
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modular approach to prepare enantioenriched cyclobutanes: synthesis of (+)-rumphellaone A†
Jordan C.
Beck‡
 ,
Caitlin R.
Lacker‡
,
Lauren M.
Chapman
and
Sarah E.
Reisman
*
,
Caitlin R.
Lacker‡
,
Lauren M.
Chapman
and
Sarah E.
Reisman
*
The Warren and Katharine Schlinger Laboratory of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: reisman@caltech.edu
First published on 19th December 2018
Abstract
A modular synthesis of enantioenriched polyfunctionalized cyclobutanes was developed that features an 8-aminoquinolinamide directed C–H arylation reaction. The C–H arylation products were derivatized through subsequent decarboxylative coupling processes. This synthetic strategy enabled a 9-step enantioselective total synthesis of the antiproliferative meroterpenoid (+)-rumphellaone A.
Introduction
The cyclobutane structural motif is present in a variety of natural products and pharmaceutical molecules (Scheme 1).1,2 Cyclobutanes are also versatile synthetic intermediates, as the ring strain inherent to these structures engenders them with unique reactivity that can be leveraged in a variety of transformations to build complex frameworks.3 [2 + 2] cycloaddition reactions represent the most extensively developed approach to construct cyclobutanes, and recent advances have given rise to elegant enantioselective reactions.4 An alternative strategy is to prepare a versatile cyclobutane building block, and then use C–H functionalization or cross-coupling chemistry to elaborate the scaffold in a modular fashion.5 In this latter approach, a single enantioenriched intermediate can quickly be converted to a variety of more functionalized structures.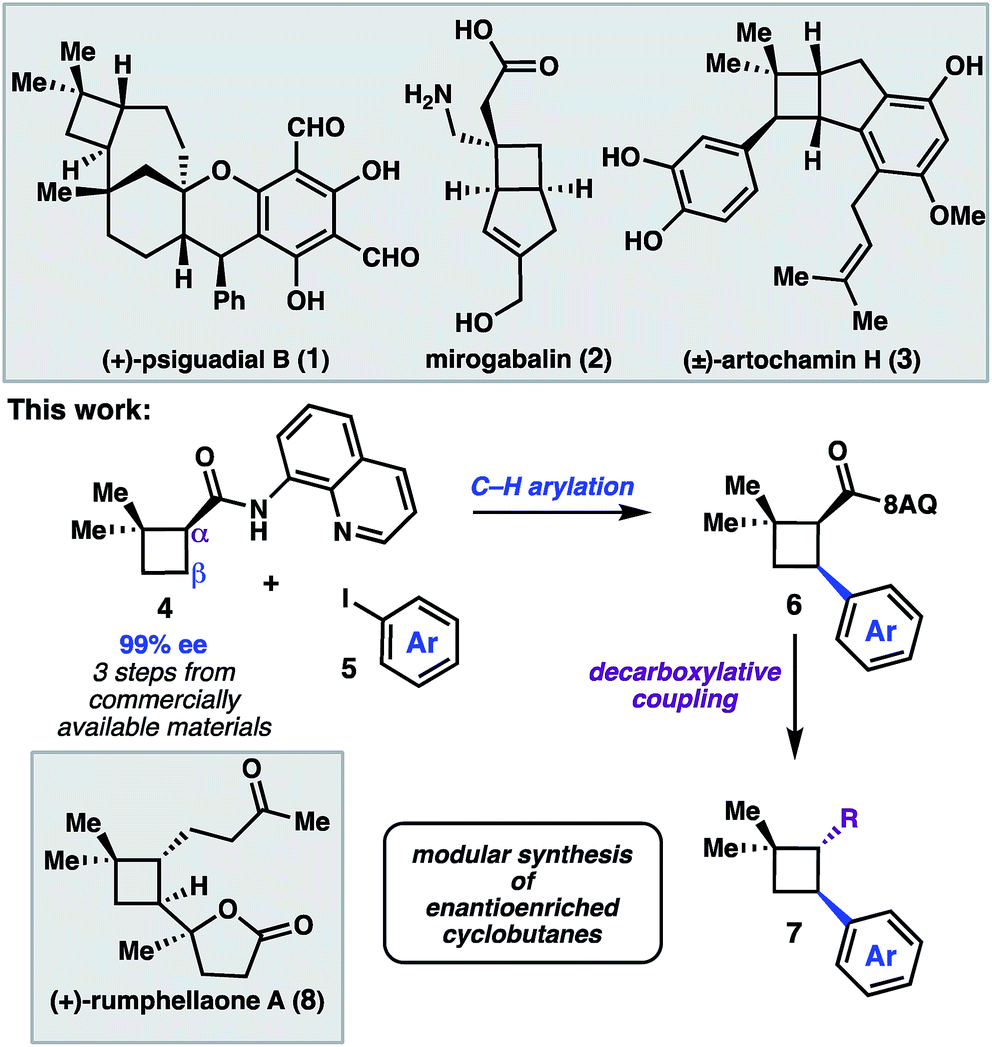 | ||
| Scheme 1 A C–H functionalization strategy to access (+)-rumphellaone A. | ||
We recently reported a synthesis of the natural product (+)-psiguadial B (1), which featured a tandem Wolff-rearrangement/asymmetric ketene addition to prepare enantioenriched 8-aminoquinolinamide 4 (Scheme 1).6 Given the short synthesis of 4 from commercial starting materials, we became interested in further applications of this chiral building block. Specifically, we envisioned that directed C–H arylation could enable diversification at the β-position,7 while hydrolysis of the 8-aminoquinolinamide followed by decarboxylative radical cross-coupling could enable diversification at the α-position. A number of powerful methods have been developed that leverage the decarboxylative formation of carbon-centered radicals for C–C and C–X bond formation.8 It was anticipated that the sequence of C–H arylation followed by decarboxylative coupling could provide access to a collection of enantioenriched polyfunctionalized cyclobutanes.
Results and discussion
We began by investigating the scope of the directed C–H arylation of 8-aminoquinolinamide 4, which was prepared in three steps and 99% ee from commercially available 2,2-dimethylcyclopentan-1-one.6 Using our previously developed conditions [Pd(OAc)2 (15 mol%), Ag2CO3 (1.0 equiv.), aryl iodide (2.0 equiv.), TBME, 90 °C], a series of cis-arylated cyclobutanes were prepared in good yields (Scheme 2).9 The reaction was compatible with both electron-rich and electron-deficient aryl iodides and tolerated substitution at the ortho, meta, and para positions. Heteroaryl iodides were also found to be competent coupling partners, allowing for incorporation of pyridines, pyrimidines, and indoles. Unfortunately, with 5-iodo-2-phenylpyridine or 5-iodo-2-methoxypyrimidine, the reaction proceeded in only modest yields (<40%). Aryl triflates failed to react under the optimized reaction conditions.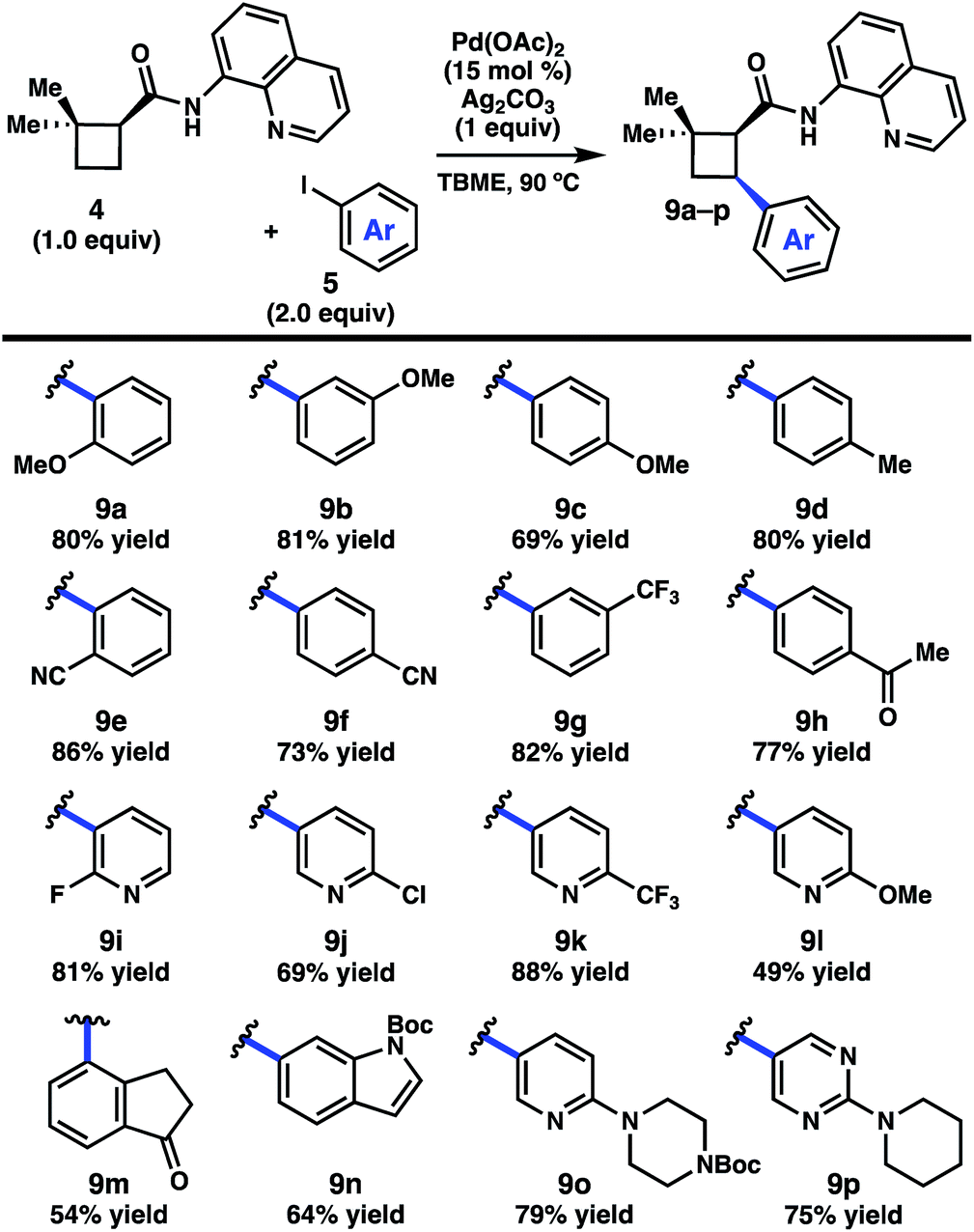 | ||
| Scheme 2 Scope of the C–H arylation of 4. Reactions were conducted on 0.20 mmol scale in a sealed 2-dram vial using Pd(OAc)2 (15 mol%), Ag2CO2 (1.0 equiv.), 5a–p (2.0 equiv.), [4] = 0.2 M in TBME. TBME = tert-butyl methyl ether. | ||
Having established the generality of the C–H arylation step, we turned our attention to diversification at the carbon bearing the 8-aminoquinolinamide through functional group interconversion or decarboxylative cross-coupling. To this end, hydrolysis of 9d proceeded with epimerization to the thermodynamically favored trans diastereomer, delivering trans-cyclobutanoic acid 10 (Scheme 3).10 Reduction of the acid delivered alcohol 12, which could be oxidized under Stahl conditions to aldehyde 13.11 Alternatively, 10 could be converted to the corresponding acid chloride and engaged in a nickel-catalyzed reductive cross-coupling with iodocyclohexane to access ketone 14.12 In order to investigate decarboxylative coupling processes, acid 10 was subjected to EDC-mediated coupling with N-hydroxyphthalimide to provide NHP ester 11.13 Ni-catalyzed coupling of 11 with arylzinc chloride 15 gave trans-diarylcyclobutane 16 in good yield as a single diastereomer.8d Similarly, NHP ester 11 underwent Ni-catalyzed reductive alkenylation with styrenyl bromide 17 to furnish cyclobutane 18 in 56% yield.8c,e Photoinduced decarboxylative borylation of 11 proceeded smoothly to afford boronic ester 20,8h and decarboxylative Minisci type arylation of 11 under photoredox catalysis delivered quinoline 19.8i
 | ||
| Scheme 3 Selected derivatizations of 10. ABNO = 9-azabicylo[3.3.1]nonane N-oxyl. NMI = N-methylimidazole. NHPI = N-hydroxyphthalimide. EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide. DMAP = 4-dimethylaminopyridine. TFA = trifluoroacetic acid. dtbbpy = di-tert-butylbipyridine. dme = dimethoxyethane. 4,4′-MeObpy = 4,4′-dimethoxybipyridine. [Ir] = Ir[dF(CF3)ppy]2(dtbbpy)PF6. | ||
To further demonstrate the utility of this cyclobutane difunctionalization strategy, we designed and executed a synthesis of the natural product (+)-rumphellaone A (8).14 (+)-Rumphellaone A (8) was isolated in 2010 from the gorgonian coral Rumphella antipathies and possesses anti-proliferative activity against human T-cell acute lymphoblastic leukemia tumor cells.2d Retrosynthetically, we envisioned disconnecting through the C1–C2 bond to give 21 (Scheme 4); in the forward sense, the ketone fragment would be incorporated through a decarboxylative Giese addition with methyl vinyl ketone. The butenolide of 21 could derive from oxidation of furan 22, which could be prepared from 4 by a directed C–H arylation. As a proof of concept, 8-aminoquinolinamide 4 was subjected to Pd-catalyzed C–H functionalization with furanyl iodide 23 to give cis-cyclobutane 24 in 90% yield.15 Hydrolysis and subsequent decarboxylative Giese reaction with methyl vinyl ketone under photoredox catalysis provided 25 in 50% yield over two steps.16
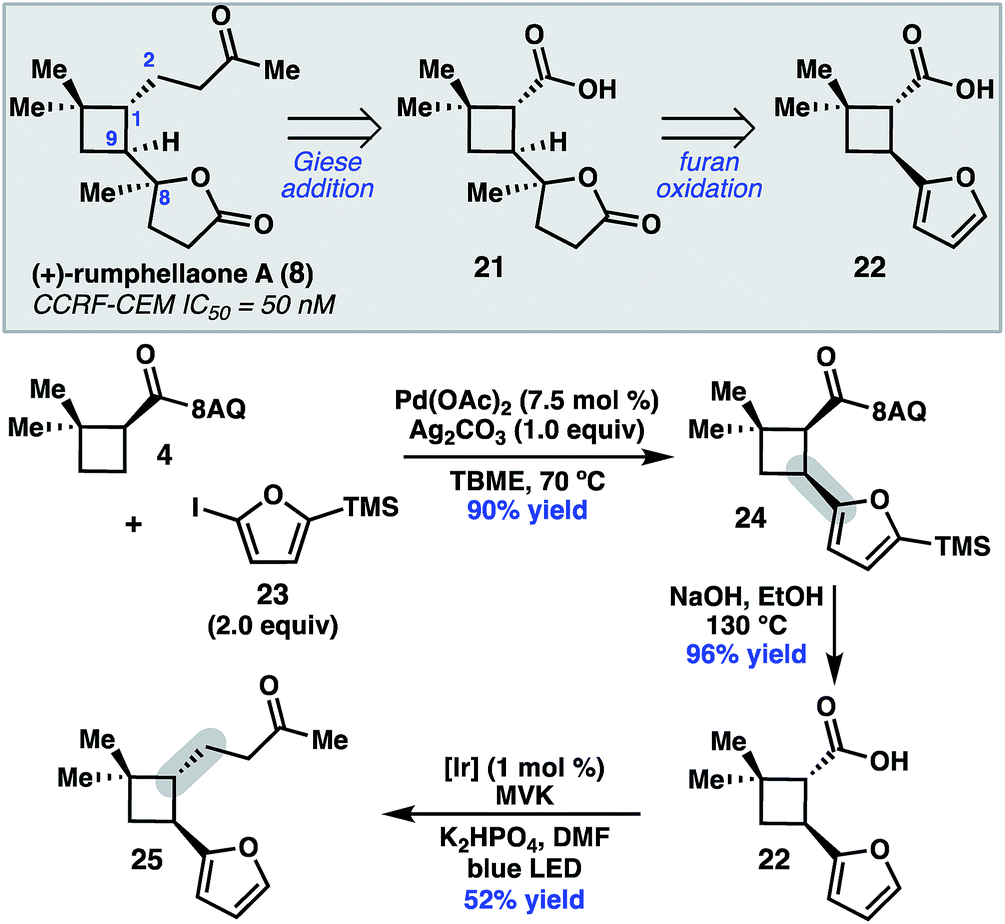 | ||
| Scheme 4 Retrosynthetic analysis of 8 and key proof-of-concept study. TMS = trimethylsilyl. MVK = methyl vinylketone. | ||
Having validated the feasibility of the two key cyclobutane functionalization reactions, attention turned to the unmasking of the butenolide functionality prior to the decarboxylative Giese reaction. Treatment of 22 with sodium chlorite under buffered conditions17 delivered 5-hydroxybutenolide 26 (Scheme 5). The remaining challenge was installation of the C8 methyl substituent with the required S-configuration. In prior syntheses of 8, this stereogenic center was set under the guidance of chiral catalyst control.14 Given that the C8 diastereomers were inseparable by column chromatography, high diastereoselectivity for this methyl addition was important.
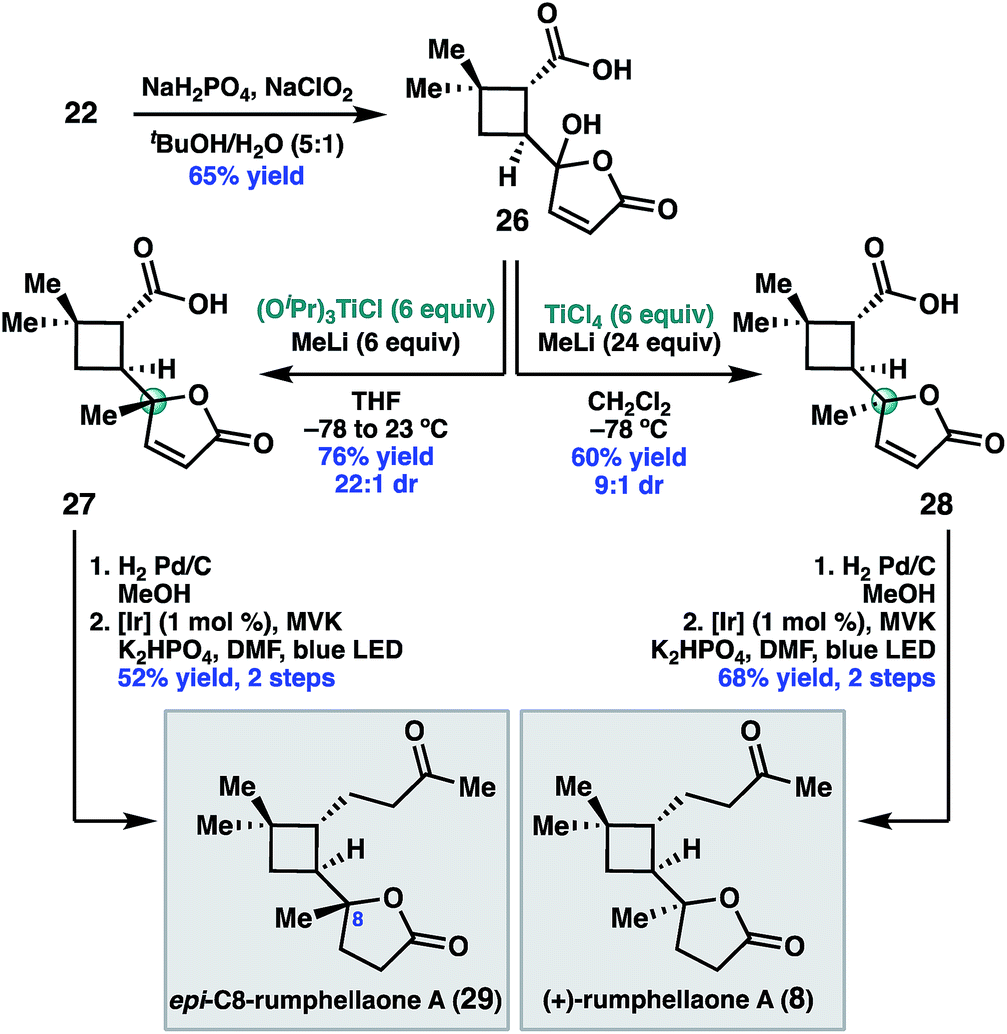 | ||
| Scheme 5 Synthesis of (+)-rumphellaone A (8). | ||
After exploring a range of conditions to effect the methylation, we were pleased to discover that either C8 diastereomer (27 or 28) could be prepared using the appropriate methyltitanium reagent. Thus, addition of 26 to a pre-formed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (iPrO)3 TiCl and MeLi at −78 °C, with warming to 23 °C, delivered the undesired C8 diastereomer, 27, in 76% yield and 22:1 dr.18 Alternatively, addition of 26 to a −78 °C solution of Ti(Me)4 in dichloromethane,19 which was prepared in situ by combining MeLi and TiCl4 in a 4:1 ratio, provided the desired diastereomer 28 in 60% yield and 9:1 dr. We hypothesize that the divergent diastereoselectivity for these two reactions resulted from the different methylating reagents, (iPrO)3TiMe or Ti(Me)4, prepared in situ. One possible explanation is that 27 formed by ligand exchange of the carboxylic acid of 26 with (iPrO)3TiMe followed by intramolecular delivery of the methyl nucleophile, while 28 resulted from addition of Ti(Me)4 without the assistance of chelation.
:1 mixture of (iPrO)3 TiCl and MeLi at −78 °C, with warming to 23 °C, delivered the undesired C8 diastereomer, 27, in 76% yield and 22:1 dr.18 Alternatively, addition of 26 to a −78 °C solution of Ti(Me)4 in dichloromethane,19 which was prepared in situ by combining MeLi and TiCl4 in a 4:1 ratio, provided the desired diastereomer 28 in 60% yield and 9:1 dr. We hypothesize that the divergent diastereoselectivity for these two reactions resulted from the different methylating reagents, (iPrO)3TiMe or Ti(Me)4, prepared in situ. One possible explanation is that 27 formed by ligand exchange of the carboxylic acid of 26 with (iPrO)3TiMe followed by intramolecular delivery of the methyl nucleophile, while 28 resulted from addition of Ti(Me)4 without the assistance of chelation.
To complete the synthesis, 28 was reduced under standard hydrogenation conditions. Decarboxylative Giese addition of 28 to methyl vinyl ketone under photoredox catalysis provided (+)-rumphellaone A (8) in good yield, completing the synthesis in 9 steps from commercially available material. Epimeric acid 27 could be analogously elaborated to (+)-epi-C8-rumphellaone A (29).
Conclusions
Through a strategy for difunctionalization, we have demonstrated that 8-aminoquinolinamide 4 can serve as a valuable building block for the synthesis of enantioenriched cyclobutanes. We further illustrated this concept in a 9-step synthesis of (+)-rumphellaone A (8). We anticipate that this general strategy could enable the expedient synthesis of additional natural products and other bioactive molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. Fellowship support was provided by an NIH Training Grant (J. C. B., Grant No. 5T32GM007616-39) and the NSF (C. R. L. and L. M. C., Grant No. DGE-1144469) and. S. E. R. is a Heritage Medical Research Foundation Investigator. Partial financial support from the NSF (CAREER-1057143) and NIH (R35GM118191-01), as well as the Research Corporation Cottrell Scholars program, is gratefully acknowledged.Notes and references
- (a) V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CrossRef CAS PubMed; (b) Y. J. Hong and D. J. Tantillo, Chem. Soc. Rev., 2014, 43, 5042–5050 RSC; (c) R. M. Ortuno, A. G. Moglioni and G. Y. Motrasio, Curr. Org. Chem., 2005, 9, 237–259 CrossRef CAS.
- (a) Psiguadial B: M. Shao, Y. Wang, Z. Liu, D.-M. Zhang, H.-H. Cao, R.-W. Jiang, C.-L. Fan, X.-Q. Zhang, H.-R. Chen and X.-S. Yao, et al. , Org. Lett., 2010, 12, 5040–5043 CrossRef CAS PubMed; (b) Mirogabalin: A. Vinik, J. Rosenstock, U. Sharma, K. Feins, C. Hsu and D. Merante, Diabetes Care, 2014, 37, 3253–3261 CrossRef CAS PubMed; (c) Artochamin H: Y.-H. Wang, A.-J. Hou, D.-F. Chen, M. Weiller, A. Wendel and R. J. Staples, Eur. J. Inorg. Chem., 2006, 15, 3457–3463 CrossRef; (d) Rumphellaone A: H.-M. Chung, Y.-H. Chen, M.-R. Lin, J.-H. Su, W.-H. Wang and P.-J. Sung, Tetrahedron Lett., 2010, 51, 6025–6027 CrossRef CAS.
- (a) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS PubMed; (b) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef CAS PubMed; (c) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1484 CrossRef CAS PubMed; (d) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254–259 RSC.
- (a) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed; (b) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed; (c) N. Vallavoju, S. Selvakumar, S. Jockusch, M. P. Sibi and J. Sivaguru, Angew. Chem., Int. Ed., 2014, 53, 5604–5608 CrossRef CAS PubMed; (d) M. L. Conner, Y. Xu and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 3482–3485 CrossRef CAS PubMed; (e) V. Pagar and T. V. RajanBabu, Science, 2018, 361, 68–72 CrossRef CAS PubMed. For reviews: (f) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (g) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS PubMed.
- (a) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS PubMed; (b) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507–7510 CrossRef CAS PubMed; (c) R. Parella, B. Gopalakrishnan and S. A. Babu, J. Org. Chem., 2013, 78, 11911–11934 CrossRef CAS PubMed; (d) W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430–2452 CrossRef CAS PubMed; (e) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS PubMed; (f) T. Chen, L. M. Barton, Y. Lin, J. Tsien, D. Kossler, I. Bastida, S. Asai, C. Bi, J. S. Chen, M. Shan, H. Fang, F. G. Fang, H.-W. Choi, L. Hawkins, T. Qin and P. S. Baran, Nature, 2018, 560, 350–354 CrossRef CAS PubMed; (g) Z. Zhuang, C.-B. Yu, G. Chen, Q.-F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 10363–10367 CrossRef CAS PubMed; (h) H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581–585 CrossRef CAS PubMed; (i) H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755–762 CrossRef CAS PubMed; (j) Q.-F. Wu, X.-B. Wang, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 2577–2581 CrossRef CAS PubMed.
- (a) L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS PubMed; (b) L. M. Chapman, J. C. Beck, C. R. Lacker, L. Wu and S. E. Reisman, J. Org. Chem., 2018, 83, 6066–6085 CrossRef CAS PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (c) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS PubMed. For a review: (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (b) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed; (c) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed; (d) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed; (e) N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150–2153 CrossRef CAS PubMed; (f) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed; (g) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 CrossRef CAS PubMed; (h) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (i) W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem.–Eur. J., 2017, 23, 2537–2541 CrossRef CAS PubMed. Reviews: (j) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (k) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS.
- Use of 1.0 equivalent of aryl iodide or lowering the catalyst loading to 7.5 mol% reduced the yield for most substrates by approximately 30%.
- Unfortunately, our efforts to derivatize the 8-aminoquinalinamide without epimerization to the trans-cyclobutane proved unfruitful.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- A. C. Wotal and D. J. Weix, Org. Lett., 2012, 14, 1476–1479 CrossRef CAS PubMed.
- Several direct decarboxylative coupling reactions using [Ir]/[Ni] catalyst systems performed poorly on this substrate. For example, cross-coupling of 10 with 3-phenyl-1-bromopropane under [Ir]/[Ni] (see ref. 8b) resulted in formation of the corresponding ester.
- Prior syntheses: (a) T. Hirokawa, T. Nagasawa and S. Kuwahara, Tetrahedron Lett., 2012, 53, 705–706 CrossRef CAS; (b) T. Hirokawa and S. Kuwahara, Tetrahedron, 2012, 68, 4581–4587 CrossRef CAS; (c) B. Ranieri, C. Obradors, M. Mato and A. M. Echavarren, Org. Lett., 2016, 18, 1614–1617 CrossRef CAS PubMed; (d) C. García-Morales, B. Ranieri, I. Escofet, L. López-Suarez, C. Obradors, A. I. Konovalov and A. M. Echavarren, J. Am. Chem. Soc., 2017, 139, 13628–13631 CrossRef PubMed.
- Under the conditions reported in Table 2, significant amounts of a bis-arylated product (by HRMS) was formed. Use of 2-iodofuran instead for 23 resulted in lower yields.
- Adapted from: L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- S. P. Annangudi, M. Sun and R. G. Salomon, Synlett, 2005, 1468–1470 CAS.
- (a) M. T. R. R. Steinbach, J. Westermann, R. Peter and B. Wenderoth, Chem. Ber., 1985, 118, 1441–1454 CrossRef; (b) W. H. Miles, D. G. Duca, J. T. Freedman, E. O. Goodzeit, K. B. Hamman, S. C. A. P. De and B. R. Selfridge, Heterocycl. Commun., 2011, 13, 195–198 Search PubMed; (c) A. Zúñiga, G. Pazos, P. Besada and Y. Fall, Tetrahedron Lett., 2012, 53, 4293–4295 CrossRef.
- M. T. Reetz, J. Westermann and S.-H. Kyung, Chem. Ber., 1985, 118, 1050–1057 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05444d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |