
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiplexed blood–brain barrier organ-on-chip†
M.
Zakharova
 *a,
M. A.
Palma do Carmo
a,
M. W.
van der Helm
a,
H.
Le-The
ab,
M. N. S.
de Graaf
c,
V.
Orlova
c,
A.
van den Berg
a,
A. D.
van der Meer
d,
K.
Broersen
d and
L. I.
Segerink
a
*a,
M. A.
Palma do Carmo
a,
M. W.
van der Helm
a,
H.
Le-The
ab,
M. N. S.
de Graaf
c,
V.
Orlova
c,
A.
van den Berg
a,
A. D.
van der Meer
d,
K.
Broersen
d and
L. I.
Segerink
a
aBIOS Lab on a Chip group, MESA+ Institute for Nanotechnology, Technical Medical Centre, Max Planck Institute for Complex Fluid Dynamics, University of Twente, The Netherlands. E-mail: m.zakharova@utwente.nl
bPhysics of Fluids, MESA+ Institute for Nanotechnology, Max Planck Institute for Complex Fluid Dynamics, University of Twente, The Netherlands
cDepartment of Anatomy and Embryology, Leiden University Medical Center, Leiden, The Netherlands
dApplied Stem Cell Technologies, Technical Medical Centre, University of Twente, The Netherlands
First published on 27th July 2020
Abstract
Organ-on-chip devices are intensively studied in academia and industry due to their high potential in pharmaceutical and biomedical applications. However, most of the existing organ-on-chip models focus on proof of concept of individual functional units without the possibility of testing multiple experimental stimuli in parallel. Here we developed a polydimethylsiloxane (PDMS) multiplexed chip with eight parallel channels branching from a common access port through which all eight channels can be addressed simultaneously without the need for extra pipetting steps thus increasing the reproducibility of the experimental results. At the same time, eight outlets provide individual entry to each channel with the opportunity to create eight different experimental conditions. A multiplexed chip can be assembled as a one-layer device for studying monocultures or as a two-layer device for studying barrier tissue functions. For a two-layer device, a ∼2 μm thick transparent PDMS membrane with 5 μm through-hole pores was fabricated in-house using a soft lithography technique, thereby allowing visual inspection of the cell-culture in real-time. The functionality of the chip was studied by recapitulating the blood–brain barrier. For this, human cerebral microvascular endothelial cells (hCMEC/D3) were cultured in mono- or coculture with human astrocytes. Immunostaining revealed a cellular monolayer with the expression of tight junction ZO-1 and adherence junction VE-cadherin proteins in endothelial cells as well as glial fibrillary acidic protein (GFAP) expression in astrocytes. Furthermore, multiplexed permeability studies of molecule passage through the cellular barrier exhibited expected high permeability coefficients for smaller molecules (4 kDa FITC–dextran) whereas larger molecules (20 kDa) crossed the barrier at a lower rate. With these results, we show that our device can be used as an organ-on-chip model for future multiplexed drug testing.
Introduction
There are different tissue barriers in our body that regulate the transport of molecules.1–4 The blood-tissue barriers, for example, play a vital role in regulating drug transport from the capillaries to the targeted organs.5 To exert functional activity, these capillaries typically consist of an endothelial cell layer and a basement membrane. More detailed, architectural, and organizational characteristics depend on organ-specific features giving rise to blood–tissue barriers variation from organ to organ.1 For instance, the capillaries in the central nervous system (CNS) are characterized by a monolayer of endothelial cells that show high expression levels of tight junction (TJ) proteins restricting the transport of various molecules and solutes.6 In addition, these capillaries are closely connected with astrocytes and pericytes that play an important role in blood–brain barrier (BBB) function, a structure that regulates the exchange of ions and nutrients between the blood and brain. The characteristic structure of the capillaries protects the brain from toxic substances and pathogens. At the same time, the transport of therapeutic compounds across the BBB is also restricted, requiring an advanced understanding of BBB transport mechanisms to effectively target the brain. For this, in vivo and in vitro models are developed that recapitulate the barrier organization and provide the ability to evaluate drug permeability. However, there is still a lack of appropriate models that can adequately resemble a complex human brain environment.In vivo studies of the BBB, performed for example in mouse models, have provided physiologically relevant information on BBB transport mechanisms within the context of the brain microenvironment.7 However, the use of animal models in lab experiments is costly and subject to ethical concerns. Moreover, drugs that were originally approved based on animal models often fail in human trials.8 In response to this, in vitro BBB models were developed which typically consist of primary cell cultures or cell lines cultured on extracellular matrix (ECM) coated filters in a transwell system.9 To date, different transwell-based BBB models have been introduced, employing a monoculture of endothelial cells or co-culture with other BBB-associated cell types.10 Even though such models reproduce some aspects of BBB physiology and allow fast and standardized evaluation of potential drugs, such cell-based static models do not reproduce some of the more complex aspects of human brain physiology.11
Recently, the urgent need for a less expensive and physiologically relevant model has led to the development of organ-on-chip (OOC) systems based on microfluidic techniques.12,13 OOC models resemble some components of the complex in vivo environment with an ECM, cell–cell interactions, and vasculature-like perfusion while requiring a minimal quantity of fluid and sample consumption due to micrometer size of the channels.14 It is possible to implement miniaturized sensors into the devices enabling real-time monitoring of tissue development and function.15 Furthermore, OOC has the potential to evaluate personalized medicine strategies when cells from a specific donor are used.16,17 Over the last decade, researchers have been able to fabricate chips for studying the BBB, and other organs demonstrating the ability of OOC to capture physiologically relevant conditions in vitro.18–22
To date, most reported microfluidic models of the BBB are fabricated using the biocompatible polydimethylsiloxane (PDMS) organic polymer. The permeability to CO2 and O2 of this polymer makes it ideal for cell studies.23 Cells from porcine, bovine and rodent origin are used in several of the reported BBB models19 in which blood endothelial cells are usually seeded in one compartment, mimicking the luminal side of the BBB, while astrocytes and pericytes are placed in a separate compartment, resembling the basal side of the BBB.10 The integrity of the barrier created inside the microfluidic device is usually confirmed by fluorescent imaging, permeability studies, or by means of trans endothelial electrical resistance (TEER) measurements.24–26
Geometry, design and functionality vary for the different in vitro models of the BBB.18,27 Depending on the research question the most appropriate design can be selected. Generally, vasculature formation and cell–cell interaction studies make use of ECM-based 3D models as they recapitulate many relevant physiological aspects of these systems.28 However specialized equipment is required to image cultured cells and the assessment of barrier formation with TEER is not possible due to 3D chip design. Alternative models consist of several channels placed in the same plane and horizontally connected through a hydrogel or microgrooves. Such models recreate the 3D structured microvessel and can be used to study cell–cell interaction, cell migration, and the diffusion of small molecules.29,30 However, this design limits the area available for visual investigation of cell–cell contact which occurs horizontally. Moreover, in the chip with micro grooves, the spacing between the two co-cultures is 50–100 μm which is not physiologically relevant.30 This limitation is circumvented in the most commonly used chip design consisting of two channels stacked on top of each other and separated by a porous membrane.31,32 In this way, a sufficient culture area is provided for two or more cell cultures in close proximity to each other. This design was previously used for mimicking the BBB as well as other biological barriers such as gastrointestinal tract, skin, liver, lung. The imaging quality can be improved by selecting a transparent membrane material.
Despite a variety of in vitro BBB models available, most of the studies using these models are focused on functional validation or toxicity screening using individual systems with low throughput workflows while being unable to test different experimental stimuli in parallel, thereby limiting readout possibilities. Parallelization and automation with higher throughput testing of variables are required to implement OOC models in preclinical studies.
Here, we present a micro-engineered multiplexed platform with eight functional units branching from the common inlet and having eight separate outlets. We show two variants of the multiplexed platform (1) a one-layer device, and (2), a two-layer device with a 2 μm thick PDMS through-hole membrane separating the two compartments. Both designs give the possibility to address all channels in each compartment simultaneously or individually by utilizing the effect of laminar flow, thus, allowing eight parallel experiments in a single chip.
