
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the location of Lewis acidic aluminum in zeolite mordenite and the role of framework-associated aluminum in mediating the switch between Brønsted and Lewis acidity†
Manoj
Ravi
 a,
Vitaly L.
Sushkevich
b and
Jeroen A.
van Bokhoven
*ab
a,
Vitaly L.
Sushkevich
b and
Jeroen A.
van Bokhoven
*ab
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
bLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institute, Villigen, 5232, Switzerland
First published on 26th January 2021
Abstract
Lewis acidic aluminum in zeolites, particularly acidity that is inherent to the framework, is an indeterminate concept. A fraction of framework aluminum changes geometry to octahedral coordination in the proton form of zeolite mordenite. Such octahedrally coordinated aluminum is the precursor of a Lewis acid site and its formation is accompanied by a loss in Brønsted acidity. Herein, we show that such Lewis acid sites have a preferred location in the pore structure of mordenite. A greater proportion of these Lewis acid sites resides in the side-pockets than in the main channel. By reverting the octahedrally coordinated aluminum back to a tetrahedral geometry, the corresponding Brønsted acid sites are restored with a concomitant loss in the ability to form Lewis acid sites. Thereby, reversible octahedral–tetrahedral aluminum coordination provides a means to indirectly switch between Lewis and Brønsted acidity. This phenomenon is unique to Lewis acidity that is inherent to the framework, thereby distinguishing it from Lewis acidity originating from extra-framework species. Furthermore, the transformation of framework aluminum into octahedral coordination is decoupled from the generation of distorted tetrahedrally coordinated aluminum, where the latter gives rise to the IR band at 3660 cm−1 in the OH stretching region.
Introduction
Zeolites are extensively used as catalyst and support materials for a wide array of industrially applied reactions.1,2 The presence of tetrahedrally coordinated aluminum in a zeolite framework necessitates a charge-compensating cation to yield a neutral material. When this cation is a proton, it results in a Brønsted acid site (Fig. 1a),3 which is at the origin of industrial application of zeolites as solid acid catalysts, replacing homogeneous mineral acids in various processes.4 Protonic zeolites are extensively used for several reactions, in particular, cracking, alkylation, and isomerization.5–7 Apart from Brønsted acid sites, certain aluminum species in zeolites possess Lewis acidic character. Lewis acidic aluminum (Fig. 1b) plays a pivotal role not only in traditional cracking reactions, but also in salient biomass valorization reactions,8,9 which include the conversion of trioses to alkyl lactates,10 and cellulose to glucose.11 Conventionally, one of the oft-used techniques to study the above-mentioned acid sites in zeolites is FTIR spectroscopy of adsorbed probe molecules.12,13 Not only can Lewis acid sites be distinguished easily from Brønsted acid ones, but finer nuances, such as acid site strength and accessibility can also be assessed. For instance, evacuating adsorbed pyridine at different temperatures provides information on the strength of adsorption, which to some extent relates to acid site strength.14–16 Likewise, using probe molecules of different sizes sheds light on the accessibility of the sites.17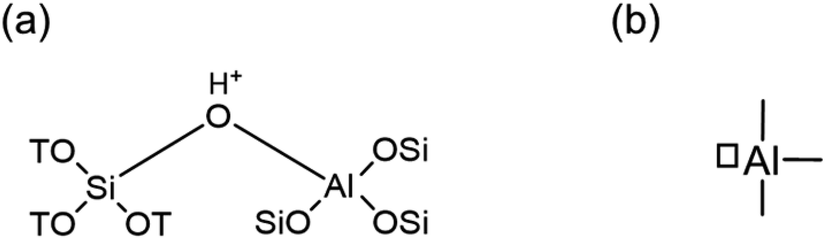 | ||
| Fig. 1 Sketch of (a) Brønsted acid site and (b) three-coordinated Lewis acid site in a zeolite. | ||
Alongside Lewis acidic aluminum species in zeolites, Brønsted acid sites engage in a cooperative fashion to catalyze multi-step ‘domino’ reactions.18 Examples of such reactions in biomass valorization chemistry include the conversion of glucose to 5-hydroxymethylfurfural (HMF) and of C3-sugars to lactic acid esters.19–21 These conversions feature consecutive reaction steps that take place over different active sites and consequently, require a precise balancing of the number of Lewis and Brønsted acid sites in the zeolite. This is experimentally evident from the existence of an optimal Lewis to Brønsted acid ratio to achieve the highest yield of the product.20,21 Likewise, in glycerol dehydration over zeolites, Brønsted acid sites are involved in activating the substrate, but the selectivity to acrolein or acetaldehyde is dictated by the Lewis acid content in the zeolite.22 Consequently, understanding the skew of these two different types of acid sites is paramount for the catalytic applications of zeolites. However, despite zeolites being used commercially on a large scale and extensive research being carried out over a number of decades now, the origin of Lewis acidity in these materials remains a nebulous concept.15,23–30 Several proposals for Lewis acidic aluminum species exist, which include framework aluminum,26,27,31 extra-framework aluminum (EFAl),25,32–34 and framework-associated aluminum.15,30 In a recently published perspective, we discussed this classification in detail and illustrated that the multiple proposals point towards a plurality of Lewis acid sites and mechanisms that generate these species.35 The uncertainty in demarcating the origin of Lewis acidic aluminum in zeolites hampers the analysis of Lewis–Brønsted acid synergy in catalytic applications.36,37
The most extensively characterized mechanism for Lewis acidity in zeolites is of that formed after dealumination via steaming.38 This route entails the hydrolysis of framework Si–O–Al bonds, translating in the removal of aluminum from the framework.32,34,39–42 In comparison to this mechanism, the proposal of Lewis acidity that is inherent to the zeolite framework is less rigorously examined, but has received greater attention of late.26,35,43,44 NMR experiments performed at high fields have enabled the characterization of a second framework Al(IV) site,45 and the elucidation of the acidic nature of tri-coordinated framework aluminum in dehydrated ZSM-5.43 The precursor to Lewis acid sites in this mechanism – unlike for the Lewis acid sites generated from EFAl – is tetrahedral framework aluminum.
Framework-associated aluminum refers to species that can undergo reversible changes in coordination from octahedral to tetrahedral.46 Through a combination of 27Al MAS NMR and FTIR spectroscopy, we demonstrated that framework-associated aluminum can be assigned Lewis acidic property in zeolite mordenite.30 It adopts an octahedral coordination in the proton form of the zeolite after calcination in static air and exposure of the sample to moisture under ambient conditions.15,30,46,47 Such an aluminum species is associated with Lewis acidity upon dehydration.30,48 The octahedrally coordinated framework-associated species can be reverted into a normal framework tetrahedral coordination, and should therefore, not be described as simply extra-framework. While the Lewis acidic character of framework-associated aluminum was evidenced, the nature of these species, its evolution as a function of conditions, and its location in the pore system of zeolite mordenite remain unaddressed. Also, the potential role of distorted tetrahedral and penta-coordinated aluminum in generating Lewis acidity was not evaluated. As per current knowledge, the extent to which the generation of these latter species are coupled to the generation of framework-associated octahedrally coordinated aluminum is unknown. By targeting these knowledge gaps, our objective is to arrive at a better understanding of the Lewis acidic property of framework-associated aluminum and of zeolites in general. We demonstrate how evolution in aluminum coordination with varying conditions influences both Lewis and Brønsted acidity in the zeolite, with the two kind of acid sites being partially interchangeable. Furthermore, we highlight the role of the side-pockets in mordenite in this phenomenon. Current knowledge on the generation of Lewis acid sites, irrespective of which kind of aluminum species shows Lewis acidic property, is that they are formed with a loss in Brønsted acidity.24,26,49 Overall, we report a novel conceptual advance on the possibility to toggle between Lewis and Brønsted acidity, founded on the basis of aluminum species that exhibit reversible octahedral–tetrahedral coordination. The Lewis acid sites generated from such aluminum species are preferentially located in the side-pockets as opposed to the main channel of mordenite.
Results and discussion
Aluminum structure from 27Al MAS NMR
Fig. 2 illustrates that the coordination of aluminum in zeolite MOR is a function of the charge-compensating cation and treatment conditions, in line with what is typically observed for zeolites.46,47,50 The details of sample nomenclature and treatment procedure are provided in Table 1. Since the aluminum structure is sensitive to many factors, such as water content and temperature,48,51 the structure determined by a conventional 27Al MAS NMR experiment is the structure that prevails after hydration at room temperature.52 The 27Al MAS NMR spectrum of NH4–MOR contains one resonance at around 56 ppm, characteristic of tetrahedral framework aluminum. Upon calcining NH4–MOR in static air at 450 °C and subsequent exposure to ambient conditions (H–MOR-450), a fraction of aluminum adopts an octahedral geometry, evidenced by the sharp resonance at 0 ppm. There is an additional change in the spectrum of the sample calcined at a higher temperature of 550 °C (H–MOR-550). Besides the presence of octahedrally coordinated aluminum, the tetrahedral feature at 56 ppm has a broad shoulder stretching to lower chemical shifts. This broadening is not present in the spectrum of NH4–MOR and is barely discernible in that of H–MOR-450. Octahedrally coordinated aluminum represents 11% of the total aluminum content in H–MOR-550 and 10% of the total aluminum content in H–MOR-450.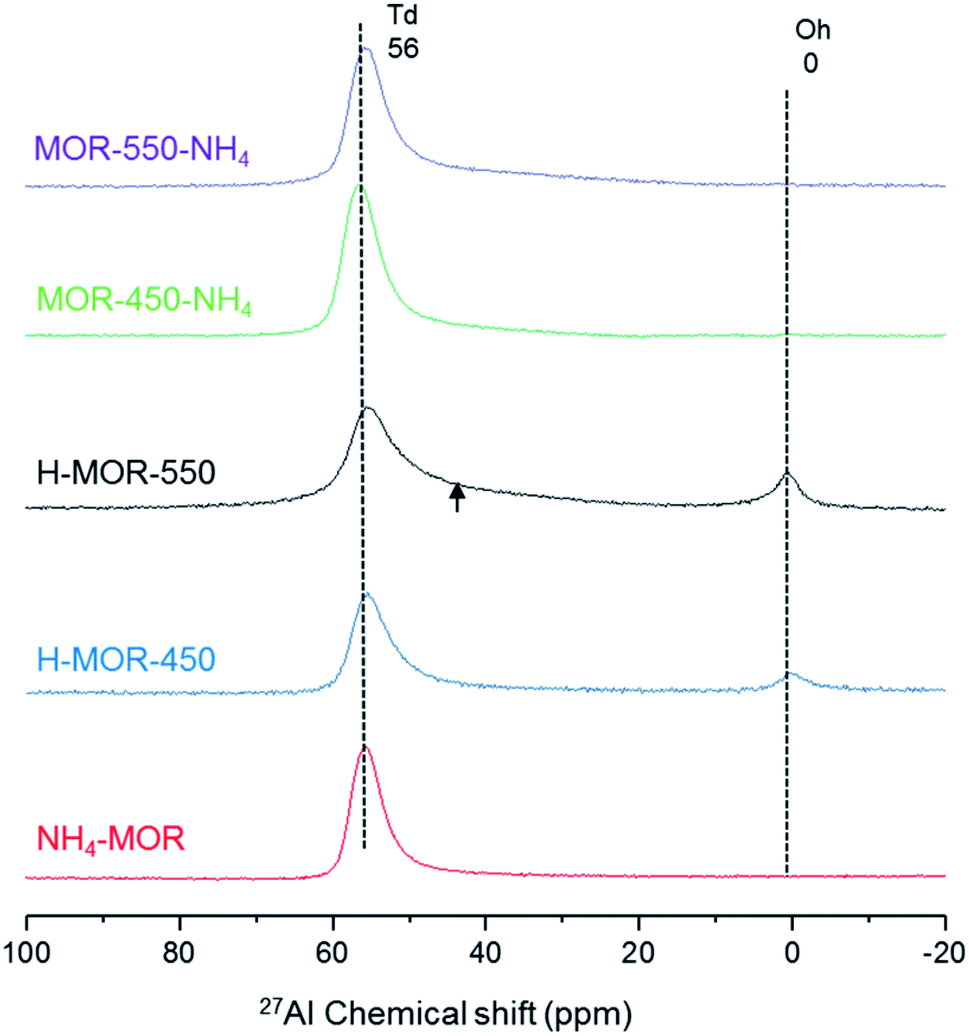 | ||
| Fig. 2 27Al MAS NMR spectra of zeolite MOR under ambient conditions of NH4–MOR, H–MOR-450, H–MOR-550, MOR-450-NH4 and MOR-550-NH4. The broad shoulder at the high field side of the peak corresponding to tetrahedrally coordinated framework aluminum in H–MOR-550 is highlighted by the black arrow mark. | ||
| Sample name | Treatment procedure |
|---|---|
| NH4–MOR | Commercial mordenite zeolite in the ammonium form, Zeolyst CBV21A, Si/Al = 11 |
| H–MOR-450 | NH4–MOR calcined at 450 °C in static air & subsequent exposure to ambient conditions |
| H–MOR-550 | NH4–MOR calcined at 550 °C in static air & subsequent exposure to ambient conditions |
| MOR-450-NH4 | H–MOR-450 after ammonium ion-exchange using aqueous solution of NH4NO3 |
| MOR-550-NH4 | H–MOR-550 after ammonium ion-exchange using aqueous solution of NH4NO3 |
Octahedrally coordinated aluminum in mordenite, which is characterized by a narrow resonance at 0 ppm by 27Al MAS NMR, can be forced back into the typical framework tetrahedral geometry on back-exchanging the zeolite to its ammonium form.30 This phenomenon of reversible octahedral–tetrahedral aluminum coordination was first observed for zeolite BEA,46 and later confirmed40 and generalized for other zeolites.15,31,48,50,53 Likewise, we observe that the octahedrally coordinated species in both, H–MOR-450 and H–MOR-550, can be forced back into a tetrahedral environment on ammonium-exchange (Fig. 2). The sharpness of the feature at 0 ppm implies that the electric field gradient surrounding this aluminum atom is small and therefore suggests a high symmetry.
The conversion of tetrahedrally coordinated framework aluminum to an octahedral coordination is driven by the adsorption of water through hydrolysis reactions.50,54,55 The original proposal for the octahedral framework-associated species was that the aluminum is connected to the framework via four oxygen atoms with a water molecule and a hydronium ion being the two other coordinations.46 However, theoretical work shows that hydrolysis of Si–O and Al–O bonds readily occur and a fourfold coordination to the framework is unlikely.42 Despite the lack of certainty about the exact structure, enough experimental evidence has emerged to suggest that such octahedrally coordinated aluminum cannot be trivially dismissed as EFAl.46–48
The 27Al MQMAS NMR spectrum of NH4–MOR shows a single feature assigned to tetrahedral framework aluminum (Fig. 3). The spectrum of H–MOR-550 shows two types of tetrahedrally coordinated aluminum species, one type of octahedrally coordinated species and no penta-coordinated aluminum. The 27Al MQMAS spectrum demonstrates that the two types of tetrahedral species have different anisotropy. Tetrahedrally coordinated framework aluminum denoted as Al(IV)a hugs onto the F1 = F2 diagonal pointing to a very low anisotropic quadrupolar interaction. In contrast, the significant quadrupolar interaction for the Al(IV)b sites is evidenced by the broad contours parallel to the F2 dimension. The absence of penta-coordinated aluminum is in line with the findings from Chen et al., who have previously detected five-coordinated aluminum in mordenite only upon calcination at temperatures in excess of 650 °C.56 The fraction of Al(IV)b is a function of calcination temperature. Such species is barely detectable in the spectrum of H–MOR-450. However, this sample does contain a substantial amount of octahedrally coordinated aluminum, suggesting that the Al(IV)b species is not a precursor to the Al(VI) sites.
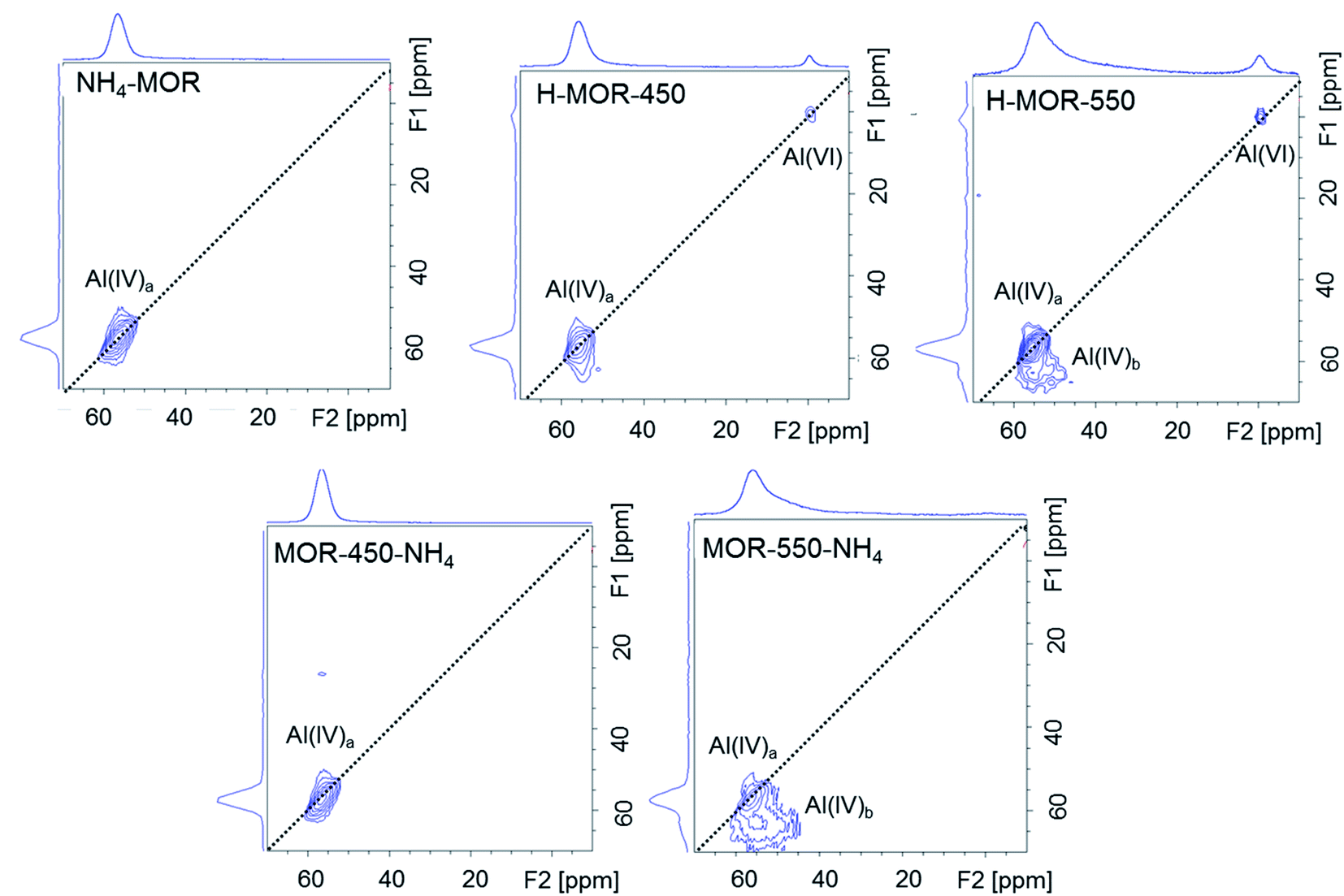 | ||
| Fig. 3 27Al MQMAS NMR spectra measured under ambient conditions for NH4–MOR, H–MOR-450, H–MOR-550, MOR-450-NH4 and MOR-550-NH4. The F1 projection shows a purely isotropic dimension and the F2 projection depicts the corresponding 27Al MAS NMR spectrum, which is acquired in a separate experiment. | ||
Fig. 3 shows that the 27Al MQMAS NMR spectrum of MOR-450-NH4 is very similar to that of NH4–MOR, but the same observation does not hold for MOR-550-NH4. In the spectrum of the latter, the resonance of the Al(VI) species disappears and that of Al(IV)a reappears but that of Al(IV)b species continues to be present. The population of distorted tetrahedral Al(IV)b sites is an important point of difference between the spectra of MOR-550-NH4 and NH4–MOR. Since the Al(IV)b sites are only formed at a calcination temperature of 550 °C, these aluminum species most likely represent irreversible thermal damage inflicted on the zeolite framework, preventing their transformation back into a framework T-site, unlike the framework-associated octahedrally coordinated aluminum.
The phenomenon of reversible tetrahedral–octahedral aluminum coordination in zeolite mordenite can be realized independent of the Si/Al ratio. To illustrate this, we calcined three ammonium–mordenite samples of different Si/Al ratio at 550 °C to obtain the corresponding proton forms (Table S1†). The percentage of octahedrally coordinated aluminum decreases from 13% for zeolite MOR with a Si/Al ratio of 8.8 to 10% for a sample of a higher Si/Al ratio of 19.7. In comparison, up to 19% of aluminum adopts an octahedral coordination in the proton form of zeolite BEA with a Si/Al ratio of 9.57 When the protonic zeolites are exchanged back into the ammonium form, we observe a quantitative transformation of octahedrally coordinated aluminum into tetrahedral coordination (Table S1†).
Relationship between aluminum coordination and Brønsted–Lewis acidity
Fig. 4a depicts the FTIR spectra of NH4–MOR, H–MOR-450, H–MOR-550, MOR-450-NH4 and MOR-550-NH4 after in situ high temperature evacuation and pyridine adsorption–desorption. The names of the samples in the figure refer to the form of the zeolite prior to heat treatment in the IR setup; high temperature activation of the samples under vacuum before pyridine adsorption–desorption converts all the zeolites to the proton form. The absorption features at 1455 and 1620 cm−1 in Fig. 4a are assigned to pyridine bound to Lewis acid sites, while those at 1545 and 1635 cm−1 correspond to protonated pyridine. Activation of NH4–MOR under vacuum results in a minor population of Lewis acid sites. In a previous study, we established that framework-associated aluminum in proton-exchanged zeolites adopts an octahedral coordination under ambient conditions and is the precursor for Lewis acid sites.30 Upon activation, both H–MOR-450 and H–MOR-550 have a lower concentration of Brønsted acid sites (BAS, 1545 cm−1) than NH4–MOR. Likewise, the spectrum of H–MOR-450 exhibits a larger peak associated with Brønsted acidity (1545 cm−1) and a smaller feature corresponding to Lewis acidity (LAS, 1455 cm−1) than that of H–MOR-550. Therefore, the generation of Lewis acid sites is accompanied by a loss of Brønsted acid sites. In other words, some of the framework aluminum that generate Brønsted acidity convert into framework-associated octahedrally coordinated species, which after dehydration show Lewis acidity. Fig. 4a and Table 2 show that MOR-450-NH4 and MOR-550-NH4 have lower Lewis acid and higher Brønsted acid concentrations after in situ activation than the corresponding proton forms, H–MOR-450 and H–MOR-550. This observation is consistent with the 27Al NMR results discussed earlier, where aluminum in octahedral coordination in the proton form of mordenite was shown to reinsert into the framework upon converting to the ammonium form (vide supra). Consequently, the conversion of the Lewis acid-forming framework-associated aluminum to a typical tetrahedral framework species results in increased Brønsted acidity upon in situ activation of the back-exchanged samples. The greater intensity of the peak corresponding to pyridinium ion in the FTIR spectra of the activated back ammonium-exchanged samples duly reflects this. | ||
| Fig. 4 FTIR spectra of (a) adsorbed pyridine over activated NH4–MOR, H–MOR-450, H–MOR-550, MOR-450-NH4 and MOR-550-NH4; (b) samples activated under vacuum at 450 °C for 4 h (solid line) and after pyridine adsorption–desorption at 150 °C (dashed line) in the OH stretching region. The names of the samples refer to the form of the zeolite prior to heat treatment in the IR setup. | ||
| Sample | BAS peak areaa [a.u.] | LAS peak area as assessed using pyridineb [a.u.] | LAS peak area as assessed using carbon monoxidec [a.u.] |
|---|---|---|---|
| a Computed by integration of the peak at 3609 cm−1 in the activated sample. b Computed by integration of the peak at 1455 cm−1 in the pyridine-adsorbed sample. c Computed by integration of the peaks at 2224 cm−1 and 2198 cm−1 in the CO-adsorbed sample. The relative error for all numbers listed in the table is ±5%. | |||
| NH4–MOR | 52 | 3.9 | 1.2 |
| H–MOR-450 | 41 | 4.1 | 1.6 |
| H–MOR-550 | 26 | 6.5 | 1.8 |
| MOR-450-NH4 | 49 | 3.5 | 1.2 |
| MOR-550-NH4 | 37 | 3.6 | 1.1 |
Fig. 4b illustrates the FTIR spectra of the same samples in the OH stretching region, before and after pyridine adsorption–desorption. The most prominent absorption in the evacuated samples is that at 3610 cm−1, which is attributed to bridging hydroxyl groups (Brønsted acid sites). The feature at 3745 cm−1 is the signature for silanols. FTIR spectra of H–MOR-550 and MOR-550-NH4 (Fig. 4b) show a shoulder at around 3660 cm−1, which is attributed to the presence of Al–OH species.58 This feature is barely visible in the spectra of NH4–MOR, H–MOR-450 and MOR-450-NH4. The peak area associated with Brønsted acid sites decreases in the order NH4–MOR ≈ MOR-450-NH4 > H–MOR-450 > MOR-550-NH4 > H–MOR-550 (Table 2). The total Brønsted acidity measured after high temperature in situ activation of MOR-450-NH4 is highly comparable to that of in situ activated NH4–MOR. However, in the case of MOR-550-NH4, the total Brønsted acidity, while being appreciably higher than in H–MOR-550, is still inferior to that of NH4–MOR. Therefore, the extent to which Brønsted acid sites are restored depends on the temperature at which the proton form is calcined. A nearly complete retrieval of Brønsted acidity is possible with the zeolite calcined at 450 °C, but not with samples calcined at higher temperatures. The presence of irreversibly formed Al–OH groups in MOR-550-NH4 results in the total Brønsted acidity being inferior to that of NH4–MOR (Table 2).
After adsorption of pyridine at 150 °C followed by desorption at the same temperature, the spectrum of activated NH4–MOR shows a feature centered at approximately 3587 cm−1 in the OH-stretching region (Fig. 4b). This corresponds to the fraction of Brønsted acid sites that are not probed by pyridine. The side-pockets in mordenite present accessibility limitations for sterically bulky molecules. While pyridine can access Brønsted acid sites in the main channel in a pristine mordenite sample, it cannot probe bridging OH groups located in the side-pockets.59–61 The above-mentioned OH-stretch frequency at 3587 cm−1 is representative of such inaccessible Brønsted acid sites. This feature continues to be present in the spectrum of H–MOR-450, but is barely observed in the spectrum of H–MOR-550 (Fig. 4b). The disappearance of this feature suggests that only a small number of bridging hydroxyl groups remain in the side-pockets of the H–MOR-550 sample and that nearly all bridging hydroxyl groups can be accessed by pyridine.
Using carbon monoxide as a probe molecule enables the distinction of Lewis acid sites in terms of their coordination number. Assuming the assignment of IR signatures from published literature, the interaction of carbon monoxide with three- and five-coordinated Lewis acidic aluminum result in FTIR absorption bands at 2224 cm−1 and 2198 cm−1 respectively.62–64 Three-coordinated Lewis acid sites are typically found in mordenite zeolites of both high and low aluminum content, but five-coordinated aluminum species are detected in appreciable amounts only in samples of low Si/Al ratio.30Fig. 5 reveals that a major portion of Lewis acid sites after activation of each sample is three-coordinated. Calcining NH4–MOR in situ under vacuum results in an almost ideal proton form with a low contribution of Lewis acid sites. While this sample is largely devoid of five-coordinated Lewis acid sites, such sites are present in non-negligible amounts in the other samples.
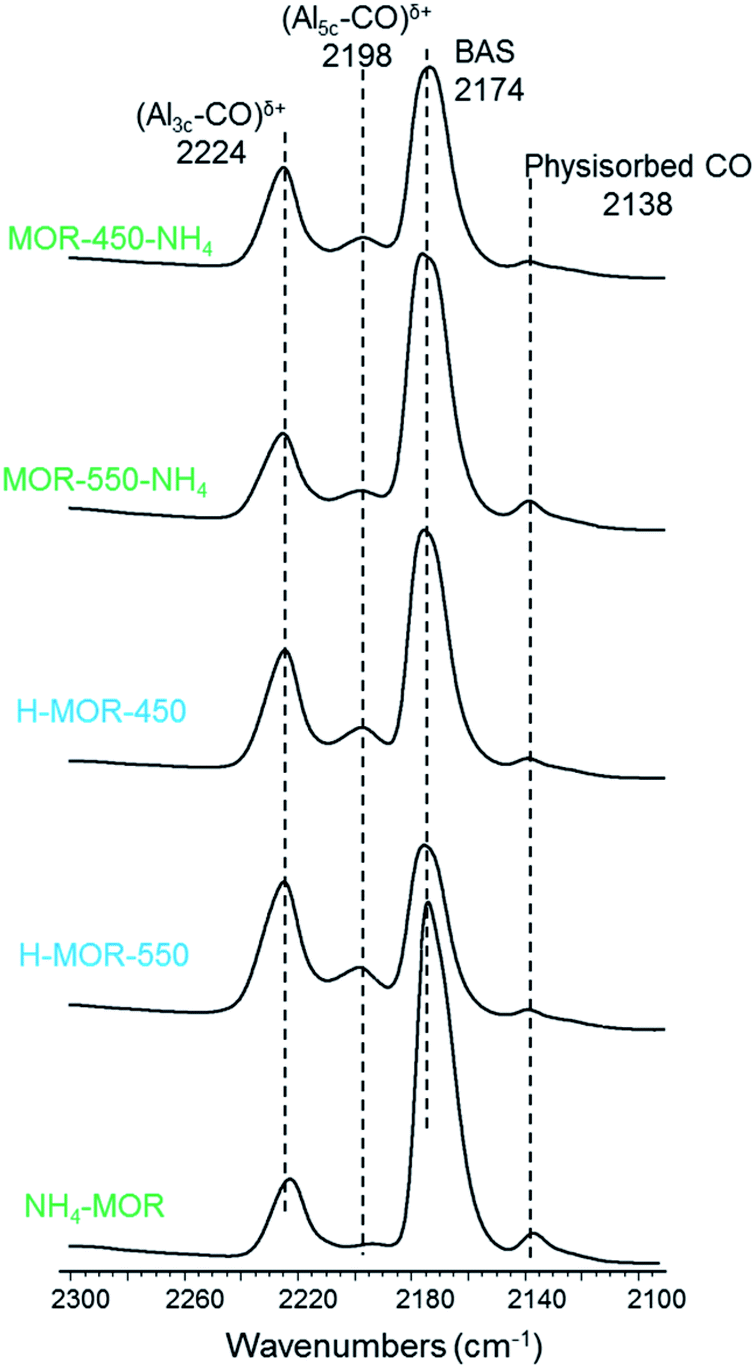 | ||
| Fig. 5 FTIR spectra of adsorbed carbon monoxide over activated parent ammonium form NH4–MOR, H–MOR-450, H–MOR-550, MOR-450-NH4 and MOR-550-NH4. The names of the samples refer to the form of the zeolite prior to heat treatment in the IR setup. | ||
The findings presented in this section constitute an important conceptual advance in the relationship between aluminum coordination and Brønsted–Lewis acidity in zeolites (Scheme 1). Framework aluminum adopts a tetrahedral geometry in the ammonium form of the zeolite. In situ activation of such sample yields Brønsted acid sites through ammonia removal. On the other hand, when NH4–MOR is calcined in static air and exposed to ambient conditions, a fraction of the aluminum described as being associated to the framework adopts octahedral geometry. Upon high-temperature activation, this aluminum species has Lewis acidic character. When the acidic form of the zeolite is back-exchanged into the ammonium form, the framework-associated aluminum reverts to a tetrahedral geometry. In situ activation of such a back-exchanged sample shows low Lewis and high Brønsted acid concentration. Therefore, the property of reversible tetrahedral–octahedral coordination exhibited by framework-associated aluminum facilitates the indirect switching between Brønsted and Lewis acidity (Scheme 1). While the switch in acidity was experimentally observed herein with mordenite, we believe that this can be extended to zeolite topologies that allow the formation of octahedrally coordinated framework-associated aluminum, which have been observed in a range of different zeolites, including BEA46 and zeolite Y.50
 | ||
| Scheme 1 Reversible tetrahedral–octahedral coordination exhibited by framework-associated aluminum and its impact on Brønsted and Lewis acidity. | ||
Location of framework-associated aluminum in the pore system of mordenite
The Lewis acid content detected with pyridine as the probe molecule in H–MOR-450 is only marginally higher than that in activated NH4–MOR (Table 2). However, there is a large difference in the Lewis acid site concentration between H–MOR-450 and H–MOR-550. From 27Al MAS NMR, we know that the fraction of Al(VI) in these acidic zeolites are not substantially different (vide supra). Therefore, the large difference in the intensity of the peak at 1455 cm−1 for these two zeolites (Fig. 4a, Table 2) might not reflect such a quantitative difference in acid site density, but instead could be caused by differences in accessibility of the Lewis acid sites for pyridine. Being a much smaller molecule, carbon monoxide can probe all acid sites in mordenite, including the ones located deep in the side-pockets. From Fig. 5 and Table 2, we infer that the Lewis acid site concentration detected with carbon monoxide in H–MOR-450 and H–MOR-550 differ minimally, while being higher than that observed in in situ activated NH4–MOR. This suggests that the much lower Lewis acid content, as measured with pyridine in H–MOR-450 compared to H–MOR-550, is due to differences in acid site accessibility for pyridine. Fig. 4b corroborates this hypothesis from the context of Brønsted acid site accessibility. While a substantial portion of bridging hydroxyl groups in H–MOR-450 remain inaccessible to pyridine, nearly all Brønsted acid sites can be probed in H–MOR-550 (dashed lines, Fig. 4b). Previous studies on zeolite mordenite of a comparable Si/Al ratio suggested that pyridine can access a part of Brønsted acid sites in the side-pockets in the proton form calcined at 550 °C, which is not the case with the parent ammonium form.59,60,65 Based on these findings, it appears that Lewis acid sites originating from framework-associated aluminum tend to be localized more in the side pockets than in the main channel of mordenite. This would explain why the Lewis acidity measured with pyridine in H–MOR-450, wherein pyridine has considerable accessibility limitations in the side-pockets, is much lower than the acidity measured in H–MOR-550, wherein these restrictions are more relaxed. And notably, the use of carbon monoxide reveals a smaller difference in Lewis acid content between these two samples.The Al–OH groups, which are present in H–MOR-550 and characterized by the IR band at 3660 cm−1, do not engage in a Lewis acidic interaction with pyridine. The corresponding back-exchanged sample, MOR-550-NH4, continues to have appreciable amounts of Al–OH moieties (Fig. 4b), but shows Lewis acidity that is comparable to activated NH4–MOR and MOR-450-NH4 (Table 2), neither of which show signatures for Al–OH. The Al–OH groups are also not Brønsted acidic, since the signature for these species in the OH stretching region remains largely unperturbed after pyridine adsorption (dashed lines, Fig. 4b). The low Lewis acid content in MOR-550-NH4, as detected by pyridine, is unlikely to be caused by accessibility limitations as most of the Brønsted acid sites in this sample can be accessed by pyridine (dashed line, Fig. 4b). Consequently, the low Lewis acid content of MOR-550-NH4 is truly reflective of the transformation of Lewis acid-forming framework-associated aluminum to tetrahedral framework aluminum, which is not Lewis acidic. This observation is further validated by measurements of Lewis acidity using carbon monoxide (Table 2).
While the concept of certain aluminum species exhibiting reversible tetrahedral-octahedral coordination has been reported in the past, we show that Lewis acid sites originating from framework-associated aluminum has preferential site occupancy in zeolites; in the case of mordenite, the side-pockets are more favored to host these sites than the main channel. This insight can have far-reaching implications for the rational synthesis of Lewis acid sites in zeolites by virtue of controlling aluminum distribution.66 Furthermore, such information on site specificity can aid in the engineering of improved catalysts, as realized in the case of Brønsted acid-catalyzed carbonylation.67 While the distribution of Brønsted acid sites can be assessed using 1H DQMAS NMR of acetonitrile-d3 adsorbed over mordenite, catalytic activity in the carbonylation of dimethyl ether correlates with the number of acidic protons in the side-pockets.68,69 Analogous to these findings on the specificity of Brønsted acid sites in carbonylation reactions, knowledge on the site specificity of Lewis acid sites in zeolites will enhance the understanding of their role(s) in catalytic applications.
As is generally observed with different zeolite types, the amount of octahedrally coordinated framework-associated aluminum in the proton form of mordenite decreases with increasing Si/Al ratio.30 The negligible amount of octahedrally coordinated aluminum in the proton form of Si-rich zeolites suggests that isolated aluminum atoms do not undergo this phenomenon, and instead, two or more aluminum atoms in close proximity might be needed for the generation of octahedral framework-associated species. Based on the FTIR results presented herein, the side-pockets in mordenite can be envisioned to provide a more favorable environment in terms of proximal aluminum location for the generation and stabilization of octahedral framework-associated aluminum. In this context, it is worth noting that the relatively stronger acidity of Brønsted acid sites in the side-pockets compared to those in the main channel of mordenite is assigned to the stronger confinement effect imposed by the zeolite framework.68
Al–OH groups and its relation to distorted tetrahedrally coordinated aluminum
The FTIR spectroscopic signature at around 3660 cm−1 for Al–OH groups correlates with the signature for distorted tetrahedrally coordinated aluminum on the 27Al MQMAS NMR. Fig. 4b shows that Al–OH species is virtually non-existent in the spectra of NH4–MOR and H–MOR-450, but present in substantial amounts in the spectrum of H–MOR-550. Stacking this against the corresponding 27Al MQMAS NMR spectra (Fig. 3) reveals that Al–OH groups are linked to the distorted tetrahedral sites (Al(IV)b). The same interpretation can also be drawn from the back-exchanged ammonium samples. MOR-450-NH4 has a negligible population of distorted tetrahedral sites, as evidenced by 27Al MQMAS NMR (Fig. 3) and correspondingly, no pronounced feature for Al–OH groups in the FTIR spectrum (Fig. 4b). MOR-550-NH4 has a far more substantial number of distorted tetrahedral sites on the 27Al MQMAS NMR and correspondingly, a distinct feature for Al–OH groups in the FTIR spectrum. Consequently, the generation of the Al–OH species upon calcination at high temperatures corresponds to a different aluminum species than the framework-associated aluminum that changes coordination. H–MOR-450 is exemplar of this hypothesis as it has appreciable amounts of octahedrally coordinated aluminum but barely any distorted tetrahedral aluminum on the 27Al MQMAS NMR (Fig. 3) or signature for Al–OH groups in the FTIR spectrum (Fig. 4b).Therefore, octahedrally coordinated aluminum in mordenite, Al(VI), which is characterized by the narrow 27Al signal at around 0 ppm under hydrated conditions - and which shows Lewis acidic behavior after dehydration – is able to revert into a framework site. However, distorted tetrahedrally coordinated aluminum, Al(IV)b, which has one or more OH coordinations and is generated by calcination at a higher temperature, behaves differently and does not change coordination upon ammonium exchange. While substantial amounts of octahedrally coordinated aluminum is present under ambient conditions in H–MOR-450 (Fig. 2), the accessibility limitations for pyridine in this sample is comparable to that in NH4–MOR (Fig. 4b). Based on this observation and on the FTIR line shapes of H–MOR-550 and MOR-550-NH4 after pyridine adsorption (Fig. 4b), it appears that it is the distorted tetrahedral sites, and not the octahedrally coordinated species, that modify accessibility for pyridine to acid sites in the side-pockets.
Evolution of aluminum structure with conditions in mordenite
Scheme S1† summarizes the key findings from this study and presents the evolution of aluminum structure in zeolite mordenite as a function of conditions. The NMR and FTIR spectroscopic signatures used for the assignment of different species are listed in a table as part of Scheme S1.† We further indicate the nature of each species, as likely being/originating from framework, extra-framework or framework-associated aluminum. Calcining the ammonium form of mordenite at 450 °C in static air and subsequent exposure of the sample to ambient conditions yields the proton form of the zeolite (H_MOR_wet in Scheme S1†) with aluminum in two different geometries: tetrahedral framework aluminum (Al(IV)a) and octahedrally coordinated framework-associated aluminum (Al(VI)). The zeolite is devoid of penta-coordinated and distorted tetrahedral aluminum moieties. Evacuating H_MOR_wet at elevated temperature converts the octahedrally coordinated aluminum species into primarily three-coordinated and some five-coordinated Lewis acidic species (H_MOR_activated in Scheme S1†) that are preferentially localized in the side-pockets of mordenite.30 Upon ammonium exchange of the H_MOR_wet sample, the coordination environment of aluminum in the zeolite reverts to being exclusively tetrahedral (H_MOR_NH4 in Scheme S1†), as present in the parent NH4_MOR. While such a change in aluminum coordination from octahedral to tetrahedral has been observed with several zeolite topologies, an elaborate mechanism on how ammonium ion-exchange, or equivalently ammonia treatment, facilitates this transformation is yet to be deduced and should be a key research direction to be explored in the future.47,55 Evacuating H_MOR_NH4 under vacuum at high temperature shows greater Brønsted acidity that in the preceding proton form. This is because the conversion of octahedrally coordinated aluminum to a tetrahedral geometry on ammonium exchange restores the typical tetrahedrally coordinated framework aluminum, which yield bridging hydroxyl groups upon ammonia removal. The evolution of aluminum structure on calcining the ammonium form of the zeolite at a higher temperature of 550 °C in static air is largely similar to the evolution observed with the sample calcined at a lower temperature of 450 °C but for one important difference. The treatment at the higher temperature generates distorted tetrahedral aluminum sites (H_MOR_highT_wet in Scheme S1†), which result in anisotropic broadening in the 27Al MAS NMR spectrum of this proton form. In the FTIR spectrum, the Al–OH species are characterized by an absorption at 3660 cm−1 in the OH stretching region and they do not interact with pyridine. While ammonium ion-exchange renders it possible to convert all octahedrally coordinated framework-associated aluminum into tetrahedral coordination, the distorted tetrahedral aluminum sites do not revert to the pristine tetrahedral framework coordination (H_MOR_highT_NH4 in Scheme S1†).Conclusions
Proton forms of zeolite mordenite, obtained by the calcination of the parent ammonium form in wet static air, possess framework-associated octahedrally coordinated aluminum under ambient conditions. After high temperature activation of such sample, the framework-associated aluminum shows Lewis acidic property. The changes in aluminum coordination and conversion of Brønsted into Lewis acidity are partially reversible. Exchanging the proton form back into the ammonium form converts octahedrally coordinated aluminum back into a framework tetrahedral coordination, with the bridging hydroxyl groups being formed upon ammonia removal. The degree of retrieval of Brønsted acidity is a function of the calcination temperature. Higher calcination temperature yields a distorted tetrahedrally coordinated aluminum species that is irreversibly formed and is characterized by a high anisotropy of chemical shift. This species contributes to the IR feature at 3660 cm−1 in the OH-stretching region, assigned to Al–OH moieties. The presence of these Al–OH species modifies pyridine accessibility to the acid sites in the side-pockets of mordenite. The attribute of framework-associated aluminum to undergo reversible transformation in coordination from tetrahedral to octahedral enables the possibility to switch between Brønsted and Lewis acidity. Aluminum in the side-pockets in mordenite are more prone to show such behavior. Therefore, Lewis acid sites formed from framework-associated aluminum are found to have a preferential site occupancy in the pore structure of mordenite. These observations are fundamentally different to the largely researched Lewis acidic extra-framework aluminum and could enable a paradigm shift in the rational design of Lewis acid sites in zeolites.Conflicts of interest
There are no conflicts of interest to declare.References
- C. Marcilly, J. Catal., 2003, 216, 47–62 CrossRef CAS
.
- G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077–1104 CrossRef CAS
- W. O. Haag, R. M. Lago and P. B. Weisz, Nature, 1984, 309, 589 CrossRef CAS
-
W. F. Hölderich and H. Van Bekkum, in Studies in Surface Science and Catalysis, Elsevier, 1991, vol. 58, pp. 631–726 Search PubMed
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC
- K. M. Minachev, V. I. Garanin, V. V. Kharlamov, T. A. Isakova and E. E. Senderov, Russ. Chem. Bull., 1969, 18, 1611–1615 CrossRef
- I. E. Maxwell, Catal. Today, 1987, 1, 385–413 CrossRef CAS
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Energy Environ. Sci., 2011, 4, 793–804 RSC
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS
- P. P. Pescarmona, K. P. F. Janssen, C. Delaet, C. Stroobants, K. Houthoofd, A. Philippaerts, C. De Jonghe, J. S. Paul, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1083–1089 RSC
- P. Lanzafame, D. M. Temi, S. Perathoner, A. N. Spadaro and G. Centi, Catal. Today, 2012, 179, 178–184 CrossRef CAS
- M. Maache, A. Janin, J. C. Lavalley, J. F. Joly and E. Benazzi, Zeolites, 1993, 13, 419–426 CrossRef CAS
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS
- S. M. Maier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 8005–8013 CrossRef CAS
- B. Gil, S. I. Zones, S.-J. Hwang, M. Bejblová and J. Čejka, J. Phys. Chem. C, 2008, 112, 2997–3007 CrossRef CAS
- M. Boronat and A. Corma, ACS Catal., 2019, 9, 1539–1548 CrossRef CAS
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouillout, V. V. Yuschenko, C. Fernandez, J. P. Gilson, F. Fajula and I. I. Ivanova, Microporous Mesoporous Mater., 2004, 71, 157–166 CrossRef CAS
- L. Bui, H. Luo, W. R. Gunther and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022–8025 CrossRef CAS
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, Chem. Soc. Rev., 2015, 44, 7025–7043 RSC
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS
- V. V. Ordomsky, V. L. Sushkevich, J. C. Schouten, J. Van Der Schaaf and T. A. Nijhuis, J. Catal., 2013, 300, 37–46 CrossRef CAS
- S. Suganuma, T. Hisazumi, K. Taruya, E. Tsuji and N. Katada, ChemistrySelect, 2017, 2, 5524–5531 CrossRef CAS
- M. A. Bañares, J. H. Cardoso, G. J. Hutchings, J. M. C. Bueno and J. L. G. Fierro, Catal. Lett., 1998, 56, 149–153 CrossRef
- B. Wichterlová, Z. Tvarůžková, Z. Sobalık and P. Sarv, Microporous Mesoporous Mater., 1998, 24, 223–233 CrossRef
- X. Pu, N.-w. Liu and L. Shi, Microporous Mesoporous Mater., 2015, 201, 17–23 CrossRef CAS
- J. Brus, L. Kobera, W. Schoefberger, M. Urbanová, P. Klein, P. Sazama, E. Tabor, S. Sklenak, A. V. Fishchuk and J. Dědeček, Angew. Chem., Int. Ed., 2015, 54, 541–545 CAS
- T. K. Phung and G. Busca, Appl. Catal., A, 2015, 504, 151–157 CrossRef CAS
- G. Busca, Microporous Mesoporous Mater., 2017, 254, 3–16 CrossRef CAS
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo and S.-B. Liu, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, J. Phys. Chem. C, 2019, 123, 15139–15144 CrossRef CAS
- G. L. Woolery, G. H. Kuehl, H. C. Timken, A. W. Chester and J. C. Vartuli, Zeolites, 1997, 19, 288–296 CrossRef CAS
- P. A. Jacobs and H. K. Beyer, J. Phys. Chem., 1979, 83, 1174–1177 CrossRef CAS
- U. Lohse, E. Löffler, M. Hunger, J. Stöckner and V. Patzelova, Zeolites, 1987, 7, 11–13 CrossRef CAS
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S.-B. Liu and A. Zheng, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS
- M. Ravi, V. L. Sushkevich and J. A. van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS
- Z. Yu, S. Li, Q. Wang, A. Zheng, X. Jun, L. Chen and F. Deng, J. Phys. Chem. C, 2011, 115, 22320–22327 CrossRef CAS
- S. Li, A. Zheng, Y. Su, H. Fang, W. Shen, Z. Yu, L. Chen and F. Deng, Phys. Chem. Chem. Phys., 2010, 12, 3895–3903 RSC
- R. A. Beyerlein, C. Choi-Feng, J. B. Hall, B. J. Huggins and G. J. Ray, Top. Catal., 1997, 4, 27–42 CrossRef CAS
- V. L. Zholobenko, L. M. Kustov, V. B. Kazansky, E. Loeffler, U. Lohser, C. Peuker and G. Oehlmann, Zeolites, 1990, 10, 304–306 CrossRef CAS
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler, H. Van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef CAS
- G. Catana, D. Baetens, T. Mommaerts, R. A. Schoonheydt and B. M. Weckhuysen, J. Phys. Chem. B, 2001, 105, 4904–4911 CrossRef CAS
- Z. Yu, A. Zheng, Q. Wang, L. Chen, J. Xu, J. P. Amoureux and F. Deng, Angew. Chem., Int. Ed., 2010, 49, 8657–8661 CrossRef CAS
- S. Xin, Q. Wang, J. Xu, Y. Chu, P. Wang, N. Feng, G. Qi, J. Trébosc, O. Lafon and W. Fan, Chem. Sci., 2019, 10, 10159–10169 RSC
- T. T. Le, A. Chawla and J. D. Rimer, J. Catal., 2020, 391, 56–68 CrossRef CAS
- K. Chen, S. Horstmeier, V. T. Nguyen, B. Wang, S. P. Crossley, T. Pham, Z. Gan, I. Hung and J. L. White, J. Am. Chem. Soc., 2020, 142, 7514–7523 CrossRef CAS
- E. Bourgeat-Lami, P. Massiani, F. Di Renzo, P. Espiau, F. Fajula and T. Des Courières, Appl. Catal., 1991, 72, 139–152 CrossRef CAS
- A. Omegna, J. A. van Bokhoven and R. Prins, J. Phys. Chem. B, 2003, 107, 8854–8860 CrossRef CAS
- J. A. van Bokhoven, A. M. J. Van der Eerden and D. C. Koningsberger, J. Am. Chem. Soc., 2003, 125, 7435–7442 CrossRef CAS
- D. Ma, F. Deng, R. Fu, X. Han and X. Bao, J. Phys. Chem. B, 2001, 105, 1770–1779 CrossRef CAS
- B. H. Wouters, T. H. Chen and P. J. Grobet, J. Am. Chem. Soc., 1998, 120, 11419–11425 CrossRef CAS
- J. A. van Bokhoven, Phys. Scr., 2005, 2005, 76–79 CrossRef
- Z. Zhao, S. Xu, M. Y. Hu, X. Bao, C. H. F. Peden and J. Hu, J. Phys. Chem. C, 2014, 119, 1410–1417 CrossRef
- P. J. Kunkeler, B. J. Zuurdeeg, J. C. Van der Waal, J. A. van Bokhoven, D. C. Koningsberger and H. Van Bekkum, J. Catal., 1998, 180, 234–244 CrossRef CAS
- L. C. De Ménorval, W. Buckermann, F. Figueras and F. Fajula, J. Phys. Chem., 1996, 100, 465–467 CrossRef
- I. J. Drake, Y. Zhang, M. K. Gilles, C. N. Teris Liu, P. Nachimuthu, R. C. C. Perera, H. Wakita and A. T. Bell, J. Phys. Chem. B, 2006, 110, 11665–11676 CrossRef CAS
- T. H. Chen, B. H. Wouters and P. J. Grobet, Eur. J. Inorg. Chem., 2000, 2000, 281–285 CrossRef
- A. Abraham, S.-H. Lee, C.-H. Shin, S. B. Hong, R. Prins and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2004, 6, 3031–3036 RSC
- I. Kiricsi, C. Flego, G. Pazzuconi, W. O. Parker Jr, R. Millini, C. Perego and G. Bellussi, J. Phys. Chem., 1994, 98, 4627–4634 CrossRef CAS
- J. Datka, B. Gil and A. Kubacka, Zeolites, 1997, 18, 245–249 CrossRef CAS
- F. Moreau, P. Ayrault, N. S. Gnep, S. Lacombe, E. Merlen and M. Guisnet, Microporous Mesoporous Mater., 2002, 51, 211–221 CrossRef CAS
- X. Li, R. Prins and J. A. van Bokhoven, J. Catal., 2009, 262, 257–265 CrossRef CAS
- L. M. Kustov, V. B. Kazanskii, S. Beran, L. Kubelkova and P. Jiru, J. Phys. Chem., 1987, 91, 5247–5251 CrossRef CAS
- S. Bordiga, E. E. Platero, C. O. Areán, C. Lamberti and A. Zecchina, J. Catal., 1992, 137, 179–185 CrossRef CAS
- F. Wakabayashi, J. N. Kondo, K. Domen and C. Hirose, J. Phys. Chem., 1995, 99, 10573–10580 CrossRef CAS
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef
- J. Dědeček, E. Tabor and S. Sklenak, ChemSusChem, 2019, 12, 556–576 CrossRef
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS
- X. Yi, Y. Xiao, G. Li, Z. Liu, W. Chen, S.-B. Liu and A. Zheng, Chem. Mater., 2020, 32, 1332–1342 CrossRef CAS
- Z. Liu, X. Yi, G. Wang, X. Tang, G. Li, L. Huang and A. Zheng, J. Catal., 2019, 369, 335–344 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06130a |
| This journal is © The Royal Society of Chemistry 2021 |