | 155 | |||||
| Bi2WO6/g-C3N4 | Mass: 5% | Decomposing (MO) under visible light | 21.1% in 3 h | g-C3N4: 81.4% | 0.26 | 31 |
| 10% | 76.0% in 3 h | 0.46 | ||||
| 30% | 45.2% in 3 h | 0.56 | ||||
| 50% | 89.6% in 3 h | 1.10 | ||||
| 70% | 99.9% in 3 h | 1.23 | ||||
| 90% | 84.4% in 3 h | 1.03 | ||||
| Ag3PO4/g-C3N4 | Mass: 16.7% | Decomposing RhB under visible light | No data | g-C3N4: no data | 3.27 | 118 |
| Ag3PO4: no data | 1.65 | |||||
| Ag3PO4/g-C3N4 | Mass: 25% | Decomposing (MO) under visible light | No data | g-C3N4: no data | 5 | 119 |
| Ag3PO4: no data | 3.5 | |||||
| ZnWO4/g-C3N4 | Mass: 5% | Decomposing MB under UV light and visible light | No data | g-C3N4: no data | 1.8 | 114 |
| No data | g-C3N4: no data | No data | ||||
| GdVO4/g-C3N4 | Mass: 90% | Decomposing (RhB) under visible light | Apparent rate constant: k: 0.0434 min−1 | g-C3N4: no data | 3.1 | 121 |
| N–TiO2: no data | 6.3 | |||||
| GdVO4: no data | 36 | |||||
| NaTaO3/g-C3N4 | Mass: 5% | Decomposing (RhB) under UV light and visible light | No data | g-C3N4: | No data | 122 |
| 10% | NaTaO3: | No data | ||||
| Degussa P25: | No data | |||||
| SrTiO3:Rh/g-C3N4 | Mass: 30% | Photocatalytic H2 evolution under visible light | RH2a: 81.0 mmol−1 | g-C3N4; RH2: 10.7 | 7.57, 1.2 | 123 |
| 20% | 223.3 | SrTiO3: Rh(0.3 mol%): 68.9 | 20.9, 3.24 | |||
| 10% | Co-catalyst: no; sacrificial reagent: not mentioned | 92.6 | 8.7, 1.3 | |||
| Bi5Nb3O15/g-C3N4 | Mass: 50% | Decomposing (MO) under visible light | 86% in 3 h | g-C3N4: 80% | 1.07; 3.1 | 124 |
| 70% | 94% in 3 h | Bi5Nb3O15: 28% | 1.2; 3.4 | |||
| 90% | 93% in 3 h | 1.2; 3.3 | ||||
| Mass: 70% | Decomposing 4-chlorophenol (4-CP) under visible light | 100% in 1h | g-C3N4: 72% | 1.4 | ||
| Bi5Nb3O15: 38% | 2.6 | |||||
| Co3(PO4)/g-C3N42 | CP-20 (UV irradiation time: 20 min) | Photocatalytic O2 evolution under visible light; co-catalyst: no; sacrificial reagent: AgNO3 | No data, 3 h | g-C3N4: 1.28 μmol | 6.8 | 110 |
| Co3(PO4)2/g-C3N4 | CP-5 | H2 evolution under visible light | No data | g-C3N4: 2.03 μmol h−1 | No data | |
| CP-10 | No data 19.48 μmol h−1 | No data | ||||
| CP-20 | No data | 9.6 | ||||
| CP-30 | No data | No data | ||||
| CP-60 | No data | |||||
| N-In2TiO5/g-C3N4 | No data | Decomposing (RhB) under visible light | Degraded completely within 20 min | g-C3N4: no data | No data | 120 |
| N-In2TiO5: no data | No data | |||||
| Ag3VO4/g-C3N4 | Mass: 10% | Decomposing basic fuchsin (BF) under visible light Malachite green (MG) Crystal violet (CV) | 60% in 2.5 h | g-C3N4: 15% | 4.0; 2.0 | 125 |
| 40% | 95% in 2.5 h | Ag3VO4: 30% | 6.3; 3.2 | |||
| 50% | 88% in 2.5 h | 5.9; 2.9 | ||||
| Ag3VO4/g-C3N4 | Mass: 10% | 97% in 1 h | g-C3N4: no data | No data | ||
| Ag3VO4: no data | No data | |||||
| Ag3VO4/g-C3N4 | Mass: 10% | 75% in 2.5 h | g-C3N4: no data | No data | ||
| Ag3VO4: no data | No data | |||||
| SmVO4/g-C3N4 | Mass: 50.3% | Decomposing (RhB) under visible light | 2.07 h−1 | g-C3N4: no data | 2.4 | 104 |
| SmVO4: no data | 6.3 | |||||
Li chose SmVO4 (band gap 2.28 eV) as a promising candidate for constructing g-C3N4 coupling heterojunction, and this g-C3N4/SmVO4 composite photocatalyst exhibited high photocatalytic activity and stability for RhB decomposition under visible light irradiation.107 Moreover, earth abundant cobalt phosphate (Co–Pi) has been demonstrated to work effectively as an oxygen-evolving electrocatalyst108–110 owing to its attractive characteristics such as the low cost and self-repairing behavior.111,112 Recently, the Co–Pi species were presented as nanoparticles were well-distributed on the g-C3N4 surface, and the Co–Pi/g-C3N4 composites exhibit stronger visible light absorption. The Co–Pi/g-C3N4 composite samples show significantly enhanced H2 and O2 evolution activities.113
It is well known that ZnWO4 itself cannot be excited by visible light. In contrast, C3N4 can absorb visible light. Wang et al. fabricated C3N4/ZnWO4 coupling composites (Fig. 5a and b), i.e. the ZnWO4, a semiconductor without visible photocatalytic performance, was successfully photo-sensitized by g-C3N4.114 These composites possessed eminent and commendably durable photocatalytic activity in removal of Rh–B, as well as enhanced photocurrent responses under visible light irradiation (Fig. 5c). More importantly, a new mechanism of ZnWO4 sensitized by carbon nitride is presented in Fig. 5d.
 | ||
| Fig. 5 HRTEM images of ZnWO4 and C3N4/ZnWO4 photocatalysts (a) ZnWO4 and (b) C3N4/ZnWO4; (c) photocurrents of ZnWO4 and C3N4/ZnWO4 electrodes with light on/off cycles under visible light irradiation (λ > 420 nm) ([Na2SO4] = 0.1 M); (d) schematic drawing of C3N4/ZnWO4 photocatalyst under visible light. (Reprinted with permission from ref. 114. Copyright (2012) The Royal Society of Chemistry.) | ||
Under visible light illumination, the excited-state electrons of the HOMO of the C3N4 would transport to the lower unoccupied molecular orbital (LUMO) of C3N4 (π–π* transition). Since the LUMO potential of C3N4 (−1.1 eV)115 is lower than the CB edge of ZnWO4 (−0.8 eV),116 a chemical interaction occurs between them. Hence the excited-state electrons on C3N4 can directly inject into the CB of ZnWO4. The electrons subsequently transfer to the photocatalyst surface to react with water and oxygen to generate hydroxyl and superoxide radicals.114 These reactive radicals are able to oxidize the pollutants. As a result, the CN polymer sensitized ZnWO4 photocatalyst exhibits enhanced visible light photocatalytic performance. To further broaden the application, many groups have gained success in development of new g-C3N4 based heterojunctions with superior performance in photocatalysis, such as C3N4/BiPO4,117 g-C3N4/Ag3PO4,118,119 g-C3N4/N-In2TiO5,120 g-C3N4/GdVO4,121 g-C3N4/NaTaO3,122 g-C3N4/Rh-SrTiO3,123 g-C3N4/Bi5Nb3O15,124 and g-C3N4/Ag3VO4.125
3.4. g-C3N4/BiOX (AgX)
Recently, as a halide, BiOX (X = Cl, Br, I) with advantages of layered structure and indirect-transition band-gaps were reported as efficient photocatalysts.126–134 The layered structure can provide large enough space to polarize the related atoms and orbitals, and then induce the presence of internal static electric fields perpendicular to the [Bi2O2] slabs and halogen anionic slabs in BiOX.135–137 It is interesting to couple BiOX with g-C3N4 to form layered nanojunctions.Among the BiOX photocatalysts, BiOBr with an appropriate band-gap (2.75 eV) exhibits high photocatalytic activity. Ye et al. synthesized BiOBr-g-C3N4 inorganic–organic composite photocatalysts by a one-step chemical bath method at low temperature.138 This heterojunction interacted by facets coupling contributes to the promoted photoinduced charge transfer between BiOBr and g-C3N4, and enhances the VLD photocatalytic activity for RhB degradation compared with bare BiOBr and g-C3N4. Furthermore, Sun et al. fabricated BiOBr-C3N4 heterojunctions (Fig. 6a) by depositing BiOBr nanosheets onto the surface of C3N4 nanosheets at room temperature.139 The heterojunction possesses intimately contacted interfaces and well-aligned straddling band-structures, which are propitious to the effective separation and transfer of photogenerated charges, bringing an excellent performance. A schematic illustration of the band gap structures for the samples is shown in Fig. 6b.
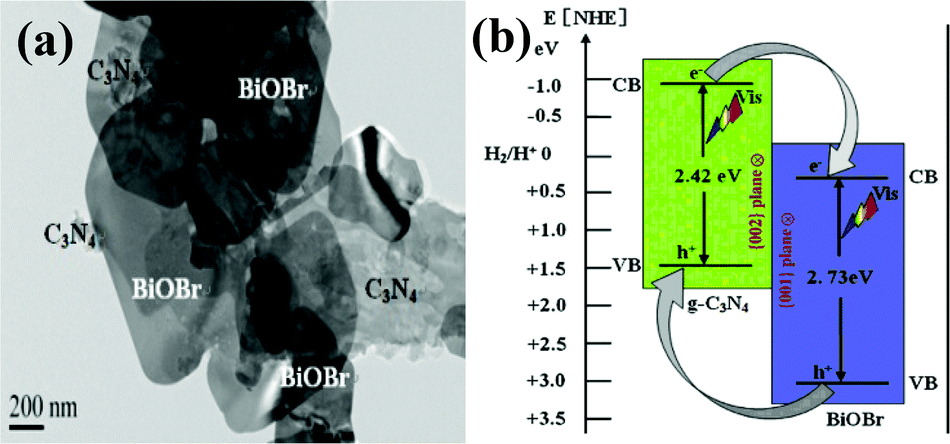 | ||
| Fig. 6 TEM of the as-synthesized BiOBr/C3N4 nanojunctions (a) and schematic illustration of the band-gap matching and the crystal-plane coupling of C3N4/BiOBr nanojunctions (b). (Reproduced with permission from ref. 139, copyright 2014, Elsevier.) | ||
The band structures of the two components are well-matched and aligned with each other (Fig. 6b). Both C3N4 and BiOBr can be excited by visible light and then generate photo-induced electrons and holes. The relative CB and VB edge positions of C3N4 nanosheets and BiOBr nanosheets imply that the well-matched band energies and crystal planes can form heterojunctions at the nanoscale level. The excited electrons in CB of C3N4 can transfer to CB of BiOBr, and excited holes in VB of BiOBr can transfer to VB of C3N4, which results in efficient separation and transport of photo-induced electrons and holes. The 2D BiOBr/C3N4 layered nanojunctions could effectively strengthen the photocatalytic activity. This work demonstrated that novel 2D nanojunctions, accompanied by high visible light activity, can be constructed by combining two visible-light-active 2D semiconductors with well-coupled crystal planes and well-matched band structures, which could provide a new approach for promoting the activity of current photocatalysts.