Materials and methods
Microdevice design
The multiplexed chip consists of eight parallel channels, 500 μm (width) × 50 μm (height), which branch from a common inlet, and have separate access ports (Fig. 1A). The common inlet allows simultaneous filling of all channels and is placed at the exact same distance from each channel assuring an even distribution of the flow and cells through the channels.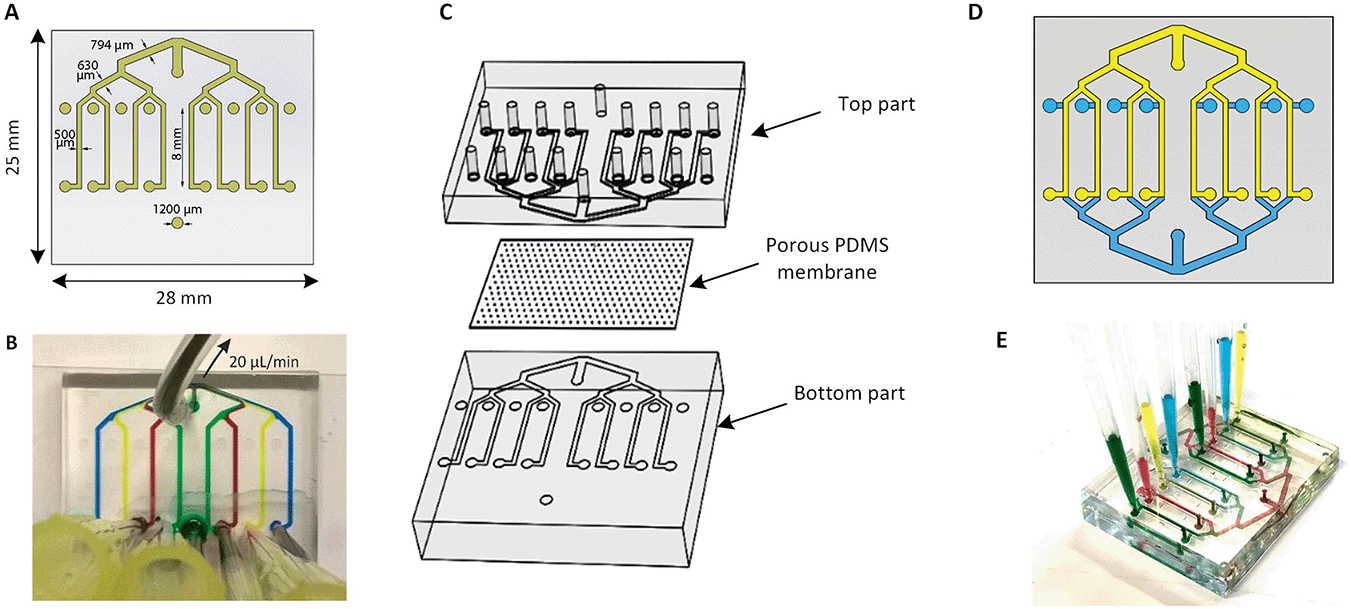 | ||
| Fig. 1 Design of one- and two-layer multiplexed chip. (A) Design of the photomask of the multiplexed chip with eight parallel channels branching from the common inlet and having separate access ports. (B) Results of creating eight different conditions in a one-layer device by inserting food dyes at each outlet. (C) Exploded view of the chip which consists of top and bottom parts with eight channels each and separated by porous PDMS membrane (D) scheme of two-layer device design. Two identical PDMS parts of which one inverted and bonded to another part (E) results of creating eight different conditions in two-layer devices. | ||
The separate outlets of eight channels, on the other hand, make them individually addressable thus allowing the performance of eight different experimental conditions at the same time by pulling liquids from eight separate reservoirs through the channels towards the common access port (Fig. 1B). In addition, the width of the channels after each junction increases consecutively to 630 μm, 794 μm and reaches 1000 μm at the inlet followed by Murray's law.33
The microchannels were fabricated using standard soft lithography.34 Briefly, the microfluidic device was made by casting PDMS prepolymer and curing agent (Sylgard 184 Silicone elastomer kit, Dow Corning) at a weight ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 onto a patterned SU-8 mold. The polymer was cured overnight at 60 °C and subsequently peeled off from the mold. The thickness of the chip was 3 mm. Next, inlets and outlets were punched using a 1 mm diameter biopsy puncher. Subsequently, the PDMS layer was bonded to the glass by exposure to the oxygen plasma (50 W, Cute, Femto Science) for 1 min. The assembly was heated at 60 °C for at least 20 min to improve the bonding between the PDMS and glass.
:1 onto a patterned SU-8 mold. The polymer was cured overnight at 60 °C and subsequently peeled off from the mold. The thickness of the chip was 3 mm. Next, inlets and outlets were punched using a 1 mm diameter biopsy puncher. Subsequently, the PDMS layer was bonded to the glass by exposure to the oxygen plasma (50 W, Cute, Femto Science) for 1 min. The assembly was heated at 60 °C for at least 20 min to improve the bonding between the PDMS and glass.
To aid barrier on-chip studies, a two-layer device was created by placing the PDMS membrane in between the identical top and bottom parts as represented in Fig. 1C. Here, the channel's height was set to 375 μm. The same procedure was performed with the top compartment by punching all inlets and outlets. The three parts of the chip were activated in oxygen plasma (50 W, Cute, Femto Science) for 2 min to enhance the bonding. The top compartment was aligned with the inversed bottom compartment using a microscope in such a way that channels overlapped with each other (Fig. 1D). By having both eight apical channels and eight corresponding basal channels, the function of each barrier can be individually assessed (Fig. 1E). The final chip was 6 mm thick and was designed to fit on a standard 25 mm wide microscope slide, making it compatible with microscopy readouts.
Membrane fabrication
In order to recreate barrier models in the two-layer chip, a thin and transparent PDMS membrane was fabricated. This membrane can be integrated into the multiplexed chip without additional glue. The fabrication process was improved from our previous work35 so that it requires only one step of photoresist (PR) spin-coating (Fig. 2). Briefly, a positive PR layer (AZ 9260, Fujifilm, Japan) was used as a sacrificial layer from which the membrane can be released by immersing it in acetone. A PR was deposited on a Si wafer (525 μm thick, Okmetic, Finland) at 2000 rpm for 60 s to obtain a 10 μm thick layer. Next, the wafer was baked on a hot plate at 110 °C for 2 min. The mask with arrays of 5 μm pore diameter and 30 μm pitch was aligned with the wafer, followed by 17 s UV-exposure at an intensity of 12 mW cm−2 using hard contact mode. To avoid bubble formation in the thick PR during a post-exposure bake, wafers were left for 1 h. This helps to evaporate N2 gas formed during UV-exposure. After the waiting step, wafers were baked at 120 °C for 2 min. Finally, the microcolumns were obtained after developing the PR for 6 min in an OPD4246 developer, followed by rinsing with de-ionized (DI) water.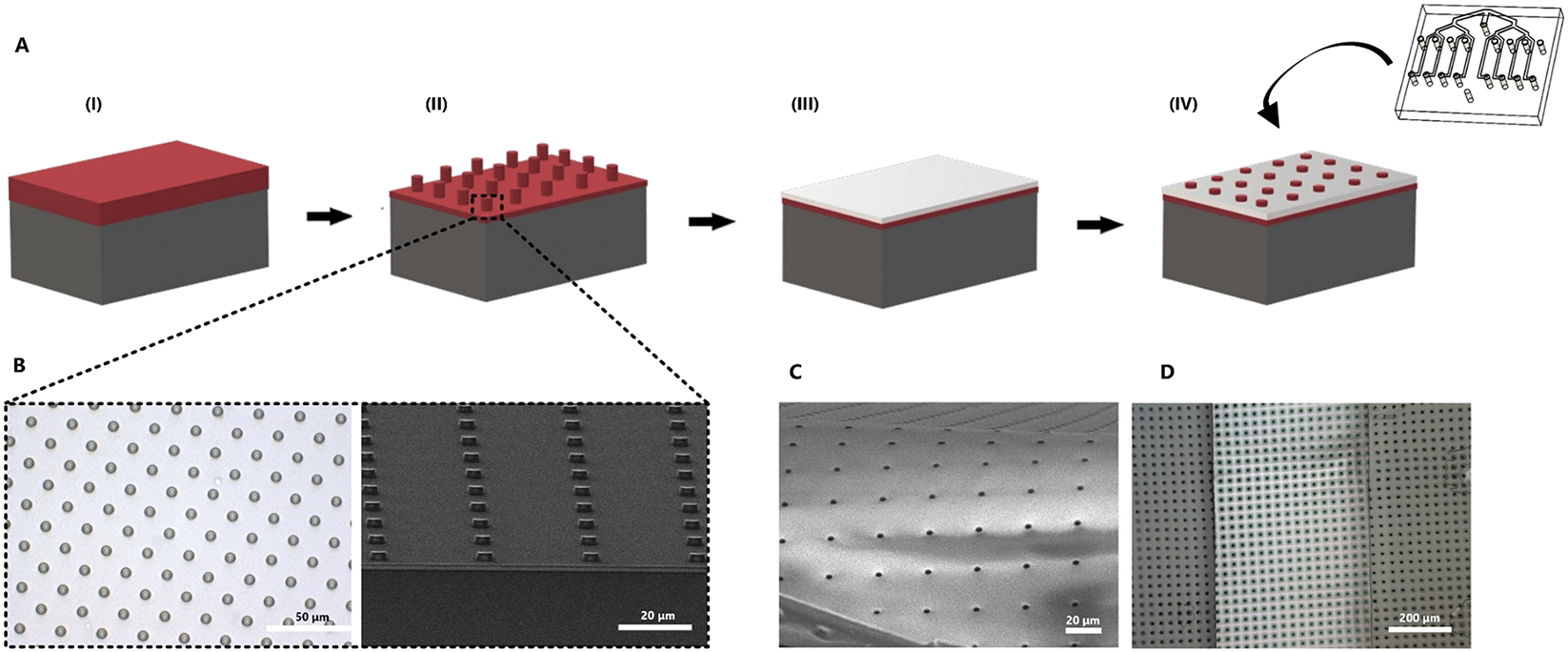 | ||
| Fig. 2 (A) Fabrication process of thin PDMS membrane. (I) A positive PR layer is spin-coated on a Si wafer followed by UV exposure. (II) A patterned array of columns after the developing process. (III) Spin coating of the PDMS–hexane (2:5 w/w) mixture on the patterned wafer. (IV) Etching of the top layer of PDMS in order to open the holes of the membrane. The PDMS through-hole membrane is released from the wafer by immersing it in acetone. For a successful release, the membrane is first plasma bonded to one layer of the chip. (B) Top and side view of the fabricated PR columns. (C) Released through-hole membrane from the wafer. (D) Freestanding PDMS membrane inside the microfluidic device. | ||
To prepare the PDMS membrane, a solution of PDMS prepolymer with a curing agent (10:1 w/w ratio) was diluted with hexane at a 2:5 w/w (PDMS:hexane) ratio to reduce viscosity. Next, the PDMS solution was spin-coated over the fabricated PR column arrays at 4000 rpm for 1 min and baked in the oven at 60 °C for at least 3 h. When spin-coating PDMS, it can fully cover the PR columns.35 To make sure that the pores are open, it is necessary to perform a plasma etching process of the cured PDMS membrane.
The etching was done using a reactive-ion etching system (TEtske, Nanolab University of Twente, The Netherlands) at 47 sccm SF6 and 17 sccm O2, 100 W and 50 mTorr for 2 min. The porosity of the resulting membrane was 2%. Scanning electron microscopy (SEM, HR-SEM, FEI Sirion microscope) at 5 kV acceleration voltage was used to evaluate the thickness and topography of the PDMS membrane.
To transfer the PDMS membrane (thickness of approximately 2 μm) to the chip, the top layer of the PDMS device and the porous PDMS surface on the Si wafer were treated with oxygen plasma and brought into contact. To guarantee a good bonding, the assembly was put into the oven at 60 °C for 10 min. Next, the sacrificial PR layer was removed by immersing it in acetone for 5 min. Finally, when the bottom part with the membrane was dried out, it was assembled with the top part using oxygen plasma treatment for 3 min. Small irregularities in the channels, dust, loose bonding or bubbles may affect the microfluidic resistance of the channels, and as a consequence, distribution of the flow may not be equal between channels. To prevent this, before starting the experiments, the device was cleaned to flush out all bubbles and fluid levels inside the reservoirs at the outlets were equalized.
Cell culture
Human cerebral microvascular endothelial cells (hCMEC/D3, Merck Millipore) were cultured in endothelial growth medium (EGM, Cell Applications, Inc) in collagen I-coated culture flasks. Human astrocytes (HAc) (Cell Applications, Inc.) were cultured in Astrocyte growth medium containing growth supplement (ScienCell Research Laboratories). Both cultures were maintained at 5% CO2 and 37 °C, refreshing the medium and subculturing every 2–3 days.Prior to cell seeding, the microfluidic chips were exposed to an oxygen plasma (50 W, Cute, Femto Science) to make the surface hydrophilic. Subsequently, the chips were rinsed with phosphate-buffered saline (PBS; Sigma) and coated with collagen type I (rat tail, Corning) (100 μg ml−1) for 2 h at 37 °C.
Endothelial cell monoculture was started by filling the chips with EGM. Next, hCMEC/D3 cells in suspension at a density of 6 × 106 cells per ml (3 × 104 cells per cm2) were pipetted into all eight channels simultaneously with a single pipetting action through the common access port. After 1 h of static incubation at 37 °C and 5% CO2, non-adherent cells were washed away with EGM.
An endothelial cell/HAc co-culture in a two-layer device was prepared by filling the channels with astrocyte medium. The HAc were seeded on the basal side of all eight channels at a concentration of 2 × 106 cells per ml (7 × 104 cells per cm2). Afterward, the chips were inverted for 4 h to allow the attachment of cells to the bottom side of the membrane. Next, the chips were flipped back and the pipette tips containing fresh astrocytes medium were inserted into each access port. After 24 h of astrocyte culturing, EGM was introduced in the top compartment and hCMEC/D3 cells were inserted at a cell density of 6 × 106 cells per ml (2 × 105 cells per cm2). The cells were allowed to adhere for 1 h. Next, EGM was refreshed in the top compartment while the astrocyte medium was refreshed in the bottom compartment twice per day. Medium inside the chips was refreshed by gravity-driven flow by inserting medium filled pipette tips in the common access ports of each device. Phase-contrast and fluorescent imaging (EVOS FL Cell Imaging System, Life Technologies and LEICA DM IRM HC, air objectives) were used for morphological observations of the cells inside the chip.
To assess the viability of hCMEC/D3 on day 5 of culture inside the multiplexed chip, Live–Dead staining was performed. A solution containing 1 μl calcein-AM, 4 μl ethidium homodimer-1 (EthD-1) (Live/Dead Viability/Cytotoxicity kit for mammalian cells, Invitrogen) and 1 drop NucBlue (Ready Probes reagent, Molecular Probes, Life Technologies) in 60 μl PBS was introduced in the chip after flushing the channels two times with PBS. The cells were imaged after 30 min of incubation (37 °C, 5% CO2) using an EVOS FL Cell Imaging System (Life Technologies); green filter (λex 470/22, λem 525/50) for calcein (product of enzymatic cleavage of calcein-AM by living cells; λex 495 nm, λem 515 nm), red filter (λex 531/40, λem 593/40) for ethidium homodimer-1 (cannot cross intact cell membranes in presence of DNA; λex 528 nm, λem 617 nm) and blue filter (λex 357/44, λem 447/60) for NucBlue (λex 359 nm, λem 463 nm). The contrast of the resulting images was equally enhanced and subsequently merged in ImageJ (Wayne Rasband, NIH, version 1.51j8). Additionally, the metabolic activity of the cells was established on day 1 and day 5 by incubation with a mitochondrial stain (100 nM, MitoTracker Red CMXRos (λex 579 nm, λem 599 nm, Thermo Fisher) for 20 min at 37 °C and 5% CO2. Subsequently, the cells were imaged with an EVOS microscope using red filter (λex 531/40, λem 593/40) for Mitotracker and blue filter (λex 357/44, λem 447/60) for NucBlue).
Chip validation
000) and the separate access ports were covered with PBS. Crosstalk between the channels in the multiplexed chip was studied by selective introduction of Trypsin 1X (Sigma) to every other channel to release adherent cells. Pipette tip reservoirs with either trypsin or EGM were inserted into the separate access ports and these solutions were pulled through for 30 min at a flow rate of 20 μl min−1 (2 dyne per cm2). Then, EGM was added to all channels and flow was applied for another 15 min to ensure all trypsin was removed from the chip.
To visualize cells upon exposure to trypsin, a solution containing 1 μl calcein-AM (Live/Dead Viability/Cytotoxicity kit for mammalian cells, Invitrogen) and one drop NucBlue (Ready Probes reagent, Molecular Probes, Life Technologies) in 60 μl PBS was introduced after flushing the channels two times with PBS. The introduction of the reagents and flushing was performed with single pipetting actions through the common access port. After incubation for 30 min at 37 °C and 5% CO2, fluorescence microscopy was performed using an EVOS FL Cell Imaging System (Life Technologies; green filter (λex 470/22, λem 510/42) for calcein and blue filter (λex 357/44, λem 447/60) for NucBlue).
:50, polyclonal, Invitrogen) and primary mouse vascular endothelial cadherin (VE-cadherin, 1:300, Santa Cruz Biotechnology) in 0.5% BSA in PBS overnight at 4 °C. Next, the cells were incubated with Alexa Fluor 647 and Alexa Fluor 488 secondary antibodies (goat anti-rabbit, and goat anti-mouse, dilution 1:500, Invitrogen) for 1 h.
The HAc were stained for Glial fibrillary acidic protein (GFAP, 1:100, monoclonal anti-mouse, Invitrogen). After the washing step, the HAc were incubated with the secondary antibody (Alexa Fluor 488, goat anti-mouse, dilution 1:500, Invitrogen) for 1 h. The nuclei of both types of cells were stained with NucBlue (Invitrogen) for 20 min. Confocal fluorescent microscopy was performed using an Andor Dragonfly® 200 spinning disk confocal on a Leica DMi8 microscope. The pinhole diameter of the disk was 40 μm. Long working distance water objectives 10× (NA 0.30), 20× (NA 0.50), 40× (NA 0.80) were used.
Fluorescence intensity was evaluated by ImageJ software. Next, fluorescence intensity values in the lower compartments were normalized to fluorescence intensity values in the upper compartments. Apparent permeability of the BBB was calculated from the normalized intensity profiles using a previously reported method:36
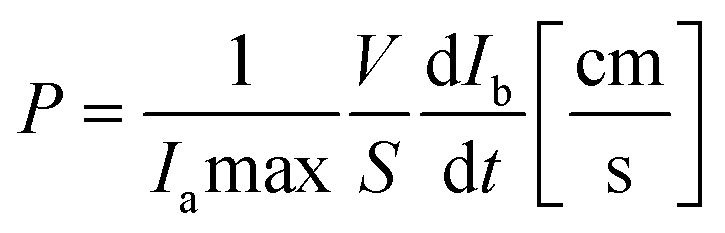 | (1) |
To obtain an actual permeability Pa of the multiplexed device, the permeability measured in the device with the cell monolayer Pc was subtracted from the permeability measured in empty chip Pm (permeability of the membrane):37
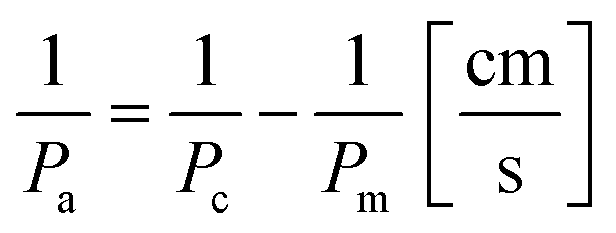 | (2) |
Results and discussion
Chip fabrication
We designed two multiplexed chips: a one-layer chip for studying cell cultures and a two-layer chip for studying barrier tissues such as BBB. When designing the multiplexed chip, several important points were taken into consideration such as ease of manipulation, the possibility of parallel cell culture with the same initial conditions and feasibility of individual collection and analyses of effluents from these parallel cultures.Fig. 3 shows the COMSOL simulation results of the flow profile in the chip, which indicate that the flow distribution in each channel is uniform, i.e. 12.2–12.9%. The fluid velocity decreases after each branching while eventually uniformly distributing in each of the eight channels. Nonetheless, the eight parallel channels are not completely separated fluidically and the risk of cross-talk between the conditions in adjacent channels has to be taken into account. Therefore the possibility of diffusion and advection of solutes into the adjacent channel has to be considered.
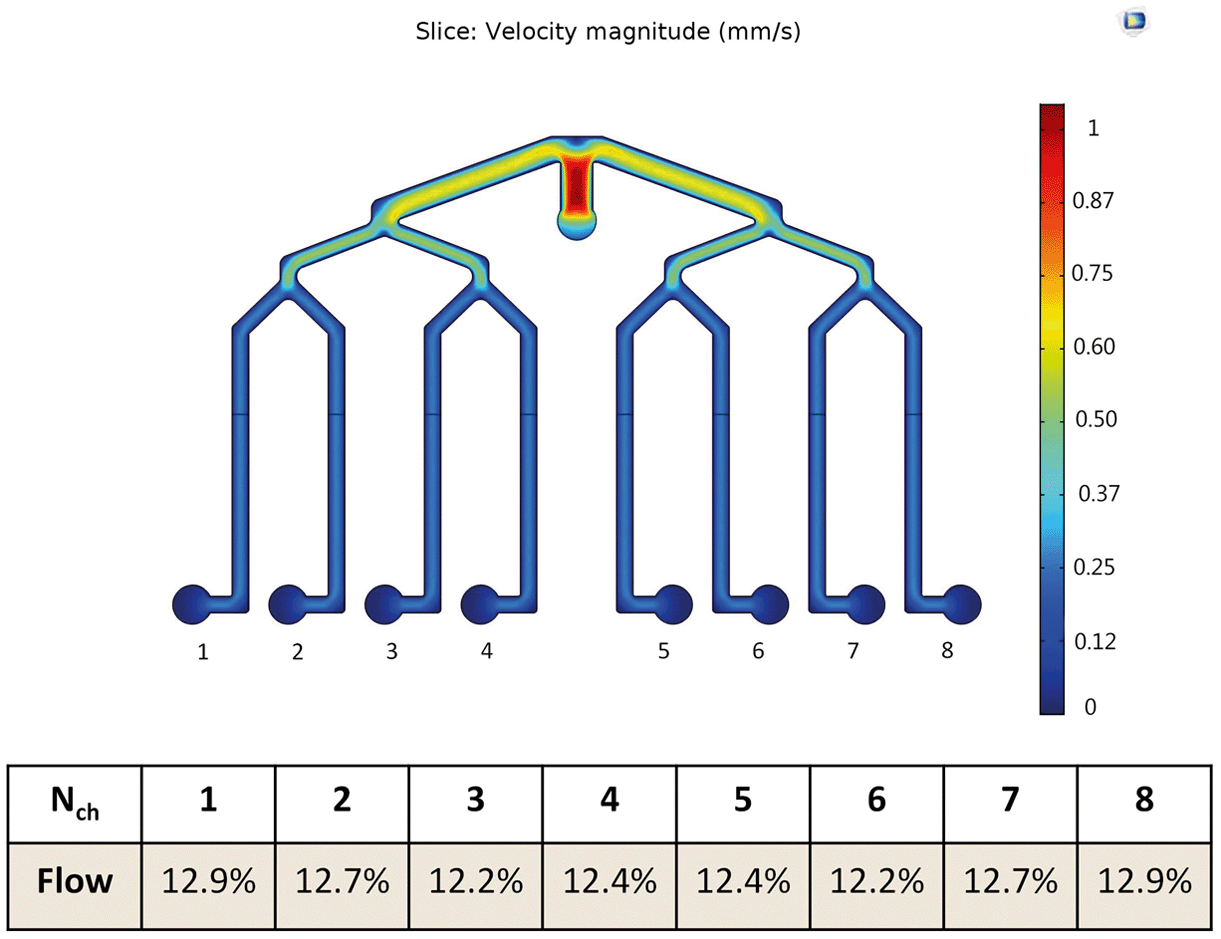 | ||
| Fig. 3 Simulated flow profile in the chip using finite element modeling. An input flow rate was distributed in all eight channels resulting in equal flow profiles with minor variation. The scale bar represents a normalized velocity range. | ||
The risk of contamination of adjacent channels by diffusion during an experiment can be determined by calculating the Péclet number for typical experimental parameters. The Péclet number (Pe)38 is defined as the ratio of the rate of advective transport of a certain solute and the rate of diffusion perpendicular to that flow, resulting in the following equation:
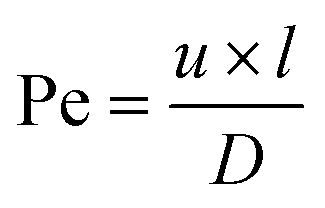 | (3) |
Next, a contribution of advection can be reviewed. The flow will equally split at each junction, under the condition that the fluid reservoirs at the eight separate access ports are identical. In that case, all fluid will move to the common access port and there will be no cross-talk between conditions. However, when the fluid levels in the reservoirs are not identical, fluid can flow into adjacent channels. To understand the risk of this advective flow during an experiment, the following equation can be used:
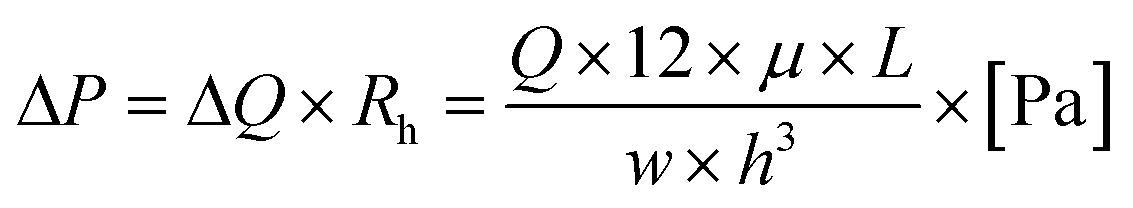 | (4) |
For equal flow distribution, the hydrodynamic resistance at each channel also has to be equal, however, this is not always the case. The difference in Rh can arise from for example a dust particle inside the channel, poor fabrication accuracy or improper bonding to the glass.39 Therefore, with a flow rate of Q = 2.5 μl min−1, a viscosity of culture medium μ = 0.7 mPa s, a distance between two adjacent access ports L = 25 mm and a channel width and height of w = 500 μm and h = 50 μm, the pressure difference is ΔP = 1.4 × 102 Pa. This corresponds to a pressure head of h = 14 mm, calculated from the equation P = ρ × g × h. Height differences of this relatively small order of magnitude could practically arise when no attention is paid to equalizing the fluid levels inside the reservoirs. Therefore, the risk of advective flow has to be taken into account when designing experiments and needs to be minimized to prevent cross-talk, especially when lower flow rates are required.
For the two-layer chip, a PDMS membrane was fabricated. Fig. 4 shows the HR-SEM images of the fabricated free-standing PDMS membrane with through-hole pores. The pore size and porosity of the membranes can be controlled by modifying the fabricated PR columns. With its transparency compared to the PC membranes,40 the fabricated PDMS membrane allowed non-invasive monitoring of cells during cell culture without additional labeling (Fig. S1†). In addition, its small thickness of ∼2 μm can allow the crosstalk between the top and bottom compartments, thus enhancing cell–cell interaction in co-culture chips.
 | ||
| Fig. 4 (A) HR-SEM image of a PDMS coated PR column array. (B) The resulted thickness of the spin-coated PDMS layer is ∼2 μm. (C and D) A bright-field microscopy image of the integrated membrane in the chip with 50 μm height (C) and 375 μm height (D). In (C), the membrane was attached to the bottom of the chip which can be seen when trying to focus both on the chip side and on the channel at the same time. (D) The channel part and the chip are at the same focus indicating no attachment of the membrane to the bottom of the channel. | ||
We found that the channels with a height of only 50 μm were clogged due to the bending of the membrane (Fig. 4C). We attribute this to the fact that the membrane is very thin and flexible, thus being easily attracted to the PDMS surface of the channel when releasing from the wafer in acetone. To prevent the binding of the PDMS membrane to the top or bottom of the channel, the height of the channels in the top and bottom compartments was increased to 375 μm (Fig. 4D).
Evaluation of the parallel functionality of the multiplexed chip
The hCMEC/D3 cells were seeded in the one-layer device by a single pipetting action through the common inlet. The distribution of cells was assessed using a phase-contrast microscope and the chips were incubated for 4 days, replacing medium every day. To verify whether the channels are individually addressable, every other channel with hCMEC/D3 monolayer was exposed to trypsin which dissociated the cells.After 30 min, the cells were visualized using a calcein-AM solution indicating intracellular esterase activity as a marker of viability. This experiment resulted in selective dissociation of the endothelial cell layer in the trypsin-treated channels while non-exposed cells remained attached as can be seen in Fig. 5. The absence of fluid cross-talk between adjacent channels was also visible in the branching area of the channels (Fig. 5C). There, cells were flushed out from the trypsinized channel and from half of the combined channel, while the cells in the EGM channel and the other half of the combined channel remained unaffected.
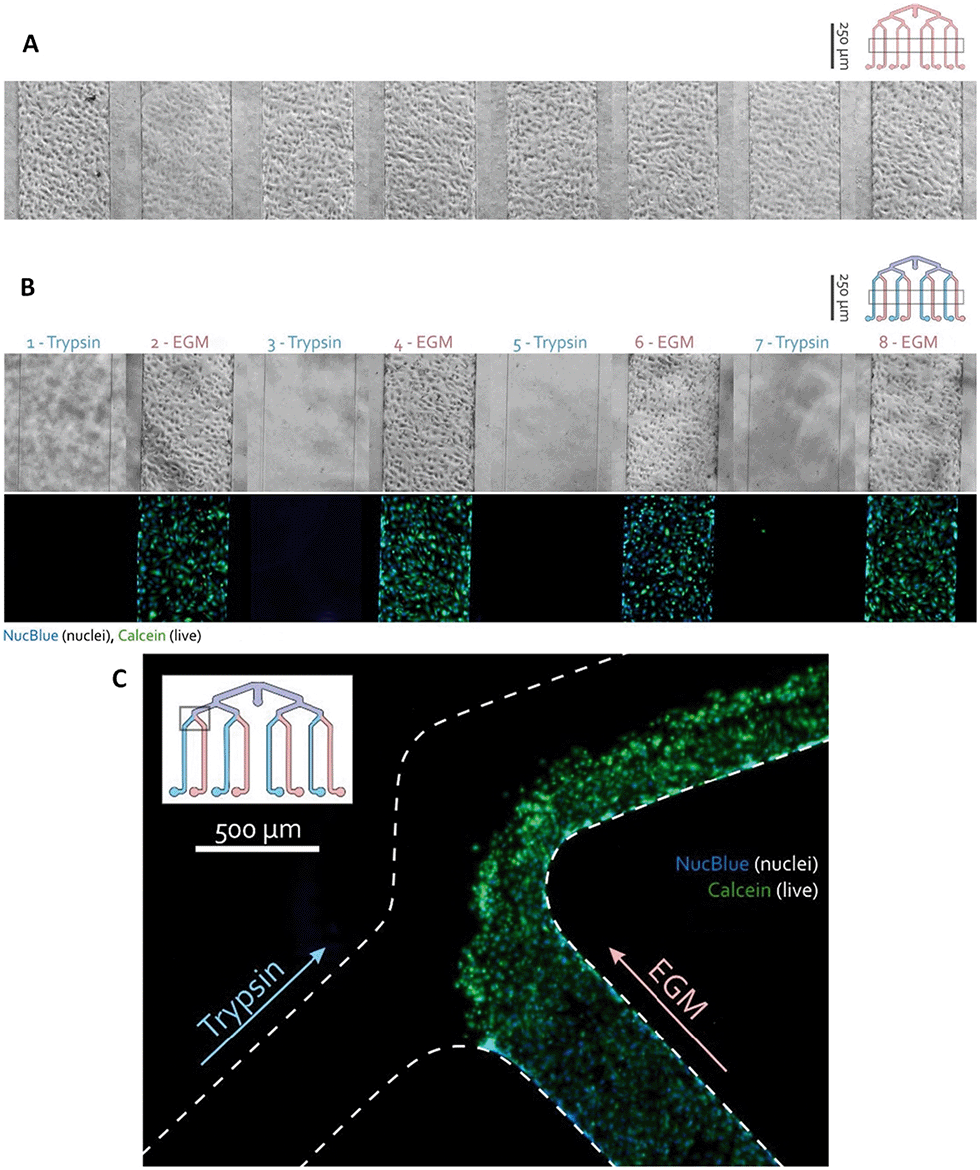 | ||
| Fig. 5 Selective trypsin exposure in a one-layer device shows that no detectable crosstalk occurs between adjacent channels. (A) After 4 days of culture, endothelial monolayers were observed in all eight parallel channels of a multiplexed chip. The inset shows where the images were approximately taken. (B) The channels were individually exposed to either trypsin or EGM culture medium. Channels were flushed after 30 min exposure, showing that trypsin-exposed cell layers fully dissociated while the cells in the EGM culture medium channels remained attached. (C) Due to the laminar flow, cells in the trypsin channel were flushed away, while the cells inside the EGM channel remained unaffected. The insert indicates where the image was approximately taken. | ||
This result indicated that the application of eight different conditions is possible on the same device. Therefore, cell culture can be selectively addressed with different assays without cross-talk between them and their effect can be studied.
Establishing BBB in the chip
After the two-layer chip was assembled and sterilized with ethanol, hCMEC/D3 endothelial cells were seeded at a cell density of 6 × 106 cells per ml in a multiplexed chip. Using a single pipetting action through the common access port, an apparent visually equal cell monolayer was obtained in all channels after 5 days of culturing (Fig. 6). Cell monolayer formation was monitored using phase-contrast microscopy. hCMEC/D3 cell adherence to the PDMS membrane was already visible using phase-contrast microscopy after 1 h of incubation (Fig. S1B†). In addition, Live–Dead staining together with mitochondrial staining confirmed viable and metabolically active cells during 5 days of culture (Fig. S3†). In a previous study, hCMEC/D3 showed to form a tight monolayer already after 3 days of culture remaining unchanged for 7 (ref. 41) or 10 (ref. 42) days. However, long culture periods can lead to the cell overgrowth compromising the physiological resemblance of BBB.43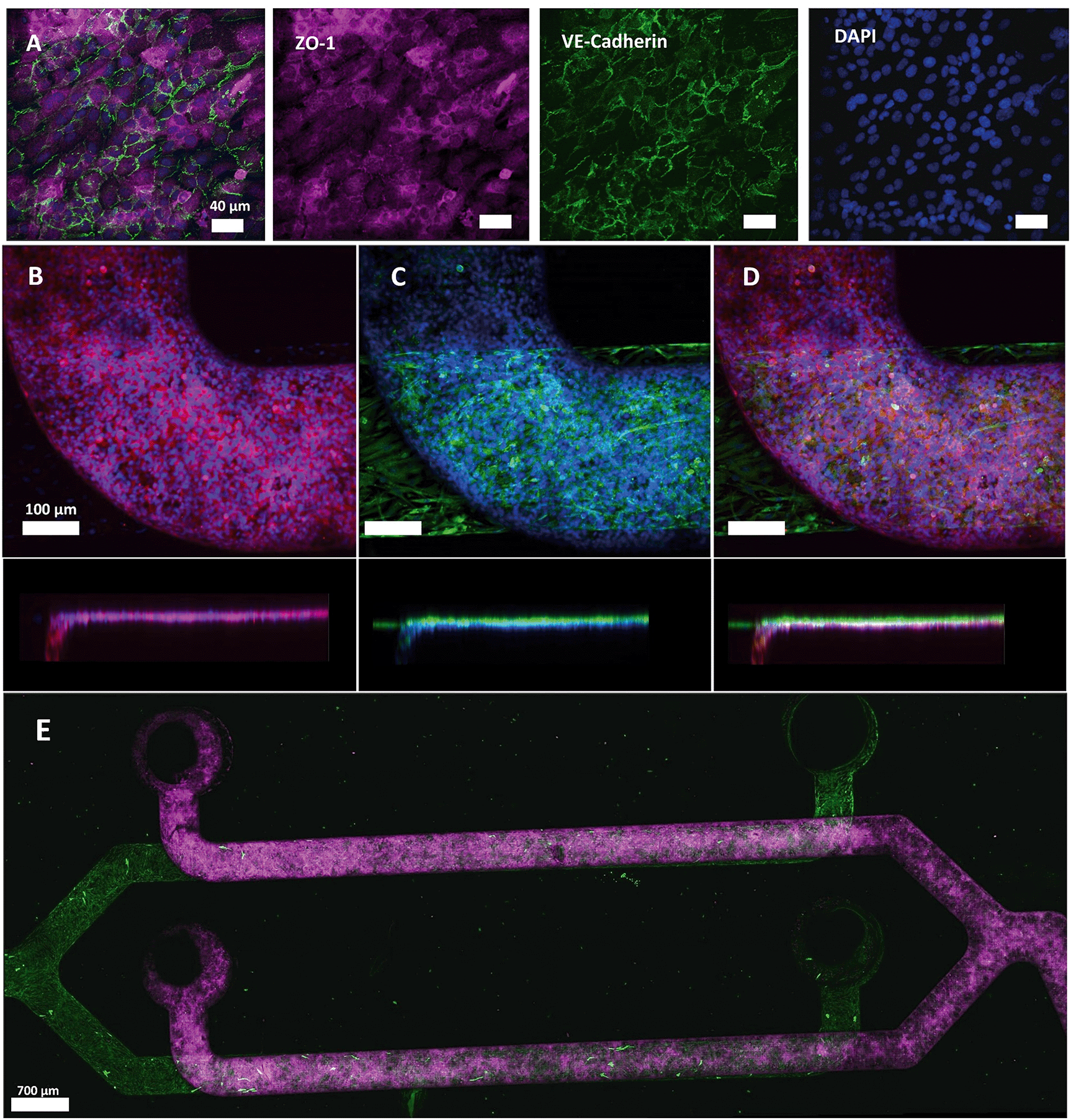 | ||
| Fig. 6 Immunostaining of tight junction protein ZO-1 and adherens junction protein VE-cadherin in hCMEC/D3 (Alexa fluor 647, red and Alexa fluor 488, green, respectively), cell nuclei (NucBlue©, blue) and GFAP in HAc (Alexa fluor 488, green) after 5 days of culture. (A) Expression of ZO-1 and VE-cadherin markers by hCMEC/D3 cells in monoculture. VE-Cadherin is localized at the cell–cell junctions. (B–D) Representative images of ZO-1 and nuclei expression (B), GFAP, and nuclei (C) and overlay of both channels (D) with a side view. The separation between the two cell types was minimized due to the PDMS membrane. (E) Confocal microscopy reveals a complete coverage of the channels with HAc and hCMEC/D3 (ESI† Video). | ||
The cells were fixed after 5 days of culture and the formation of tight junctions was investigated. Immunostaining of hCMEC/D3 cells revealed cytoplasmic expression of tight junction component ZO-1 with peripheral localization of VE-cadherin, an important endothelial adherens junction marker, (Fig. 6A and B and S4A†) indicating that hCMEC/D3 cells were able to form intracellular interactions required for barrier formation and showed cobblestone morphology when cultured in the multiplexed chips. Interestingly, in different studies, hCMEC/D3 cells were reported to show either cobblestone44,45 or spindle-like morphology.46 This difference in cell shape can be attributed to several factors together with the culture support material used. PDMS, the material of choice in our chip design, has different stiffness properties from PC membranes and polystyrene culture flasks. Therefore, hCMEC/D3 had more elongated morphology when cultured in a flask compared to the more dense layer of cells with cobblestone-like morphology observed when cultured on PDMS (Fig. S5†). In addition, bovine aortic endothelial cells (BAEC) were shown before to attain a more rounded morphology on a PDMS substrate than on a stiffer membrane as a function of integrin and cadherin receptor sensing leading to cytoskeletal remodeling.47,48 Also, ZO-1 localization appeared dependent on the cellular substrate as it has been demonstrated that ZO-1 shows higher levels of cytoplasmic expression when cultured on glass compared to transwell inserts.49 Thus, in our experiment, the PDMS membrane on which the cells were cultured, as well as physiological conditions the cells were exposed to could have affected cell morphology and ZO-1 expression pattern.
Physiologically, the perivascular endfeet of astrocytes are physically associated with the microvasculature and astrocytes and endothelial cells were shown to interact intensively at the site of the BBB.50 To add physiological relevance to the BBB model HAc were seeded on the abluminal side (Fig. 6C) of the chip while endothelial cells were cultured on the luminal side. Both cell types were separately imaged using specific antibodies and an overlay of two images for each channel was generated (Fig. 6D and E and S4†). The membrane thickness of 2 μm minimized the distance between two cell types (Fig. 6D), which, together with a membrane pore size of 5 μm, contributes to the establishment of intercellular interactions that were previously shown to influence the growth and development of the BBB.50 The confocal images revealed a confluent monolayer of hCMEC/D3 cell and full coverage of the channels with both cell types (ESI† Video).
The acquired images reveal a limitation of the presented multiplexed two-layer design. Since the thickness of the bottom part of the chip was 3 mm with a channel height of 375 μm, the limited working distance of high magnification objectives hampered image acquisition. This problem can be solved by reducing the thickness of the bottom compartment or by creating a bottom-open chip attached to a glass substrate. This will significantly reduce the distance from the objective to the culture area and higher resolution imaging can be performed.
Permeability
To assess whether the BBB model generated in our chip formed a physiological barrier, a permeability assay was performed on the 5th day of hCMEC/D3 cell culture. When applying flow to both common access ports, a counter-current flow occurs which can form a transmembrane pressure gradient. The advection resulting from these pressure differences is therefore expected to influence the permeability measurements in a two-layer device. Moreover, even a small pressure difference in the upper or bottom channels can affect the flow, giving rise to backflow making it more complicated to collect the same amount of liquid from each channel. Therefore, here the permeability experiment was performed statically.In the absence of cells, 4 kDa and 20 kDa fluorescent molecules could diffuse through the PDMS membrane from the top channels as the fluorescence intensity in the bottom channels increased over time (Fig. 7B). The measured intensity in the bottom channels was plotted as a function of time for all eight channels (Fig. 7C and D) and the average permeability coefficient was determined. Using eqn (1) and (2) the calculated permeability coefficient for 4 kDa decreased from 2.23 × 10−5 ± 5.75 × 10−6 cm s−1 (Pm) in the absence of cell to 8.92 × 10−6 ± 5.8 × 10−6 cm s−1 (Pa) when hCMEC/D3 cells were present in the device. The same trend was seen for 20 kDa FITC dextran molecules; the permeability coefficient in the empty device Pm = 4.69 × 10−6 ± 5.54 × 10−7 cm s−1 was significantly lower than permeability values in the presence of cells Pa = 1.06 × 10−6 ± 5.13 × 10−7 cm s−1 (Fig. 7E). This significant difference suggests that the permeability to fluorescent molecules across the membrane was decreased in the presence of the cell monolayer while fabricated PDMS membrane allowed free diffusion of FITC dextran.
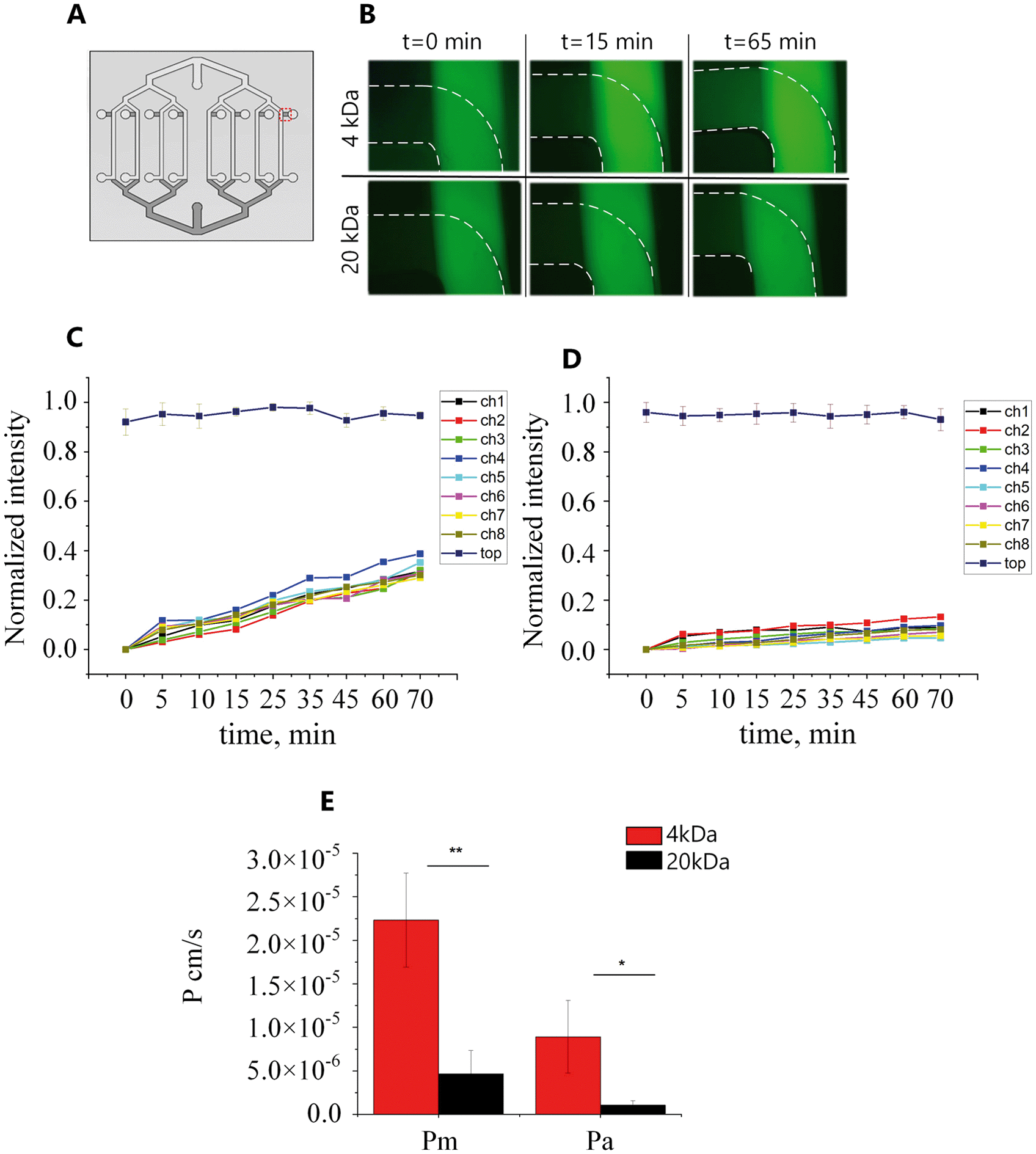 | ||
| Fig. 7 Permeability assay of fluorescently labeled FITC dextran of 4 and 20 kDa through hCMEC/D3 monolayer. (A) A representative schematic of the chip with an indication where images were taken during the permeability assay. All eight channels were analyzed. (B) Fluorescent images of 4 kDa (top) and 20 kDa (bottom) FITC dextran penetration from top to bottom channel within the time. (C) Normalized intensity profiles of the transport of 4 kDa and (D) 20 kDa molecules through the multiplexed chip within 70 min. (E) Calculated permeability in the empty chips (Pm) and actual permeability (Pa) of various molecular weight dextrans through the microfluidic BBB model. The presence of hCMEC/D3 monolayer significantly reduced the permeability to both fluorescently labeled molecules. Significance determined by student's t-test; *P < 0.05, **P < 0.01; n = 8. | ||
The reported permeability values correlate well in both trend and magnitude with those found in other in vitro microfluidic models of the BBB. Reported values for 4 kDa FITC dextran were in the range of 5–13 × 10−6 cm s−1 and 0.45–∼2 × 10−6 cm s−1 for 20 kDa molecules.31,45,51–53 However, these reported permeability values are still higher than those reported in vivo,54 where the permeability was 0.92 × 10−6 cm s−1 and 0.24 × 10−6 cm s−1 for 4 kDa and 20 kDa, respectively.
In vivo, the BBB is composed of multiple cell types that were shown to play a role in establishing the selective permeability of the BBB.55 The addition of BBB specific cell types, such as astrocytes and pericytes is expected to reduce the permeability of the BBB as has been shown before56 but requires specific investigation in our system in follow-up studies. Moreover, hCMEC/D3 cells are known to form a less tight barrier with higher permeability and TEER values compared to primary human brain microvascular endothelial cells (hBMEC).57 Therefore the permeability values could be reduced in this model by culturing hBMEC cells which will be beneficial for in vivo predictability.
While permeability of PDMS to O2 and CO2 is highly beneficial in our system, one important limitation of PDMS is its ability to absorb small hydrophobic molecules which can influence drug studies.58 PDMS mediated absorption can reportedly be minimized in future by performing a surface modification as was shown before.59
Conclusions
We fabricated a functional multiplexed OOC which provides the opportunity to simultaneously address eight channels through a common inlet. In addition, the separate access ports allow the independent collection of effluents from each culture for their individual study. Our fabricated chips can be used as a one-layer chip, or with an integrated thin PDMS membrane as a two-layer chip. The small thickness and transparency of the fabricated PDMS membrane improve the optical visualization of cells while decreasing spacing between two cultured cell types compared to commonly used PC membranes. The two-layer device can be fabricated with a plasma activation step, replacing the longer gluing process of membranes in between the PDMS parts.60 Computed simulations of the fluid flow and experimental results indicated that the proposed chip design allows an equal distribution of flow from the common inlet, thus equal conditions in each channel are formed. Additionally, we demonstrated that eight independent conditions can be created using separate access ports. We also showed that this fabricated chip is suitable for maintaining a viable and metabolically active cell culture. The BBB was recapitulated in eight channels with tight junction and adherent junction expression in hCMEC/D3 cells and GFAP expression in HAc. The permeability assay showed a size-dependent trend for Pa coefficients where our BBB model allowed faster passage of small molecules (4 kDa) compared to larger molecules (20 kDa), which is in accordance with other BBB models.We believe that our device can be used for the characterization of barrier tissues that allows better reproducibility and identical conditions, e.g. temperature, seeding density, and pH, within one single chip.
TEER is another important parameter that can provide information on the barrier function. While TEER allows non-invasive, real-time monitoring of barrier formation, the electrodes require embedding in the model in close proximity to the culture channels involving specific design considerations which remains challenging in the OOC field. In the future, the possibility to integrate the electrodes in the multiplexed chip will be examined.
Noteworthy, the reported device is not limited to only eight parallel channels placed on the top and bottom. Alternatively, a multiplexed design approach can involve placement of channels in the same plane for the higher resolution imaging, as been shown previously30 and the number of channels can be increased for higher throughput of the device, however, certain aspects have to be taken into account.
The channels have to be at the same distance from the inlet so that the same hydraulic resistance builds up in all channels leading to equal distribution of the flow. For this, the channel number has to be equal to 2n. Therefore it is possible to fabricate a device where a common inlet is symmetrically branching to 2, 4, 8, 16, 32, 64, 128 and so on chambers. The fabrication process has to be also automated to prevent fabrication inaccuracy (precise bonding of two-layer device on top of each other) and to avoid cross-contamination by advection and diffusion as discussed previously. Finally, the high-throughput systems require the capacity of standardized tools to operate, monitor, and analyze the cultures inside the device which are yet to be developed.61
Development of the microfluidic high-throughput OOCs will help to transit from academia to the industry, opening up new possibilities for personalized drug screening and testing.
Author contributions
M. A. P and M. W. H developed the design of the chip. H. L. T and M. Z. fabricated thin PDMS membranes. M. Z., M. W. H and M. A. P. performed experimental work. M. N. G and V. O. performed imaging of the chips. M. Z. wrote the manuscript and L. S., K. B., A. M., A. B., H. L. T., M. G., V. O. contributed to writing, review, and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This version replaces the version published on 5th August 2020 which contained an error in Fig. 3.Acknowledgements
The authors acknowledge the funding received from the Dutch Science Foundation (NWO) under the Gravitation Grant “NOCI” Program (Grant No. 024.003.001). The authors would also like to acknowledge the Light and Electron Microscopy Facility at Leiden University Medical Center for the use of their services and facilities.References
- L. Hein, K. Mohr and D. Bieger, Color Atlas of Pharmacology, ed. H. Lullmann, 3rd edn, Thieme, 2005 Search PubMed.
- A. M. Marchiando, W. V. Graham and J. R. Turner, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 119–144 CrossRef CAS PubMed.
- F. Rezaee and S. N. Georas, Am. J. Respir. Cell Mol. Biol., 2014, 50, 857–869 CrossRef PubMed.
- M. C. Dias, J. A. Mapunda, M. Vladymyrov and B. Engelhardt, Int. J. Mol. Sci., 2019, 20(21), 5372 CrossRef CAS PubMed.
- P. S. Caceres, I. Benedicto, G. L. Lehmann and E. J. Rodriguez-Boulan, Cold Spring Harbor Perspect. Biol., 2017, 9, a027847 CrossRef PubMed.
- R. Daneman and A. Prat, Cold Spring Harbor Perspect. Biol., 2015, 7, a020412 CrossRef PubMed.
- F. L. Cardoso, D. Brites and M. A. Brito, Brain Res. Rev., 2010, 64, 328–363 CrossRef CAS PubMed.
- M. B. Bracken, J. R. Soc. Med., 2009, 102, 120–122 CrossRef PubMed.
- I. Wilhelm and I. A. Krizbai, Mol. Pharmaceutics, 2014, 11, 1949–1963 CrossRef CAS PubMed.
- H. C. Helms, N. J. Abbott, M. Burek, R. Cecchelli, P.-O. Couraud, M. A. Deli, C. Förster, H. J. Galla, I. A. Romero, E. V. Shusta, M. J. Stebbins, E. Vandenhaute, B. Weksler and B. Brodin, J. Cereb. Blood Flow Metab., 2016, 36, 862–890 CrossRef CAS PubMed.
- P. Horvath, N. Aulner, M. Bickle, A. M. Davies, E. Del Nery, D. Ebner, M. C. Montoya, P. Östling, V. Pietiäinen, L. S. Price, S. L. Shorte, G. Turcatti, C. Von Schantz and N. O. Carragher, Nat. Rev. Drug Discovery, 2016, 15, 751–769 CrossRef CAS PubMed.
- Q. Wu, J. Liu, X. Wang, L. Feng, J. Wu, X. Zhu, W. Wen and X. Gong, Biomed. Eng. Online, 2020, 19, 1–19 CrossRef PubMed.
- D. E. Ingber, Cell, 2016, 164, 1105–1109 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- J. R. Soucy, A. J. Bindas, A. N. Koppes and R. A. Koppes, iScience, 2019, 21, 521–548 CrossRef PubMed.
- A. Cochrane, H. J. Albers, R. Passier, C. L. Mummery, A. van den Berg, V. V. Orlova and A. D. van der Meer, Adv. Drug Delivery Rev., 2019, 140, 68–77 CrossRef CAS PubMed.
- Y. A. Jodat, M. G. Kang, K. Kiaee, G. J. Kim, A. F. H. Martinez, A. Rosenkranz, H. Bae and S. R. Shin, Curr. Pharm. Des., 2019, 24, 5471–5486 CrossRef PubMed.
- M. W. van der Helm, A. D. van der Meer, J. C. T. Eijkel, A. van den Berg and L. I. Segerink, Tissue Barriers, 2016, 4, e1142493 CrossRef PubMed.
- C. M. Sakolish, M. B. Esch, J. J. Hickman, M. L. Shuler and G. J. Mahler, EBioMedicine, 2016, 5, 30–39 CrossRef PubMed.
- I. Raimondi, L. Izzo, M. Tunesi, M. Comar, D. Albani and C. Giordano, Front. Bioeng. Biotechnol., 2020, 7 Search PubMed.
- A. Bhalerao, F. Sivandzade, S. R. Archie, E. A. Chowdhury, B. Noorani and L. Cucullo, Fluids Barriers CNS, 2020, 17 Search PubMed.
- M. I. Teixeira, M. H. Amaral, P. C. Costa, C. M. Lopes and D. A. Lamprou, Pharmaceutics, 2020, 12, 1–37 Search PubMed.
- S. Halldorsson, E. Lucumi, R. Gómez-Sjöberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- O. Y. F. Henry, R. Villenave, M. J. Cronce, W. D. Leineweber, M. A. Benz and D. E. Ingber, Lab Chip, 2017, 17, 2264–2271 RSC.
- J. Yeste, M. García-Ramírez, X. Illa, A. Guimerà, C. Hernández, R. Simó and R. Villa, Lab Chip, 2018, 18, 95–105 RSC.
- Y. B. Arık, M. W. van der Helm, M. Odijk, L. I. Segerink, R. Passier, A. van den Berg and A. D. van der Meer, Biomicrofluidics, 2018, 12, 042218 CrossRef PubMed.
- A. Oddo, B. Peng, Z. Tong, Y. Wei, W. Y. Tong, H. Thissen and N. H. Voelcker, Trends Biotechnol., 2019, 37, 1295–1314 CrossRef CAS PubMed.
- M. N. S. de Graaf, A. Cochrane, F. E. van den Hil, W. Buijsman, A. D. van der Meer, A. van den Berg, C. L. Mummery and V. V. Orlova, APL Bioeng., 2019, 3, 026105 CrossRef PubMed.
- G. Adriani, D. Ma, A. Pavesi, R. D. Kamm and E. L. K. Goh, Lab Chip, 2017, 17, 448–459 RSC.
- J. B. Sheffield, B. Wang, M. F. Kiani, B. Krynska, S. P. Deosarkar and B. Prabhakarpandian, PLoS One, 2015, 10, e0142725 CrossRef PubMed.
- R. Booth and H. Kim, Lab Chip, 2012, 12, 1784–1792 RSC.
- G. D. Vatine, R. Barrile, M. J. Workman, S. Sances, B. K. Barriga, M. Rahnama, S. Barthakur, M. Kasendra, C. Lucchesi, J. Kerns, N. Wen, W. R. Spivia, Z. Chen, J. Van Eyk and C. N. Svendsen, Cell Stem Cell, 2019, 24, 995–1005.e6 CrossRef CAS PubMed.
- K. Zografos, R. W. Barber, D. R. Emerson and M. S. N. Oliveira, Microfluid. Nanofluid., 2015, 19, 737–749 CrossRef.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS PubMed.
- H. Le-The, M. Tibbe, J. Loessberg-Zahl, M. P. Do Carmo, M. Van Der Helm, J. Bomer, A. Van Den Berg, A. Leferink, L. Segerink and J. Eijkel, Nanoscale, 2018, 10, 7711–7718 RSC.
- H. Yuan, M. W. Gaber, T. McColgan, M. D. Naimark, M. F. Kiani and T. E. Merchant, Brain Res., 2003, 969, 59–69 CrossRef CAS PubMed.
- M.-P. Dehouck, P. Jolliet-Riant, F. Brée, J.-C. Fruchart, R. Cecchelli and J.-P. Tillement, J. Neurochem., 1992, 58, 1790–1797 CrossRef CAS PubMed.
- T. M. Squires, R. J. Messinger and S. R. Manalis, Nat. Biotechnol., 2008, 26, 417–426 CrossRef CAS PubMed.
- B. Gale, A. Jafek, C. Lambert, B. Goenner, H. Moghimifam, U. Nze and S. Kamarapu, Inventions, 2018, 3, 60 CrossRef.
- H. H. Chung, M. Mireles, B. J. Kwarta and T. R. Gaborski, Lab Chip, 2018, 18, 1671–1689 RSC.
- L. M. Griep, F. Wolbers, B. De Wagenaar, P. M. Ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. Van Der Meer and A. Van Den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- S. Hinkel, K. Mattern, A. Dietzel, S. Reichl and C. C. Müller-Goymann, Int. J. Pharm., 2019, 566, 434–444 CrossRef CAS PubMed.
- E. Urich, S. E. Lazic, J. Molnos, I. Wells and P.-O. Freskgård, PLoS One, 2012, 7, e38149 CrossRef CAS PubMed.
- A. Noack, B. Gericke, M. Von Köckritz-Blickwede, A. Menze, S. Noack, I. Gerhauser, F. Osten, H. Y. Naim and W. Löscher, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9590–E9599 CrossRef CAS PubMed.
- E. De Jong, D. S. Williams, L. K. E. A. Abdelmohsen, J. C. Van Hest and I. S. Zuhorn, J. Controlled Release, 2018, 289, 14–22 CrossRef CAS PubMed.
- B. B. Weksler, E. A. Subileau, N. Perrière, P. Charneau, K. Holloway, M. Leveque, H. Tricoire-Leignel, A. Nicotra, S. Bourdoulous, P. Turowski, D. K. Male, F. Roux, J. Greenwood, I. A. Romero and P. O. Couraud, FASEB J., 2005, 19, 1872–1874 CrossRef CAS PubMed.
- F. Ataollahi, S. Pramanik, A. Moradi, A. Dalilottojari, B. Pingguan-Murphy, W. A. B. Wan Abas and N. A. Abu Osman, J. Biomed. Mater. Res., Part A, 2015, 103, 2203–2213 CrossRef CAS PubMed.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver and P. A. Janmey, Cell Motil. Cytoskeleton, 2005, 60, 24–34 CrossRef PubMed.
- E. A. L. M. Biemans, L. Jäkel, R. M. W. de Waal, H. B. Kuiperij and M. M. Verbeek, J. Neurosci. Res., 2017, 95, 1513–1522 CrossRef CAS PubMed.
- N. J. Abbott, L. Rönnbäck and E. Hansson, Nat. Rev. Neurosci., 2006, 7, 41–53 CrossRef CAS PubMed.
- C. Förster, M. Burek, I. A. Romero, B. Weksler, P. O. Couraud and D. Drenckhahn, J. Physiol., 2008, 586, 1937–1949 CrossRef PubMed.
- S. Bang, S. R. Lee, J. Ko, K. Son, D. Tahk, J. Ahn, C. Im and N. L. Jeon, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- A. Thomas, S. Wang, S. Sohrabi, C. Orr, R. He, W. Shi and Y. Liu, Biomicrofluidics, 2017, 11, 024102 CrossRef PubMed.
- W. Yuan, Y. Lv, M. Zeng and B. M. Fu, Microvasc. Res., 2009, 77, 166–173 CrossRef CAS PubMed.
- H. C. Bauer, I. A. Krizbai, H. Bauer and A. Traweger, Front. Neurosci., 2014, 8, 392 Search PubMed.
- T.-E. Park, D. E. Ingber, J. J. F. Sleeboom, A. D. van der Meer, A. Herland and E. A. FitzGerald, PLoS One, 2016, 11, e0150360 CrossRef PubMed.
- D. E. Eigenmann, G. Xue, K. S. Kim, A. V. Moses, M. Hamburger and M. Oufir, Fluids Barriers CNS, 2013, 10, 33 CrossRef PubMed.
- B. J. van Meer, H. de Vries, K. S. A. Firth, J. van Weerd, L. G. J. Tertoolen, H. B. J. Karperien, P. Jonkheijm, C. Denning, A. P. IJzerman and C. L. Mummery, Biochem. Biophys. Res. Commun., 2017, 482, 323–328 CrossRef CAS PubMed.
- A. Gokaltun, M. L. Yarmush, A. Asatekin and O. B. Usta, Technology, 2017, 05, 1–12 CrossRef PubMed.
- M. W. van der Helm, M. Odijk, J.-P. Frimat, A. D. van der Meer, J. C. T. Eijkel, A. van den Berg and L. I. Segerink, J. Visualized Exp., 2017, 56334 Search PubMed.
- C. Probst, S. Schneider and P. Loskill, Curr. Opin. Biomed. Eng., 2018, 6, 33–41 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00399a |
| This journal is © The Royal Society of Chemistry 2020 |