
Silicon monophosphides with controlled size and crystallinity for enhanced lithium anodic performance†
Huanhuan
Yang‡
ab,
Binlu
Yu‡
ac,
Shuang
Gu
a,
Hao
Huang
ab,
Yanli
Zhang
a,
Danni
Liu
a,
Xue
Zhang
a,
Yihong
Kang
ab,
Jiahong
Wang
 *abd,
Paul K.
Chu
d and
Xue-Feng
Yu
*ab
*abd,
Paul K.
Chu
d and
Xue-Feng
Yu
*ab
aShenzhen Engineering Center for the Fabrication of Two-Dimensional Atomic Crystals, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P. R. China. E-mail: xf.yu@siat.ac.cn; jh.wang1@siat.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cSchool of Microelectronics, Southern University of Science and Technology, Shenzhen 518055, China
dDepartment of Physics and Department of Materials Science and Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China
First published on 23rd November 2020
Abstract
New electrode materials are crucial to high-performance lithium-ion batteries (LIBs). Silicon monophosphides (SiPs), composed of silicon and phosphorus, have a very high theoretical capacity (3060 mA h g−1), which is more than 8 times that of graphite (372 mA h g−1). The two-dimensional structure of SiPs also benefits ion transportation and diffusion. In this work, the chemical vapor transport (CVT) method is employed to synthesize SiPs for LIB anodes, and the lithium storage capacity co-affected by size and crystallinity is investigated using controllably synthesized thin belts and bulk crystals. The SiPs prepared by the high-temperature iodine-assisted CVT method have a belt-like morphology about 72 nm thick. After 200 cycles, the stable capacity is about 615 mA h g−1 at 100 mA g−1, and a reversible capacity of ∼320 mA h g−1 is achieved at a high current density of 5.0 A g−1. In contrast, the micrometer-thick bulk SiP crystals cannot provide efficient lithium ion extraction. Moreover, the smaller and thinner SiPs obtained at a lower temperature show abnormally high mass transport resistance and low lithium ion diffusivity. These results demonstrate that SiPs are promising LIB anode materials, and the size and crystallinity are closely related to the anodic performance. This new knowledge is valuable for the development of high-performance LIBs.
1. Introduction
Lithium-ion batteries (LIBs) are being widely used due to their portability, rechargeable nature, and high energy density. Since electrode materials directly affect the capacity, a higher-performance lithium anode and cathode are required for next-generation LIBs. Among the various types of anode materials,1 graphite with a low specific capacity of 372 mA h g−1 does not meet the increasing demand for high-performance LIBs today. Introducing silicon into carbon can significantly improve the specific capacity.2–4 Silicon and phosphorus are desirable anode materials because of high theoretical specific capacities, such as 4200 mA h g−1 for Li22Si5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and 2596 mA h g−1 for Li3P,6,7 but both materials are restricted by low lithium ion diffusion and conductivity.8 Hence, it is important to achieve a high capacity while avoiding disadvantages such as the large volume expansion for anode materials. Layered materials composed of strong in-plane covalent bonds and weak interlayer van der Waals forces provide a possible solution.9 The large interlayer space in layered materials can accommodate the intercalation of guest ions and serve as fast ion diffusion channels,10 and the in-plane covalent bonds are available for charge transfer,11,12 consequently benefiting the reversibility in LIBs. Therefore, a layered compound containing silicon and phosphorus is studied in this work from the perspective of high-performance anodes.
5 and 2596 mA h g−1 for Li3P,6,7 but both materials are restricted by low lithium ion diffusion and conductivity.8 Hence, it is important to achieve a high capacity while avoiding disadvantages such as the large volume expansion for anode materials. Layered materials composed of strong in-plane covalent bonds and weak interlayer van der Waals forces provide a possible solution.9 The large interlayer space in layered materials can accommodate the intercalation of guest ions and serve as fast ion diffusion channels,10 and the in-plane covalent bonds are available for charge transfer,11,12 consequently benefiting the reversibility in LIBs. Therefore, a layered compound containing silicon and phosphorus is studied in this work from the perspective of high-performance anodes.
The orthogonal silicon monophosphide (SiP) with silicon–silicon and silicon–phosphorus interactions has a layered structure.13 The theoretical specific capacity of Li15Si4 and Li3P is up to 3060 mA h g−1 (ref. 14), and a large interlayer distance of 3.076 Å bodes well for ion diffusion in the gap15 (Fig. 1a). Chemical vapor deposition (CVD),16 flux,17,18 and chemical vapor transport (CVT)19 have been proposed for the synthesis of SiPs; the massive amount of liquid Sn used in the flux method results in cumbersome processing and the conversion efficiency of traditional CVD and CVT is quite low. Besides, the reported initial coulombic efficiency (ICE), stable delithiation capacity, and rate capability of SiPs prepared by low-temperature CVT are much lower than the theoretically predicted values.19 Nevertheless, CVT is a mature technique for the growth of single crystals with different structures by modulating the growth kinetics.20 An iodine-assisted CVT method has been used in the synthesis of crystalline red phosphorus,21 monolayer MoS2,20 octahedral silicon,22etc. Therefore, considering that the size and crystallinity can influence ion and charge transfer, a more facile and controllable synthesis is highly desired in order to incorporate SiPs into high-performance lithium anodes.