BiOI is an attractive p-type semiconductor with a strong photoresponse in the visible light region due to its narrow band gap energy (1.78 eV) and could act as a potential sensitizer to sensitize wide band gap semiconductors. Jiang et al. obtained a novel p–n heterojunction photocatalyst constructed by porous graphite-like C3N4 and nanostructured BiOI.140 In fact, the coupling of p- and n-type semiconductors is believed to be helpful because an internal electric field is built up between them. As a result, the coupling of n-type g-C3N4 with a p-type narrow band gap BiOI semiconductor is a good strategy to enhance the visible light absorption capability and the photocatalytic performance of g-C3N4 under visible light irradiation. Besides, Wang and his group successfully used BiOCl as an alternative to prepare BiOCl/C3N4 heterojunction photocatalysts via an ionic-liquid-assisted solvent-thermal route, and these nanocomposites demonstrated significantly enhanced visible light performance in degradation of MO due to the formation of well-matched heterostructures.141
AgX based photocatalysts have attracted great attention as promising candidates for the development of highly efficient visible light photocatalysts. Some efforts were made to explore a new type of g-C3N4 based heterojunction by coupling silver halide. Silver halide AgX (X = Br, I) is a photosensitive material extensively used in the photography field.142–144 Up to now, Xu et al. have successfully applied AgX (X = Br, I) developing novel visible-light-driven AgX/graphite-like C3N4 (X = Br, I) hybrid materials, and the photocatalytic activity dramatically improved using methyl orange (MO) as a target pollutant.145 The high photocatalytic activity of the hybrid materials could be ascribed to the strong coupling between g-C3N4 and AgX, which facilitated interfacial charge transfer and inhibited electron–hole recombination. Table 4 summarizes the photocatalytic properties of different g-C3N4/BiOX (AgX) composite photocatalysts.
| Composite photocatalyst | Mass or molar fraction of g-C3N4 | Typical parameters of photocatalytic experiments | Photocatalytic activity | Reference photocatalyst; photocatalytic activity | Enhancement factor over the reference photocatalyst | Reference |
|---|---|---|---|---|---|---|
| BiOBr/g-C3N4 | Mass: 50% | Decomposing rhodamine B (RhB) under visible light | 95% degradation in 30 min | g-C3N4: 15% | 6.3 | 138 |
| BiOBr: 35% | 2.7 | |||||
| BiOBr/g-C3N4 | Mass: 50% | NO removal under visible light | 32.7% degradation in 30 min | g-C3N4: 22.9% | 1.4 | 139 |
| BiOBr: 21.2% | 1.5 | |||||
| BiOCl/g-C3N4 | Molar: 50% | Decomposing methyl orange (MO) under visible light | 95% degradation in 80 min | g-C3N4: 11% | 8.6 | 141 |
| BiOCl: 58% | 1.6 | |||||
| BiOI/g-C3N4 | Mass: 77.5% | Decomposing methyl blue (MB) under visible light | 99% degradation in 3 h | g-C3N4: 64% | 1.5 | 140 |
| BiOI: 51% | 1.9 | |||||
| AgBr/g-C3N4 | Molar: 70% | Decomposing MO under visible light | 91% degradation in 10 h | g-C3N4: low | No data | 145 |
| AgBr: 23% | 4 | |||||
| AgI/g-C3N4 | Molar: 97.5% | Decomposing MO under visible light | 28% in 3.5 h | g-C3N4: no data | No data | |
| 95% | 44% in 3.5 h | AgI: no data | No data | |||
| 90% | 79% in 3.5 h | |||||
| 70% | 81% in 3.5 h | |||||
| 50% | 62% in 3.5 h | |||||
| AgBr/g-C3N4 | Molar: 70% | Decomposing 4-chlorophenol under visible light | 30% in 6 h | g-C3N4: 6.1% | 4.9 | |
| AgBr: no data | No data | |||||
| AgI/g-C3N4 | Molar: 70% | Decomposing 4-chlorophenol under visible light | 53% in 6 h | g-C3N4: 6.1% | 8.7 | |
| AgI: no data | No data |
3.5. g-C3N4/noble metal
The semiconductor–metal junction is widely used to create a space–charge separation region (called the Schottky barrier). At the interface of the two components, electrons transfer from one component to the other to align the Fermi energy levels, preventing the charge recombination and enhancing the photocatalytic performance. On the other hand, the surface plasmon resonance (SPR) endowed in noble metals could increase the visible light utilization and probably show special synergistic effects with g-C3N4.146–154 It is therefore feasible to couple noble metals with g-C3N4 in order to enhance the photocatalytic activity.155–161 The work of Di et al., for example, preferentially demonstrated that the deposition–precipitation method was successfully applied to prepare Au(III) nanoparticles on the surface of a structured polymeric g-C3N4.155 The as-synthesized nanoscopic Au–semiconductor heterojunctions effectively accelerate charges transfer on the intimate interface between g-C3N4 and Au nanoparticles, enabling high photocatalytic hydrogen production. In addition, given that gold nanoparticles (AuNPs) can serve as photocatalysts for the degradation of dyes under visible-light irradiation through the surface plasmon effect,156 it is expected that AuNPs/g-C3N4 nanohybrids could show improved photocatalytic performance. Cheng et al. proposed that Au nanoparticles (AuNPs) were successfully loaded on graphitic carbon nitride (g-C3N4), and the nanohybrids show superior photocatalytic activities for the decomposition of methyl orange under visible-light irradiation due to the facilitated separation of photogenerated electron–hole pairs, and the surface plasmon resonance excitation in AuNPs.157It is well-documented that Ag, a famous noble metal, was regarded as a decent co-catalyst in constructing heterojunctions with g-C3N4 in photocatalytic utilizations.158,159 Yang et al. reported that an efficient visible-light plasmonic photocatalyst, Ag/g-C3N4 heterostructure, is facilely fabricated by a simple polymerization–photodeposition route.158 Compared with individual g-C3N4, the photocatalytic activity of Ag/g-C3N4 was sharply enhanced toward the degradation of MO and p-nitrophenol, which may be ascribed to the enhanced visible-light utilization efficiency due to the SPR absorption of silver nanoparticles as well as fast generation, separation and transportation of the photogenerated carriers. Bai and his group have developed novel core–shell Ag@g-C3N4 plasmonic nanocomposites by the facile reflux treating method, exhibiting a superb capability in photodegradation of MB and H2 evolution.159 Moreover, by embedding Pd nanoparticles on the g-C3N4, a Pd/g-C3N4 metal–semiconductor heterojunction was constructed which demonstrated highly strengthened photocatalytic performance in bisphenol removal.160 Meanwhile, note that Li et al. have successfully obtained g-C3N4/noble metal by directly loading with Pt, Au and Pd NPs respectively via a conventional solution impregnation method.161 The functional catalyst/support system can unambiguously act as a tandem catalyst to effectively trigger the water reduction reaction to form H2 and activation of the as-formed H2 for further reduction of 4-nitrophenol to 4-aminophenol.
g-C3N4 could function as an effective solid stabilizer for chaperoning various ultrafine noble metal NPs.161 It is worthwhile to elucidate the functional mechanisms of g-C3N4/noble metal nanohybrids with improved photocatalytic performance. As an example of Au/g-C3N4 nanohybrids,157 a schematic diagram of the photocatalytic mechanism of Au/g-C3N4 is presented in Fig. 7a. Owing to surface plasmon resonance of noble metal,4 the as-prepared Au/g-C3N4 nanocomposites exhibit highly enhanced visible-light photocatalytic activity. On the one hand, the SPR effect of Au nanoparticle causes intense local electromagnetic fields, which can speed up the formation rate of holes and electrons within g-C3N4.158–161 Additionally, the favorable Fermi level of noble metal facilitates the separation of electrons and holes, which in turn enhances the quantum efficiency of g-C3N4, due to the intimate combination of noble metal/g-C3N4 nanohybrids (Fig. 7b).162 Moreover, the transfer of electrons shifts the Fermi level to more negative potential, thereby improving the reducibility of electrons in the Fermi level close to the CB of g-C3N4.163 On the other hand, the efficient utilization of sunlight can be realized due to SPR absorption in the visible-light region as well as UV light response of the interband transition of noble metal nanoparticles. Table 5 shows the photocatalytic properties of different g-C3N4/noble metal nanocomposite photocatalysts.
