
An AIE molecule featuring changeable triplet emission between phosphorescence and delayed fluorescence by an external force†
Lili
Huang‡
a,
Xue
Wen‡
a,
Jianwei
Liu
a,
Mingxing
Chen
b,
Zhiyong
Ma
 *a and
Xinru
Jia
b
*a and
Xinru
Jia
b
aBeijing State Key Laboratory of Organic–Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: mazhy@mail.buct.edu.cn
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of the Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 22nd August 2019
Abstract
In this article, we report a newly designed molecule ODFRPTZ with an asymmetric D–A–D′ type structure. ODFRPTZ is endowed with aggregation-induced emission (AIE), intriguing mechanochromism and changeable emission between delayed fluorescence and phosphorescence. In the THF/H2O system, the emission efficiency of ODFRPTZ is greatly improved by increasing the water fraction due to the restriction of the intramolecular rotation and vibration in the aggregation states. Intriguingly, ODFRPTZ shows mechanochromic properties under both UV light and visible light. The emission color switches from green to yellow and the color under visible light changes from white to yellow after grinding due to the changeable conformations of phenothiazine from quasi-axial to quasi-equatorial. The crystalline powder shows prompt fluorescence and phosphorescence characteristics. The lifetime of phosphorescence continuously increases from 0.4 ms at 300 K to 121.1 ms at 77 K. The emission of ground powder is composed of prompt fluorescence and delayed fluorescence with lifetimes of 18.7 ns and 0.9 μs, respectively. To the best of our knowledge, it is a rarely reported AIE example that the triplet state emission can be altered from phosphorescence to delayed fluorescence by an external force.
Introduction
To overcome the notorious aggregation-caused quenching (ACQ),1 a large number of luminescent materials with aggregation-induced emission (AIE) properties have been rapidly developed as they can be widely applied in organic light-emitting diodes (OLED),2–4 biological probes,5 fluorescent sensors,6etc. In addition to the two well-known AIE molecules silole7–9 and tetraphenylethene (TPE),10 phenothiazine (PTZ) has also attracted much attention in AIE systems.11–14 In the past few years, PTZ has largely served as the electron donor in intramolecular charge transfer (ICT) systems and the highly twisted structure of PTZ effectively avoids the quenching caused by strong intermolecular interactions in the aggregation state.15–18 The process of ICT is hard to proceed in solutions due to the free rotation of the molecular bond.19 However, emission efficiency can be greatly improved due to the restriction of intramolecular motions (RIM) in the solid state.20–22 Hence, employing PTZ as the electron donor part in donor–acceptor (D–A) or donor–acceptor–donor (D–A–D) type molecules is another effective way to achieve good AIE properties.13Besides, it has been revealed that PTZ derivatives combining delayed fluorescence (DF) with AIE properties are promising candidates for next generation emitters of OLEDs.11,23,24 For those AIE-DF materials, the twisted structures of phenothiazine played a crucial role in separating the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), thus generating a small singlet–triplet energy gap (ΔEST) and furthermore promoting the triplet excitons to return to excited singlet states (S1) through reverse intersystem crossing (RISC).14,25 Ma and his co-workers realized simultaneous DF and room-temperature phosphorescence in their designed molecule and could determine the dominant emission through the utilization of thermal-controlled triplet excitons.26 The generation of triplet excitons is crucial for both DF and phosphorescence and we speculated that DF-phosphorescence AIE emitters could be obtained if triplet excitons were appropriately utilized in PTZ derivatives.26 Last several years have witnessed the rise of metal-free organic phosphorescent materials by methods like introducing aromatic carbonyls27 or heavy atoms into molecules,28,29 incorporating halogen bonding into crystals,30 molecular H-aggregation31 and embedding organic phosphors into other matrices.32–34 Previous work has demonstrated that triplet excitons could be easily captured in benzophenone derivatives.35–38 Therefore, alternative emissions are supposed to be achieved if phenothiazine and benzophenone are introduced as the electron donor and acceptor, respectively.
