
Challenges and opportunities for adsorption-based CO2 capture from natural gas combined cycle emissions
Rebecca L.
Siegelman†
 ab,
Phillip J.
Milner†‡
ab,
Eugene J.
Kim
a,
Simon C.
Weston
*c and
Jeffrey R.
Long
*abd
ab,
Phillip J.
Milner†‡
ab,
Eugene J.
Kim
a,
Simon C.
Weston
*c and
Jeffrey R.
Long
*abd
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: jrlong@berkeley.edu
bMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cCorporate Strategic Research, ExxonMobil Research and Engineering Company, Annandale, NJ 08801, USA. E-mail: simon.c.weston@exxonmobil.com
dDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA
First published on 8th May 2019
Abstract
In recent years, the power sector has shown a growing reliance on natural gas, a cleaner-burning fuel than coal that emits approximately half as much CO2 per kW h of energy produced. This rapid growth in the consumption of natural gas has led to increased CO2 emissions from gas-fired power plants. To limit the contribution of fossil fuel combustion to atmospheric CO2 levels, carbon capture and sequestration has been proposed as a potential emission mitigation strategy. However, despite extensive exploration of solid adsorbents for CO2 capture, few studies have examined the performance of adsorbents in post-combustion capture processes specific to natural gas flue emissions. In this perspective, we emphasize the importance of considering gas-fired power plants alongside coal-fired plants in future analyses of carbon capture materials. We address specific challenges and opportunities related to adsorptive carbon capture from the emissions of gas-fired plants and discuss several promising candidate materials. Finally, we suggest experiments to determine the viability of new CO2 capture materials for this separation. This broadening in the scope of current carbon capture research is urgently needed to accelerate the deployment of transformational carbon capture technologies.
Broader contextGlobal climate change is driven in part by the growing concentration of CO2 in the atmosphere and is predicted to lead to continued increases in temperature and sea level. The CO2 emitted from fossil fuel combustion is currently responsible for approximately 60% of global anthropogenic greenhouse gas emissions, and thus the energy sector is a natural candidate for mitigation efforts. Post-combustion CO2 capture from large point sources, such as fossil fuel-fired power plants, has received significant attention as a promising strategy to limit the influence of human activity on climate change. However, research efforts to date have largely focused on coal-fired plants. In recent years, natural gas has supplied an increasing share of global primary energy, with continued growth supported by its lower greenhouse gas footprint compared to coal and its lower emission of other pollutants such as SOx, NOx, and Hg. As a result, new research efforts are needed to design efficient capture systems to target the emissions of gas-fired plants. To that end, greater awareness of the specific challenges and opportunities in this area will be critical in advancing the field of CO2 capture to best serve the changing energy landscape. |
Introduction
Rising atmospheric levels of greenhouse gases, particularly carbon dioxide (CO2), are contributing to global climate change.1 The combustion of fossil fuels such as coal, natural gas, and oil for energy production is currently responsible for the release of approximately 32 Gt per year of CO2 into the atmosphere, or 60–65% of all anthropogenic greenhouse gas emissions.1,2 One strategy that has been proposed to reduce global CO2 emissions is carbon capture and sequestration (CCS), in which CO2 from the flue gas streams of fossil fuel-fired power plants is captured and stored underground—or used as a chemical feedstock—instead of being released into the atmosphere.1,3–7 Considering sequestration, the Intergovernmental Panel on Climate Change (IPCC) has estimated the technical potential for global storage capacity in geological formations to be at least 2000 Gt of CO2.8 This would be sufficient to store the entirety of global emissions for at least 50 years, assuming constant or declining emissions.To date, the vast majority of CCS studies have focused on coal-fired power plants,7,9 as the combustion of coal is currently responsible for approximately 27% of the global energy supply and 44% of anthropogenic energy-related CO2 emissions.2,6,7 However, natural gas is the fastest-growing fossil fuel source in terms of worldwide consumption (increasing with a compound average annual growth rate of ∼1.9% per year, compared to ∼0.1% per year for coal), and it is projected to overtake coal in terms of contribution to global energy by 2030 (2018 New Policy Scenario, International Energy Agency)10 or 2032 (U.S. Energy Information Administration, Fig. 1a).11 Although global gas-related emissions are not predicted to surpass those from the combustion of coal until after 2040,10 this transition has already occurred in select important regional markets where natural gas is already a major fuel source for energy production, such as the United States (Fig. 1b), the United Kingdom, and Russia.2,12,13
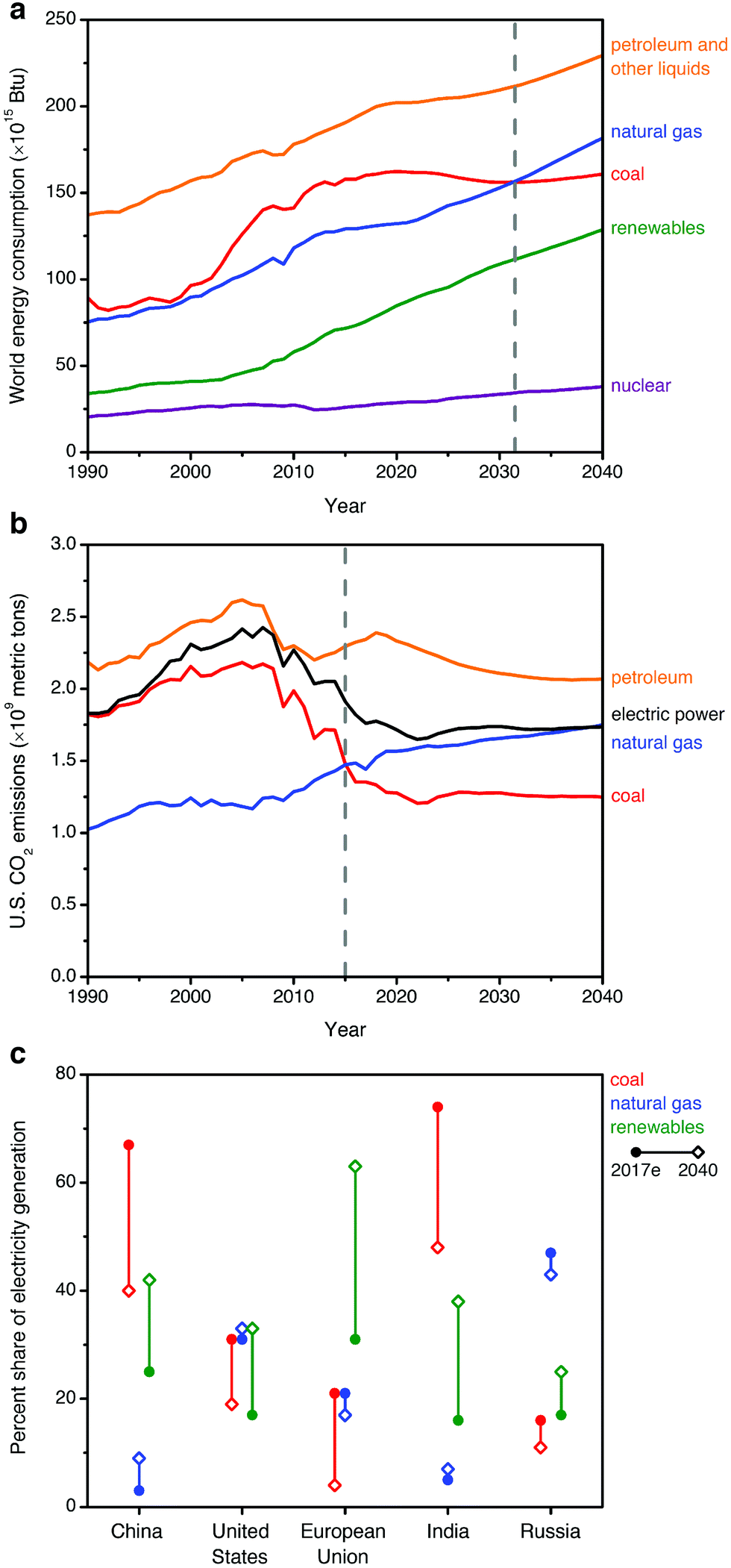 | ||
| Fig. 1 (a) Historical and projected worldwide consumption of energy sources from 1990–2040, suggesting that consumption of natural gas will match that of coal by ∼2032 (dashed gray line).11 (b) Historical and projected CO2 emissions from the combustion of fossil fuels and the overall electric power sector from 1990–2040 in the United States, a representative natural gas-centric energy economy. Natural gas overtook coal in contributing to anthropogenic U.S. CO2 emissions after 2015 (dashed gray line).12 (c) Estimated current (2017e) and projected 2040 shares of electricity generation (percent of TW h) by fuel for selected regions, following the 2018 New Policy Scenario of the International Energy Agency.10 | ||
The growing worldwide consumption of natural gas is due to increasingly available reserves and its low greenhouse gas footprint relative to coal, since the combustion of natural gas produces approximately half as much CO2 as the combustion of coal per kW h of energy produced.14 Indeed, recent declining emissions in the electric power sector for natural gas-dominant regions such as the United States correlate closely with a shift in the energy landscape from coal to renewable sources and natural gas (Fig. 1b), although overall CO2 emissions remain substantial.12 Considering the role of natural gas in electric power generation for other major energy stakeholders, forecasts remain highly sensitive to region-specific policy, market, and technology factors. In Fig. 1c, we show the current and projected fuel distribution10 for regions responsible for the largest portions of global CO2 emissions from fuel combustion: China (28.0% of global CO2 emissions), the United States (15.0%), the European Union (9.9%), India (6.4%), and Russia (4.5%).2 In 2040, natural gas will likely continue to constitute a major portion of electricity generation in the United States and Russia, the largest current markets. In China and India, coal will likely continue to supply a major fraction of electric power in the near-term, but modest increases are anticipated in the share of gas-fired electricity generation in these countries.10 In the European Union, the overall use of fossil fuels for electric power continues to decline, but coal use is projected to decay at an accelerated rate compared to natural gas use.10 In light of these trends, near- and long-term research is urgently needed to address the unique challenges of CCS from natural gas-fired power plants, in conjunction with ongoing efforts toward CCS from coal-fired power plants.6,7,13,15,16
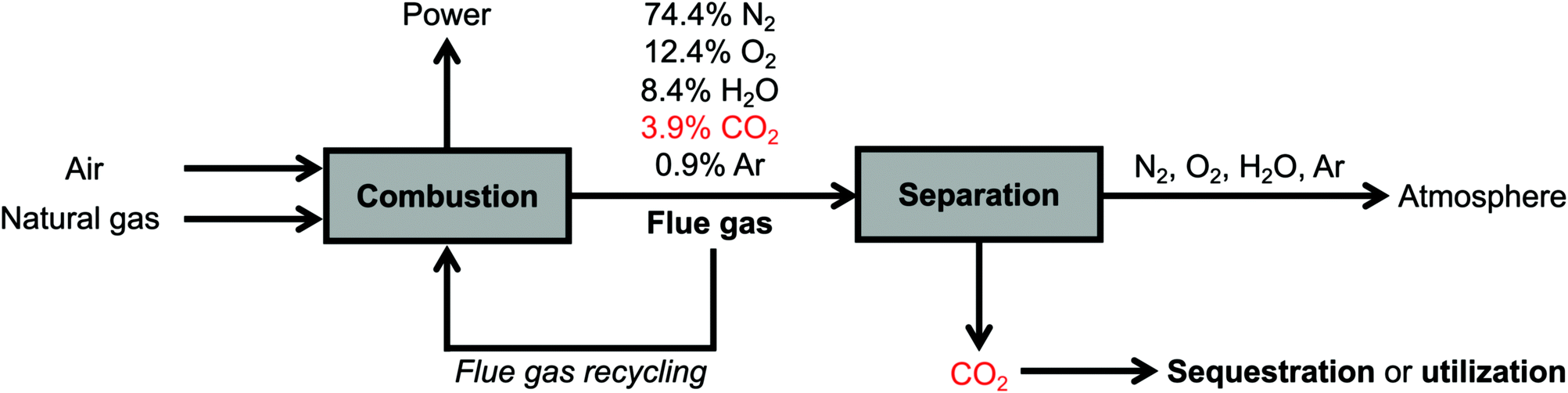
The conditions for carbon capture from natural gas and coal flue gases are relatively similar, with several key differences. Both flue gas streams are released at atmospheric pressure, are generated at high temperatures (>80 °C) but typically cooled to 40–60 °C to enable effective CO2 capture, and consist primarily of CO2, O2, H2O, and N2.6,13,17,18 However, while coal flue gas streams typically contain relatively high CO2 concentrations (∼15%), the typical flue gas from a natural gas combined cycle (NGCC) power plant contains 3.9% CO2 with a balance of 74.4% N2, 12.4% O2, 8.4% H2O, and 0.9% Ar,18 rendering CO2 capture from NGCC flue emissions more technically challenging.13,14,16,17,19 (Simple-cycle power plants with gas-fired boilers, which emit flue gas streams containing higher CO2 concentrations of 7–8% but operate with lower efficiencies,8 are not addressed in this perspective.) A second challenge is the much higher concentration of O2 in NGCC flue gas (12.4%) compared to that in coal flue gas (∼4%).13,16 However, natural gas flue emissions contain a much lower concentration of other corrosive components, such as SOx, and other contaminants, such as NOx, heavy metals, and particulate matter, resulting in an overall cleaner stream than coal flue gas.18 Thus, effective materials for CO2 capture from NGCC power plants must be thermally and oxidatively stable while demonstrating strong, selective adsorption of CO2 at low concentrations (≤3.9%) under humid conditions, but their stability to SOx and NOx, a limiting challenge for CO2 capture from coal-fired power plants, is less critical.
Many of the design principles developed for coal flue gas capture should translate to the development of new materials for capture from natural gas flue emissions.7,13,14,16 Designing efficient capture systems is critical, because up to 80% of the cost of CCS from a natural gas-fired power plant accrues during the CO2 capture step.20 The most technology-ready materials for CCS are aqueous amine solutions, due to their low cost and selective reaction with CO2.21,22 Indeed, a pilot-scale demonstration (1991–2005) of CO2 capture from the Bellingham NGCC power plant in the United States employed the Econamine FG PlusSM amine-based technology, and the use of several other advanced amine scrubbers has been demonstrated at the Test Centre Mongstad facility in Norway.13 Despite their advanced state of development, aqueous amine solutions continue to suffer from a number of drawbacks, including low working capacities (<2 wt%), high regeneration temperatures (>120 °C), and oxidative and thermal degradation during long-term cycling.23–28 These challenges are exacerbated in a natural gas post-combustion capture process, where the high regeneration temperatures required to desorb strongly-bound CO2 and the high O2 content of the target stream lead to increased thermal and oxidative amine degradation (discussed further below).13,16
Due to these drawbacks, significant research efforts have been directed toward the development of new strategies to reduce CO2 emissions from fossil fuel-fired power plants. Several engineering solutions have been explored to integrate CO2 capture with plant operation, including the use of molten carbonate fuel cells for simultaneous concentration of CO2 (up to ∼75%) and excess power production,29 as well as the Allam cycle, in which CO2 is used as the working fluid in an oxy-fuel process to yield high-pressure, pipeline-quality CO2.30,31 In addition, new materials have been designed for post-combustion CO2 capture,6,7,32,33 including water–lean liquid absorbents34,35 and membranes.7,36
Recently, porous solids such as zeolites, carbons, silicas, porous polymers, and metal–organic frameworks have also been studied extensively for carbon capture applications, due to their potential to achieve larger CO2 working capacities and improved cycling stabilities compared to aqueous amine solutions.7,37–65 However, nearly all studies to date have focused on CO2 scrubbing from coal flue gas, direct capture from air, and removing CO2 from crude natural gas, with very few studies investigating adsorptive capture from natural gas flue emissions.13,16,66 Therefore, this perspective focuses on the unique challenges and opportunities inherent to the application of adsorbents for CCS from NGCC power plants, which remains an underdeveloped but important area of research. Specifically, we will examine (i) the challenges unique to adsorptive CO2 capture from natural gas flue emissions, (ii) promising materials for this process that warrant further study, and (iii) recommended experiments to assess the suitability of new carbon capture materials.
Challenges associated with capture from natural gas flue emissions
Low partial pressure of CO2
The low concentration of CO2 in natural gas flue emissions is due to the large excess of air that is used as a thermal diluent during combustion (200–250% of the stoichiometric O2 requirement).19 This low concentration (3.9%) renders CO2 capture from natural gas flue gas more energetically demanding than from coal flue gas.13,16–19,51,67 Therefore, adsorbents for the target process must possess high adsorption capacities (ideally >2–3 mmol g−1) at low CO2 concentrations (≤3.9%) with adsorption temperatures of ≥40 °C.44,68,69 In order to achieve the U.S. Department of Energy's target of 90% CO2 capture from the flue gas stream,18 materials would need to demonstrate adsorption at CO2 concentrations as low as 0.39%. Many adsorbents explored to date for CCS from coal-fired power plants are unable to adsorb appreciable quantities of CO2 at such dilute levels. Furthermore, such strong adsorption typically necessitates large CO2 adsorption enthalpies,70 which in turn can lead to large temperature increases in the adsorbent bed—and significant losses in CO2 working capacity—if the heat release is not properly managed.71 An additional consequence of this requirement is a high theoretical minimum work for a CCS process (0.14–0.20 MJ per kg CO2),7 leading to high regeneration temperatures and energies in a temperature swing adsorption (TSA) process or very low desorption pressures in a vacuum swing adsorption (VSA) process.44Despite the higher theoretical minimum work for CO2 capture from NGCC power plants, CCS from gas-fired plants may be less costly than from coal plants when considering the levelized cost of electricity (LCOE), the average lifetime unit cost of electricity generation of a power plant.72,73 Recent estimates suggest that the LCOE for NGCC power plants would increase from 42–83 USD per MW h without CCS to 63–122 USD per MW h with CCS, while that of supercritical pulverized coal-fired plants would increase from 61–79 USD per MW h without CCS to 95–150 USD per MW h with CCS (costs are in constant 2013 USD and include capture, transport, and geological storage).73 The potentially lower LCOE for NGCC power plants with CCS is favored in the case of low gas prices and reflects the lower emission intensity of natural gas, because a post-combustion capture system at a NGCC plant would process only approximately half as much CO2 as a capture system at a coal plant.8,72,73 However, specific cost estimates should be interpreted with caution, due to the large number of region-dependent technical, economic, and financial assumptions required, as well as remaining uncertainty resulting from a lack of empirical data from full-scale demonstrations.74 As capture systems progress toward full-scale deployment, continued refinement of techno-economic models will help narrow the wide range of projected costs for specific projects. In addition, advanced capture technologies may enable more favorable techno-economic analyses, but additional data and larger-scale demonstrations are needed to reduce the even greater degree of uncertainty for cost predictions with these systems.
Recently, engineering strategies have been developed to increase the partial pressure of CO2 in NGCC flue gas and thus allow for more efficient CO2 capture. One such strategy is exhaust gas recycling, in which recycled flue gas (∼4% CO2, ∼12% O2) is used in place of air (400 ppm CO2, ∼21% O2) during the fuel combustion process (Fig. 2).15,17,67,75–77 This strategy produces flue gases with enriched CO2 concentrations of up to ∼8%, making CO2 capture less thermodynamically challenging.17 Indeed, recent techno-economic analyses have suggested that coupling up to 50% flue gas recycling with the use of aqueous monoethanolamine (MEA) for CO2 capture should lead to a lower energy penalty on a natural gas-fired power plant than the use of aqueous MEA alone.15,17,67,78,79 However, the use of >35% exhaust gas recycling requires significant capital investment with respect to the combustion unit and is unlikely to be a retrofit option for current power plants, and therefore an optimal balance exists between the extent of exhaust gas recycling and the energy penalty of the carbon capture step.17 In a related strategy under development, membranes can be used to recycle CO2 selectively from flue gas to the combustor, increasing the concentration of CO2 in the final flue gas stream up to ∼19% with almost no thermodynamic penalty.19 Although the use of membranes for pre-concentrating CO2 would lead to increased capital and maintenance costs, the benefit of the subsequently more favorable CO2 capture step may ultimately make this strategy more feasible. Overall, the use of flue gas recycling or membrane pre-concentration strategies simplifies the carbon capture step but requires additional capital costs compared to a direct capture configuration, which mandates significant techno-economic analysis to determine the ideal CCS strategy for a given power plant.
 | ||
| Fig. 2 Schematic of a natural gas-fired power plant with post-combustion CO2 capture, including optional flue gas recycling to increase the concentration of CO2 in the flue gas up to ∼8%. | ||
High partial pressure of O2
The large excess of air used as a thermal diluent during the combustion of natural gas also contributes to a high O2 content (12.4%) in the flue gas stream,18,19 which can accelerate the oxidative degradation of aqueous amine solutions.13,16 Although the mechanisms by which aqueous amines degrade in the presence of O2 have been well studied,25,26,80 significantly less attention has been paid to the oxidative stability of porous solids for CO2 capture, such as metal–organic frameworks and porous organic networks.81 Overcoming this barrier will be a primary challenge in the commercialization of CO2 capture adsorbents specific to NGCC flue gas, and further discussion of oxidative stability is included in subsequent evaluation of individual adsorbent classes. For amine-functionalized adsorbents, recent studies have demonstrated that materials functionalized with secondary amines, such as polyethylenimine, are subject to CO2 capacity loss upon extended exposure to O2, but materials functionalized with primary amines possess significantly improved oxidative stability.27,81–86 Spectroscopic studies have suggested that the main oxidation products of amine-functionalized materials are imines and carbonyl-containing species such as amides, imides, and carboxylic acids.82,87–89One potential engineering solution to the oxidative degradation of carbon capture materials is flue gas recycling, which has the added benefit of depleting the O2 content of the flue gas stream (down to ∼4% O2 with 50% recycling), in addition to increasing the CO2 content.17 When exhaust gas recycling is combined with stoichiometric combustion, excess O2 may be essentially eliminated from the flue gas, and CO2 exhaust concentrations as high as approximately 20% can be achieved.90 Nonetheless, the current lack of information on the oxidative stability of porous materials hinders further development of adsorbents for CO2 capture from NGCC flue emissions. Beyond oxidative stability, the CO2/O2 selectivity and resulting influence on the purity of captured CO2 will also need to be assessed.
Saturation with water
A critical challenge shared by adsorption-based CO2 capture from coal and natural gas flue gas is that both streams are saturated in H2O (up to 10% by volume), which mandates that a prospective material not only capture CO2 selectively in the presence of H2O, but also possess long-term stability under humid conditions.18,39,44,46,48 Furthermore, parasitic energy costs associated with cycling of any co-adsorbed water must be minimized.69 Challenges related to water saturation are particularly onerous for CO2 removal from NGCC flue gas, which contains more than twice as much water (8.4%) as CO2 (3.9%), in contrast to coal flue gas, which contains nearly twice as much CO2 as water.18 Although the flue gas stream can be dried prior to CO2 removal, a dehydration unit would likely be extremely costly and impractical on large scale.91 The presence of water in the flue gas stream is problematic for materials relying on equilibrium selectivity for CO2 at exposed metal ions, such as certain zeolites and metal–organic frameworks, as these sites have been shown to adsorb water preferentially over CO2 in both multicomponent measurements and theoretical calculations.46,92–100 In contrast, amine-functionalized materials often show improved CO2 adsorption capacities under humid conditions.41,92,101,102 Amine-functionalized adsorbents may also benefit from humidity through the mitigation of deleterious urea formation.41,103,104 Nonetheless, a dearth of information persists for a range of promising materials regarding competitive CO2 adsorption under humid conditions and the costs associated with the desorption of co-adsorbed water.92,105 Overall, the requirement for selective adsorption of CO2 in the presence of water is often the primary barrier that precludes the application of a new adsorbent for CO2 removal from flue gas.Key opportunities and promising adsorbents for capture from natural gas flue emissions
Despite the above-mentioned challenges, gas-fired power plants offer a number of advantages over coal plants in post-combustion capture processes beyond the overall reduction in CO2 emissions before capture. First, the combustion of natural gas typically generates significantly fewer contaminants. While coal can contain high levels of mercury (up to 0.15 ppm, dry weight) and sulfur (up to ∼3 wt%), gas-fired plants generate no mercury waste and emit only trace amounts of SOx.18 Furthermore, the generation of particulate matter is significantly reduced for NGCC plants compared to coal plants.18 This cleaner emission stream relaxes the need for adsorbents that are stable to trace SOx or for extensive pre-treatment to protect the CO2 adsorbent. Considering the experimentally-observed poisoning of a wide array of adsorbents by SOx and NOx,85,106–112 CCS efforts on cleaner NGCC flue gas streams may lead to extended adsorbent lifetimes and thereby increase the techno-economic favorability of adsorptive CO2 capture. Second, the installation of innovative capture technology may be more straightforward at gas plants than at coal plants, where the CO2 capture system must be integrated with upstream pollutant control systems.13 Finally, while the cost of capture is typically higher for natural gas plants due to the more dilute stream, lifecycle analyses have suggested that the cost of integrating CCS into a new natural gas plant may approach that of a retrofit capture system at an aging coal plant.113Targeted exploration of adsorbents for carbon capture from natural gas power stations has been relatively limited to date, with the majority of post-combustion capture studies instead focused on coal flue emissions. Although adsorbents capable of direct capture of CO2 from air59 may prove effective for capture from NGCC flue gas, these materials will likely require excessively high regeneration energies due to the stronger binding enthalpies typically needed to capture CO2 at ppm levels. Therefore, adsorbents previously investigated for CO2 capture from coal flue gas and air provide a basis for our discussion of promising candidate materials for CCS from natural gas flue emissions, but further research is required to design materials and capture systems specifically for this challenging capture process. In general, the most promising adsorbents share a common feature of strong binding sites, such as amines, that enable selective adsorption of CO2 at low partial pressures from humid flue gas (Fig. 3).
 | ||
| Fig. 3 Promising adsorbents for post-combustion capture from NGCC flue gas require strong and selective binding sites, such as amines, to enable CO2 adsorption at low partial pressures under humid conditions. Promising material classes and specific examples include (a) zeolites, such as an ethylenediamine-grafted Y zeolite93 (silver, red, and yellow spheres represent Al/Si, O, and Na atoms, respectively); (b) amine-functionalized silicas, such as PEI-MCM-41101 (PEI = polyethylenimine); (c) permanently porous organic polymers, such as polyamine-grafted PPN-6;137,138 and (d) metal–organic frameworks, such as diamine-functionalized variants of Mg2(dobpdc)148 (green, red, and grey spheres represent Mg, O, and C atoms, respectively; H atoms are omitted for clarity). | ||
Zeolites
Zeolites are crystalline, microporous aluminosilicates that can be extracted as minerals or produced synthetically. These materials have already achieved commercialization in industrial gas separations, and a wealth of knowledge is available regarding their incorporation into engineered forms within full-scale processes.114 For carbon capture applications, the mechanism of separation typically relies upon preferential interaction of the quadrupole moment of CO2 with the electric field of cations within the zeolite pores, enabling strong CO2 binding at low partial pressures.115,116 The crystalline nature of these materials and the comprehensive library of known and predicted structures make zeolites particularly well-suited for computational evaluation; indeed, previous studies have leveraged these properties to predict the optimal zeolites for CO2 capture from both coal117 and natural gas51 flue emissions.Initial studies of zeolites specific to CO2 capture from NGCC flue gas included an evaluation of zeolite 13X in an electric swing adsorption (ESA) process, where an electrical regeneration energy of 2.04 GJ per ton CO2 was reported, excluding compression energy and water removal.118 However, zeolites such as 13X that operate via equilibrium adsorption mechanisms suffer from a critical limitation: water typically outcompetes CO2 at cationic binding sites, leading to loss of selectivity for CO2 in humid flue gases.92,119,120 Future research in zeolite-based processes for CO2 capture from natural gas flue emissions must take this limitation into account by designing innovative materials or processes to overcome water passivation or by incorporating increased capital and operating costs to pre-dry the flue gas stream.91
Despite these limitations, zeolites remain attractive candidates for separations under harsh conditions due to their high thermal, oxidative, and hydrolytic stability. This stability arises from the strong tetrahedral Al–O and Si–O bonds of the inorganic backbone, as well as the relatively small increase in energy for permanently porous phases compared to analogous condensed structures.121 Moving forward, amine-functionalized zeolites (Fig. 3a) merit further study for CO2 capture from natural gas flue emissions due to their strong CO2 binding at low partial pressures and their selectivity for CO2 in the presence of water.93,122–125 However, incorporation of amines may result in lower stability for the resulting organic–inorganic hybrid materials. For example, composite materials formed by physical impregnation of zeolite NaX with MEA or tetraethylenepentamine (TEPA) were found to undergo significant amine volatilization upon 24 h of exposure to flowing N2 at 130 °C, a realistic desorption temperature for TSA cycling with NGCC flue gas.93 As a notable improvement, a recently reported ethylenediamine-Y zeolite bearing chemically grafted amines was shown to possess thermal stability up to 180 °C.93 Chemical grafting of amines to the zeolite scaffold was also found to suppress urea formation as compared to a PEI-impregnated silica (PEI = polyethylenimine), a result the authors attributed to strongly co-adsorbed water within the hydrophilic zeolite pores.93 Critically, the oxidative stability of these organic–inorganic hybrid materials and their resulting suitability for CO2 capture from NGCC emissions remain untested.
Amine-functionalized silicas
The impregnation (Class I) or covalent grafting (Class II) of (poly)amines within porous silicas has been demonstrated as a strategy to generate highly selective CO2 capture adsorbents.41,64,126,127 These materials typically bind CO2 by chemisorption to form ammonium carbamate and/or carbamic acid species depending on the identity and proximity of amine functionalities.128 Importantly, chemisorption of CO2 enables many amine-functionalized silicas to preserve selectivity for CO2 in the presence of water, typically with improved capacities, due to the formation of bicarbonate species.41,101Despite the dearth of information specific to NGCC post-combustion CO2 capture for other adsorbent classes, a few reports have already discussed the application of amine-functionalized silicas for this separation. In breakthrough experiments with simulated flue gas containing 7.4–7.7% CO2, 14.6% H2O, and ∼4.45% O2 (similar to what would be expected for a simple-cycle gas-fired process or an NGCC process with flue gas recycling), PEI-MCM-41 (Fig. 3b) was shown to capture CO2 selectively in two consecutive adsorption/desorption cycles.129 A techno-economic study benchmarking this material against a conventional MEA solvent process (following the U.S. Department of Energy's NGCC base case18) indicated a potential 21% reduction in capital costs as well as a lower cost of electricity and cost of CO2 avoided compared to the MEA process.130 More recently, a combined experimental and theoretical study of a PEI-functionalized silica reported a regeneration heat of 5.0 GJ per ton CO2 captured for a NGCC plant (compared to 4.2 GJ per ton for a pulverized coal plant).69 In a sensitivity analysis, this study suggested that development of optimized adsorbents with large CO2 capacities (>4 mmol g−1) could dramatically reduce the regeneration heat, as would the use of a direct contact cooler to limit the water content of the flue gas.69 Another process simulation study with a PEI-functionalized silica adsorbent likewise found that the net plant efficiency could be increased with enhanced CO2 working capacities and reduced water co-adsorption, and that improved heat recovery and the use of steam for desorption could lead to further gains in efficiency.131
Despite these promising initial studies, key challenges remain for this class of materials, including amine oxidation, urea formation, leaching of amines from Class I materials, and slow adsorption/desorption kinetics in certain diffusion-limited variants.37,41 Recent work has demonstrated that the incorporation of longer alkyl spacers between amines,132 integration of hydrogen bonding groups,89 and addition of small amounts of chelators to remove oxidation-catalyzing trace metal impurities133 can dramatically improve the oxidative stability of these materials. These recent findings merit further study toward the application of amine-functionalized silicas for CO2 capture from natural gas flue emissions.
Porous organic networks
Permanently porous purely organic materials can be prepared via crosslinking of multitopic organic monomers to achieve amorphous porous organic polymers or crystalline covalent–organic frameworks. These materials offer a number of advantages in carbon capture processes, including modular pore size and surface functionality, high stability, and the potential for high gravimetric capacities through the use of light elements in the polymer backbone.134–136 However, experimental evaluation of porous organic polymers specific to post-combustion capture from NGCC power stations has been extremely limited to date. A computational study of CO2 capture from dry NGCC flue gas with several materials, including zeolites, metal–organic frameworks, porous organic polymers, and hypothetical adsorbents, found a group of amine-functionalized porous polymer networks (PPNs) to be the most promising candidates for this separation.51 These materials feature a biphenyl-based, diamond-like polymeric support (PPN-6, also known as PAF-1) that is post-synthetically covalently functionalized with polyamines, enabling strong and selective CO2 binding at low partial pressures (Fig. 3c).137,138 A series of PPN-6 materials functionalized with triethylenetetramine (TETA), tris(2-aminoethyl)amine (TAEA), and diethylenetriamine (DETA) groups were found to offer parasitic energies of 807, 858, and 880 kJ per kg CO2, respectively, with all other examined materials requiring parasitic energies in excess of 900 kJ per kg CO2.51 Importantly, the same computational study identified the DETA-containing derivative as the top-performing adsorbent for direct capture of CO2 from air (400 ppm CO2, 1215 kJ per kg CO2) and the TETA-containing derivative as the second-best candidate for capture from coal flue gas (14% CO2 in N2, 742 kJ per kg CO2).51 These results highlight the overlap in materials effective for each of these separations.Due to the constituent covalent bonds, many porous organic networks possess significant hydrothermal stability.139 While the oxidative stability of polymers has been studied more broadly over the past several decades,140 much remains unknown regarding the stability of porous organic networks in the context of CO2 capture. To date, exploration of O2 adsorption by these materials has largely been limited to application in direct air capture (DAC) of CO2. Room-temperature O2 isotherms for a series of amine-functionalized porous polymers showed low O2 uptake, leading to a high selectivity predicted for CO2 over N2 and O2 on the basis of ideal adsorbed solution theory calculations and transient breakthrough simulations.138 Nonetheless, further work is needed to probe the oxidative stability of these materials under the gas compositions and higher temperatures relevant to post-combustion CO2 capture from gas-fired power plants.
Moving forward, materials bearing amine functionalities or other CO2 chemisorption sites should be a primary focus of porous organic network development for CO2 capture from low partial pressure streams.136 Kinetics data, cycling tests, and multicomponent experiments incorporating H2O and O2 are also needed to support further evaluation of porous polymers for post-combustion capture applications. In addition, the ability of these materials to be synthesized at scale and formulated into industrially relevant structured forms remains to be demonstrated.
Metal–organic frameworks
Metal–organic frameworks consist of inorganic ions or clusters connected by bridging organic ligands, enabling control of pore size, shape, and surface chemistry to target strong and selective binding of specific adsorbates.141,142 As with zeolites, metal–organic frameworks possess crystalline structures that facilitate precise characterization of framework–guest interactions. The multitude of possible structures accessible with metal–organic frameworks has led to a proliferation of synthetic and computational reports on these materials for carbon capture, with a particular emphasis on post-combustion capture from coal-fired power plants.42,61,62,143,144Many studies have focused on the use of metal–organic frameworks bearing metals with open coordination sites to bind CO2 selectively over N2. However, these binding sites can suffer from the same water passivation issues encountered with cationic binding sites in zeolites.92,96–98 As an alternative approach, amine-functionalized metal–organic frameworks have been found to combine the advantages of crystalline materials with the selective reactivity toward CO2 demonstrated by amine-grafted silicas, amine-functionalized porous polymers, and amine solutions.145,146 Amine functionalities can be incorporated pre- or post-synthetically within the organic bridging ligands or post-synthetically onto open metal coordination sites within the framework. The latter strategy recently led to the development of adsorbents that bind CO2 cooperatively and reversibly through CO2 insertion into metal–amine bonds to form ammonium carbamate chains (Fig. 3d).147 These materials feature step-shaped CO2 adsorption isotherms, and the threshold pressure for cooperative adsorption can be tailored to match the conditions of post-combustion capture from NGCC power stations.105,148 Certain cooperative, amine-appended frameworks have shown stable temperature-swing cycling performance under humid coal flue gas streams,105,149 as well as high predicted CO2/O2 selectivities,149,150 but more work is needed to assess the thermodynamics and kinetics of CO2 capture from streams relevant to NGCC plants, including lower CO2 concentrations, higher O2 and H2O concentrations, and higher desorption temperatures. In a related approach, framework surfaces bearing metals bound by a monodentate hydroxide ligand were recently demonstrated to have large CO2 capacities under simulated coal flue gas conditions through the reversible formation of metal-bound bicarbonate—a strategy that could also prove effective for capture from natural gas flue emissions, pending further evaluation of the oxidative and hydrothermal stability of these materials.151
Despite these promising advances in fundamental materials design, several major challenges remain in advancing metal–organic frameworks toward industrial application. First, as with porous polymers, the majority of frameworks are synthesized as powders, but industrial application will require formulation of shaped particles, monoliths, or fibers. Additionally, limited hydrothermal stability remains a barrier for many frameworks due to the lability of metal–ligand coordination bonds.152 Oxidative stability is likewise a challenge for many metal–organic frameworks, with oxidation possible at the metal node153 and/or the organic linker,154 particularly in the presence of both O2 and H2O.155–157 Furthermore, development of metal–organic frameworks at a scale relevant to post-combustion capture will require thoughtful selection of earth-abundant metals and inexpensive organic bridging units as well as exploration of alternatives to classic solvothermal synthetic routes, which rely on costly and environmentally harmful organic solvents. Considering the early stage of development of metal–organic frameworks, large-scale demonstrations are needed within relatively short timeframes to encourage continued investment in their commercialization for carbon capture.
Evaluating adsorbents for capture from natural gas flue emissions
Effective deployment of adsorbents for post-combustion carbon capture at NGCC power plants will require rapid down-selection of the most promising materials for testing at the pilot scale and beyond. As the number of reported carbon capture adsorbents continues to increase, standardized performance metrics are needed to determine the viability of new capture materials and identify the top performers. Here, we propose a series of experiments to evaluate materials at the laboratory scale (i.e. <1 g). This series of tests will require only a small set of additional experiments for adsorbents already under evaluation for CO2 capture from air or coal flue gas. While these suggestions are intended primarily for experimentalists, we stress the critical role that computation can play in evaluating known and possible adsorbent structures.51,143In experimental or computational research, it is important to evaluate adsorbents under realistic conditions, particularly with respect to the high water and O2 content in natural gas flue emissions. Practicing scientists and engineers in the carbon capture community have long appreciated these concerns; however, with researchers from more diverse backgrounds continuing to bring valuable expertise to the field, it is important to review these considerations and associated experiments here. We also emphasize that all research on adsorbents for carbon capture would benefit immensely from closer collaboration between materials scientists and process engineers from the earliest stages of development. The ultimate success of adsorption-based processes hinges not only on fundamental material design, but also on the optimization of a structured adsorbent (e.g., pellets, monoliths, or fibers) within a full-scale process.158,159 To that end, toolsets such as those developed by the Carbon Capture Simulation Initiative (CCSI) will be invaluable in dramatically reducing overall development timelines by optimizing process design concepts and guiding sensitivity analysis within techno-economic models.160,161
Specific challenges associated with carbon capture from natural gas flue emissions are summarized and paired with corresponding suggested experiments in Table 1. We discuss each of these approaches in greater detail in the subsequent sections.
| Challenge | Experiment(s) |
|---|---|
| Low partial pressure of CO2 | Equilibrium studies to probe adsorption capacity for CO2 under 40 mbar or 4% CO2 at 40 °C |
| High partial pressure of O2 | Characterization of oxidative stability under harshest anticipated cycling conditions |
| Saturation with water | Multicomponent experiments to probe cycling stability and CO2 capacity under humid simulated flue gas |
| Selective CO2 adsorption | Multicomponent experiments to probe selectivity for CO2 over other contaminants (O2, N2) |
| Slow kinetics | Thermogravimetric analysis, breakthrough, and/or zero-length column experiments with 4% CO2 in N2 |
CO2 adsorption capacity, working capacity, and thermodynamics
To minimize the energy penalty associated with CO2 capture from natural gas flue emissions—here taken as a stream of 4% CO2 in N2 at ∼1 bar total pressure—adsorption capacities of at least 2–3 mmol g−1 at 40 mbar of CO2 are desirable.44,68,69 This is because larger CO2 adsorption capacities typically correlate with lower energy requirements, particularly for TSA processes.51 Flue gas typically exits the heat recovery/steam generation unit at 80–120 °C and can be sent to a direct contact cooler to reduce the temperature further to a minimum of ∼40 °C.79 Because Langmuir-type adsorbents typically show the largest adsorption capacities at the lowest possible adsorption temperature, we suggest that a temperature of 40 °C be used for standardized comparisons of adsorption capacity. However, adsorbents capable of operating with similar or improved performance at higher adsorption temperatures should be highlighted for their potential to decrease cycle times in TSA processes and enhance the overall cycling efficiency. In addition, higher-temperature adsorption data are valuable to inform process models and predict the effect of thermal fronts on the adsorbent efficiency.The single-component adsorption capacity of a material can be readily extracted from adsorption isobars or isotherms under the appropriate conditions (4% CO2 at ≥40 °C, or 40 mbar of CO2 assuming adsorption at 1 bar). Beyond adsorption capacity, projected working capacities are needed to assess the viability of new adsorbents. Many different cycling configurations may be considered, including TSA, pressure or vacuum swing adsorption (PSA or VSA), electrical swing adsorption (ESA), steam stripping, or a combination thereof.7 Note that desorption with an inert purge gas such as Ar or N2 is not viable for large-scale application, as this will compromise the CO2 product purity necessary for subsequent compression, transportation, and sequestration steps.
To design the optimal capture process for a particular adsorbent and calculate the resulting regeneration or parasitic energy, an understanding of the thermodynamics of CO2 adsorption (and, ideally, of H2O, N2, and O2 adsorption) is also needed. For early-stage materials research, knowledge of the adsorption enthalpy of each flue gas component is valuable and can be calculated from the single-component adsorption isotherms. These values are particularly critical for NGCC capture processes, where the large adsorption enthalpies required to capture CO2 at low partial pressures tend to increase the regeneration energy, as highlighted above.70 Importantly, the CO2 adsorption enthalpy is typically already reported for materials designed with direct air capture and/or post-combustion capture from coal flue gas applications in mind, and therefore expanding the application scope of these adsorbents to include capture from natural gas flue emissions should pose no added experimental burden.
Effect of impurities on CO2 capacity and selectivity
Competitive models and/or multicomponent measurements are needed to determine the effect of flue gas impurities on the CO2 capacity and selectivity of an adsorbent. For natural gas flue emissions, the primary contaminants of interest are O2 and H2O, which are both present at higher partial pressures than CO2. Therefore, extensive analyses are needed to evaluate the stability and performance of adsorbents under the specific conditions relevant to natural gas flue emissions.18Simple qualitative experiments can enable rapid assessment of the stability of candidate adsorbents to impurities. For example, changes in the single-component CO2 adsorption capacity after exposure to hot O2- and/or H2O-containing streams can reveal oxidative or hydrolytic instability in a simulated temperature-swing process. Secondary analysis techniques, such as infrared spectroscopy, can then be used to probe the mechanism of degradation. Importantly, while adsorbent exposure to individual impurities can help identify specific degradation pathways, the combination of O2 and H2O is known to cause enhanced degradation of many porous materials and should therefore also be considered.155–157
Beyond validation of stability, several techniques may be used to probe competitive adsorption. Selectivity can rapidly be calculated from single-component isotherms over a broad parameter space using ideal adsorbed solution theory (IAST), given that the assumptions of the theory hold or that appropriate corrections are applied.162–164 Experimental characterization under simulated flue gas, while more complex and labor-intensive, is ultimately necessary to evaluate the performance of new materials. When instrumentation permits, multicomponent equilibrium measurements coupled with residual gas analysis are ideal for quantitative characterization of competitive adsorption.92 Breakthrough experiments with simulated flue gas are likewise informative and more routine, although larger sample sizes (∼1 g or greater) are preferable. As an alternative technique, temperature-programmed desorption (TPD) coupled with spectroscopy or mass spectrometry can also be used to quantify co-adsorbed species after exposure to a simulated flue gas stream.
Long-term cycling stability
Adsorbents for post-combustion capture must be able to withstand extended cycling to be economically feasible. Cycling experiments should be conducted using realistic desorption conditions and the most realistic flue gas mixture possible with the given instrumentation, with a particular emphasis on the O2 and H2O content of the stream. When available, automated cycling systems with residual gas analysis are ideal for this purpose, as the CO2 purity and recovery can be extracted directly from the data. Alternatively, rapid cycling can be performed under realistic conditions using a TGA. Post-cycling analysis of the material can then provide insight into the mechanisms of any thermal, oxidative, or hydrolytic degradation.Kinetics of adsorption
The low partial pressure of CO2 in natural gas flue emissions may result in kinetic barriers to effective adsorption/desorption cycling.44,66,165,166 Therefore, characterization of adsorption and desorption kinetics is needed to assess the viability of new adsorbents and inform the selection of cycle times and bed sizes in a capture process. From a process standpoint, the ultimate kinetics of interest will need to be measured using structured materials, for which kinetics behavior can deviate from the as-synthesized adsorbents. However, at the laboratory scale, thermogravimetric analysis, zero-length column measurements, or breakthrough experiments can be used to assess the rate of CO2 capture from low partial pressure streams.Conclusions
Although natural gas releases half as much CO2 as coal per kW h energy produced, rapid growth in the consumption of natural gas and resulting CO2 emissions necessitates accelerated research into carbon capture from NGCC power plants. Most adsorbents for carbon capture applications are currently at technology readiness levels (TRLs) of 3–5, indicating that few adsorbents have yet advanced beyond laboratory testing to deployment in slipstreams or full-scale power plants.167 Given that the use of natural gas is increasing rapidly, it is critical that work to bring these materials to TRL 9, which involves deployment in a 500 MW power plant, be given a high research priority. Although a number of prospective materials are under development for CO2 capture from coal flue gas, air, and crude natural gas, the suitability of these materials for CO2 capture from NGCC power plants must be established. Here, we have identified the key challenges encountered in the capture of CO2 from NGCC power plants, as well as some of the most promising materials for this application. We have also proposed a series of characterization methods aimed at guiding adsorbent design and informing process models to assess the viability of new materials for this important process. Overall, more thorough characterization of new materials under realistic conditions is needed in order to advance materials toward deployment in full-scale post-combustion capture processes. Finally, as new capture processes advance toward commercialization, techno-economic and life-cycle analyses should be undertaken to identify optimal cost/benefit opportunities in terms of CO2 capture rate, total CO2 emissions avoided, and cost of electricity increase across the power sector as a whole.Conflicts of interest
The authors declare the following competing financial interests: J. R. L. has a financial interest in Mosaic Materials, Inc., a start-up company working to commercialize metal–organic frameworks for gas separations, including CO2 capture applications.Acknowledgements
We gratefully acknowledge ExxonMobil Research and Engineering Company for financial support of this work. We thank the National Institute of General Medical Sciences of the National Institutes of Health for a postdoctoral fellowship for P. J. M. (F32GM120799). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We further thank Dr Tim Barckholtz and Dr Joseph Falkowski of ExxonMobil Research and Engineering Company for helpful discussions, and Dr Katie Meihaus of the University of California, Berkeley for editorial assistance.Notes and references
- R. K. Pachauri, M. R. Allen, V. R. Barros, J. Broome, W. Cramer, R. Christ, J. A. Church, L. Clarke, Q. Dahe and P. Dasgupta, Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the fifth assessment report of the Intergovernmental Panel on Climate Change, IPCC, 2014.
- CO 2 Emissions from Fuel Combustion 2018, International Energy Agency, Paris, France, 2018.
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed.
- J. Wilcox, Carbon Capture, Springer, New York, 2012 Search PubMed.
- The Global Status of CCS 2015: Summary Report, Global Carbon Capture and Storage Institute, Docklands, Australia, 2015.
- 20 Years of Carbon Capture and Storage – Accelerating Future Deployment, International Energy Agency, Paris, France, 2016.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1076 RSC.
- B. Metz, O. Davidson, H. de Coninck, M. Loos and L. Meyer, IPCC Special Report on Carbon Dioxide Capture and Storage, Intergovernmental Panel on Climate Change, 2005.
- P. H. Stauffer, G. N. Keating, R. S. Middleton, H. S. Viswanathan, K. A. Berchtold, R. P. Singh, R. J. Pawar and A. Mancino, Environ. Sci. Technol., 2011, 45, 8597–8604 CrossRef CAS PubMed.
- World Energy Outlook 2018, International Energy Agency, 2018.
- International Energy Outlook 2018, Energy Information Administration, U.S. Department of Energy, 2018.
- Annual Energy Outlook 2018 with Projections to 2050, U.S. Energy Information Administration, 2018.
- Carbon Capture Opportunities for Natural Gas Fired Power Systems, U.S. Department of Energy, 2017.
- E. S. Rubin and H. Zhai, Environ. Sci. Technol., 2012, 46, 3076–3084 CrossRef CAS PubMed.
- CO2 Capture at Gas Fired Power Plants, IEAGHG, 2012.
- Carbon Dioxide Capture for Natural Gas and Industrial Applications, U.S. Department of Energy, 2015.
- M. Vaccarelli, R. Carapellucci and L. Giordano, Energy Procedia, 2014, 45, 1165–1174 CrossRef CAS.
- Cost and Performance Baseline for Fossil Energy Plants. Volume 1a: Bituminous Coal (PC) and Natural Gas to Electricity. Revision 3, U.S. Department of Energy, National Energy Technology Laboratory, 2015.
- T. C. Merkel, X. Wei, Z. He, L. S. White, J. G. Wijmans and R. W. Baker, Ind. Eng. Chem. Res., 2013, 52, 1150–1159 CrossRef CAS.
- The Costs of CO 2 Capture, Transport and Storage, European Technology Platform for Zero Emission Fossil Fuel Power Plants (ZEP), 2011.
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS PubMed.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- S. Chi and G. T. Rochelle, Ind. Eng. Chem. Res., 2002, 41, 4178–4186 CrossRef CAS.
- M. S. Jassim, G. Rochelle, D. Eimer and C. Ramshaw, Ind. Eng. Chem. Res., 2007, 46, 2823–2833 CrossRef CAS.
- C. Gouedard, D. Picq, F. Launay and P.-L. Carrette, Int. J. Greenhouse Gas Control, 2012, 10, 244–270 CrossRef CAS.
- S. B. Fredriksen and K.-J. Jens, Energy Procedia, 2013, 37, 1770–1777 CrossRef CAS.
- F. Vega, A. Sanna, B. Navarrete, M. M. Maroto-Valer and V. J. Cortés, Greenhouse Gases: Sci. Technol., 2014, 4, 707–733 CrossRef CAS.
- S. A. Mazari, B. Si Ali, B. M. Jan, I. M. Saeed and S. Nizamuddin, Int. J. Greenhouse Gas Control, 2015, 34, 129–140 CrossRef CAS.
- M. Spinelli, S. Campanari, S. Consonni, M. C. Romano, T. Kreutz, H. Ghezel-Ayagh and S. Jolly, J. Electrochem. Energy Convers. Storage, 2018, 15, 031001 CrossRef.
- R. J. Allam, J. E. Fetvedt, B. A. Forrest and D. A. Freed, in Proceedings of ASME Turbo Expo 2014: Turbine Technical Conference and Exposition, ASME, 2014, p. V03BT36A016.
- J. D. Laumb, M. J. Holmes, J. J. Stanislowski, X. Lu, B. Forrest and M. McGroddy, Energy Procedia, 2017, 114, 573–580 CrossRef CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- T. P. Senftle and E. A. Carter, Acc. Chem. Res., 2017, 50, 472–475 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- P. Luis and B. Van der Bruggen, Greenhouse Gases: Sci. Technol., 2013, 3, 318–337 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- N. Hedin, L. Chen and A. Laaksonen, Nanoscale, 2010, 2, 1819 RSC.
- S. Keskin, T. M. van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879–891 CrossRef CAS PubMed.
- Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- P. Bollini, S. A. Didas and C. W. Jones, J. Mater. Chem., 2011, 21, 15100–15120 RSC.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 RSC.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- Y. Liu, Z. U. Wang and H.-C. Zhou, Greenhouse Gases: Sci. Technol., 2012, 2, 239–259 CrossRef CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- C.-H. Yu, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC.
- J. M. Huck, L.-C. Lin, A. H. Berger, M. N. Shahrak, R. L. Martin, A. S. Bhown, M. Haranczyk, K. Reuter and B. Smit, Energy Environ. Sci., 2014, 7, 4132–4146 RSC.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- R. Sabouni, H. Kazemian and S. Rohani, Environ. Sci. Pollut. Res., 2014, 21, 5427–5449 CrossRef CAS PubMed.
- P. A. Webley, Adsorption, 2014, 20, 225–231 CrossRef CAS.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868 RSC.
- J. C. Abanades, B. Arias, A. Lyngfelt, T. Mattisson, D. E. Wiley, H. Li, M. T. Ho, E. Mangano and S. Brandani, Int. J. Greenhouse Gas Control, 2015, 40, 126–166 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2015, 23, 1–11 CrossRef CAS.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- Q. Wang, J. Bai, Z. Lu, Y. Pan and X. You, Chem. Commun., 2016, 52, 443–452 RSC.
- E. González-Zamora and I. A. Ibarra, Mater. Chem. Front., 2017, 1, 1471–1484 RSC.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Adv. Energy Mater., 2017, 7, 1601296 CrossRef.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- E. E. Ünveren, B. Ö. Monkul, Ş. Sarıoğlan, N. Karademir and E. Alper, Petroleum, 2017, 3, 37–50 CrossRef.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- J. A. A. Gibson, E. Mangano, E. Shiko, A. G. Greenaway, A. V. Gromov, M. M. Lozinska, D. Friedrich, E. E. B. Campbell, P. A. Wright and S. Brandani, Ind. Eng. Chem. Res., 2016, 55, 3840–3851 CrossRef CAS.
- H. Li, G. Haugen, M. Ditaranto, D. Berstad and K. Jordal, Energy Procedia, 2011, 4, 1411–1418 CrossRef CAS.
- R. Veneman, H. Kamphuis and D. W. F. Brilman, Energy Procedia, 2013, 37, 2100–2108 CrossRef CAS.
- J. W. Dijkstra, S. Walspurger, G. D. Elzinga, J. A. Z. Pieterse, J. Boon and W. G. Haije, Ind. Eng. Chem. Res., 2018, 57, 1245–1261 CrossRef CAS.
- A. H. Berger and A. S. Bhown, Energy Procedia, 2014, 63, 2092–2099 CrossRef CAS.
- F. Rezaei and M. Grahn, Ind. Eng. Chem. Res., 2012, 51, 4025–4034 CrossRef CAS.
- H. S. Kheshgi, H. Thomann, N. A. Bhore, R. B. Hirsch, M. E. Parker and G. Teletzke, SPE Econ. Manag., 2012, 4, 24–31 CrossRef.
- E. S. Rubin, J. E. Davison and H. J. Herzog, Int. J. Greenhouse Gas Control, 2015, 40, 378–400 CrossRef CAS.
- E. S. Rubin, Int. J. Greenhouse Gas Control, 2012, 10, 181–190 CrossRef.
- A. T. Evulet, A. M. ELKady, A. R. Branda and D. Chinn, Energy Procedia, 2009, 1, 3809–3816 CrossRef CAS.
- D. Dillon, D. Grace, A. Maxson, K. Børter, J. Augeli, S. Woodhouse and G. Aspelund, Energy Procedia, 2013, 37, 2397–2405 CrossRef CAS.
- Y. Tanaka, M. Nose, M. Nakao, K. Saitoh, E. Ito and K. Tsukagoshi, Mitsubishi Heavy Ind. Tech. Rev., 2013, 50, 1–6 Search PubMed.
- A. Kyle, J. Black, M. Woods, N. Kuehn, W. Shelton and W.-C. Yang, Carbon Capture Approaches for Natural Gas Combined Cycle Systems: Revision 2, U.S. Department of Energy, National Energy Technology Laboratory, 2010 Search PubMed.
- U. Ali, E. O. Agbonghae, K. J. Hughes, D. B. Ingham, L. Ma and M. Pourkashanian, Appl. Therm. Eng., 2016, 103, 747–758 CrossRef CAS.
- H. Lepaumier, D. Picq and P.-L. Carrette, Ind. Eng. Chem. Res., 2009, 48, 9068–9075 CrossRef CAS.
- P. Bollini, S. Choi, J. H. Drese and C. W. Jones, Energy Fuels, 2011, 25, 2416–2425 CrossRef CAS.
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Microporous Mesoporous Mater., 2011, 145, 146–149 CrossRef CAS.
- A. Heydari-Gorji and A. Sayari, Ind. Eng. Chem. Res., 2012, 51, 6887–6894 CrossRef CAS.
- S. Bali, T. T. Chen, W. Chaikittisilp and C. W. Jones, Energy Fuels, 2013, 27, 1547–1554 CrossRef CAS.
- A. P. Hallenbeck and J. R. Kitchin, Ind. Eng. Chem. Res., 2013, 52, 10788–10794 CrossRef CAS.
- S. A. Didas, R. Zhu, N. A. Brunelli, D. S. Sholl and C. W. Jones, J. Phys. Chem. C, 2014, 118, 12302–12311 CrossRef CAS.
- A. Ahmadalinezhad, R. Tailor and A. Sayari, Chem. – Eur. J., 2013, 19, 10543–10550 CrossRef CAS PubMed.
- A. Ahmadalinezhad and A. Sayari, Phys. Chem. Chem. Phys., 2014, 16, 1529–1535 RSC.
- C. S. Srikanth and S. S. C. Chuang, ChemSusChem, 2012, 5, 1435–1442 CrossRef CAS PubMed.
- N. Sundaram, R. Gupta, H. Thomann, H. S. Caram, L. K. Starcher, F. F. Mittricker, S. C. Weston and S. J. Weigel, US Pat., US9476356B2, 2016 Search PubMed.
- K. B. Lee and S. Sircar, AIChE J., 2008, 54, 2293–2302 CrossRef CAS.
- J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- C. Kim, H. S. Cho, S. Chang, S. J. Cho and M. Choi, Energy Environ. Sci., 2016, 9, 1803–1811 RSC.
- W. R. Woerner, A. M. Plonka, X. Chen, D. Banerjee, P. K. Thallapally and J. B. Parise, J. Phys. Chem. C, 2016, 120, 360–369 CrossRef CAS.
- Y. Wang and M. D. LeVan, J. Chem. Eng. Data, 2010, 55, 3189–3195 CrossRef CAS.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301–14307 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- J. Yu and P. B. Balbuena, J. Phys. Chem. C, 2013, 117, 3383–3388 CrossRef CAS.
- L. Joos, J. A. Swisher and B. Smit, Langmuir, 2013, 29, 15936–15942 CrossRef CAS PubMed.
- L.-C. Lin, K. Lee, L. Gagliardi, J. B. Neaton and B. Smit, J. Chem. Theory Comput., 2014, 10, 1477–1488 CrossRef CAS PubMed.
- X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113–8119 CrossRef CAS.
- J. Yu, Y. Zhai and S. S. C. Chuang, Ind. Eng. Chem. Res., 2018, 57, 4052–4062 CrossRef CAS.
- A. Sayari and Y. Belmabkhout, J. Am. Chem. Soc., 2010, 132, 6312–6314 CrossRef CAS PubMed.
- D. Wang, X. Wang and C. Song, ChemPhysChem, 2017, 18, 3163–3173 CrossRef CAS PubMed.
- P. J. Milner, J. D. Martell, R. L. Siegelman, D. Gygi, S. C. Weston and J. R. Long, Chem. Sci., 2018, 9, 160–174 RSC.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514–1520 CrossRef CAS.
- Y. Belmabkhout and A. Sayari, Energy Fuels, 2010, 24, 5273–5280 CrossRef CAS.
- S. Sjostrom and H. Krutka, Fuel, 2010, 89, 1298–1306 CrossRef CAS.
- S. Han, Y. Huang, T. Watanabe, Y. Dai, K. S. Walton, S. Nair, D. S. Sholl and J. C. Meredith, ACS Comb. Sci., 2012, 14, 263–267 CrossRef CAS PubMed.
- F. Rezaei and C. W. Jones, Ind. Eng. Chem. Res., 2013, 52, 12192–12201 CrossRef CAS.
- J. S. Yeon, W. R. Lee, N. W. Kim, H. Jo, H. Lee, J. H. Song, K. S. Lim, D. W. Kang, J. G. Seo, D. Moon, B. Wiers and C. S. Hong, J. Mater. Chem. A, 2015, 3, 19177–19185 RSC.
- S. Bhattacharyya, R. Han, W.-G. Kim, Y. Chiang, K. C. Jayachandrababu, J. T. Hungerford, M. R. Dutzer, C. Ma, K. S. Walton, D. S. Sholl and S. Nair, Chem. Mater., 2018, 30, 4089–4101 CrossRef CAS.
- K. Gerdes, Incentivizing Carbon Capture Retrofits of the Existing PC and NGCC Fleet, U.S. Department of Energy, National Energy Technology Laboratory, Pittsburgh, PA, USA, 2014 Search PubMed.
- S. Kulprathipanja, Zeolites in Industrial Separation and Catalysis, John Wiley & Sons, 2010 Search PubMed.
- T.-H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 RSC.
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. W. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- C. A. Grande, R. P. P. L. Ribeiro and A. E. Rodrigues, Energy Fuels, 2009, 23, 2797–2803 CrossRef CAS.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339–8344 CrossRef CAS.
- G. Li, P. Xiao, P. Webley, J. Zhang, R. Singh and M. Marshall, Adsorption, 2008, 14, 415–422 CrossRef CAS.
- A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268–3292 CrossRef CAS PubMed.
- P. D. Jadhav, R. V. Chatti, R. B. Biniwale, N. K. Labhsetwar, S. Devotta and S. S. Rayalu, Energy Fuels, 2007, 21, 3555–3559 CrossRef CAS.
- J. C. Fisher, J. Tanthana and S. S. C. Chuang, Environ. Prog. Sustainable Energy, 2009, 28, 589–598 CrossRef CAS.
- F. Su, C. Lu, S.-C. Kuo and W. Zeng, Energy Fuels, 2010, 24, 1441–1448 CrossRef CAS.
- T. H. Nguyen, S. Kim, M. Yoon and T.-H. Bae, ChemSusChem, 2016, 9, 455–461 CrossRef CAS PubMed.
- C. Chen, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2014, 31, 1919–1934 CrossRef CAS.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- G. S. Foo, J. J. Lee, C.-H. Chen, S. E. Hayes, C. Sievers and C. W. Jones, ChemSusChem, 2017, 10, 266–276 CrossRef CAS PubMed.
- X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Fuel Process. Technol., 2005, 86, 1457–1472 CrossRef CAS.
- A. Seif El Nasr, T. Nelson, A. Kataria and M. R. M. Abu-Zahra, Int. J. Greenhouse Gas Control, 2015, 42, 583–592 CrossRef CAS.
- W. Zhang, C. Sun, C. E. Snape, R. Irons, S. Stebbing, T. Alderson, D. Fitzgerald and H. Liu, Int. J. Greenhouse Gas Control, 2017, 58, 276–289 CrossRef CAS.
- S. H. Pang, L.-C. Lee, M. A. Sakwa-Novak, R. P. Lively and C. W. Jones, J. Am. Chem. Soc., 2017, 139, 3627–3630 CrossRef CAS PubMed.
- K. Min, W. Choi, C. Kim and M. Choi, Nat. Commun., 2018, 9, 726 CrossRef PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H.-C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480–7484 CrossRef CAS PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna and H.-C. Zhou, J. Phys. Chem. C, 2013, 117, 4057–4061 CrossRef CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- M. C. Celina, Polym. Degrad. Stab., 2013, 98, 2419–2429 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- D. G. Madden, H. S. Scott, A. Kumar, K.-J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry and M. J. Zaworotko, Philos. Trans. R. Soc., A, 2017, 375, 20160025 CrossRef PubMed.
- Y. Lin, C. Kong and L. Chen, RSC Adv., 2016, 6, 32598–32614 RSC.
- A. J. Emerson, A. Chahine, S. R. Batten and D. R. Turner, Coord. Chem. Rev., 2018, 365, 1–22 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS PubMed.
- A. F. Cozzolino, C. K. Brozek, R. D. Palmer, J. Yano, M. Li and M. Dincă, J. Am. Chem. Soc., 2014, 136, 3334–3337 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, X. Jia, J. Yang and J. Li, Dalton Trans., 2017, 46, 15573–15581 RSC.
- B. Tu, Q. Pang, H. Xu, X. Li, Y. Wang, Z. Ma, L. Weng and Q. Li, J. Am. Chem. Soc., 2017, 139, 7998–8007 CrossRef CAS PubMed.
- J. G. Vitillo and S. Bordiga, Mater. Chem. Front., 2017, 1, 444–448 RSC.
- F. Rezaei and P. Webley, Chem. Eng. Sci., 2009, 64, 5182–5191 CrossRef CAS.
- S. J. A. DeWitt, A. Sinha, J. Kalyanaraman, F. Zhang, M. J. Realff and R. P. Lively, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 129–152 CrossRef PubMed.
- D. C. Miller, M. Syamlal, D. S. Mebane, C. Storlie, D. Bhattacharyya, N. V. Sahinidis, D. Agarwal, C. Tong, S. E. Zitney, A. Sarkar, X. Sun, S. Sundaresan, E. Ryan, D. Engel and C. Dale, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 301–323 CrossRef CAS PubMed.
- D. C. Miller, J. T. Litynski, L. A. Brickett and B. D. Morreale, AIChE J., 2015, 62, 2–10 CrossRef.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- K. S. Walton and D. S. Sholl, AIChE J., 2015, 61, 2757–2762 CrossRef CAS.
- C. M. Simon, B. Smit and M. Haranczyk, Comput. Phys. Commun., 2016, 200, 364–380 CrossRef CAS.
- E. R. Monazam, L. J. Shadle, D. C. Miller, H. W. Pennline, D. J. Fauth, J. S. Hoffman and M. L. Gray, AIChE J., 2013, 59, 923–935 CrossRef CAS.
- L. Darunte, T. Sen, C. Bhawanani, K. S. Walton, D. S. Sholl, M. J. Realff and C. W. Jones, Ind. Eng. Chem. Res., 2019, 58, 366–377 CrossRef CAS.
- A. S. Bhown, Energy Procedia, 2014, 63, 542–549 CrossRef CAS.
Footnotes |
| † These authors contributed equally. |
| ‡ Current address: Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY, 14853, USA. |
| This journal is © The Royal Society of Chemistry 2019 |