
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic hydrogen evolution with gallium hydride and ligand-centered reduction†
Ni
Wang
a,
Haitao
Lei
a,
Zongyao
Zhang
b,
Jianfeng
Li
 c,
Wei
Zhang
a and
Rui
Cao
*ab
c,
Wei
Zhang
a and
Rui
Cao
*ab
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: ruicao@ruc.edu.cn
bDepartment of Chemistry, Renmin University of China, Beijing 100872, China
cCollege of Materials Science and Optoelectronic Technology, University of Chinese Academy of Science, Beijing 101408, China
First published on 27th December 2018
Abstract
GaIII chloride tetrakis(pentafluorophenyl)porphyrin (1) was synthesized and shown to be a highly active and stable post-transition metal-based electrocatalyst for the hydrogen evolution reaction (HER). Electrochemical and spectroscopic studies indicate that both the first and second reduction events of 1 are ligand-centered. The 2e-reduced form 12− reacts with a proton to give GaIII–H species (1–H), which undergoes protonolysis with Brønsted acids to produce H2. The identification of key intermediates 1−, 12−, and 1–H leads to a catalytic cycle, which is valuable for the fundamental understanding of the HER. This study presents a rare but highly active HER electrocatalyst with unusual features, including ligand-centered electron transfer and formation of post-transition metal hydride.
Introduction
Hydrogen is a promising energy carrier and is largely required in chemical and petroleum industries.1–3 With energy supplies from renewable sources, H2 can be produced through the reduction of protons in the electrolysis of water.4,5 Thus, the electrocatalytic proton reduction provides an attractive approach for producing H2, a clean and sustainable fuel.1–3 Extensive efforts have been made to develop molecular catalysts of earth-abundant transition metals, such as Fe,6–10 Co,11–24 Ni,25–32 and Cu,33,34 for the HER. For these transition metal catalysts, reduced metal ions are proposed to react with protons to form metal-hydride intermediates,25,33,35,36 which can then evolve H2 through heterolytic or homolytic pathways. Consequently, tuning both the electron and proton transfer abilities of transition metal ions is a rational strategy to improve catalytic performance.Several ligand design strategies have been demonstrated to improve the performance of these catalysts. Multidentate phosphine ligands can stabilize low-valent metal ions and increase metal ion basicity through the π-back-donating interaction.37,38 Pendant amine groups in Ni complexes of Dubois and co-workers function as proton relays to increase activities.39–41 Similarly, appending intramolecular proton transfer sites has been shown to be successful in other catalyst systems.42–46 For these prevailing transition metal-based catalysts, metal ions are the major sites for both electron and proton transfer. As a consequence, the reduced metal ions with a low valence state usually become weaker in coordination. This will lead to possible demetalation especially under acidic conditions, which is one of the main catalyst deactivation routes in the HER.
As an alternative strategy, ligands that can mediate both electron and proton transfer have been explored recently. Catalysts of this series include AlIII bis(imino)pyridine,47–49 ZnII thiosemicarbazone,50 NiII pyrazinedithiolate,51 CuII diacetyl-bis(N-4-methyl-3-thiosemicarbazonato),52 and ReII phosphinobenzenethiolate.53 For these catalysts, the ligands are proposed to play essential roles in both electron transfer and proton transfer. Active X–H (X = C, N, and O) units with relatively strong covalent bond features rather than metal hydrides are considered to be involved in the H–H bond formation.
Herein, we report GaIII porphyrin 1 (Fig. 1a) as a highly active and stable post-transition metal HER catalyst with the porphyrin ligand acting as the electron transfer site and the GaIII ion as the hydride-binding site. Porphyrin ligands are well known to have redox-active features, which play crucial roles in several electrocatalytic processes, including the HER, and O2 and CO2 reduction reactions.3,25,54–57 Recent studies from Wang and co-workers by using a Zn porphyrin for CO2 reduction highlight the role of porphyrin ligands in redox catalysis.58 On the other hand, although GaIII ions have a strong Lewis acid character and are promising in binding and transferring hydride atoms, no GaIII complexes have been shown to be active for the HER. Significantly, by identifying 1−, 12−, and 1–H with spectroscopic methods and studying their reactivity, we are able to draw a catalytic cycle. Mechanistic studies provide valuable insights into the fundamental knowledge for the design of efficient post-transition metal redox catalysts.
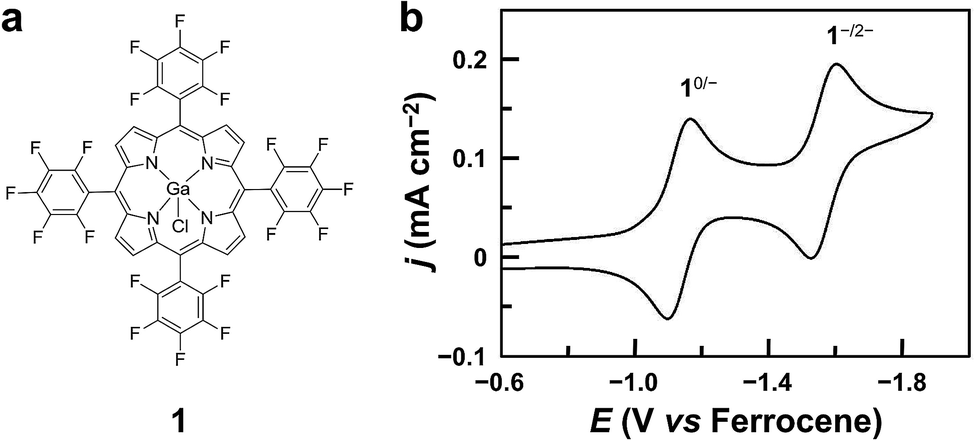 | ||
| Fig. 1 (a) Molecular structure of GaIII porphyrin 1. (b) CV of 1.0 mM 1 in acetonitrile, showing two reversible 1e reduction waves at E1/2 = −1.13 and −1.57 V. Conditions: 0.1 M Bu4NPF6, a GC working electrode, 100 mV s−1 scan rate, and 20 °C. | ||
Results and discussion
Synthesis and electrochemistry
Complex 1 was prepared by reacting GaCl3 with a free-base porphyrin in acetic acid. The details of synthesis and characterization are described in the Experimental section. The cyclic voltammogram (CV) of 1 using a glassy carbon (GC) electrode in acetonitrile with 0.1 M Bu4NPF6 displays two reversible 1e reduction waves at E1/2 = −1.13 and −1.57 V versus ferrocene (Fig. 1b, all potentials reported in this work are versus ferrocene). These two reduction waves are diffusion-controlled as suggested by the linear correlation between their peak currents and the square root of scan rates (Fig. S3†). For simplicity, the 1e- and 2e-reduced forms of 1 are denoted as 1− and 12−, respectively.Electrocatalytic HER studies
The CV of 1 with the addition of trifluoroacetic acid (TFA, pKa = 12.7 in acetonitrile59,60) shows a pronounced catalytic wave (Fig. 2a). The peak currents increase with the first-order dependence on the concentrations of both TFA (Fig. 2a, inset) and 1 (Fig. S4†), indicating catalytic proton reduction. Control experiments using GaCl3 (Fig. S5†) and the free-base porphyrin (Fig. S6†) gave very small currents under the same conditions. This result confirms that the catalytic activity is from 1 rather than from the demetalated species. It is necessary to note that this free-base porphyrin is active for the electrocatalytic HER if a stronger p-toluenesulfonic acid is used as the proton source with much more negative potentials applied.61 The HER activity of 1 reached an acid-independent region with more than 97 equivalents of TFA (Fig. S7†). The catalytic current icat value showed a scan rate ν-independence with ν > 1.0 V s−1 (Fig. S8†), and the icat/ip value (ip is the peak current of the first reduction wave) also displayed an inflection point at ν = 1.0 V s−1 (Fig. S9†). Therefore, under these pure kinetic conditions, we can estimate the turnover frequency (TOF) of 1 for the HER to be up to 9.4 × 104 s−1 using the reported foot of the wave analysis (FOWA, Fig. S10†).62,63 This value is comparable to those of transition metal-based HER catalysts,25,33,63–66 although it is obtained at relatively large overpotentials. In addition, we examined the electrocatalytic HER activity of GaIII complexes of 5,15-bis(pentafluorophenyl)-10,20-diphenylporphyrin (Fig. S11†) and 5,10,15,20-tetrakisphenylporphyrin (Fig. S12†). By replacing meso-C6F5 groups with phenyl groups, the onset of the catalytic HER wave shifts to the negative direction by more than 370 mV.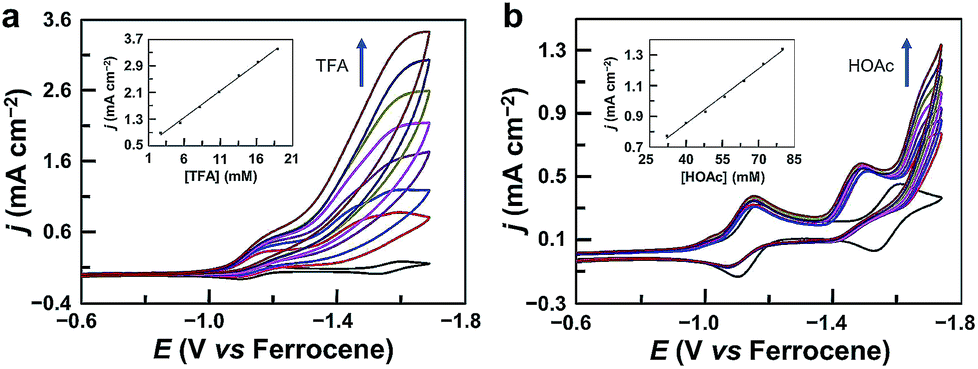 | ||
| Fig. 2 (a) CVs of 1.0 mM 1 in acetonitrile with increasing TFA. Inset: plot of catalytic peak current versus TFA concentration. (b) CVs of 1.0 mM 1 in acetonitrile with increasing acetic acid (HOAc). Inset: plot of catalytic current at −1.75 V versus HOAc concentration. Conditions: 0.1 M Bu4NPF6, a GC working electrode, and 20 °C. | ||
The catalytic stability of 1 was verified by using controlled potential electrolysis. The UV-vis spectrum was first collected before electrolysis, showing that 1 remains unchanged in 50 mM TFA acetonitrile solution (Fig. S13†). Electrolysis was then conducted at −1.70 V in a three-compartment cell. During 5 h electrolysis using a 0.07 cm2 GC working electrode, substantial and stable currents at ca. 13.1 mA cm−2 could be maintained (Fig. S14†), and H2 gas bubbles were observed on the GC electrode surface. The amount of produced H2 was determined by gas chromatography, giving a faradaic efficiency of 97% for H2 generation (Fig. S15†). The turnover number (TON) was 18 with respect to the total amount of catalyst in the solution. As a control, electrolysis with the same concentration of TFA but without 1 gave much smaller currents at 0.7 mA cm−2. After electrolysis, the solution of 1 showed a UV-vis spectrum almost identical to that before electrolysis (Fig. S16†). The GC electrode after electrolysis showed almost the same current as a freshly cleaned GC electrode did in a TFA solution without 1 (Fig. S17†). In addition, surface analysis by scanning electron microscopy and energy-dispersive X-ray spectroscopy (Fig. S18†) excluded the formation of any heterogeneous phase on the GC electrode.
We also examined the proton reduction activity of 1 by using weak acetic acid (pKa = 22.3 in acetonitrile59,60). As shown in Fig. 2b and S19,† in the presence of acetic acid, the first redox couple shows a tiny decrease in the reverse oxidation wave, while the second redox couple completely loses the reversibility in the reverse oxidation scan. This result indicates that 12− is the active species for proton reduction to form GaIII–H species. Significantly, the second reduction peak of 1 still exists, and the catalytic wave appears well behind the second reduction wave, indicating that GaIII–H needs to be further reduced to drive the catalysis with acetic acid for H2 generation.
The different electrochemical behaviors of 1 with TFA and acetic acid are likely due to the difference in reactivity of 1–H with the two acids. We can get insights from the thermodynamic analysis. As shown in eqn (1)–(7), the driving force for the 12−/0 redox couples to reduce TFA to generate H2 is 0.57 V, but this value is only 0.01 V to reduce acetic acid to produce H2. This result is consistent with the observation that more negative potentials are required to drive the catalysis using acetic acid. Notably, the value of −0.028 V was used for calculating the standard potential of the solvated H+/H2 couple in acetonitrile.67 If the value of −0.14 V was used,59 then the driving forces for the 12−/0 redox couples to reduce TFA and acetic acid to produce H2 were 0.45 and −0.11 V, respectively.
| 1− → 1 + e−, −E° = 1.13 V | (1) |
| 12− → 1− + e−, −E° = 1.57 V | (2) |
| 12− → 1 + 2e−, −E° = 1.35 V | (3) |
| 2TFAH + 2e− → H2 + 2TFA−, E° = −0.78 V | (4) |
| 2HOAc + 2e− → H2 + 2OAc−, E° = −1.34 V | (5) |
| 12− + 2TFAH → 1 + H2 + 2TFA−, ΔE° = 0.57 V | (6) |
| 12− + 2HOAc → 1 + H2 + 2OAc−, ΔE° = 0.01 V | (7) |
In the case of acetic acid as the proton source, the icat at −1.75 V increases with the first-order dependence on the concentrations of both acetic acid (Fig. 2b, inset) and 1 (Fig. S20†), and it shows ν-independence when ν is larger than 2.5 V s−1 (Fig. S21†), indicating catalytic proton reduction. Electrolysis of 1 with acetic acid was also performed. During 5 h electrolysis at −1.70 V using a 1.0 cm2 GC working electrode, a current of ca. 0.8 mA cm−2 could be maintained (Fig. S22†) with a TON of 14 and a faradaic efficiency of 96% for H2 generation (Fig. S23†). The stability of 1 during the electrolysis with acetic acid was also confirmed: the UV-vis spectrum showed negligible changes after electrolysis (Fig. S24†); the GC electrode after electrolysis gave no catalytic current in acetic acid solution without 1 (Fig. S25†); surface analysis also excluded the formation of any heterogeneous phase on the GC electrode.
Mechanism studies
In order to get more insights into the reaction mechanism, we synthesized and characterized 1−, 12−, and 1–H (Fig. 3a). Reduction of 1 with one equivalent of cobaltocene (Cp2Co) gave 1− under a N2 atmosphere. The CV of Cp2Co is depicted in Fig. S26,† showing a reversible 1e reduction wave at −1.33 V. This value is between the two reduction waves of 1. Further reduction of 1− by one equivalent of potassium graphite (KC8, Ep,c < −2.0 V) leads to 12−. Moreover, 12− can be prepared separately from 1 with two equivalents of KC8. Reaction of 12− with one equivalent of acetic acid leads to 1–H. This reaction can be monitored by 1H NMR, showing the building up of the hydride signal at −6.45 ppm with increasing acetic acid concentration (Fig. 3b). The substantial upfield shift of this GaIII–H signal by more than 13 ppm compared to the reported GaIII–H signals68,69 is consistent with its location at the center of the aromatic porphyrin macrocycle. Notably, these 1H NMR data exclude the formation of phlorin species, which can possibly be generated by the protonation at the meso-position of porphyrin units and have typical chemical shifts of the meso-H at 3–5 ppm.70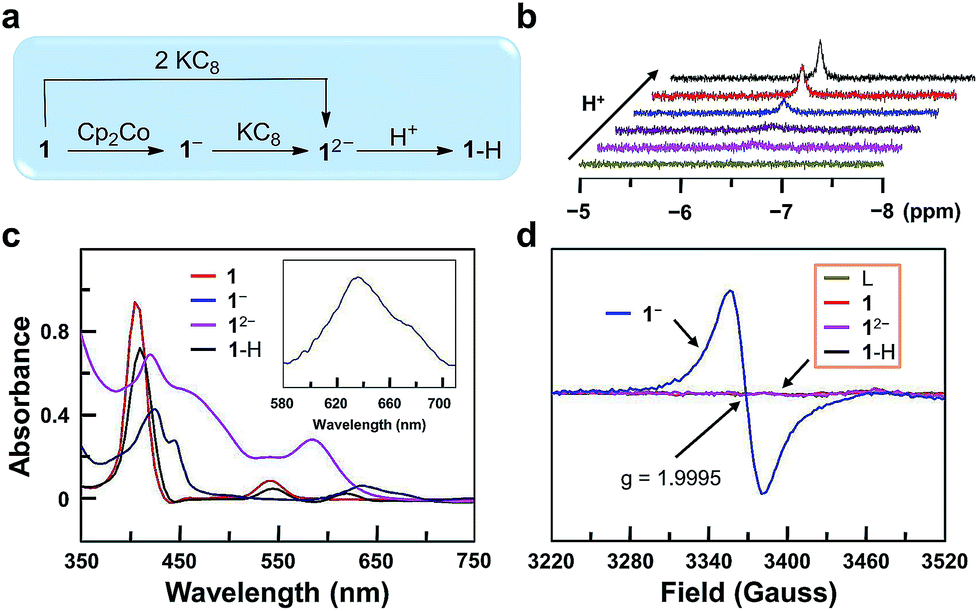 | ||
| Fig. 3 (a) Synthetic route to 1−, 12−, and 1–H. (b) 1H NMR spectra for the formation of 1–H in CDCl3 with increasing HOAc. The GaIII–H peak is found at −6.45 ppm. (c) UV-vis spectra of 1, 1−, 12−, and 1–H. (d) EPR spectra of the free-base porphyrin (L), 1, 1−, 12−, and 1–H. | ||
UV-vis spectroscopic studies confirm this reaction sequence (Fig. 3c). Upon reduction, the absorption in the 560–700 nm range increases significantly. This indicates ligand-centered reduction to give 1e-reduced porphyrin anion radical 1− and 2e-reduced porphyrin dianion 12−. After the reaction of 12− with acids, the resulting complex 1–H shows weak absorption in the 560–700 nm range, which suggests the reduction of a proton by 12−. Notably, the UV-vis spectrum of 1–H further confirms the protonation at the GaIII ion rather than at the meso-position of porphyrin, since for the latter case, the resulting phlorin species will have the characteristic very broad and intense absorption in the range of 650–800 nm.71
Electron paramagnetic resonance (EPR) analysis provides additional evidence for the ligand-centered reduction processes. Complexes 1, 12−, 1–H and the free-base porphyrin are EPR-silent at room temperature (Fig. 3d). Complex 1− shows a single signal at g = 1.9995. This result suggests a ligand-centered reduction, because hyperfine splitting signals, which are indicators of GaII (I = 3/2, 4s1), are not observed in the EPR spectrum of 1−. Furthermore, we synthesized the GaIII complex of 1,4,7,10-tetraazacyclotetradecane (Fig. S27†). The CV of this GaIII complex with a redox-inactive ligand does not show any reduction wave under the same conditions by scanning to −1.95 V (Fig. S28†). In addition, the CV of the free-base porphyrin displayed two reversible 1e reduction waves at E1/2 = −1.17 and −1.63 V (Fig. S29†), which are close to those of complex 1 (−1.13 and −1.57 V). Based on these results, we can conclude that the two reduction events are both ligand-centered.
Attempts to obtain high quality crystals of 1–H suitable for X-ray analysis were not successful. As an alternative, the reactions of 1–H with different Brønsted acids were studied, and the products were structurally characterized, providing further evidence for this GaIII–H hydride species. The reaction of 1–H with excess benzoic acid in acetonitrile gives 2, which contains a benzoate at the axial position of GaIII with a short Ga–O bond length of 1.872(2) Å (Fig. 4).
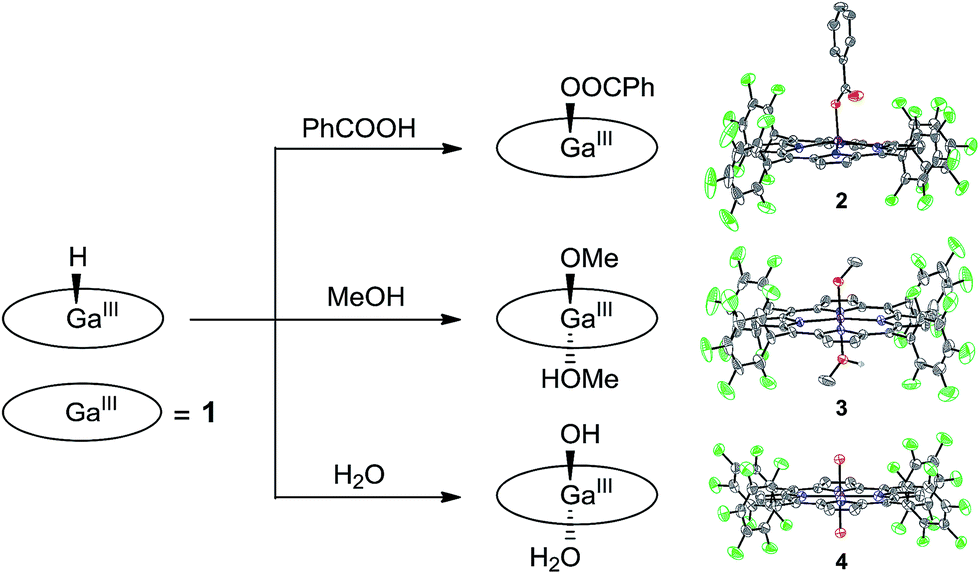 | ||
| Fig. 4 Schematic representation showing the reaction of 1–H with different Brønsted acids and the corresponding products with thermal ellipsoid plots (50% probability) of their X-ray structures. | ||
Crystal growth of 1–H in methanol produces 3, having the two axial positions occupied by a methoxide anion with a short Ga–O bond length of 1.882(3) Å and a methanol molecule with a long Ga–O bond length of 2.085(3) Å. The reaction of 1–H with excess water gives 4. The Ga atom in 4 is located at the crystallographically required inversion center, leading to the symmetry equivalence of the two axial O atoms with a Ga–O bond length of 1.9827(12) Å. Notably, Mayer and co-workers reported the Ga–OH and Ga–OH2 bond lengths of GaIII porphyrin analogues as 1.810(8) and 2.091(3) Å, respectively.72 The Ga–O bond length as observed in 4 is exactly at the middle of these values. Therefore, we assign the two axial groups as one hydroxide and one aqua group, which are positionally disordered due to the crystallographically imposed symmetry. All these results are consistent with the protonolysis of GaIII–H with Brønsted acids to produce H2 and the corresponding GaIII–OR species (R = PhCO for 2, Me for 3, and H for 4).
It is necessary to note that although water and methanol are weaker acids than acetic acid, their reactions with GaIII–H are still observed. This can be explained by the following two reasons. First, water and methanol are used in large excess, and methanol is even used as the solvent. Consequently, the equilibrium will be moved to the product’s direction. Second, the thermodynamic analysis is only used for estimating the reaction of 12− + 2ROH → 1 + H2 + 2RO−. However, the reaction for the formation of 3 and 4 is 1–H + ROH → 1–OR + H2. The binding of the methoxide anion and hydroxide anion to GaIII will certainly lower the energy of the product, which leads to different thermodynamic results. Moreover, the reaction of GaIII–H with water and methanol evolves H2, which is an entropy-increased process. In addition, this process happens on a long time scale (i.e., in days) during the growth of crystals. These factors together may explain the observation of complexes 3 and 4 in this study.
On the basis of these studies, we can draw a catalytic cycle for the HER with 1. As shown in Fig. 5, GaIII porphyrin dianion 12− is generated upon 2e reduction of 1. It then reacts with a proton to form the GaIII–H intermediate. Under electrocatalytic conditions, this hydride can undergo rapid protonolysis with a strong acid like TFA to drive the catalysis for the HER, or it needs to be further reduced by one electron to drive the catalysis with weak acetic acid. Importantly, the reaction of GaIII–H and deuterated TFA or acetic acid gave HD, which was detected by gas chromatography–mass spectrometry (Fig. S30†). The reaction with TFA completed instantly upon the addition of the acid (Fig. 3c), while the reaction with acetic acid completed on a much longer time scale (Fig. S31†). This difference is consistent with the relatively larger driving force of the protonolysis with TFA than that with acetic acid.
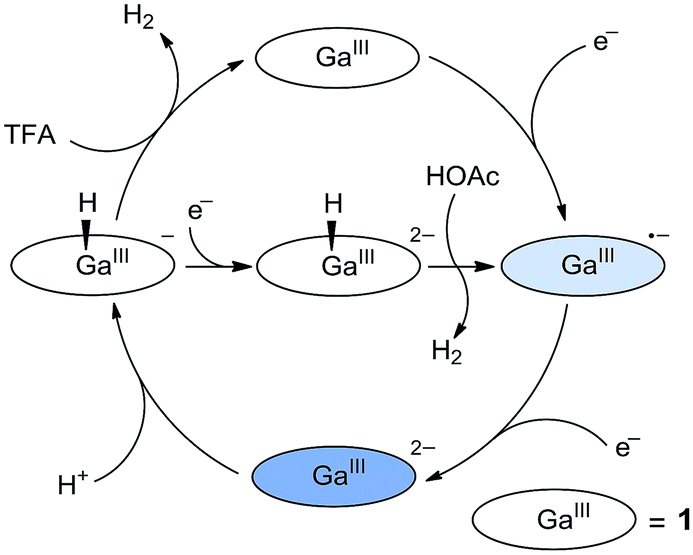 | ||
| Fig. 5 Proposed catalytic cycles for the HER by 1 with the use of either TFA or acetic acid as the proton source. | ||
According to this mechanism, we reanalyzed the CV of 1 with TFA. As shown in Fig. 2a, when the concentration of TFA is low, there are actually two plateaus at ca. −1.48 and −1.62 V in the catalytic wave. This result indicates the involvement of two catalytic mechanisms. We compared the CVs of 1 with low-concentration TFA and with acetic acid (Fig. S32†). The first plateau potential as observed in the CV of 1 with TFA is similar to the second reduction wave potential of 1 in the presence of acetic acid. The second plateau potential in the CV of 1 with TFA is close to the catalytic wave of 1 with acetic acid. Importantly, with one equivalent of TFA, we can see a quasi-reversible redox couple at E1/2 = −1.66 V (Fig. S33†). As we demonstrated above, after 2e reduction, 12− can react with a proton to generate GaIII–H. This leads to the proposal that this redox couple is due to the GaIII–H intermediate. Based on these results, we propose that at the first plateau, the generated GaIII–H intermediate will drive the catalysis with TFA to evolve H2. At more negative potentials, the GaIII–H intermediate can be further reduced by one electron and can then drive the catalysis with TFA, leading to the observation of the second plateau.
Very few post-transition metal complexes are demonstrated as HER catalysts. Recently, Crabtree and co-workers reported SbV porphyrins for the electrocatalytic HER.73 Comparison of the HER mechanisms of 1 and SbV porphyrins shows three differences. First, ligand-centered reduction is suggested in the HER with 1, but both the porphyrin ligand and the Sb ion are redox active for Sb porphyrins. Second, 1 is the real HER catalyst, but the SbIII form converted from starting SbV porphyrins is the real catalyst. Third, GaIII–H can react with TFA in acetonitrile to produce H2, while SbV–H needs to be further reduced to undergo catalytic protonolysis with TFA. This difference in reactivity is likely due to the fact that Ga has a smaller electronegativity than Sb, and thus Ga–H is a stronger hydride donor than Sb–H.74,75 These differences are notable, highlighting the significance of post-transition metals in controlling the activity and mechanism of redox catalysis.
Conclusions
In conclusion, we reported GaIII porphyrin 1 as a highly active and stable post-transition metal-based electrocatalyst for the HER. On the basis of the mechanistic studies, we are able to draw a catalytic proton reduction cycle with 1. Identifying the key intermediates 1−, 12−, and 1–H suggests that the porphyrin ligand acts as the electron transfer site and the GaIII ion acts as the hydride-binding site. This ligand-centered reduction feature enables redox-inert metal ions to become catalytically active in redox reactions, and more importantly, provides a simple but general route for tuning the catalytic activity by modifying ligand substituents. This work presents the potential uses of post-transition metal complexes in redox catalysis and provides insights into the fundamental knowledge for designing efficient post-transition metal-based catalysts.Experimental section
General materials and methods
All reagents were purchased from commercial suppliers and were used as received unless otherwise noted. Acetonitrile was dried by distillation with calcium hydride. Porphyrin ligands were prepared according to the methods reported previously.25 Tetrabutylammonium hexafluorophosphate (Bu4NPF6) was recrystallized from absolute ethanol. 1H NMR spectra were acquired on a Brüker spectrometer operating at 400 MHz. UV-vis absorption spectra were measured on a Hitachi U-3310 spectrophotometer. High-resolution mass spectra (HRMS) were acquired using a Brüker MAXIS. The isotopically labelled HD gas was detected by gas chromatography–mass spectrometry using a Micromeritics AutoChem-2920. X-band continuous wave electron paramagnetic resonance (EPR) measurements were carried out on a Bruker E500 EPR spectrometer at a microwave frequency of 9.45 GHz. The EPR spectrum was recorded at 298 K. The H2 produced during the controlled potential electrolysis was determined by using an SP-6890 gas chromatograph.Synthesis of GaIII porphyrin 1
The free-base porphyrin 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (300 mg, 0.31 mmol), sodium acetate (80 mg, 0.98 mmol) and excess GaIII chloride (546 mg, 3.1 mmol) were added into a flask with 40 mL of dry acetic acid under a N2 atmosphere. The mixture was refluxed overnight, and the solvent was then removed using a rotary evaporator. The purple-red solid of 1 was acquired by silica gel column chromatography (CH2Cl2/CH3OH = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) with a yield of 83.8%. 1H NMR (400 MHz, CDCl3): δ = 9.16 (s, 8H) (Fig. S1†). High-resolution ESI-MS for [C44H8F20N4Ga]+: calcd 1040.9685; found, 1040.9671 (Fig. S2†). Anal. calcd for [GaCl(C44H8F20N4)]: C, 49.04; H, 0.75; N, 5.20. Found: C, 49.23; H, 0.81; N, 5.32.
:1 v/v) with a yield of 83.8%. 1H NMR (400 MHz, CDCl3): δ = 9.16 (s, 8H) (Fig. S1†). High-resolution ESI-MS for [C44H8F20N4Ga]+: calcd 1040.9685; found, 1040.9671 (Fig. S2†). Anal. calcd for [GaCl(C44H8F20N4)]: C, 49.04; H, 0.75; N, 5.20. Found: C, 49.23; H, 0.81; N, 5.32.
Synthesis of GaIII chloride 5,15-bis(pentafluorophenyl)-10,20-diphenylporphyrin
The synthetic procedure of this GaIII porphyrin is identical to that of 1 except for the use of 5,15-bis(pentafluorophenyl)-10,20-diphenylporphyrin. 1H NMR (400 MHz, CDCl3): δ = 9.17 (d, J = 5.2 Hz, 4H), 9.00 (d, J = 5.2 Hz, 4H), 8.25 (dd, J = 7.8 Hz, 1.4 Hz, 4H), 7.70–7.90 (m, 6H). High-resolution ESI-MS for [C44H18F10N4Ga]+: calcd 861.0627; found, 861.0630.Synthesis of GaIII chloride 5,10,15,20-tetrakisphenylporphyrin
The synthetic procedure of this GaIII porphyrin is identical to that of 1 except for the use of 5,10,15,20-tetrakisphenylporphyrin. 1H NMR (400 MHz, CDCl3): δ = 9.05 (s, 8H), 8.11 (dd, J = 7.8 Hz, 1.4 Hz, 8H), 7.90–8.04 (m, 12H). High-resolution ESI-MS for [C44H28N4Ga]+: calcd 681.1569; found, 681.1575.Synthesis of GaIII chloride 1,4,7,10-tetraazacyclotetradecane
Excess GaIII chloride (100 mg, 0.58 mmol) and 1,4,7,10-tetraazacyclotetradecane (510 mg, 2.9 mmol) were added to acetonitrile under a N2 atmosphere. The mixture was refluxed overnight, and the solvent acetonitrile was then removed using a rotary evaporator. The product was acquired by slow vapor diffusion of ethyl ether to an acetonitrile solution of this GaIII complex (yield 89.1%). High-resolution ESI-MS for [C8H20N4GaCl2]+: calcd 311.0320; found, 311.0313.Crystallographic studies
Complete data sets for 2 (CCDC 1855035), 3 (CCDC 1855036), 4 (CCDC 1855037), and GaIII chloride 1,4,7,10-tetraazacyclotetradecane (CCDC 1855038) were collected. Single crystals suitable for X-ray analysis were coated with Paratone-N oil, suspended in a small fiber loop, and placed in a cooled gas stream at 153(2) K on a Bruker D8 Venture X-ray diffractometer. Diffraction intensities were measured using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data collection, indexing, data reduction and final unit cell refinements were carried out using APEX2;76 absorption corrections were applied using the program SADABS.77 The structure was solved by direct methods using SHELXS78 and refined against F2 on all data by full-matrix least squares with SHELXL,79 following the established refinement strategies. In all crystal structures, all non-hydrogen atoms were refined anisotropically. All hydrogen atoms bound to carbon were included into the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms they are linked to (1.5 times for methyl groups). Details of the data quality and a summary of the residual values of the refinements are listed in Table S1.† The CheckCIF report for the structure of 4 shows one level A alert (Short Inter D⋯A Contact O1⋯O1). This indicates the presence of strong hydrogen bonding interactions between these oxygen atoms.Electrochemical studies
All electrochemical experiments were carried out using an electrochemical analyzer (CH Instruments, model CHI660D) at 20 °C, and the solution was bubbled with N2 gas for at least 30 min before analysis. Cyclic voltammograms (CVs) were acquired in 5 mL of dry acetonitrile containing 1.0 mM catalyst and 0.1 M n-Bu4NPF6 and using a three-compartment cell with a 0.07 cm2 glassy carbon (GC) electrode as the working electrode, Ag/Ag+ as the reference electrode, and a graphite rod as the auxiliary electrode. The working electrode was polished with α-Al2O3 of decreasing size (1.0 μm to 50 nm) and washed with distilled water and absolute ethanol. Ferrocene was used as an internal standard, and all potentials reported in this work are referenced to the ferrocenium/ferrocene (Fc+/Fc) couple. Notably, the reported Fc+/Fc potential values in acetonitrile can be converted to versus saturated calomel electrode (SCE) by adding +0.38 V as shown in previous studies.80 Addition of TFA (1.0 M solution in acetonitrile) or acetic acid (1.0 M solution in acetonitrile) was done using a microsyringe. Controlled potential electrolysis in 5 mL of acetonitrile solution containing 50 mM TFA with or without 1.0 mM 1 was measured at −1.70 V in a three-compartment cell with the same electrodes as that for CV measurements. H2 gas produced during the electrolysis was measured using an SP-6890 gas chromatograph.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for support from the “Thousand Talents Program” of China, Fok Ying-Tong Education Foundation for Outstanding Young Teachers in University, National Natural Science Foundation of China (Grants 21101170, 21573139, and 21773146), Fundamental Research Funds for the Central Universities, and Research Funds of Shaanxi Normal University.Notes and references
- T. B. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- W. Zhang, W. Z. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- V. S. Thoi, Y. J. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Angew. Chem., Int. Ed., 2010, 49, 8033–8036 CrossRef CAS PubMed.
- M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310–8313 CrossRef CAS PubMed.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS PubMed.
- J. Noth, J. Esselborn, J. Güldenhaupt, A. Brünje, A. Sawyer, U.-P. Apfel, K. Gerwert, E. Hofmann, M. Winkler and T. Happe, Angew. Chem., Int. Ed., 2016, 55, 8396–8400 CrossRef CAS PubMed.
- T.-W. Chiou, T.-T. Lu, Y.-H. Wu, Y.-J. Yu, L.-K. Chu and W.-F. Liaw, Angew. Chem., Int. Ed., 2015, 54, 14824–14829 CrossRef CAS PubMed.
- X. L. Li, H. T. Lei, J. Y. Liu, X. L. Zhao, S. P. Ding, Z. Y. Zhang, X. X. Tao, W. Zhang, W. C. Wang, X. H. Zheng and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 15070–15075 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- W. M. Singh, T. Baine, S. Kudo, S. L. Tian, X. A. N. Ma, H. Y. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed.
- A. Mahammed, B. Mondal, A. Rana, A. Dey and Z. Gross, Chem. Commun., 2014, 50, 2725–2727 RSC.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381–3387 CrossRef CAS PubMed.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS PubMed.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef CAS PubMed.
- D. Basu, S. Mazumder, X. T. Shi, H. Baydoun, J. Niklas, O. Poluektov, H. B. Schlegel and C. N. Verani, Angew. Chem., Int. Ed., 2015, 54, 2105–2110 CrossRef CAS PubMed.
- M. van der Meer, E. Glais, I. Siewert and B. Sarkar, Angew. Chem., Int. Ed., 2015, 54, 13792–13795 CrossRef CAS PubMed.
- A. G. Maher, G. Passard, D. K. Dogutan, R. L. Halbach, B. L. Anderson, C. J. Gagliardi, M. Taniguchi, J. S. Lindsey and D. G. Nocera, ACS Catal., 2017, 7, 3597–3606 CrossRef CAS.
- J. W. Jurss, R. S. Khnayzer, J. A. Panetier, K. A. El Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano and C. J. Chang, Chem. Sci., 2015, 6, 4954–4972 RSC.
- A. Call, F. Franco, N. Kandoth, S. Fernández, M. González-Béjar, J. Pérez-Prieto, J. M. Luis and J. Lloret-Fillol, Chem. Sci., 2018, 9, 2609–2619 RSC.
- H. L. Sun, Y. Z. Han, H. T. Lei, M. X. Chen and R. Cao, Chem. Commun., 2017, 53, 6195–6198 RSC.
- X. L. Li, H. T. Lei, X. J. Guo, X. L. Zhao, S. P. Ding, X. Q. Gao, W. Zhang and R. Cao, ChemSusChem, 2017, 10, 4632–4641 CrossRef CAS PubMed.
- Y. Z. Han, H. Y. Fang, H. Z. Jing, H. L. Sun, H. T. Lei, W. Z. Lai and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 5457–5462 CrossRef CAS PubMed.
- S. Ramakrishnan, S. Chakraborty, W. W. Brennessel, C. E. D. Chidsey and W. D. Jones, Chem. Sci., 2016, 7, 117–127 RSC.
- W. A. Hoffert, J. A. S. Roberts, R. M. Bullock and M. L. Helm, Chem. Commun., 2013, 49, 7767–7769 RSC.
- K. Meyer, S. Bashir, J. Llorca, H. Idriss, M. Ranocchiari and J. A. van Bokhoven, Chem.–Eur. J., 2016, 22, 13894–13899 CrossRef CAS PubMed.
- J. B. Hou, M. Fang, A. J. P. Cardenas, W. J. Shaw, M. L. Helm, R. M. Bullock, J. A. S. Roberts and M. O'Hagan, Energy Environ. Sci., 2014, 7, 4013–4017 RSC.
- A. J. P. Cardenas, B. Ginovska, N. Kumar, J. B. Hou, S. Raugei, M. L. Helm, A. M. Appel, R. M. Bullock and M. O'Hagan, Angew. Chem., Int. Ed., 2016, 55, 13509–13513 CrossRef CAS PubMed.
- C. M. Klug, A. J. P. Cardenas, R. M. Bullock, M. O'Hagan and E. S. Wiedner, ACS Catal., 2018, 8, 3286–3296 CrossRef CAS.
- Z.-Y. Wu, T. Wang, Y.-S. Meng, Y. Rao, B.-W. Wang, J. Zheng, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 5953–5961 RSC.
- H. T. Lei, H. Y. Fang, Y. Z. Han, W. Z. Lai, X. F. Fu and R. Cao, ACS Catal., 2015, 5, 5145–5153 CrossRef CAS.
- P. L. Zhang, M. Wang, Y. Yang, T. Y. Yao and L. C. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- O. A. Ulloa, M. T. Huynh, C. P. Richers, J. A. Bertke, M. J. Nilges, S. Hammes-Schiffer and T. B. Rauchfuss, J. Am. Chem. Soc., 2016, 138, 9234–9245 CrossRef CAS PubMed.
- N. Elgrishi, D. A. Kurtz and J. L. Dempsey, J. Am. Chem. Soc., 2017, 139, 239–244 CrossRef CAS PubMed.
- C. Tsay and J. Y. Yang, J. Am. Chem. Soc., 2016, 138, 14174–14177 CrossRef CAS PubMed.
- R. Tatematsu, T. Inomata, T. Ozawa and H. Masuda, Angew. Chem., Int. Ed., 2016, 55, 5247–5250 CrossRef CAS PubMed.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- A. M. Appel, D. H. Pool, M. O'Hagan, W. J. Shaw, J. Y. Yang, M. R. DuBois, D. L. DuBois and R. M. Bullock, ACS Catal., 2011, 1, 777–785 CrossRef CAS.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS PubMed.
- E. S. Wiedner, J. A. S. Roberts, W. G. Dougherty, W. S. Kassel, D. L. DuBois and R. M. Bullock, Inorg. Chem., 2013, 52, 9975–9988 CrossRef CAS PubMed.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS PubMed.
- A. Rana, B. Mondal, P. Sen, S. Dey and A. Dey, Inorg. Chem., 2017, 56, 1783–1793 CrossRef CAS PubMed.
- N. D. Loewen, E. J. Thompson, M. Kagan, C. L. Banales, T. W. Myers, J. C. Fettinger and L. A. Berben, Chem. Sci., 2016, 7, 2728–2735 RSC.
- E. J. Thompson and L. A. Berben, Angew. Chem., Int. Ed., 2015, 54, 11642–11646 CrossRef CAS PubMed.
- T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed.
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
- A. Z. Haddad, B. D. Garabato, P. M. Kozlowski, R. M. Buchanan and C. A. Grapperhaus, J. Am. Chem. Soc., 2016, 138, 7844–7847 CrossRef CAS PubMed.
- K. Koshiba, K. Yamauchi and K. Sakai, Angew. Chem., Int. Ed., 2017, 56, 4247–4251 CrossRef CAS PubMed.
- A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS PubMed.
- A. Z. Haddad, D. Kumar, K. O. Sampson, A. M. Matzner, M. S. Mashuta and C. A. Grapperhaus, J. Am. Chem. Soc., 2015, 137, 9238–9241 CrossRef CAS PubMed.
- M. Natali, A. Luisa, E. Lengo and F. Scandola, Chem. Commun., 2014, 50, 1842–1844 RSC.
- S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS PubMed.
- S. Bhunia, A. Rana, P. Roy, D. J. Martin, M. L. Pegis, B. Roy and A. Dey, J. Am. Chem. Soc., 2018, 140, 9444–9457 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- Y. S. Wu, J. B. Jiang, Z. Weng, M. Y. Wang, D. L. J. Broere, Y. R. Zhong, G. W. Brudvig, Z. X. Feng and H. L. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef CAS PubMed.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS PubMed.
- V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS PubMed.
- Y. Y. Wu, N. Rodríguez-López and D. Villagrán, Chem. Sci., 2018, 9, 4689–4695 RSC.
- C. Costentin and J.-M. Savéant, J. Am. Chem. Soc., 2017, 139, 8245–8250 CrossRef CAS PubMed.
- V. Artero and J. M. Savéant, Energy Environ. Sci., 2014, 7, 3808–3814 RSC.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pécaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054–1060 CrossRef CAS PubMed.
- B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133, 18070–18073 CrossRef CAS PubMed.
- F. Möller, S. Piontek, R. G. Miller and U. P. Apfel, Chem.–Eur. J., 2018, 24, 1471–1493 CrossRef PubMed.
- J. A. S. Roberts and R. M. Bullock, Inorg. Chem., 2013, 52, 3823–3835 CrossRef CAS PubMed.
- A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem.–Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed.
- G. Robinson, C. Y. Tang, R. Köppe, A. R. Cowley and H. J. Himmel, Chem.–Eur. J., 2007, 13, 2648–2654 CrossRef CAS PubMed.
- M. O. Senge, W. W. Kalisch and I. Bischoff, Chem.–Eur. J., 2000, 6, 2721–2738 CrossRef CAS.
- B. H. Solis, A. G. Maher, D. K. Dogutan, D. G. Nocera and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 485–492 CrossRef CAS PubMed.
- A. G. DiPasquale and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 1812–1813 CrossRef CAS PubMed.
- J. B. Jiang, K. L. Materna, S. Hedström, K. R. Yang, R. H. Crabtree, V. S. Batista and G. W. Brudvig, Angew. Chem., Int. Ed., 2017, 56, 9111–9115 CrossRef CAS PubMed.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- R. H. Morris, Chem. Rev., 2016, 116, 8588–8654 CrossRef CAS PubMed.
- APEX2 v2009, Bruker AXS, Madison, WI, 2009 Search PubMed.
- G. M. Sheldrick, SADABS, 2008/1, University of Göttingen, Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 1990, A46, 467–473 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data as CIF files; Fig. S1–S34; and Table S1. CCDC 1855035–1855038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05247f |
| This journal is © The Royal Society of Chemistry 2019 |