
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe intramolecular hydrogen bonded–halogen bond: a new strategy for preorganization and enhanced binding†
Asia Marie S.
Riel‡
,
Daniel A.
Decato‡
,
Jiyu
Sun
,
Casey J.
Massena
,
Morly J.
Jessop
and
Orion B.
Berryman
 *
*
University of Montana, 32 Campus Drive, Missoula, MT, USA. E-mail: orion.berryman@umontana.edu
First published on 21st June 2018
Abstract
Natural and synthetic molecules use weak noncovalent forces to preorganize structure and enable remarkable function. Herein, we introduce the intramolecular hydrogen bonded–halogen bond (HB–XB) as a novel method to preorganize halogen bonding (XBing) molecules, while generating a polarization-enhanced XB. Positioning a fluoroaniline between two iodopyridinium XB donors engendered intramolecular hydrogen bonding (HBing) to the electron-rich belt of both XB donors. NMR solution studies established the efficacy of the HB-XB. The receptor with HB–XBs (G2XB) displayed a nearly 9-fold increase in halide binding over control receptors. Gas-phase density functional theory conformational analysis indicated that the amine stabilizes the bidentate conformation. Furthermore, gas-phase interaction energies showed that the bidentate HB–XBs of G2XBme2+ are more than 3.2 kcal mol−1 stronger than the XBs in a control without the intramolecular HB. Additionally, crystal structures confirm that HB–XBs form tighter contacts with I− and Br− and produce receptors that are more planar. Collectively the results establish the intramolecular HB–XB as a tractable strategy to preorganize XB molecules and regulate XB strength.
Introduction
Preorganization is a central tenet of supramolecular chemistry that facilitates precise molecular function and higher order self-assembly.1−3§ Preorganized biomolecular structures rely on noncovalent interactions for their critical role in natural processes (e.g. catalysis, ion channels, signaling, nutrient transport, and antibodies). For example, the exceptional catalytic activity of phosphatase is aided by the secondary and tertiary structure imparted by noncovalent interactions. The rate of phosphate hydrolysis is enhanced by 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 over the uncatalyzed reaction.4 Nature's proficiency continues to inspire synthetic studies “beyond the molecule,” as principles governing protein–ligand and protein–protein interactions are the same in synthetic systems.5–9 Thus, developing new preorganization methods is valuable to all fields impacted by supramolecular chemistry. In this article, we introduce a novel strategy to preorganize structure and enhance halogen bonding (XBing)—the hydrogen bonded-halogen bond (HB–XB).
21 over the uncatalyzed reaction.4 Nature's proficiency continues to inspire synthetic studies “beyond the molecule,” as principles governing protein–ligand and protein–protein interactions are the same in synthetic systems.5–9 Thus, developing new preorganization methods is valuable to all fields impacted by supramolecular chemistry. In this article, we introduce a novel strategy to preorganize structure and enhance halogen bonding (XBing)—the hydrogen bonded-halogen bond (HB–XB).
Preorganization of small molecules has largely focused on macrocycles since the seminal cation binding reports of Cram, Lehn, and Pedersen.1,2,10–13 More recently, preorganized macrocycles,14–18 rotaxanes,19,20 and catenanes21 have also successfully sequestered anionic and neutral guests. The utility of macrocyclization is unmistakable, yet synthetic challenges have encouraged the development of acyclic molecules that are preorganized by noncovalent interactions (e.g. hydrogen bonding,22–27 steric effects,28,29 ion-pairing,30,31 and π-π stacking32–34). In particular, intramolecular hydrogen bonds (HBs) have been effective at preorganizing structure, including helicies,35,36 cavitands37–39 and ion transporters.40 Internal41 and external22,26 intramolecular HBing are the two predominate strategies used for HB preorganization.42 Internal intramolecular HBs (proximal to the binding site) usually share the HB donor between the receptor and the guest, which typically weakens the HB interaction to the guest. Alternatively, external intramolecular HBs (away from the binding site) can compete with the guest binding to the desired location. In contrast, the unique characteristics of a XB donor provides an opportunity for internal intramolecular HB preorganization that also enhances binding.
The XB—an attractive and highly directional noncovalent interaction between a polarizable electron-deficient halogen and an electron rich Lewis base43—is finding diverse applications in chemistry44–46 and biochemistry.47,48 The strict linearity of the XB arises from electronic anisotropy of the halogen donor. Polarization produces a partial positive σ-hole at the distal end of the atom and an electron-rich region circling the halogen.49¶|| XB directionality has been exploited in selective anion binding,44,50 organocatalysis,29,51,52 crystal engineering,53–55 and self-assembly.56–59 Considering the required linear directionality of the XB interaction, XB based molecules should benefit more from preorganization than other less directional interactions. However, few XB receptors are noncovalently preorganized (Fig. 1).29,60–63**
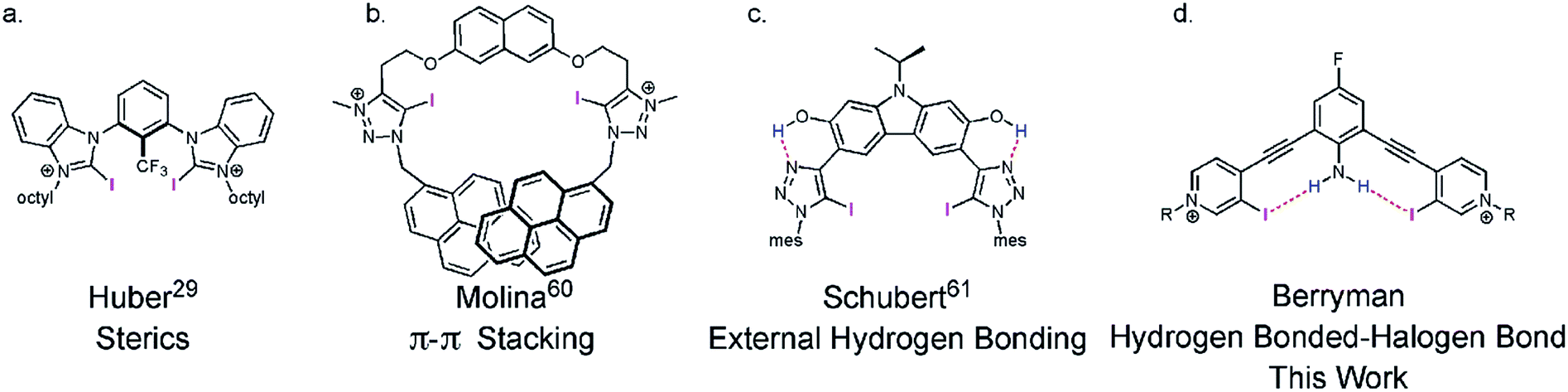 | ||
| Fig. 1 Representative noncovalent preorganization strategies for XB receptors. | ||
Polarization-enhanced hydrogen bonding—where the heteroatom of a HB donor concurrently accepts a HB—is an established method to strengthen HBs.64,65 However, despite many studies comparing the XB and HB, few have evaluated how they influence each other. Simultaneous XBing and HBing to a single Lewis base has been the primary focus, with evaluations in the gas, solution, and solid phases. Collectively the diverse findings—including both competitive and cooperative effects, geometric and energetic orthogonality, and unique anion binding selectivity—necessitate further study.66–74 Studies evaluating the direct influences on each other—electron-rich belt of a XB donor or XBing to an electronegative region of a HB donor heteroatom—have been largely limited to intermolecular structural database and computational evaluations.75,76†† Computational reports of intermolecular HBing to XB donors suggest that HBs can have both cooperative and noncoooperative effects on XB strength.66,77–86 More recently, a computational and protein data bank analysis found that ligand binding is improved by up to 250-fold when XB donors on the ligand accept HBs from the protein.87 Remarkably, solution studies of HBing to XB donors are nonexistent.‡‡ Herein, we introduce a polarization-enhanced XB. Findings suggest that the intramolecular HB–XB can be used to noncovalently preorganize a molecule while simultaneously enhancing the XB interaction in solution, solid, and in silico.
Results and discussion
Design and synthesis of 1,3-bis(4-ethynylpyridinium) receptors
Recently our lab developed a bisethynyl receptor that binds anions and neutral Lewis bases with two iodopyridinium XB donors.88,89 The alkynes promote rigidity and directionality, but their low rotational barrier allows the receptors to adopt three planar binding conformations (Fig. 2). Our interest in bidentate XB interactions prompted us to preorganize the receptor. Macrocyclization and external intramolecular HBing (away from the binding site) were not synthetically tractable. However, we hypothesized that preorganization could be accomplished by HBing to the electron-rich belt of the XB donors with an electron-deficient aniline (Fig. 1d). The fluorinated core was introduced to increase the HB strength of the –NH2 as halogens are regarded as poor acceptors. Initially, we pursued a trifluorinated derivative but synthetic difficulties prevented this possibility. We refer to these second-generation receptors as G2XB and G2HB (Scheme 1).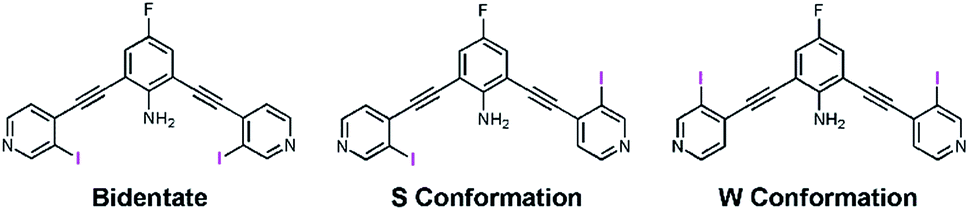 | ||
| Fig. 2 ChemDraw representations of the three planar conformations obtained by rotating about the alkyne bonds: the bidentate conformation (left), where both XB donors are convergent; the S conformation (middle), where the XB donors are on opposite sides of the molecule; and the W conformation (right), where both XBs are directed away from the amine. | ||
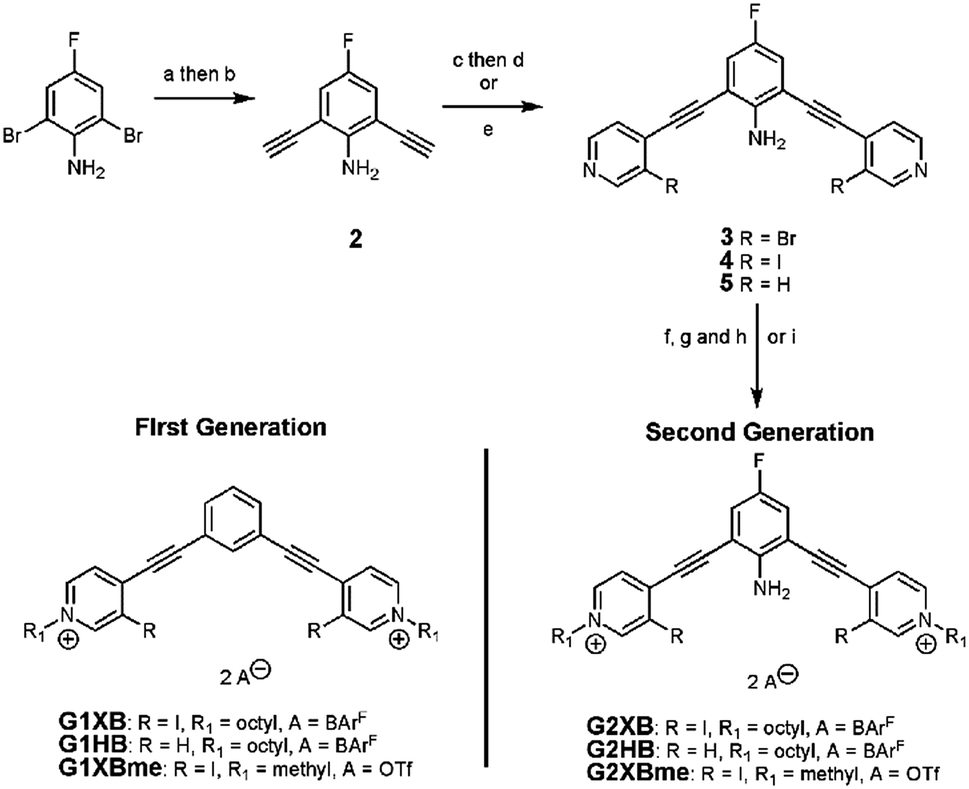 | ||
| Scheme 1 Synthesis of G2XB, G2HB and G2XBme receptors. Reagents and conditions: (a) TMS-acetylene, Pd(PPh3)2Cl2, Cu(I)I, DIPEA, DMF, overnight, N2, 60 °C, 90%; (b) K2CO3, MeOH/DCM (1:1 v/v), 4 hours, rt, 73%; (c) 3-bromo-4-iodopyridine, Pd(PPh3)2Cl2, Cu(I)I, DIPEA, DMF, overnight, N2, rt, 90%; (d) NaI, Cu(I)I, 1,4-dioxane, trans-N,N′-dimethylcyclohexane-1,2-diamine, microwave reactor, 150 °C, 5.5 hours, 81%; (e) 4-iodopyridine, Pd(PPh3)2Cl2, Cu(I)I, DIPEA, DMF, overnight, N2, 60 °C, 58%; (f) octyl triflate, DCM, rt, overnight, 54%; (g) TBA+Cl−, MeCN, overnight, 77%; (h) Na+BArF−, DCM, rt, overnight, 63%; (i) methyl triflate, DCM, rt, overnight, 89%. | ||
The synthesis of G2XB, G2HB and G2XBme is outlined in Scheme 1. 2,6-bis(ethynyl)-4-fluoroaniline (2) was synthesized by Sonogashira90 cross-coupling 2,6-dibromo-4-fluoroaniline with trimethylsilylacetylene followed by removal of the trimethylsilyl protecting groups. Precursors 3 and 5 were synthesized by Sonogashira cross coupling 2 at the iodo-functionality of 3-bromo-4-iodopyridine or 4-iodopyridine, respectively. The XB donor iodines of 4 were installed by microwave assisted halogen exchange of 3. Alkylation of the pyridines with octyl triflate activated the XB and HB donors of 4 and 5 respectively and enabled organic solubility. Anion metathesis of the triflate (OTf−) counteranions for noncoordinating tetrakis (3,5-bis(trifluoromethyl)phenyl)borate (BArF−)91 anions produced G2XB and G2HB (see ESI†). The methyl derivative, G2XBme, was similarly synthesized for X-ray diffraction studies. G1XB, G1HB and G1XBme were synthesized as previously reported.88
Solution assessment of HB–XB preorganization and enhanced XB
1H NMR titrations were conducted to probe intramolecular HB–XB preorganization and XB enhancement in G2XB (Table 1). Association constants were determined by HypNMR 200892 for each receptor and the tetra-n-butylammonium (TBA+) halides (Cl−, Br− and I−). All titrations were performed in 60% CD3NO2/40% CDCl3 at 25 °C to ensure solubility and to prevent the binding constants from exceeding the limit of NMR. Both XB receptors, G2XB and G1XB, exhibit anion-induced upfield pyridinium proton shifts, characteristic of XBing in solution.93 In contrast, the pyridinium protons of the HB receptors, G2HB and G1HB, only shifted downfield, consistent with HBing in solution (see ESI†).| G2XB | G1XB | G2HB | G1HB | |||||
|---|---|---|---|---|---|---|---|---|
| K 11 (M−1) | K 12 (M−1) | K 11 (M−1) | K 12 (M−1) | K 11 (M−1) | K 12 (M−1) | K 11 (M−1) | K 12 (M−1) | |
|
a The K11 and K12 values are reported as the average of three titration experiments. All titrations were performed in 40% CDCl3/60% CD3NO2; errors are estimated at 10%. Tetra-n-butylammonium halides were used and titrations were performed at 25 °C. HypNMR 2008 was used to fit changes in chemical shift to a stepwise 1:1 and a 1:2 host-guest binding model. Continuous refinements of multiple isotherms provided stability constants (Ka) for all host-guest complexes in solution.
|
||||||||
| Cl− | 23700 |
25 | 2630 | 37 | 2500 | 47 | 9040 | 38 |
| Br− | 32900 |
35 | 4690 | 32 | 2110 | 44 | 1150 | 38 |
| I− | 36900 |
28 | 4380 | 28 | 1750 | 44 | 1030 | 33 |
Intramolecular HB–XBs enhance halide binding by nearly 9-fold over G1XB, which lacks intramolecular HB–XBs. The halide K11 values for G2XB are 23700 M−1 for Cl−, 32900 M−1 for Br− and 36900 M−1 for I−. In contrast, G1XB binds halides much more weakly with K11 values of 2630 M−1 for Cl−, 4690 M−1 for Br−, and 4380 M−1 for I−. Additionally, both G2XB and G1XB prefer larger halides (I− ≈ Br− > Cl−) which could be attributed to the size selective binding pocket and HSAB complementarity of the XB.89,94 The second binding event (K12) is quite weak for all receptors and likely represents nonspecific ion pairing to balance the charge.
To verify that the amine forms intramolecular HBs instead of HBing directly with the anions, the binding of G2XB was compared with three control receptors. G2XB binds halides more than an order of magnitude greater than G2HB, which contains the amine but lacks XB donors. The K11 values for G2HB are 2500 M−1 for Cl−, 2110 M−1 for Br−, and 1750 M−1 for I−. Furthermore, the amine of G2HB marginally increases binding for both Br− and I− and even decreases Cl− binding when compared to G1HB, which lacks both the amine and fluorine (G1HBK11 values of 9040 for Cl−, 1150 for Br−, and 1030 M−1 for I−). Thus, the amine does not significantly HB to the halides in this system.
Comparing the binding of G2HB to G1XB establishes that the XB is more effective at binding Br− and I− than the HBs in G2HB. The K11 values of G1XB (4690 M−1 for Br− and 4380 M−1 for I−) are more than double the K11 values of G2HB (2110 M−1 for Br− and 1750 M−1 for I−). Collectively these studies support that intramolecular HB–XBs preorganizes the receptor and significantly enhances halide binding.
NMR analysis of chemical shifts, hydrogen/deuterium (H/D) exchange rates, and rotational barriers can provide solution evidence of intramolecular HBs to strong acceptors.95,96§§ In contrast, there is a deficiency of NMR studies on HBing to weak organic halogen acceptors.97 Chemical shift analysis and H/D exchange experiments on this system support that intramolecular HBing occurs in G2XB. The NH21H NMR resonance in G2XB was shifted downfield by 0.52 ppm (5.30 ppm, CDCl3) as compared to G2HB (4.78 ppm, CDCl3). The downfield resonance in G2XB is indicitive of deshielded protons which suggest that intramolecular HBing is occuring. H/D exchange experiments were conducted on G2XB and G2HB. G2XB had a slower H/D exchange than G2HB, which is attributed to intermolecular HBing and increased steric interactions in G2XB (see ESI†) 98,99 Additionally, variable temperature (VT) 1H NMR was used to evaluate intramolecular HBing. Upon warming, the NH2 signals for both G2XB and G2HB shift upfield.35,36¶¶ However, the shift in G2XB is slightly more upfield (see ESI†), consistent with intramolecular HBing. Together, the solution studies demonstrate that intramolecular HB-XB preorganization is operable and contributes to the improved halide recognition.
Computational evaluations of receptor conformations and XB enhancement
Gas-phase density functional theory (DFT) calculations further quantified the role of intramolecular HB–XBing in preorganization and XB augmentation. To simplify calculations, three planar conformations of G2XBme2+ were evaluated. Calculations were performed with the B3LYP functional |||| using the Gaussian 09 suite of programs.100 The 6-31+G (d,p) basis set was employed for all atoms except nitrogen and iodine. To appropriately account for the HB donor the aug-cc-pVTZ basis set was used for nitrogen.101 To model the polarizable iodine the LANL2DZdp and effective core potential (ECP) was used.102 The LANL2DZdp ECP basis set was downloaded from the EMSL Basis Set Exchange.103 Crystal structures were used as starting positions for all calculations. Optimized geometries and frequencies were calculated to confirm molecules were at local minima (details in ESI†).Single point energy calculations of G2XBme2+ illustrate that intramolecular HB–XBs stabilize the receptor (Fig. 3). The number of intramolecular HB–XBs directly correlates with receptor stability. The bidentate conformation, with two intramolecular HBs, is more stable than the W conformation (no HB–XB) by 1.29 kcal mol−1. The S conformation, with one intramolecular HB contact, is less stable than the bidentate conformation by 0.61 kcal mol−1. The W conformation, lacking intramolecular HB–XBs, is the least stable and is 0.68 kcal mol−1 higher in energy than the S conformation.
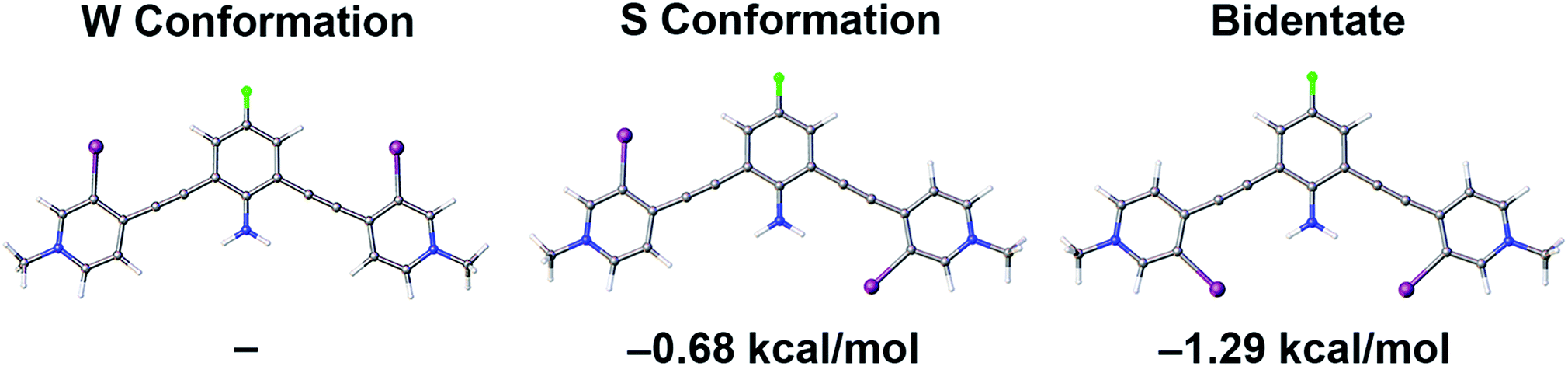 | ||
| Fig. 3 Gas-phase DFT single point energy calculations of the three planar conformations of G2XBme2+ highlight intramolecular HB–XB stabilization. Ball and stick models were produced from lowest energy conformations. | ||
To further analyze how intramolecular HBs stabilize G2XB, conformational analysis about the alkynes of G2XBme2+ and G1XBme2+ were examined. G2XBme2+ favors the bidentate conformation over the S conformer by over 0.60 kcal mol−1 which is comparable to the single point energy calculations. In contrast, G1XBme2+ favors the S conformer by 0.16 kcal mol−1 over the bidentate conformation, highlighting the critical proximity of the HBing amine (see ESI†). Notably, addition of the intramolecular HBs increases the rotational barrier between the bidentate and S conformation by 1.64 kcal mol−1. Both single point energy and alkyne conformational driving studies further support the hypothesis that HB–XB preorganizes bidentate XB conformations of G2XB.
Computations were also used to model how the intramolecular HB influences the strength of the XB. The effect is illustrated by electrostatic potential (ESP) maps of G1XBme2+, G2XBme2+, and two derivatives of G2XBme2+ (Fig. 4). G2XBme2+ (Fig. 4b) has a larger, more electrophilic region at the σ-hole than G1XBme2+ (Fig. 4a). This augmentation is attributed to the additional polarization by the intramolecular HB. The HB further polarizes the electron density around the halogen which strengthens the XB interaction. *** Additional calculations verify that the fluorine atom is not the cause of this effect. The derivative of G2XBme2+ without the fluorine (Fig. 4c) has a similar σ-hole to G2XBme2+. However, when the amine is removed but the fluorine is retained, the σ-hole is markedly reduced (Fig. 4d).
 | ||
| Fig. 4 ChemDraw and ESP maps of G1XBme2+ (a), G2XBme2+ (b), G2XBme2+ with no fluorine (c) and G2XBme2+ with no amine (d). All ESP maps are displayed on the same scale. Electron-deficient regions are blue and electron-rich regions are red. | ||
The magnitude by which HBing enhances bidentate XBing in this system was evaluated by computing gas-phase interaction energies with Br−. The interaction energy is computed as the difference between the complex and the isolated consituents in the same geometry as the complex (see ESI†). G2XBme2+ and the derivative of G2XBme2+ with no amine (no HB–XBs), were compared. The presence of the amine in G2XBme2+ strengthened the bidentate XB interaction by more than 3.23 kcal mol−1 over the receptor with no HB–XB. Together, these calculations corroborate the solution data and the dual role of the HB–XB to enhance the σ-hole and promote preorganization.
HB–XB impact on solid-state features
The intramolecular HB–XBs in G2XBme2+ promote the bidentate conformation. In contrast, previous solid-state studies of G1XBme2+ produced structures with multiple conformations. Here, we present crystal structures of G2XBme2+, G2XB2+, and G1XBme2+ that illustrate intramolecular HB–XBs facilitate planar conformations with stronger XB contacts for anion recognition. The receptors crystallize in the bidentate conformation with every halide, allowing direct comparison of the XB contacts and geometries of each receptor (crystal growth conditions and details in ESI†).Bidentate XB structures
Both G2XBme2+ and G1XBme2+ crystallized with Br− (Fig. 5) forming bidentate XB interactions with one Br−. As designed, G2XBme2+ forms intramolecular HBs that influence both the receptor conformation and the strength of the XB. The intramolecular HBs in G2XBme2+·2Br− have N–H⋯I distances and angles of 2.96 (10) Å, 165 (6)° and 3.14 (14) Å, 155 (5)° that correlate well with a CSD search. ††† The amine protons preorganize the complex of G2XBme2+·2Br− resulting in pyridnium rings that twist out of coplanarity with the fluoroaniline core by only 4.8 (2) and 7.8 (2)°. In contrast, G1XBme2+·2Br− which lacks intramolecular HBs, is more distorted, with the pyridinium rings twisting out of coplanarity with the benzene core by 15.18 (12) and 14.13 (12)°. Additionally, the intramolecular HBs strengthen the XB. The XB distances and angles in G2XBme2+·2Br− are shorter and more linear with values of 3.2358 (11) Å, 176.86 (19)° and 3.2787 (11) Å, 166.78 (19)° (RIBr values of 0.82 and 0.83). The distances and angles of G1XBme2+·2Br− are longer at 3.3938 (5) Å, 169.9 (1)° and 3.3905 (5) Å, 171.13 (10)° (RIBr values of 0.87 and 0.87). The second Br− anion in both structures interacts with other receptor molecules through C–H HB and ion-pairing to balance the two positive charges associated with the receptor. The significant reduction (up to 0.158 Å) in HB–XB (G2XBme2+·2Br−) distance over the non-HB–XB (G1XBme2+·2Br−) supports the HB–XB enhancement observed in the computational and solution studies.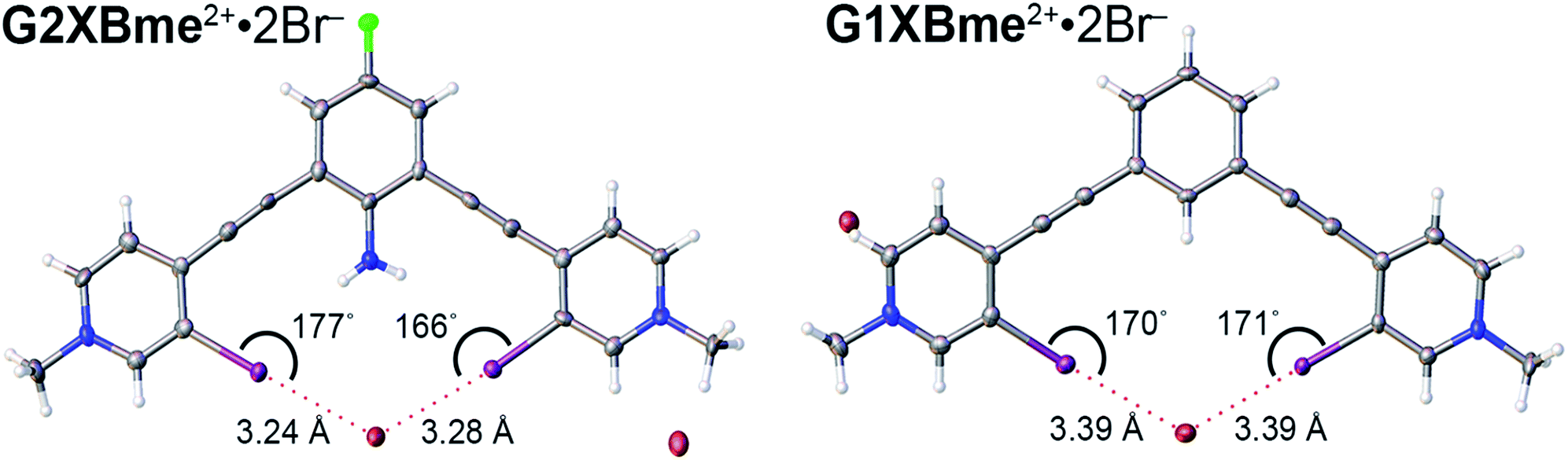 | ||
| Fig. 5 Bidentate XBing conformations of G2XBme2+·2Br− (left) and G1XBme2+·2Br− (right). XB distances and angles are displayed. Thermal ellipsoids are drawn at the 50% probability level. G1XBme2+·2Br− crystallized with a methanol molecule which HBs with noncoordinating Br− and is omitted for clarity. | ||
Crystals of G2XB2+ and G1XBme2+ were also obtained with I− (Fig. 6) and the structural features parallel the Br− complexes. Both G1XBme2+ and G2XB2+ form bidentate XB interactions with one I−. Again, G2XB2+ forms intramolecular HBs to both XB iodine donors in G2XB2+·2I− with N–H⋯I distances and angles of 2.94 (13) Å, 168 (8)° and 3.00 (7) Å, 170 (8)°. These HBs preorganize the complex of G2XB2+·2I− resulting in pyridinium rings that twist out of coplanarity with the fluoroaniline core by only 2.4 (3) and 2.8 (3)°. In comparison, the pyridinium rings of G1XBme2+·2I− deviate from planarity with the core benzene by 14.0 (2) and 14.8 (2)°. Again, the HB–XBs produce closer, stronger XB contacts with the anion. The XB distances and angles in G2XB2+·2I− are 3.363 (11) Å, 168.7 (3)° and 3.3865 (11) Å, 176.9 (2)° (RII values are 0.82 and 0.83). The weaker XBs in G1XBme2+2I− have distances and angles of 3.5235 (6) Å, 169.82 (19)° and 3.5085 (7) Å, 170.70 (16)° (RII values are 0.87 and 0.86). Also, the second I− anion in both systems interacts with other receptor molecules through C–H HB and ion-pairing to balance the two positive charges associated with the receptor. The nearly 0.16 Å reduction in the XB contacts and more planar conformations in G2XB2+ over G1XBme2+ support that intramolecular HB–XBs facilitate preorganization and XB enhancement.
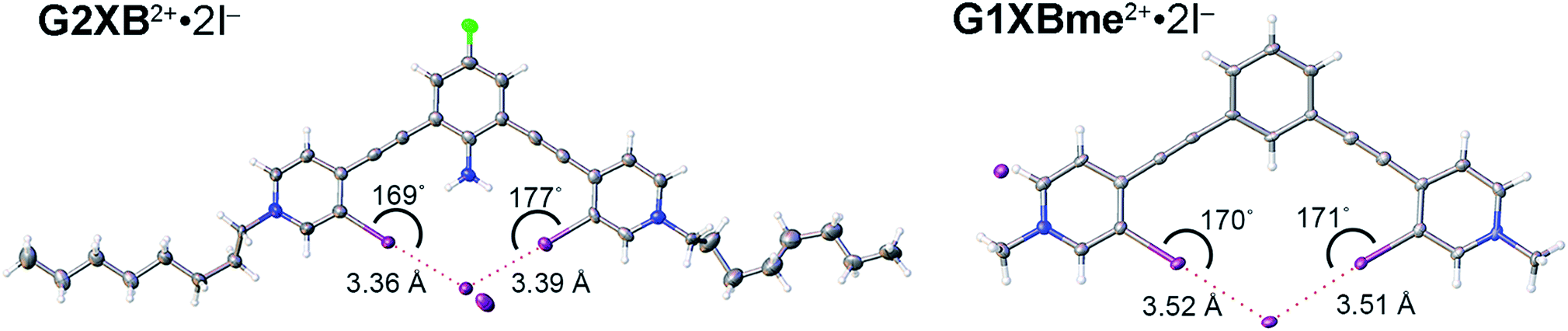 | ||
| Fig. 6 Bidentate XB conformations of G2XB2+·2I− (left) and G1XBme2+·2I− (right). XB distances and angles are displayed. Thermal ellipsoids are drawn at the 50% probability level. G1XBme2+·2I− crystallized with a methanol molecule which exhibits HBing with noncoordinating I− and is omitted for clarity. | ||
A bidentate structure with Cl− was also obtained for G1XBme2+ but not G2XBme2+ (Fig. 7). The XB distances and angles are 3.1530 (9) Å, 171.08 (9)° and 3.3527 (9) Å, 162.19 (9)° (RICl values of 0.82 and 0.87). The second anion is held above the complex through C–H HB and ion-pairing that help balance the two positive charges associated with the receptor. The pyridinium rings of G1XBme2+·2Cl− are twisted out of planarity with the benzene core by 3.83 (11) and 8.33 (11)°. This structure is more planar than the Br− and I− structures of G1XBme2+. In contrast, the XBs of G1XBme2+·2Cl− are not as linear as G2XB2+ and G2XBme2+.
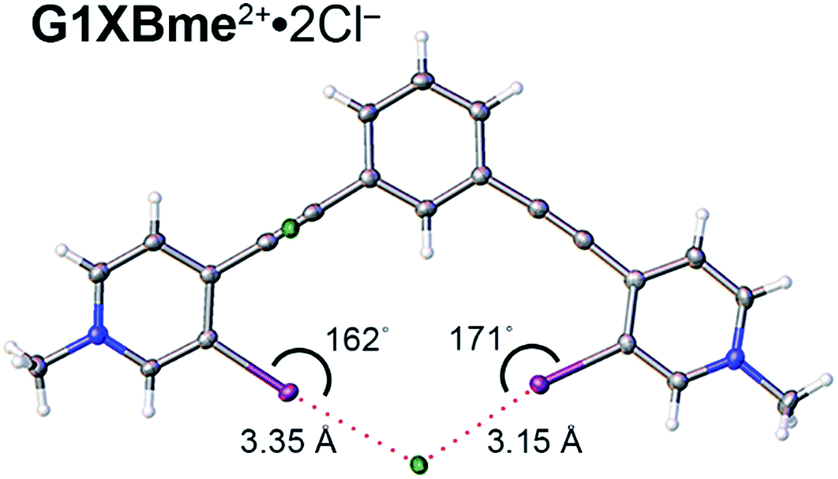 | ||
| Fig. 7 Bidentate binding conformation of G1XBme2+·2Cl−. XB distances and angles are displayed. Thermal ellipsoids are drawn at the 50% probability level. | ||
Monodentate XB structures
Crystallization with the OTf− anion induces monodentate XBing for both G2XBme2+ and G1XBme2+. However, G2XBme2+·2OTf−, with intramolecular HBs crystallizes in both the bidentate (Fig. 8a) and monodentate S conformation (Fig. 8b) (≈50/50 disorder). The bidentate conformer of G2XBme2+·2OTf− contains one OTf− molecule in the binding pocket that accepts two XBs to two separate oxygen atoms. The amine HBs to the XB donors with N–H⋯I distances and angles of 2.9672 (6) Å, 168.1 (2)° and 3.0546 (3) Å, 166.5 (3)°. The bidentate XB distances and angles are 3.195 (10) Å, 172.7 (3)° and 3.280 (9) Å, 148.4 (2)° (RIO values of 0.90 and 0.93). | ||
| Fig. 8 3D prints generated from the crystal structures (vdW radii) of disordered G2XBme2+·2OTf− the bidentate (a) and S conformations (b) and G1XBme2+·2OTf− in the W conformation (c). | ||
The monodentate S conformer of G2XBme2+·2OTf− provides a unique comparison of two types of XB donors within the same structure, one that accepts an amine HB (HB–XB) and one that does not. The HB–XB donor forms a stronger XB to the OTf− with a distance and angle of 2.908 (8) Å, 175.91 (18)° (RIO value of 0.82). Whereas the other XB has a distance and angle of 3.089 (6) Å, 168.1 (2)° (RIO value of 0.87). The greater than 0.18 Å reduction in XB distance (between the HB–XB and non-HB–XB donors) is comparable to the Br− and I− structures discussed previously.
The crystal of G1XBme2+·2OTf−—lacking the preorganizing amine—adopts the W conformation (Fig. 8c). Crystallographic symmetry of this structure dictates a single unique XB contact with a distance and angle of 2.886 (3) Å and 169.28 (10)° (RIO value of 0.82). This contact is shorter than those observed in G2XBme2+·2OTf−, likely a result of each OTf− anion accepting only one XB. Unfortunately, the structural differences prevent a direct comparison of XB strength between G2XBme2+·2OTf− and G1XBme2+·2OTf−.
The crystals of G2XBme2+ and G2XB2+ confirm that intramolecular HB–XBs can preorganize a receptor while simultaneously enhancing the XB. The Br−and I− comlexes show a marked reduction in XB distance, signifying an increase in XB strength. Furthermore, G2XBme2+ and G2XB2+ are more planar due to intramolecular HB–XB preorganization. The amine limits the monodentate conformation observed in G2XBme2+·2OTf−, which further supports preorganization. These crystallographic studies demonstrate the dual function of the intramolecular HB–XB to simultaneously preorganize and strengthen the XB.
Conclusions
In this work we have introduced a polarization-enhanced XB, the intramolecular HB–XB, as a practical strategy to preorganize halogen containing molecules and augment XB strength. Solution studies of a series of four receptors demonstrated that HB–XBing enhances halide binding by nearly 9-fold over the control receptors. Simultaneous preorganization and enhancement of the XB was confirmed by gas-phase DFT calculations of single point energies, ESP maps, interaction energies and alkyne conformational analyses. In the solid-state, contracted HB–XB distances with I− and Br− and more planar HB–XB receptors supported the gas-phase and solution findings. These results highlight that the intramolecular HB–XB is a new method to both preorganize structure and regulate XB strength.The unique characteristics of the XB continue to inspire new approaches in chemistry and biochemistry. We hypothesize several benefits of the HB–XB such as: (i) gentle (non-rigid) preorganization, halogens are relatively weak HB acceptors and we envision that molecules employing HB–XBs would enable both preorganization and induced fit binding; (ii) hydrophobicity, the size and lipophilic nature of the halogen will sterically shield intramolecular HBs without drastically increasing the hydrophilicity of the host. These properties will benefit numerous applications including those related to self-assembly, drug development and synthetic ion channels. Studies to understand the factors that influence intramolecular HB–XB strength (e.g., solvent, electron donating/withdrawing effects and HSAB complementarity between HB donor and XB donor) are currently underway in our lab.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Foundation (NSF) CAREER CHE-1555324, the Center for Biomolecular Structure and Dynamics CoBRE (NIH NIGMS grant P20GM103546), Montana University System MREDI 51030-MUSRI2015-02, and the University of Montana (UM). The X-ray crystallographic data were collected using a Bruker D8 Venture, principally supported by NSF MRI CHE-1337908. We thank Earle Adams for general NMR guidance and Nicholas Wageling for insightful computational discussions.Notes and references
- D. J. Cram, Angew. Chem., Int. Ed., 1986, 25, 1039–1057 CrossRef
.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef PubMed
-
J. B. Wittenberg and L. Isaacs, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons Ltd, Chichester, UK, 2012, vol. 1, ch. 3, pp. 25–44 Search PubMed
- C. Lad, N. H. Williams and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5607–5610 CrossRef PubMed
- M. H. M. Olsson, W. W. Parson and A. Warshel, Chem. Rev., 2006, 106, 1737–1756 CrossRef PubMed
- A. Gutteridge and J. Thornton, J. Mol. Biol., 2005, 346, 21–28 CrossRef PubMed
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef PubMed
- P. T. R. Rajagopalan and S. J. Benkovic, Chem. Rec., 2002, 2, 24–36 CrossRef PubMed
- A. Pohorille, M. A. Wilson and G. Shannon, Life, 2017, 7, 23 CrossRef PubMed
- D. J. Cram and G. M. Lein, J. Am. Chem. Soc., 1985, 107, 3657–3668 CrossRef
- D. J. Cram, T. Kaneda, R. C. Helgeson, B. Brown, C. B. Knobler, E. Maverick and K. N. Trueblood, J. Am. Chem. Soc., 1985, 107, 3645–3657 CrossRef
- J.-M. Lehn, Angew. Chem., Int. Ed., 1990, 29, 1304–1319 CrossRef
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef PubMed
- J. J. Christensen, D. J. Eatough and R. M. Izatt, Chem. Rev., 1973, 351–384 Search PubMed
- A. Caballero, N. G. White and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 1845–1848 CrossRef PubMed
- K. Choi and A. D. Hamilton, Coord. Chem. Rev., 2003, 240, 101–110 CrossRef
- B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed., 1996, 35, 1838–1840 CrossRef
- T. J. Wedge and M. F. Hawthorne, Coord. Chem. Rev., 2003, 240, 111–128 CrossRef
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039–1043 CrossRef PubMed
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364–15365 CrossRef PubMed
- M. A. Kline, X. Wei, I. J. Horner, R. Liu, S. Chen, S. Chen, K. Y. Yung, K. Yamato, Z. Cai, F. V. Bright, X. C. Zeng and B. Gong, Chem. Sci., 2015, 6, 152–157 RSC
- P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sánchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886–1887 CrossRef PubMed
- K. P. McDonald, B. Qiao, E. B. Twum, S. Lee, P. J. Gamache, C.-H. Chen, Y. Yi and A. H. Flood, Chem. Commun., 2014, 50, 13285–13288 RSC
- S. Lee, Y. Hua, H. Park and A. H. Flood, Org. Lett., 2010, 12, 2100–2102 CrossRef PubMed
- J. Shang, W. Si, W. Zhao, Y. Che, J. L. Hou and H. Jiang, Org. Lett., 2014, 16, 4008–4011 CrossRef PubMed
- D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83–93 RSC
- S. H. Jungbauer, S. Schindler, E. Herdtweck, S. Keller and S. M. Huber, Chem. - Eur. J., 2015, 21, 13625–13636 CrossRef PubMed
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef PubMed
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784 RSC
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef PubMed
- J. Fernández-Lodeiro, C. Núñez, C. S. de Castro, E. Bértolo, J. S. Seixas de Melo, J. L. Capelo and C. Lodeiro, Inorg. Chem., 2013, 52, 121–129 CrossRef PubMed
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef PubMed
- Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528–15533 CrossRef PubMed
- B. Gong, Y. Yan, H. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. Ickes, J. Am. Chem. Soc., 1999, 121, 5607–5608 CrossRef
- X. Yang, A. L. Brown, M. Furukawa, S. Li, W. E. Gardinier, E. J. Bukowski, F. V Bright, C. Zheng, X. C. Zeng and B. Gong, Chem. Commun., 2003, 56–57 RSC
- S. M. Biros and J. Rebek Jr., Chem. Soc. Rev., 2007, 36, 93–104 RSC
- B. W. Purse and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10777–10782 CrossRef PubMed
- D. M. Rudkevich, G. Hilmersson and J. Rebek, J. Am. Chem. Soc., 1997, 119, 9911–9912 CrossRef
- A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef PubMed
- Y. Hamuro, S. J. Geib and A. D. Hamilton, Angew. Chem., Int. Ed., 1994, 33, 446–448 CrossRef
- D. W. Zhang, X. Zhao, J. L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef PubMed
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 Search PubMed
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef PubMed
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004 CrossRef PubMed
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef PubMed
- R. K. Rowe and P. S. Ho, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 255–264 CrossRef PubMed
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef PubMed
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef PubMed
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef PubMed
- S. M. Walter, F. Kniep, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2011, 50, 7187–7191 CrossRef PubMed
- P. Metrangolo and G. Resnati, Chem.–Eur. J., 2001, 7, 2511–2519 CrossRef
- P. Metrangolo, T. Pilati, G. Terraneo, S. Biella and G. Resnati, CrystEngComm, 2009, 11, 1187 RSC
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC
- O. Dumele, B. Schreib, U. Warzok, N. Trapp, C. A. Schalley and F. Diederich, Angew. Chem., Int. Ed., 2017, 56, 1152–1157 CrossRef PubMed
- O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339–12344 CrossRef PubMed
- N. K. Beyeh, F. Pan and K. Rissanen, Angew. Chem., 2015, 127, 7411–7415 CrossRef
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef PubMed
- F. Zapata, A. Caballero, P. Molina, I. Alkorta and J. Elguero, J. Org. Chem., 2014, 79, 6959–6969 CrossRef PubMed
- R. Tepper, B. Schulze, H. Görls, P. Bellstedt, M. Jäger and U. S. Schubert, Org. Lett., 2015, 17, 5740–5743 CrossRef PubMed
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef PubMed
- F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2012, 134, 11533–11541 CrossRef PubMed
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef
-
G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer Berlin Heidelberg, Berlin, Heidelberg, 1991 Search PubMed
- C. B. Aakeröy, C. L. Spartz, S. Dembowski, S. Dwyre and J. Desper, IUCrJ, 2015, 2, 498–510 CrossRef PubMed
- C. B. Aakeröy, P. D. Chopade and J. Desper, Cryst. Growth Des., 2011, 11, 5333–5336 CrossRef
- P. Politzer, J. S. Murray and P. Lane, Int. J. Quantum Chem., 2007, 107, 3046–3052 CrossRef
- A. R. Voth, P. Khuu, K. Oishi and P. S. Ho, Nat. Chem., 2009, 1, 74–79 CrossRef PubMed
- M. G. Chudzinski, C. A. McClary and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 10559–10567 CrossRef PubMed
- B. R. Mullaney, A. L. Thompson and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11458–11462 CrossRef PubMed
- C. B. Aakeröy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772–13773 CrossRef PubMed
- A. Takemura, L. J. McAllister, S. Hart, N. E. Pridmore, P. B. Karadakov, A. C. Whitwood and D. W. Bruce, Chem.–Eur. J., 2014, 20, 6721–6732 CrossRef PubMed
- V. Vasylyeva, S. K. Nayak, G. Terraneo, G. Cavallo, P. Metrangolo and G. Resnati, CrystEngComm, 2014, 16, 8102–8105 RSC
- P. Zhou, J. Lv, J. Zou, F. Tian and Z. Shang, J. Struct. Biol., 2010, 169, 172–182 CrossRef PubMed
- Y. Lu, Y. Wang, Z. Xu, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Phys. Chem. B, 2009, 113, 12615–12621 CrossRef PubMed
- Q. Zhao, D. Feng and J. Hao, J. Mol. Model., 2011, 17, 2817–2823 CrossRef PubMed
- P.-P. Zhou, W.-Y. Qiu, S. Liu and N.-Z. Jin, Phys. Chem. Chem. Phys., 2011, 13, 7408 RSC
- A. Kovács and Z. Varga, Coord. Chem. Rev., 2006, 250, 710–727 CrossRef
- M. Domagała and M. Palusiak, Comput. Theor. Chem., 2014, 1027, 173–178 CrossRef
- M. Domagała, A. Lutyńska and M. Palusiak, Int. J. Quantum Chem., 2017, 117, e25348 CrossRef
- L. Albrecht, R. J. Boyd, O. Mó and M. Yáñez, J. Phys. Chem. A, 2014, 118, 4205–4213 CrossRef PubMed
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef PubMed
- M. Solimannejad, M. Malekani and I. Alkorta, Mol. Phys., 2011, 109, 1641–1648 CrossRef
- S. A. C. McDowell and H. K. Yarde, Phys. Chem. Chem. Phys., 2012, 14, 6883 RSC
- H. Sun, C. Navarro and C. A. Hunter, Org. Biomol. Chem., 2015, 13, 4981–4992 RSC
- F. Y. Lin and A. D. Mackerell, J. Phys. Chem. B, 2017, 121, 6813–6821 CrossRef PubMed
- C. J. Massena, A. M. S. Riel, G. F. Neuhaus, D. a. Decato and O. B. Berryman, Chem. Commun., 2015, 51, 1417–1420 RSC
- A. M. S. Riel, M. J. Jessop, D. A. Decato, C. J. Massena, V. R. Nascimento and O. B. Berryman, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 203–209 CrossRef PubMed
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef
- D. L. Reger, T. D. Wright, C. A. Little, J. J. S. Lamba and M. D. Smith, Inorg. Chem., 2001, 40, 3810–3814 CrossRef PubMed
- C. Frassineti, S. Ghelli, P. Gans, A. Sabatini, M. S. Moruzzi and A. Vacca, Anal. Biochem., 1995, 231, 374–382 CrossRef PubMed
- M. Cametti, K. Raatikainen, P. Metrangolo, T. Pilati, G. Terraneo and G. Resnati, Org. Biomol. Chem., 2012, 10, 1329 RSC
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC
- R. D. Parra, H. Zeng, J. Zhu, C. Zheng, X. C. Zeng and B. Gong, Chem.–Eur. J., 2001, 7, 4352–4357 CrossRef
- E. Peris, J. C. J. Lee, J. R. Rambo, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 1995, 117, 3485–3491 CrossRef
- Y.-Y. Zhu, H.-P. Yi, C. Li, X.-K. Jiang and Z.-T. Li, Cryst. Growth Des., 2008, 8, 1294–1300 CrossRef
- L. R. Steffel, T. J. Cashman, M. H. Reutershan and B. R. Linton, J. Am. Chem. Soc., 2007, 129, 12956–12957 CrossRef PubMed
- P. Froimowicz, K. Zhang and H. Ishida, Chem.–Eur. J., 2016, 22, 2691–2707 CrossRef PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- J. E. Del Bene, J. Phys. Chem., 1993, 97, 107–110 CrossRef
- M. G. Chudzinski and M. S. Taylor, J. Org. Chem., 2012, 77, 3483–3491 CrossRef PubMed
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Titration data, gas-phase DFT calculations and coordinates, NMR spectroscopic data, crystallographic refinement details. CCDC 1825325–1825331. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01973h |
| ‡ These authors contributed equally. |
| § Preorganization is the notion that it is energetically favorable to arrange binding sites that minimize conformational change upon guest binding. E. V. Anslyn, and D. A. Dougherty, Modern Physical Organic Chemistry, University Science, Sausalito, CA, 2006. |
| ¶ The XB is complex in nature with multiple contributions from electrostatics, charge-transfer and polar flattening. Specifically, electrostatic investigations do not always offer a complete description of the XB. |
| || Traditionally, XB strength is strengthened by introducing electron withdrawing groups and/or exchanging the halogen with a more polarizable halogen. |
| ** Beer and coworkers highlight another example of preorganization by metal coordination (Mole et al. J. Organomet. Chem. 2015, 792, 206–210). |
| †† Elegant macrocycles and foldamers have employed fluorine and chlorine as HB acceptors to encourage higher order structures. See ref. 42. However, HBing is clearly the dominate force in facilitating function. As expected, XBing is not mentioned as contributing to the function of these systems as fluorine and chlorine are very poor XB donors when compared to iodine and bromine XB donors. |
| ‡‡ Only the more capable XB donors of iodine and bromine are considered here. In another report, Zhu et al. provide solution data demonstrating intramolecular HBs can be formed with fluorinated, chlorinated and brominated aromatic amides. However, these would not be considered good XB donors and XBing was not mentioned. Zhu et al. Cryst. Growth Des. 2008, 8, 1294–1300. |
| §§ G2XB and G2HB were investigated by IR spectroscopy to lend support to intramolecular hydrogen bonding, however no conclusive shifting was observed. The effects of weak hydrogen bonds are not as straightforward as strong hydrogen bonds. Specifically, simple systems can produce spectral complexity which can prevent proper interpretation. However, lack of chemical shift does not indicate absence of hydrogen bonding. More can be found on this topic in Baker et al. J. Am. Chem. Soc. 1958, 80, 5358; A. W. Baker, J. Am. Chem. Soc. 1958, 80, 3598; G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999. |
| ¶¶ G2HB exhibits an upfield shift due to the relative acidity of the amine protons. The relatively weak iodine HB acceptors could contribute to the relatively small differences observed between G2XB and G2HB. |
| |||| The B3LYP functional does not accurately estimate the XB. However, this functional correctly estimates the HB interaction to the Lewis base sites of the XB donor. |
| *** It is known that the electrostatic potential of a molecule is influenced by neighboring atoms, and a decrease in σ-hole electron density does not necessarily correlate with a more positive electrostatic potential. However, our collective findings indicate that the electron density and the electrostatic potential for this system tract. Further discussion on ESP in σ-hole interactions is found here: P. Politzer, and J. Murray, Crystals, 2017, 7, 212, DOI: 10.3390/cryst7070212. |
| ††† A CSD search of HBs to organic iodine atoms was compared to G2XBme2+·2Br−, G2XBme2+·2I−, and G2XBme2+·2OTf−. See ESI† for details. |
| This journal is © The Royal Society of Chemistry 2018 |