
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplified circularly polarized phosphorescence from co-assemblies of platinum(II) complexes†
Gyurim
Park
a,
Hyungchae
Kim
b,
Hoichang
Yang
 c,
Kyung Ryoul
Park
b,
Inho
Song
de,
Joon Hak
Oh
d,
Changsoon
Kim
b and
Youngmin
You
*a
c,
Kyung Ryoul
Park
b,
Inho
Song
de,
Joon Hak
Oh
d,
Changsoon
Kim
b and
Youngmin
You
*a
aDivision of Chemical Engineering and Materials Science, Ewha Womans University, Seoul 03760, The Republic of Korea. E-mail: odds2@ewha.ac.kr
bGraduate School of Convergence Science and Technology, Inter-University Semiconductor Research Center, Seoul National University, Seoul 08826, The Republic of Korea
cDepartment of Chemical Engineering, Inha University, Incheon 22212, The Republic of Korea
dSchool of Chemical and Biological Engineering, Seoul National University, Seoul 08826, The Republic of Korea
eDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 37673, The Republic of Korea
First published on 29th November 2018
Abstract
Molecules capable of producing zero-field circularly polarized phosphorescence (CPP) are highly valuable for chiroptoelectronic applications that rely on triplet exciton. However, the paucity of tractable molecular design rules for obtaining CPP emission has inhibited full utilization. We report amplification of CPP by the formation of helical co-assemblies consisting of achiral square planar cycloplatinated complexes and small fractions of homochiral cycloplatinated complexes. The latter has a unique Pfeiffer effect during the formation of superhelical co-assemblies, enabling versatile chiroptical control. Large dissymmetry factors in electronic absorption (gabs, 0.020) and phosphorescence emission (glum, 0.064) are observed from the co-assemblies. These values are two orders of magnitude improved relative to those of individual molecules. In addition, photoluminescence quantum yields (PLQY) also increase by a factor of ten. Our structural, photophysical, and quantum chemical investigations reveal that the chiroptical amplification is attributable to utilization of both the magnetically allowed electronic transition and asymmetric coupling of excitons. The strategy overcomes the trade-off between glum and PLQY which has frequently been found for previous molecular emitters of circularly polarized luminescence. It is anticipated that our study will provide new insight into the future research for the exploitation of the full potential of CPP.
Introduction
Circularly polarized luminescence (CPL) refers to photoemission that is enriched with light having a polarization axis rotating either right-handedly or left-handedly.1 The circular enrichment is quantitated using a luminescence dissymmetry factor glum = 2(ILCPL − IRCPL)/(ILCPL + IRCPL), where ILCPL and IRCPL are the light intensities of the left- and right-handed CPL, respectively. Circularly polarized luminescence is valuable for its utility in elucidating various photofunctions. For example, 3D displays with a high luminance are possible using CP electroluminescence,2–4 because the two handedness directions can be used to create binocular disparities with minimal losses in the light intensity.5 CPL is anticipated to find a broader range of photonic applications, including quantum encryption and security-enhanced optical communications.6,7Exploiting the photonic utility of CPL requires zero-field emitters that produce high values of |glum| and photoluminescence quantum yield (PLQY). Lanthanide complexes, such as [Eu(facam)3] (facam = 3-(trifluoromethylhydroxymethylene)-(+)-camphorate), have been most successful, but their applicability is limited by inflexible syntheses and a lack of tunability.8 Two strategies have been pursued for the development of molecular CPL emitters. One strategy is based on asymmetric coupling between excitons within a molecular dyad; for example, CPL can be obtained through the asymmetric Davydov splitting of fluorophores, such as perylene diimide,9,10 5,6-biscarbazolylphthalimide,11 terthiophene,12 pyrene,13 anthracene,14 and azaacene,15 in chiral arrangements. Exciton coupling is reinforced in aggregate states, leading to improvements in |glum|.16 The other strategy used to develop molecular CPL emitters employs molecules bearing helical π-conjugation. Derivatives of helicene,3,17–27 1,1′-binaphthalene,28–31 paracyclophane,32 chiral boron dipyrromethene,33,34 cyclic polyaromatic compounds,35 and chiral complexes of Ir(III)23,36–38 or Pt(II)39–43 have been found to display CPL activity. Although molecular factors that control the CPL behaviors of these molecules require further resolution, the helical structures may confer magnetic allowance to the electronic transition resulting in CPL emission.
Despite advances in the field, the development of molecular emitters of CPL has remained a significant challenge. The apparently necessary trade-off between |glum| and PLQY presents a major obstacle to the full utilization of CPL photofunctions (see ESI, Table S1† for a list of |glum| and PLQY values reported previously for CPL emitters). In addition, the two strategies tested thus far have required labor-intensive syntheses with poor flexibility for further elaboration. Therefore, there is a strong need for tractable strategies for obtaining CPL with high values of |glum| and PLQY.
Herein, we report the generation and amplification of circularly polarized phosphorescence from helical assemblies of a simple, achiral molecule. Achiral monocycloplatinated complexes (soldiers) co-assemble into helical nanoribbons with a smaller fraction of chiral monocycloplatinated complexes (sergeants). The helical sense of the assembly is dictated by the sergeant (Fig. 1). The sergeants-and-soldiers co-assembly strategy improves |glum| by two orders of magnitude and PLQY by one order of magnitude relative to the |glum| of the chiral sergeant and the PLQY of the soldier, respectively. Our approach uniquely combines two previous strategies in that both asymmetric exciton coupling and the magnetic allowance in the electronic transition are facilitated in the co-assemblies.
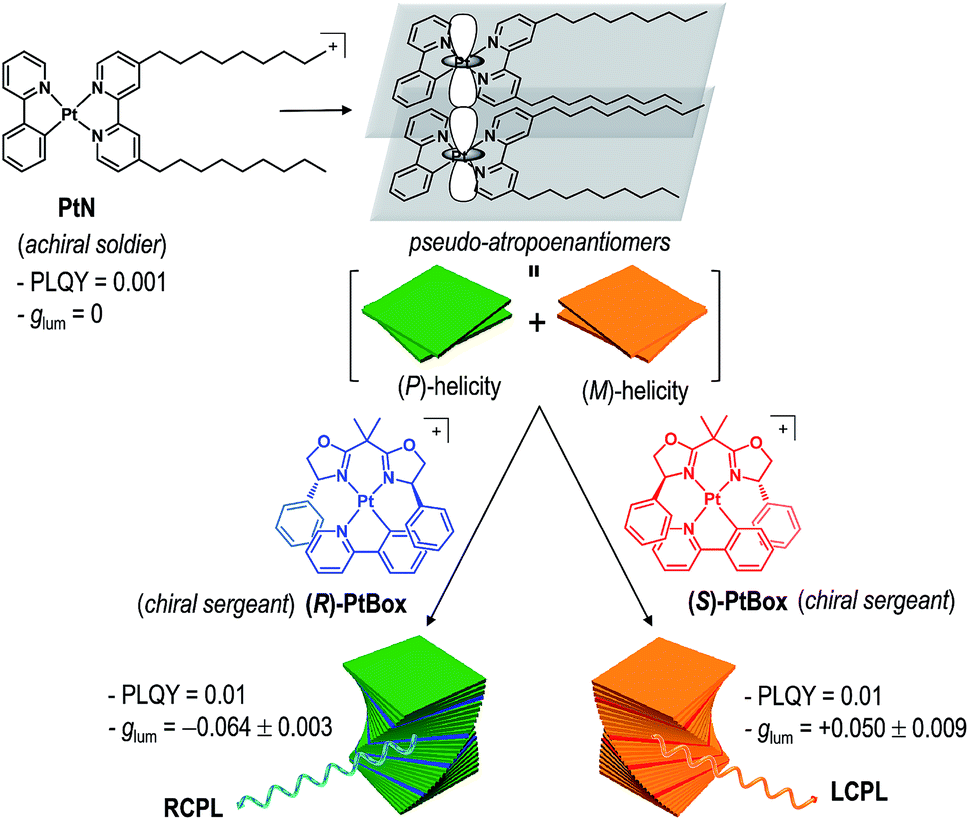 | ||
| Fig. 1 Molecular strategy to obtaining circularly polarized phosphorescence from superhelical co-assemblies consisting of PtN soldiers and PtBox sergeants. | ||
Results and discussion
Chiroptical induction by controlling torsional displacement
Our co-assembly strategy exploits the strong propensity of square planar Pt(II) complexes to form co-facial stacks. Pioneering studies by the groups of Che, Yam, De Cola, Kato, and Castellano established that the spontaneous assembly is driven by the synergetic effects of a Pt⋯Pt metallophilic interaction and a π–π interaction among the cyclometalating ligands.44–52 The resulting Pt(II) assemblies are useful in a variety of applications.53–57 We employed a Pt(II) complex having two nonyl chains, [Pt(2-phenylpyridinato)(4,4′-bis(nonyl)-2,2′-bipyridine)]ClO4 (PtN), as a soldier molecule (see Fig. 1 for the structure of PtN). The nonyl chains provided torque that induced torsional displacement along the axis orthogonal to the molecular planes, thereby generating a helical PtN stack. The resulting PtN stacks were pseudo-atropo-enantiomeric because the formation of the right-handed (i.e., (P)-helical) and left-handed (i.e., (M)-helical) stacks was equally probable. Therefore, self-assemblies of PtN alone did not display any chiroptical activities (vide infra).We anticipated that the addition of a chiral analogue of PtN (solider) would induce a Pfeiffer effect and enrich the enantiomeric excess in the helical PtN stacks.58 The initial PtN helical stack would serve as a homochiral seed for the hierarchical growth of superhelical assemblies because helical inversion is thermodynamically improbable.59 This strategy is advantageous over previous assembly approaches that employed co-assemblies of chiral gelators and an achiral fluorophore,60 as it improves both |glum| and PLQY. In addition, the use of a simple achiral molecule as a CPL unit offers unique benefits over the self-assembly of homochiral CPL emitters.10,16,17,19,40,41,60–70
As a first step in our study, we performed quantum chemical calculations of a dimeric stack of truncated PtN molecules at the CAM-B3LYP level of theory. The optimized PtN stack possessed the lowest energy metal–metal-to-ligand charge-transfer (MMLCT) singlet and triplet states, indicative of electronic coupling between the two PtN molecules (Fig. 2 and ESI, Fig. S1†). The MMLCT transition carried a non-negligible magnetic allowance. The electric (μ) and magnetic (m) transition dipole moments of the MMLCT transition were parallel each other with an angle (θ) of 4.54°, yielding a rotatory strength (R, R = |μ|·|m|cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ) as large as 4.8 × 10−39 erg esu cm per Gauss (ESI, Fig. S2†). The parallel disposition between μ and m markedly contrasts with the cases of single molecular CPL emitters. As expected, the R of the MMLCT transition of the PtN stack oscillated between positive and negative values as the top PtN molecule rotated relative to the bottom PtN molecule while maintaining a fixed Pt–Pt distance of 3.7 Å (Fig. 2). Although this calculation was limited to a dimer,71 it supported our claim that the chiroptical activities could be controlled by dihedral rotation within the PtN assemblies.
θ) as large as 4.8 × 10−39 erg esu cm per Gauss (ESI, Fig. S2†). The parallel disposition between μ and m markedly contrasts with the cases of single molecular CPL emitters. As expected, the R of the MMLCT transition of the PtN stack oscillated between positive and negative values as the top PtN molecule rotated relative to the bottom PtN molecule while maintaining a fixed Pt–Pt distance of 3.7 Å (Fig. 2). Although this calculation was limited to a dimer,71 it supported our claim that the chiroptical activities could be controlled by dihedral rotation within the PtN assemblies.
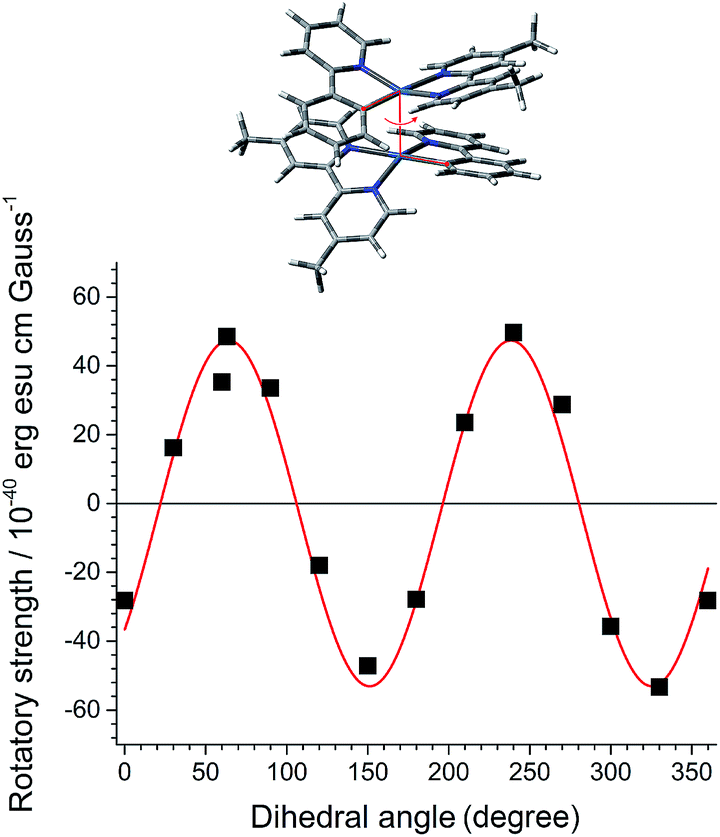 | ||
| Fig. 2 Torsional control of chiroptical properties. Simulation of the rotatory strength (R) of the MMLCT transition as a function of the dihedral angle of a dimer comprising a truncated form of the PtN molecules (top structure). The red oscillatory curve provides a visual guide. Calculation conditions: TD-CAM-B3LYP/LANL2DZ:6-311+G(d,p)//CAM-B3LYP/LANL2DZ:6-311+G(d,p). Twenty states were considered for the TD-DFT calculations. | ||
Formation of superhelical co-assemblies by Pfeiffer effect
To enable our study, we synthesized an enantiomeric pair of sergeant Pt(II) complexes having chiral bisoxazoline ligands in place of 4,4′-bis(nonyl)-2,2′-bipyridine, [Pt(2-phenylpyridinato)((+)-2,2-isopropylidenebis((4R)-4-phenyl-2-oxazoline))]ClO4 ((R)-PtBox) and [Pt(2-phenylpyridinato)((−)-2,2-isopropylidenebis((4S)-4-phenyl-2-oxazoline))]ClO4 ((S)-PtBox). These complexes were prepared from a dinuclear cycloplatinated precursor, following the standard chloride abstraction method that employed AgClO4 (Scheme 1). The synthetic procedures are described in the ESI.† Structural identification data, obtained using 1H and 13C{1H} NMR spectroscopy, high-resolution mass spectrometry, and elemental analyses, were fully consistent with the proposed structures (ESI, Fig. S3–S8†).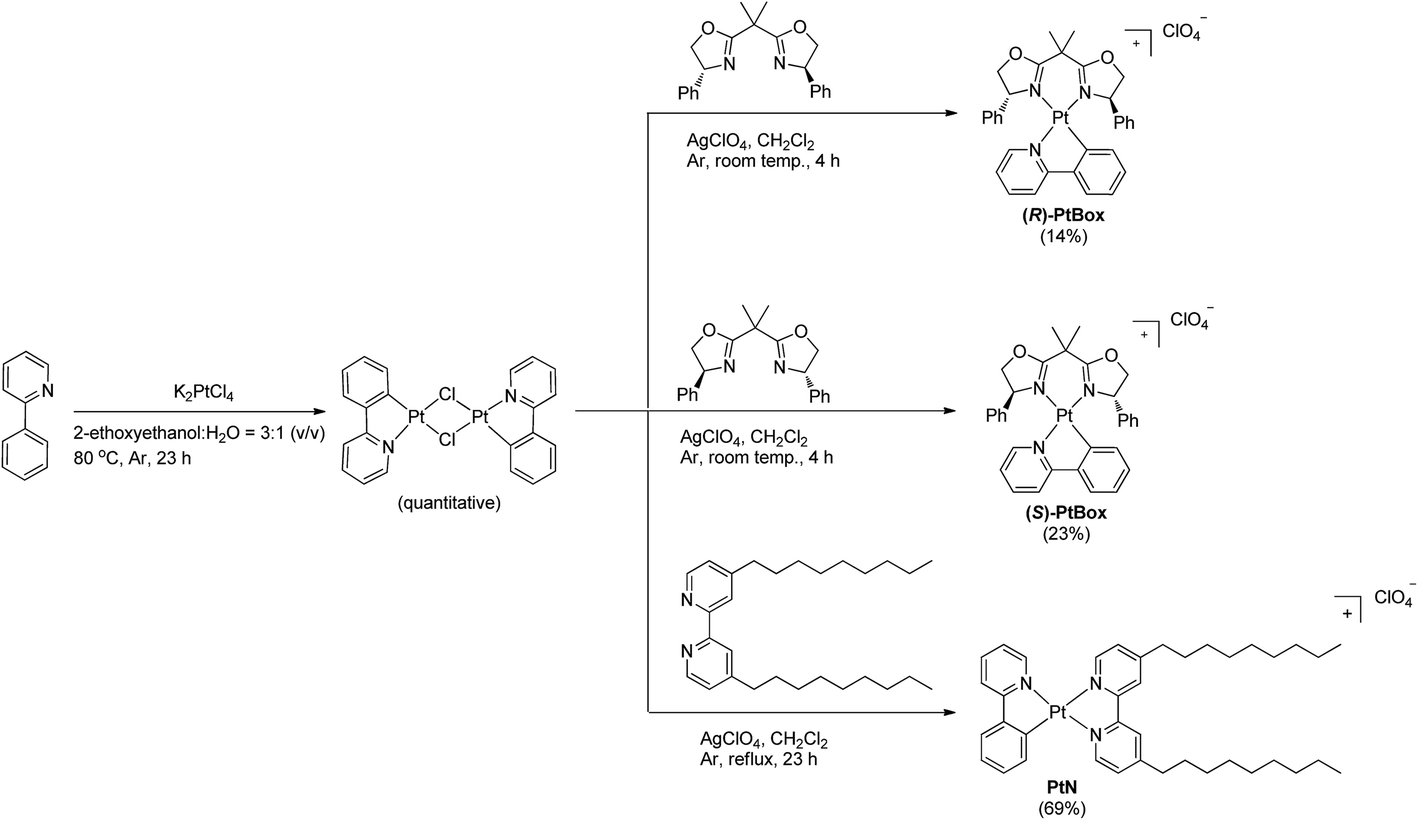 | ||
| Scheme 1 Syntheses of (R)-PtBox, (S)-PtBox and PtN. | ||
Co-assemblies were grown on quartz substrates by slow evaporation of a 1,2-dichloroethane solution containing PtBox and PtN (2.0 wt% total solute; PtBox:PtN = 2:5, w/w) under atmospheric pressure at room temperature. As shown in Fig. 3a, confocal laser-scanning micrographs (λex = 405 nm, λobs = 470–700 nm) revealed the formation of homohelical nanoribbons of the co-assemblies. The helical sense was determined exclusively by the identity of the added PtBox; (R)-PtBox and (S)-PtBox furnished (P)-helical and (M)-helical nanoribbons, respectively. By contrast, PtN self-assemblies did not contain any helical structures. Helical control over the co-assemblies was further supported by field-emission scanning electron microscopy (FE-SEM) images (Fig. 3b).
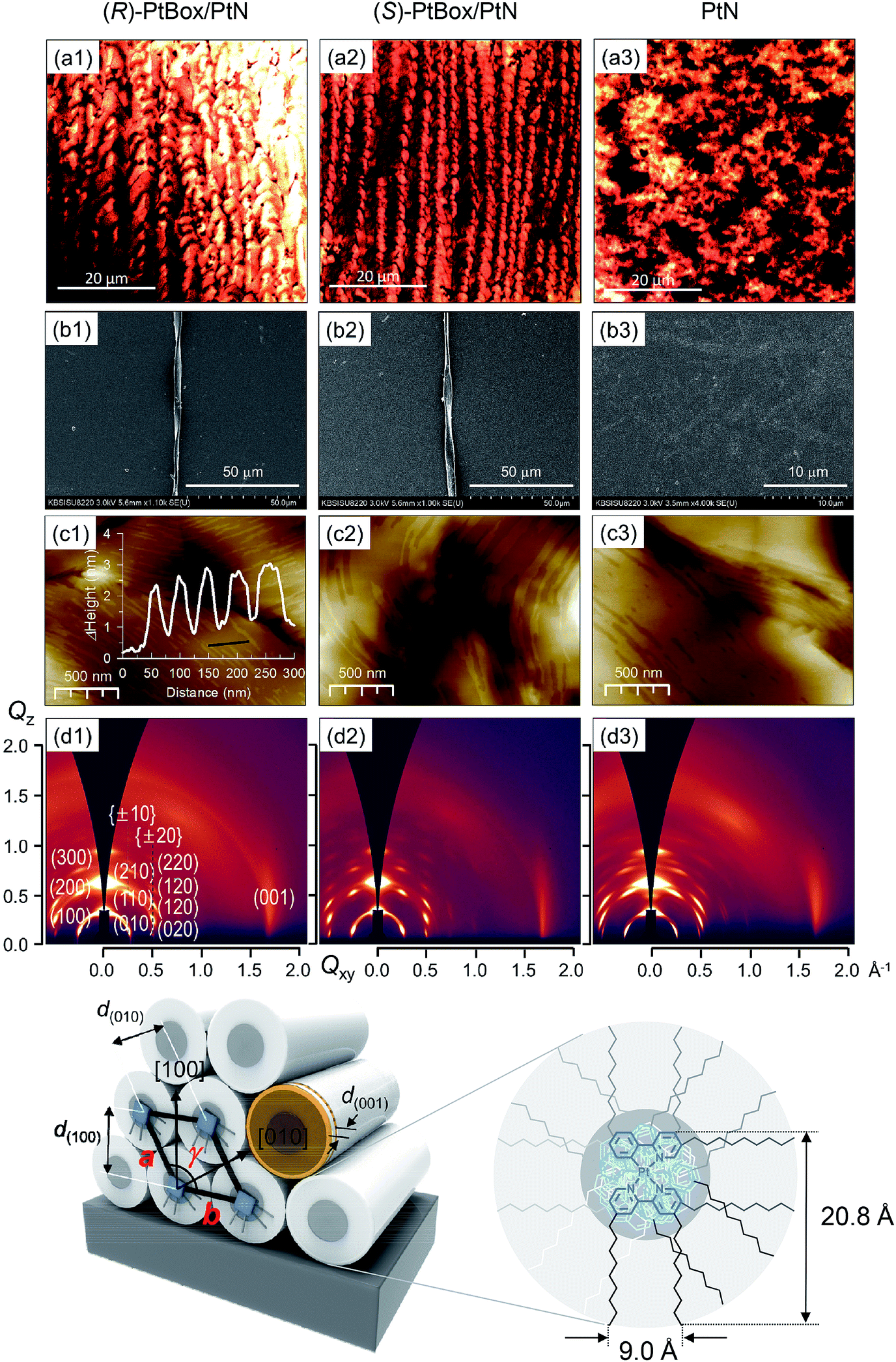 | ||
| Fig. 3 Chiral amplification in the co-assemblies of PtBox/PtN. The co-assemblies were grown by drop-casting a 1,2-dichloroethane solution (2 wt% total solute) containing PtN and (R)- (a1, b1, c1, and d1) or (S)-PtBox (a2, b2, c2, and d2) (PtBox:PtN = 2:5, w/w). Self-assemblies of PtN were grown identically (a3, b3, c3, and d3). (a) Confocal laser-scanning micrographs of the PtN assemblies prepared in the presence of (R)-PtBox (a1) or (S)-PtBox (a2) and in the absence of PtBox (a3). λex = 405 nm. λem = 470–700 nm. (b) FE-SEM images and (c) AFM topographies of the co-assemblies prepared in the presence of (R)-PtBox (b1, c1) or (S)-PtBox (b2, c2) and in the absence of PtBox (b3, c3). The inset graph depicts the step profile along the black line. (d) 2D GIXD patterns obtained from assemblies of PtN prepared in the presence of (R)-PtBox (d1) or (S)-PtBox (d2) and in the absence of PtBox (d3). The bottom structures show a monoclinic structure of the PtBox/PtN assemblies established from the 2D GIXD results. The lattice parameters, a, b and γ, are marked in red. | ||
Note that the widths of the nanoribbons (2–5 μm) were much greater than the calculated molecular dimensions of PtN (20.8 Å × 9.0 Å). The AFM topographies indicated the formation of assemblies with a step height of 2.2 nm (Fig. 3c). Obviously, this height was longer than the longest dimension of PtN (i.e., 20.8 Å), which suggested the interdigitation of PtN molecules within the assemblies. The packing structures were elucidated using 2D grazing-incident X-ray diffraction (2D GIXD) studies. As shown in Fig. 3d, 2D GIXD patterns of the PtN and PtBox/PtN assemblies produced highly oriented X-ray reflections, corresponding to the (h00) reflections along the Qz axis and Braggs peaks vertically along the Qz direction at a given Qxy. These reflection patterns were indicative of pseudo hexagonal close packing of PtN cylinders. Analyses of the 2D GIXD patterns revealed a monoclinic lattice structure (a = 24.66 Å, b = 27.09 Å, γ = 117°), where the PtN cylinders were oriented parallel to the surface (see the bottom structures in Fig. 3). On the contrary, an orthorhombic structure developed in the PtN self-assemblies. The X-ray reflection of (001) at Qxy = 1.708 Å−1 (d = 3.680 Å) corresponded to the intermolecular distance between PtN molecules within the cylinder. This distance is slightly longer than the lengths of typical Pt⋯Pt metallophilic interactions (<3.5 Å),49 indicating weak dz2–dz2 orbital overlap. However, the distance was consistent with the red-shifted MMLCT emission (vide infra).
Amplification of chiroptical properties
The co-assemblies included Pt⋯Pt metallophilic interactions, as was evident by the emergence of the weak MMLCT transition at a wavelength of 500 nm in the UV-vis absorption spectra (Fig. 4a). 1H NMR experiments corroborated this notion (see the results and discussion in ESI, Fig. S9†). The association constant (Ka) for the PtN assembly was estimated to be <100 M−1, based on the measured concentration-dependent increase in the MMLCT absorbance (ESI, Fig. S10†). Although the MMLCT transition made non-negligible contribution, as shown in Fig. 4b, the electronic circular dichroism (ECD) spectra of the co-assemblies were dominated by the strong bisignate signals of the metal-to-ligand charge-transfer (MLCT) band of PtN at a wavelength of 450 nm. The chiral exciton theory dictates that (P)-helical co-assemblies displayed a positive Cotton effect, whereas (M)-helical co-assemblies displayed a negative Cotton effect.1 Therefore, judging from the bisignate signals in Fig. 4b, it was concluded that (R)-PtBox/PtN and (S)-PtBox/PtN co-assemblies were in (P)- and (M)-helical sense, respectively. Note that the MLCT transition was unimolecular, whereas the MMLCT transition was supramolecular. These chiroptical behaviors, thus, suggested that the asymmetric coupling among MLCT excitons dominated the magnetically allowed MMLCT transition in the chiral photon absorption process. The ECD signals were devoid of linear dichroism or birefringence because the ECD spectra were invariant under the rotation of the azimuthal angle of the sample (ESI, Fig. S11†). | ||
| Fig. 4 Induction and amplification of electronic circular dichroism (ECD). (a) UV-vis absorption spectra of 10 μM PtN (THF, black) and co-assemblies of (R)-PtBox/PtN (2:5, w/w; green), and (S)-PtBox/PtN (2:5, w/w; orange). (b) ECD spectra obtained from self-assembled PtN (black), 50 μM (R)-PtBox (THF, blue), 50 μM (S)-PtBox (THF, red), or co-assemblies of (R)-PtBox/PtN (green) and (S)-PtBox/PtN (orange). Absorption dissymmetry factor (gabs): solution and self-assembly of PtN, 0; (R)-PtBox, +3.7 × 10−5 (308 nm); (S)-PtBox, −4.3 × 10−5 (308 nm); co-assembly of (R)-PtBox/PtN, +0.020 (500 nm); co-assembly of (S)-PtBox/PtN, −0.020 (500 nm). | ||
The Kuhn dissymmetry factor gabs, defined as gabs = 2(εL − εR)/(εL + εR) (εL and εR are the ellipticity of the left- and right-handed circularly polarized absorption), in the ECD spectra increased in proportion to the amount of PtBox added to form the co-assemblies, and it reached a plateau at PtBox mole fraction ([PtBox]/([PtBox] + [PtN])) of 0.2 (ESI, Fig. S12†). This result indicated chiral amplification based on at least four PtN solider molecules following one PtBox sergeant during the growth of the homohelical co-assemblies. The largest gabs values were found to be +0.020 and −0.020 at 500 nm for co-assemblies of (R)-PtBox/PtN and (S)-PtBox/PtN (PtBox:PtN = 2:5, w/w), respectively. These values were three orders of magnitude greater than the largest gabs for (R)-PtBox (gabs = +3.7 × 10−5) and (S)-PtBox (gabs = −4.3 × 10−5), indicating the effectiveness of the asymmetric exciton coupling of PtN molecules in the co-assemblies (ESI, Fig. S13 and Table S2†). As expected, self-assemblies of PtN (i.e., no PtBox) did not produce any ECD signals (Fig. 4b).
The PtBox/PtN co-assemblies produced a broad phosphorescence spectrum with an emission peak wavelength (λem) of 615 nm and a phosphorescence lifetime (τobs) of 0.24 ± 0.05 μs (Table 1). The phosphorescence emission resulted from the triplet MMLCT transition in the PtN assemblies, which was identical to the emission observed in PtN self-assemblies, and differed from the phosphorescence emission observed from a dilute solution of PtN (λem = 480 nm; 10 μM in 1,2-dichloroethane) (ESI, Fig. S14†). The photoluminescence excitation (PLE) spectrum corresponding to the 615 nm phosphorescence emission overlapped with the MMLCT absorption band, supporting this notion (ESI, Fig. S15†). In addition, the transient photoluminescence traces were devoid of a delayed rise-and-decay profile, excluding the possibility that an excimer was responsible for the 615 nm phosphorescence emission (ESI, Fig. S16†).72 Neither (R)-PtBox nor (S)-PtBox molecules displayed chromic shifts upon self-aggregate formation, eliminating the possibility that excitonic interactions among sergeants contributed to the 615 nm phosphorescence emission.
| λ abs (nm) | λ em (nm) | PLQYc | τ avg (μs) | k r (104 s−1) | k nr (106 s−1) | g abs | g lum | |
|---|---|---|---|---|---|---|---|---|
|
a Absorption peak wavelength.
b Photoluminescence peak wavelength. Excitation wavelengths = 370 nm (PtN) and 326 nm ((R)- and (S)-PtBox).
c Absolute photoluminescence quantum yields determined with employing an integrating sphere.
d Photoluminescence lifetime obtained after pulsed laser excitation at 377 nm (temporal resolution = 1.6 ns; observation wavelength (λobs) = 615 nm) (PtN self-assemblies) and 615 nm (PtBox/PtN co-assemblies). A biexponential decay model was used: photoluminescence intensity = A1exp(−t/τ1) + A2exp(−t/τ2) + A0. τavg = (∑Aiτi2)/(∑Aiτi) (i = 1–2).
e Radiative rate constant, kr = PLQY/τavg.
f Non-radiative rate constant, knr = (1 − PLQY)/τavg.
g Absorption dissymmetry factor. λobs = 500 nm.
h Luminescence dissymmetry factor. λobs = 576 nm.
|
||||||||
| PtN self-assemblies | 490 | 615 | 0.006 | 0.61 | 0.98 | 1.62 | N.A. | N.A. |
| (R)-PtBox/PtN co-assemblies | 490 | 615 | 0.01 | 0.24 | 4.2 | 4.1 | +0.02 | −0.064 ± 0.003 |
| (S)-PtBox/PtN co-assemblies | 490 | 615 | 0.01 | 0.25 | 4.0 | 4.0 | −0.02 | +0.050 ± 0.009 |
The enhancement in the PLQY was notable. The absolute PLQY values obtained from both (R)-PtBox/PtN and (S)-PtBox/PtN co-assemblies under air-equilibrated conditions were 0.01, one order of magnitude greater than the value (<0.001) recorded in a deaerated solution of PtN. This increase resulted from the acceleration of the radiative process by the MMLCT transition. Indeed, the radiative rate (kr) of the MMLCT phosphorescence of the co-assemblies (λem = 615 nm) was 9.8 × 103 s−1, whereas a one order of magnitude slower kr (0.83 × 103 s−1) was obtained for the MLCT phosphorescence of a PtN molecule. The non-radiative rate (knr) values of the MMLCT and MLCT transitions did not vary significantly (knr = 1.62 × 106 s−1 (MMLCT) vs. 0.83 × 106 s−1 (MLCT)), indicating radiative control.
In addition to the radiatively boosted PLQY, ultrafast energy transfer from PtBox to PtN contributed to the phosphorescence emission of the co-assemblies. As shown in Fig. 5a, (R)-PtBox and (S)-PtBox exhibited long-lived luminescence with τobs values of 3.4 μs and 3.7 μs, respectively. Incorporating PtBox into the co-assemblies significantly shortened τobs by enabling energy transfer to PtN: τobs decreased to 0.011 μs in both (R)- and (S)-PtBox/PtN. The energy-transfer rates (kETs) were estimated to be 9.1 × 107 s−1 for both (R)-PtBox and (S)-PtBox, approximately four and two orders magnitude greater than kr (∼103 s−1) and knr (∼105 to 106 s−1), respectively (see ESI, Table S2† for the kr and knr values). The ultrafast intra-assembly energy transfer was further supported by the observation of increased phosphorescence intensities from co-assemblies containing increased concentrations of PtBox (ESI, Fig. S17†).
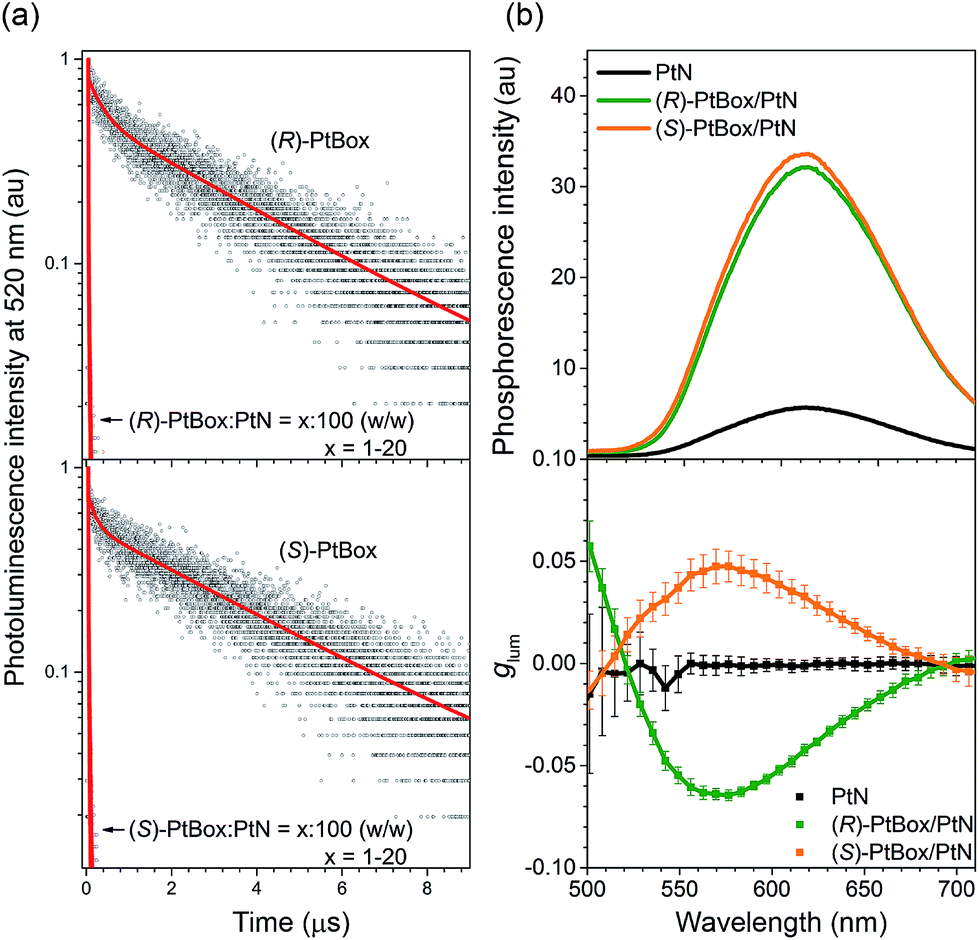 | ||
| Fig. 5 Amplified circularly polarized phosphorescence. (a) Comparisons of the photoluminescence decay traces of (R)- or (S)-PtBox in Ar-saturated THF (50 μM) or of the co-assemblies (PtBox:PtN = x:100, w/w; x = 1–20), observed at 520 nm after pulsed laser photoexcitation at 377 nm (temporal resolution = 1.6 ns). Top, (R)-PtBox; bottom, (S)-PtBox. The red curves show fits to a biexponential decay model. The fit results are summarized in ESI, Table S3.† (b) Steady-state phosphorescence emission (top, λex = 363 nm) and plots of glum values as a function of the emission wavelength (bottom) for the self-assemblies of PtN (black) and the co-assemblies of (R)-PtBox/PtN (green) or (S)-PtBox/PtN (orange). The CPL measurements were repeated 50 times for each fresh sample, and the average and standard deviations are shown. | ||
The MMLCT phosphorescence disappeared upon heating. An abrupt decrease in the MMLCT phosphorescence emission was observed at 343 K, suggesting the presence of cooperative assemblies of PtN molecules (ESI, Fig. S18†).73 The endothermic phosphorescence decrease accompanied losses in the bisignate Cotton effect in the ECD spectra and in the MMLCT absorption in the UV-vis absorption spectra of the (R)-PtBox/PtN co-assemblies (ESI, Fig. S19†). These results suggested thermally activated disassembly. The disassembly temperature could be shifted by varying the length of the alkyl chains in PtN.
As a final phase of our study, circularly polarized phosphorescence was recorded from the co-assemblies over the emission range 500–710 nm using a linear polarizer and a rotating quarter-wave retarder. The validity of our detection method was examined using [Eu(facam)3] as a standard. Perfect sinusoidal oscillations and identical phosphorescence intensities were obtained from (R)-PtBox/PtN and (S)-PtBox/PtN assemblies when the axis between the polarizer and the retarder was rotated (ESI, Fig. S20†), suggesting that linearly polarized emission contributed minimally to the CPL. As shown in Fig. 5b, the circularly polarized phosphorescence dissymmetry spectra overlapped with the arbitrary phosphorescence spectra obtained without a polarizer and a retarder. The coincidence indicated that the triplet MMLCT state was responsible for the circularly polarized phosphorescence. The glum values of the (R)-PtBox/PtN and (S)-PtBox/PtN co-assemblies were −0.064 ± 0.003 and +0.050 ± 0.009, respectively. These values were at least one order of magnitude greater than those obtained from (R)-PtBox (|glum| < 0.001) and (S)-PtBox (|glum| < 0.001).
Conclusions
We devised and demonstrated the molecular strategy to overcoming the trade-off problem in circularly polarized luminescence. The formation of hierarchical helix assemblies of cycloplatinated complexes improved both the luminescence dissymmetry factor and the quantum yield. The sergeants-and-soldiers principle was employed in the creation of the superhelical assemblies of cycloplatinated complexes. The principle is reminiscent of that employed in chiral nematic liquid crystals;74 however, the strong Pfeiffer effect associated with the generation and amplification of circularly polarized phosphorescence is unprecedented. The chiroptical amplification was ascribed to both the magnetically allowed MMLCT state for phosphorescence emission and enantiomeric enrichment of the circular polarization via absorption through the asymmetrically coupled MLCT states prior to populating the emitting MMLCT state (vide supra). Therefore, our co-assemblies were characterized by a unique three level system: Absorption of circularly polarized photons occurred through a transition to the asymmetrically exciton-coupled MLCT state, whereas circularly polarized phosphorescence resulted from the magnetically allowed MMLCT transition (see Fig. 6). Note that glum and gabs values for the co-assemblies had opposite signs. The reversal in the dissymmetry factors may support our claim that the MLCT and MMLCT transitions associated with absorption and emission of CPL, respectively, functioned independently of one another. It is envisioned that decoupling the chiroptical absorption and emission processes could expand our ability to control circularly polarized light.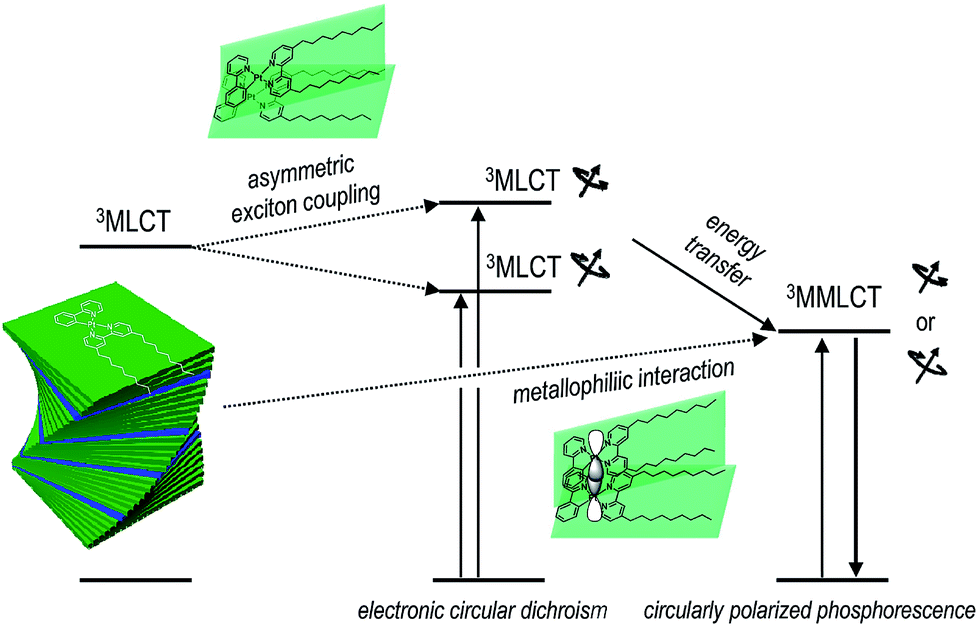 | ||
| Fig. 6 Plausible mechanism underlying the photophysical processes that produce circularly polarized phosphorescence in the co-assemblies. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Samsung Research Funding Center for Future Technology (SRFC-MA1602-03). H. Y. acknowledges the grant provided by the National Research Foundation of Korea (NRF2017R1A2B4009313).Notes and references
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS
.
- F. Zinna, U. Giovanella and L. Di Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed
- E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909–9910 CrossRef CAS
- T. Kim, S. Kim, M.-J. Gim and S.-W. Choi, J. Korean Phys. Soc., 2013, 62, 713–717 CrossRef CAS
- C. Wagenknecht, C.-M. Li, A. Reingruber, X.-H. Bao, A. Goebel, Y.-A. Chen, Q. Zhang, K. Chen and J.-W. Pan, Nat. Photonics, 2010, 4, 549–552 CrossRef CAS
- J. F. Sherson, H. Krauter, R. K. Olsson, B. Julsgaard, K. Hammerer, I. Cirac and E. S. Polzik, Nature, 2006, 443, 557–560 CrossRef CAS PubMed
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC
- H. Tsumatori, T. Nakashima and T. Kawai, Org. Lett., 2010, 12, 2362–2365 CrossRef CAS PubMed
- J. Kumar, H. Tsumatori, J. Yuasa, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2015, 54, 5943–5947 CrossRef CAS PubMed
- M. Li, S.-H. Li, D. Zhang, M. Cai, L. Duan, M.-K. Fung and C.-F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS PubMed
- B. M. W. Langeveld-Voss, D. Beljonne, Z. Shuai, R. A. J. Janssen, S. C. J. Meskers, E. W. Meijer and J.-L. Bredas, Adv. Mater., 1998, 10, 1343–1348 CrossRef CAS
- Y. Mimura, T. Nishikawa, R. Fuchino, S. Nakai, N. Tajima, M. Kitamatsu, M. Fujiku and Y. Imai, Org. Biomol. Chem., 2017, 15, 4548–4553 RSC
- J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef CAS PubMed
- Y. Inoue, D. Sakamaki, Y. Tsutsui, M. Gon, Y. Chujo and S. Seki, J. Am. Chem. Soc., 2018, 140, 7152–7158 CrossRef CAS PubMed
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed
- K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899–11907 CrossRef CAS PubMed
- J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808–11809 CrossRef CAS PubMed
- T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684–3687 CrossRef CAS PubMed
- K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555–5558 CrossRef CAS PubMed
- H. Sakai, S. Shinto, J. Kumar, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2015, 119, 13937–13947 CrossRef CAS
- D.-Q. He, H.-Y. Lu, M. Li and C.-F. Chen, Chem. Commun., 2017, 53, 6093–6096 RSC
- N. Hellou, M. Srebro-Hooper, L. Favereau, F. Zinna, E. Caytan, L. Toupet, V. Dorcet, M. Jean, N. Vanthuyne, J. A. G. Williams, L. Di Bari, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2017, 56, 8236–8239 CrossRef CAS PubMed
- T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS PubMed
- M. Li, H.-Y. Lu, C. Zhang, L. Shi, Z. Tang and C.-F. Chen, Chem. Commun., 2016, 52, 9921–9924 RSC
- L. Fang, M. Li, W.-B. Lin, Y. Shen and C.-F. Chen, J. Org. Chem., 2017, 82, 7402–7409 CrossRef CAS PubMed
- M. Li, C. Zhang, L. Fang, L. Shi, Z. Tang, H.-Y. Lu and C.-F. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 8225–8230 CrossRef CAS PubMed
- S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed
- R. Aoki, R. Toyoda, J. F. Kogel, R. Sakamoto, J. Kumar, Y. Kitagawa, K. Harano, T. Kawai and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 16024–16027 CrossRef CAS PubMed
- J. Han, S. Guo, J. Wang, L. Wei, Y. Zhuang, S. Liu, Q. Zhao, X. Zhang and W. Huang, Adv. Opt. Mater., 2017, 5, 1700359 CrossRef
- K. Takaishi, M. Yasui and T. Ema, J. Am. Chem. Soc., 2018, 140, 5334–5338 CrossRef CAS PubMed
- Y. Morisaki, M. Gon, T. Sasamori, N. Tokitoh and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 3350–3353 CrossRef CAS PubMed
- E. M. Sanchez-Carnerero, F. Moreno, B. L. Maroto, A. R. O. Agarrabeitia, J. María, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef CAS PubMed
- R. B. Alnoman, S. Rihn, D. C. O'Connor, F. A. Black, B. Costello, P. G. Waddell, W. Clegg, R. D. Peacock, W. Herrebout, J. G. Knight and M. J. Hall, Chem.–Eur. J., 2016, 22, 93–96 CrossRef CAS PubMed
- S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13097–13101 CrossRef CAS PubMed
- C. Schaffner-Hamann, A. von Zelewsky, A. Barbieri, F. Barigelletti, G. Muller, J. P. Riehl and A. Neels, J. Am. Chem. Soc., 2004, 126, 9339–9348 CrossRef CAS PubMed
- F. J. Coughlin, M. S. Westrol, K. D. Oyler, N. Byrne, C. Kraml, E. Zysman-Colman, M. S. Lowry and S. Bernhard, Inorg. Chem., 2008, 47, 2039–2048 CrossRef CAS PubMed
- T.-Y. Li, Y.-M. Jing, X. Liu, Y. Zhao, L. Shi, Z. Tang, Y.-X. Zheng and J.-L. Zuo, Sci. Rep., 2015, 5, 14912 CrossRef CAS PubMed
- C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915–1927 RSC
- X.-P. Zhang, V. Y. Chang, J. Liu, X.-L. Yang, W. Huang, Y. Li, C.-H. Li, G. Muller and X.-Z. You, Inorg. Chem., 2015, 54, 143–152 CrossRef CAS PubMed
- S. Tanaka, K. Sato, K. Ichida, T. Abe, T. Tsubomura, T. Suzuki and K. Shinozaki, Chem.–Asian J., 2016, 11, 265–273 CrossRef CAS PubMed
- T. R. Schulte, J. J. Holstein, L. Krause, R. Michel, D. Stalke, E. Sakuda, K. Umakoshi, G. Longhi, S. Abbate and G. H. Clever, J. Am. Chem. Soc., 2017, 139, 6863–6866 CrossRef CAS PubMed
- J. Song, M. Wang, X. Zhou and H. Xiang, Chem.–Eur. J., 2018, 24, 7128–7132 CrossRef CAS PubMed
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed
- B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28–29 CrossRef CAS PubMed
- C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136–12143 CrossRef CAS PubMed
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS PubMed
- M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952–2955 CrossRef CAS PubMed
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed
- J. Moussa, K. M.-C. Wong, L.-M. Chamoreau, H. Amouri and V. W.-W. Yam, Dalton Trans., 2007, 3526–3530 RSC
- H. Sesolis, J. Dubarle-Offner, C. K. M. Chan, E. Puig, G. Gontard, P. Winter, A. L. Cooksy, V. W. W. Yam and H. Amouri, Chem.–Eur. J., 2016, 22, 8032–8037 CrossRef CAS PubMed
- H. Sesolis, C. K.-M. Chan, G. Gontard, H. L.-K. Fu, V. W.-W. Yam and H. Amouri, Organometallics, 2017, 36, 4794–4801 CrossRef CAS
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC
- M. Yoshida and M. Kato, Coord. Chem. Rev., 2018, 355, 101–115 CrossRef CAS
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed
- F. N. Castellano and C. E. McCusker, Dalton Trans., 2015, 44, 17906–17910 RSC
- C.-M. Che and S.-W. Lai, Coord. Chem. Rev., 2005, 249, 1296–1309 CrossRef CAS
- P. Pfeiffer and K. Quehl, Ber. Dtsch. Chem. Ges. B, 1931, 64, 2667–2671 CrossRef
- S. Lifson, M. M. Green, C. Andreola and N. C. Peterson, J. Am. Chem. Soc., 1989, 111, 8850–8858 CrossRef CAS
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS PubMed
- F. K.-W. Kong, A. K.-W. Chan, M. Ng, K.-H. Low and V. W.-W. Yam, Angew. Chem., Int. Ed., 2017, 56, 15103–15107 CrossRef CAS PubMed
- S. Tanaka, K. Sato, K. Ichida, T. Abe, T. Tsubomura, T. Suzuki and K. Shinozaki, Chem.–Asian J., 2016, 11, 265–273 CrossRef CAS PubMed
- T. Ikeda, T. Masuda, T. Hirao, J. Yuasa, H. Tsumatori, T. Kawai and T. Haino, Chem. Commun., 2012, 48, 6025–6027 RSC
- H. L.-K. Fu, C. Po, H. He, S. Y.-L. Leung, K. S. Wong and V. W.-W. Yam, Chem.–Eur. J., 2016, 22, 11826–11836 CrossRef CAS PubMed
- J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737–2747 RSC
- T. Ikeda, M. Takayama, J. Kumar, T. Kawai and T. Haino, Dalton Trans., 2015, 44, 13156–13162 RSC
- A. Satrijo, S. C. J. Meskers and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 9030–9031 CrossRef CAS PubMed
- J. Kumar, T. Nakashima, H. Tsumatori and T. Kawai, J. Phys. Chem. Lett., 2014, 5, 316–321 CrossRef CAS PubMed
- Z. Shen, T. Wang, L. Shi, Z. Tang and M. Liu, Chem. Sci., 2015, 6, 4267–4272 RSC
- T. Ikeda, K. Hirano and T. Haino, Mater. Chem. Front., 2018, 2, 468–474 RSC
- Q. Wan, W.-P. To, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 3089–3093 CrossRef CAS PubMed
- T. Kayano, S. Takayasu, K. Sato and K. Shinozaki, Chem.–Eur. J., 2014, 20, 16583–16589 CrossRef CAS PubMed
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed
- K. Akagi, Chem. Rev., 2009, 109, 5354–5401 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; Fig. S1–S20, displaying the quantum chemical calculation results, 1H and 13C{1H} NMR spectra, the 1H NMR and the UV-vis absorption spectra obtained with increasing the concentration of PtN, the ECD spectra of the co-assemblies recorded upon rotating the sample, the ECD spectra for the co-assemblies containing increased concentrations of PtBox, steady-state photophysical and chiroptical behaviors of (R)-/(S)-PtBox, phosphorescence and photoluminescence excitation spectra of PtN, phosphorescence decay traces for the self-assemblies of PtN, phosphorescence spectra of the co-assemblies obtained with increasing the concentration of PtBox, temperature-dependent phosphorescence, UV-vis absorption and ECD spectra of the co-assemblies, and the circularly polarized phosphorescence spectra; Tables S1–S3, listing chiroluminescence data reported in literatures, photophysical data of PtN and (R)-/(S)-PtBox, and biexponential decay fit results of the traces shown in Fig. 5a. See DOI: 10.1039/c8sc04509g |
| This journal is © The Royal Society of Chemistry 2019 |