
Covalent functionalization of carbon materials with redox-active organic molecules for energy storage
Rizwan
Khan
*ab and
Yuta
Nishina
 *ab
*ab
aGraduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushimanaka, Kita-ku, Okayama 700-8530, Japan. E-mail: nisina-y@cc.okayama-u.ac.jp
bResearch Core for Interdisciplinary Sciences, Okayama University, 3-1-1 Tsushimanaka, Kita-ku, Okayama 700-8530, Japan
First published on 24th November 2020
Abstract
Carbon-based materials (CBMs) have shown great versatility because they can be chemically combined with other materials for various applications. Chemical modification of CBMs can be achieved via covalent or non-covalent interactions. Non-covalent interactions are weak and fragile, causing structural change and molecule dissociation. Therefore, in this review, we summarize the covalent modification of CBMs via organic chemistry techniques, aiming at forming more robust and stable CBMs. Besides, their application as electrode materials in energy storage systems is also within the scope of this review. Covalent binding of redox-active organic molecules with CBMs improves the transfer rate of electrons and prevents the dissolution of redox-active molecules, resulting in good conductivity and cycle life. Numerous papers on the functionalization of CBMs have been published to date, but some of them lack scientific evidence and are unable to understand from chemistry viewpoint. Reliable articles with adequate evidence are summarized in this review from a synthetic chemistry viewpoint.
1. Introduction
CBMs have attracted considerable interest in electrochemical applications due to their abundance, processability, stability, and relatively environmentally friendly characteristics.1 CBMs have been widely used as electrode materials. Some of the useful properties of carbon-based electrodes include electrochemical stabilities and wide potential windows for repetitive redox reactions.2 CBMs exist in different allotropic forms, such as 3-D diamond, 3-D graphite, 2-D graphene, 1-D carbon nanotubes (single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs)), and 0-D fullerenes; each of them has different physical and electrochemical properties. The sp2 and/or sp3 hybridized carbons and some of the oxygenated functional groups provide reactive sites for surface functionalization, which is beneficial for electrochemical applications, especially for electrode materials.Covalent functionalization could significantly change the electrical structure and properties of the CBMs.3,4 In particular, acid (e.g., nitric acid or sulfuric acid)-promoted oxidation is one of the most commonly used methods for covalent functionalization.5,6 The carbon atoms in CBMs can form robust covalent bonds with organic molecules, giving rise to a variety of composites with distinguished properties. Functionalization can be applicable for classic materials such as diamond, graphite, carbon black, and porous carbon, as well as for nanocarbons, which came into play in the last two decades. Nowadays, in addition to the nano and porous CBMs, surface functionalized CBMs have also been used for a number of applications such as catalysis,7,8 energy conversion,9,10 sensing,11,12 separation media,13–17 and biomedicine.18–20 However, most of the applications rely on the concept of electrochemistry.21,22
The fundamental components in electrochemical processes are the electrodes. CBMs show high chemical stability,23 good mechanical properties,24,25 and high electrical conductivity.26,27 However, despite the potential of CBMs, a common problem is their low surface wettability,28 which leads to a low utilization rate of the specific surface area and low energy storage. The change of the surface chemistry by functionalization is a common route to optimize the interaction of the carbon surface with the external environment. The modification of the surface chemistry of CBMs strongly improves their interaction with aqueous and organic environments, showing improved performance as electrode materials in energy storage systems.
Covalent modification with organic molecules is expected to allow fine control of the function and physical properties at the atomic or molecular level. The covalent functionalization of CBMs with redox-active materials is achieved by nitrene and diazonium chemistry for sp2 carbons, the ring-opening reaction of epoxide groups, etherification, esterification, amidation and cyclization reaction for oxidized carbon.
The introduction of an aziridine adduct on the CBMs is achieved via a nitrene intermediate.29–31 The nitrene is generated from thermal- or photodecomposition of an azide group by eliminating a gaseous nitrogen molecule and is a highly reactive singlet species which subsequently undergoes a cycloaddition reaction on the sp2 network of CBMs to form an aziridine adduct (Fig. 1a–A).
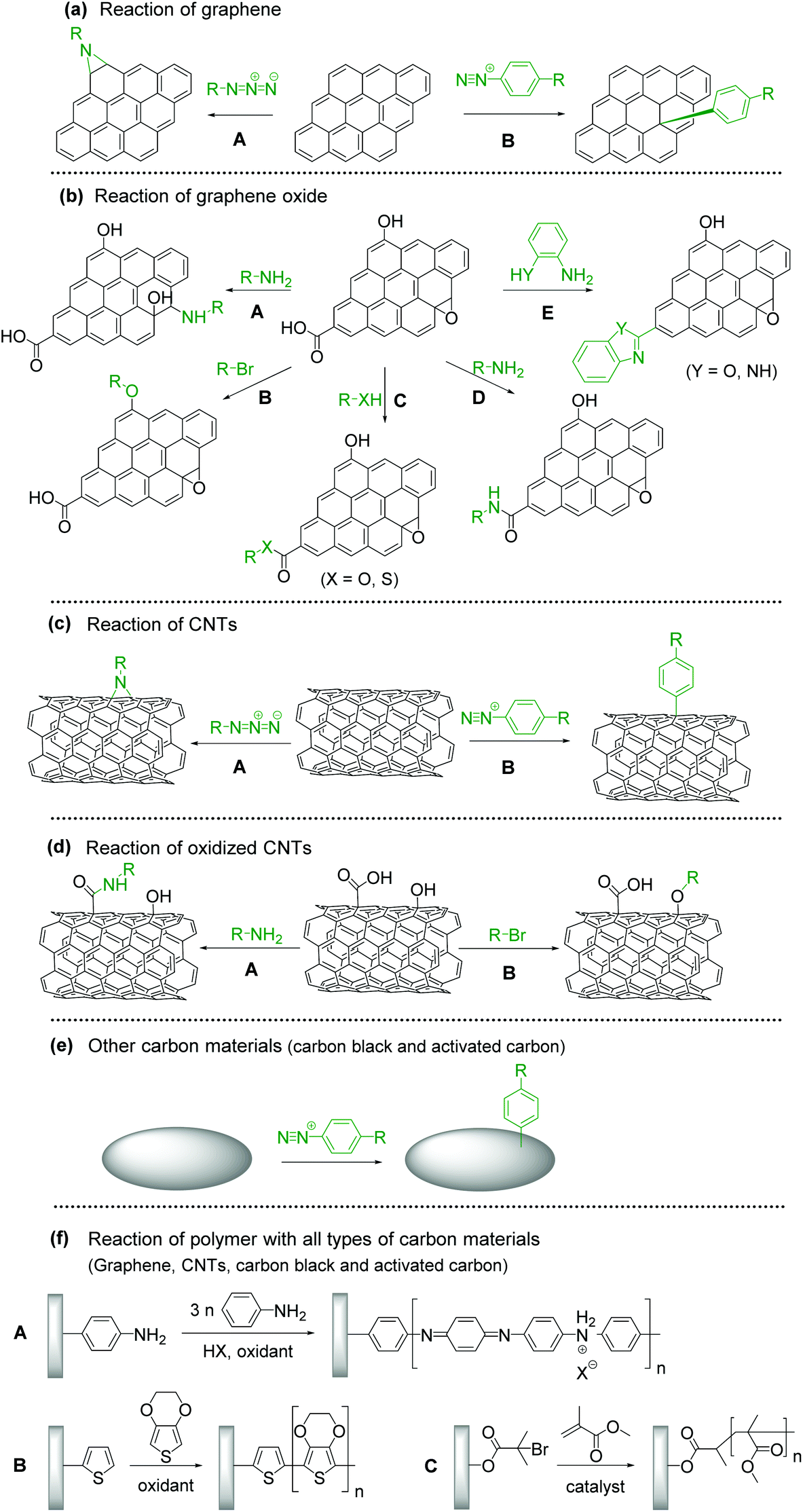 | ||
| Fig. 1 Various functionalization routes of CBMs with redox-active molecules (a) toward graphene, (b) graphene oxide, (c) carbon nanotubes, (d) oxidized carbon nanotubes, and (e) other carbon materials, (f) reaction of polymer with all types of carbon materials. | ||
Surface modification of CBMs with diazonium chemistry has been achieved initially using an isolated diazonium salt and later using in situ-generated one. Although diazonium salt is unstable, it can be easily synthesized with an amine and NaNO2 in an acidic aqueous solution,32tert-butyl nitrite, or NOBF4.33 Diazonium salt generates a radical with the elimination of nitrogen, and then covalently reacts with CBMs (Fig. 1a–B).34–36 The radical formation can be initiated by electrochemical, thermal, or pH-dependent methods. Diazonium salts are important intermediates in organic chemistry, and thus their derivatives are widely available.
Epoxide groups can be functionalized with nucleophilic molecules, which is well known as epoxy curing for the synthesis of epoxy resins. Oxidized CBMs, such as graphene oxide (GO), contain a large amount of epoxides.37 Amine, thiol, or sodium azide can be used as a nucleophile for the ring-opening reaction (Fig. 1b–A).38–40 The reaction proceeds under neutral and mild reaction conditions without any additives or reagents; therefore, this is suitable for bonding unstable molecules such as biomolecules.41,42
The hydroxy groups can undergo etherification. The modified Claisen reaction allows the functionalization of the hydroxy groups of CBMs through an ether bond.43,44 The Williamson reaction enables the formation of an ether bond from an organohalide and a deprotonated alcohol (alkoxide ion) via a substitution reaction (Fig. 1b–B).45 The Williamson synthesis gives the best yields with methyl or primary halides because the reaction occurs by SN2 displacement in which a halide ion is the leaving group. The yield is lower for secondary alkyl halides because they also react with the alkoxide ion in a competing elimination reaction.46
The esterification reaction is a method to functionalize the carboxylic acids on the surface of CBMs (Fig. 1b–C).47,48 For the activation of carboxylic acid, thionyl chloride is employed to obtain acyl chloride. Then, alcohol is added in combination with a base, resulting in the formation of an ester bond.49 Another approach for esterification is a combination of N,N′-dicyclohexylcarbodiimide and N,N-dimethyl-4-aminopyridine, known as the Steglich esterification.50 Similarly, a carboxylic acid group can also be functionalized by a thiol group using thiol–carboxylic-acid esterification.51
Amides are usually prepared by condensation of carboxylic acids and amines using dehydration reagents,52,53 or by prior conversion of carboxylic acids into reactive derivatives.54,55 Alternative procedures include the Staudinger ligation,56 aminocarbonylation of aryl halides,57 and oxidative amidation of aldehydes.58 For the functionalization of CBMs, thionyl chloride activation, coupling reagents, or simple heating is employed (Fig. 1b–D).
Cyclization reactions are promoted by the electrons moving in a circular manner, which includes bond cleavage and bond formation processes simultaneously (Fig. 1b–E).59
CBM–polymer composites are generally prepared through initiation sites on CBMs. In the case of polyaniline (PANI) (Fig. 1f–A) or polythiophene (Fig. 1f–B), an aniline or thiophene group is introduced by diazonium chemistry, epoxy ring-opening, amidation, or esterification, and then oxidative polymerization is performed. Atom transfer radical polymerization is performed for the synthesis of an acrylate-type polymer (Fig. 1f–C).
1.1. Redox-active organic molecules and their operating mechanism
To solve the problem of global energy consumption, the development of high-performance and low-cost clean energy devices is required. According to the growth of renewable energy utilization, demands for advanced energy storage devices with high efficiency are increasing. Lithium-ion batteries (LIBs) and supercapacitors (SCs) are two of the most promising energy storage devices due to their notable characteristics, such as high energy density,60 high power density,61 and long cycle life.62 For instance, widespread success has been achieved for LIBs since their commercialization in the 1990s.63 Current commercial LIBs are constructed with metal-based materials, such as LiMn2O4,64 LiCoO2,65 and LiFePO4,66 on the cathode side and CBMs such as graphite on the anode side. In the case of SCs, pseudocapacitive materials, such as metal oxides, sulfides, selenides and phosphides, are widely explored to enhance the energy density.67 However, the concerns of high cost, limited availability, and electronic hazards of transition metals, as well as poor cycling stability, would hinder their practical use.68 Thus, metal-free redox-active materials for the fabrication of electrodes have been widely studied and can be potential candidates for next-generation energy storage.The first organic electrode was introduced by Williams using dichloroisocyanuric acid as an active material for a primary battery in 1969.69 After this study, a variety of redox-active organic molecules have been studied, including small organic molecules such as quinones and dianhydrides and conjugated polymers such as polypyrrole and polyacetylene. The reported redox-active organic molecules can be categorized into carbonyl compounds,70 organic radicals,71 organosulfur compounds,72 phenazine derivatives,73 and conductive polymers (Fig. 2).74
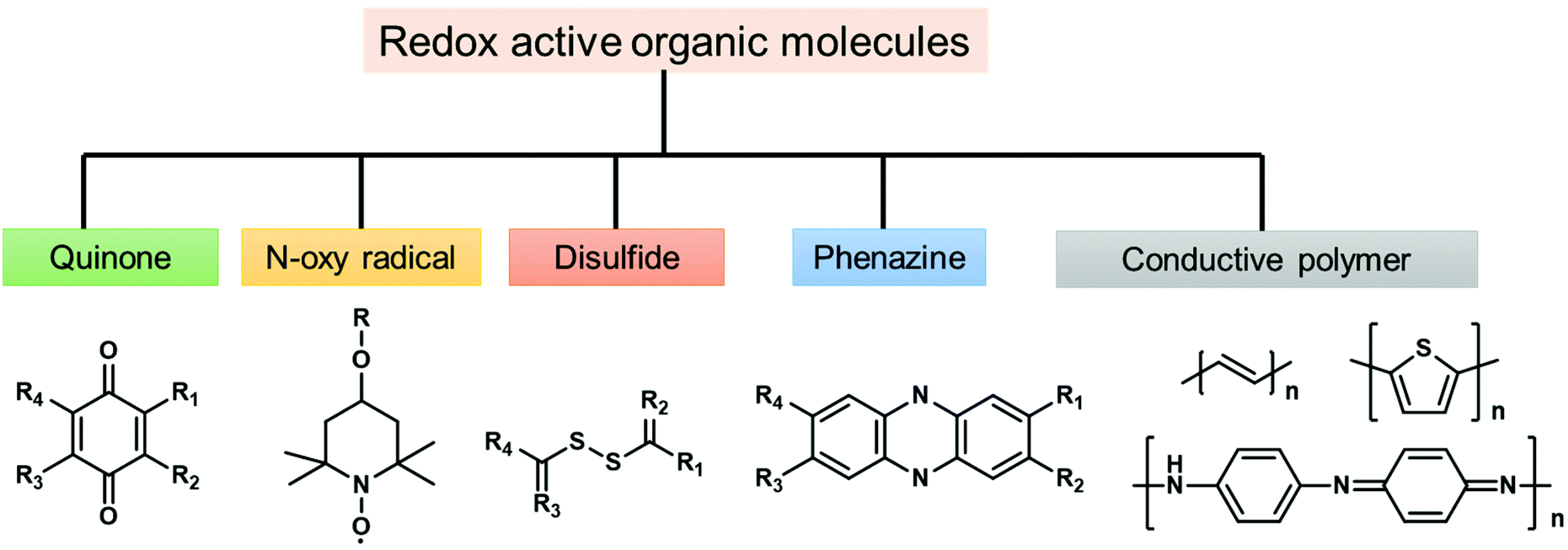 | ||
| Fig. 2 Classification of organic redox-active molecules. | ||
Redox-active organic molecules have attracted more and more interest recently, due to their intrinsic and reliable redox behavior, which can be readily utilized for charge-storage–memory applications. A redox-active molecule contains a redox component acting as the charge-storage center surrounded by insulators/barriers. The electrons tunnel through the barrier during the oxidation and reduction processes. Typically, the application of an oxidation voltage will cause electron loss in the redox molecules; reversely, the electrons will be driven back to the molecules by applying a reduction voltage. Generally, redox molecules have multiple stable states. The switching between these states is dynamically reversible through the loss or capture of charge, that is, oxidation and reduction of the redox centers (Fig. 3).
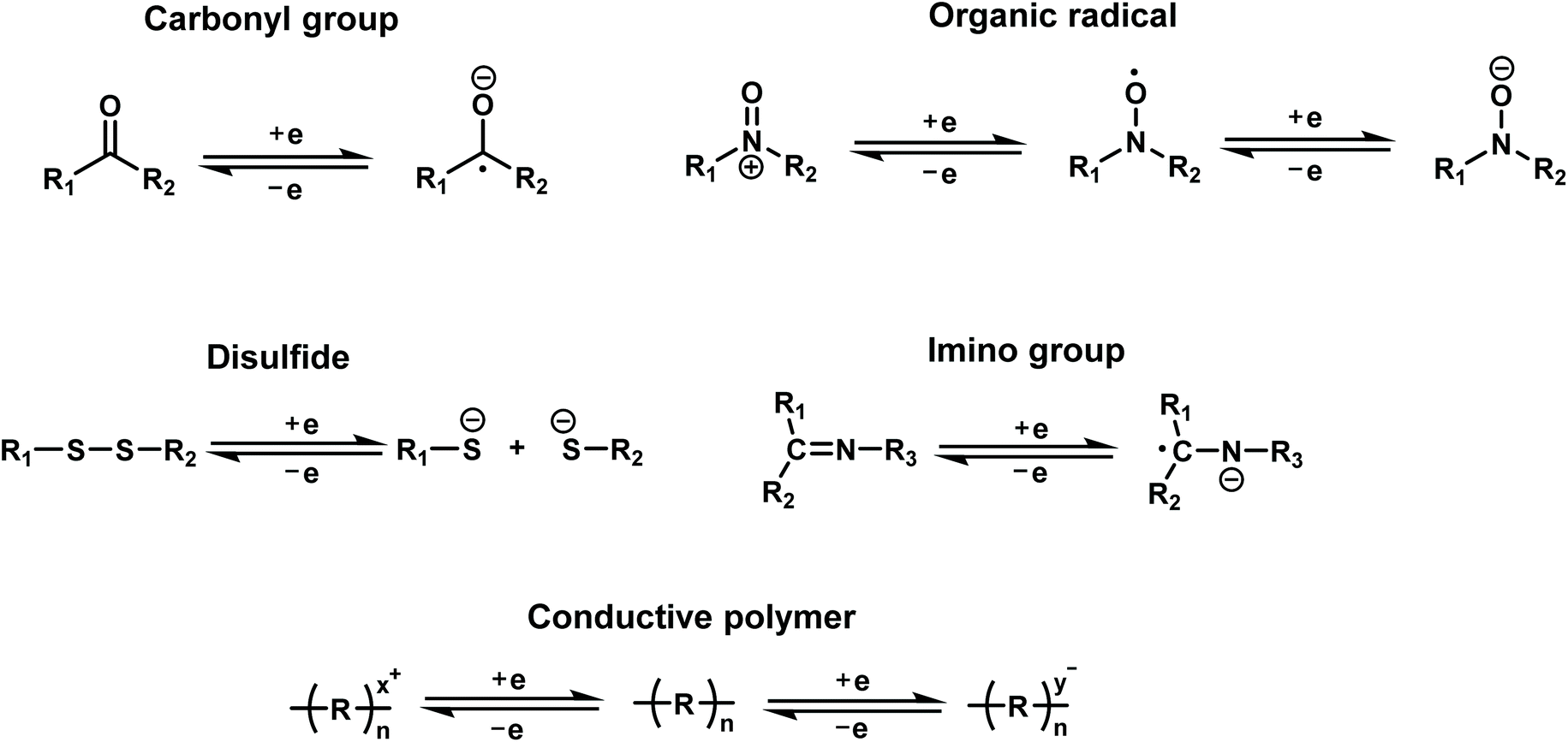 | ||
| Fig. 3 Redox mechanisms of various types of organic redox-active molecules. | ||
To date, several reviews have been published on organic electrodes for energy devices; Schon,75 Shanmukaraj,76 Shea,77 Han,78 and Song79 independently reported the progress of organic electrodes for SCs and LIBs, and all of them have faced difficulties in the development of organic electrodes. First, the organic molecules on electrodes tend to dissolve into the electrolytes, which results in a poor cycle life.80 Second, their poor conductivity results in limited electrochemical performance; as a result, their potential cannot be withdrawn completely due to the insulation effect.81 A covalent functionalization strategy is expected to solve these problems.
1.2. Scope of this review
The organic modification of nanocarbons can be classified into covalent,82 non-covalent,83 and ionic84 bond formations. Organic molecules have been functionalized on nanocarbons through non-covalent interactions and ionic bonding; as a result, high capacity, energy density, and power density were obtained.85 Li,86 Yuan,87 Notarianni,88 and Iqbal89 independently reviewed the progress of carbon-based composites as electrode materials for SCs and LIBs (Fig. 4), focusing mainly on physical (non-covalent) interactions. However, since physical interactions are weak, molecular dissociation occurs readily.90,91 Also, rapid fading of the electrode performance due to the volume change during charge–discharge cycles is a problem that needs to be addressed. The covalent functionalization of CBMs results in the formation of durable materials.92 The electrochemical performance of redox-active organic molecules can be properly regulated by introducing redox-active molecules via diverse organic reactions. Covalent binding of redox-active organic molecules with CBMs improves the transfer rate of electrons and prevents the dissolution of the redox-active materials, resulting in good conductivity and long cycle life.93,94 In this review, we will summarize and discuss the reported literature based on the covalent modification of redox-active organic molecules on CBMs and their electrochemical stability as electrode materials in SCs and LIBs.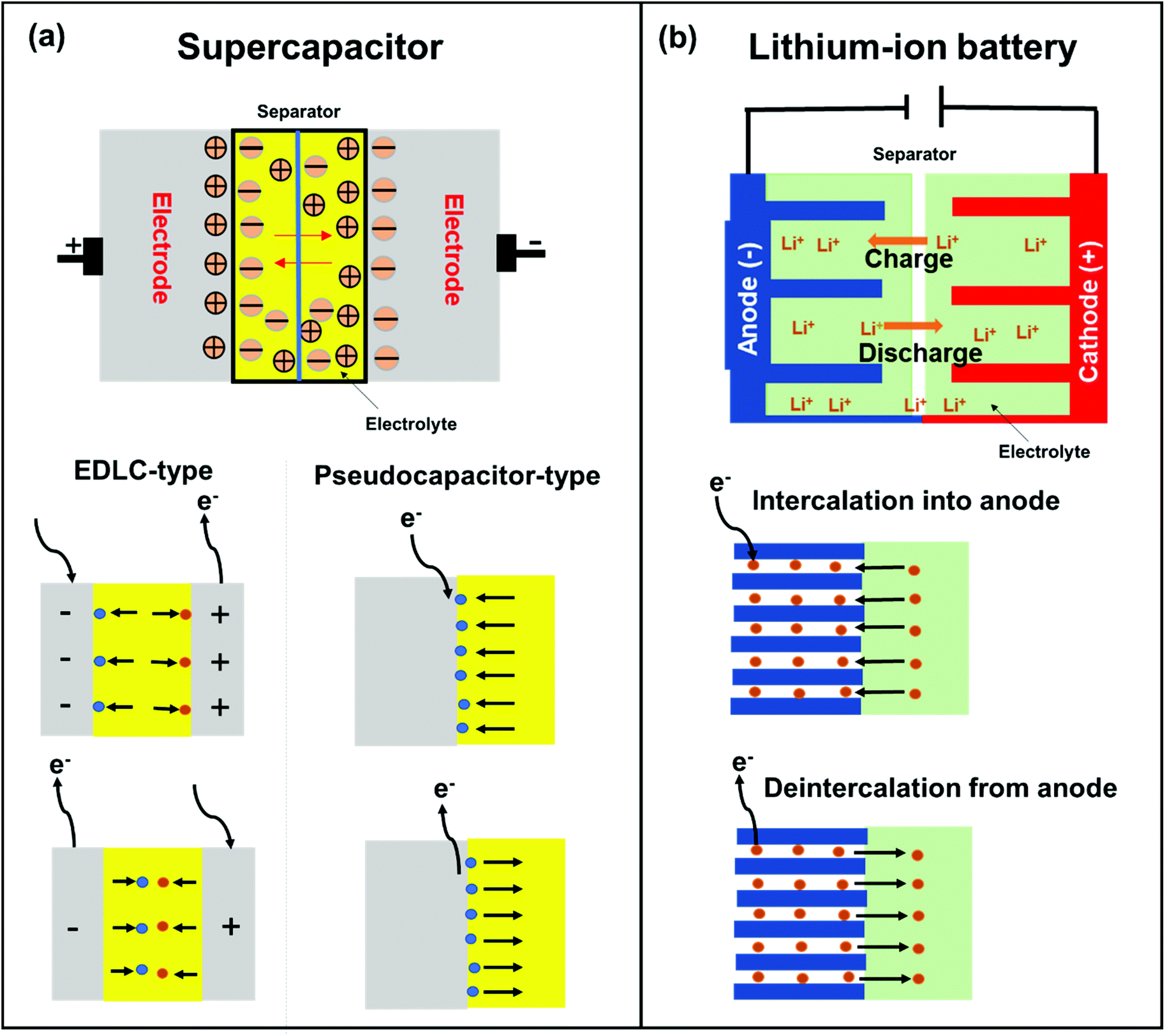 | ||
| Fig. 4 Schematic representation of the working principle of (a) SCs and (b) LIBs. | ||
2. Covalent functionalization of graphene with redox-active organic molecules for SCs
2.1. Small redox-active molecule-functionalized graphene for SCs
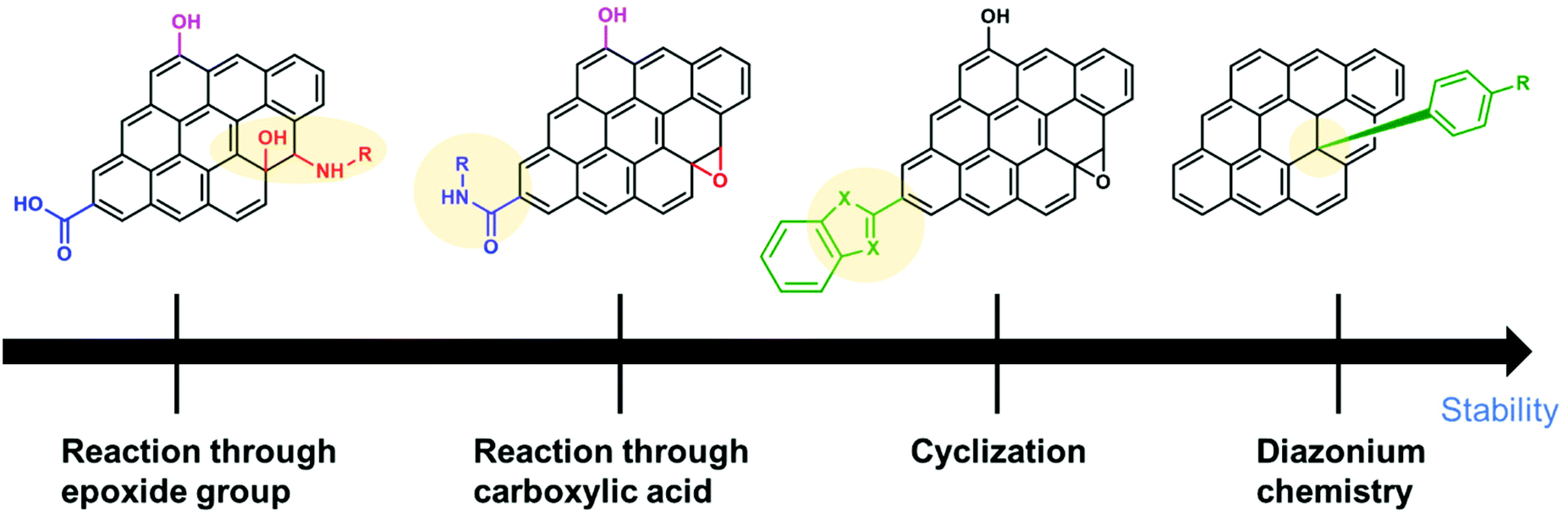
SCs are attractive for high power applications because of their ability to rapidly store/release electrical energies.95–97 High-performance SCs should have large specific capacitances, high-power capabilities, and ultra-long cycle lives. Among the proposed applications of graphene-based materials, SCs, also known as electrochemical capacitors, have attracted much attention over the past decade. Compared with secondary batteries, graphene-based SCs are electrochemical energy storage devices that promise outstanding power density, charge/discharge rate, cycling stability, and operational safety.98 SCs are often utilized individually or in tandem with batteries for energy storage and supply. Based on their energy storage mechanisms, SCs are mainly classified into electrical double-layer capacitors (EDLCs) and pseudocapacitors.99,100 EDLCs store energy by electrostatic charge accumulation at electrode/electrolyte interfaces, while the pseudocapacitance is mainly attributed to the redox reactions at electrode surfaces (Fig. 4a).The covalent attachment of redox-active molecules to graphene has been proven to be a viable means of increasing electron transfer rates and preventing the dissolution of redox-active materials, leading to a good cycle life.101 When redox-active molecules are covalently attached to graphene, they can represent reversible reactions; therefore, electrodes can have both pseudocapacitance and EDLC mechanisms simultaneously. Thus, the energy density of the electrode enhances and results in a stable behavior without any structural disturbance. Selected examples are summarized in Table 1. Reduced graphene oxide (RGO) was functionalized with phenylenediamine (RGO-PD),102 2-aminoanthraquinone (RGO-AAQ),103 and 2-aminopyrene-3,4,9,10-tetraone (RGO-PYT)104 through the nucleophilic ring-opening reaction of the epoxide group and applied for SCs. The resulting composite materials showed capacitance retention values of 92%, 100%, and 100%, respectively. The covalently bonded redox-active moieties contribute to pseudocapacitance. Graphene provides a large specific surface area for forming electrical double layers, works as a host material for redox-active molecules, and promotes charge transfer and electrolyte diffusion. The edges of graphene can be functionalized by a cyclization reaction at carboxylic groups.59,105 The prepared composite material shows a capacitance retention of 98% after 9000 cycles. The cyclization reaction does not destroy the in-plane sp2 framework of graphene, allowing better electrical conductivity. However, this functionalization method requires a phosphoric acid catalyst and multistep reactions. Thiourea functionalized GO (RGO-TU) was prepared through an amide bond.106 The specific capacitance of the material increases slightly in the initial cycles and then becomes stable with no obvious loss in specific capacitance after prolonged cycling. We assume that sulfur-containing thiourea is activated in the first few cycles and undergoes a redox reaction. In another report, adenine (AD) was functionalized on GO (GO-AD) through an amide bond.107 The GO-AD electrode exhibited an excellent cycling retention of 100% after 1000 cycles. However, the reaction was performed at room temperature; therefore, the functionalization might occur only at the epoxide groups instead of carboxyl groups. Another approach for graphene functionalization is diazonium chemistry. Anthraquinone (AQ) molecules were grafted on graphene through diazonium chemistry, forming a covalently linked graphene framework (G-AQ). Covalently linked AQ molecules worked as pillars to construct the graphene framework and prevented the restacking of graphene sheets during fabrication processes. The capacitance retention of G-AQ was 96% after 5000 cycles.108 Similarly, 2-amino-3-chloro-1,4-naphthoquinone (ACNQ) molecules were introduced onto graphene via diazonium chemistry (G-CNQ).109 The electrode exhibited a long cycling life, with nearly no loss after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Diazonium chemistry is simple, fast, and efficient. The key species of the reaction is an aryl radical, which readily adds to carbon frameworks at the basal planes and edges, achieving sufficient introduction of functional groups. To summarize this section, the order of the desirable functionalization is diazonium chemistry > cyclization > amidation reaction > reaction through the epoxide group (Fig. 5), although the investigation may not be comprehensively performed.
000 cycles. Diazonium chemistry is simple, fast, and efficient. The key species of the reaction is an aryl radical, which readily adds to carbon frameworks at the basal planes and edges, achieving sufficient introduction of functional groups. To summarize this section, the order of the desirable functionalization is diazonium chemistry > cyclization > amidation reaction > reaction through the epoxide group (Fig. 5), although the investigation may not be comprehensively performed.
| Material | Functionalization reaction | Cycle number | Capacity retention | Ref. |
|---|---|---|---|---|
| RGO-PD | Ring opening of epoxide | 1000 | 92% | 102 |
| RGO-AAQ | Ring opening of epoxide | 1000 | 100% | 103 |
| RGO-PYT | Ring opening of epoxide | 25000 |
100% | 104 |
| G-BBO | Cyclization (phosphoric acid-catalyzed cyclization reaction) | 9000 | 98% | 59 |
| G-BO | Cyclization (phosphoric acid-catalyzed cyclization reaction) | 2000 | 100% | 105 |
| RGO-TU | Amidation and ring opening of epoxide | 10000 |
100% | 106 |
| GO-AD | Amidation (mixing and heating) | 1000 | 100% | 107 |
| G-AQ | Diazonium chemistry | 5000 | 96% | 108 |
| G-CNQ | Diazonium chemistry | 10000 |
100% | 109 |
 | ||
| Fig. 5 Comparison of different covalent functionalization methods toward electrochemical stability. | ||
2.2. Conductive polymer functionalized graphene for SCs
Polymer-based SCs offer some unique properties such as controllable solubility, precise optimization of potentials, a variety of counter ions, and flexible or even bendable electrodes and, subsequently, devices can be prepared.110 Various graphene–polymer composites with non-covalent and covalent interactions have been synthesized, and high capacitive performances have been achieved. The non-covalent interaction between graphene and polymers limits the charge transfer at the interface and causes volumetric changes during the charge–discharge cycles, reducing the cycling stability when applied as electrode materials.111–114 To overcome this issue, polymers were introduced on graphene through covalent bonds. At the same time, the polymer structure plays important roles in both the stability and capacitance. Various types of polymers have been introduced on graphene through covalent bonds and applied for SCs (Table 2). PANI was functionalized on RGO (RGO-PANI) through introduction of a carboxyl group on RGO and amidation, followed by oxidative polymerization. Diazonium chemistry is widely employed for the covalent functionalization of RGO because it can prevent the graphene sheets from aggregating during functionalization. In addition, RGO can provide an excellent conducting path to the grafted PANI and improve the charge transport of PANI. The cycling stability of the nanocomposites after 1000 cycles was 72% of the initial capacitance.115 Similarly, poly(3,4-ethylenedioxythiophene) was covalently grafted onto RGO, which was prefunctionalized with thiophene by diazonium chemistry (RGO-PEDOT), where thiophene acts as a linker, followed by chemical polymerization. The thiophene unit on GO promotes selective polymerization with EDOT at the 2- and 5-positions of thiophene, forming covalently grafted GO-PEDOT. The prepared material showed a capacitance retention of 80% after 1000 cycles.116 Also, PANI was introduced on RGO using PD as a linker. The capacitance retention was achieved in the range of 82%–90%.117–119 These results suggest that the type of linker affects the electrochemical stability. Using the cyclization reaction, 1,3-bis(2-benzimidazolyl)-5-aminobenzene was functionalized on RGO (RGO-BOA). The capacitance retention of electrode materials was 87% after 5000 cycles.120 Through amide linkage, melamine was functionalized on RGO (RGO-M), and then the nanocomposite with polyorthoaminophenol was prepared (RGO-M-PAP). The prepared material was applied as an electrode, with a capacity retention of 90% after 1000 cycles.121 This result suggests that nitrogen-containing RGO has the potential to improve the electrochemical behavior of SCs. In another work, PPD was functionalized on RGO (RGO-PPD) through an amide bond, using the thionyl chloride activation method, followed by polymerization. The composite exhibits excellent cycling stability, maintaining 90% of its initial capacitance after 1000 cycles.122 The excellent supercapacitor properties rely on the polymer nanoparticles wrapped within or on the graphene surface, providing RGO with a large surface area and a high pore volume. To further improve the electrochemical performance, an organic dopant was introduced into the structure of PANI which is covalently functionalized on the surface of graphene (G-PANI) through diazonium chemistry. The resulting material showed high electrochemical cycling stability, with a capacitance retention of 95% after 1000 cycles.123 The synthesized composites showed a unique hierarchical morphology, which increased the accessible surface area for the redox reaction and allowed faster ion diffusion for excellent electrochemical performance. Recently, we reported a graphene–polymer composite through the ring opening reaction of epoxides (RGO-SBP). The composite was synthesized using a three-step reaction involving a cross-linker, an initiator, and a monomer. The prepared material showed a cycling stability of 98% after 1000 cycles.124 Furthermore, RGO was functionalized with a redox-active thiourea–formaldehyde polymer (TF), yielding a multifunctional hybrid system (RGO-TF). The functionalization of RGO was achieved by thiol–carboxylic-acid esterification. The presence of multiple functional groups such as sulfur, nitrogen, and oxygen provides an additional contribution to the faradaic redox reaction in SCs, leading to effective pseudocapacitances. The prepared material exhibited good cycling stability with a capacitance retention of 100% after 5000 cycles.125 The literature reveals that the most important point for the electrochemical stability is the structure of the polymer along with the covalent connection. The introduction of a suitable linker and construction of a multifunctional polymer increase the stability and specific capacitance of polymer–graphene composites.| Materials | Functionalization reaction | Cycle number | Capacity retention | Ref. |
|---|---|---|---|---|
| RGO-PANI | Diazonium chemistry | 1000 | 72% | 115 |
| RGO-PEDOT | Diazonium chemistry | 1000 | 80% | 116 |
| RGO-PANI | Diazonium chemistry | 2000 | 83% | 117 |
| RGO-PANI | Ring opening of epoxide | 10000 |
82% | 118 |
| RGO-PANI | Ring opening of epoxide | 1000 | 81% | 119 |
| RGO-BOA | Cyclization | 5000 | 88% | 120 |
| RGO-M-PAP | Amidation | 1000 | 90% | 121 |
| RGO-PPD | Amidation (SOCl2 activation) | 1000 | 90% | 122 |
| G-PANI | Diazonium chemistry | 1000 | 95% | 123 |
| RGO-SBP | Ring opening of epoxide | 1000 | 98% | 124 |
| RGO-TF | Esterification | 5000 | 100% | 125 |
3. Covalent functionalization of graphene with redox-active organic molecules for LIBs
3.1. Small redox-active molecule-functionalized graphene for LIBs
Small organic molecules covalently grafted onto the surface of graphene can result in high capacity and maintain excellent cycling stability in LIBs. To date, several approaches for the covalent functionalization of graphene have been reported (Table 3). AQ was grafted on RGO (RGO-AQ) through nitrene chemistry. The material showed a capacity retention of 70% after ten cycles in LIBs.126 The cycling stability of the material decreases significantly, indicating that the formation of RGO-AQ via nitrene is not ideal. To further improve the electrochemical stability, graphene was functionalized with an organic radical, 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (4-hydroxy-TEMPO), through an esterification reaction. This composite has an electrically conductive network of graphene sheets with abundant electrochemically active nitroxide radical functionalities. However, when the prepared material was applied as an electrode, the capacity decreases in the first few cycles, suggesting the instability of the ester bond.127 Esterification was also investigated for the functionalization of tetrahydroxybenzoquinone (THBQ) on GO (GO-THBQ). GO-THBQ was applied as an electrode for LIBs and demonstrated a cycling stability of 68% after 2000 cycles.128 Similarly, carboxyl-enriched RGO was functionalized with a nitroxide radical (RGO-NO) via one-step esterification. The capacity retention of RGO-NO was 89% after 2000 cycles, as derived from the folded RGO-NO structure that shortens the distance of electron transport between radicals.129 These results suggest that ester bonding is not stable. In another report, N,N′-diamino-1,4,5,8-naphthalenetetracarboxylic bisimide (DNTCB)-functionalized GO (GO-DNTCB) was prepared by amidation. The resulting material showed a capacity retention of 85% after 50 cycles in the LIB electrode.130 Similarly, GO was functionalized with naphthalenediimide diamine (NDIDA) through an amide bond (GO-NDIDA), using a conjugated linker. GO-NDIDA exhibits a capacity retention of 100% after 50 cycles with the aid of a conjugated linker (aniline).131 These results suggested that the introduction of a conjugated linker between graphene and redox-active molecules can improve the electrochemical stability. The functionalization of graphene through the amide bond is a better approach for achieving stability in electrochemical reactions. This is because amide is the most stable form of carboxylic acid derivatives.| Material | Functionalization reaction | Cycle number | Capacity retention | Ref. |
|---|---|---|---|---|
| RGO-AQ | Cyclization (nitrene chemistry) | 10 | 70% | 126 |
| GO-TEMPO | Esterification (SOCl2 activation) | 400 | 65% | 127 |
| GO-THBQ | Esterification (simple mixing) | 2000 | 68% | 128 |
| RGO-NO | Esterification (SOCl2 activation) | 200 | 89% | 129 |
| GO-DNTCB | Amidation (coupling reagent) | 50 | 85% | 130 |
| GO-NDIDA | Amidation (coupling reagent) and conductive linker | 50 | 100% | 131 |
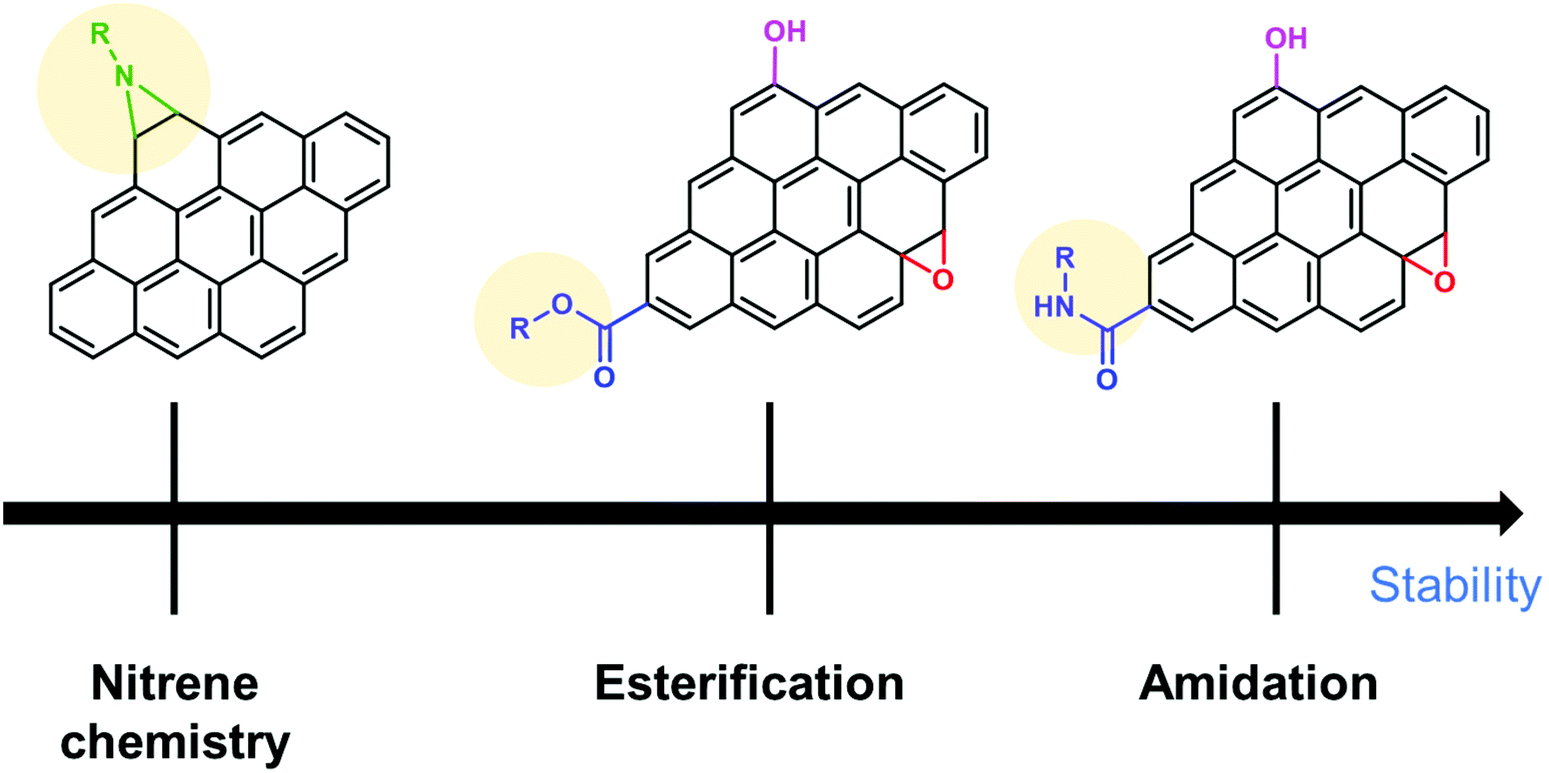
To summarize this section, the order of the desirable functionalization is amidation > esterification > nitrene chemistry (Fig. 6), although there is still room for consideration.
 | ||
| Fig. 6 Comparison of different covalent functionalization methods of graphene toward electrochemical stability. | ||
3.2. Conductive polymer functionalized graphene for LIBs
Surface covalent functionalization of graphene sheets by polymers has been confirmed as an effective approach for maintaining the sheets from restacking in the composite materials, which is a different solution to allow the utilization of the surface groups of graphene sheets.132,133 Covalently grafted graphene with polysulfur (G-PS) was synthesized through inverse vulcanization and applied for ultra-high loading Li-polyS batteries.134 The prepared material showed a capacity retention of 67% after 100 cycles. Graphene sheets improve the conductivity and stability of the polysulfide matrix. The nitroxide radical polymer, poly(2,2,6,6-tetramethylpiperidinyloxy-4-ylmethacrylate) (PTMA), attracts attention as a promising cathode material for high-rate, organic radical batteries, which are also called organic radical polymer batteries.135 Graphene is covalently functionalized with a radical polymer (G-PTMA), via surface-initiated atom transfer radical polymerization, and applied for LIBs. After 250 cycles, the capacity retention of the prepared material was almost 70%. The covalently bonded PTMA could interact with graphene sheets at the molecular level, improving electron and ion transportation. G-PTMA could also inhibit the restacking of graphene sheets during the electrode fabrication process.136 However, this method requires several preparation steps, which is time-consuming, and a metallic catalyst.To summarize this section, the atom transfer radical polymerization reaction showed relatively better electrochemical stability than the synthesis and grafting of polymers through inverse vulcanization (Table 4).
4. Covalent functionalization of other CBMs with redox-active organic molecules for SCs
4.1. Small redox-active molecules functionalized on other CBMs for SCs
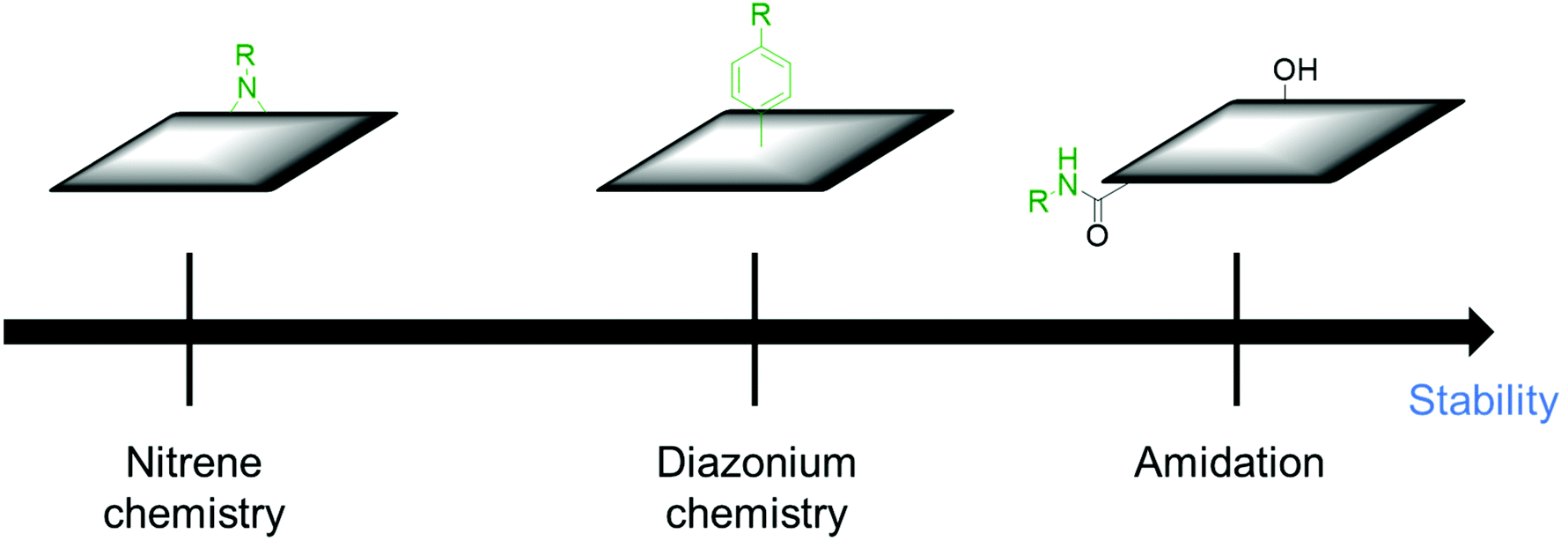
Sections 2 and 3 dealt with the covalent functionalization of graphene. In this section, the covalent functionalization of other CBMs is summarized. The covalent functionalization of other CBMs with redox-active molecules improved the stability and the electrochemical performance of electrode materials. MWCNTs were functionalized with aromatic azides via nitrene chemistry and applied for SCs. The capacitive retention was above 85% after 1000 charge/discharge cycles in different electrolytes.137 Organic molecules having specific functional groups enhance the dispersion stability of MWCNTs in different electrolytes and device performance. Catechol was grafted on high surface area Black Pearls 2000 carbon using spontaneous reduction of catechol diazonium salt in situ prepared in aqueous solution from the corresponding amine (BP-cat). The capacitance retention of the prepared composite was 89% after 2000 cycles.138 Ali et al. functionalized MWCNTs with 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) (MWCNTs-TEMPO), using oxidized MWCNTs and followed by amide bond formation with amino-TEMPO. The capacity retention of the composite was 90% after 4000 cycles in symmetrical SCs.139 The covalent functionalization of MWCNTs with redox-active materials ensures good cycling stability. Carbon nano-onions (CNOs) were functionalized through an amidation reaction that occurs between the oxidized CNOs and 4-(pyren-4-yl)butanehydrazide (Pyr) and used as electrode materials for SCs. The CNO–Pyr electrodes exhibited 99% retention from its initial value after 1000 cycles.140 The functionalization of carbon materials through an amidation reaction is stable under electrochemical conditions. The reason for the high stability of amide bonds is also discussed in section 3.1 (Table 5).To summarize this section, the order of the desirable functionalization in the viewpoint of electrochemical stability is amidation reaction > diazonium chemistry > nitrene chemistry (Fig. 7).
 | ||
| Fig. 7 Comparison of different covalent functionalization methods of other CBMs toward electrochemical stability. | ||
4.2. Conductive polymers functionalized on other CBMs for SCs (Table 6)
Physical mixing of polymers tends to degrade during the long-term charge–discharge process and leads to peeling off from CBMs, deteriorating the electrodes’ performance. The physical contact between CBMs and polymers may also slow down the charge transfer, causing a decrease in the overall performance of the electrodes. In addition, phase-separation generally occurs for polymers, which hampers the formation of uniform composites. Covalent connections of polymers with CBMs are expected to solve these problems. PANI was grafted on MWCNTs through a multi-amino dendrimer (PAMAM) linker, affording MWCNT–PANI electrode composites for SCs. During the successive charging–discharging cycles, the specific capacitance of MWCNT–PANI remained 85% after 50 cycles.141 Covalently functionalized PAMAM on MWCNTs provides more reaction sites for in situ polymerization of ordered PANI, which can shorten the ion diffusion length and lead to the full use of redox-active materials. In order to further improve the electrochemical stability, poly(benzodithiophenylcarbazole) (PBDTC) was grafted on MWCNTs through the Williamson reaction. The capacitance retention of the prepared composite (MWCNTs–PBDTC) was 96% after 5000 cycles.142 However, this method employed several preparation steps and metallic catalysts, which make this process less efficient. In another report, a crosslinked C-CNTs/PANI composite was prepared by diazonium chemistry. In the first step, PD was functionalized with CNTs through diazonium chemistry. In the second step 1,4-benzoquinone was introduced as a spacer between the functionalized CNTs. Finally, the aniline monomer was added and oxidative polymerization was performed on cross-linked CNTs. The composite retains 95% of its initial specific capacitance after 1000 cycles.143 This result suggests that the introduction of a redox-active molecule as a spacer improves the electrochemical stability and electrochemical performance. This also avoids the aggregation of CNTs and contributes to the electrochemical reaction at the same time. The order of the desirable functionalization in the viewpoint of electrochemical stability is etherification ≥ diazonium chemistry > amidation reaction (Fig. 8).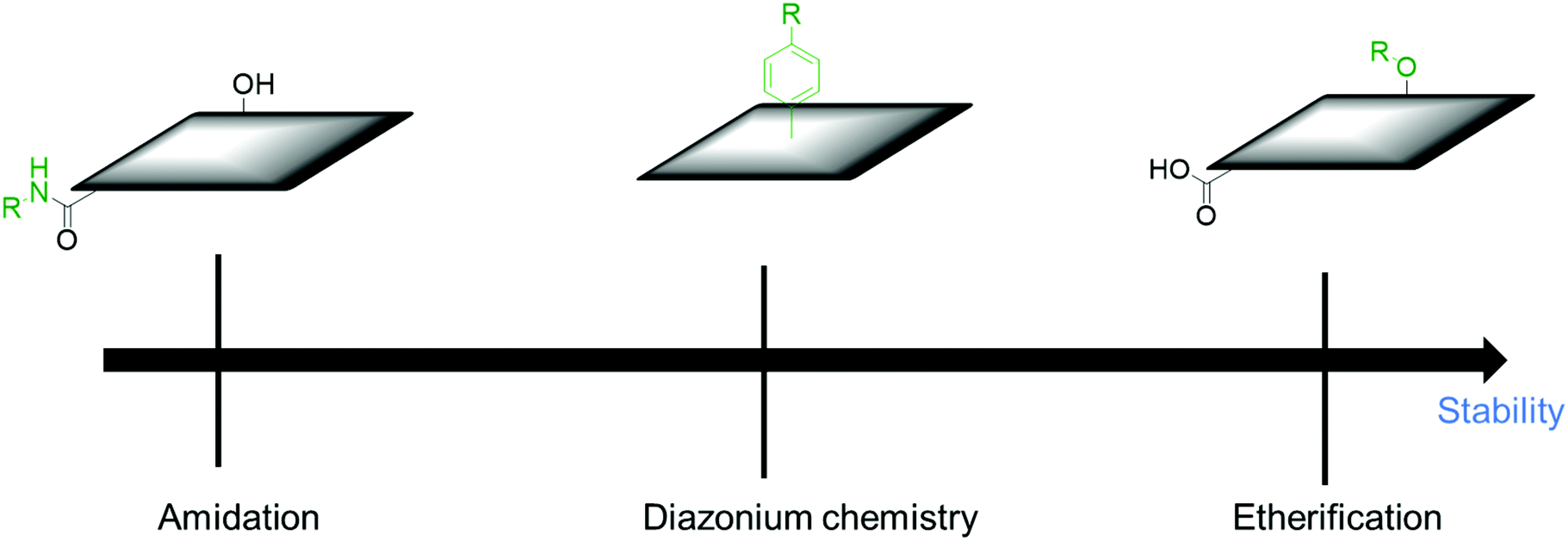 | ||
| Fig. 8 Comparison of different covalent functionalization methods of other CBMs toward electrochemical stability. | ||
5. Covalent functionalization of other CBMs with redox-active organic molecules for LIBs (Table 7)
CBMs show high electronic conductivity and serve as support materials for covalent immobilization of redox-active molecules for LIB applications; however, only a few examples have been reported. 9,10-Phenanthrenequinone (PAQ) was functionalized on Ketjen black (KB-PAQ) through diazonium chemistry and applied for LIBs. The electrode is capable of charging and discharging for 500 cycles, preserving 71% of the initial capacity.94 Covalent functionalization improves the cycling stability of the electrode. MWCNTs were functionalized with a disulfide molecule (DS) through diazonium chemistry (MWCNTs-DS). The electrode material includes disulfide bonds that can react with lithium during the discharge of the batteries. The MWCNTs play the double role of a scaffold for functionalization and of an electronic conductor in the electrodes. The covalently functionalized system presents 95% of the initial capacity after 50 cycles.144 By grafting anthraquinone diazonium salts to MWCNTs (MWCNTs-AQ), 98% of the initial capacity was retained after 50 cycles.145 Besides covalent functionalization, the structure of redox-active molecules also plays a great role in the stability of the electrodes.6. Conclusive remarks in the viewpoint of electrochemical stability
6.1. Covalent interactions vs. non-covalent interactions
The stability and electrochemical performance of the CBM electrodes rely on the functionalization methods. The chemical functionalization of CBMs has been achieved via covalent or non-covalent (including π–π, ionic, and hydrophobic interactions, etc.) bonding to improve the performance of the electrode materials. Here, we take PANI as an example. The covalently grafted GO-PANI by diazonium chemistry showed a capacitance retention as high as 83% over 2000 charge–discharge cycles, while non-grafted (physically mixed) PANI/GO only retained 68% of capacitance after 2000 cycles.117 Furthermore, PANI covalently functionalized with MWCNTs through an amide bond, which retained a capacitance of 85% after 2000 cycles, while non-covalently bonded PANI on MWCNTs retained only 61% of capacitance after 2000 cycles.141 These results suggest that covalent binding of redox-active organic molecules with CBMs improves stability.6.2. Covalent functionalization of small redox-active molecules on CBMs
The dissolution of redox-active components into an electrolyte results in low cycling stability and inadequate electrochemical performance, which have limited the practical use of organic electrode materials. To solve the problem, several strategies have been proposed to alleviate the dissolution, such as using the solid-state electrolytes, optimizing the molecular structure, and polymerization of small molecules into insoluble materials. Covalent binding of redox-active organic molecules to CBMs has been demonstrated to be a promising strategy, which improves the transfer rate of electrons and prevents the dissolution of the redox-active materials. For example, GO was functionalized with THBQ through a covalent bond and applied for LIBs. The covalently functionalized material (GO-THBQ) showed a capacity retention of 68% after 2000 cycles. The cycling stability of THBQ was decreased significantly after a few cycles due to the dissolution in the electrolyte.128 Similarly, the GO-DNTCB composite was prepared through a covalent bond and applied for LIBs. The covalent composite demonstrated a capacity retention of 85% after 50 cycles. In comparison, the capacity retention of the DNTCB electrode without covalent binding was 16%, after 50 cycles, due to significant dissolution in the electrolyte.130 Thus, organic molecules covalently grafted onto the surface of CBMs show excellent cycling stability.6.3. Covalent functionalization of conducting polymers on CBMs
Similarly, conductive polymers suffer from the retention of specific capacitance because of their large volume change and ion dissolution during the charge–discharge processes. To address these shortcomings, polymers are combined with CBMs through covalent bonds, to improve its cycle life. For example, GO-PEDOT was prepared through covalent bonding using diazonium chemistry. Electrochemical analysis suggested that GO-PEDOT showed relatively better cycling stability with a capacitance retention of 80% after 1000 cycles than PEDOT without grafting (69%).116 MWCNTs were functionalized with polymers through covalent bonds and applied for SCs. The covalently functionalized MWCNTs (MWCNTs-PANI) showed a capacity retention of 85% after 2000 cycles. The capacitance of PANI without covalent grafting continued to decrease throughout the cycling process; the value decreased to 35% of its initial value.141 Covalent functionalization provides a high surface area, uniform distribution, and unique structure, which compensate for the swelling and shrinking of the polymer network during cycling.7. Conclusions and future prospects
We have summarized the covalent functionalization of CBMs with redox-active small molecules and polymers and compared their performances for use in SCs and LIBs. CBMs have been functionalized by diazonium chemistry, amidation, the ring-opening reaction of epoxide groups, cyclization, esterification, nitrene chemistry, etherification and a radical reaction (Table 8). Among these reactions, diazonium chemistry has been mostly employed for the functionalization of CBMs because many types of aromatic amines, which are the precursors of diazonium, are commercially available, and the synthesis and use of diazonium salts only necessitate one or a few steps. Amidation is another useful functionalization method of CBMs. The reason may be the well-established carboxyl group transformations; however, amide bonds can be cleaved under acidic conditions by hydrolysis.| Methods for the functionalization of CBMs | Characteristics | Issues |
|---|---|---|
| Diazonium chemistry | Simple, stable, basal-plane functionalization | Disrupt the sp2 framework, side reactions can occur |
| Amidation | Preserve the sp2 framework, well-established chemistry, stable | Functionalization only at the edge, harsh reaction conditions, hydrolysis can occur |
| Ring opening of epoxide | Simple, mild conditions, basal-plane functionalization | Disrupt the sp2 framework, less stable |
| Cyclization | Stable, preserve the sp2 framework | Harsh reaction conditions (phosphoric acid), multistep reaction |
| Esterification | Preserve the sp2 framework, strong | Harsh reaction conditions (SOCl2 activation), less stable (hydrolysis), functionalization only at the edge |
| Nitrene chemistry | Simple, basal-plane functionalization | Harsh reaction conditions (180 °C), less stable |
| Etherification | Stable, basal-plane functionalization | Harsh reaction conditions, disrupt the sp2 framework |
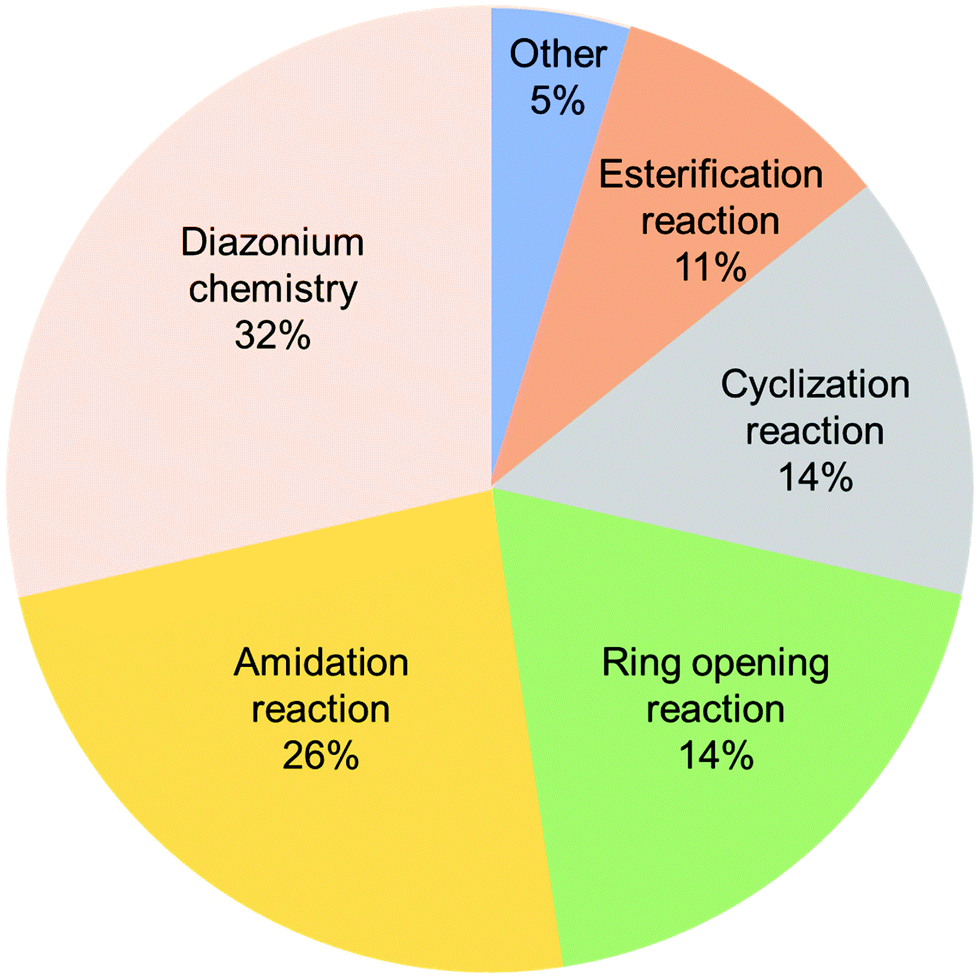
The covalently bonded composites prevent the dissolution of small redox-active molecules, stabilize the polymer structure, and show better electrochemical stability than non-covalently bonded ones. The literature survey reveals that comprehensive research has not been done on the covalent functionalization of CBMs for SC and LIB applications, despite the development of various types of organic transformations in recent years. Only a few traditional reactions have been employed for the functionalization of CBMs (Fig. 9). Interestingly, quite a limited number of papers on the covalent functionalization of classic carbons, such as carbon black, Ketjen black, and activated carbon, have been reported; most of the studies focused on the functionalization of novel nanocarbons, such as graphene and CNTs. Therefore, further investigation is needed combining the traditional and latest chemical transformations and classical carbons and nanocarbons for developing advanced electrochemical electrode materials.
 | ||
| Fig. 9 Methods for the covalent functionalization of CBMs with redox-active molecules. | ||
Abbreviations
| CBMs | Carbon-based materials |
| GO | Graphene oxide |
| RGO | Reduced graphene oxide |
| CNTs | Carbon nanotubes |
| SWCNTs | Single-walled carbon nanotubes |
| MWCNTs | Multi-walled carbon nanotubes |
| SCs | Supercapacitors |
| EDLCs | Electrical double-layer capacitors |
| LIBs | Lithium ion batteries |
| PANI | Polyaniline |
| PD | Phenylenediamine |
| PPD | Poly(phenylenediamine) |
| AAQ | Amino-anthraquinone |
| AQ | Anthraquinone |
| ACNQ | 2-Amino-3-chloro-1,4-naphthoquinone |
| CNQ | 3-Chloro-1,4-naphthoquinone |
| PYT | 2-Aminopyrene-3,4,9,10-tetraone |
| TU | Thiourea |
| AD | Adenine |
| BBO | Benzobisoxazole |
| BO | Benzoxazole |
| PEDOT | Poly(3,4-ethylene dioxythiophene) |
| BOA | 1,3-Bis(2-benzimidazolyl)-5-aminobenzene |
| PAP | Polyorthoaminophenol |
| SAB | N,N′-Bissulphinyl-m-benzenediamine-p-phenylenediamine |
| TF | Thiourea-formaldehyde |
| TEMPO | 2,2,6,6-Tetramethylpiperidin-1-oxyl |
| THBQ | Tetrahydroxybenzoquinone |
| NO | Nitroxide |
| NDIDA | Naphthalenediimide diamine |
| DNTCB | N,N′-Diamino-1,4,5,8-naphthalenetetracarboxylic bisimide |
| PS | Polysulfur |
| PTMA | Poly(2,2,6,6-tetramethylpiperidinyloxy-4-ylmethacrylate |
| CNO | Carbon nano-onions |
| Pyr | 4-(Pyren-4-yl)butanehydrazide |
| PBDTC | Poly(benzodithiophenylcarbazole) |
| PAQ | 9,10-Phenanthrenequinone |
| KB | Ketjen black |
| DS | Disulfide |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Our research was financially supported by JST CREST (JPMJCR18R3) and JSPS KAKENHI (19H02718).References
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672 RSC
.
- P. R. Unwin, A. G. Güell and G. Zhang, Acc. Chem. Res., 2016, 49, 2041–2048 CrossRef CAS
- A. Kasprzak, K. Fateyeva, M. Bystrzejewski, W. Kaszuwara, M. Fronczak, M. Koszytkowska-Stawinska and M. Poplawska, Dalton Trans., 2018, 47, 11190–11202 RSC
- T. J. M. Fraga, M. N. Carvalho, M. G. Ghislandi, M. A. D. M. Sobrinho, T. J. M. Fraga, M. N. Carvalho, M. G. Ghislandi and M. A. da Motta Sobrinho, Braz. J. Chem. Eng., 2019, 36, 1–31 CrossRef CAS
- M. N. Tchoul, W. T. Ford, G. Lolli, D. E. Resasco and S. Arepalli, Chem. Mater., 2007, 19, 5765–5772 CrossRef CAS
- C. Li, X. Chen, L. Shen and N. Bao, ACS Omega, 2020, 5, 3397–3404 CrossRef CAS PubMed
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS
- M. J. Lázaro, S. Ascaso, S. Pérez-Rodríguez, J. C. Calderón, M. E. Gálvez, M. J. Nieto, R. Moliner, A. Boyano, D. Sebastián, C. Alegre, L. Calvillo and V. Celorrio, C. R. Chim., 2015, 18, 1229–1241 CrossRef
- W.-J. Liu, H. Jiang and H. Q. Yu, Energy Environ. Sci., 2019, 12, 1751–1779 RSC
- C. Hu, Y. Xiao, Y. Zou and L. Dai, Electrochem. Energy Rev., 2018, 1, 84–112 CrossRef CAS
- M. D. Angione, R. Pilolli, S. Cotrone, M. Magliulo, A. Mallardi, G. Palazzo, L. Sabbatini, D. Fine, A. Dodabalapur, N. Cioffi and L. Torsi, Mater. Today, 2011, 14, 424–433 CrossRef CAS
- M. Pan, Z. Yin, K. Liu, X. Du, H. Liu and S. Wang, Nanomaterials, 2019, 9, 1330 CrossRef CAS PubMed
- E. Kanao, T. Kubo and K. Otsuka, Bull. Chem. Soc. Jpn., 2020, 93, 482–489 CrossRef CAS
- X. Yang, Y. Wan, Y. Zheng, F. He, Z. Yu, J. Huang, H. Wang, Y. S. Ok, Y. Jiang and B. Gao, Chem. Eng. J., 2019, 366, 608–621 CrossRef CAS
- A. Azari, R. Nabizadeh, S. Nasseri, A. H. Mahvi and A. R. Mesdaghinia, Chemosphere, 2020, 250, 126238 CrossRef CAS PubMed
- R. Gusain, N. Kumar and S. S. Ray, Coord. Chem. Rev., 2020, 405, 213111 CrossRef CAS
- M. Selvaraj, A. Hai, F. Banat and M. A. Haija, J. Water Process Eng., 2020, 33, 100996 CrossRef
- C. Cha, S. R. Shin, N. Annabi, M. R. Dokmeci and A. Khademhosseini, ACS Nano, 2013, 7, 2891–2897 CrossRef CAS PubMed
- A. M. Monaco and M. Giugliano, Beilstein J. Nanotechnol., 2014, 5, 1849–1863 CrossRef
- L. S. Porto, D. N. Silva, A. E. F. de Oliveira, A. C. Pereira and K. B. Borges, Rev. Anal. Chem., 2020, 38 DOI:10.1515/revac-2019-0017
- Y. E. Jeun, B. Baek, M. W. Lee and H. S. Ahn, Chem. Commun., 2018, 54, 10052–10055 RSC
- M. Starowicz, B. Stypuła and J. Banaś, Electrochem. Commun., 2006, 8, 227–230 CrossRef CAS
- L. Fagiolari and F. Bella, Energy Environ. Sci., 2019, 12, 3437–3472 RSC
- X. Zhang, L. Ge, Y. Zhang and J. Wang, Materials, 2019, 12, 2019 CrossRef PubMed
- A. Takakura, K. Beppu, T. Nishihara, A. Fukui, T. Kozeki, T. Namazu, Y. Miyauchi and K. Itami, Nat. Commun., 2019, 10, 3040 CrossRef PubMed
- I. M. Alarifi, J. Mater. Res. Technol., 2019, 8, 4863–4893 CrossRef CAS
- S. Zhang, W. Xiao, Y. Zhang, K. Liu, X. Zhang, J. Zhao, Z. Wang, P. Zhang and G. Shao, J. Mater. Chem. A, 2018, 6, 22555–22565 RSC
- J. Feng and Z. Guo, Nanoscale Horiz., 2019, 4, 339–364 RSC
- W. Yang, P. Thordarson, J. J. Gooding, S. P. Ringer and F. Braet, Nanotechnology, 2007, 18, 412001 CrossRef
- J. Han and C. Gao, Nano-Micro Lett., 2010, 2, 213–226 CrossRef
- S. K. Yadav, I. J. Kim, H. J. Kim, J. Kim, S. M. Hong and C. M. Koo, J. Mater. Chem. C, 2013, 1, 5463–5470 RSC
- M. He and T. M. Swager, Chem. Mater., 2016, 28, 8542–8549 CrossRef CAS
- A. Bensghaïer, F. Mousli, A. Lamouri, P. S. Postnikov and M. M. Chehimi, Chem. Afr., 2020, 1–35 Search PubMed
- A. A. Golosova, C. M. Papadakis and R. Jordan, MRS Online Proc. Libr., 2011, 1362, 1114–1120 Search PubMed
- J. C. Padilha, J.-M. Noël, J. F. Bergamini, J. Rault-Berthelot and C. Lagrost, ChemElectroChem, 2016, 3, 572–580 CrossRef
- M. Flores-Guerrero, L. E. Elizalde, A. Elías-Zuñiga, R. Ledezma, G. D. L. Santos and C. Avila-Orta, Des. Monomers Polym., 2014, 17, 416–424 CrossRef CAS
- D. S. Shin, H. G. Kim, H. S. Ahn, H. Y. Jeong, Y.-J. Kim, D. Odkhuu, N. Tsogbadrakh, H. B. R. Lee and B. H. Kim, RSC Adv., 2017, 7, 13979–13984 RSC
- J. M. Jebaranjitham, C. Mageshwari, R. Saravanan and N. Mu, Composites, Part B, 2019, 171, 302–309 CrossRef
- I. A. Vacchi, C. Spinato, J. Raya, A. Bianco and C. Ménard-Moyon, Nanoscale, 2016, 8, 13714–13721 RSC
- A. de Leon, M. Mellon, J. Mangadlao, R. Advincula and E. Pentzer, RSC Adv., 2018, 8, 18388–18395 RSC
- N. D. Q. Chau, G. Reina, J. Raya, I. A. Vacchi, C. Ménard-Moyon, Y. Nishina and A. Bianco, Carbon, 2017, 122, 643–652 CrossRef CAS
- G. Reina, N. D. Q. Chau, Y. Nishina and A. Bianco, Nanoscale, 2018, 10, 5965–5974 RSC
- W. Zheng, B. Shen and W. Zhai, New Prog. Graphene Res., 2013 DOI:10.5772/50490
- K.-C. Mei, N. Rubio, P. M. Costa, H. Kafa, V. Abbate, F. Festy, S. S. Bansal, R. C. Hider and K. T. Al-Jamal, Chem. Commun., 2015, 51, 14981–14984 RSC
- I. A. Vacchi, J. Raya, A. Bianco and C. Ménard-Moyon, 2D Mater., 2018, 5, 035037 CrossRef
- P. J. Boratyński, M. Zielińska-Błajet and J. Skarżewski, Chem. Biol., 2019, 82, 29–145 Search PubMed
- K. Balasubramanian and M. Burghard, Small, 2005, 1, 180–192 CrossRef CAS PubMed
- Y. Li, J. Li, Y. Li, Y. Li, Y. Song, S. Niu and N. Li, Ultrason. Sonochem., 2018, 40, 798–807 CrossRef CAS PubMed
- A. Kasprzak, A. Zuchowska and M. Poplawska, Beilstein J. Org. Chem., 2018, 14, 2018–2026 CrossRef CAS PubMed
- C. Bonneaud, M. Decostanzi, J. Burgess, G. Trusiano, T. Burgess, R. Bongiovanni, C. Joly-Duhamel and C. M. Friesen, RSC Adv., 2018, 8, 32664–32671 RSC
- M. Sayes and A. B. Charette, Green Chem., 2017, 19, 5060–5064 RSC
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS
- C. C. Caliman, A. F. Mesquita, D. F. Cipriano, J. C. C. Freitas, A. A. C. Cotta, W. A. A. Macedo and A. O. Porto, RSC Adv., 2018, 8, 6136–6145 RSC
- B. K. Zambroń, S. R. Dubbaka, D. Marković, E. Moreno-Clavijo and P. Vogel, Org. Lett., 2013, 15, 2550–2553 CrossRef
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC
- R. R. A. Rios, D. E. Alves, I. Dalmázio, S. F. V. Bento, C. L. Donnici and R. M. Lago, Mater. Res., 2003, 6, 129–135 CrossRef CAS
- S. Jamalifard, J. Mokhtari and Z. Mirjafary, RSC Adv., 2019, 9, 22749–22754 RSC
- M. Xuan, C. Lu, M. Liu and B.-L. Lin, J. Org. Chem., 2019, 84, 7694–7701 CrossRef CAS PubMed
- W. Ai, W. Zhou, Z. Du, Y. Du, H. Zhang, X. Jia, L. Xie, M. Yi, T. Yu and W. Huang, J. Mater. Chem., 2012, 22, 23439–23446 RSC
- Y. Qiao, K. Jiang, H. Deng and H. Zhou, Nat. Catal., 2019, 2, 1035–1044 CrossRef CAS
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef
- Y. Zhao, C. Wu, J. Li and L. Guan, J. Mater.
Chem. A, 2013, 1, 3856–3859 RSC
- J. B. Goodenough, Nat. Electron., 2018, 1, 204–204 CrossRef
- J. Drews, R. Wolf, G. Fehrmann and R. Staub, J. Power Sources, 1997, 65, 129–132 CrossRef CAS
- J. Qian, L. Liu, J. Yang, S. Li, X. Wang, H. L. Zhuang and Y. Lu, Nat. Commun., 2018, 9, 1–11 CrossRef
- C.-T. Hsieh, C.-T. Pai, Y.-F. Chen, P.-Y. Yu and R.-S. Juang, Electrochim. Acta, 2014, 115, 96–102 CrossRef CAS
- J. H. Kim, S. Lee, J. Choi, T. Song and U. Paik, J. Mater. Chem. A, 2015, 3, 20459–20464 RSC
- M. E. Bhosale, S. Chae, J. M. Kim and J.-Y. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911 RSC
- D. L. Williams, J. J. Byrne and J. S. Driscoll, J. Electrochem. Soc., 1969, 116, 2 CrossRef CAS
- L. I. U. Mengyun, G. U. Tiantian, Z. Min, W. Kangli, C. Shijie and J. Kai, Energy Storage Sci. Technol., 2018, 7, 1171 Search PubMed
- C. Friebe and U. S. Schubert, Top. Curr. Chem., 2017, 375, 19–19 CrossRef
- B. Boateng, Y. Han, C. Zhen, G. Zeng, N. Chen, D. Chen, C. Feng, J. Han, J. Xiong, X. Duan and W. He, Nano Lett., 2020, 20, 2594–2601 CrossRef CAS PubMed
- H. Wang, J. Lin and Z. X. Shen, J. Sci. Adv. Mater. Dev., 2016, 1, 225–255 Search PubMed
- M. Lee, J. Hong, B. Lee, K. Ku, S. Lee, C. B. Park and K. Kang, Green Chem., 2017, 19, 2980–2985 RSC
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC
- D. Shanmukaraj, P. Ranque, H. B. Youcef, T. Rojo, P. Poizot, S. Grugeon, S. Laruelle and D. Guyomard, J. Electrochem. Soc., 2020, 167, 070530 CrossRef CAS
- J. J. Shea and C. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 5361–5380 CrossRef CAS PubMed
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, J. Mater. Chem. A, 2019, 7, 23378–23415 RSC
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed
- D. Tuncel, Nanoscale, 2011, 3, 3545–3554 RSC
- Z. Liu, T. Rios-Carvajal, M. P. Andersson, M. Ceccato, S. L. S. Stipp and T. Hassenkam, Environ. Sci.: Nano, 2019, 6, 2281–2291 RSC
- F. Tournus, S. Latil, M. I. Heggie and J.-C. Charlier, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 075431 CrossRef
- Q. Li, M. Horn, Y. Wang, J. MacLeod, N. Motta and J. Liu, Materials, 2019, 12, 703 CrossRef CAS PubMed
- W. Yuan, Y. Zhang, L. Cheng, H. Wu, L. Zheng and D. Zhao, J. Mater. Chem. A, 2016, 4, 8932–8951 RSC
- M. Notarianni, J. Liu, K. Vernon and N. Motta, Beilstein J. Nanotechnol., 2016, 7, 149–196 CrossRef CAS PubMed
- S. Iqbal, H. Khatoon, A. H. Pandit and S. Ahmad, Mater. Sci. Energy Technol., 2019, 2, 417–428 Search PubMed
- C. A. Dyke and J. M. Tour, J. Phys. Chem. A, 2004, 108, 11151–11159 CrossRef CAS
- G. Speranza, J. Carbon Res., 2019, 5, 84 CrossRef
- I. A. Vacchi, C. Ménard-Moyon and A. Bianco, Phys. Sci. Rev., 2017, 2, 035037 Search PubMed
- A. Bakandritsos, P. Jakubec, M. Pykal and M. Otyepka, FlatChem, 2019, 13, 25–33 CrossRef CAS
- A. Jaffe, A. S. Valdes and H. I. Karunadasa, Chem. Mater., 2015, 27, 3568–3571 CrossRef CAS
- N. A. Elessawy, J. E. Nady, W. Wazeer and A. B. Kashyout, Sci. Rep., 2019, 9, 1129 CrossRef PubMed
- R. Vellacheri, A. Al-Haddad, H. Zhao, W. Wang, C. Wang and Y. Lei, Nano Energy, 2014, 8, 231–237 CrossRef CAS
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623–1632 RSC
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983–5992 RSC
- H. Ji, X. Zhao, Z. Qiao, J. Jung, Y. Zhu, Y. Lu, L. L. Zhang, A. H. MacDonald and R. S. Ruoff, Nat. Commun., 2014, 5, 3317 CrossRef PubMed
- Y. M. Volfkovich, A. A. Mikhailin, D. A. Bograchev, V. E. Sosenkin and V. S. Bagotsky, Recent Trend Electrochem. Sci. Technol., 2012, 159 DOI:10.5772/1891
- S. Seo, M. Min, S. M. Lee and H. Lee, Nat. Commun., 2013, 4, 1920 CrossRef PubMed
- M. M. Sk and C. Y. Yue, RSC Adv., 2014, 4, 19908–19915 RSC
- Q. Wu, Y. Sun, H. Bai and G. Shi, Phys. Chem. Chem. Phys., 2011, 13, 11193–11198 RSC
- J. Shi, Z. Zhao, J. Wu, Y. Yu, Z. Peng, B. Li, Y. Liu, H. Kang and Z. Liu, ACS Sustainable Chem. Eng., 2018, 6, 4729–4738 CrossRef CAS
- W. Ai, X. Cao, Z. Sun, J. Jiang, Z. Du, L. Xie, Y. Wang, X. Wang, H. Zhang, W. Huang and T. Yu, J. Mater. Chem. A, 2014, 2, 12924–12930 RSC
- W. S. V. Lee, M. Leng, M. Li, X. L. Huang and J. M. Xue, Nano Energy, 2015, 12, 250–257 CrossRef CAS
- D. M. El-Gendy, N. A. A. Ghany, E. E. F. E. Sherbini and N. K. Allam, Sci. Rep., 2017, 7, 43104 CrossRef CAS PubMed
- Y. Qin, J. Li, X. Jin, S. Jiao, Y. Chen, W. Cai and R. Cao, Ceram. Int., 2020, 46, 15379–15384 CrossRef CAS
- L. Hou, Z. Hu, H. Wu, X. Wang, Y. Xie, S. Li, F. Ma and C. Zhu, Dalton Trans., 2019, 48, 9234–9242 RSC
- J. Kim, J. H. Kim and K. Ariga, Nanoarchitectonics, Joule, 2017, 1, 739–768 CrossRef CAS
- D. Majumdar, Innov. Energy Res., 2016, 5, 1–9 Search PubMed
- Q. Zhao, J. Chen, F. Luo, L. Shen, Y. Wang, K. Wu and M. Lu, J. Appl. Polym. Sci., 2017, 134, 44808 Search PubMed
- Y. Teng, S. Li, C. Xue, H. Zhang, L. Zhu and Y. Tang, Adv. Polym. Mater., 2017, 134 DOI:10.1155/2020/8730852
- X. Liu, Y. Zheng and X. Wang, Chem. – Eur. J., 2015, 21, 10408–10415 CrossRef CAS PubMed
- R. Li, Y. Yang, D. Wu, K. Li, Y. Qin, Y. Tao and Y. Kong, Chem. Commun., 2019, 55, 1738–1741 RSC
- M. Wang, R. Jamal, Y. Wang, L. Yang, F. Liu and T. Abdiryim, Nanoscale Res. Lett., 2015, 10, 370 CrossRef PubMed
- Z.-F. Li, H. Zhang, Q. Liu, Y. Liu, L. Stanciu and J. Xie, Carbon, 2014, 71, 257–267 CrossRef CAS
- Y. Zou, R. Liu, W. Zhong and W. Yang, J. Mater. Chem. A, 2018, 6, 8568–8578 RSC
- C. Wang, Y. Yang, R. Li, D. Wu, Y. Qin and Y. Kong, Chem. Commun., 2020, 56, 4003–4006 RSC
- A. Roy, S. Dhibar, S. Kundu and S. Malik, RSC Adv., 2019, 9, 24646–24653 RSC
- F. B. Ajdari, E. Kowsari, A. Ehsani, L. Chepyga, M. Schirowski, S. Jäger, O. Kasian, F. Hauke and T. Ameri, Appl. Surf. Sci., 2018, 459, 874–883 CrossRef CAS
- Z. Liu, H. Zhou, Z. Huang, W. Wang, F. Zeng and Y. Kuang, J. Mater. Chem. A, 2013, 1, 3454–3462 RSC
- A. Mohammadi, S. J. Peighambardoust, A. A. Entezami and N. Arsalani, J. Mater. Sci.: Mater. Electron., 2017, 28, 5776–5787 CrossRef CAS
- R. Khan and Y. Nishina, J. Mater. Chem. A, 2020, 8, 13718–13724 RSC
- S. Witomska, Z. Liu, W. Czepa, A. Aliprandi, D. Pakulski, P. Pawluć, A. Ciesielski and P. Samorì, J. Am. Chem.
Soc., 2019, 141, 482–487 CrossRef CAS PubMed
- S. B. Sertkol, B. Esat, A. A. Momchilov, M. B. Yılmaz and M. Sertkol, Carbon, 2017, 116, 154–166 CrossRef
- Z. Du, W. Ai, L. Xie and W. Huang, J. Mater. Chem. A, 2014, 2, 9164–9168 RSC
- Y. Wang, X. Li, L. Chen, Z. Xiong, J. Feng, L. Zhao, Z. Wang and Y. Zhao, Carbon, 2019, 155, 445–452 CrossRef CAS
- C. Lu, G. Pan, Q. Huang, H. Wu, W. Sun, Z. Wang and K. Sun, J. Mater. Chem. A, 2019, 7, 4438–4445 RSC
- Y. Song, Y. Hu, Y. Sha, H. Rong, H. Wen, H.-J. Liu and Q. Liu, Ionics, 2019, 25, 2987–2995 CrossRef CAS
- Y. Song, Y. Gao, H. Rong, H. Wen, Y. Sha, H. Zhang, H.-J. Liu and Q. Liu, Sustainable Energy Fuels, 2018, 2, 803–810 RSC
- A. Maio, R. Fucarino, R. Khatibi, S. Rosselli, M. Bruno and R. Scaffaro, Compos. Sci. Technol., 2015, 119, 131–137 CrossRef CAS
- J. L. Suter, R. C. Sinclair and P. V. Coveney, Adv. Mater., 2020, 32, 2003213 CrossRef CAS PubMed
- C. H. Chang and A. Manthiram, ACS Energy Lett., 2018, 3, 72–77 CrossRef CAS
- J.-K. Kim, G. Cheruvally, J.-H. Ahn, Y.-G. Seo, D. Choi, S.-H. Lee and C. Song, J. Ind. Eng. Chem., 2008, 14, 371–376 CrossRef CAS
- Y. Li, Z. Jian, M. Lang, C. Zhang and X. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 17352–17359 CrossRef CAS PubMed
- S. K. Ujjain, R. Bhatia, P. Ahuja and P. Attri, PLoS One, 2015, 10, e0131475 CrossRef PubMed
- G. Pognon, C. Cougnon, D. Mayilukila and D. Bélanger, ACS Appl. Mater. Interfaces, 2012, 4, 3788–3796 CrossRef CAS PubMed
- G. A. M. Ali, E. Megiel, J. Romański, H. Algarni and K. F. Chong, J. Mol. Liq., 2018, 271, 31–39 CrossRef CAS
- J. D. Velásquez, M. Tomczykowa, M. E. Plonska-Brzezinska and M. N. Chaur, Materials, 2020, 13, 1141 CrossRef PubMed
- L. Jin, Y. Jiang, M. Zhang, H. Li, L. Xiao, M. Li and Y. Ao, Sci. Rep., 2018, 8, 6268 CrossRef PubMed
- T. Ye, Y. Sun, X. Zhao, B. Lin, H. Yang, X. Zhang and L. Guo, J. Mater. Chem. A, 2018, 6, 18994–19003 RSC
- D. Liu, X. Wang, J. Deng, C. Zhou, J. Guo and P. Liu, Nanomaterials, 2015, 5, 1034–1047 CrossRef CAS PubMed
- G. Charrier, H. Kamaleddine, C. Barchasz, R. Cornut, B. Jousselme and S. Campidelli, ChemElectroChem, 2018, 5, 1732–1737 CrossRef CAS
- G. Charrier, A. Desrues, C. Barchasz, J. Leroy, R. Cornut, B. Jousselme and S. Campidelli, J. Mater. Chem. A, 2016, 4, 15036–15040 RSC
| This journal is © The Royal Society of Chemistry 2021 |