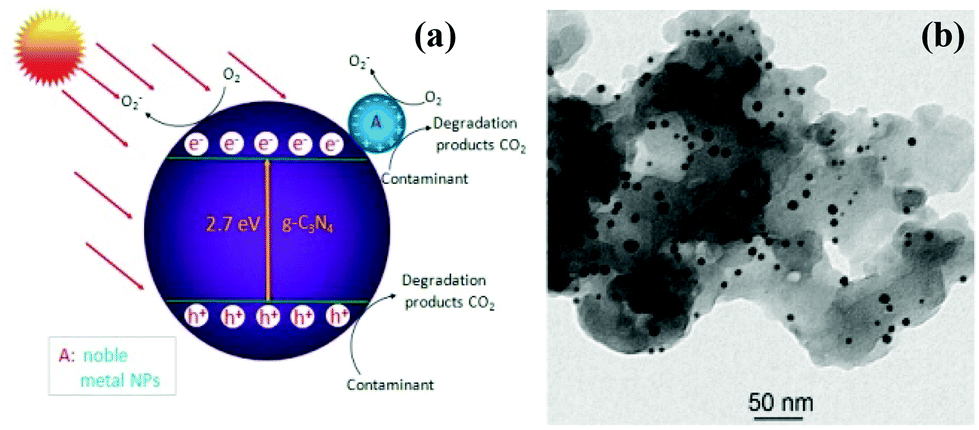 | ||
| Fig. 7 (a) Schematic diagram illustrating the photocatalytic degradation of organic contaminants over noble metal NPs/g-C3N4 hybrid under visible-light irradiation; (b) TEM image of AuNP/g-C3N4 nanohybrids. (Reprinted with permission from ref. 157. Copyright (2013) American Chemical Society.) | ||
| Composite photocatalyst | Mass fraction or mass of g-C3N4 | Typical parameters of photocatalytic experiments | Photocatalytic activity | Reference photocatalyst; photocatalytic activity | Enhancement factor over the reference photocatalyst | Reference |
|---|---|---|---|---|---|---|
| Au/g-C3N4 | Mass: 4.5% | Decomposing methyl orange (MO) under visible light | 92.6% degradation in 150 min | g-C3N4: 28.7% | 3.2 | 157 |
| Au: no data | No data | |||||
| Au/g-C3N4 | No data | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: triethanolamine | RH2: no data | Pt/g-C3N4; RH2: | No data | 15 |
| Pt-Au/g-C3N4 | No data | RH2: no data | No data | |||
| Pd-Au/g-C3N4 | No data | RH2: no data | ||||
| Ru-Au/g-C3N4 | No data | RH2: no data | ||||
| Ag-Au/g-C3N4 | No data | RH2: no data | ||||
| m-CNR-Au(2 nm) | No data | Reduction of 4-nitrophenol to 4-aminophenol under visible light | >96% in 5 min | No data | No data | 161 |
| m-CNR-Pd(3 nm) | No data | |||||
| m-CNR-Pt(2 nm) | No data | |||||
| m-CNR-Pt(2 nm) | No data | Photocatalytic H2 evolution under a 300 W Xe lamp; co-catalyst: no; sacrificial reagent: triethanolamine | >85% H2O conversion | No data | No data | |
| Ag/g-C3N4 | Ag: 0.1 g | Decomposing MO under visible light | MO of 74% | g-C3N4: 70%; | 1; 1.3 | 158 |
| Ag: 0.5 g | MO of 78% | P25 TiO2: 56% | 1.1; 1.4 | |||
| Ag: 1 g | MO of 86% | 1.2; 1.5 | ||||
| Ag: 2 g | MO of 91% | 1.3; 1.6 | ||||
| Ag: 5 g | MO of 92% | 1.3; 1.6 | ||||
| Ag/g-C3N4 | Ag: 2 g | Decomposing p-nitrophenol (PNP) under visible light | PNP of 98% | g-C3N4: 83%; | 1.2 | |
| P25 TiO2: <10% | >9.8 | |||||
| Ag/g-C3N4 | Mass: 99.5% | Decomposing MO methyl blue (MB) phenol under visible light | No data | g-C3N4: no data | 2.1 | 159 |
| No data | g-C3N4: no data | 1.8 | ||||
| g-C3N4: no data | 2.2 | |||||
| Ag/g-C3N4 | Mass: 90% | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: triethanolamine | No data | g-C3N4: no data | 30 | |
| Pd/g-C3N4 | No data | Decomposing bisphenol A under solar light | 93.9% degradation in 180 min | Bulk g-C3N4: 6% | 15.65 | 160 |
| P25 TiO2: 6.7% | 14.01 | |||||
| g-C3N4: 52.1% | 1.8 |
3.6. g-C3N4 based complex system
Although a number of g-C3N4-based two-component nanocomposites have been developed for enhanced visible light photocatalysis, there are still some shortages that need to be further addressed. With this aim, multicomponent complex heterojunction systems have been developed, in which two or more visible-light active components are integrated.164–167 For example, novel Ag/AgBr/g-C3N4 ternary composite photocatalysts were successfully constructed via the deposition–precipitation method, presenting a dramatically improved photocatalytic performance for degradation of MO as well as good stability under visible light.164The high photocatalytic activity and stability of Ag/AgBr/g-C3N4 are mainly attributed to the synergetic effect of the AgBr/g-C3N4 interface and metallic Ag (Fig. 8a and 8b). Namely, the matching energy band structure of AgBr/g-C3N4 and the excellent electron trapping role of metallic Ag ensured the efficient separation of electron–hole pairs, i.e. metallic Ag quickly evacuated the electrons on the side of AgBr which guaranteed the stability of Ag/AgBr/g-C3N4, shown in Fig. 8c. Moreover, Chai et al. designed and prepared g-C3N4–Pt-TiO2 three component nanocomposites via a facile chemical adsorption–calcination process, accompanied by remarkably enhanced performance for hydrogen production under visible light irradiation.165 The cocatalyst Pt deposited on TiO2 surfaces in the g-C3N4-TiO2 heterojunction is beneficial for exerting the synergistic effect existing between TiO2 and g-C3N4, which can strengthen the photogenerated carrier separation in space.
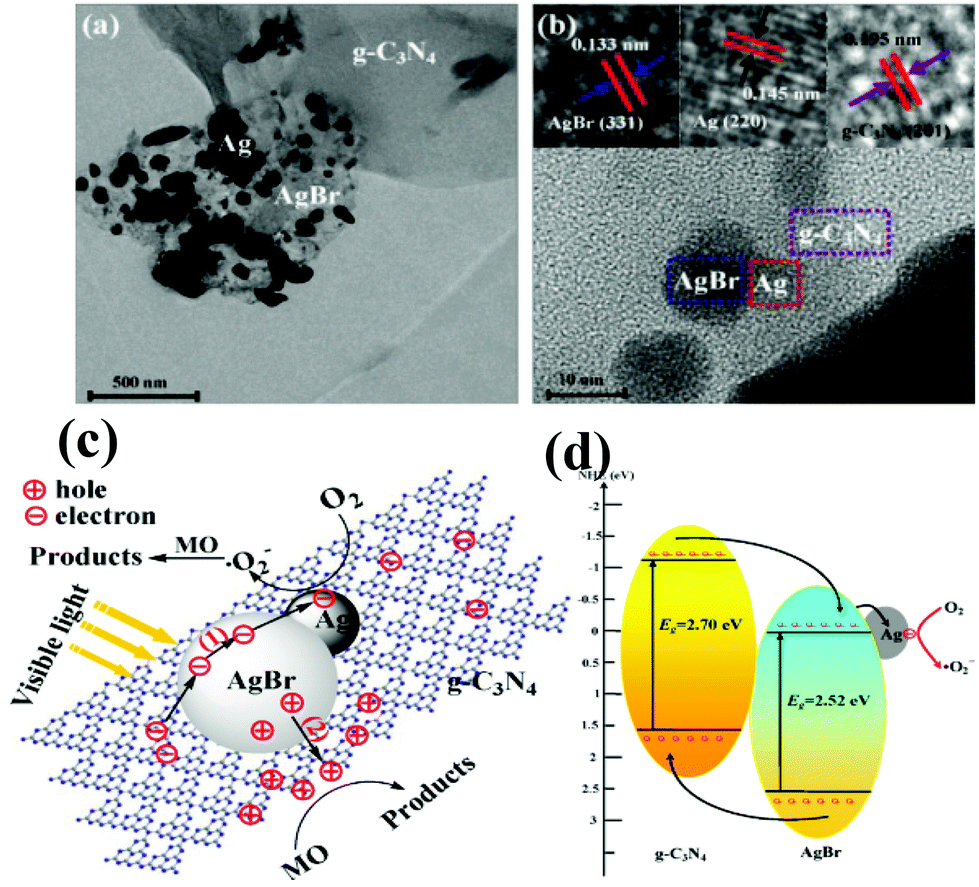 | ||
| Fig. 8 (a) TEM image and (b) HRTEM image of 50% Ag/AgBr/g-C3N4; (c) schematic diagram of electron–hole pairs separation and the possible reaction mechanism over Ag/AgBr/g-C3N4 photocatalyst under visible light irradiation. (Reproduced with permission from ref. 164, copyright 2013, Elsevier.) | ||
Inspiringly, a new type of Pt-TiO2/g-C3N4–MnOx quaternary composites was also fabricated by an impregnation method with an extraordinarily enhanced high H2 production capacity, firstly reported by S. Obregón and G. Colón.166 Such a marked increase in the photoactivity might be related to the incorporation of Pt and g-C3N4–MnOx into the charge separation process, which is in favor of retarding the charge recombination and improving the photoactivity for H2 production. A novel complex g-C3N4/CoO/graphene hybrid, with g-C3N4 embedded CoO particles covalently supported on a two-dimensional graphene sheet, is synthesized through a facile and scalable method by Jin and his co-workers.167 Above all, these significant achievements can pave the way for constructing g-C3N4-based multi-component heterojunctions and it is highly possible to optimize the visible light absorption, the charge carriers separation and transfer by adequately tailoring the structure of the photocatalysts. Recent progress on g-C3N4 based complex nanocomposites is summarized in Table 6.
| Composite photocatalyst | Mass or molar fraction of g-C3N4 | Typical parameters of photocatalytic experiments | Photocatalytic activity | Reference photocatalyst; photocatalytic activity | Enhancement factor over the reference photocatalyst | Reference |
|---|---|---|---|---|---|---|
| Ag/AgBr/g-C3N4 | Molar: 50% | Decomposing methyl orange (MO) under visible light | 95.3% degradation in 30 min | g-C3N4: 2.5% | 38.1 | 164 |
| Ag/AgBr: 62.3% | 1.5 | |||||
| Ag/AgBr/g-C3N4 | Molar: 50% | Decomposing 2-chlorophenol under visible light | 70.51% degradation in 4 h | No data | No data | |
| g-C3N4/Pt/TiO2 | Mass: 70% | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: triethanolamine | RH2: 0.178 mmol h−1 | Pt-loaded g-C3N4–TiO2; RH2: 0.124 mmol h−1 | 1.4 | 165 |
| Pt-TiO2/g-C3N4-MnOx | No data | Decomposing phenol under UV light | 96.4% degradation | Pt- TiO2: 96.5% | 0.9 | 167 |
| Time: no data | Pt-TiO2/g-C3N4: 96% | 1.0 | ||||
| Pt-TiO2/g-C3N4-MnOx | No data | Photocatalytic H2 evolution under visible light; co-catalyst: no; sacrificial reagent: isopropanol | RH2: 7.5 mmol h−1 g−1 | Pt- TiO2; RH2: 5.5 mmol h−1 g−1 | 1.36 | |
| Pt- TiO2/g-C3N4; RH2: 6.5 mmol h−1 g−1 | 1.15 |
4. Multifunctional applications
Semiconductor-mediated photocatalysis has attracted worldwide attention for its potential in environmental and energy-related applications.45,117–125 Nevertheless, the rapid recombination rate of photogenerated electron–hole pairs within photocatalytic materials results in low efficiency, thus limiting its practical applications. Therefore, the retardation of recombination of charge carriers is the key for the enhancement of photocatalytic performance of semiconductor photocatalysts. g-C3N4-semiconductor hybrid materials as a new class of photocatalysts have recently attracted wide research interest.138–141 In this regard, nanocomposites that combine g-C3N4 and other components could potentially provide desirable efficiency for separating electron–hole pairs. As documented above, the g-C3N4-based semiconductor photocatalysts have been widely used for the degradation of pollutants, photocatalytic hydrogen generation and photocatalytic conversion of carbon dioxide to methane fuel, etc. In this section the multiple applications of g-C3N4-based nanocomposites are briefly summarized.4.1. Photocatalytic degradation of pollutants
In recent years, a large quantity of efforts has been devoted to solving the widespread pollution of effluents and gaseous pollutants from urban and agricultural industries. Various catalytic techniques have been applied in environmental conservation. Photocatalysis has been widely used in environmental applications such as air purification,168–170 water disinfection,171,172 hazardous waste remediation173,174 and water decontamination.175Graphene carbon nitrogen, as a “rising star” material, has many exceptional properties, such as environmentally benign, stable physicochemical properties, large specific surface area and low bad-gap, etc.18–20 Hence, g-C3N4-based semiconductor photocatalysts have been extensively applied to photocatalytic degradation of environmental pollutants.89,90 These composites possess a high dye adsorption capacity, extended light absorption boundary, and accelerated charge transportation and separation properties.
For instance, a novel, multi-walled carbon nanotubes (CNT) modified white C3N4 composite (CNT/white C3N4) was fabricated by Xu et al.77 The CNT/C3N4 nanocomposite was prepared by the hydrothermal method through electrostatically-driven self-assembly. The CNT/C3N4 composite shows a significantly enhanced photocatalytic performance in MB dye removal. A possible photocatalytic mechanism of CNT/white C3N4 on the enhancement of visible light performance is proposed (Fig. 9a). Firstly, the electrons are motivated from the VB to the CB of C3N4 under visible light irradiation. Secondly, the photo-excited electrons are effectively collected by CNT, hindering the recombination process of the electron–hole pairs. Thirdly, the O2 adsorbed on the surface of the catalyst can be reduced to active species. The holes and the generated ˙OH can react with the organic dye and generate degradation products. Furthermore, the photocatalytic stability of CNT/white C3N4 was investigated through the repeated MB degradation experiments as shown in Fig. 9b. It is clear that the MB dye can be completely bleached after each photocatalytic run, and CNT/C3N4 is stable enough during the repeated experiments without significant loss of photocatalytic activity. Therefore, CNT/white C3N4 can be used as an effective photocatalyst for organic compound degradation with good stability.
 | ||
| Fig. 9 (a) Proposed photocatalytic mechanism of the CNT/white C3N4; (b) recycling in the repeated MB degradation experiments with CNT/white C3N4 under visible light irradiation. (Reproduced with permission from ref. 77. Copyright (2013) The Royal Society of Chemistry.) | ||
Moreover, WO3, a decent visible light catalyst, was also selected to combine with g-C3N4.88–90 Zang et al.88 reported that the g-C3N4/WO3 composite exhibited a significant enhancement of photocatalytic degradation of MO in water under visible light irradiation compared to the bare g-C3N4 and WO3. As illustrated in section 3 of g-C3N4/metal oxide (metal sulfide), in the g-C3N4/WO3 composite, holes activated on the VB of the WO3 would transfer to the VB of g-C3N4, while the electrons on CB of g-C3N4 may transfer to the CB of WO3 under light irradiation due to the well-matched energy band structure. Therefore, the photo-generated electrons and holes can be separated effectively in this way, and the recombination rate of the photo-generated electrons and holes would be hampered, which contributed to the superior photocatalytic reactivity. Moreover, this coupling heterojunction could also be used to remove the poisonous gaseous pollutant acetaldehyde gas (CH3CHO), which is a typical volatile organic compound (VOC) and exerts adverse effects on the health of humans. By incorporating g-C3N4 with different contents of WO3, Katsumata et al. studied the photocatalytic performance of this composite in acetaldehyde gas removal.90 As a result, they found that as the WO3 content increases, the photodegradation effect became more conspicuous, and the optimum mass ratio of g-C3N4 to WO3 for photocatalysis is two to eight.90 What is more, Huang et al. further utilized this novel composite to treat another two organic pollutants of MB and 4-CP.89 The highest degradation efficiency was achieved for the WO3/g-C3N4 (9.7%) sample, which induced 97% degradation of MB within 2 h and 43% degradation of 4-CP within 6 h under visible light irradiation, while pristine g-C3N4 only induced 81% degradation of MB within 3 h and 3% degradation of 4-CP within 6 h.89
Apart from CNT/C3N4 and WO3/g-C3N4 nanocomposites, composites of g-C3N4 and other semiconductor photocatalysts such as g-C3N4/TiO2,83,85 CeO2/g-C3N4,93 g-C3N4/CdS,42 g-C3N4/TaON,30 g-C3N4/Bi2WO6,105,106 BiOBr/g-C3N4,138,139 BiOI/g-C3N4140 and Pd/g-C3N4160 have been reported as efficient photocatalysts for decomposition of pollutants in water. Among these coupling systems, TiO2 as a famous UV light-driven and g-C3N4 as a robust visible-light-driven photocatalysts are favourably selected to be coupled for enhanced photocatalysis. Zhao et al. evaluated both the UV and visible-light photocatalytic activities of g-C3N4/TiO2 for degradation of phenol in water, and found that these composites exhibited excellent photocatalytic performance.83 The pseudo-first-order kinetic constant of phenol degradation on g-C3N4/TiO2 was 2.41 and 3.12 times higher than those on pristine g-C3N4 and TiO2 respectively, which could be ascribed to the wide absorption wavelength range and effective photogenerated charge separation. Moreover, Fu and co-workers reported that the g-C3N4 hybridized CdS composite is an efficient photocatalyst.42 The optimum activity of 0.7C3N4–0.3CdS photocatalyst is almost 20.5 and 3.1 times higher than those of individual C3N4 and CdS for methyl orange degradation and 41.6 and 2.7 fold higher for 4-aminobenzoic acid removal, respectively.42 Moreover, its activity is also much higher than those of C3N4–TiO2 and CdS–TiO2 composites, as well as famous N-modified TiO2. Of particular significance is that the present C3N4–CdS composites also demonstrate high stabilities under illumination, in contrast with CdS.42 The improvement in both performance and stability should be assigned to the effective separation and transfer of photogenerated charges originating from the well-matched overlapping band-structures and closely contacted interfaces.
Recently, the g-C3N4–Bi2WO6 composite photocatalysts were studied by Tian and co-workers for photocatalytic degrading MO under visible light irradiation.106 Encouragingly, the resulting g-C3N4–Bi2WO6 heterojunctions have a strong absorption in the visible light region and have apparently enhanced photocatalytic performances. This superior activity may be ascribed to the electronic interactions and charge equilibration between g-C3N4 and Bi2WO6, leading to the shift in the Fermi level and decreasing the conduction band potential of Bi2WO6. Simultaneously, the negative shift in the Fermi level of g-C3N4–Bi2WO6 and the high migration efficiency of photoinduced electrons can suppress the charge recombination effectively, resulting in the enhanced photocatalytic degradation of pollutants. Moreover, with bismuth subcarbonate ((BiO)2CO3) as a novel photocatalyst, we achieved new g-C3N4/(BiO)2CO3 nanojunctions by an in situ strategy of depositing (BiO)2CO3 nanoflakes onto the surface of g-C3N4 nanosheets through the efficient capture of atmospheric CO2 (crude material of (BiO)2CO3) at room temperature.176 These nanojunctioned photocatalysts showed excellent visible-light capability by degrading RhB and phenol, accompanied by good stability.
Hitherto, arrays of successful efforts of g-C3N4-based heterojunctions have also been made to degrade NOx, which have been considered worldwide as gaseous pollutants. Our groups have investigated the photocatalytic capability of novel 2D BiOBr/C3N4 nanojunctions in NOx removal.139 The NO removal ratios of BiOBr/C3N4 can be achieved to 32.7%, which is much higher than that of the pristine BiOBr (21.2%) and C3N4 (21.2%). The enhancement of photocatalytic performance of BiOBr/C3N4 could be ascribed to the synergistic effect of well-coupled crystal planes and band structure. To further boost the photocatalytic activities, we constructed the g-C3N4/g-C3N4 metal-free isotype heterojunction by a facile in situ method.81 The CN-TU dramatically exhibited an improved NO removal ratio of 47.6%, compared to host substrates of CN-U and CN-T. Besides, this metal-free isotype heterojunction demonstrated good stability as well. The strengthened photocatalytic performance of the g-C3N4/g-C3N4 isotype heterojunction can be directly ascribed to the efficiently accelerated charge separation across the heterojunction interface as well as prolonged lifetime of charge carriers.