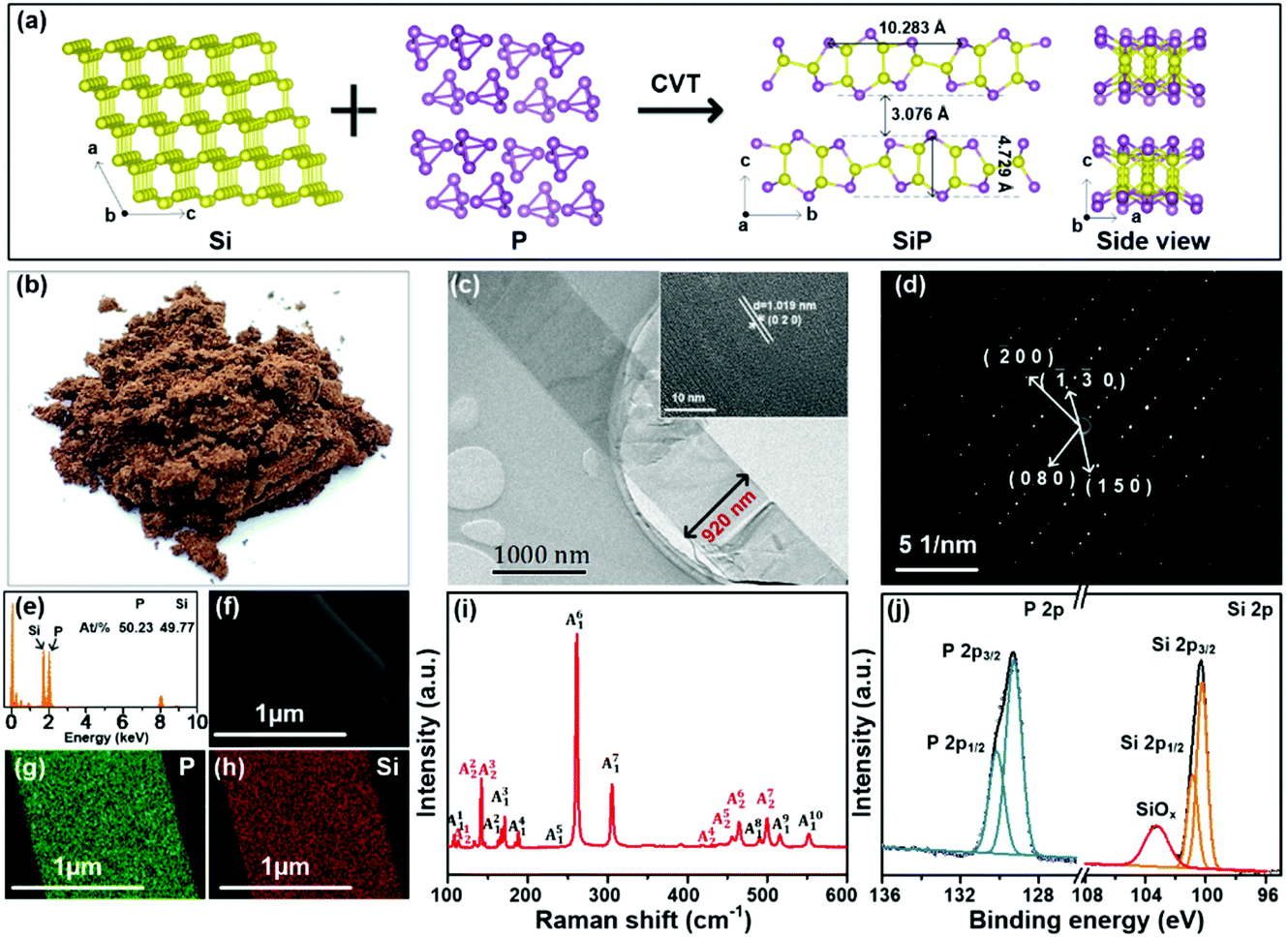 | ||
| Fig. 1 (a) Schematic diagram of the CVT method and the crystal structure of SiP; morphological characteristics of the HT-SiPs: (b) photograph of the crystals after grinding; (c) TEM bright-field image with the HR-TEM image in the inset; (d) SAED pattern with indexed diffraction spots along the (001) zone axis; (e) STEM-EDS analysis and composition of HT-SiPs; (f) STEM image; (g, h) STEM-EDS element maps of P and Si; (i) Raman scattering spectrum; (j) P 2p and Si 2p XPS spectra of HT-SiPs. | ||
Herein, an efficient high-temperature iodine-assisted CVT method is developed to synthesize SiPs with a high rate capacity of 320 mA h g−1 (5000 mA g−1), and stable capacities of 747 mA h g−1 (100 mA g−1) after 50 cycles and 615 mA h g−1 (100 mA g−1) after 200 cycles. In order to explore the effects of the size and crystallinity on the lithium anodic performance, samples are prepared by adjusting the temperature and additives, and electrochemical analysis and characterization reveal the correlation between the lithium storage capacity, crystal size and crystallinity.
2. Results and discussion
In a typical synthesis, silicon powder, red phosphorus lumps, and a carrier of iodine are sealed in a quartz ampoule and inserted into a horizontal two-zone furnace. The product is prepared in the high-temperature zone. The SiP sample produced at a higher temperature of 1060 °C (designated as HT-SiPs) is a brownish-yellow powder after grinding (Fig. 1b). As shown in the transmission electron microscopy (TEM) image in Fig. 1c, the HT-SiPs have a belt-like structure about ten micrometers long and one micrometer wide. The lattice spacing determined by high-resolution TEM (HR-TEM) is 1.019 nm, which can be ascribed to the (020) crystal plane of HT-SiPs. In the selected-area electron diffraction (SAED) pattern in Fig. 1d, the regular dots arising from the (080), (−200), (−1−30), and (150) planes confirm the single crystallinity. The energy-dispersive X-ray spectroscopy (EDS) elemental maps of HT-SiPs in Fig. 1e–h reveal that phosphorus and silicon are evenly distributed in the belt and the atomic concentrations of P and Si are 50.23% and 49.77%, respectively, showing a P/Si ratio of almost 1:1. The sharp Raman scattering peaks of HT-SiPs in Fig. 1i are consistent with previous results,18 corroborating the successful synthesis of SiPs. The elemental chemical states of HT-SiPs are determined by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1j, the P 2p XPS spectrum can be fitted by doublets at 129.3 eV (P 2p3/2) and 130.1 eV (P 2p1/2). The Si 2p XPS peaks at 100.2 eV (Si 2p3/2) and 100.9 eV (Si 2p1/2) stem from Si–Si and Si–P and the peak at 103.4 eV arises from the natural oxidization and production of SiOx.23
To elucidate the relationship between the SiP structure and electrochemical characteristics, two control samples are prepared, one of which is the SiP sample produced at a lower temperature of 900 °C (labeled as LT-SiPs) and the other is prepared by sulfur–iodine co-assisted high-temperature CVT (named bulk SiPs). The three techniques we report have good reproducibility, and consistent morphologies for each method can be obtained in repeated results. Even if the input of raw materials with the same proportion is increased, complete conversion can be achieved (Fig. S1†). Scanning electron microscopy (SEM) is carried out to investigate the different morphologies. As shown in Fig. 2a, the HT-SiPs have a belt-like shape and flat surface. As for the LT-SiPs, although they have a similar morphology to HT-SiPs, they have a smaller size and rougher surface (Fig. 2b). Additionally, as sulfur and iodine can transport silicon efficiently at a high temperature, the bulk SiPs have a much larger size and brick-like bulk structure (Fig. 2c). The width and thickness of the HT-SiPs, LT-SiPs, and bulk SiPs are summarized in Fig. 2d. The dimensions of HT-SiPs, LT-SiPs, and bulk SiPs are about 0.62 μm × 72 nm, 0.31 μm × 21 nm, and 26 μm × 8.8 μm, respectively (the sample size is measured according to the SEM and AFM images; see the corresponding statistics graphs in Fig. S2 and S3†). The X-ray diffraction (XRD) patterns in Fig. 2e show that the three SiP samples have similar characteristic peaks in accordance with the standard card (ICSD-PDF#89-5922). The salient diffraction peaks at 13° and 26° correspond to the crystal planes of (002) and (004), and the preferred orientations confirm the 2D layered structure of SiP, which is thus easy to assemble on the substrate in the same Z-axis direction. The diffraction peaks of 40–60° reflect the different exposure degrees of the high index crystal plane, in which the HT-SiPs have the highest normalization intensity. The peaks at 43.5°, 47.6°, 52.1°, 53.9°, 55.1°, 56.3° and 59.1° correspond to the crystal planes of (046), (190), (136), (200), (008), (194), (1 11 1), (223) and (1 11 3), respectively (Fig. S4†). The full-width at half-maximum (FWHM) of HT-SiPs, LT-SiPs, and bulk SiPs at (002) plane are 0.095°, 0.147° and 0.062° (Fig. 2f), respectively, indicating that the crystalline quality is improved by a high calcination temperature and the introduction of additive sulfur. Furthermore, although the three samples have the same crystal structure, there are major differences in the size and crystallinity.
 | ||
| Fig. 2 (a) SEM image of HT-SiPs with the AFM image in the inset; (b) SEM image of LT-SiPs with the AFM image in the inset; (c) SEM image of bulk SiPs with the cross-section in the inset; (d) average widths, thicknesses and error bars of the three samples; (e); XRD patterns of HT-SiPs, LT-SiPs and bulk SiPs; (f) full-width at half-maximum (FWHM) of the differential peak at 13°. | ||
To investigate the SiP growth mechanism in iodine-assisted CVT, raw materials of a stoichiometric mixture of silicon and phosphorus (1:1) without transport agents are used in the control experiments. The products of SiPs cannot be synthesized at a lower temperature of 800–900 °C but only once the temperature rises to 1000 °C, which is because the orthogonal phase SiP can be synthesized at a temperature higher than 1134 °C according to the Si–P phase diagram.24 Since no iodine is added, the shape of the product calcined at 1000 °C is irregular (Fig. S5†). The introduction of iodine in the reaction reduce the synthesis temperature and promote the formation of belt-like products (LT-SiPs) at lower temperatures. Besides, the addition of iodine is beneficial in lowering the annealing time of SiPs, speeding up the reaction, and promoting the complete conversion of the raw materials, and the crystallinity and size increase as the temperature increases. Co-assisted by iodine and sulfur,25 the crystallinity is further improved and the size increases almost exponentially (the FWHM and grain size of different SiPs are listed in Table S1†). Overall, iodine and sulfur play an important role in adjusting the morphology, size, and crystallinity of SiPs.
To determine the electrochemical properties of the three samples, coin-type half-cells (CR2032) are assembled with lithium foil as the counter electrode. The cyclic voltammetry (CV) profiles of the initial three cycles for HT-SiPs (red), LT-SiPs (orange), and bulk SiPs (blue) electrodes in Fig. 3a exhibit similar features including three reduction peaks and two oxidation peaks during discharging and charging, respectively. The corresponding differential capacity plots (dQ/dV) for the SiP electrodes in the first two cycles are presented in Fig. S6.† The reduction peak at 0.7 V during lithiation only appears in the first cycle due to the solid electrolyte interphase (SEI) formation or irreversible phase transition.26–28 The reduction peaks at 0.37 V and 0.09 V are associated with the formation of intermediate LixP and alloying of LixSi, respectively.29 The two oxidation peaks at 0.28 V and 0.6 V appearing during the extraction of lithium correspond to the de-alloying of LixSi and Li removal from LixP, which is similar to the lithium de-intercalation mechanism of SiPx and GePx.30–33 A small oxidation peak at ∼0.44 V is observed in the dQ/dV plots, and relates to the dealloying of crystalline Li15Si4 to crystalline Si.34 The initial discharge capacity of SiP is about 2600–2800 mA h g−1, which is similar to the theoretical capacity of 2753 mA h g−1 (based on the final phases of Li13Si4 and Li3P).33 The first cycle of the speculative reaction mechanism of the SiP electrode included the following possible steps:
Discharge: (1) SiP → LixSiP → LixP + LixSi, (2) LixP + Li+ + e− → LiP, (3) LiP + LixSi + Li+ + e− → Li3P + Li13Si4; Charge: (4) Li13Si4 + Li3P → LiP + Si + Li3P → SiP.
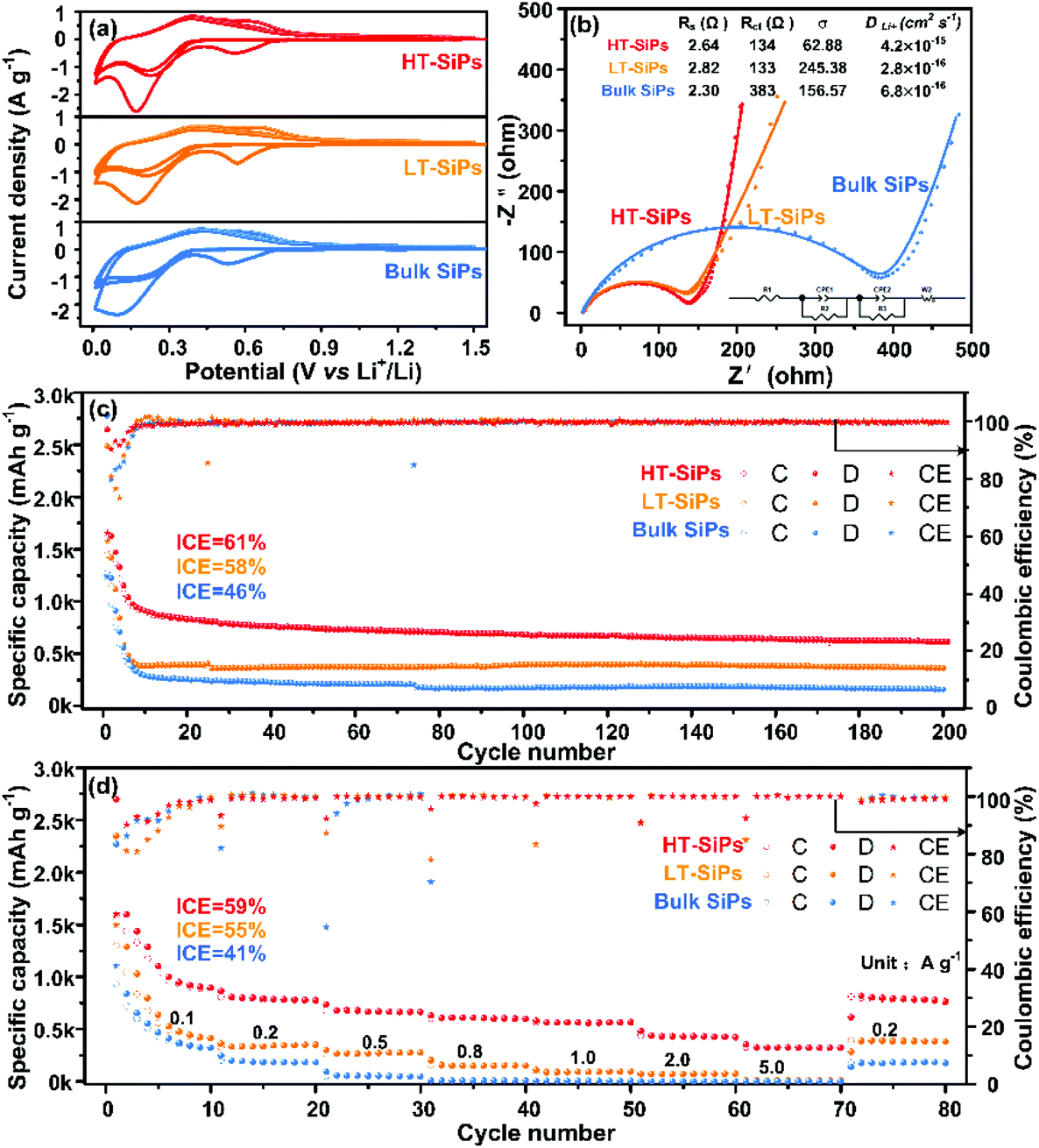 | ||
| Fig. 3 Electrochemical characteristics of HT-SiPs, LT-SiPs, and bulk SiPs in the voltage range of 0–1.5 V: (a) CV curves at a scanning rate of 0.1 mV s−1; (b) EIS results of the fresh cells for the different electrodes with the inset showing the corresponding equivalent circuit and fitted results: (c) cycling performance of the different samples at a current density of 100 mA g−1; (d) rate capabilities of the SiP electrodes at different current densities ranging from 0.1 to 5 A g−1 (C stands for charge, D stands for discharge, and CE stands for coulombic efficiency). | ||
The electrochemical impedance spectra (EIS) and the corresponding fits are presented in Fig. 3b.35 In the fresh cell, the internal resistances (Rs) of HT-SiPs, LT-SiPs, and bulk SiPs are 2.64 Ω, 2.82 Ω, and 2.30 Ω, respectively, and meanwhile, the charge transfer resistances (Rct) are 134 Ω (HT-SiPs), 133 Ω (LT-SiPs), and 383 Ω (bulk SiPs), respectively. The higher Rct value of bulk SiPs indicates a weaker charge transfer ability. The Warburg coefficients (σ) at low frequencies for HT-SiPs, LT-SiPs, and bulk SiPs are calculated to be 62.88, 245.38, and 156.57, respectively, indicating that the Li+ diffusivities (DLi+) are 4.226 × 10−15 cm2 s−1 (HT-SiPs), 2.775 × 10−16 cm2 s−1(LT-SiPs), and 6.816 × 10−16 cm2 s−1(bulk SiPs), respectively36,37 (Fig. S7†). The Li+ diffusivity (DLi+) of HT-SiPs is higher than that of monocrystalline silicon, but less than that of graphite (Table S2†). After 200 cycles (Fig. S8†), Rs of the HT-SiPs, LT-SiPs and bulk SiPs increased to 11.7 Ω, 25.5 Ω and 24.7 Ω, respectively, due to the formation of a thick SEI film on the anode surface.38,39 The smallest Rs value of HT-SiPs demonstrates that they can show a better rate performance and long life stability.
In order to investigate the lithium anodic properties of the three samples, galvanostatic charging/discharging tests are carried out. As shown in Fig. 3c, all the anodes prepared using HT-SiPs, LT-SiPs, and bulk SiPs exhibit high initial discharge capacities of 2644, 2485, and 2778 mA h g−1, respectively (the initial areal capacities of the three samples (HT-SiPs, LT-SiPs and bulk SiPs) are 2.644, 2.485 and 2.778 mA h cm−2, respectively), and the corresponding ICE values are 61%, 58%, and 46%, respectively. The nano-thick SiPs with lower crystallinity show a higher ICE than micro-thick SiPs with high crystallinity due to their large volume change resulting in the formation of a fresh surface with mass Li+ ions consumed and the increasing side reaction between the anodes and the electrolyte. After 200 cycles, HT-SiPs maintained a discharge capacity of 615 mA h g−1, whereas the discharge capacities of LT-SiPs and bulk SiPs decrease to 365 and 160 mA h g−1, respectively. The discharging capacities change significantly in the first 10 cycles and the capacities of HT-SiPs, LT-SiPs, and bulk SiPs are 908, 380, and 290 mA h g−1 (Fig. S9 and Table S3†), respectively, which is related to the irreversible formation of lithium silicate. The difference in capacity may be related to the initial sizes and crystallinity of the SiPs. The rate capabilities of the three electrodes obtained at current densities between 0.1 and 5.0 A g−1 are shown in Fig. 3d. The reversible discharging capacities of HT-SiPs are 780, 560, 420, and 320 mA h g−1 at 0.2, 1.0, 2.0, and 5.0 A g−1, respectively. When the current density returned to 0.2 A g−1, the discharging capacity also reverted to about 780 mA h g−1, confirming excellent capacity retention. In contrast, the discharging capacities of LT-SiPs and bulk SiPs decrease quickly with increasing current, disclosing poor rate capabilities.
The charge transport kinetics of the different samples is analyzed by the galvanostatic intermittent titration technique (GITT),40 which can provide information about the lithium ion diffusivity during charging/discharging.41 In the first discharging/charging cycle from 0.01 to 1.5 V, Din-Li+ of HT-SiPs fluctuates from 10−12 to 10−13 cm2 s−1 and Dex-Li+ decreases from 10−10 to 10−13 cm2 s−1 (Fig. 4a and Fig. S10a, b†). At the beginning of charging, Dex-Li+ increases to 10−10 cm2 s−1 due to substantial lithium ion intercalation, and then Dex-Li+ decreases to 10−12–10−13 cm2 s−1 because of the difficult lithium ion extraction. Similar behavior is observed in the other two samples. Din-Li+ of LT-SiPs and bulk SiPs decreased to 10−13–10−14 cm2 s−1 and Dex-Li+ fluctuates within 10−12–10−11 cm2 s−1 (Fig. 4b and c). Among the three samples, HT-SiPs have the highest Din-Li+ at the discharge plateau of 0.55 V and a relatively low Dex-Li+. After 200 cycles (Fig. 4d–f and Fig. S10c, d†), owing to the stable SEI film, Din-Li+ of HT-SiPs increases to 10−11 cm2 s−1, but Dex-Li+ decreases from 10−9 cm2 s−1 to 10−11 cm2 s−1 as the voltage increases to 1.5 V. All in all, Li+ ions can be extracted more readily from HT-SiPs than LT-SiPs or bulk SiPs. The difficult Li+ ion extraction observed from LT-SiPs and bulk SiPs may cause serious pulverization of the materials during long-term cycling.42,43 A specific path could be obtained through theoretical calculations,44–47 and it will be further studied. HT-SiPs show higher exposure in the crystal planes of (190), (200), and (1 11 1) which may be the reason for the fastest lithium ion diffusion among the three samples.
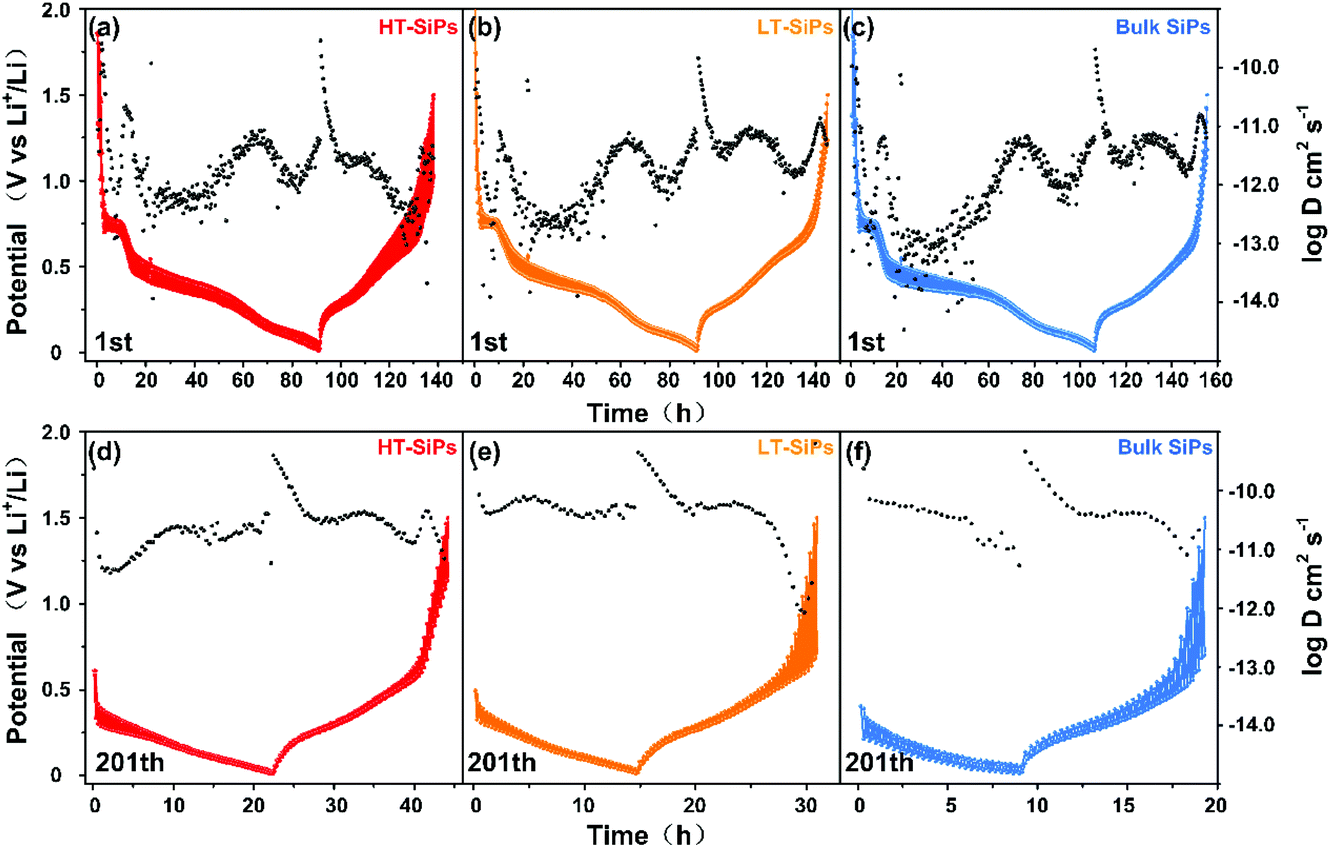 | ||
| Fig. 4 1st cycle of the GITT plots of (a) HT-SiP, (b) LT-SiP, and (c) bulk SiP anodes; 201st cycle of the GITT plots of (d) HT-SiP, (e) LT-SiP, and (f) bulk SiP anodes. | ||
In general, there are three reasons for the deterioration of the lithium anodic performance:48,49 (a) a thick SEI layer obstructing ion exchange, (b) irreversible structural transformation, expansion, and pulverization of the electrode materials and delaminating from the current collector (Fig. S11–S14†), and (c) incomplete lithium intercalation caused by the formation of LiP with a low ion conductivity50 (Fig. S15†). Reducing the size of the materials and the amorphous structure has been reported to alleviate the huge volume expansion during the charge and discharge process.51 The relationship between the thickness of SiPs and their final capacity is shown in Fig. S16.† The lithium anodic performance of nano-thick SiPs is 2 to 4 times that of micro-thick SiPs, which may be related to the shorter diffusion lengths for Li+ ions.52 Highly crystalline materials are more susceptible to greater structural or mechanical strain and pulverization due to lattice volume changes.53 The larger size and the higher crystallinity of the bulk anodic materials impose great internal stress during lithium insertion/extraction and Li+ cannot de-intercalate efficiently; furthermore, the thin nano-belts exhibit high mass transport resistance and affect the rate capability. It is apparent that the size and crystallinity together affect the anodic characteristics.
Fig. 5a summarizes the anodic characteristics of HT-SiPs, LT-SiPs, and bulk SiPs. HT-SiPs deliver the best performance as exemplified by an ICE of 61%, a discharge capacity of 615 mA h g−1 after 200 cycles, the fastest reaction kinetics with the largest DLi+, and a good rate capability at a high current density (320 mA h g−1 at 5 A g−1). Moreover, as shown in Fig. 5b and Table S4,† the lithium anodic performance of HT-SiPs is much better than that of SiP-based anodes in the literature. In contrast, the small LT-SiPs have worse LIB properties possibly because of nanobelt agglomeration and irreversible consumption of Li+ caused by the SEI film with a large volume fraction.54 Large bulk SiPs with higher crystallinity have the worst cycling and rate capability due to severe pulverization of the materials induced by the high internal stress changes during Li+ de-intercalation. Accordingly, the size and crystallinity of SiPs can synergistically affect the lithium anodic performance.
 | ||
| Fig. 5 (a) Electrochemical performance of the three materials and (b) comparison of the capacities for different cycles with previously reported SiPx for LIBs. | ||
3. Conclusion
A high-temperature iodine-assisted CVT method is developed to prepare high-quality layered HT-SiP belts to meet the demanding storage requirement with high rate performance and excellent cycling stability. In the process of CVT, iodine helps to reduce the growth temperature and annealing time of SiPs, speed up the reaction, and promote the complete conversion of raw materials. The HT-SiPs with a low charge transfer resistance exhibit fast lithium ion diffusion and a high rate capability of 320 mA h g−1 at a high current density of 5.0 A g−1. Meanwhile, failure analyses show that HT-SiPs experience the smallest structural deformation during the cycling process. HT-SiPs with excellent structural stability also show a steady capacity of 615 mA h g−1 after 200 cycles. By analyzing the samples with different sizes and crystallinities prepared using different temperatures and additives, both the micro-sized bulk crystal SiPs and thin LT-SiPs have lower capacities than HT-SiPs, confirming that the size and crystallinity together affect the anodic characteristics. The size-dependent phenomenon reveals that the lithium anodic performance could be improved by modulating the size and crystallinity. In addition, the CVT method can also effectively control the semiconductor type, color, and other characteristics of the material. The flexible CVT method for preparing high-quality single crystal materials can not only be suitable for LIB anode materials, but can also be used for photodetection, photocatalysis, and absorption.Author contributions
J. Wang, B. Yu and H. Yang conceived the idea and designed the experiments. B. Yu conducted the material growth and characterization. H. Yang carried out the electrochemical measurements and analysis. S. Gu, H. Huang, X. Zhang and Y. Kang participated in data analysis and discussion. D. Liu and Y. Zhang conducted the SEM and AFM tests. H. Yang, J. Wang and B. Yu wrote the manuscript, and then J. Wang, Y. Zhang, X.-F. Yu and P. K. Chu edited and polished the manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (51702352, 21975280, 51821229), the Key Research Program of Frontier Sciences, CAS (QYZDB-SSW-SLH034), the Youth Innovation Promotion Association Chinese Academy of Sciences (2020354), the Guangdong Special Support Program (2017TX04C096), the Leading Talents of Guangdong Province Program (00201520), the Shenzhen Science and Technology Research Funding (JCYJ20180507182530279, JCYJ20180507182047316), and the City University of Hong Kong Strategic Research Grant (SRG) No. 7005105.References
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2013, 31, 317–336 CrossRef.
- W. F. Ren, J. T. Li, S. J. Zhang, A. L. Lin, Y. H. Chen, Z. G. Gao, Y. Zhao, L. Deng, L. Huang and S. G. Sun, J. Energy Chem., 2020, 48, 160–168 CrossRef.
- R. J. C. Dubey, P. V. W. Sasikumar, N. Cerboni, M. Aebli, F. Krumeich, G. Blugan, K. V. Kravchyk, T. Graule and M. V. Kovalenko, Nanoscale, 2020, 12, 13540 RSC.
- J. Zhang, S. Fang, X. Qi, Z. Yu, Z. Wu, J. Yang and S. Lu, J. Energy Chem., 2020, 40, 171–179 CrossRef.
- F. Li, J. Xu, Z. Hou, M. Li and R. Yang, ChemNanoMat, 2020, 6, 720–738 CrossRef CAS.
- L.-Q. Sun, M.-J. Li, K. Sun, S.-H. Yu, R.-S. Wang and H.-M. Xie, J. Phys. Chem. C, 2012, 116, 14772–14779 CrossRef CAS.
- C. M. Park and H. J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS.
- K. Ogata, E. Salager, C. J. Kerr, A. E. Fraser, C. Ducati and A. J. Morris, Nat. Commun., 2014, 5, 3217–3228 CrossRef CAS PubMed.
- K. S. Chen, I. Balla, N. S. Luu and M. C. Hersam, ACS Energy Lett., 2017, 2, 2026–2034 CrossRef CAS.
- F. Shojaei and H. S. Kang, J. Phys. Chem. C, 2016, 120, 23842–23850 CrossRef CAS.
- G. A. Tritsaris, E. Kaxiras, S. Meng and E. Wang, Nano Lett., 2013, 13, 2258–2263 CrossRef CAS PubMed.
- L. Kou, Y. Ma, X. Tan, T. Frauenheim, A. Du and S. Smith, J. Phys. Chem. C, 2015, 119, 6918–6922 CrossRef CAS.
- A. Q. Cheng, Z. He, J. Zhao, H. Zeng and R.-S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 5133–5139 CrossRef CAS PubMed.
- D. Duveau, S. S. Israel, J. Fullenwarth, F. Cunin and L. Monconduit, J. Mater. Chem. A, 2016, 4, 3228–3232 RSC.
- H. B. Shu and J. Y. Guo, Mater. Res. Express, 2018, 5, 036302–036311 CrossRef.
- C. G. Beck and R. Stickler, J. Appl. Physiol., 1966, 37, 4683–4687 CrossRef CAS.
- C. Li, S. Wang, X. Zhang, N. Jia, T. Yu, M. Zhu, D. Liu and X. Tao, CrystEngComm, 2017, 19, 6986–6991 RSC.
- C. Li, S. Wang, C. Li, T. Yu, N. Jia, J. Qiao, M. Zhu, D. Liu and X. Tao, J. Mater. Chem. C, 2018, 6, 7219–7225 RSC.
- R. Reinhold, D. Mikhailova, T. Gemming, A. B. Missyul, C. Nowka, S. Kaskel and L. Giebeler, J. Mater. Chem. A, 2018, 6, 19974–19978 RSC.
- D. K. Hu, G. C. Xu, L. Xing, X. X. Yan, J. Y. Wang, J. Y. Zheng, Z. X. Lu, P. Wang, X. Q. Pan and L. Y. Jiao, Angew. Chem., Int. Ed., 2017, 56, 3611–3615 CrossRef CAS PubMed.
- Q. Liu, X. Zhang, J. Wang, Y. Zhang, S. Bian, Z. Cheng, N. Kang, H. Huang, S. Gu, Y. Wang, D. Liu, P. K. Chu and X. F. Yu, Angew. Chem., Int. Ed., 2020, 59, 14383–14387 CrossRef CAS PubMed.
- Z. Cheng, H. Cui, Q. Xiao, H. Huang, Y. Kang, Q. Liu, J. Wang, P. K. Chu and X. F. Yu, Small, 2020, 16, 2003594 CrossRef CAS PubMed.
- R. Reinhold, U. Stoeck, H. J. Grafe, D. Mikhailova, T. Jaumann, S. Oswald, S. Kaskel and L. Giebeler, ACS Appl. Mater. Interfaces, 2018, 10, 7096–7106 CrossRef CAS PubMed.
- S. M. Liang and R. J. Schmid-Fetzer, Phase Equilib. Diffus., 2014, 35, 24–35 CrossRef CAS.
- A. F. Armington, J. Cryst. Growth, 1967, 1, 47–48 CrossRef CAS.
- B. Peng, Y. Xu, X. Wang, X. Shi and F. M. Mulder, Sci. China: Phys., Mech. Astron., 2017, 60, 064611 Search PubMed.
- C. Peng, H. Chen, G. Zhong, W. Tang, Y. Xiang, X. Liu, J. Yang, C. Lu and Y. Yang, Nano Energy, 2019, 58, 560–567 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15–41 CrossRef.
- G. Coquil, B. Fraisse, N. Dupré and L. Monconduit, ACS Appl. Energy Mater., 2018, 1, 3778–3789 CrossRef CAS.
- X. Li, W. Li, P. Shen, L. Yang, Y. Li, Z. Shi and H. Zhang, Ceram. Int., 2019, 45, 15711–15714 CrossRef CAS.
- H. Shen, Y. Huang, Y. Chang, R. Hao, Z. Ma, K. Wu, P. Du, B. Guo, Y. Lyu, P. Wang, H. Yang, Q. Li, H. Wang, Z. Liu and A. Nie, ACS Appl. Mater. Interfaces, 2020, 12, 17466–17473 CrossRef CAS PubMed.
- W. Li, X. Li, J. Yu, J. Liao, B. Zhao, L. Huang, A. Ali, H. Zhang, J. H. Wang, Z. Guo and M. Liu, Nano Energy, 2019, 61, 594–603 CrossRef CAS.
- H.-T. Kwon, C. K. Lee, K.-J. Jeon and C.-M. Park, ACS Nano, 2016, 10, 5701–5709 CrossRef CAS PubMed.
- J. Wang, X. Wang, B. Liu, H. Lu, G. Chu, J. Liu, Y. Guo, X. Yu, F. Luo, Y. Ren, L. Chen and H. Li, Nano Energy, 2020, 78, 105101–105110 CrossRef CAS.
- X. Liu, G. B. Xu, T. T. Cheng, L. W. Yang and J. X. Cao, ChemElectroChem, 2020, 7, 846–854 CrossRef CAS.
- R. Ruffo, S. S. Hong, C. K. Chan, R. A. Huggins and Y. Cui, J. Phys. Chem. C, 2009, 113, 11390–11398 CrossRef CAS.
- B. Long, Y. Zou, Z. Li, Z. Ma, W. Jiang, H. Zou and H. Chen, ACS Appl. Energy Mater., 2020, 3, 5572–5580 CrossRef CAS.
- Y. Zhang, O. I. Malyi, Y. Tang, J. Wei, Z. Zhu, H. Xia, W. Li, J. Guo, X. Zhou, Z. Chen, C. Persson and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 14847–14852 CrossRef CAS PubMed.
- N. Ding, J. Xu, Y. X. Yao, G. Wegner, X. Fang, C. H. Chen and I. Lieberwirth, Solid State Ionics, 2009, 180, 222–225 CrossRef CAS.
- Z. Li, F. Du, X. F. Bie, D. Zhang, Y. M. Cai, X. R. Cui, C. Z. Wang, G. Chen and Y. J. Wei, J. Phys. Chem. C, 2010, 114, 22751–22757 CrossRef CAS.
- Y. Son, M. Park, Y. Son, J.-S. Lee, J.-H. Jang, Y. Kim and J. Cho, Nano Lett., 2014, 14, 1005–1010 CrossRef CAS PubMed.
- P. Kumar, C. Berhaut, D. Zapata Dominguez, E. De Vito, S. Tardif, S. Pouget, S. Lyonnard and P. H. Jouneau, Small, 2020, 16, 1906812 CrossRef CAS PubMed.
- M. M. Kabir and D. E. Demirocak, Int. J. Energy Res., 2017, 41, 1963–1987 CrossRef CAS.
- B. Lang, B. Ziebarth and C. Elsässer, Chem. Mater., 2015, 27, 5040–5048 CrossRef CAS.
- F. Shojaei and H. S. Kang, J. Phys. Chem. C, 2016, 120, 23842–23850 CrossRef CAS.
- F. Yao, F. Gunes, H. Q. Ta, S. M. Lee, S. J. Chae, K. Y. Sheem, C. S. Cojocaru, S. S. Xie and Y. H. Lee, J. Am. Chem. Soc., 2012, 134, 8646–8654 CrossRef CAS PubMed.
- J. Tian, Y. Su, F. Wu, S. Xu, F. Chen, R. Chen, Q. Li, J. Li, F. Sun and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 582–587 CrossRef CAS PubMed.
- H. Li, X. Huang, L. Chen, G. Zhou, Z. Zhang, D. Yu, Y. J. Mo and N. Pei, Solid State Ionics, 2000, 135, 181–191 CrossRef CAS.
- H. Li, L. Shi, Q. Wang, L. Chen and X. Huang, Solid State Ionics, 2002, 148, 247–258 CrossRef CAS.
- G. Nazri, Solid State Ionics, 1989, 34, 97–102 CrossRef CAS.
- X. Li, X. Meng, J. Liu, D. Geng, Y. Zhang, M. Banis, Y. Li, R. Li, X. Sun, M. Cai and M. Verbrugge, Adv. Funct. Mater., 2012, 22, 1647–1654 CrossRef CAS.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294–1305 RSC.
- L. Cui, R. Ruffo, C. Chan, H. Peng and Y. Cui, Nano Lett., 2009, 9, 491–495 CrossRef CAS PubMed.
- S. P. V. Nadimpalli, V. A. Sethuraman, S. Dalavi, B. Lucht, M. J. Chon, V. B. Shenoy and P. R. Guduru, J. Power Sources, 2012, 215, 145–151 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: AFM, SEM, TEM, and XPS measurement results of the SiPs and the supplementary electrochemical characterization. See DOI: 10.1039/d0nr07386e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |