
Boosting electrochemical CO2 reduction to formate using SnO2/graphene oxide with amide linkages†
Zhongjie
Yang
ab,
Caoyu
Yang
ab,
Jianyu
Han
a,
Wenshi
Zhao
ab,
Shengxian
Shao
ab,
Siyu
Li
a,
Huiwen
Gao
ab,
Haijiao
Xie
c and
Xiaofei
Zhang
 *a
*a
aCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: stone623717@outlook.com
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cHangzhou Yanqu Information Technology Co., Ltd., Zhejaing 310003, China
First published on 7th June 2021
Abstract
Selective electrochemical CO2 reduction to formic acid is an appealing strategy to achieve efficient utilization of CO2. Modification of the catalytic interface with organic linkers has been proven to promote the conversion of CO2 to CO, but the highly selective generation of formic acid is still a challenge. Herein, modification of the interface of graphene oxide (GO) with amide linkages to anchor SnO2 nanoparticles is presented to catalyze the CO2 reduction reaction (CO2RR). As expected, the as-synthesized SnO2/tert-GO shows a competitive high cathodic faradaic efficiency (FE) of 84.4% for formate at −0.96 V and shows long-term stability for the CO2RR. Further, density functional theory (DFT) calculations indicated that amide linkages facilitate the conversion of CO2 to formate.
Introduction
Excessive consumption of fossil fuels not only poses a huge challenge to the sustainable development of society, but also has a negative impact on the global climate with increasing CO2 emissions.1–4 However directly using carbon dioxide as a chemical reactant will turn undesired waste into treasure. To alleviate these problems, electrocatalytic reduction of CO2 to valuable hydrocarbons is considered as a promising solution due to mild conditions and conversion efficiency.5–10 Nevertheless, the practical production of liquid fuels (e.g., C2H5OH and HCOOH) is greatly impeded by the large overpotential and byproducts derived from the hydrogen evolution reaction (HER).11–14Among the various products, HCOOH occupies a pivotal position in industry. As an important chemical intermediate, formic acid is undoubtedly strongly needed and widely utilized in textile manufacturing, antibacterial agents, fuel cells and livestock feed.15–17 Recently, immense efforts have been made to produce formic acid with high selectivity.18–21 Homogeneous and heterogeneous catalysts have been widely used.22–26 However some obvious flaws still exist. The separation of homogeneous catalysts requires high energy consumption and a complicated process. Compared with their homogeneous counterparts, heterogeneous catalysts overcome the problem of separation. Commonly used materials including Pd, Pb, Hg, In, and Cd exhibit good catalytic activity for HCOOH, but sometimes their application is hindered by the shortcomings of scarcity and toxicity.27
Among reported emerging catalysts, Sn containing catalysts are of particular interest due to the advantages of cost-effectiveness and nontoxicity. As a result, different catalytic systems including Sn containing alloys and SnO2 with specific crystal planes and composites are developed to improve the yield of HCOOH.28–30 Although some progress has been made, the utilization efficiency of Sn is still very low. Furthermore, the stability of the catalysts and the synergistic effect of multiple components are often low. Therefore, it is necessary to construct a new catalytic system to meet the requirements of high efficiency and stability for the CO2RR.
In recent years, two dimensional (2D) materials have been widely used in catalysis and greatly promote the development of heterogeneous catalysts.31,32 Whether the 2D materials themselves have active sites, or have active components loaded on their surface, they can maximize the utilization of metal atoms and help to increase the material transfer rate. However, only a few cases of Sn based 2D materials have so far been reported for catalyzing the CO2RR, which are subject to limitations of synthetic methods and poor stability. For instance, Li et al. reported a 2D SnO2 nanosheet catalyst for the CO2RR with a faradaic efficiency of more than 80%.30 However low conductivity hindered the transfer of electrons to the carbon black support. More recently, Tsujiguchi et al. explored Sn nanoparticles (NPs) loaded on reduced graphene oxide (rGO) for the CO2RR.28 The synthesized Sn/rGO composites showed stronger CO2 adsorption ability than bare Sn catalysts. Thus, the synergy effect between the active centers and support promotes the catalytic conversion of carbon dioxide. Obviously, the chosen supports not only can stabilize the active sites but also provide additional opportunities to enhance the catalytic activity.
The surface molecular approach has emerged as an effective means of regulating catalytic activity due to straightforwardness and controllability.33 The interaction between the catalysts and reactants determines the overall catalytic activity and selectivity of products. In our previous work, we demonstrated that the introduced amide linkages on the surface of graphene oxide can effectively capture CO2 and further accelerate the reaction of terminal alkyne with CO2 to generate acetylenic acid with Ag active sites.34 Motivated by the above exploration, we constructed SnO2/amide graphene oxide named SnO2/tert-GO, in which the amide group enhances the enrichment of carbon dioxide around tin oxide in ambient atmosphere (Fig. 1). To the best of our knowledge, although the ligand effect is widely studied in the electroreduction of carbon dioxide to carbon monoxide,35 its role in promoting the production of formic acid with high activity has been rarely explored.
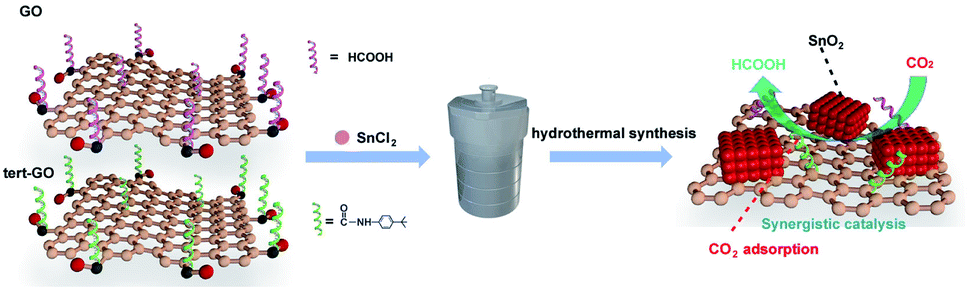 | ||
| Fig. 1 Synthesis scheme of SnO2/GO and SnO2/tert-GO. | ||
Herein, SnO2/tert-GO was synthesized by a simple amide linkage functionalization of graphite oxide powder and hydrothermal transformation of SnCl2 to form SnO2. The as-obtained SnO2/tert-GO showed superior CO2RR catalytic performance compared to SnO2/GO without amide linkages and bare SnO2 NPs, and the FEHCOO− reached 84.4% at – 0.96 V vs. RHE.
Experimental section
Chemical materials
Tin dichloride dihydrate (SnCl2·2H2O) was bought from Alfa Aesar and 4-tert-butylaniline (98%) was purchased from TCI. N,N-Dimethylformamide (DMF, 99.8%) was purchased from Ailan (Shanghai) Chemical Technology Co., Ltd. Graphite oxide was bought from Aladdin. Graphene oxide was bought from Yuanye (Shanghai) Bio-Technology Co., Ltd. Ultrapure water (18 MΩ) was supplied by a Millipore System (Millipore Q). All the chemicals were used without further purification.Synthesis of tert-GO
Graphite oxide (150 mg) was dispersed in 15 mL ethanol. Then, 2,2′-azobis(2-methylpropionitrile) (30 mg, 0.183 mmol) and 4-(t-butyl)aniline (1.5 mL, 9.42 mmol) were added into the above solvent. Subsequently, the mixture was subjected to sonication for 3 h. Afterwards, the product was collected by centrifugation, followed by washing with DMF, ethanol and ultrapure water in turn. Finally, the product of tert-GO was successfully obtained by freeze drying.In this process, the amide bond is formed by the condensation reaction between the carboxyl group on the surface of the graphene oxide and 4-tert-butylaniline.
Synthesis of SnO2/GO, SnO2/tert-GO and SnO2
In a typical procedure, 85 mg of SnCl2·2H2O and 200 mg of tert-GO were mixed with 40 mL of deionized water and stirred for 1 h. Then the suspension was transferred into an autoclave and heated at 200 °C for 12 h. After cooling down to room temperature, the precipitate was collected by centrifugation, followed by washing with water and alcohol several times. The final product of SnO2/tert-GO was obtained after drying for 12 h at 80 °C in a vacuum oven. Keeping other conditions unchanged, with the addition of 200 mg or no GO, SnO2/GO and SnO2 were obtained.36Materials characterization
SEM images were taken using a Hitachi S-8220 Scanning Electron Microscope (SEM) operated at 8 kV. Transmission electron microscopy (TEM), high-resolution TEM (HRTEM), high-angle annular dark-field scanning TEM (HAADF-STEM) imaging and X-ray energy dispersive spectroscopy (X-EDS) were performed using a Tecnai G2 F20 S-TWIN at 200 kV. X-ray photoelectron spectroscopy (XPS) measurements were carried out on an ESCALAB250Xi spectrometer at a pressure of ∼3 × 10−9 mbar (1 mbar = 100 Pa) using Al Kα as the excitation source (hν = 1486.6 eV) and operated at 15 kV and 20 mA. Powder X-ray diffraction (XRD) patterns were recorded on a D/MAX-TTRIII (Cross Beam Optics) with Cu Kα radiation (λ = 1.542 Å) operating at 40 kV and 200 mA. Atomic force microscopy (AFM) was carried out by using a Bruker multimode-8 scanning probe microscope.Electrochemical measurements
Electrochemical experiments were carried out in a two-compartment electrochemical cell with a Nafion 117 proton-exchange membrane as the separator. All electrochemical measurements were performed with a CHI 660E (CHI Instrument Co.) potentiostat using carbon paper as the working electrode, a platinum wire as the counter electrode and Ag/AgCl (3.5 M KCl) as the reference electrode, respectively. All potentials were displayed versus the reversible hydrogen electrode (RHE) values using the equation: E (vs. RHE) = E (vs. Ag/AgCl) + 0.205 V + 0.059 V × pH. A 0.1 M KHCO3 aqueous solution saturated with CO2 was (pH = 6.8) used as the electrolyte. During the electrochemical test, CO2 bubbles entering the cathode portion of the cell were kept at 20 mL min−1. For the preparation of the catalyst ink, 10 mg of catalyst and 30 μL of 5 wt% Nafion solution were dispersed into 220 μL water and 750 μL isopropanol, and vigorously sonicated for 30 min to form a uniform catalyst ink. Each working electrode was prepared by uniformly dropping 100 μL of catalyst ink onto the 1 × 1 cm2 carbon paper electrode. Linear sweep voltammetry (LSV) measurements were performed at a scan rate of 10 mV s−1. In order to determine the reduction products and their faradaic efficiency (FE), bulk electrolysis was conducted at selected potentials (−0.66 V to −1.26 V vs. RHE). All electrochemical curves were obtained without iR corrections. The gaseous products from the CO2 reduction were measured by on-line gas chromatography. The liquid products were determined by 1H-NMR spectroscopy (Bruker Advance 400 spectrometer, 400 MHz) via water suppression using a presaturation method. 500 μL of electrolyte was mixed with 100 μL of D2O, and 0.7 ppm dimethyl sulfoxide (DMSO) was added as the internal standard.Computational method
The Vienna Ab initio Simulation Package (VASP)37,38 is employed to perform all density functional theory (DFT) calculations within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE)39 functional. The projected augmented wave (PAW) potentials40,41 were selected to describe the ionic cores together taking valence electrons into account by using a plane wave basis set with a kinetic energy cutoff of 400 eV. A Monkhorst–Pack k-point mesh of 2 × 2 × 1 was applied for all the calculations. The DFT-D3 empirical correction method was carried out to describe van der Waals interactions.42 Geometry optimizations were performed with the force convergency smaller than 0.05 eV Å−1. All the atoms in models are relaxed in all the calculations. The reaction steps for the electrochemical reduction of CO2 to formic acid and the hydrogen evolution reaction were considered as follows:| CO2 + H + e− + * → OCHO* |
| OCHO* + H + e− + * → HCOOH |
The free energy change ΔG of the reaction was calculated as the difference between the free energies of the initial and final states as shown below:
| ΔG = ΔE + ΔZPE − TΔS | (1) |
Results and discussion
The morphology of pristine graphite oxide shows three-dimensional bulk in SEM and TEM images (Fig. S1a and 1b†). After being treated with tert-butylaniline, the morphology of synthesized graphene oxide (tert-GO) turns into ultrathin nanosheets. The thickness of the obtained nanosheets is about 4 nm which was measured by AFM (Fig. S2†). The resulting tert-GO is further utilized as a support to disperse SnCl2·2H2O in water under stirring. The SnO2/tert-GO composites are obtained under hydrothermal conditions at 200 °C for 16 h (Fig. 1). The SEM image reveals that SnO2 nanoparticles are evenly dispersed on the tert-GO substrate (Fig. S4†). The abundant oxygen-containing groups provide uniform rivet sites to anchor SnO2 NPs. The TEM image further confirms the successful preparation of 2D composite nanomaterials (Fig. 2a and S3a†). The SnO2(110) with a lattice constant of 0.33 nm can be observed in the high-resolution transmission electron microscopy (HRTEM) image of SnO2/tert-GO (Fig. 2c and S3b†). In order to further confirm the composition of elements, high-angle annular dark-field scanning TEM (HAADF-STEM) and energy-dispersive X-ray (EDS) elemental mapping analysis are carried out (Fig. 2b). The results show that all the C, N, O and Sn elements are evenly distributed on the tert-GO (Fig. 2d–g). Next, the SnO2/tert-GO composite is characterized by X-ray diffraction (XRD). The XRD pattern of SnO2/tert-GO exhibits diffraction peaks that can be assigned to the planes of 110, 101, 200, 211, 220, 301, 310 respectively and the standard diffraction data of SnO2 (COD 5000224) are provided for comparison (Fig. 2h). The valence is further investigated by X-ray photoelectron spectroscopy (XPS). The Sn 3d XPS spectra show a peak at 487.44 eV for SnO2/tert-GO (Fig. 2i).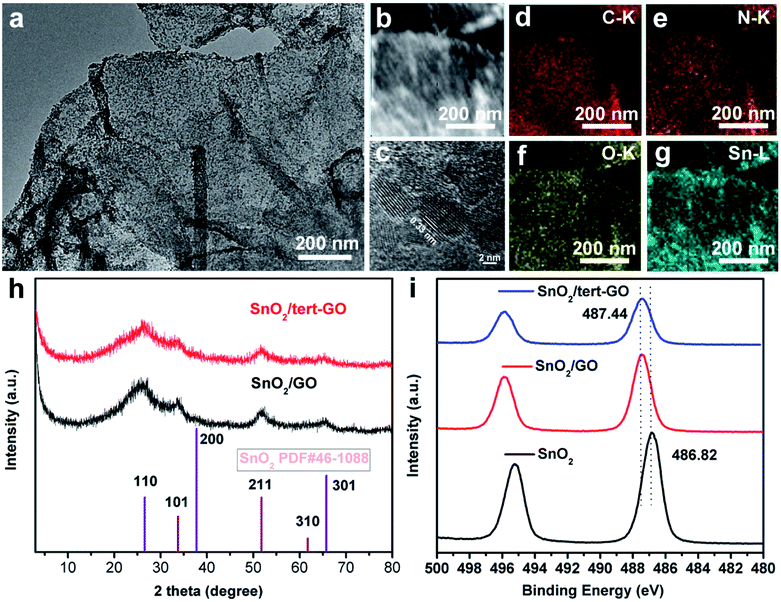 | ||
| Fig. 2 Characterization of SnO2/GO and SnO2/tert-GO catalysts. (a) TEM bright-field image of SnO2/tert-GO catalysts. (b) A higher magnification STEM image of SnO2/tert-GO catalysts. (c–g) The X-EDS mappings of SnO2/tert-GO catalysts. (h) XRD pattern of SnO2/GO and SnO2/tert-GO catalysts. (i) High-resolution XPS spectra of SnO2/GO, SnO2/tert-GO and SnO2. | ||
Furthermore, SnO2/graphene oxide composites denoted as SnO2/GO and bare SnO2 NPs are synthesized under similar conditions as a comparison. The difference is that only GO or no substrate is added. The morphology of SnO2/GO remained almost the same as that of SnO2/tert-GO, which can be confirmed in the SEM and TEM images (Fig. S5 and S6a†). Further, the XRD pattern of SnO2/GO and SnO2 reveals the successful synthesis of SnO2 nanoparticles (Fig. 2h and S7†). Elemental mapping analysis shows that all the C, O and Sn elements are evenly distributed on the GO (Fig. S6c–e†). Meanwhile, the structure and morphology of SnO2 NPs are characterized by XRD, SEM and TEM (Fig. S7, S8a, and S8b†). Next, the electronic structures of SnO2/GO and SnO2 are further explored by XPS. The XPS spectra of Sn 3d (Fig. 2i) show peaks at 487.44 eV and 486.82 eV, and what needs to be emphasized is that the XPS peak deviation of SnO2 and SnO2/tert-GO comes from coordination unsaturated Sn–O bonds caused by the graphene oxide substrate. The tin atoms in SnO2/tert-GO have a higher oxidation state compared with SnO2, which helps to increase reactivity.
The electrochemical CO2RR performances of SnO2, SnO2/GO and SnO2/tert-GO were evaluated in CO2-saturated 0.1 M KHCO3 (pH = 6.8) electrolyte in an H-type cell. All catalysts were uniformly drop cast onto carbon papers and subjected to electrolysis. All potentials reported here were measured versus the Ag/AgCl (3.5 M KCl) electrode and then converted to values versus the reversible hydrogen electrode (RHE) without iR compensation. The liquid and gaseous products were detected by 1H NMR spectroscopy and on-line GC, respectively. The electrochemical activities of SnO2, SnO2/GO and SnO2/tert-GO were initially investigated by linear sweep voltammetry (LSV) which was performed at a scan speed of 10 mV s−1 in a CO2-saturated solution of 0.1 M KHCO3 (Fig. 3a). All three catalysts showed obvious catalytic activity indicating that SnO2 was the original active site for catalytic electroreduction of carbon dioxide. Among all three samples, the current density of SnO2 is significantly lower than that of SnO2/GO and SnO2/tert-GO. This indicated that the 2D GO substrate can effectively increase the conductivity for SnO2, which is beneficial for the fast transfer of electrons during the CO2RR. Notably, SnO2/tert-GO displays the largest current density in the potential range −0.6 to −1.3 V vs. RHE. Compared with the carboxyl functional group in SnO2/GO, the amide linkages in tert-GO show stronger CO2 adsorption ability, thus increasing the catalytic activity.31
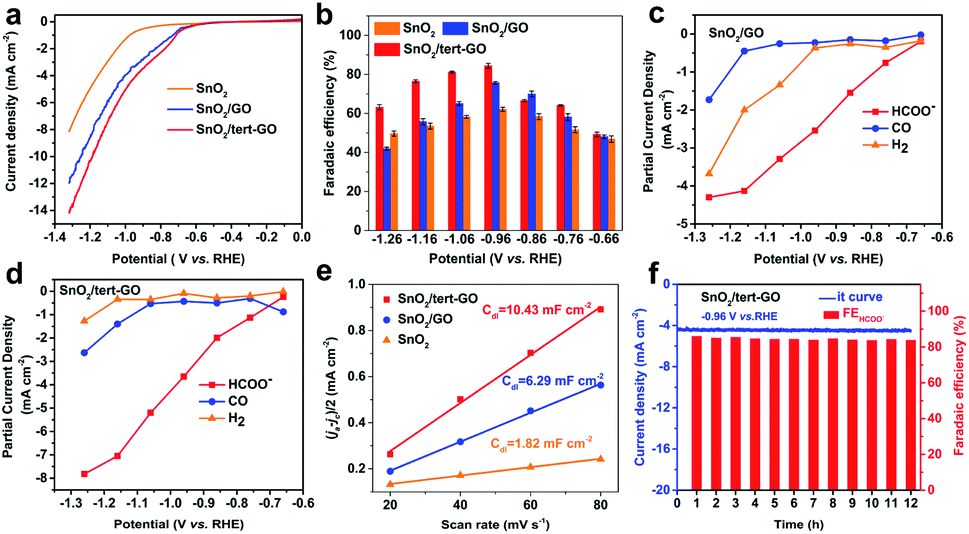 | ||
| Fig. 3 (a) LSV curves (a) of SnO2, SnO2/GO and SnO2/tert-GO at a scan rate of 10 mV s−1. (b) Faradaic efficiencies of formate when using SnO2, SnO2/GO and SnO2/tert-GO. (c and d) The partial current densities of formate, CO and H2 of SnO2/GOand SnO2/tert-GO. (e) ECSA of SnO2, SnO2/GO and SnO2/tert-GO,where ja and jc are the anodic and cathodic current density, respectively. (f) The durability test of SnO2/tert-GO at −0.96 V vs. RHE. All the tests were done in CO2-saturated 0.1 M KHCO3 aqueous solution. | ||
Product selectivity of HCOOH is a vital evaluation index in the CO2RR. The gaseous products and liquid products were quantified by online GC and 1H-NMR, respectively. In order to obtain suitable electrolytic potentials, SnO2, SnO2/GO and SnO2/tert-GO modified electrodes were electrolyzed at a gradually increasing applied voltage range of −0.66 to −1.26 V vs. RHE for 3600 s, respectively (Fig. S9†). As shown in Fig. 3b and S10,† the maximum FEHCOO− is 62.2%, 75.7% and 84.4% for SnO2, SnO2/GO and SnO2/tert-GO at −0.96 V vs. RHE, respectively. Obviously, SnO2/GO and SnO2 showed lower FEHCOO− than SnO2/tert-GO. The hydrophilicity test is carried out to explore the effect of different linkers on products. The contact angle of SnO2/GO and SnO2/tert-GO is 13.7° and 42.2°, respectively. The weakening of hydrophilicity in SnO2/tert-GO can inhibit the HER reaction to a certain extent, thus enhancing the selectivity of formate (Fig. S12†). To achieve high CO2RR production rates, the concentrations of CO2 at the surface of obtained catalysts should be promoted at the molecular level. However, CO2 molecules are originally dissolved in the liquid electrolyte and hardly supplied to the surface of catalysts due to the low CO2 solubility. The introduced amide linkages on graphene oxide build an effective fence around the active center to capture carbon dioxide.
We also summarized the FEH2 and FECO of the three catalysts, as shown in Fig. S9.† For SnO2/tert-GO, the FEHCOO− and FECO added up to stay above 80% at the applied voltage range of −0.76 to −1.26 V vs. RHE. For SnO2 and SnO2/GO, the FEH2 was obviously higher than that of SnO2/tert-GO. The partial current densities of formate, hydrogen and carbon monoxide were calculated according to the FEs of the products and the LSV curves for SnO2/GO and SnO2/tert-GO. As shown in Fig. 3c, for SnO2/tert-GO, the partial current density of formate is higher than that of hydrogen and carbon monoxide, indicating that the reactions of both carbon monoxide and hydrogen are inhibited, which is also attributed to the GO surface modified by the hydrophobic group. For SnO2/GO, as shown in Fig. 3d, the partial current density of hydrogen is higher than that of carbon monoxide. This result indicated that SnO2 loaded on GO had improved electrical conductivity, and some active sites of GO for the HER were activated.
Furthermore, the electrochemical surface areas (ECSAs) of the cathodes were examined to understand the reaction mechanism. The double layer capacitance values of SnO2, SnO2/GO and SnO2/tert-GO were calculated as 1.82, 6.29 and 10.43 mF cm−2, respectively. By estimating the double-layer capacitance measured via cyclic voltammetry (CV, Fig. S11† and Fig. 3e), it can be inferred that the ECSA of SnO2/tert-GO is larger than that of SnO2/GO and SnO2. It can be concluded that both GO and tert-GO substrates could effectively increase the ECSA and the number of CO2RR active sites. Moreover, the durability of SnO2/tert-GO is obtained at an applied potential of −0.96 V vs. RHE (Fig. 3f). The cathodic current density was maintained at −4.5 mA cm−2 for 12 h with negligible activity decay and the corresponding FEHCOO− was kept at around 84% for the whole electrolysis period, suggesting that SnO2/tert-GO exhibited prominent stability under the test conditions. In addition, Fourier Transform Infrared Spectroscopy (FTIR) is utilized to prove the presence of amide groups and stability of SnO2/tert-GO (Fig. S13†). The signal of bending vibration of N–H in the amide bond appears at 1567 cm−1, and a small amount of reduced signal of carbonyl can be distinguished at 1726 cm−1 in the FITR spectrum of SnO2/tert-GO. After the CO2RR process, the signal of the amide bond still exists, indicating the good stability of amide groups in catalysis. As a result, compared with the reported literature, SnO2/tert-GO shows a competitive performance for CO2-to-formate (Table S1†).
To further understand the origin of catalytic activity of the SnO2/tert-GO catalyst, the calculation models in which a small SnO2 cluster was loaded on GO and tert-GO are established. The SnO2 could be stabilized by the abundant oxygen-containing groups on the surface of graphene oxide. Thus, the interface of SnO2/tert-GO and SnO2/GO is composed of SnO2 and different organic linkers, which could reasonably illustrate the effect of ligand on catalytic activity. The Gibbs free energies of converting CO2 into formic acid were calculated with SnO2 loaded on tert-GO and GO (Fig. 4). Furthermore, the OCHO* intermediate has been regarded as the key intermediate in HCOOH production. We discovered that SnO2/tert-GO is beneficial to form the OCHO* intermediate (ΔG = 0.63 eV). In contrast, SnO2/GO shows a lower value (ΔG = 0.98 eV) for the OCHO* intermediate. These results indicated that the amide linkages in surfaces of tert-GO could accelerate the rate-determining step of *COOH formation, thus, HCOOH formation was enhanced. In order to understand the reason of high selectivity for formate, the theoretical calculations including the competitive pathway of the HER and CO2 to CO are carried out. The free energy (ΔG) of SnO2/tert-GO and SnO2/GO in the HER is 0.95 eV and 1.43 eV, respectively. Meanwhile, the ΔG of SnO2/tert-GO and SnO2/GO in production of CO is 1.53 eV and 1.85 eV, respectively (Fig. S14†). Obviously, both of them show a higher energy barrier compared with production of formate and prove that SnO2/tert-GO facilitates the conversion of CO2 to formate.
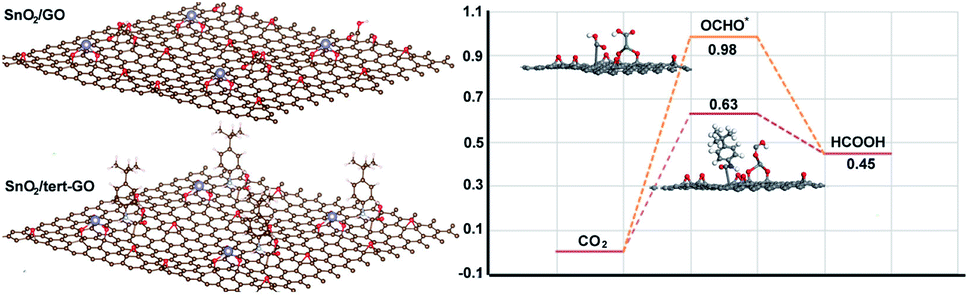 | ||
| Fig. 4 Theoretical calculation of conversion of CO2 to HCOOH. | ||
Conclusion
In summary, we develope graphene oxide functionalized with amide linkages to support SnO2 to elucidate the role of catalytic interface modification in electrochemical CO2 reduction. The as-synthesized SnO2/tert-GO shows a competitive high cathodic FE of 84.4% for formate at −0.96 V vs. RHE and retains long-term stability for the CO2RR. Further, the DFT calculations illustrate that the introduced amide linkages in tert-GO facilitate the adsorption and further promote the conversion of carbon dioxide by decreasing the activation energy of the rate-determining step. Therefore, this approach by regulating the catalytic interface to activate CO2 can be applied to other catalytic systems involving CO2.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the support provided by the National Center for Nanoscience and Technology and professor Zhiyong Tang.References
- P. Prabhu, V. Jose and J. M. Lee, Adv. Funct. Mater., 2020, 30, 1910768 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632 RSC.
- H. B. Zhang, W. R. Cheng, D. Y. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 2–22 CrossRef.
- N. Q. Zhang, X. X. Zhang, L. Tao, P. Jiang, C. L. Ye, R. Lin, Z. W. Huang, A. Li, D. W. Pang, H. Yan, Y. Wang, P. Xu, S. F. An, Q. H. Zhang, L. C. Liu, S. X. Du, X. D. Han, D. S. Wang and Y. D. Li, Angew. Chem., 2021, 133, 6235–6241 CrossRef.
- A. Vasileff, Y. P. Zhu, X. Zhi, Y. Q. Zhao, L. Ge, H. M. Chen, Y. Zheng and S. Z. Qiao, Angew. Chem., 2020, 132, 19817–19821 CrossRef.
- C. Peng, G. Luo, J. B. Zhang, M. H. Chen, Z. Q. Wang, T. K. Sham, L. J. Zhang, Y. F. Li and G. F. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- C. Chen, J. F. K. Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- Q. Y. Wang, C. Cai, M. Y. Dai, J. W. Fu, X. D. Zhang, H. J. W. Li, H. Zhang, K. J. Chen, Y. Y. Lin, H. M. Li, J. H. Hu, M. Miyachi and M. Liu, Small Science, 2021, 1, 2000028 CrossRef.
- P. Ding, H. T. Zhao, T. S. Li, Y. S. Luo, G. Y. Fan, G. Chen, S. Y. Gao, X. F. Shi, S. Y. Lu and X. P. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- P. D. An, L. Wei, H. J. W. Li, B. P. Yang, K. Liu, J. W. Fu, H. M. Li, H. Liu, J. H. Hu, Y.-R. Liu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC.
- F. H. Li and Q. Tang, J. Mater. Chem. A, 2019, 7, 19872–19880 RSC.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS.
- S. B. Liu, X. F. Lu, J. Xiao, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS.
- F. C. Lei, W. Liu, Y. F. Sun, J. Q. Xu, K. T. Liu, L. Liang, T. Yao, B. C. Pan, S. Q. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS.
- S. Liu, J. Xiao, X. Feng Lu, J. Wang, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS.
- Y. Zhong, Y. Xu, J. Ma, C. Wang, S. Y. Sheng, C. T. Cheng, M. X. Li, L. Han, L. L. Zhou, Z. Cai, Y. Kuang, Z. Liang and X. M. Sun, Angew. Chem., 2020, 132, 19257–19263 CrossRef.
- Y. X. Duan, K. H. Liu, Q. Zhang, J. M. Yan and Q. Jiang, Small Methods, 2020, 4, 1900846 CrossRef CAS.
- M. Liu, M. G. Liu, X. M. Wang, S. M. Kozlov, Z. Cao, P. D. Luna, H. M. Li, X. Q. Qiu, K. Liu, J. H. Hu, C. K. Jia, P. Wang, H. M. Zhou, J. He, M. Zhong, X. Z. Lan, Y. S. Zhou, Z. Q. Wang, J. Li, A. Seifitokaldani, C. T. Dinh, H. Y. Liang, C. Q. Zou, Y. Yang, T.-S. Chan, Y. Han, L. Cavallo, T.-K. Sham, B.-J. Hwang and E. H. Sargent, Joule, 2019, 3, 1703–1718 CrossRef CAS.
- Y. Yu, Z. J. Wang, C. Yang, A. X. Guan, L. M. Shang, A. M. Al-Enizi, L. J. Zhang and G. F. Zheng, J. Mater. Chem. A, 2018, 6, 20121–20127 RSC.
- J. D. E. Linn, R. B. King and J. A. D. King, J. Mol. Catal., 1993, 80, 151–163 CrossRef.
- C. Liang, B. Kim, S. Yang, Y. Liu, C. F. Woellner, Z. Li, R. Vajtai, W. Yang, J. Wu and P. J. Kenis, J. Mater. Chem. A, 2018, 6, 10313 RSC.
- Z. J. Yang, X. F. Zhang, C. Long, S. H. Yan, J. Y. Han, J. Zhang, P. F. An, L. Chang and Z. Y. Tang, CrystEngComm, 2020, 22, 1619–1624 RSC.
- C. Long, X. Li, J. Guo, Y. N. Shi, S. Q. Liu and Z. Y. Tang, Small Methods, 2019, 3, 1800369 Search PubMed.
- N. Han, P. Ding, L. He, Y. Y. Li and Y. G. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- T. Tsujiguchi, Y. Kawabe, S. Jeong, T. Ohto, S. Kukunuri, H. Kuramochi, Y. Takahashi, T. Nishiuchi, H. Masuda, M. Wakisaka, K. L. Hu, G. Elumalai, J. I. Fujita and Y. Ito, ACS Catal., 2021, 11, 3310–3318 CrossRef CAS.
- L. X. Liu, Y. Zhou, Y. C. Chang, J. R. Zhang, L. P. Jiang, W. L. Zhu and Y. H. Lin, Nano Energy, 2020, 77, 105296 CrossRef CAS.
- J. Li, J. Q. Jiao, H. C. Zhang, P. Zhu, H. F. Ma, C. Chen, H. Xiao and Q. Lu, ACS Sustainable Chem. Eng., 2020, 8, 4975–4982 CrossRef CAS.
- X. F. Zhang, H. T. Liu, P. F. An, Y. N. Shi, J. Y. Han, Z. J. Yang, C. Long, J. Guo, S. L. Zhao, K. Zhao, H. J. Yin, L. R. Zheng, B. H. Zhang, X. P. Liu, L. J. Zhang, G. D. Li and Z. Y. Tang, Sci. Adv., 2020, 6, eaaz4824 CrossRef CAS.
- J. Y. Han, P. F. An, S. H. Liu, X. F. Zhang, D. W. Wang, Y. Yuan, J. Guo, X. Y. Qiu, K. Hou, L. Shi, Y. Zhang, S. L. Zhao, C. Long and Z. Y. Tang, Angew. Chem., 2019, 131, 12841–12846 CrossRef.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- X. F. Zhang, H. T. Liu, Y. N. Shi, J. Y. Han, Z. J. Yang, Y. Zhang, C. Long, J. Guo, Y. F. Zhu, X. Y. Qiu, G. X. Xue, L. J. Zhang, B. H. Zhang, L. Chang and Z. Y. Tang, Matter, 2020, 3, 558–570 CrossRef.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS.
- K. N. Zhao, L. Zhang, R. Xia, Y. F. Dong, W. W. Xu, C. J. Niu, L. He, M. Y. Yan, L. B. Qu and L. Q. Mai, Small, 2016, 12, 588–594 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- Z. Z. Du, X. J. Chen, W. Hu, C. H. Chuang, S. Xie, A. J. Hu, W. S. Yan, X. H. Kong, X. J. Wu, H. X. Ji and L. J. Wan, J. Am. Chem. Soc., 2019, 141(9), 3977–3985 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02780h |
| This journal is © The Royal Society of Chemistry 2021 |