4.2. Photocatalytic hydrogen generation
Hydrogen energy is considered as an ultimate clean fuel in the near future because of its high-energy capacity, environmental benignancy, and recycling utilization.96,177,178 Photocatalytic splitting of water into hydrogen and oxygen using semiconductor photocatalysts has been considered to be an attractive and promising approach to produce hydrogen energy. A range of semiconductor photocatalysts have been reported to catalyze the evolution of hydrogen from water. However, the practical applications of this strategy are limited on account of the prompt recombination of photogenerated electron–hole pairs within photocatalysts. Considering its visible-light-driven properties and high specific surface area, g-C3N4 can be considered as an efficient photocatalyst for enhancing the visible light utilization and accelerating the photo-induced charge transfer hampering the backward reaction, which will highly promote the photocatalytic H2-production activity.179,180With Ni(OH)2/g-C3N4 composites loaded with different contents of Ni(OH)2, Yu and co-workers96 studied their water splitting performance in triethanolamine aqueous solutions. The results demonstrated that Ni(OH)2 was an efficient co-catalyst for g-C3N4 photocatalytically producing H2. The optimal Ni(OH)2 loaded on g-C3N4 was found to be 0.5 mol%, giving a H2-production rate of 7.6 mmol h−1 with a quantum efficiency (QE) of 1.1% at 420 nm, depicted in Fig. 10a. The fact that the potential of Ni2+/Ni is lower than that of the CB of g-C3N4 and more negative than that of H+/H2 helps the electron to transfer from CB of g-C3N4 to Ni(OH)2 and then reduce H+ to H2 with a decent performance (Fig. 10(b)). This work proved that Ni(OH)2 is a very promising co-catalyst to combine with g-C3N4 for visible-light photocatalytic hydrogen production. Very recently, Hong et al.99 systematically studied the efficiency of H2 evolution for the NiS/g-C3N4 composite constructed by a simple hydrothermal method. This photocatalyst showed efficient hydrogen evolution (48.2 mmol h−1) under visible light when using triethanolamine as a sacrificial reagent. The optimal ratio was 1.1 wt% of NiS loaded on g-C3N4 and the rates of H2 production can be enhanced by about 250 times compared with the native C3N4. The highest apparent quantum efficiency was recorded at 1.9%, induced by 440 nm light irradiation. In order to further explore the possible application of g-C3N4/single metal oxide (metal sulfide) composites in hydrogen production, Hou and co-workers98 constructed MoS2/g-C3N4 nanojunctions and found that visible-light performance of H2 production of mpg-CN is significantly improved compared with bare MoS2 and g-C3N4. Inspiringly, the 0.5 wt% MoS2/mpg-CN performed better than 0.5 wt% Pt/mpg-CN under identical reaction conditions. The geometric similarity (layered morphology) of MoS2 and g-CN (Fig. 11), accompanied by the mesoporous architectures of mpg-CN, help in forming an intimate planar MoS2/mpg-CN interface, which can significantly promote the photoactivity of MoS2/g-C3N4. They also discovered that other layered transition metal dichalcogenides (such as WS2) are also efficient promoters of hydrogen production combined with g-C3N4. Herein they have presented not only an example of a catalyst made of abundant C, N, Mo and S elements for efficient H2 photosynthesis, but also a conceptual advance to rationally design and fabricate thin, effective interfacial 2D junctions between co-catalysts and semiconductors that have similar layered geometric architectures.
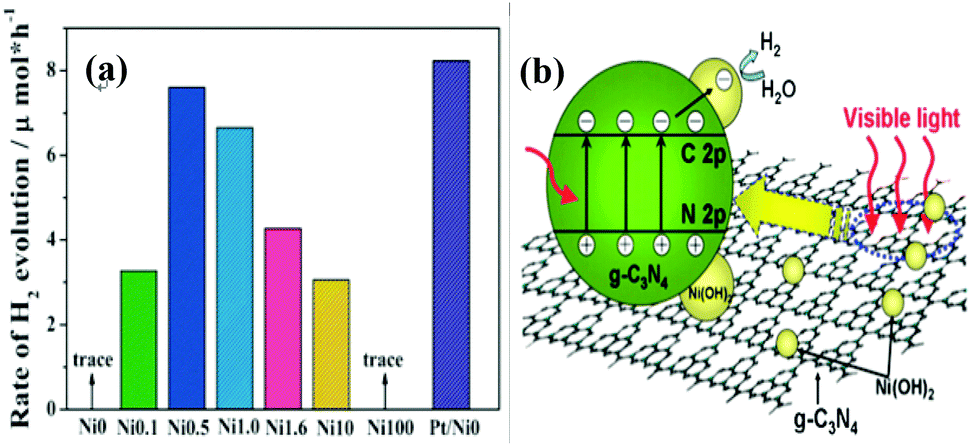 | ||
| Fig. 10 (a) Comparison of the photocatalytic activity of the Ni0, Ni0.1, Ni0.5, Ni1.0, Ni1.6, Ni10, Ni100 and Pt-deposited g-C3N4 samples for the photocatalytic H2 production from triethanolamine aqueous solution; (b) schematic illustration for the charge transfer and separation in the Ni(OH)2-modified g-C3N4 system under visible light irradiation. (Reprinted with permission from ref. 96. Copyright (2013) The Royal Society of Chemistry.) | ||
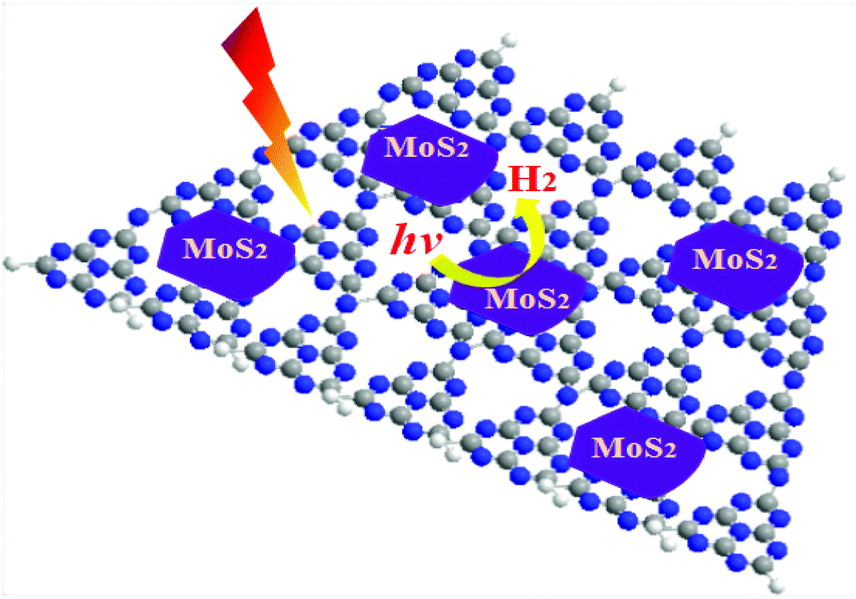 | ||
| Fig. 11 Schematic illustration of the idealized structural model of the resulting MoS2/g-CN layered junctions. | ||
Apart from the g-C3N4/single metal oxide (sulfide) composites, research studies have coupled C3N4 with other decent photocatalysts to further strengthen the H2 generation efficiency.123,155,165 Taking noble metals as an example, Di et al. reported that gold (Au) nanoparticles were successfully deposited on the surface of g-C3N4 by the deposition–precipitation method.155 The as-synthesized nanoscopic Au-semiconductor heterojunctions effectively accelerated the transfer of charges between g-C3N4 and Au, which can enable Au-g-C3N4 with excellent visible-light activity for hydrogen production. Besides, surface modifying Au/g-C3N4 with a second metal Pt (low over-potential for water reduction) further improved the activity of the photocatalytic system. This can be explained by the simultaneous optimization of electron transfer induced by the gold and chemical reactivity afforded by the secondary metal Pt. Moreover, Kang and his group systematically studied the g-C3N4–SrTiO3:Rh photocatalyst for improved H2 evolution under visible light irradiation.123 A high hydrogen evolution rate of 223.3 mmol h−1 was achieved using 0.1 g of as-prepared photocatalyst powder comprised of 20 wt% g-C3N4 and 80% SrTiO3:Rh (0.3 mol%), measured in aqueous methanol solution. A mechanism for the electron–hole separation of the g-C3N4–SrTiO3:Rh composite under visible light irradiation was proposed. The Rh ions doped into the SrTiO3 lattice structure formed a donor level from the VB to CB, which eased the transfer of photo-induced holes from the SrTiO3:Rh surface to the g-C3N4 surface and photo-induced electrons from the g-C3N4 surface to the SrTiO3:Rh surface, thereby preventing the recombination of electron–hole pairs. Also, Chai et al. investigated a ternary nanocomposite g-C3N4–Pt-TiO2 in photocatalytic hydrogen evolution.165 The visible-light-induced photocatalytic hydrogen evolution rate can be remarkably strengthened by coupling TiO2 with g-C3N4, and the g-C3N4–(Pt-TiO2) composite with a mass ratio of 70:30 shows the maximum photocatalytic H2 production rate (178 mmol h−1), as well as excellent photostability. Moreover, the cocatalyst Pt deposited on TiO2 surfaces is beneficial for exerting the synergistic effect existing between TiO2 and g-C3N4. Thus, the photogenerated electrons of g-C3N4 can directionally migrate to Pt–TiO2 due to the close interfacial connections and the synergistic effect existing between Pt–TiO2 and g-C3N4. The photogenerated electrons and holes can be efficiently separated in space, beneficial for retarding the charge recombination and improving the photoactivity. This study not only shows that g-C3N4, as an effective sensitizer of TiO2, can be widely used to improve photocatalytic H2 production, but also affords various methods for developing novel, steady and visible-light-responsive photocatalysts.
Also, graphene, a two-dimensional macromolecular sheet of carbon atoms with a honeycomb structure, extremely high specific surface area and thermal conductivity, showed a good photocatalytic performance for hydrogen or oxygen production.181 After the introduction of graphene sheets, g-C3N4 is immobilized to form layered composites, which will largely increase the BET and catalytic performance.32 In this system, graphene sheets act as conductive channels efficiently separating the photogenerated charge carriers as well as enhancing the visible-light capability of H2-production. The optimal graphene content was found to be 1.0 wt%, and the corresponding H2-production rate was 451 μmol h−1 g−1, which exceeded that of pure g-C3N4 more than 3.07 times. Normally, when g-C3N4 is immobilized on the surface of graphene sheets to form the layered composites, these photogenerated electrons on the CB of g-C3N4 tend to transfer to graphene sheets due to their excellent electronic conductivity, favoring the hole–electron separation. Meanwhile, Sui et al. reported that they have successfully loaded highly-dispersed conductive polymer polypyrrole (PPy) nanoparticles on the surface of g-C3N4.78 Surprisingly, they found that the H2 evolution rate of g-C3N4 loaded with 1.5 wt% PPy can increase up to 50 times compared with that of pristine g-C3N4, and the reaction proceeded in a pure water system excluding the need for sacrificial agents. We believe that this work can provide an effective route for developing efficient photocatalysts for photocatalytic hydrogen evolution from pure water systems.
4.3. Other applications
Solar fuels have been considered to be one of the most important renewable clean energy resources and photocatalytically converting CO2 into a valuable hydrocarbon fuel (CH4) is known to be a challenging but promising application for sustainable energy resources.79,182–186 In g-C3N4 based composites, Yuan et al. significantly obtained the red phosphor/g-C3N4 heterojunction with enhanced photocatalytic activities for solar fuels production.79 The introduction of g-C3N4 into the red phosphor surface led to considerable improvement of the photocatalytic activity for CO2 conversion into CH4 in the presence of water vapor. The enhancement could be attributed to the effective separation of photogenerated electrons and holes across the red phosphor/g-C3N4 heterojunction. Owing to the advantages of non-toxicity, low cost and abundance in nature, this active heterostructural red phosphor/g-C3N4 would have great potential for efficient solar fuels production. Very recently, In2O3/g-C3N4 hybrid photocatalysts were fabricated by Cao and his group, and the resulting In2O3–g-C3N4 hybrid structures exhibited considerable photocatalytic activities for CO2 reduction.102 After 4 h irradiation, the optimal In2O3/g-C3N4 (10 wt% In2O3) exhibited a CH4 production yield of 76.7 ppm (over 20 mg samples) without any cocatalyst, which is more than three times higher than that of individual g-C3N4 and more than four times higher than that of pure In2O3. The enhanced activities were attributed to the enhanced interfacial transfer of photogenerated electrons and holes between g-C3N4 and In2O3, leading to effective charge separation on both parts.Moreover, g-C3N4 based composites were also considered to be a promising candidate in photocatalytic disinfection.75 A novel class of metal-free ternary heterojunction photocatalysts was prepared by wrapping reduced graphene oxide and g-C3N4 sheets on crystals of cyclooctasulfur (α-S8), reported by Wang et al.75 Two distinctive structures were fabricated by wrapping reduced graphene oxide (RGO) and CN sheets in different orders. The first was RGO sheets sandwiched in a heterojunction of CN sheets and α-S8 (i.e., CNRGOS8), while the second structure was the other way around (i.e., RGOCNS8). Fascinatingly, both structures exhibited outstanding antibacterial activity under aerobic or anaerobic conditions irradiated with visible light (Fig. 12), which made us choose colibacillus as a representative microorganism to evaluate the photocatalytic water disinfection performances. This work not only provided new inroads into the exploration and understanding of the photocatalytic bacterial inactivation mechanisms, but also afforded to develop suitable protocols for solar-driven photocatalytic water disinfection under different conditions.
 | ||
| Fig. 12 Schematic illustration of the VLD photocatalytic bacterial inactivation mechanisms of (a) CNRGOS8 and (b) RGOCNS8 under aerobic conditions, and (c) CNRGOS8 and (d) RGOCNS8 under anaerobic conditions. CNRGOS8 was RGO sheets sandwiched in a heterojunction of CN sheets and α-S8, while RGOCNS8 was RGO wrapped in the heterojunction of CN sheets and α-S8. (Reprinted with permission from ref. 75. Copyright (2013) American Chemical Society.) | ||
In addition, g-C3N4-based heterojunctions are also utilized to perform as supercapacitors. Xu et al. mentioned that the g-C3N4/a-Fe2O3 hollow microspheres have been successfully prepared in the presence of a metal ion-containing reactable ionic liquid under the solvothermal conditions.91 The capacitance of the g-C3N4/a-Fe2O3 composites as electrode materials was tested by cyclic voltammetry and chronopotentiometry measurements, demonstrating that the g-C3N4/a-Fe2O3 composites were capable of delivering the largest specific capacitance and the highest coulombic efficiency. It can be assumed that the enhanced specific capacitance of the g-C3N4/a-Fe2O3 hollow microspheres could be attributed to hollow sphere-like structures, high BET surface area, incorporation of g-C3N4, and low electronic resistance. Thus, the g-C3N4/a-Fe2O3 hollow microspheres are promising candidates for high-performance supercapacitors.
We have made a summary of the design strategy, categories and their multifunctional applications of g-C3N4 based nanocomposites as illustrated in Fig. 13.
 | ||
| Fig. 13 Schematic illustration of the g-C3N4 based heterojunction/heterostructure and their multifunctional applications. | ||
5. Conclusion and perspectives
In summary, various types of g-C3N4-based nanocomposite systems have been designed and constructed. The incorporation of g-C3N4 into these nanocomposites can impart them with unique properties and induce enhanced performance, such as a high adsorption capacity, an extended light absorption range, and accelerated charge separation and transportation, which strengthens the overall photocatalytic performance. A wide range of heterostructures, including metal-free, metal/semiconductor, semiconductor/semiconductor, molecule/semiconductor, and multi-component heterostructures, have been explored for the improved photocatalysis by increasing the light absorption, improving the charge separation and transportation, enhancing the catalytic activity and prolonging the charge carrier lifetime. These nanocomposite photocatalysts have been widely used for photocatalytic degradation of pollutants, photocatalytic hydrogen generation, carbon dioxide storage and disinfection. The present review depicts the fabrication, microstructure and photocatalytic performance, as well as the photocatalytic mechanisms when g-C3N4 is coupled with an array of materials such as metal-free, single metal oxides (metal sulfides), composite oxides, halides, noble metals, etc. It is anticipated that the g-C3N4-based nanocomposites will receive ever increasing research attention in the future.Although considerable progress has been achieved, the studies in this field are still at the primary stage and further systematic investigations are needed. Rational design of complex heterostructures that could simultaneously facilitate efficient optical absorption, carrier generation, separation, transportation and utilizations is central for a new generation of highly efficient and robust photocatalysts. Significant challenges remain in the synthesis of such complex nanostructures with well-designed architectures and optimized charge cascading processes.
First, a fundamental understanding of the charge transport process in the multi-heterostructure photocatalysts is critical for the optimization of the charge cascading process to maximize the utilization of photogenerated charge carriers for desirable redox chemistry. While it is relatively straightforward to understand electron–hole separation and transportation in a two component heterostructure, the understanding and control of charge transport in multi-component heterostructures have become increasingly more complex, particularly on the nanometer scale. The interface properties of the photocatalysts determine the eventual efficiency of the photocatalytic system. To understand the charge generation, separation and transportation across these nanoscale interfaces is critical. Current investigations mainly focus on the overall apparent efficiency of the entire photocatalytic system. Detailed mechanism studies of the charge transfer process in photocatalysts integrating three or more components are scarce and will be necessary to further advance the field.
Secondly, the stability of g-C3N4-based nanocomposites is less addressed and will be the main challenge for photocatalyst development. A photocatalyst with very high efficiency is still useless if the lifetime of the catalyst is too short. For the majority of systems, chemical corrosion and/or photodegradation of photocatalysts are difficult to avoid. The promoted photocatalyst stability has only been achieved in a few examples to date, which usually however require highly complicated synthetic processes. To develop a stable photocatalytic system with high efficiency at low cost will be the central task to realize a practically viable photocatalyst for potential practical applications.
Thirdly, the mechanisms of photocatalytic enhancement by the g-C3N4-based semiconductor composite systems are partly unclear. The explanation of photocatalytic activity by the g-C3N4 content in the composites is still controversial. Therefore, more studies are required for promoting the general understanding of the enhancement mechanism of g-C3N4 based nanocomposites, especially employing advanced in situ techniques. Also, it is necessary to develop a uniform method to assess the photocatalytic performance, as the current evaluation methods are diverse. A photocatalyst working better in water treatment may show poor performance in air purification.
Finally, the rapid development of materials science and nanotechnology in the past few years has resulted in the creation of various advanced photocatalytic materials. Interesting properties may be explored by combining novel photocatalysts with g-C3N4. With a better understanding of the fundamental photocatalysis mechanism, assisted by the rapid development of advanced new nanomaterials, the bottleneck of environmental and energy-related global issues could be overcome in the near future. Overall, it is certain that there will be numerous exciting opportunities on g-C3N4 based nanocomposites, which require the combined efforts of scientists all over the world.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (51478070 and 51108487), the Natural Science Foundation Project of CQ CSTC (cstc2013jcyjA20018), the Science and Technology Project from Chongqing Education Commission (KJ1400617 and KJ130725), and the Innovative Research Team Development Program in University of Chongqing (KJTD201314).Notes and references
- C. C. Chen, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 206–4219 Search PubMed
.
- G. Liu, L. Z. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC
- D. Q. Zhang, G. S. Li and J. C. Yu, J. Mater. Chem., 2010, 20, 4529–4536 RSC
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 CAS
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed
- M. L. de Souza and P. Corio, Appl. Catal., B, 2013, 136–137, 325–333 CrossRef CAS PubMed
- M. Radoičić, Z. Šaponjić, I. A. Janković, G. Ćirić-Marjanović, S. P. Ahrenkiel and M. I. Comor, Appl. Catal., B, 2013, 136–137, 133–139 CrossRef PubMed
- M. B. Radoičić, I. A. Janković, V. N. Despotović, D. V. Šojić, T. D. Savić, Z. V. Šaponjić, B. F. Abramović and M. I. Čomor, Appl. Catal., B, 2013, 138/139, 122–127 CrossRef PubMed
- J. X. Wang, P. X. Wang, Y. T. Cao, J. Chen, W. J. Li, Y. Shao and D. Z. Li, Appl. Catal., B, 2013, 136–137, 94–102 CrossRef CAS PubMed
- T. T. Li, Y. M. He, H. J. Lin, J. Cai, L. Z. Dong, X. X. Wang, M. F. Luo, L. H. Zhao, X. D. Yi and W. Z. Weng, Appl. Catal., B, 2013, 138–139, 95–103 CrossRef CAS PubMed
- W. D. Shi, J. Q. Shi, S. Yu and P. Liu, Appl. Catal., B, 2013, 138–139, 184–190 CrossRef CAS PubMed
- D. G. Wang, H. F. Jiang, X. Zong, Q. Xu, Y. Ma, G. L. Li and C. Li, Chem. – Eur. J., 2011, 17, 1275–1282 CrossRef CAS PubMed
- Y. N. Guo, L. Chen, X. Yang, F. Y. Ma, S. Q. Zhang, Y. X. Yang, Y. H. Guo and X. Yuan, RSC Adv., 2012, 24, 656–4663 Search PubMed
- D. Q. He, L. L. Wang, D. D. Xu, J. L. Zhai, D. J. Wang and T. F. Xie, ACS Appl. Mater. Interfaces, 2011, 3, 3167–3171 CAS
- K. Maeda, Phys. Chem. Chem. Phys., 2013, 15, 10537–10548 RSC
- B. Siritanaratkul, K. Maeda, T. Hisatomi and K. Domen, ChemSusChem, 2011, 4, 7–78 CrossRef PubMed
- J. G. Hou, Z. Wang, W. B. Kan, S. Q. Jiao, H. M. Zhu and R. V. Kumar, J. Mater. Chem., 2012, 22, 7291–7299 RSC
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- K. Takanabe, K. Kamata, X. Wang, M. Antonietti, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 13020–13025 RSC
- F. Dong, L. W. Wu, Y. J. Sun, M. Fu, Z. B. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC
- B. Chai, T. Y. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC
- J. H. Liu, Y. W. Zhang, L. H. Lu, G. Wu and W. Chen, Chem. Commun., 2012, 48, 8826–8828 RSC
- Y. W. Zhang, J. H. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC
- J. D. Hong, X. Y. Xia, Y. S. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006–15012 RSC
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed
- Y. J. Wang, Z. X. Wang, S. Muhammad and J. He, CrystEngComm, 2012, 14, 5065–5070 RSC
- G. G. Zhang, J. S. Zhang, M. W. Zhang and X. C. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC
- J. S. Zhang, M. W. Zhang, R. Q. Sun and X. C. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC
- L. Ge, C. C. Han and J. Liu, Appl. Catal., B, 2011, 108, 100–107 CrossRef PubMed
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS
- F. Z. Su, S. C. Mathew, G. Lipner, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed
- L. Ge, C. C. Han, J. Liu and Y. F. Li, Appl. Catal., A, 2011, 409–410, 215–222 CrossRef CAS PubMed
- Y. Di, X. C. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834–838 CrossRef CAS
- C. Chang, Y. Fu, M. Hu, C. Y. Wang, G. Q. Shan and L. Y. Zhu, Appl. Catal., B, 2013, 142–143, 553–560 CrossRef CAS PubMed
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed
- Y. J. Zhang, T. Mori, J. H. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed
- H. J. Yan and H. Yan, Chem. Commun., 2011, 47, 4168–4170 RSC
- J. Fu, B. B. Chang, Y. L. Tian, F. N. Xi and X. P. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 CAS
- Y. Y. Wang, X. J. Bai, C. S. Pan, J. He and Y. F. Zhu, J. Mater. Chem., 2012, 22, 11568–11573 RSC
- D. L. Jiang, L. L. Chen, J. J. Zhu, M. Chen, W. D. Shi and J. M. Xie, Dalton Trans., 2013, 42, 15726–15734 RSC
- X. S. Zhou, B. Jin, L. D. Li, F. Peng, H. J. Wang, H. Yu and Y. P. Fang, J. Mater. Chem., 2012, 22, 17900–17905 RSC
- W. Liu, M. L. Wang, C. X. Xu and S. F. Chen, Chem. Eng. J., 2012, 209, 386–393 CrossRef CAS PubMed
- Y. J. Wang, Z. X. Wang, S. Muhammad and J. He, CrystEngComm, 2012, 14, 5065–5070 RSC
- H. McDaniel, P. E. Heil, C. L. Tsai, K. K. Kim and M. Shim, ACS Nano, 2011, 5, 7677–7683 CrossRef CAS PubMed
- M. Shim, H. McDaniel and N. Oh, J. Phys. Chem. Lett., 2011, 2, 2722–2727 CrossRef CAS
- X. C. Wang, W. B. Mi, E. Y. Jiang and H. L. Bai, Appl. Surf. Sci., 2009, 255, 4005–4010 CrossRef CAS PubMed
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC
- S. Polarz and M. Antonietti, Chem. Commun., 2002, 2593–2604 RSC
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS
- C. D. Liang, K. L. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 116, 5909–5913 CrossRef
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS
- M. Antonietti and C. Opin, J. Colloid Interface Sci., 2001, 6, 244–248 CrossRef CAS
- D. H. Chen, Z. Li, Y. Wan, X. J. Tu, Y. F. Shi, Z. X. Chen, W. Shen, C. Z. Yu, B. Tu and D. Y. Zhao, J. Mater. Chem., 2006, 16, 1511–1519 RSC
- C. D. Liang, Z. J. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 120, 3754–3776 CrossRef
- N. Y. Cheng, J. Q. Tian, Q. Liu, C. J. Ge, A. H. Qusti, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, Appl. Mater. Interfaces, 2013, 5, 6815–6819 CrossRef CAS PubMed
- Y. J. Zhang, A. Thomas, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 50–51 CrossRef CAS PubMed
- Y. G. Xu, H. Xu, L. Wang, J. Yan, H. M. Li, Y. H. Song, L. Y. Huang and G. B. Cai, Dalton Trans., 2013, 42, 7604–7613 RSC
- Y. Wang, Y. Di, M. Antonietti, H. R. Li, X. F. Chen and X. C. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS
- Y. Wang, H. R. Li, J. Yao, X. C. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC
- G. Liu, P. Niu, C. H. Sun, S. C. Smith, Z. G. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed
- M. Kawaguchi and K. Nozaki, Chem. Mater., 1995, 7, 257–264 CrossRef CAS
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 122, 451–454 CrossRef
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed
- Y. Q. Sun, C. Li, Y. X. Xu, H. Bai, Z. Y. Yao and G. Q. Shi, Chem. Commun., 2010, 46, 4740–4742 RSC
- J. G. Yu, L. F. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CAS
- J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394–16401 CAS
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed
- Y. B. Li, H. M. Zhang, P. R. Liu, D. Wang, Y. Li and H. J. Zhao, Small, 2013, 9, 3336–3344 CAS
- W. J. Wang, J. C. Yu, D. H. Xia, P. K. Wong and Y. C. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CAS
- L. Ge and C. C. Han, Appl. Catal., B, 2012, 117–118, 268–274 CrossRef CAS PubMed
- Y. G. Xu, H. Xu, L. Wang, J. Yan, H. M. Li, Y. H. Song, L. Y. Huang and G. B. Cai, Dalton Trans., 2013, 42, 7604–7613 RSC
- Y. Sui, J. H. Liu, Y. W. Zhang, X. K. Tian and W. Chen, Nanoscale, 2013, 5, 9150–9155 RSC
- Y. P. Yuan, S. W. Cao, Y. S. Liao, L. S. Yin and C. Xue, Appl. Catal., B, 2013, 140–141, 164–168 CrossRef CAS PubMed
- J. S. Zhang, M. W. Zhang, R. Q. Sun and X. C. Wang, Angew. Chem., Int. Ed., 2012, 124, 10292–10296 CrossRef
- F. Dong, Z. W. Zhao, T. Xiong, Z. L. Ni, W. D. Zhang, Y. J. Sun and W. K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS
- B. Chai, X. Liao, F. K. Song and H. Zhou, Dalton Trans., 2014, 43, 982–989 RSC
- S. S. Zhao, S. Chen, H. T. Yu and X. Quan, Sep. Purif. Technol., 2012, 99, 50–54 CrossRef CAS PubMed
- K. Sridharan, E. Jang and T. J. Park, Appl. Catal., B, 2013, 142–143, 718–728 CrossRef CAS PubMed
- J. G. Yu, S. H. Wang, J. X. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC
- J. Ran, J. Yu and M. Jaroniec, Green Chem., 2011, 13, 2708–2713 RSC
- L. A. Silva, S. Y. Ryu, J. Choi, W. Choi and M. R. Hoffmann, J. Phys. Chem. C, 2008, 112, 12069–12073 CAS
- Y. P. Zang, L. P. Li, Y. Zuo, H. F. Lin, G. S. Li and X. F. Guan, RSC Adv., 2013, 3, 13646–13650 RSC
- L. Y. Huang, H. Xu, Y. P. Li, H. M. Li, X. N. Cheng, J. X. Xia, Y. G. Xu and G. B. Cai, Dalton Trans., 2013, 42, 8606–8616 RSC
- K. Katsumata, R. Motoyoshi, N. Matsushita and K. Okada, J. Hazard. Mater., 2013, 260, 475–482 CrossRef CAS PubMed
- L. Xu, J. X. Xia, H. Xu, S. Yin, K. Wang, L. Y. Huang, L. G. Wang and H. M. Li, J. Power Sources, 2014, 245, 866–874 CrossRef CAS PubMed
- S. Ye, L. G. Qiu, Y. P. Yuan, Y. J. Zhu, J. Xia and J. F. Zhu, J. Mater. Chem. A, 2013, 1, 3008–3015 CAS
- L. Y. Huang, Y. P. Li, H. Xu, Y. G. Xu, J. X. Xia, K. Wang, H. M. Li and X. N. Cheng, RSC Adv., 2013, 3, 22269–22279 RSC
- L. Y. Huang, H. Xu, R. X. Zhang, X. N. Cheng, J. X. Xia, Y. G. Xu and H. M. Li, Appl. Surf. Sci., 2013, 283, 25–32 CrossRef CAS PubMed
- X. S. Zhou, B. Jin, R. Q. Chen, F. Peng and Y. P. Fang, Mater. Res. Bull., 2013, 48, 1447–1452 CrossRef CAS PubMed
- J. G. Yu, S. H. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Technol., 2013, 3, 1782–1789 CAS
- M. Xu, L. Han and S. H. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 12533–12540 CAS
- Y. D. Hou, A. B. Laursen, J. S. Zhang, G. G. Zhang, Y. S. Zhu and X. C. Wang, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed
- J. D. Hong, Y. S. Wang, Y. B. Wang, W. Zhang and R. Xu, ChemSusChem, 2013, 6, 2263–2268 CrossRef CAS PubMed
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC
- W. Liu, M. L. Wang, C. X. Xu and S. f. Chen, Chem. Eng. J., 2012, 209, 386–393 CrossRef CAS PubMed
- S. W. Cao, X. F. Liu, Y. P. Yuan, Z. Y. Zhang, Y. S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef CAS PubMed
- Z. Y. Jin, N. Y. Murakami, T. Tsubota and T. Ohno, Appl. Catal., B, 2014, 150–151, 479–485 CrossRef CAS PubMed
- M. Shalom, S. Inal, D. Neherb and M. Antonietti, Catal. Today, 2014, 225, 185–190 CrossRef CAS PubMed
- Y. J. Wang, X. J. Bai, C. S. Pan, J. He and Y. F. Zhu, J. Mater. Chem., 2012, 22, 11568–11573 RSC
- Y. L. Tian, B. B. Chang, J. L. Lu, J. Fu, F. N. Xi and X. P. Dong, ACS Appl, 2013, 5, 7079–7085 CrossRef CAS PubMed
- T. T. Li, L. H. Zhao, Y. M. He, J. Cai, M. F. Luo and J. J. Lin, Appl. Catal., B, 2013, 129, 255–263 CrossRef CAS PubMed
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed
- E. M. P. Steinmiller and K. S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20633–20636 CrossRef CAS PubMed
- Y. Surendranath, M. Dinca and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326–6336 CrossRef CAS PubMed
- L. Ge, C. C. Han, X. L. Xiao and L. L. Guo, Appl. Catal., B, 2013, 142–143, 414–422 CrossRef CAS PubMed
- Y. J. Wang, Z. X. Wang, S. Muhammad and J. He, CrystEngComm, 2012, 14, 5065–5070 RSC
- S. Zhu, T. Xu, H. Fu, J. Zhao and Y. Zhu, Environ. Sci. Technol., 2007, 41, 6234 CrossRef CAS
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS
- C. S. Pan, J. Xu, Y. J. Wang, D. Li and Y. F. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS
- F. J. Zhang, F. Z. Xie, S. F. Zhu, J. Liu, J. Zhang, S. F. Mei and W. Zhao, Chem. Eng. J., 2013, 228, 435–441 CrossRef CAS PubMed
- S. Kumar, T. Surendar, A. Baruah and V. Shanker, J. Mater. Chem. A, 2013, 1, 5333–5340 CAS
- Y. Liu, G. Chen, C. Zhou, Y. D. Hu, D. G. Fu, J. Liu and Q. Wang, J. Hazard. Mater., 2011, 190, 75–80 CrossRef CAS PubMed
- Y. M. He, J. Cai, T. T. Li, Y. Wu, H. J. Lin, L. H. Zhao and M. F. Luo, Chem. Eng. J., 2013, 215–216, 721–730 CrossRef CAS PubMed
- S. Kumar, B. Kumar and S. T. V. Shanker, Mater. Res. Bull., 2014, 49, 310–318 CrossRef CAS PubMed
- H. W. Kang, S. N. Lim, D. S. Song and S. B. Park, Int. J. Hydrogen Energy, 2012, 37, 11602–11610 CrossRef CAS PubMed
- S. Q. Zhang, Y. X. Yang, Y. N. Guo, W. Guo, M. Wang, Y. H. Guo and M. X. Huo, J. Hazard. Mater., 2013, 261, 235–245 CrossRef CAS PubMed
- S. M. Wang, D. L. Li, C. Sun, S. G. Yang, Y. Guan and H. He, Appl. Catal., B, 2014, 144, 885–892 CrossRef CAS PubMed
- X. Zhang, Z. Ai, F. Jia and L. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CAS
- J. Henle, P. Simon, A. Frenzel, S. Scholz and S. Kaskel, Chem. Mater., 2007, 19, 366–373 CrossRef CAS
- M. A. Gondala, X. Chang, M. A. Ali, Z. H. Yamania, Q. Zhou and G. Ji, Appl. Catal., A, 2011, 397, 192–200 CrossRef PubMed
- L. Ye, J. Liu, C. Gong, L. Tian, T. Peng and L. Zan, ACS Catal., 2012, 2, 1677–1683 CrossRef CAS
- L. Ye, C. Gong, J. Liu, L. Tian, T. Peng, K. Deng and L. Zan, J. Mater. Chem., 2012, 22, 8354–8360 RSC
- L. Ye, L. Tian, T. Peng and L. Zan, J. Mater. Chem., 2011, 21, 12479–12484 RSC
- Y. Feng, L. Li, J. Li, J. Wang and L. Liu, J. Hazard. Mater., 2011, 192, 538–544 CrossRef CAS PubMed
- Y. Fan, Y. Huang, J. Yang, P. Wang and G. Cheng, Environ. Sci. Technol., 2011, 45, 1593–1600 CrossRef PubMed
- J. Xu, W. Meng, Y. Zhang, L. Li and C. Guo, Appl. Catal., B, 2011, 107, 355–362 CrossRef CAS PubMed
- W. L. Huang and Q. Zhu, Comput. Mater. Sci., 2008, 43, 1101–1108 CrossRef CAS PubMed
- L. Ye, L. Zan, L. Tian, T. Peng and J. Zhang, Chem. Commun., 2011, 47, 6951–6953 RSC
- J. Jiang, K. Zhao, X. Xiao and L. Zhang, J. Am. Chem. Soc., 2012, 134, 4473–4476 CrossRef CAS PubMed
- L. Q. Ye, J. Y. Liu, Z. Jiang, T. Y. Peng and L. Zan, Appl. Catal., B, 2013, 142–143, 1–7 CAS
- Y. J. Sun, W. D. Zhang, T. Xiong, Z. W. Zhao, F. Dong, R. Q. Wang and W. K. Ho, J. Colloid Interface Sci., 2014, 418, 317–323 CrossRef CAS PubMed
- D. L. Jiang, L. l. Chen, J. J. Zhu, M. Chen, W. D. Shi and J. M. Xie, Dalton Trans., 2013, 42, 15726–15734 RSC
- X. J. Wang, Q. Wang, F. T. Li, W. Y. Yang and Y. Zhao, Chem. Eng. J., 2013, 234, 361–371 CrossRef CAS PubMed
- H. Wang, J. Gao, T. Guo, R. Wang, L. Guo, Y. Liu and J. Li, Chem. Commun., 2012, 48, 275–277 RSC
- Z. Lou, B. Huang, X. Qin, X. Zhang, Z. Wang, Z. Zheng, H. Cheng, P. Wang and Y. Dai, Catal. Today, 2011, 175, 256–263 CrossRef PubMed
- L. Liu, H. Xu, H. Li, Y. Xu, J. Xia and S. Yin, J. Phys. Chem. Solids, 2012, 73, 523–529 CrossRef CAS PubMed
- H. Xu, J. Yan, Y. G. Xu, Y. H. Song, H. M. Li, J. X. Xia, C. J. Huang and H. L. Wan, Appl. Catal., B, 2013, 129, 182–193 CrossRef CAS PubMed
- C. R. Crowell and S. M. Sze, Solid-State Electron., 1966, 9, 1035–1048 CrossRef CAS
- Y. Di, X. C. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834–838 CrossRef CAS
- J. S. Jang, H. G. Kim and J. S. Lee, Catal. Today, 2012, 185, 270–277 CrossRef CAS PubMed
- P. V. Kamat, J. Phys. Chem. Lett., 2012, 3(5), 663–672 CrossRef CAS
- N. Serpone and A. V. Emeline, J. Phys. Chem. Lett., 2012, 3, 673–677 CrossRef CAS
- K. Y. Lee, R. Hahn, M. Altomare, E. Selli and P. Schmuki, Adv. Mater., 2013, 25, 6133–6137 CrossRef CAS PubMed
- H. Chen, S. Chen, X. Quan, H. T. Yu, H. M. Zhao and Y. B. Zhang, J. Phys. Chem. C, 2008, 112(25), 9285–9290 CAS
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS PubMed
- S. N. Basahel, K. Y. Lee, R. Hahn, P. Schmuki, S. M. Bawakeda and S. A. Al-Thabaiti, Chem. Commun., 2014, 48, 6123–6125 RSC
- Y. Di, X. C. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834–838 CrossRef CAS
- H. Y. Zhu, X. Chen, Z. F. Zheng, X. B. Ke, E. Jaatinen, J. C. Zhao, C. Guo, T. F. Xie and D. J. Wang, Chem. Commun., 2009, 7524–7526 RSC
- N. Y. Cheng, J. Q. Tian, Q. Liu, C. J. Ge, A. H. Qusti, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6815–6819 CAS
- Y. X. Yang, Y. N. Guo, F. Y. Liu, X. Yuan, Y. H. Guo, S. Q. Zhang, W. Guo and M. X. Huo, Appl. Catal., B, 2013, 142–143, 828–837 CrossRef CAS PubMed
- X. J. Bai, R. L. Zong, C. X. Li, D. Liu, Y. F. Liu and Y. F. Zhu, Appl. Catal., B, 2014, 147, 82–91 CrossRef CAS PubMed
- C. Chang, Y. Fu, M. Hu, C. y. Wang, G. q. Shan and L. Y. Zhu, Appl. Catal., B, 2013, 142–143, 553–560 CrossRef CAS PubMed
- X. H. Li, X. C. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170–2174 RSC
- D. B. Ingram, P. Christopher, J. L. Bauer and S. Linic, ACS Catal., 2011, 1, 1441–1447 CrossRef CAS
- S. W. Cao, Z. Yin, J. Barber, F. Y. C. Boey, S. C. J. Loo and C. Xue, ACS Appl. Mater. Interfaces, 2011, 4, 418–423 Search PubMed
- J. Cao, Y. J. Zhao, H. L. Lin, B. Y. Xu and S. F. Chen, Mater. Res. Bull., 2013, 48, 3873–3880 CrossRef CAS PubMed
- B. Chai, T. Y. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC
- S. Obregón and G. Colón, Appl. Catal., B, 2014, 144, 775–782 CrossRef PubMed
- J. T. Jin, X. G. Fu, Q. Liu and J. Y. Zhang, J. Mater. Chem. A, 2013, 1, 10538–10545 CAS
- J. G. Yu, J. C. Yu, M. K. P. Leung, W. K. Ho, B. Cheng, X. J. Zhao and J. C. Zhao, J. Catal., 2003, 217, 69–78 CAS
- J. G. Yu, W. G. Wang, B. Cheng and B. L. Su, J. Phys. Chem. C, 2009, 113, 6743–6750 CAS
- J. G. Yu, L. J. Zhang, B. Cheng and Y. R. Su, J. Phys. Chem. C, 2007, 111, 10582–10589 CAS
- J. C. Yu, W. K. Ho, J. G. Yu, H. Yip, P. K. Wong and J. C. Zhao, Environ. Sci. Technol., 2005, 39, 1175–1179 CrossRef CAS
- J. G. Yu, Y. R. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984–1990 CrossRef CAS
- B. Cheng, Y. Le and J. G. Yu, J. Hazard. Mater., 2010, 177, 971–977 CrossRef CAS PubMed
- J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394–16401 CAS
- J. G. Yu, Q. J. Xiang, J. R. Ran and S. Mann, CrystEngComm, 2010, 12, 872–879 RSC
- W. D. Zhang, Y. J. Sun, F. Dong, W. Zhang, S. Duan and Q. Zhang, Dalton Trans., 2014, 43, 12026–12036 RSC
- J. G. Yu and J. R. Ran, Energy Environ. Sci., 2011, 4, 1364–1371 CAS
- J. G. Yu, Y. Hai and M. Jaroniec, J. Colloid Interface Sci., 2011, 357, 223–228 CrossRef CAS PubMed
- H. J. Yan, Y. Chen and S. M. Xu, Int. J. Hydrogen Energy, 2012, 37, 125–133 CrossRef CAS PubMed
- S. W. Cao and J. G. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS
- S. Patchkovskii, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Heine and G. Seifert, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10439–10444 CrossRef CAS PubMed
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed
- H. F. Cheng, B. B. Huang, Y. Y. Liu, Z. Y. Wang, X. Y. Qin, X. Y. Zhang and Y. Dai, Chem. Commun., 2012, 48, 9729–9731 RSC
- X. Y. Chen, Y. Zhou, Q. Liu, Z. D. Li, J. G. Liu and Z. G. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 3372–3377 CAS
- H. Xu, S. X. Ouyang, P. Li, T. Kako and J. H. Ye, ACS Appl. Mater. Interfaces, 2013, 5, 1348–1354 CAS
- X. L. Liang, X. Dong, G. D. Lin and H. B. Zhang, Appl. Catal., B, 2009, 88, 315–322 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |