
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFatty acid based transient nanostructures for temporal regulation of artificial peroxidase activity†
Sahnawaz
Ahmed
,
Ayan
Chatterjee
,
Krishnendu
Das
and
Dibyendu
Das
 *
*
Department of Chemical Sciences, Centre for Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, India. E-mail: dasd@iiserkol.ac.in
First published on 1st July 2019
Abstract
Natural systems access transient high energy self-assembled structures for temporal regulation of different biological functions through dissipative processes. Compartmentalization within self-assembled structures is used by living systems to organize vital biochemical reactions that define cellular metabolism. Herein, we demonstrate a simple fatty acid based system where a redox active base (dimethylaminomethyl ferrocene, Fc-NMe2) acts as a countercation to access unique hexagonal compartments resulting in the formation of a self-supporting gel. An oxidizing environment helps in the dissipation of energy by converting Fc-NMe2 to oxidized waste and the gel autonomously undergoes transition to a sol. Hence, the system requires the addition of the fuel Fc-NMe2 to access the temporal gel state. Notably, these transient compartments were able to temporally upregulate and downregulate hemin-catalyzed oxidation reactions mimicking peroxidase, a ubiquitous enzyme in extant biology. An order of magnitude variation in kcat values was observed with time and the chemical reaction persists as long as the gel state was present.
Introduction
Natural systems have exceptional capabilities to access transient non-equilibrium nanostructures by energy consuming processes.1–4 Through remarkable spatiotemporal control over the size and conformation in the nanoscale domain, biological systems access these high-energy transient morphologies which are utilized to perform work. For instance, vital functions such as cell motility are achieved by transient length changes of microtubules and actin filaments in the presence of nucleoside triphosphates as a chemical fuel.5,6 Elegant strategies to access out of equilibrium artificial synthetic systems have utilized energy from light or chemical energy stored in high-energy, kinetically stable molecules which are termed as chemical fuels.7–27 As such, there is mounting interest to develop synthetic transient assemblies that are capable of displaying temporal control of functions foreshadowing the pathways followed by living systems. To this end, we were interested to develop fuel driven temporal generation of compartments or confinements which are capable of controlling the chemical reactivity of another reaction. Nature uses the strategy of compartmentalization to organize essential chemical reactions that define cellular metabolism.28 Many of these reactions if performed in a single environment without compartments would produce futile chemical cycles, undesired by-products, or suffer from unfavourable thermodynamics from the bulk environment.29 Compartments are also used to store and regulate enzymes in extant biology.30 Interestingly, reports have argued the importance of transient compartmentalization in early Earth to protect RNA replicators from parasitic mutants.31 We asked whether a transient compartment of natural fatty acids can be achieved under dissipative conditions and if the transient changes of the microenvironment would lead to temporal control of the entrapped guest, as seen in organelles in living systems.Results and discussion
To achieve the transient formation of compartments, the challenge is to find a subtle balance of activation–deactivation rates, where the rate of the reaction which will create the proposed compartment should be faster than the deactivation reaction leading to its degradation.7–9 For the fast activation step, we envisaged to use a redox responsive metastable base which can react with the fatty acid to form a salt that undergoes thermodynamically driven assembly (Fig. 1). For the slow deactivation step, experimental conditions can be set up which promote dissipation by gradual oxidation of the countercation, leading to transient accumulation of the proposed compartment as shown in the reaction scheme (Fig. 1a and b and the representative SEM image of the fibre like morphology is shown in Fig. 1c). Thus the redox responsive base will work as the fuel to drive the transient assembly. To test our hypothesis, we mixed stearic acid (C18A) and dimethylaminomethyl ferrocene (Fc-NMe2) at varying molar ratio in DMSO/water (10%, v/v). Although C18A and Fc-NMe2 are separately not soluble in 10% v/v DMSO/water, their mixtures dissolved in molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75, 1:1 and 1:1.25 (C18A:Fc-NMe2), suggesting colloidal stability of the organic salt. The mixtures with molar ratios of ≈ 1:1 gradually turned more viscous suggesting assembly (see the ESI† for details). Notably, when we increased the concentration of the acid and counterion (ca. 210 mM, 1:1), rapid gelation was observed within 25 s with viscoelastic behavior (Fig. S1–S3, ESI†). Since ferrocene is known to acutely respond to changes in the redox environment, to design the deactivation step, the acid–base reaction was done under conditions promoting its oxidation (Fig. 1). Orthogonal routes of activation (salt formation) and deactivation (oxidation of the counterion) would give us the opportunity to manipulate the kinetics of the competing reactions, leading to accumulation of the transient assemblies under dissipative conditions (Fig. 1).7–9,22 Clearly, since both the acid–base neutralization and subsequent gelation are fast steps, the transient accumulation of the assembled structures will be determined by the rate limiting oxidation of the cation by the environment. In order to find out the suitable rate of deactivation, the acid and base were mixed in a 1:1 ratio (245 mM each) in the presence of varying concentrations of Fe(ClO4)3, a common oxidizing agent for ferrocene (Fig. S4, ESI†). The pH of the system was measured to be in the range of 8.1 to 8.3. From 2 mM to 12.5 mM, gels were formed but interestingly showed autonomous disassembly (Fig. 1e–j). Due to faster oxidation in the presence of a higher amount of the oxidant, the lifetimes of these transient gels showed a gradual decrease from ca. 24 h to 2 h (Fig. S4, ESI†). Also, the time required for gel formation increased with the increase in the concentration of the oxidant, from about a minute to up to ca. 16 min (Fig. S4, ESI†). With the increase of the oxidant concentration, the oxidation of the fuel and thus the deactivation kinetics, i.e. the disassembly process, are expedited as waste is generated before the formation of the organic salt. This eventually retards the gelation process as reflected from slow gelation. Finally, when the concentration of the oxidant reaches 15 mM or beyond, no gelation was observed. Rheology done in the presence of different oxidant concentrations showed a decrease of the storage modulus (G′) with an increase in the Fe(ClO4)3 concentration (Fig. S5, ESI†).
:0.75, 1:1 and 1:1.25 (C18A:Fc-NMe2), suggesting colloidal stability of the organic salt. The mixtures with molar ratios of ≈ 1:1 gradually turned more viscous suggesting assembly (see the ESI† for details). Notably, when we increased the concentration of the acid and counterion (ca. 210 mM, 1:1), rapid gelation was observed within 25 s with viscoelastic behavior (Fig. S1–S3, ESI†). Since ferrocene is known to acutely respond to changes in the redox environment, to design the deactivation step, the acid–base reaction was done under conditions promoting its oxidation (Fig. 1). Orthogonal routes of activation (salt formation) and deactivation (oxidation of the counterion) would give us the opportunity to manipulate the kinetics of the competing reactions, leading to accumulation of the transient assemblies under dissipative conditions (Fig. 1).7–9,22 Clearly, since both the acid–base neutralization and subsequent gelation are fast steps, the transient accumulation of the assembled structures will be determined by the rate limiting oxidation of the cation by the environment. In order to find out the suitable rate of deactivation, the acid and base were mixed in a 1:1 ratio (245 mM each) in the presence of varying concentrations of Fe(ClO4)3, a common oxidizing agent for ferrocene (Fig. S4, ESI†). The pH of the system was measured to be in the range of 8.1 to 8.3. From 2 mM to 12.5 mM, gels were formed but interestingly showed autonomous disassembly (Fig. 1e–j). Due to faster oxidation in the presence of a higher amount of the oxidant, the lifetimes of these transient gels showed a gradual decrease from ca. 24 h to 2 h (Fig. S4, ESI†). Also, the time required for gel formation increased with the increase in the concentration of the oxidant, from about a minute to up to ca. 16 min (Fig. S4, ESI†). With the increase of the oxidant concentration, the oxidation of the fuel and thus the deactivation kinetics, i.e. the disassembly process, are expedited as waste is generated before the formation of the organic salt. This eventually retards the gelation process as reflected from slow gelation. Finally, when the concentration of the oxidant reaches 15 mM or beyond, no gelation was observed. Rheology done in the presence of different oxidant concentrations showed a decrease of the storage modulus (G′) with an increase in the Fe(ClO4)3 concentration (Fig. S5, ESI†).
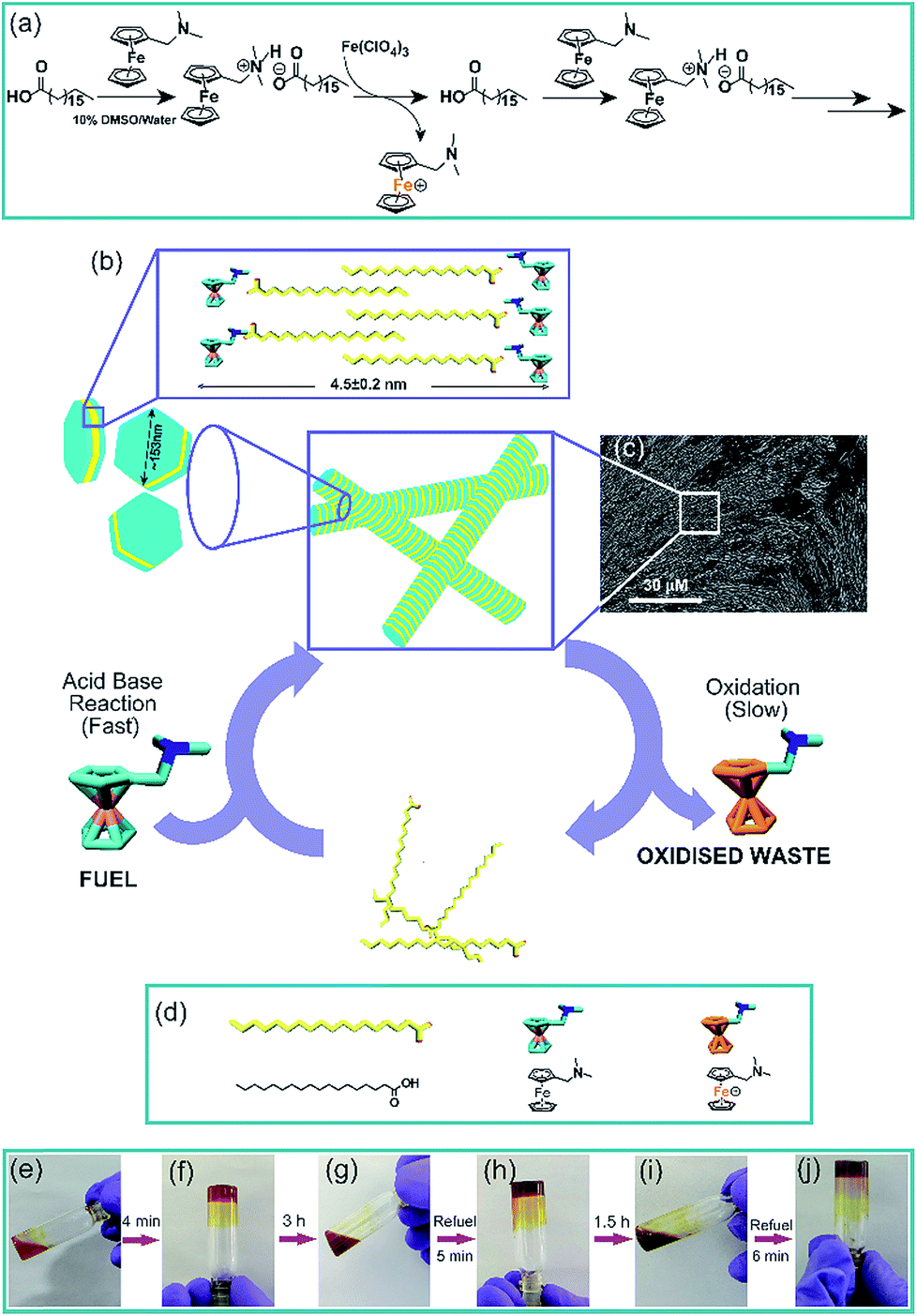 | ||
| Fig. 1 (a) Reaction scheme used in the present work and (b) schematic representation of a typical reaction cycle. Fuel driven transient assembly is shown along with the model of packing of interdigitated fatty acids, predicted from atomic force microscopy (AFM). The average of height profiles obtained from 15 randomly chosen hexagon like morphologies is calculated as 4.5 ± 0.2 nm. The average size of the hexagon is about 153 nm (distance between the apexes) as obtained from transmission electron microscopy (TEM). (c) SEM image of the transient assembly showing fibre like morphology at 30 min, (d) chemical structures of the molecules involved and (e–j) representative images of temporal sol–gel–sol transition cycles prepared with C18A (245 mM) with Fc-NMe2 (245 mM) and Fe(ClO4)3 (10 mM) in 10% (v/v) DMSO/water. | ||
We were intrigued to investigate the morphology of the transient fatty acid assemblies directed in the presence of an aromatic countercation. Transmission electron microscopy (TEM) performed at different time intervals revealed distinct hexagonal morphologies at t ≈ 0 h with an average diameter of ca. 153 nm (Fig. 2a, histograms of distance between the apexes in Fig. 2d). Interestingly, TEM done at t = 3 h showed deformed hexagons along with few intact hexagonal structures, underpinning the transient nature of the assemblies (Fig. 2b). Scanning electron microscopy (SEM) also showed similar morphologies of stacks of hexagonal nanostructures at t = 0 h (Fig. 2g and S6, ESI†) and autonomous deformation at t = 3 h (Fig. 2e and f). Atomic force microscopy (AFM, Fig. 2h and i) at t ≈ 0 h showed uniform height profiles in the range of 4.5 ± 0.2 nm, indicating the interdigitation of two fatty acid chains (Fig. 1b). These hexagons formed from the mixing of Fc-NMe2 and C18A acid underwent further higher order assembly leading to the formation of fibre like intertwined structures (Fig. 1c and S6, ESI†).
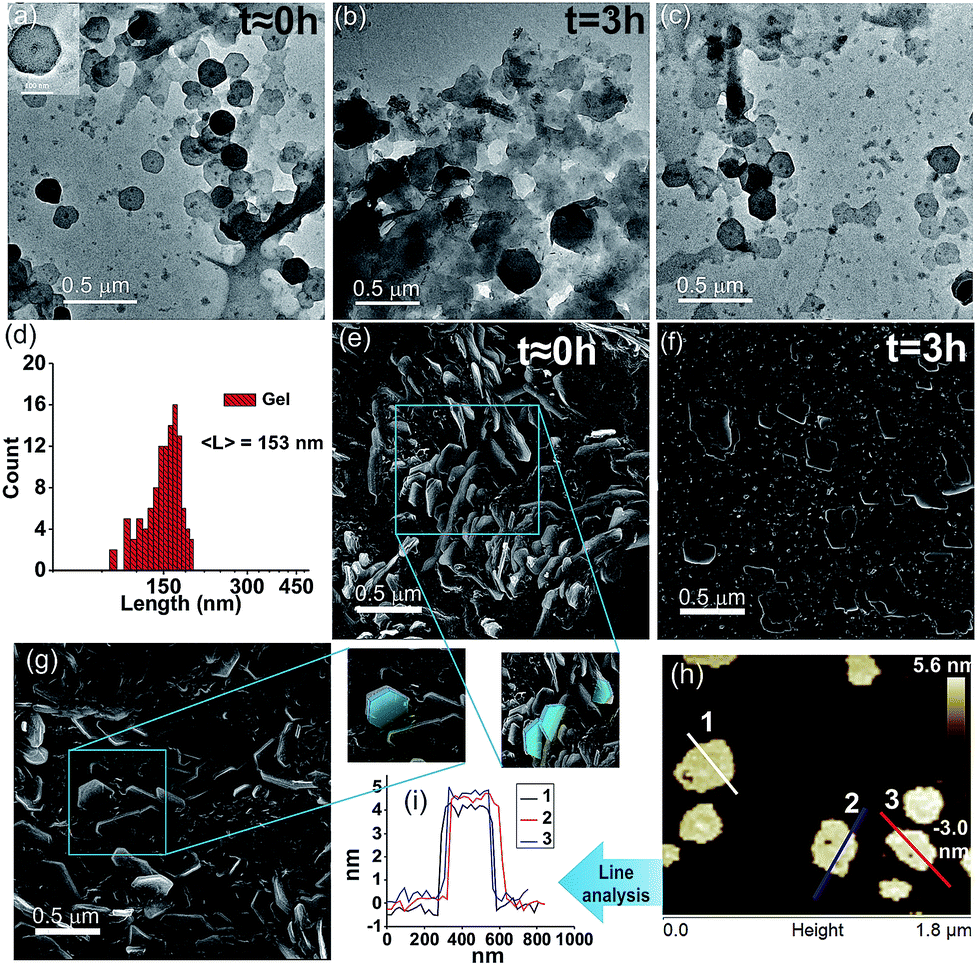 | ||
| Fig. 2 TEM images of (a) C18A–Fc-NMe2 for t ≈ 0 h gel (inset: enlarged view), (b) t = 3 h sol, and (c) refueled gel (4 mM Fc-NMe2). (d) Histogram of hexagon dimensions from the TEM results at t ≈ 0 h. SEM images (e) at t ≈ 0 h with blue traces showing the hexagonal structures, (f) t = 3 h sol, (g) refueled gel (5 min), and (h) AFM image at t ≈ 0 h and (i) the corresponding height profiles. | ||
We monitored the rate of conversion of Fc-NMe2 to the oxidized waste Fc(+)-NMe2 by HPLC (Fig. 3a and b, with 25 times diluted samples, see the ESI† for details). A gradual decrease in the Fc-NMe2 area was observed with time. At t = 3 h, when the gel melts autonomously, the extent of decrease corresponded to 6 mM Fc-NMe2 (calculated from the standard plot, Fig. S7, ESI†), thus suggesting the presence of 239 mM Fc-NMe2. Despite the presence of a substantially larger amount (239 mM) of residual Fc-NMe2, the collapse of the gel is in contrast to the finding that 210 mM was sufficient for gelation in the absence of an oxidising environment. Hence, these results underpin the role of oxidation from the assembled state. As expected, when we added a 1:1 mixture of C18A–Fc(+)-NMe2, no gel formation was observed. To find out the role of self-assembled state, HPLC of the C3A salt of Fc-NMe2 was done. C3A–Fc-NMe2 did not show any viscous behavior and did not assemble (TEM, Fig. S8, ESI†). Intriguingly, the consumption after 60 min in the case of C3A–Fc-NMe2 salt was ca. 9.2 mM while it was only 2.9 mM for C18A–Fc-NMe2 (Fig. 3a). These data suggested that the formation of the assembled hexagons reduces the kinetics of oxidation and increases the lifetime of the gel. To further understand the role of the transient assembled state, we simulated the disassembly by premixing different ratios of waste Fc(+)-NMe2 with the fuel Fc-NMe2, keeping the total concentration of Fc(+)-NMe2 and Fc-NMe2 fixed at 245 mM (Fig. 3c and S9, ESI†). These were added to C18A in the absence of the oxidizing agent. The concentration of Fc(+)-NMe2 was kept from 0–15 mM as suggested from HPLC. As expected, only viscous liquids (VLs) or free flowing solutions were observed. This result indicated that the instability of the assembled structures arises due to the presence of oxidized cations as the ion pair of C18A. Interestingly, when the same experiment was carried out by adding the oxidized waste Fc(+)-NMe2 after the formation of the gel of C18A and Fc-NMe2, the gels remained intact in the entire concentration range (Fig. 3c). Hence, when the assembled structures are already formed, the oxidized waste is unable to exchange with the fuel to be an ion pair of C18A. This result along with the slower oxidation kinetics for the assembled state (Fig. 3a), underpinned the fact that the oxidative conversion of the fuel to waste occurs from the assembled state in the C18A–Fc-NMe2 system which subsequently results in instability.7–9 This was further supported from SEM images where ordered structures were seen when 5 mM of Fc(+)-NMe2 (Fig. 3d) was added after assembly. In contrast, when the same amount of Fc(+)-NMe2 was added before assembly, less ordered structures were seen (Fig. 3e). Since the fatty acid exhibited fidelity towards the fuel over the oxidized waste for assembly (Fig. 3c and S9, ESI†), we expected the system to display multiple sol to gel to sol cycles upon refuelling. Indeed, when Fc-NMe2 (4 mM) was added again to a representative system (245 mM, 1:1 with 10 mM oxidant), reformation of the gel was observed (Fig. 1g–j). Remarkably, TEM and SEM showed the reappearance of hexagonal nanostructures (Fig. 2c and g, histogram in Fig. S10, ESI†). The pH remained in the range of pH 8.3 ± 0.1. The sol–gel–sol transition could be performed three times before the accumulation of waste and consumption of the oxidant resulted in no further gelation. This autonomous sol–gel–sol transition could also be seen from the viscoelastic behaviour. The samples with different ages and with refuelling batches were subjected to rheology which displayed temporal changes in the storage modulus, G′ (Fig. 3f).
 | ||
| Fig. 3 (a) Comparison of the consumption kinetics of Fc-NMe2 in salts with C18A and C3A, (b) representative HPLC chromatogram of the consumption of Fc-NMe2 with C18A at t ≈ 0 and t = 4 h, and (c) effect of the addition of Fc(+)-NMe2 in varying ratios to the pre-assembled and post-assembled mixture of C18A and Fc-NMe2 (total concentration of Fc-NMe2 and Fc(+)-NMe2 kept constant at 245 mM). Representative SEM images of (d) gel state when oxidized waste (5 mM) was added after assembly and (e) when oxidized waste (5 mM) was added before assembly. (f) Time resolved rheology of gel–sol–gel cycles. The red arrow shows the refueling step with Fc-NMe2 (4 mM). The error bars are the result of triplicate measurements. | ||
The temporal existence of the hexagonal fatty acid assemblies prompted us to investigate their capability of entrapping small molecules and hosting guests. Fluorophore pyrene suffered rapid quenching when added to the gel of C18A–Fc-NMe2 (Fig. S11, ESI†) due to the known capability of ferrocene to quench pyrene's fluorescence.32,33 Interestingly, the extent of quenching was substantially less for the C3A–Fc-NMe2 system which does not assemble, suggesting closer packing and entrapment in the case of the assembled structures (Fig. S11, ESI†). We asked whether these hexagonal compartments can entrap hemin, the prosthetic group of extant peroxidases and metalloproteins, to mimic and possibly regulate temporally the artificial peroxidase activity.32–34
Temporal enzyme regulation through compartmentalization is a feature seen in extant biology.30,34–36 To investigate the interaction of hemin in transient systems, UV-vis spectroscopy was employed (Fig. 4a). In 10% DMSO/water, free hemin showed a Soret band at 392 nm and a shoulder at 365 nm along with a low intensity band at around 610 nm, indicating the presence hemin dimers connected through μ-oxo bridges.35,37 In the C18A–Fc-NMe2 gel at t = 0 h, the Soret band red shifted to 401 nm indicating the existence of monomeric hemin and thus supporting the entrapment of hemin.35,37 In the sol state at t = 3 h, hemin still retained the Soret peak at 401 nm (Fig. 4a).
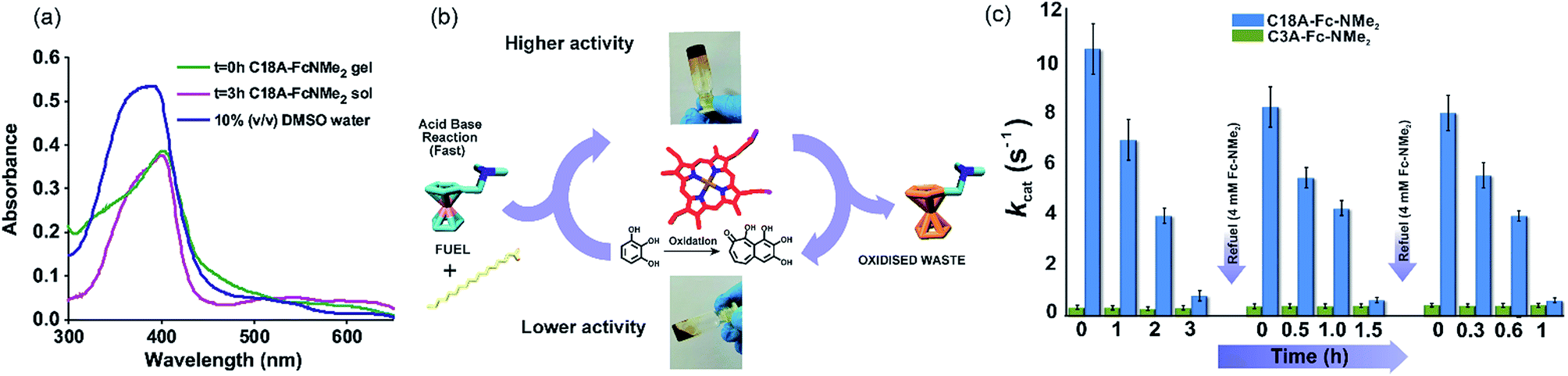 | ||
| Fig. 4 (a) UV-vis spectra of free hemin and hemin entrapped C18A–Fc-NMe2 gel at ≈0 h and 3 h. All samples had histidine (1 mM). (b) Schematic representation of the temporal regulation of artificial peroxidase activity of hemin entrapped in the transient system. (c) kcat values of hemin (0.1 mM) catalyzed oxidation of pyrogallol (varying concentration) by H2O2 (30 mM) in the presence of 1 mM histidine for C18A–Fc-NMe2 and C3A–Fc-NMe2 systems. The error bars are the result of triplicate measurements. | ||
For the investigation of hemin activity, pyrogallol was used as a standard substrate in the presence of 1 mM histidine and H2O2 as the oxidant.35 Intriguingly, hemin mixed with the C18A–Fc-NMe2 gel showed temporal changes in the oxidation rates, and displayed autonomous upregulation and downregulation of peroxidase like activity (Fig. 4b and c and the ESI† for details). The upregulated activity at t ≈ 0 h (kcat = 10.6 ± 1 s−1, Fig. S12, Table S1, ESI†) was significantly higher than the activity of free hemin.35 This activity showed autonomous decrease and dropped to kcat = 0.84 ± 0.2 in 3 h (Fig. S13, Table S1, ESI†). Specific constants were found to decrease from 0.35 ± 0.03 in the gel state to 0.19 ± 0.02 mM−1 s−1 in the sol state (Table S1, ESI†). The reason behind the temporal increase of the kcat values can be attributed to the transient formation of amphiphilic self-assembled nanostructures. Nanostructured gels have been previously shown to activate hemin and proteins such as peroxidases and hydrolases.34,35,38–40 On autonomous gel to sol transformation, the density of amphiphilic nanostructures decreased, resulting in less activity in the sol state (Fig. 2b and f).34,35,38–40 Although, the temporal upregulation and downregulation of activity showed a modest range of an order of magnitude in terms of kcat values, this result underpins the subtle but significant role of the transiently assembled compartments in the artificial peroxidase activity. Furthermore, after each refueling, the kcat values showed two spikes of 8.3 ± 0.8 and 8.06 ± 0.7 s−1 respectively (Fig. 4c and S12, ESI†). Interestingly, the specific constants after refueling also registered a recovery up to 0.32 ± 0.04 mM−1 s−1 (Table S1, ESI†).The kcat values subsequently decrease again to 0.67 ± 0.1 and 0.65 ± 0.1 s−1 within 1.5 and 1 h respectively (Fig. 4c). Controls studied with transient gels in the absence of H2O2 but in the presence of Fe(ClO4)3 showed formation of miniscule amounts of oxidation products under the experimental conditions. This suggested that H2O2 is the only oxidant under the experimental conditions. Furthermore, the control system of C3A–Fc-NMe2 studied in this range did not show such temporal variation of activity (Fig. 4c and Table S1, ESI†). The pH increase of ∼0.1 units after each refueling step (4 mM Fc-NMe2) did not have an impact on the activity of hemin in the assembled systems.
Conclusions
In conclusion, we have developed fatty acid based transient hexagonal nanostructured assemblies which dissipate energy from the conversion of chemical fuels to oxidized waste. In an oxidizing environment, the high-energy self-supporting gel state is stable only when the fuel is supplied. Furthermore, these hexagonal nanostructures with transient lifetimes were able to host hemin and temporally upregulate and downregulate the peroxidase mimicking activity up to an order of magnitude, thus foreshadowing the compartmentalized biochemical processes seen in extant biology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DD is thankful to the SERB (EMR/2017/005126), DST, GOI, for financial assistance. SA is thankful to the NPDF (PDF/2017/000782), SERB, AC is thankful to the CSIR and KD is thankful to the Nano Mission (SR/NM/NS-1082/2015) for fellowships. The authors gratefully acknowledge Dr S. Roy and Pooja Sharma for their help.Notes and references
- J. H. van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC
.
- B. A. Grzybowski and W. T. S. Huck, Nat. Nanotechnol., 2016, 11, 585–592 CrossRef CAS PubMed
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed
- H. Hess and J. L. Ross, Chem. Soc. Rev., 2017, 46, 5570–5587 RSC
- M. Caplow and J. Shanks, J. Biol. Chem., 1990, 265, 8935–8941 CAS
- R. D. Astumian, Nat. Nanotechnol., 2012, 7, 684–688 CrossRef CAS PubMed
- C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491–5507 RSC
- C. Cheng, P. R. McGonigal, J. F. Stoddart and R. D. Astumian, ACS Nano, 2015, 9, 8672–8688 CrossRef CAS PubMed
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS
- S. De and R. Klajn, Adv. Mater., 2018, 30, 1706750 CrossRef PubMed
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82–88 CrossRef CAS PubMed
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC
- J. K. Sahoo, C. G. Pappas, I. R. Sasselli, Y. M. Abul-Haija and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 6828–6832 CrossRef CAS PubMed
- J. Boekhoven, A. Brizard, K. Kowlgi, G. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed
- N. Singh, M. P. Conte, R. V. Ulijn, J. F. Miravet and B. Escuder, Chem. Commun., 2015, 51, 13213–13216 RSC
- J. Boekhoven, W. Hendriksen, G. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed
- P. S. Muñana, G. Ragazzon, J. Dupont, C. Z.-J. Ren, L. J. Prins and J. L.-Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 16469–16474 CrossRef PubMed
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS PubMed
- S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS PubMed
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. L. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed
- J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef CAS PubMed
- S. Dhiman, A. Jain and S. J. George, Angew.
Chem., Int. Ed., 2017, 56, 1329–1333 CrossRef CAS PubMed
- S. Dhiman, A. Jain, M. Kumar and S. J. George, J. Am. Chem. Soc., 2017, 139, 16568–16575 CrossRef CAS PubMed
- A. Sorrenti, J. Leira-Iglesias, A. Sato and T. M. Hermans, Nat. Commun., 2017, 8, 15899 CrossRef CAS PubMed
- D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56, 15461–15465 CrossRef CAS PubMed
- J. Leira-Iglesias, A. Tassoni, T. Adachi, M. Stich and T. M. Hermans, Nat. Nanotechnol., 2018, 13, 1021–1027 CrossRef CAS PubMed
- F. Hinzpeter, U. Gerland and F. Tostevin, Biophys. J., 2017, 112, 767–779 CrossRef CAS PubMed
- R. P. Goodman, S. E. Calvo and V. K. Mootha, J. Biol. Chem., 2018, 293, 7508–7516 CrossRef CAS PubMed
- M. S. Cuesta, S. A. Rahman, N. Furnham and J. M. Thornton, Biophys. J., 2015, 109, 1082–1086 CrossRef PubMed
- S. Matsumura, Á. Kun, M. Ryckelynck, F. Coldren, A. Szilágyi, F. Jossinet, C. Rick, P. Nghe, E. Szathmáry and A. D. Griffiths, Science, 2016, 354, 1293–1296 CrossRef CAS PubMed
- S. Basak, I. Singh, A. Ferranco, J. Syed and H. B. Kraatz, Angew. Chem., Int. Ed., 2017, 56, 13288–13292 CrossRef CAS PubMed
- Q. Wang, Z. Yang, L. Wang, M. Ma and B. Xu, Chem. Commun., 2007, 1032–1034 RSC
- Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS PubMed
- P. T. T. Nguyen, A. J. Conley, J. Sneyd, R. S. F. Lee, T. K. Soboleva and P. R. Shorten, J. Theor. Biol., 2013, 332, 52–64 CrossRef CAS PubMed
- P. K. Shantha, G. S. S. Saini, H. H. Thanga and A. L. Verma, J. Raman Spectrosc., 2001, 32, 159 CrossRef CAS
- Q. Liu, H. Wang, X. H. Shi, Z. G. Wang and B. Q. Ding, ACS Nano, 2017, 11, 7251–7258 CrossRef CAS PubMed
- T. Bhattacharyya, Y. P. Kumar and J. Dash, ACS Biomater. Sci. Eng., 2017, 3, 2358–2365 CrossRef CAS
- N. Kapil, A. Singh and D. Das, Angew. Chem., Int. Ed., 2015, 54, 6492 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02648g |
| This journal is © The Royal Society of Chemistry 2019 |