Moreover, PTZ derivatives, whose conformations can be changed from quasi-axial to quasi-equatorial, are also commendable mechanical-stimuli-responsive materials.39 Chi et al. reported a symmetric donor–acceptor–donor (D–A–D) type molecule which contained sulfobenzide as a donor and phenothiazine as an electron acceptor, exhibiting AIE and two-color switching mechanochromic luminescence (MCL) properties (2014).40 Then in 2017, Takeda et al. found that molecules comprising dibenzo[a,j]phenazine and PTZ showed both tricolor MCL and efficient DF.25 Recently, Li and his group designed a D–A structure molecule with the electron acceptor part benzophenone and donor part PTZ possessing dynamic conformations and showing continuous blue–white–yellow triplet mechanoluminescent (ML) emissions.41 Tian and co-workers have revealed that the change of the intramolecular geometrical conformation could influence the locally excited (LE) state and the ICT state; as a result, luminescent color could be tuned through mechanical stimuli.42 Undeniably, a series of ML and MCL fluorescent materials have been developed; nevertheless, mechanical-responsive AIE emitters with delayed fluorescence and phosphorescence are still quite rare.43
Herein, we report our newly designed molecule ODFRPTZ, displaying multiple properties of AIE, delayed fluorescence (DF), room-temperature phosphorescence and mechanochromism. One pivotal factor for the phenomena of DF and phosphorescence is the generation of triplet excitons and adding O, N and P heteroatoms into the molecule is one effective way to enhance ICT and generate triplet excitons.44 Therefore, dibenzofuran and phenothiazine are introduced to act as the electron donor and acceptor, respectively (Fig. 1). As expected, the AIE effect is observed in THF/water mixtures. Interestingly, ODFRPTZ shows remarkable mechanochromic properties. The emission color can change from green to yellow and the color under visible light can be altered from white to yellow by adjusting the molecular conformation through grinding. The transition direction of the triplet excitons is conformation-dependent, which determines the emission type. In the crystal state, PTZ adopts the highly constrained quasi-axial conformation and phosphorescence dominates. After grinding, the conformation of PTZ changes to quasi-equatorial, promoting DF. As far as we know, stimuli-responsive AIE molecules with changeable emission between DF and phosphorescence under an external force have rarely been reported. We believe this work may provide deep insight into the emission mechanism and broaden the applications of luminescent materials.
 | ||
| Fig. 1 Molecular structure of ODFRPTZ. | ||
Results and discussion
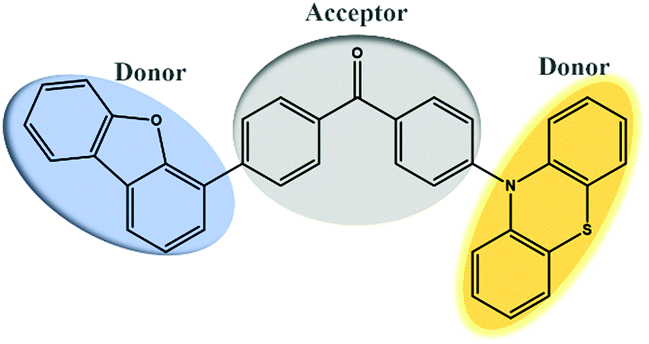
ODFRPTZ features a D–A–D′ type structure with dibenzofuran and phenothiazine as donors and benzophenone as the acceptor. ODFRF was obtained by typical Suzuki coupling, starting from (4-bromophenyl) (4-fluorophenyl) methanone and dibenzo[b,d]thiophen-4-ylboronic acid. The targeted molecule ODFRPTZ was afforded with a yield of 83% via the substitution reaction between ODFRF and phenothiazine. It is very interesting that the rate of reduced pressure evaporation made a difference in the appearance of the color of the final powder. Quick evaporation could lead to a yellow powder while slow evaporation could result in a white powder. This unique phenomenon inspired us to deeply study arrangement-dependent photophysical properties of ODFRPTZ. The molecular strucutre and purity were verified by 1H NMR, 13C NMR and HR-MS. Furthermore, the single crystal of ODFRPTZ was obtained and the chemical structure and purity were further confirmed. The details of synthetic procedures and the related characterization are described in the ESI† (Scheme S1 and Fig. S1–S8).ODFRPTZ shows typical AIE properties due to the existence of the highly twisted structure of phenothiazine. We prepared eleven mixed solutions containing THF/water with different water fractions (fw) and studied their AIE properties. As shown in Fig. 2a, only a weak emission band could be observed in THF solution, owing to the free intramolecular motion. However, AIE appeared when the water fraction reached 70%. An emission peak red-shifted to 550 nm ascribed to the ICT emission and intensified yellow fluorescence could be observed when fw reached 95% as the aggregation states changed. Simultaneously, the absorption band around 280 nm decreased as fw increased and a new broad absorption band appeared in the range of 320–370 nm (Fig. 2b), further confirming the aggregation process. In aggregation states, the restriction of the intramolecular rotation and vibration reduced the non-radiative relaxation, contributing to dramatically increased emission intensity.
 | ||
| Fig. 2 (a) Fluorescence spectra and (b) absorption spectra in different THF/water (v/v) mixtures. (c) Optical images of ODFRPTZ in the mixed solution (20 μM, λex = 365 nm). | ||
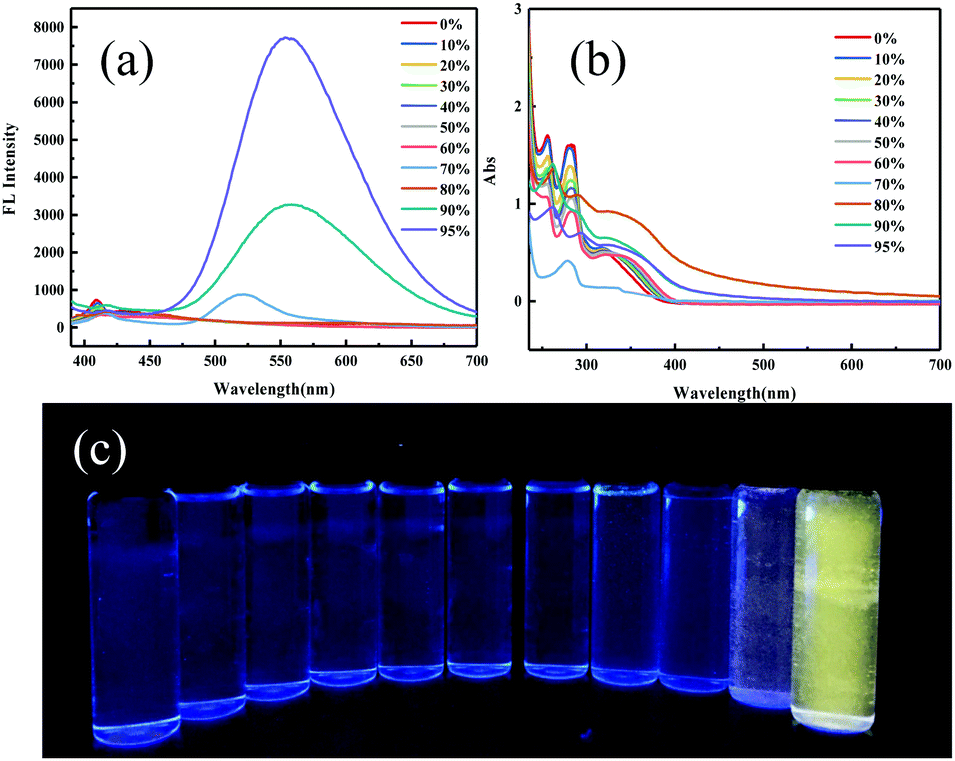
Regulating the luminescent behavior by external stimuli in the solid state has been attracting tremendous attention. ODFRPTZ showed intriguing mechanochromic properties under UV light and room light in the solid state. Originally, ODFRPTZ afforded green emission under UV irradiation with two emission peaks at 428 nm and 540 nm, attributed to quasi-axial phenothiazine (verified by single crystal data and discussed later). The emission color switched to yellow under an external force (Fig. 3b and d). The emission peak at 428 nm disappeared and the emission peak at 540 nm red-shifted by 41 nm to 581 nm (Fig. 3e). The emission red-shift was due to the conformation change from the quasi-axial conformer to a quasi-equatorial one, whose structures were relatively planar and thus facilitated the intermolecular electron transition from phenothiazine to carbonyl. Rather interestingly, the color under visible light also changed from pure white to yellow after grinding (Fig. 3a and c). The absorption of the ground powder was highly enhanced and a new absorption band emerged in the visible region (over 400 nm) compared to the pristine powder (Fig. S9, ESI†), inferring that the energy gap of ICT was altered by the external force. The mechanochromic process was well reversible. The microcrystals could re-form after fuming with dichloromethane and the color could return to the original under both visible and UV light (Fig. S10, ESI†).
 | ||
| Fig. 3 (a)–(d) Optical images of the original crystalline and ground powder under visible light and UV light, respectively. (e) Steady-state emission spectra of the pristine powder and ground powder (λex = 365 nm). (f) XRD patterns of the original powder and the ground one. | ||
Wide-angle X-ray diffraction (WAXD) was employed to further understand the microstructure change by applying force (Fig. 3f). The XRD patterns showed that the pristine ODFRPTZ powder had multiple sharp peaks, agreeing well with the single crystal result, whereas they became weak and even disappeared after the mechanical stimulus, indicating the transition from the well-ordered crystalline structure to the amorphous state. Additionally, differential scanning calorimetry (DSC) was carried out to explore the thermal properties of the original one and ground powder. For the pristine powder, a sharp endothermic peak appeared at 235.5 °C. After grinding, the sharp peak became broad and a new exothermic peak emerged at 93.8 °C, further verifying the force-induced phase transition from the crystalline state to amorphous state (Fig. S11, ESI†).
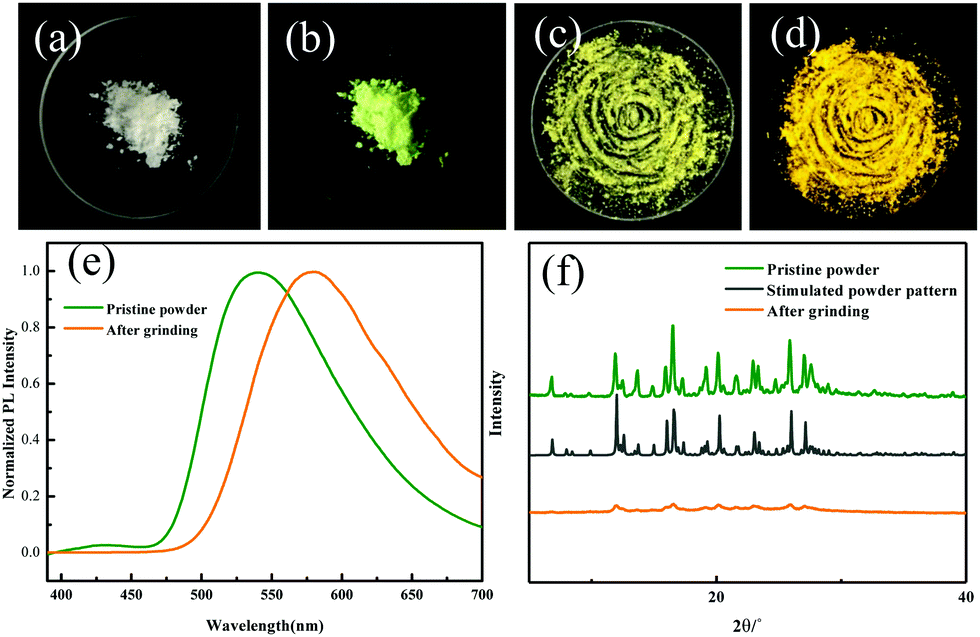
To gain deep insight into the mechanism for the mechanochromism of ODFRPTZ, detailed photophysical properties in different solid states were studied. Crystalline powder of ODFRPTZ showed prompt fluorescence and triplet excited state emission characteristics. The pristine crystalline powder showed highly green emission under 365 nm UV light and no afterglow was observed at room temperature after removing the UV radiation. In the steady-state photoluminescence spectra, the emission band centered at 540 nm with a small shoulder at 428 nm. Delayed photoluminescence spectra were also measured and the main emission band ranging from 480 nm to 700 nm was identical to the 540 nm emission in the steady-state one, manifesting the nature of triplet excited state emission (Fig. 4a). DF and phosphorescence are dependent on oxygen as triplet excited states are involved in these emission pathways; thus, an oxygen dependence experiment was carried out to deconvolute emission processes. Fig. 4b shows steady-state emission spectra in the absence of oxygen and in the presence of oxygen. The emission intensity was remarkably enhanced for the pristine powder of ODFRPTZ under an argon atmosphere and it had a Idegas/IO2 of 2.2, further confirming the triplet excited state emissions.
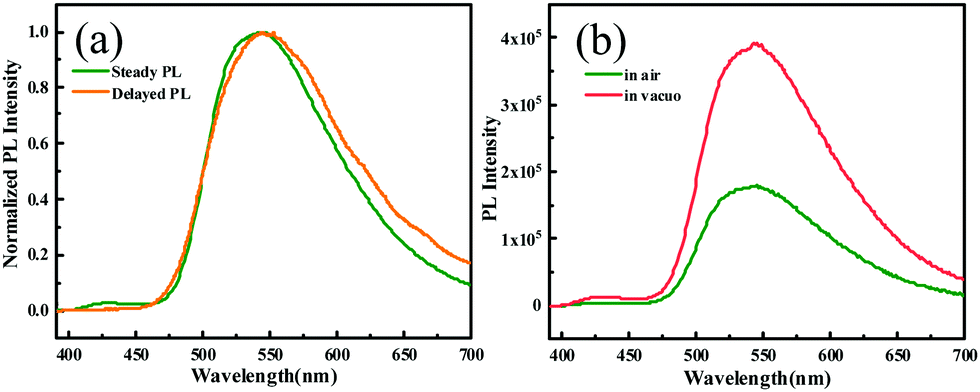 | ||
| Fig. 4 (a) Steady-state photoluminescence spectra and delayed photoluminescence spectra of the ODFRPTZ crystalline powder at room temperature. (b) Steady-state photoluminescence spectra of the crystalline powder of ODFRPTZ in the presence and absence of oxygen. | ||
To discriminate prompt fluorescence, delayed fluorescence and phosphorescence, decay spectra were recorded using a different light source. The lifetime of prompt fluorescence at 540 nm was 8.9 ns (Fig. S12, ESI†), assigned to a typical singlet excited state relaxation channel. The lifetime of the 540 nm emission at room temperature was also tested using a VPL light source and a long lifetime of 0.4 ms was acquired (Fig. 5e), implying the existence of phosphorescence. Afterwards, phosphorescence was further validated by placing the crystalline powder in liquid nitrogen. After removing the UV radiation, a distinct green afterglow was observed by the naked eye, which was similar to its fluorescent color. At low temperatures, it's difficult for the triplet excitons to return to the excited singlet states through RISC. Thus, T1 excitons directly went back to the ground state, resulting in phosphorescence. Moreover, temperature-dependent emission spectra and lifetime measurements were performed subsequently. The emission band gradually blue-shifted upon lowering the temperature (Fig. 5f) and the lifetime continuously increased from 0.4 ms at 300 K to 121.1 ms at 77 K (Fig. 5e), agreeing well with the characteristics of phosphorescence and verifying the existence of phosphorescence. The above-mentioned results reveal that the emission of the ODFRPTZ crystalline powder is composed of prompt fluorescence and phosphorescence and the lifetime of phosphorescence is prolonged by lowering the temperature.
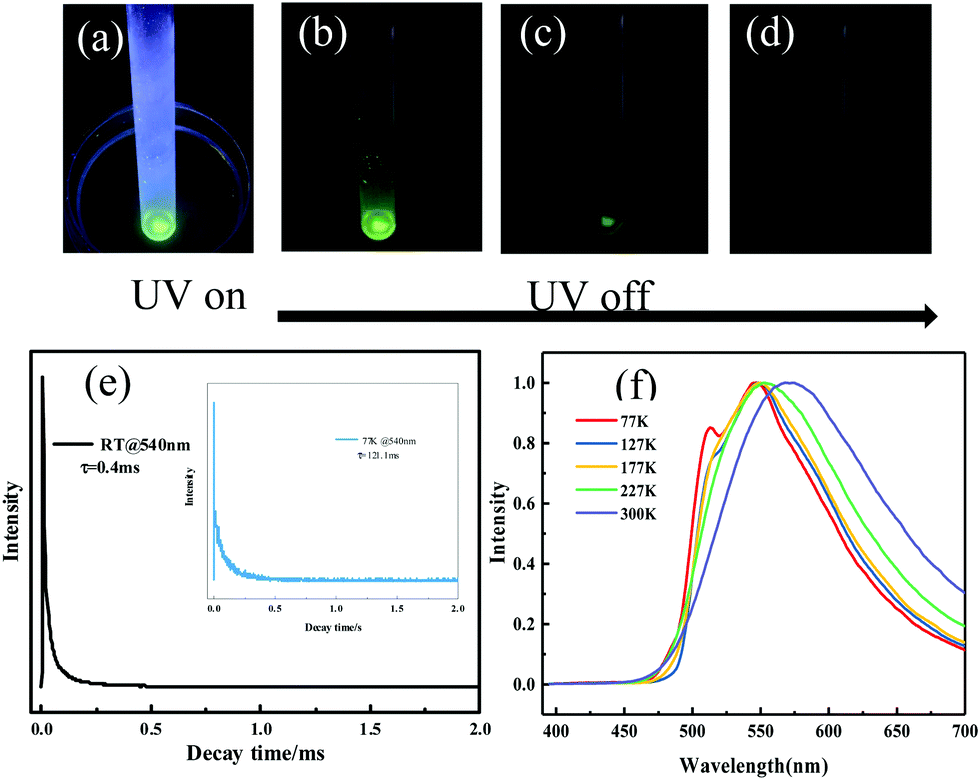 | ||
| Fig. 5 Optical images of the pristine ODFRPTZ powder (a) under 365 nm UV light and (b–d) the afterglow after ceasing the UV light in liquid nitrogen. (e) Decay spectra of the prompt fluorescence. (e) Decay spectra and (f) delayed photoluminescence spectra of the pristine crystalline powder at different temperatures (from 300 K to 77 K). | ||
To shed light on intermolecular interactions for singlet–triplet orbital coupling, the single crystal of ODFRPTZ was developed successfully by slowly evaporating the solution in a mixed solvent of dichloromethane/petroleum ether at room temperature and the crystallographic data were analysed (Table S1, ESI†). In the single crystal, ODFRPTZ molecules arranged according to the space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2) in the triclinic crystal system. Each unit cell contains two molecules. The whole molecule is highly twisted and the phenothiazine moiety adopts the “quasi-axial” conformation with a dihedral angle of 110.7° (Fig. S13, ESI†). Expanding the molecular stacking, the single crystal was in a layer-by-layer structure with a considerable offset constrained by strong intermolecular interactions (Fig. S14, ESI†), which effectively decrease the nonradiative decay rates of deactivation by oxygen and heat, facilitating the triplet excited state emission. The dibenzofuran groups and carbonyl groups were coplanar while the phenothiazine moieties were embedded in the interlamination. There existed strong interactions between any two of dibenzofuran, phenothiazine and carbonyl subunits (Fig. 6 and Fig. S15, ESI†), which effectively promoted the intersystem cross process.
(No. 2) in the triclinic crystal system. Each unit cell contains two molecules. The whole molecule is highly twisted and the phenothiazine moiety adopts the “quasi-axial” conformation with a dihedral angle of 110.7° (Fig. S13, ESI†). Expanding the molecular stacking, the single crystal was in a layer-by-layer structure with a considerable offset constrained by strong intermolecular interactions (Fig. S14, ESI†), which effectively decrease the nonradiative decay rates of deactivation by oxygen and heat, facilitating the triplet excited state emission. The dibenzofuran groups and carbonyl groups were coplanar while the phenothiazine moieties were embedded in the interlamination. There existed strong interactions between any two of dibenzofuran, phenothiazine and carbonyl subunits (Fig. 6 and Fig. S15, ESI†), which effectively promoted the intersystem cross process.
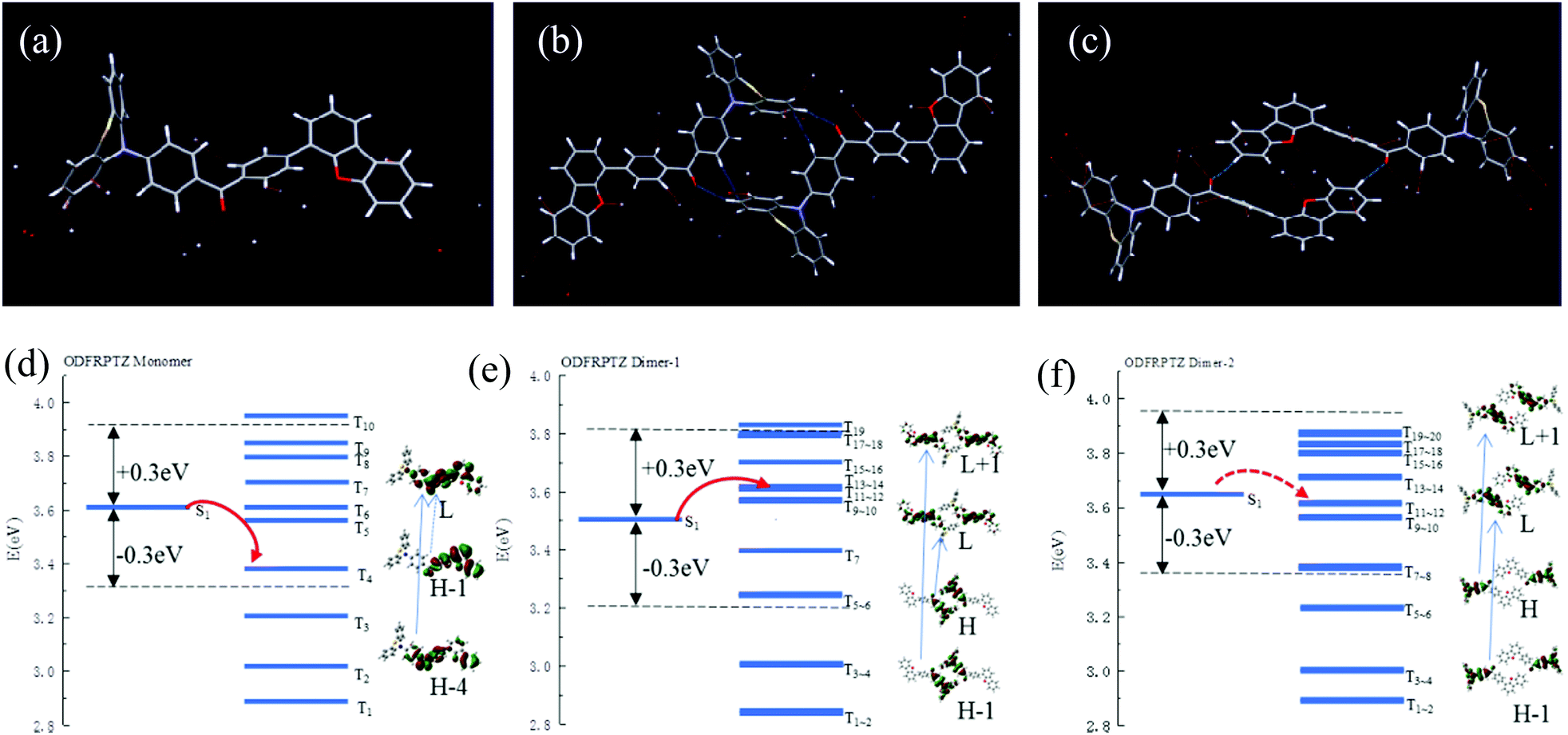 | ||
| Fig. 6 Molecular geometries of (a) the ODFRPTZ monomer, (b) Dimer-1 and (c) Dimer-2 in the unit cell; diagrams of the TD-DFT calculated energy levels and possible ISC channels of a monomer (d), Dimer-1 (e) and Dimer-2 (f) at the singlet (S1) and triplet (Tn) states. H and L represent the HOMO and the LUMO, respectively. | ||
A time-dependent density functional theory (TDDFT) calculation based on B3LYP using 6-311+G(d,p) as the basis set was then implemented to explore the mechanism of the observed triplet excited state emission. Firstly, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the ODFRPTZ monomer in the single crystal were obtained. The HOMO was mainly distributed on the phenothiazine subunit and most electron density of the LUMO was located on the benzophenone subunit, suggesting a typical D–A structure (Fig. S16, ESI†). In the single crystal, there are two kinds of dimers with distinct intermolecular interactions (called Dimer-1 and Dimer 2). According to the calculation result, the dimers play vital roles in populating triplet excited states (Table S2, ESI†). For the isolated monomer, there are merely three main transition channels from S1 to Tn (S1 to T4, T5 and T6) for intersystem crossing. However, for the coupled dimers, the numbers of energy transition channels increase quite a lot (for Dimer-1, S1 to T5, T6, T7, T8, T9, T10, T11, T12, T13 and T14; for Dimer-2, S1 to T7, T8, T9, T10, T11 and T12), enhancing the intersystem crossing. The calculated ΔEST values of the isolated monomer, Dimer-1 and Dimer-2 are 0.73 eV, 0.66 eV and 0.76 eV, respectively. The large ΔEST is not beneficial to the reverse intersystem crossing (RISC) and the phosphorescence is dominated in the triplet state emission, agreeing well with our experimental results.
As for the ground powder of ODFRPTZ, the emission was observed at 581 nm in the steady-state photoluminescence spectra. In the delayed PL spectra, a similar emission band also emerged, indicating that the emission of the ground powder was composed of prompt fluorescence and delayed fluorescence (Fig. 7a). Then, the lifetime test verified the above-mentioned hypothesis. The prompt fluorescence has a short lifetime of 18.7 ns (Fig. S17, ESI†), ascribed to the CT(eq) emission. The delayed fluorescence has a long lifetime of 0.9 μs (Fig. 7b). As documented, the quasi-equatorial conformer of PTZ was conducive to the generation of delayed fluorescence.45 The emerging DF reversely evidenced the PTZ conformation changing from quasi-axial to quasi-equatorial. It is worth noting that the triplet state emission can be altered from phosphorescence to DF by an external force, which has rarely been reported previously. Moreover, the quantum yield increased from 3.24% to 18.88% after grinding. We speculated that the highly twisted geometry in the restrained crystal was destroyed after grinding, promoting the planarization of the molecular structure. The planarization facilitated the ICT process, accordingly resulting in the higher emission efficiency. The quantum yield data also confirmed the PTZ conformation change.
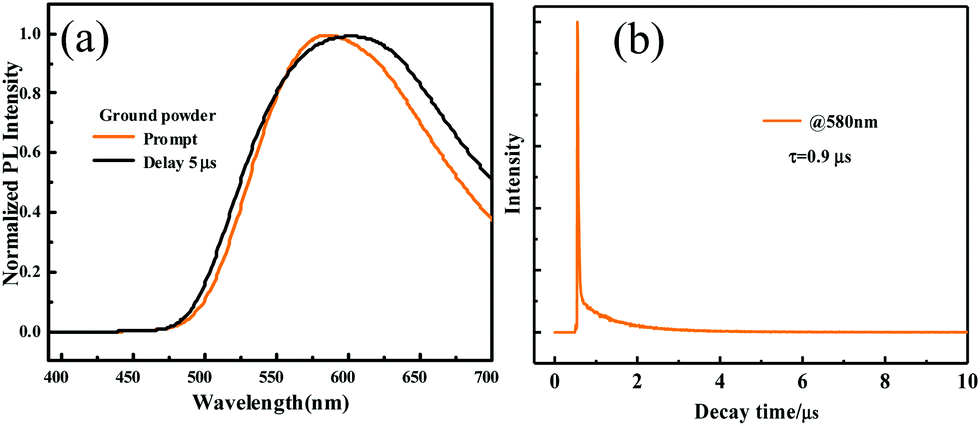 | ||
| Fig. 7 (a) Prompt and delayed (5 μs) photoluminescence spectra of the ground powder of ODFRPTZ. (b) The decay curve of the delayed fluorescence. | ||
Based on all the above-mentioned data and results, we can have a deep discussion on the assignments of the emissions at 428 nm, 540 nm and 581 nm. As documented,46–48 the highly constrained quasi-axial phenothiazine enhances emission from 1LE, suppresses the CT state and activates 3LE phosphorescence generated through the intersystem crossing (ISC) from the 1CT state. It can be concluded that RTP was boosted in the quasi-axial conformer and delayed fluorescence was dominant in the quasi-equatorial conformer,49 which agrees well with our results. The emission at 428 nm is assigned to the 1LE emission of phenothiazine. Only when the phenothiazine adapts the highly constrained quasi-axial conformation are the HOMO and LUMO of ODFRPTZ totally separated, and the 1LE emission of PTZ will appear. After grinding, the highly constrained conformation is destroyed and the HOMO and LUMO of ODFRPTZ will be delocalized, leading to the disappearance of the 1LE emission and enhancement of the CT emission. The emission at 540 nm comprises the CT(ax) emission and 3LE phosphorescence. The emissions with a short lifetime of 8.9 ns and long lifetime of 0.4 ms are assigned to the CT(ax) emission and the 3LE phosphorescence, respectively. When lowering the temperature from 300 K to 77 K, the emission wavelength shift evidences that the 540 nm emission is made of different components, and the prolonged lifetime verifies that the delayed emission is indeed phosphorescence. The TDDFT calculation results also show that the CT(ax) emission and the 3LE phosphorescence coexist. For the ground powder, the PTZ conformation is changed from quasi-axial to quasi-equatorial and the emission at 581 nm is composed of prompt fluorescence with a lifetime of 18.7 ns and DF with a lifetime of 0.9 μs, assigned to the CT(eq) emission.
Conclusions
In summary, photophysical properties of the D–A–D′ molecule ODFRPTZ were investigated and we have discussed that specific emission can be obtained by altering the temperature or molecular conformation. Mechanochromism under both visible light and UV light was realized by changing its conformation from quasi-axial to quasi-equatorial. Carbonyl and the twisted structure of phenothiazine are crucial for ISC and RISC. The quasi-axial conformation facilitated phosphorescence while the quasi-equatorial conformation promoted DF, thus making it possible for the emission switch between DF and phosphorescence by an external force. AIE molecules with stimuli-responsive and multi-emission properties are still scarce and more research studies will focus on multi-functional materials to enrich applications of luminescent materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21704002), the Natural Science Foundation of Beijing Municipality (2182054), the Big Science Project from BUCT (XK180301) and the Fundamental Research Funds for the Central Universities (buctrc201723&ZY1701) to Z. Y. Ma.Notes and references
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed
.
- H. Jing, S. Ning, Y. Dong, R. Tang, L. Ping, C. Ping, Q. Li, D. Ma, J. Qin and L. Zhen, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC
- Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner, Angew. Chem., Int. Ed., 2012, 51, 3964–3968 CrossRef CAS PubMed
- D. D. La, S. V. Bhosale, L. A. Jones and S. V. Bhosale, ACS Appl. Mater. Interfaces, 2018, 10, 12189–12216 CrossRef CAS PubMed
- M. Gao and B. Z. Tang, ACS Sens., 2017, 2, 1382–1399 CrossRef CAS PubMed
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 CAS
- J. Mei, J. Wang, J. Z. Sun, H. Zhao, W. Yuan, C. Deng, S. Chen, H. H. Y. Sung, P. Lu and A. Qin, Chem. Sci., 2012, 3, 549–558 CAS
- D. Ding, R. T. K. Kwok, Y. Yuan, G. Feng, B. Z. Tang and B. Liu, Mater. Horiz., 2015, 2, 100–105 RSC
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed
- J. Yang, Z. Ren, B. Chen, M. Fang, Z. Zhao, B. Z. Tang, Q. Peng and Z. Li, J. Mater. Chem. C, 2017, 5, 9242–9246 RSC
- J. Guo, X.-L. Li, H. Nie, W. Luo, R. Hu, A. Qin, Z. Zhao, S.-J. Su and B. Z. Tang, Chem. Mater., 2017, 29, 3623–3631 CrossRef CAS
- B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu and J. Xu, Chem. Sci., 2016, 7, 2201–2206 RSC
- R. Huang, J. S. Ward, N. A. Kukhta, J. Avó, J. Gibson, T. Penfold, J. C. Lima, A. S. Batsanov, M. N. Berberan-Santos, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2018, 6, 9238–9247 RSC
- B. Huang, W.-C. Chen, Z. Li, J. Zhang, W. Zhao, Y. Feng, B. Z. Tang and C.-S. Lee, Angew. Chem., Int. Ed., 2018, 57, 12473–12477 CrossRef CAS PubMed
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 57, 16407–16411 CrossRef CAS PubMed
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2009, 1345 Search PubMed
- R. Hu, N. L. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC
- K. Wang, Y.-Z. Shi, C.-J. Zheng, W. Liu, K. Liang, X. Li, M. Zhang, H. Lin, S.-L. Tao, C.-S. Lee, X.-M. Ou and X.-H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 31515–31525 CrossRef CAS PubMed
- P. Data and Y. Takeda, Chem. – Asian J., 2019, 14, 1613–1636 CrossRef CAS PubMed
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC
- Y. Ling, Z. Wu, Z. Cheng, G. Xie, Z. Zhu, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700588 CrossRef
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W.-w. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed
- S. Pan, Z. Chen, X. Zheng, D. Wu, G. Chen, J. Xu, H. Feng and Z. Qian, J. Phys. Chem. Lett., 2018, 9, 3939–3945 CrossRef CAS PubMed
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed
- X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 10854–10858 CrossRef CAS PubMed
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed
- S. Aloïse, C. Ruckebusch, L. Blanchet, J. Réhault, A. Guy Buntinx and J. P. Huvenne, J. Mater. Chem. A, 2008, 112, 224–231 Search PubMed
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed
- J. Yang, X. Gao, Z. Xie, Y. Gong, M. Fang, Q. Peng, Z. Chi and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 15299–15303 CrossRef CAS PubMed
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4171 CrossRef
- Y. Takeda, T. Kaihara, M. Okazaki, H. Higginbotham, P. Data, N. Tohnai and S. Minakata, Chem. Commun., 2018, 54, 6847–6850 RSC
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, J. A. Aguilar, D. Hall, A. S. Batsanov, Z. Ren, F. B. Dias and M. R. Bryce, J. Org. Chem., 2019, 84, 3801–3816 CrossRef CAS PubMed
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, J. Santos, F. B. Dias and M. R. Bryce, J. Org. Chem., 2018, 83, 14431–14442 CrossRef CAS PubMed
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC
- P. Data, M. Okazaki, S. Minakata and Y. Takeda, J. Mater. Chem. C, 2019, 7, 6616–6621 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Details of Experimental sections, including synthesis, characterization, spectra, SAXSS patterns and optical images. CCDC 1902888. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00509a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |