
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Particulate nitrate photolysis in the atmosphere
Masao
Gen
 a,
Zhancong
Liang
bc,
Ruifeng
Zhang
bc,
Brix Raphael
Go
bc and
Chak K.
Chan
*bcd
a,
Zhancong
Liang
bc,
Ruifeng
Zhang
bc,
Brix Raphael
Go
bc and
Chak K.
Chan
*bcd
aFaculty of Frontier Engineering, Institute of Science and Engineering, Kanazawa University, Kanazawa, 920-1192, Japan
bSchool of Energy and Environment, City University of Hong Kong, Hong Kong, China. E-mail: chak.k.chan@cityu.edu.hk
cCity University of Hong Kong Shenzhen Research Institute, Shenzhen, China
dGuy Carpenter Asia-Pacific Climate Impact Center, City University of Hong Kong, Hong Kong, China
First published on 13th January 2022
Abstract
Multiphase and heterogeneous photochemistry is an emerging component of atmospheric and air pollution research. It is primarily driven by reactions of photochemically produced free radicals in the particle phase with dissolved gaseous species. It has significant implications to promote the oxidation of aerosol particles, one of the most important atmospheric processes for secondary inorganic and organic aerosol formation. Nitrate is an increasingly important component in atmospheric aerosol particles with the trend of dominating over sulfate. Nitrate photolysis has long been known to produce highly reactive oxidants such as hydroxyl radicals in both gas and bulk or cloud phases. Recent studies have found that nitrate photolysis in the particle phase (i.e., particulate nitrate photolysis) proceeds faster than bulk solutions or cloud droplets by many orders of magnitude. Factors and mechanisms affecting particulate nitrate photolysis include the formation of solvent cages, pH, and co-existing species, but they remain controversial. Hence, the impact of nitrate photolysis in atmospheric chemistry is still uncertain. This paper reviews the current status of knowledge about the effects of particulate nitrate photolysis, instead of relatively well-known gas- and bulk-phase nitrate photolysis, in the atmosphere. Recommendations for future research directions on the mechanistic understanding of particulate nitrate photolysis and its parameterizations in air quality models are also made.
Environmental significanceAtmospheric particulate matter or aerosol particles, directly and indirectly, impact climate, regional air quality, and human health. The atmosphere is a giant and strongly oxidizing chemical reactor, and the atmospheric oxidizing capacity is closely associated with the chemical evolution of aerosol particles. Inorganic nitrate photolysis can contribute to the atmospheric oxidizing capacity by generating strong oxidants such as hydroxyl radical. Nitrate photolysis in aerosol particles is accelerated by many orders of magnitude relative to bulk solution reactions, but factors and mechanisms affecting particulate nitrate photolysis remain controversial. As a result, the impact of particulate nitrate photolysis on the atmospheric oxidizing capacity is still uncertain. We review the current status of knowledge about particulate nitrate photolysis in the atmosphere. |
1. Introduction
Atmospheric particulate matter (PM) or aerosol particles have significant impacts on climate, regional air quality, and human health.1 PM is emitted from many diverse anthropogenic and biogenic sources.2 During its atmospheric lifetime (∼a week), PM is subjected to many processes leading to physical and chemical transformations such as changes in its size, morphology, and chemical composition.3 The Earth's atmosphere is a giant and strongly oxidizing chemical reactor, and hence the atmospheric oxidizing capacity is closely associated with the evolution of PM's composition and properties. Despite the crucial importance of PM in the atmospheric environment, our understanding of the physical and chemical transformation of PM is far from complete. In this paper, we will use the terms PM and aerosol particles interchangeably.Sunlight, especially in the ultraviolet spectral region, is a source of atmospheric free radicals that drive the chemical changes in the atmosphere.2 The sunlight reaching low altitudes has a wavelength longer than 290 nm.4 One of the essential atmospheric photochemical processes is the generation of free radicals through gas-phase photochemistry.5,6 The gas-phase reactions are crucial in ozone depletion in the stratosphere and tropospheric oxidant production and organic oxidation, relevant to the abundance of climate forcing agents.7 However, the chemistry occurring within or on aerosol particles and cloud droplets is much less known. In this paper, we will focus on the photochemical processes of PM or aerosol particles. Solar radiation gives the energy to initiate photochemical reactions of aerosol particles and gaseous species. Multiphase and heterogenous photochemistry is an emerging field in atmospheric and air pollution research, and it has the potential to promote atmospheric oxidation greatly.8
The importance of multiphase and heterogeneous photochemistry in the atmosphere has been demonstrated in many laboratory studies. For example, multiphase photolysis of aerosols containing a trace amount of photosensitive compounds (e.g., humic acid) produces strong oxidants (e.g., superoxide, hydroxyl, nitrate, and organic radicals) in the particle phase.9 These in-particle oxidants can lead to fast uptake of non-condensable volatile organic compounds (VOCs) of limonene and isoprene without gas-phase oxidation. That study challenges the traditional view that such non-condensable VOCs need to be oxidized in the gas phase before partitioning into the particle phase to form secondary organic aerosol.3,10 Another example is the reactive uptake of sulfur dioxide (SO2) by irradiated particulate nitrate. Nitrate photolysis can produce in-particle hydroxyl (OH), nitrogen dioxide (NO2) radicals, and nitrite, promoting the oxidation of dissolved SO2 in the particle phase for sulfate production.11,12 This photochemistry can potentially reconcile the difference between field measurements and model estimations of sulfate formation during highly polluted episodes in China.13 Thus, multiphase and heterogenous photochemistry has significant implications in atmospheric chemistry.
Among the photolytic sources of strong oxidants such as irradiated mineral dust,14,15 iron–organic complexes,16,17 nitrate/nitrite,18,19 hydrogen peroxide,20 and hypochlorous acid,21,22 inorganic nitrate anion (NO3−) is an increasingly important component in atmospheric aerosol particles as sulfate concentrations decrease. Sulfate was the dominant inorganic constituent in atmospheric aerosol particles and is mainly formed from SO2 oxidation. SO2 emission has reduced globally while there is a modest increase in ammonia emission due to intensified agricultural activity livestock farming following population growth.23 For instance, SO2 emissions in China have decreased by 75% since 2007, while India is surpassing China as the world's largest emitter of anthropogenic SO2.24 Across the United States, SO2 emissions have decreased at ∼6% per year from 2001 to 2010.25 This SO2 reduction elevates aerosol pH and facilitates nitrate partitioning into the aerosol phase, leading to the growing nitrate dominance over sulfate. Nitrate-dominated aerosols have been observed in many locations, including the USA,26 Europe,27,28 and East Asia.29–32
Inorganic nitrate photolysis18,33 has an influence on NOx, OH, and O3 mass burdens in the atmosphere.34 It can be also used as a photolytic source of OH radicals to remove hazardous organic pollutants in environmental waters in advanced oxidation technologies.35,36 The mechanism of nitrate photolysis has been extensively studied in bulk solutions,18,33 but that of particulate nitrate photochemistry is not fully assessed despite increasing evidence of the nitrate-dominated aerosols as mentioned above. When nitrate photolysis is confined in a small droplet or a deliquesced aerosol particle, which is referred to as particulate nitrate photolysis in this review, it is accelerated by order(s) of magnitude relative to bulk solution reactions.11,37–40 Several factors/mechanisms affecting particulate nitrate photolysis such as solvent cages,41 pH,42 and co-existing species43 have been suggested in the literature, but they remain controversial. As a result, the impact of nitrate photolysis in atmospheric chemistry is still uncertain. Similarly, organic nitrates play important roles in the atmosphere because their fate including photolysis could affect the NOx recycling and O3 production.44,45 Photolysis of organic nitrates can produce NO2 and HO2,46 which may have impacts on subsequent reactions in the particle phase.47 However, there are very few studies on photolysis of particulate organic nitrates, and thus we limit our focus to that of particulate inorganic nitrate.
This paper reviews the current status of knowledge about the effects of particulate inorganic nitrate photolysis, instead of relatively well-known gas- and bulk-phase nitrate photolysis, in the atmosphere. We begin with an overview of nitrate photolysis mechanisms, followed by discussing factors affecting the product yields of particulate nitrate photolysis. We then review the quantum yields and nitrate photolysis rate constants reported in the literature for quantifying the impacts of nitrate photolysis in the atmosphere and used in air quality models. Finally, we summarize chemical reactions related to nitrate photolysis in the particle phase. Particulate nitrate photolysis generates gas-phase oxidants such as NO2 and HONO, which can tremendously affect ozone and halogen chemistry.34 However, the gas-phase reactions are not addressed in this review. Critical issues and recommendations for future research directions will be presented at the end of this review paper.
2. Nitrate photolysis mechanism
Nitrate anion (NO3−) is a crucial chromophore in environmental waters. Nitrate can be photolyzed in both aqueous48 and crystalline states.49 In this section, we briefly review nitrate photolysis mechanisms relevant to the atmosphere. For more detailed mechanisms of nitrate photolysis, we refer readers to the previous reviews.18,33As a result of the absorption of UV photons, electrons of nitrate anions can move from their ground state to an unoccupied or partially occupied molecular orbital of higher energy. Because the energy level of UV light is of the same order as the enthalpies of the covalent bonds, this additional energy results in a bond cleavage, which splits the excited nitrate, [NO3−]*, into two fragments, a process known as photolysis (R1). Nitrate photolysis produces oxidants such as hydroxyl (OH) and nitrogen dioxide (NO2) radicals and nitrite ions (NO2−) ((R2) and (R3); Fig. 1).50 Furthermore, [NO3−]* can isomerize (R4). These photoproducts can have potentially significant impacts on subsequent reactions in aerosol particles, as will be discussed in Section 5. The absorption spectrum of NO3− is dominant by a weak n → π* band around 302 nm (ε = 7.2 M−1 cm−1) and a much stronger π → π* band at 200 nm (ε = 9900 M−1 cm−1).19,48 Excitation in the n → π* band (λ > 280 nm) mainly proceeds through (R2) and (R3), whereas excitation in the π → π* band (λ < 280 nm) proceeds via the two primary photo-processes (R2) and (R4).19
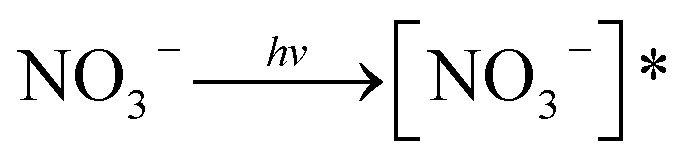 | (R1) |
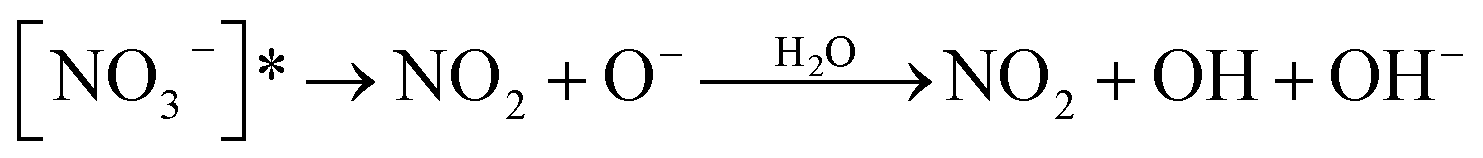 | (R2) |
| [NO3−]* → NO2− + O(3P) | (R3) |
| [NO3−]* → ONOO− | (R4) |
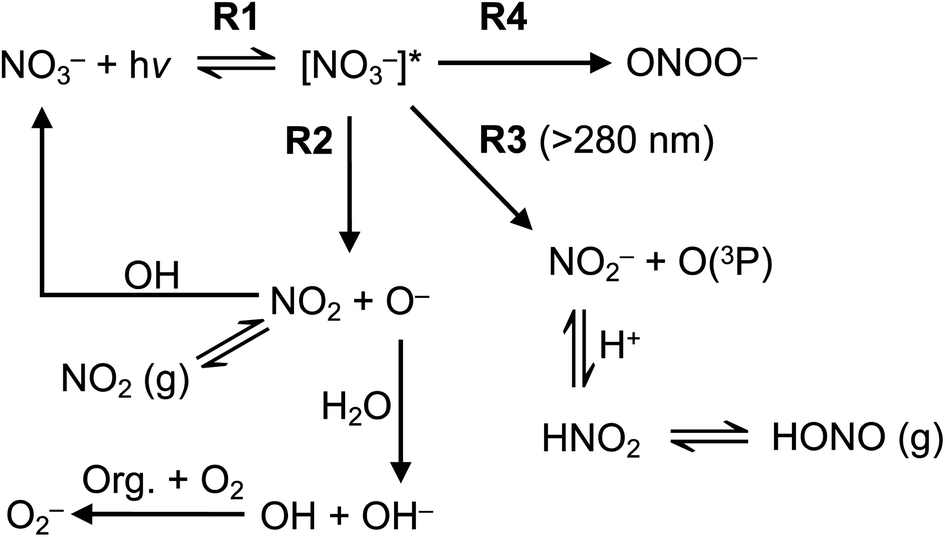 | ||
| Fig. 1 Simplified nitrate photolysis mechanisms.50 | ||
Under atmospherically relevant irradiation (i.e., >300 nm), (R2) and (R3) are more relevant, and (R4) is negligible at >280 nm. Goldstein et al. reported the quantum yield of ONOO− is lower than 0.2% under 300 nm illumination.19 The pKa (O−/OH) is around 12, and the primary fragment O− is immediately protonated in water to yield OH radicals.33 OH radical is also a precursor for H2O2, another important oxidant in the atmosphere. However, H2O2 has not been detected as a significant photoproduct from nitrate photolysis at λ > 200 nm, likely due to the very low concentration and short lifetime of OH.18
(R3) is a potentially important source of nitrite (NO2−).18 Photolysis of NO2− and its protonated form, nitrous acid (HNO2), produces OH and nitric oxide (NO), which subsequently react with NO2 or OH radicals to reproduce NO2−/HNO2 (Table 1). Oxygen atoms (O(3P)) react with dissolved O2 to form O3, which can further react with OH radicals to produce HO2 radicals and can also react with NO2− to reproduce NO3−. Organic compounds scavenge OH radicals, and the reaction generally produces superoxide (O2− in Fig. 1), following subsequent reactions (Table 1). The photolysis quantum yield of (R2) for OH production is 1.35 ± 0.3% at 298 K under 302 nm irradiation.20,36,51 The quantum yield of (R3) for NO2− production is 1.1 ± 0.2% under 313 nm irradiation at pH > 5,50 indicating that these two channels ((R2) and (R3)) may be of comparable importance in nitrate photolysis. These photolysis quantum yields are affected by many factors that will be discussed in Section 3.
| Reactions | Rate constant | Reference |
|---|---|---|
| a Depending on conditions such as irradiation wavelength and intensity. b Reverse reaction. c Generalized rate constant in atmospheric waters. | ||
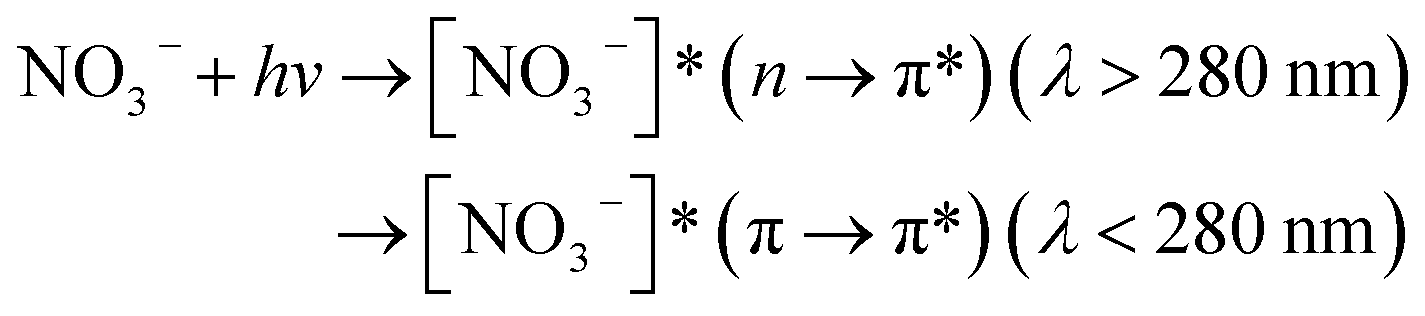
|
18 | |
| [NO3−]* → NO2− + O(3P) | 18 | |
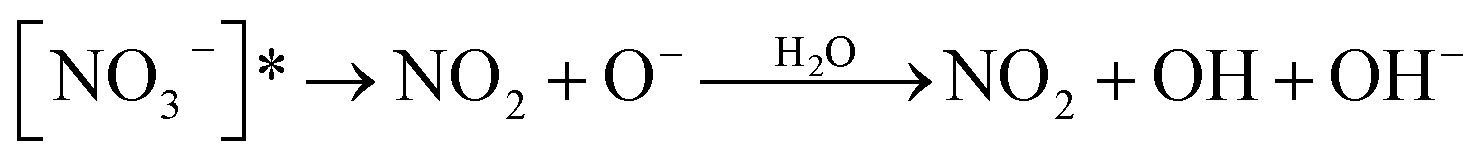
|
18 | |
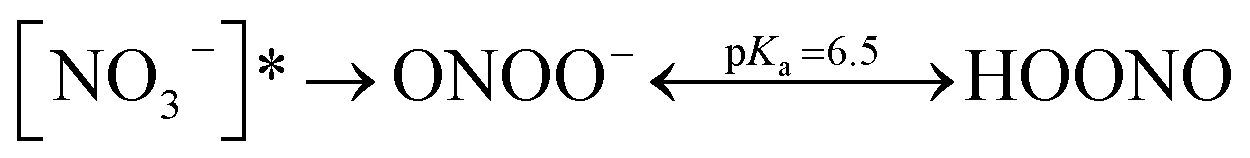
|
18 | |
| NO2− + hv → [NO2−]* | 18 | |
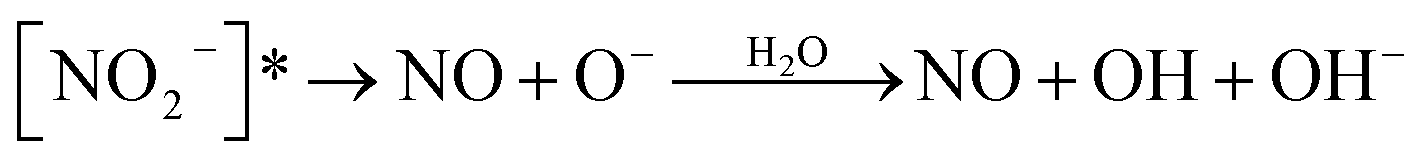
|
18 | |
| O− + H2O ↔ OH + OH− | 1.7 × 106 M−1 s−1/1.2 × 1010 M−1 s−1b | 18 |
| OH + NO2 ↔ HOONO | 1.3 × 109 M−1 s−1/0.35 s−1b | 18 and 19 |
| HOONO → NO3− + H+ | 1.4 s−1 | 18 |
| OH + NO2− → NO2 + OH− | 1.0 × 1010 M−1 s−1 | 18 |
| OH + HNO2 → NO2 + H2O | 3.0 × 109 M−1 s−1 | 42 |
| OH + NO → HNO2 | 1.0 × 1010 M−1 s−1 | 18 |
| OH + OH → H2O2 | 5.5 × 1010 M−1 s−1 | 21 |
| NO + NO2 → N2O3 | 1.1 × 109 M−1 s−1 | 18 |
| N2O3 + H2O → 2H+ + 2NO2− | 5.3 × 102 s−1 | 18 |
| NO2 + NO2 → N2O4 | 4.5 × 108 M−1 s−1 | 18 |
| N2O4 + H2O → NO2− + NO3− + 2H+ | 1.0 × 103 s−1 | 18 |
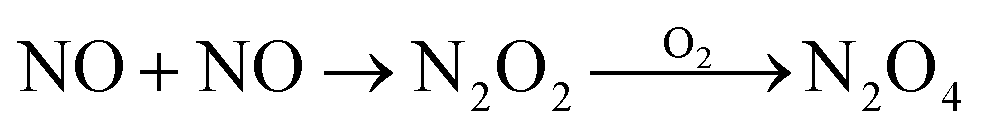
|
N.A. | 18 |
| O(3P) + NO3− → O2NOO− → O2 + NO2− | 3.0 × 108 M−1 s−1 | 18 |
| O(3P) + NO2− → NO3− | 3.0 × 109 M−1 s−1 | 18 |
| O(3P) + O2 → O3 | 4.0 × 109 M−1 s−1 | 21 |
| NO2− + O3 → NO3− + O2 | 5.0 × 105 M−1 s−1 | 21 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Presence of organic compounds (Org.) | ||
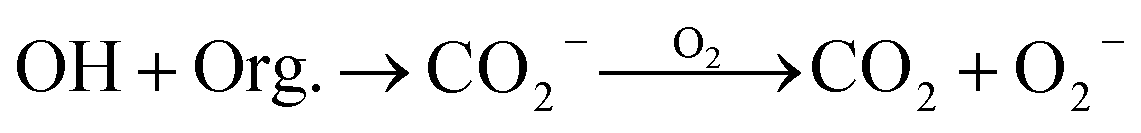
|
3.8 × 108 M−1 s−1c | 52 |
| HO2 ↔ O2− + H+ | 7.9 × 105 s−1/5.0 × 1010 M−1 s−1b | 53 |
| HO2 + O2− + H2O → H2O2 +O2 + OH− | 9.7 × 107 M−1 s−1 | 54 |
| OH + HO2 → H2O + O2 | 9.9 × 107 M−1 s−1 | 53 |
| OH + O2− → OH− + O2 | 1.1 × 1010 M−1 s−1 | 53 |
| O2− + NO → ONOO− | 4.3 × 109 M−1 s−1 | 55 |
| ONOO− + CO2 → ONOOCO2− | 3.0 × 104 M−1 s−1 | 56 |
| ONOOCO2− → NO3− + CO2 | 6.7 × 105 s−1 | 53 |
| ONOO− + H+ ↔ ONOOH | 5.0 × 1010 M−1 s−1/1.2 × 104 s−1b | 53 |
| ONOOH → NO3− + H+ | 9.0 × 10−1 s−1 | 53 |
3. Factors affecting particulate nitrate photolysis
While nitrate photolysis has been investigated for decades in laboratory experiments, theoretical studies, and field measurements, understanding how physical and environmental conditions affect particulate nitrate photolysis is far from complete. Most of the previous experimental works were performed in diluted bulk solutions, which are useful to reveal the chemistry in aqueous solutions such as in cloud droplets. However, reactions in atmospheric aerosol particles with significantly higher nitrate concentrations and surface area to volume ratios can differ from those in bulk solutions. In this section, we discuss how various factors alter particulate nitrate photochemistry.3.1 Bulk versus interface: solvent cage and surface propensity
The absorption cross section of NO3− in aqueous solution at 310 nm is 25 times that of gas phase HNO3 due to symmetry breaking of NO3− by hydration.57 However, the water molecules surrounding NO3− form a solvent cage and retard nitrate photolysis. Specifically, the fragments generated from nitrate photolysis are initially surrounded by a cage of solvent (water) molecules. Their diffusion out of the cage competes with the regeneration of nitrate anions by recombining the fragments. The recombination accounts for the reduced quantum yields of nitrate photolysis in the aqueous phase compared to the gas phase.41The solvent cages near the air–water interface are less complete. As a result, the recombination processes are inhibited, increasing the quantum yield of nitrate photolysis. Aerosol particles have a much larger surface area to volume ratio than bulk solutions.8 Hence, the relative contribution of interface reactions to the overall reaction can be higher for smaller particles.58 Furthermore, several studies have reported the surface propensities of nitrate anions at the air–water interface,59–63 while some others reported that nitrate anions favorably reside at bulk solvation or exhibit nearly uniform distribution.59,60,64–67 This is still a controversial topic. Therefore, more work on the surface propensity of nitrate in aerosol particles should be warranted in the future. Our experimental results showed that the surface propensity of nitrate can be enhanced by the presence of co-existing species (i.e., halide ions),37 which will be covered later.
3.2 pH
Aerosol acidity plays a critical role in atmospheric processes.68 It affects chemical compositions, gas-particle partitioning, and toxicity68 through various oxidation reactions, either directly or indirectly.69,70 Here we refer acidity to as the activity of hydrogen ions or pH of aerosol particles. The inherent nitrate photolysis rate constant is not pH-dependent, and the molar light absorptivity of nitrate in aqueous solution is not sensitive to pH in the range of 2 to 6.42 The rate of NO2 production from nitrate thin-film photolysis is pH-independent under the atmospherically relevant pH range (pH = 0.5–6).71 OH production from nitrate photolysis requires the protonation of O−, but it is also insensitive at pH < 9,20 due to its high pKa (O−/OH) of 12.72Nonetheless, pH has significant impacts on the effective quantum yield for production of NO2−/HNO2, a particular category of products with emerging oxidative potential.11,50 Since the pKa of HNO2 is around 3, the speciation of NO2−/HNO2 can vary; with NO2− dominating at pH > 3 and HNO2 dominating at pH < 3.73–76 Furthermore, HNO2 or HONO is volatile with Henry's law constant of 49 ± 3 M atom−1 at 25 °C77 and hence can partition from the aqueous phase into the gas phase. Scharko et al. found that gaseous HONO production from nitrate photolysis is the highest at the lowest pH they studied (∼2) and decreases with pH, reaching almost zero at pH higher than 4, whereas gaseous NO2 production remains constant in the pH range of 2 to 6.42 Furthermore, Benedict et al. measured nitrite production and found that its quantum yield increases with pH and remains constant at pH higher than 4.5.50 Thus, aerosol pH is the determining factor in the distribution of NO2−/HNO2 in the gas phase or aqueous phase.
3.3 RH
Nitrate is hygroscopic, and the amount of water uptake is sensitive to the counter cation, particle size (Kelvin effect), and relative humidity (RH).78 Particularly, RH is an important parameter to determine the phase of nitrate-containing particles.79 In this section, we cover RH effects on nitrate photolysis in aqueous solutions or droplets followed by those in the solid phase.In general, lower RH increases nitrate concentration in aqueous particles due to reduced liquid water content in the particles, leading to higher nitrate photolysis rates. However, the quantum yield of nitrate photolysis also depends on nitrate concentration. Concentrated nitrate solutions have lower quantum yields for nitrite production than diluted ones have.80 At 310 nm irradiation and pH = 4, the quantum yield of calcium nitrate solution for nitrite production significantly decreases from (1.4 ± 0.1) × 10−2 to (4.2 ± 0.3) × 10−3 as nitrate concentration increases from 0.01 to 15 M. In contrast, a similar decrease was not observed in sodium nitrate solutions. This ∼30 folds decrease in the nitrite quantum yield of calcium nitrate solution is attributable to the blue shift of the n–π* absorption (i.e., away from actinic wavelengths) with increasing nitrate concentration. Specifically, the absorption peak of calcium nitrate solutions blue-shifts from 302 to 294 and 289 nm as the concentration increases from 0.01 to 6.0 and 14.9 M, respectively. In contrast, the blue shift for sodium nitrate solution was minimal: from 303 nm at 0.01 M to only 301 nm at 6.2 M.80,81 In summary, higher nitrate concentration reduces the quantum yield due to the blue shift and therefore the photolysis rate constant at a given photon flux. However, it may also increase the overall nitrate photolysis rate, which is the product of nitrate concentration and nitrate photolysis rate constant.
At low RH, nitrate can exist as crystalline solids. The quantum yields of solid-phase nitrate photolysis are four orders of magnitude lower than those of aqueous phase one.82 Nonetheless, emissions of gaseous NO2 at >1 ppb min−183 and HONO at < 0.06 ppt min−184 are possible from solid-phase nitrate photolysis when thin water films or so-called surface adsorbed water (SAW) are present on the solid surface. While the exact role of SAW in nitrate photolysis remains poorly understood, the presence of SAW likely promotes the dissolution of solid nitrate to form aqueous nitrate in SAW. Because RH regulates the amount of SAW,85 the formation of nitrate photolysis products is expected to be RH-sensitive. The increased amount of SAW might increase the availability of nitrate in its dissociated form and then enhance the quantum yields.
3.4 Temperature
Although the molar absorptivity of nitrate anion almost remains unchanged in the range of 273–298 K,86 the quantum yields of nitrate photolysis is affected by temperature.86–89 Specifically, the quantum yields of nitrate photolysis for production of OH, Φ(OH), and NO2−, Φ(NO2−), increase with temperature (T). Anastasio and co-workers have found the temperature dependence of the quantum yields of the two channels as ln(Φ(OH)) = −(2400 ± 480)(1/T) + (3.6 ± 0.8)86 and ln(Φ(NO2−)) = −(1330 ± 100)(1/T) + (0.09 ± 0.39).87 According to the temperature dependence, the quantum yields for OH production can be ∼0.0171 at 313 K, nearly three times that at 273 K (∼0.0056). The quantum yield for nitrite production is 0.0156 at 313 K and decreases to ∼0.0084 at 273 K.3.5 Ice and snow
Nitrate photolysis on ice and snow has significant implications.49,90,91 Nitrate can be embedded in snow pack and ice via deposition, heterogeneous dissolution of HNO3(g), and freezing the water contained nitrate (e.g. sea and lake). Gaseous products from nitrate photolysis are observed in much higher quantities from snow packs than that from aqueous solutions.91,92 A comprehensive review on nitrate photolysis in ice/snow is available elsewhere.93 Here we briefly introduce the main features of nitrate photolysis on ice and snow.Similar to the situation in the aqueous phase, nitrate photolysis on ice and snow proceeds faster at the air–snow interface than that in the bulk.88,92 In addition to the partial solvation at the interface which allows gas phase products to easily escape, intramolecular geometrical distortion of nitrate anions at interface resulting in an increase in the absorption cross section of nitrate enhances nitrate photolysis.92 In snow, nitrate photolysis likely occurs in the liquid like region on the surface of ice grains, or in cracks between ice grains.94 Highly variable quantum yields of nitrate photolysis on ice and snow are reported because they are strongly influenced by the location of nitrate anions in an ice grain,95,96 and the co-existing species (e.g., Cl−).97 Meusinger et al. have proposed two photochemical domains of nitrate photolysis: photolabile nitrate anion and nitrate anion buried within the ice grain.95 Photoproducts produced from photolabile nitrate anion can escape the ice grain and hence the quantum yields are higher than those of nitrate anion buried within the ice grain. In contrast, photoproducts from buried nitrate anions are likely to undergo a recombination reaction to regenerate nitrate anion (Fig. 1).
3.6 Effect of co-existing chemical species
Atmospheric aerosol particles consist of a myriad of different components, with diverse spatial differences, temporal variations, and distinctive source dependence.10,98 Most laboratory studies on nitrate photolysis have used only nitrate salts without other atmospherically relevant species. The presence of co-existing chemical species has been reported to influence aqueous nitrate photolysis through (1) affecting the solvent cage effects, (2) participating in chemical reactions directly or indirectly, and (3) regulating nitrate concentration because of their hygroscopic properties determining the liquid water content. Because the last factor is reasonably well covered in the wealth of literature,79,99 we will focus on the first two issues in this review.With enormous contributions from sea spray and anthropogenic sources, halide ions are ubiquitously found in atmospheric particles.105–107 Nitrate are often internally mixed with chloride ions in the atmosphere through the chloride depletion reactions of sea spray particles. According to field measurements, the molar ratio of halides to nitrate in fresh sea spray aerosol is usually higher than 1.0,108–112 whereas that in urban aerosol or aged marine aerosol falls in the range of 0–1.1.113–117 Wingen et al. reported that the coexistence of chloride ions results in an enhanced gaseous NO2 production from deliquesced nitrate aerosol particles under illumination by a factor of 1.6 to 2.4.43 Zhang et al. found that the particulate nitrate photolysis rate constant increases by a factor of 2.0, 1.7, and 2.1 in the presence of Cl−, Br−, and I−, respectively, leading to enhanced sulfate production from heterogenous oxidation of SO2 by a factor of 1.4, 1.3, and 2.0.37 A linear relation was found between the nitrate photolysis rate constant, jNO3−, and the initial molar ratio of Cl− to NO3−, [Cl−]0/[NO3−]0, as jNO3− = 9.7 × 10−5 × [Cl−]0/[NO3−]0 + 1.9 × 10−5 at [Cl−]0/[NO3−]0 below 0.2. No further enhancement of nitrate photolysis rate constant was observed when [Cl−]0/[NO3−] > 0.2, where jNO3− can be considered the same as that at [Cl−]0/[NO3−]0 = 0.2.
Compared with chloride ions, bromide and iodide ions have higher intrinsic surface propensities,102–104 and therefore their potential impacts on the enhanced nitrate photolysis on a per molecule of halide basis are expected to be comparable or greater. However, the concentrations of bromide and iodide ions are many orders of magnitude lower than that of chloride ([Cl−]:[Br−]:[I−] = 1000000:∼1515:1), making them insignificant in enhancing nitrate photolysis in typical tropospheric environments.118–120
Organic compounds are highly reactive toward OH radicals and increase the effective quantum yields for NO2 production by suppressing the NO2 consuming reaction between NO2 and OH radicals.50 Scavenging of OH radicals can also increase the quantum yields for N(III) production in two ways. Firstly, organic scavengers reduce N(III) oxidation loss by OH radicals by consuming them. Second, some organics such as ethylene glycol and glyoxal react with OH radicals to form O2−/HO2 radicals, which can further lead to secondary formation of N(III) from the NO2 + O2−/HO2 reaction.11,42,128
It is found that HONO emissions from nitrate photolysis are enhanced by dissolved aliphatic organic matter through enhanced production of superoxide.128 Wang et al. demonstrated the importance of solvated electrons produced from photosensitizing reactions in enhanced nitrite production from nitrate photolysis.158 They suggested that the solvated electrons are mainly scavenged by nitrate, leading to more NO2 production for further conversion to nitrite.
Highly viscous organic materials could hinder reactions in the particle phase. Liang et al. examined nitrate photolysis in mixed sucrose–nitrate–sulfate particles as a proxy of viscous aerosol particles.129 They found the suppressed nitrate crystallization by the presence of sucrose and the high photolysis rate constants (∼10−5 s−1), irrespective of the RH. They observed the formation of enlarged hollow semisolid particles at high sucrose content and low RH, likely due to the release of gaseous species like NO2/HONO pushing the viscous materials radially outward. Thus, particulate nitrate photolysis may affect the microphysics of aerosol particles.
3.7 Mie resonances of droplets
Light intensity is a crucial parameter in determining nitrate photolysis rates. In the photochemistry of micrometer-sized spherical droplets, the actinic flux in the droplet can be enhanced due to (i) the Mie resonances, also known as the whispering gallery mode resonances, or the morphology-dependent resonances (MDRs), and (ii) the increased light pathlengths in the droplets.41,130,131 MDRs, characterized in terms of the size parameter (i.e., particle diameter × π/wavelength), have been studied in the physical and chemical characterization of aerosols, especially in laboratory studies including elastic scattering, fluorescence, and Raman spectroscopy.132 Although MDRs can yield the orders of magnitudes increase in the internal actinic flux, their contribution to the actinic flux enhancement is not profound when averaged over typical droplet size distributions.130,133 Under broadband solar irradiation (290–600 nm), MDRs and the increased light pathlengths in ∼2 μm droplet can produce a ∼2-fold intensity enhancement (2.06 in 1-decene; 1.76 in pure water) in spherical aqueous droplets relative to bulk-liquid solutions.130,131 However, the role of Mie resonances in enhancing nitrate photolysis has not been experimentally ascertained.3.8 Mineral dusts
Mineral dusts are one of the most significant contributors to aerosol mass, with an estimated annual emission of 1000–3000 Tg.134 Recent work reported a synergistic effect of iron–organic complexes and nitrate photolysis in nitrite/nitrous acid generation.159 Previous studies explored nitrate photolysis on the surface of mineral oxides: (1) non-photoactive oxides (NPO; e.g., Al2O3, SiO2) and (2) photoactive semiconductive oxides (PSO; e.g., TiO2). Generally speaking, both NPO and PSO provide numerous surface reactive sites for the adsorption of nitrate.136–138 Spectroscopic analysis revealed that the interactions between HNO3/nitrate and reactive surface sites could distort the molecular structure of HNO3/nitrate, which results in a red shift in n → π* absorption and an increase of light absorption cross section relative to gas-phase HNO3.136,139,140 Additionally, PSO have excellent photocatalytic capacity via an electron–hole conductive mechanism.8,14 In ambient environment, the adsorbed oxygen (O2) accepts an electron to produce highly reactive O2−, facilitating nitrate adsorption and subsequent photoreactions.8While nitrate photochemistry on oxide surfaces has been widely investigated, aluminosilicates, which can account for >70% of dust mass, are rarely explored.141 Using NaY zeolite as a model system of aluminosilicates, Gankanda and Grassian found that its photoactivity may be significantly different from the non-photoactive and photoactive oxides.142,143 N(III) produced from nitrate photolysis can stably exist as the primary product inside the zeolite cage during nitrate photolysis, whereas nitrate photolysis on oxide surfaces mainly produces gaseous NO2. Hence, porous materials in mineral dust can potentially act as a platform for producing daytime gaseous HONO.
4. Quantum yields and photolysis rate constants
The impacts of particulate nitrate photolysis in atmospheric chemistry rely highly on its quantum yields or photolysis rate constants. They are the parameters required for implementing particulate nitrate photolysis mechanisms in air quality modeling.13 They are relatively well constrained for the gas phase and aqueous (bulk) phase photolysis,6,144 but not for particulate nitrate. This section summarizes the reported quantum yields and photolysis rate constants of nitrate (Tables 2 and 3, respectively) to discuss the current understanding of nitrate photolysis rate constants.| Sample | Nitrate concentration | Φ (%) for NO2− production | pH | OH scavenger | Irradiation wavelength | Note/reference |
|---|---|---|---|---|---|---|
| a Quantum yields for ONOO− production. b Quantum yields for OH production. | ||||||
| NaNO3 aqueous solution | 50 μM | 0.93 ± 0.1 | 5.2 | 500 μM 2-propanol | 313 nm | 88 |
| NaNO3 aqueous solution | 50 μM | 1.1 ± 0.2 | ≥5 | None | 313 nm | 50 |
| <0.26a | 50 μM formate | |||||
| 1.18 ± 0.14 | 50 μM cysteine | |||||
| 1.16 ± 0.04 | ||||||
| Ca(NO3)2 | 50 μM | 1.01–1.20 | 7.3–7.52 | None | 313 nm | 50 |
| Mg(NO3)2 | 0.99–1.06 | 7.2–7.58 | ||||
| NH4NO3 | 0.9 ± 0.1 | 7.14–7.39 | ||||
| KNO3 | 1.16 ± 0.13 | 7.35–7.4 | ||||
| NaNO3 aqueous solutions | 0.01 M | 0.45 | 4 | None | 310 nm | 80 |
| 0.1 M | 0.35 | |||||
| 1.4 M | 0.62 | |||||
| 5.1 M | 0.25 | |||||
| NaNO3 aqueous solutions | 0.01 M | 0.80 | 4 | 10 mM formate | 310 nm | 80 |
| 0.1 M | 1.13 | |||||
| 1.4 M | 0.92 | |||||
| 5.1 M | 1.25 | |||||
| KNO3 aqueous solution | 0.1 M | 0.65 ± 0.04 | N.A. | 0.5 M formate | 310 nm | 160 |
| 0.60 ± 0.04 | 0.34 M EtOH | 313 nm | ||||
| NaNO3 aqueous solution | 0.02–1 M | 0.94 ± 0.02 | 4.2–4.5 | 10 mM formate | 300 nm | 19 |
| NaNO3 aqueous solution | 0.01 M | 0.72 ± 0.09b | 3.0 | 0.13 M 2-propanol | 305 nm | 51 |
| 1.00 ± 0.04 | 4.0 | |||||
| 1.02 ± 0.07 | 5.6 | |||||
| 1.24 ± 0.14 | 9.0 | |||||
| 1.22 ± 0.07 | 11.0 | |||||
| KNO3 aqueous solution | 3 mM | 1.7 ± 0.3b | 4–9 | 0.3 M thiocyanate | 308 nm | 20 |
| Sample type (particle size)a | j NO3− (s−1) | Species used for estimating jNO3− | Irradiation wavelengths | Note/reference |
|---|---|---|---|---|
| a AN: ammonium nitrate; Gly: glyoxal; OA: oxalic acid; SBC: sodium bicarbonate. | ||||
| Ambient aerosol | (6.1 ± 4.2) × 10−5 | Gaseous NO2 and HONO | >290 nm | Albany, NY(urban)/38 |
| Ambient aerosol | (1.5 ± 1.2) × 10−4 | Gaseous NO2 and HONO | >290 nm | Delmar, NY (suburban)/38 |
| Ambient aerosol | (2.3 ± 2.4) × 10−4 | Gaseous NO2 and HONO | >290 nm | Whiteface mountain summit, NY (remote areas)/38 |
| Ambient aerosol | (1.9 ± 1.2) × 10−4 | Gaseous NO2 and HONO | >290 nm | Aircraft measurements in southeast US/38 |
| HNO3/nitrate on building material surfaces | (6.0 ± 5.3) × 10−5 | Gaseous NO2 and HONO | Natural sun light or Hg lamp | 147 |
| HNO3/nitrate on plant leaf surfaces | (6.0 ± 8.7) × 10−5 | Gaseous NO2 and HONO | Natural sun light or Hg lamp | 147 |
| HNO3/nitrate on urban grime | (2.7 ± 1.0) × 10−4 to (5.4 ± 2.7) × 10−4 | Nitrate | >290 nm | 148 |
| Nitrate aerosol (80 nm) | 3 × 10−6 for UVA; 2 × 10−5 for UVB | Gaseous NO2, NO, and HONO | UVA and UVB | Laboratory generated/39 |
| Ambient aerosol | 1.0 × 10−4 | Gaseous NO2, and HONO | >300 nm Xenon lamp | Clean marine boundary layer/40 |
| AN (25 μm) | 1.0 × 10−7 | Sulfate as oxidation product | 250 nm mercury lamp | Laboratory generated droplets/12 |
| AN (∼50 μm) | 7.4 × 10−6 | Sulfate as oxidation product | 300 nm LED | Laboratory generated droplets/11 |
| AN/Gly | 2.7 × 10−5 | |||
| AN/OA | 3.0 × 10−5 | |||
| AN/SBC | 2.7 × 10−6 | |||
| AN (∼50 μm) | 2.0 × 10−5 | Sulfate as oxidation product | 300 nm LED UV lamp | Laboratory generated droplets/37 |
| AN/chloride | 4.0 × 10−5 | |||
| AN/bromide | 3.4 × 10−5 | |||
| AN/iodide | 7.5 × 10−5 | |||
| Nitrate solution | (1.23 ± 0.04) × 10−7 | Reaction products of benzoic acid and OH radicals | >290 nm Xenon lamp | 161 |
| Nitrate solution | ∼3.0 × 10−7 | Reaction products of benzoic acid and OH radicals | Natural sunlight | 144 |
4.1 Nitrate photolysis rate constants of ambient aerosol particles
Most works reported that nitrate photolysis rate constants of ambient particles ranged from 10−5 to 10−4 s−1,38,145,146 which are 100–1000 times that of aqueous solution and gaseous HNO3.18 Ye et al. measured nitrate photolysis rate constants of ambient particles collected at different locations in North America. They reported the mean values of 6.1 (±4.2) × 10−5 s−1 for samples collected in Albany, NY (urban area), 1.5 (±1.2) × 10−4 s−1 in Delmar, NY (rural area), 2.3 (±2.4) × 10−4 s−1 from Whiteface Mountain summit (remote area), and 1.9 (±1.2) × 10−4 s−1 from flight sampling.38 Bao et al. reported the nitrate photolysis rate constants of ambient particles sampled in Beijing from 1.2 × 10−5 to 4.8 × 10−4 s−1.145 In contrast, Romer suggested the particulate nitrate photolysis rate constants of 7 × 10−6 to 2.1 × 10−5 s−1, 10–30 times higher than that of gas-phase HNO3, based on the aircraft observations over South Korea.146 On the other hand, Shi et al. found a limited role in the photolysis of particulate nitrate for gaseous NOx and HONO production.39 Nitrate photolysis rate constants on building material surfaces, plant leaf surfaces, and urban grime have been reported to be 6.0 (±5.3) × 10−5 s−1, 6.0 (±8.7) × 10−5 s−1 and 1.2 × 10−3 s−1, respectively.144,147,148 Furthermore, Laufs and Kleffmann reported a very low HNO3 photolysis rate constant on quartz surfaces for HONO formation, implying the negligible contribution of nitrate photolysis to the daytime HONO sources.149 The contradictory results related to HNO3 surface photolysis were also reported, highlighting the importance of HNO3 coverage on solid surfaces in the absence versus in the presence of water vapor.150 Thus, the rate constants in the atmosphere are highly variable and uncertain.4.2 Estimation of particulate nitrate photolysis rate constant
There is a growing body of research on the enhancement of particulate nitrate photolysis. Accurate estimation of nitrate photolysis rate constant is key to quantifying atmospheric relevance of enhanced particulate nitrate photolysis. The particulate nitrate photolysis rate constant, jpNO3−, is a first order decay rate constant: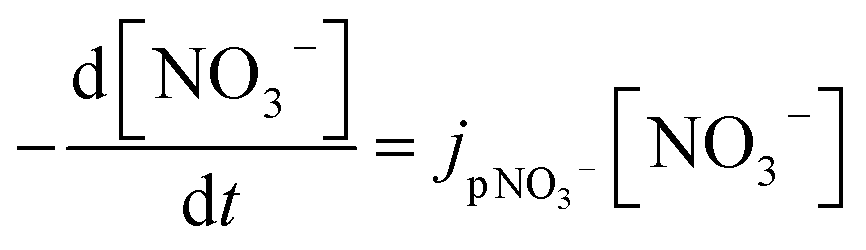 | (1) |
One way to measure jpNO3− is to quantify the decay of nitrate.148 However, it is challenging due to its small value of reported jpNO3−: 10−6 ∼ 10−4 s−1. For instance, it takes about 12 days to see the nitrate decrease by 1 M at jpNO3− of 10−6 s−1. Hence, studies on the direct measurements of nitrate decay are scarce (Table 3). In addition, this estimation may be complicated by regeneration reactions of nitrate during nitrate photolysis (Table 1), but the effect of the regeneration reaction (OH + NO2) on the quantification of jpNO3− would be minimized in the presence of OH scavengers. Another estimation method measures the gas phase photoproducts of nitrate photolysis such as NO2, and HONO,39 assuming low concentrations in the particle phase given their low Henry's law constants:151
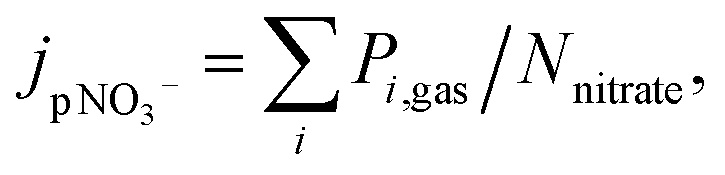 | (2) |
UV irradiance fluctuates daily and seasonally as a function of latitude, solar zenith angle, cloud cover, and stratospheric ozone and particle concentrations.144jpNO3− is related to the wavelength-dependent photon fluxes received by particulate nitrate, IpNO3−(λ), the molar absorptivity, εNO3−(λ), and the quantum yield, ϕNO3−(λ):
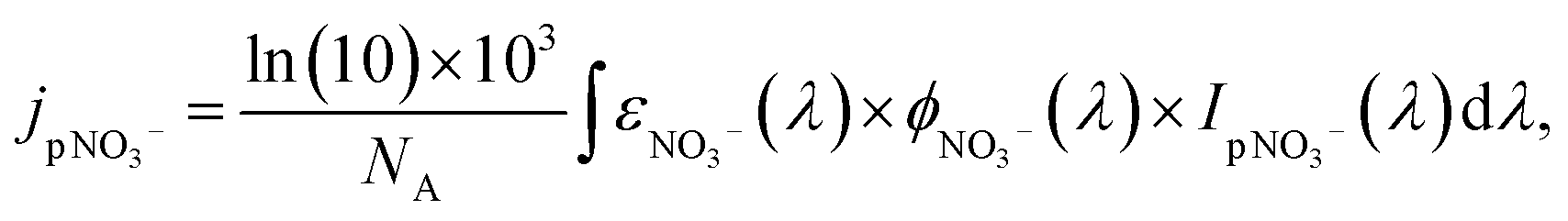 | (3) |
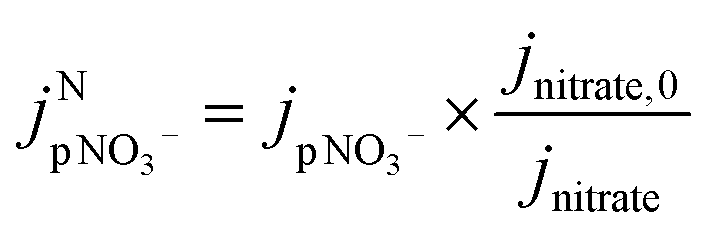 | (4) |
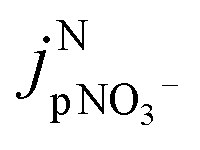 is the jpNO3− normalized to the typical tropical summer condition; jnitrate,0, and jnitrate are the photolysis rate constants of an aqueous solution under the typical tropical summer conditions and that exposed to the experimental photon fluxes, respectively. A value of 3.0 × 10−7 s−1 can be used for jnitrate,0.6 Reporting
is the jpNO3− normalized to the typical tropical summer condition; jnitrate,0, and jnitrate are the photolysis rate constants of an aqueous solution under the typical tropical summer conditions and that exposed to the experimental photon fluxes, respectively. A value of 3.0 × 10−7 s−1 can be used for jnitrate,0.6 Reporting 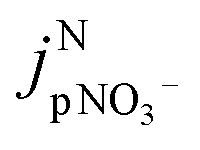 based on jpNO3− is recommended for a quantitative comparison of experimental results obtained under different conditions.
based on jpNO3− is recommended for a quantitative comparison of experimental results obtained under different conditions.
Besides nitrate, many other light-absorbing species such as black carbon152 and brown carbon153 exist, and they can be internally mixed with nitrate in atmospheric particles.154,155 The incident photon flux, I(λ), is then absorbed by nitrate as well as those light-absorbing species in the particle phase. The spectral photon fluxes absorbed by a component (i), Iai(λ), in light-absorbing multicomponent mixtures is written by
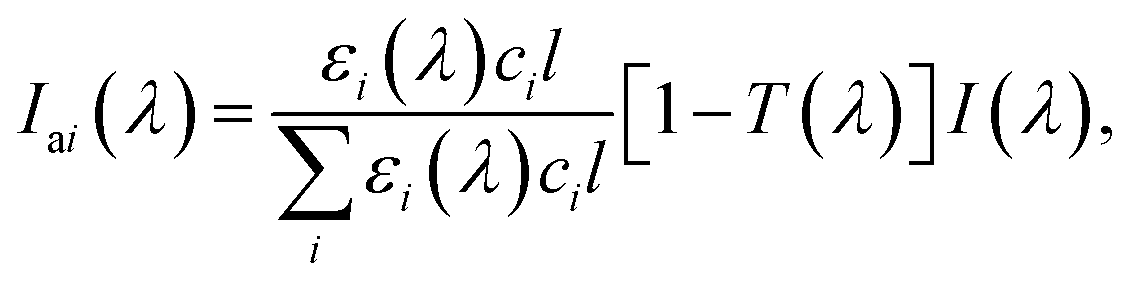 | (5) |
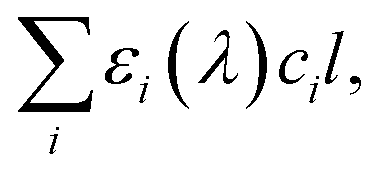 is crucial to constrain jpNO3−. Given that the sources and chemical compositions of brown carbon remain to be understood,153,156,157 the total adsorbed photon fluxes,
is crucial to constrain jpNO3−. Given that the sources and chemical compositions of brown carbon remain to be understood,153,156,157 the total adsorbed photon fluxes, 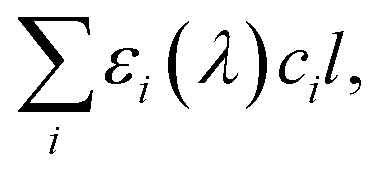 are highly uncertain. Studies of brown carbon in association with particulate nitrate photolysis are warranted.135,179 Note that eqn (5) assumes homogeneous mixing. If a nitrate particle was covered by light-absorbing species (e.g., through liquid–liquid phase separation129), such a screening effect would be intensified.
are highly uncertain. Studies of brown carbon in association with particulate nitrate photolysis are warranted.135,179 Note that eqn (5) assumes homogeneous mixing. If a nitrate particle was covered by light-absorbing species (e.g., through liquid–liquid phase separation129), such a screening effect would be intensified.
5. Nitrate-photolysis-initiated reactions
As discussed in Section 2, particulate nitrate photolysis produces strong in-particle oxidants of OH, NO2, NO2−/HNO2, and O3, and they will initiate a series of reactions in the particle phase.162–164 Note that reactions induced by OH radicals are not only specific to nitrate photolysis, but also the other OH sources such as phase transfer from gas phase and H2O2 photolysis.165 This section reviews studies of the following reactions promoted by particulate nitrate photolysis: multiphase oxidation of (1) SO2 and (2) organic compounds, and (3) the formation of nitrated products in the aqueous phase of deliquesced aerosols and cloud droplets.5.1 Multiphase oxidation of SO2
Particulate nitrate photolysis has recently been found to promote multiphase SO2 oxidation by generating OH, NO2, and NO2−/HONO (Fig. 2).11,12,37 SO2 is dissolved in aerosol liquid water and is present as bisulfite or/and sulfite depending on the pH of the particle. Under typical acidic conditions (pH < 6), dissolved SO2 mainly exists as bisulfite.2 Bisulfite can react with all OH, NO2, and NO2−/HONO for sulfate production. O3 is also possible to oxidize bisulfite,2 but this oxidation mechanism was not efficient in our previous study.11 The reaction of bisulfite and OH radical forms the sulfite radical anion, which initiates the chain reactions involving SO5−, HSO5−, and SO4− in the presence of dissolved O2 to produce multiple sulfate ions from each attack of OH on dissolved SO2.2 The oxidation by dissolved NO2, one of the most feasible mechanisms during the haze events,166 is a one-step process. During particulate nitrate photolysis at 300 nm, the highest sulfate production is found from the oxidation by NO2−/HONO, compared to the other oxidation mechanisms such as OH and NO2 radicals. A simple parameterization of sulfate production results using the reactive uptake coefficient of SO2, γSO2, and nitrate photolysis rate, PNO3−, gives the following relation: γSO2 = 1.64 × PNO3−.11 Given that nitrate concentration is as high as 10 M under highly polluted episodes and much faster particulate nitrate photolysis than that in aqueous solution,8,38,167γSO2 can become >10−5, which is comparable to the values necessary for explaining the observations in the haze events in China.168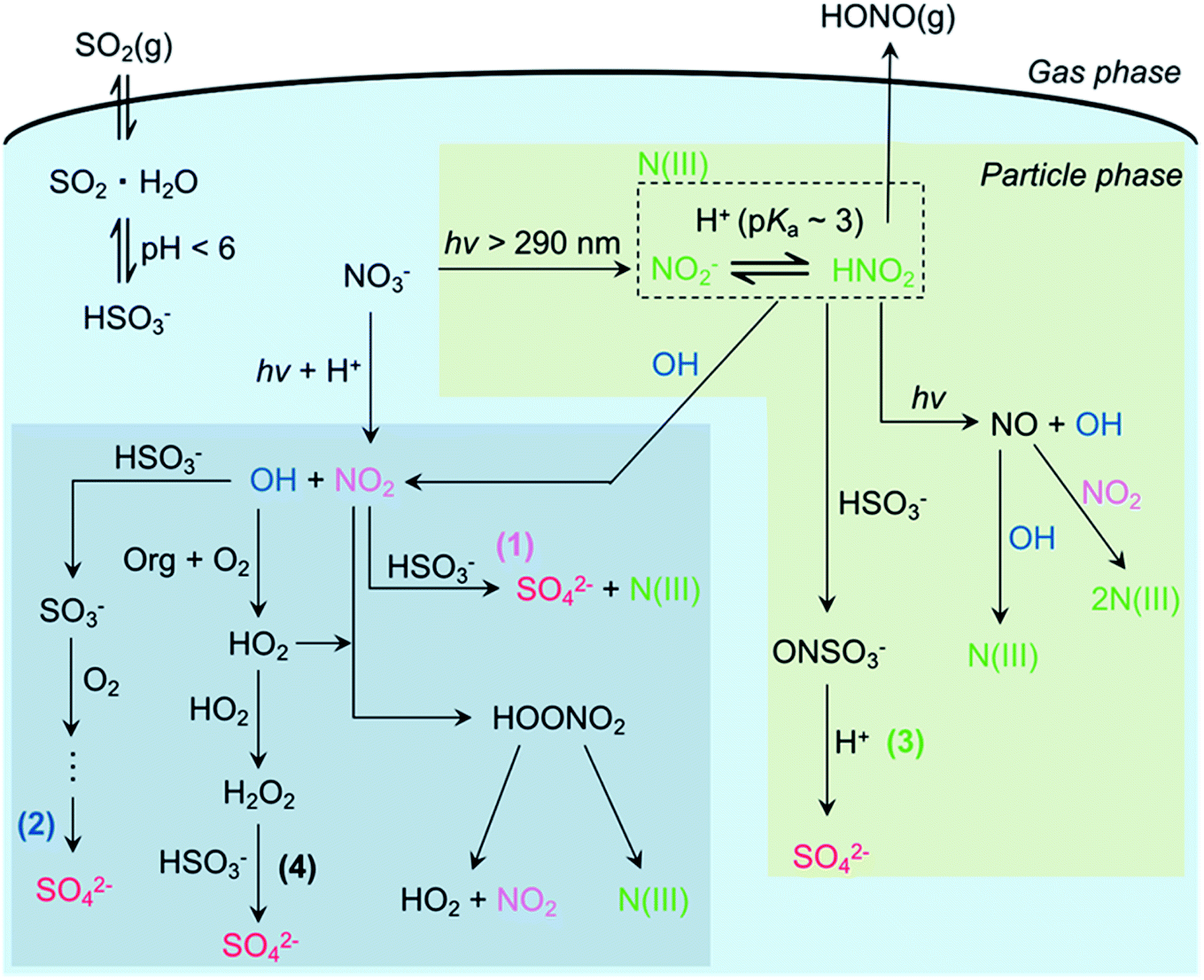 | ||
| Fig. 2 Proposed multiphase oxidation of SO2 promoted by particulate nitrate photolysis. Reprinted with permission from ref. 11 Copyright 2019 American Chemical Society. | ||
5.2 Multiphase oxidation of organics in aqueous phase secondary organic aerosol formation
Organic aerosol accounts for about 20–90% of the total particulate matter on a global scale.10 A significant fraction of this organic matter is secondary, i.e., formed in the atmosphere by converting gases into the condensed matter.169 The in-particle OH radicals produced from nitrate photolysis can promote the formation of aqueous-phase secondary organic aerosol (SOA). In aqueous aerosol particles or cloud droplets, OH radicals oxidize dissolved organic compounds such as glyoxal and methylglyoxal, pyruvic acid, glycolaldehyde, methacrolein, methyl vinyl ketone, and acetone, yielding both low-molecular-weight products (e.g., dicarboxylic acids) and high-molecular-weight compounds (e.g., oligomers).170–174 Aqueous SOA yields from the photo-oxidation of phenolic carbonyls in nitrate solution are twice as high as those in sulfate solution due to the efficient generation of OH through nitrate photolysis.175 Our recent work examined the role of particulate nitrate photolysis in the formation of SOA from particle-phase oxidation of glyoxal by OH radicals.176 Interestingly, we did not observe typical oxidation products such as oxalic acid, glyoxylic acid, and higher-molecular-weight products previously reported in the literature. Instead, formic acid/formate was the main oxidation product. In the presence of ammonium as a source of dissolved ammonia, light-absorbing species are formed177,178 and trigger the photosensitization reactions to promote glyoxal oxidation.179 Particulate nitrate photolysis can alter major reaction pathways of glyoxal oxidation.5.3 Nitration for browning atmospheric aerosol
Nitration is a chemical process that introduces a nitro group into an organic compound. The nitration of aromatic compounds has gained attention as an emerging process to produce light-absorbing organic matter or brown carbon (BrC) in the atmosphere.153,180–190 It can also chemically modify allergenic proteins present in the atmosphere (e.g., amino-acid tyrosine) and enhance their allergenicity.191–195 Nitrate photolysis is a potential contributor to the nitration process for browning atmospheric aerosol175,181,182,189 and increasing the allergenicity196 by producing nitrating agents of NO2/N2O4, nitrite (NO2−), nitrous acid (HNO2), and peroxynitrous acid (HOONO).197Nitration by NO2 has been reported for aromatic compounds such as phenols,198–200 methoxyphenols,181,182,189 benzene,201 toluene,202 and catechols.203 Phenols are important precursors for SOA formation, including BrC.204,205 Among the major nitrated aromatic compounds (NACs) found in the atmosphere are nitrophenols, nitrocatechols, nitrosalicylic acids, and nitroguaiacols.180,184,185 NACs and their derivatives contribute to 50–80% of the total visible light absorption by BrC emitted from biomass burning,206 with mass absorption coefficients (MAC) ranging from 0.05 to 4 m2 g−1.206–208 NACs can be directly emitted into the atmosphere such as by traffic exhaust,209 and biomass burning,210 and secondarily formed by the nitration of aromatic precursors in both the aqueous and the gas phases.189,201,211
Nitrophenol is one of the most abundant nitrated organic species in the atmosphere.206 The most prominent atmospheric process for nitrophenol formation is the nitration of phenol.212Fig. 3 summarizes the proposed reaction mechanisms for the nitration of phenol.213,214 Nitration is initiated by the reaction between phenol and two NO2 (or N2O4) (Fig. 3) via H-atom abstraction or electrophilic addition to the ring, resulting in a radical intermediate which is either a phenoxyl (I in Fig. 3)215 or hydroxynitrocyclohexadienyl (II).216 The phenoxyl (I) can react with either OH radicals to form hydroxyderivatives (e.g., catechol and hydroquinone, resorcinol to a lesser extent) or with NO2 to yield nitrophenols.213 Contrastingly, hydroxynitrocyclohexadienyl (II) can undergo H-atom abstraction by O2 or another NO2 to form nitrophenols.200,216 While other pathways of nitrophenol formation are possible,199,200,215,217 the nitration of phenol via nitrate photolysis in the atmosphere (the presence of oxygen) primarily proceeds through the hydroxynitrocyclohexadienyl (II in Fig. 3).
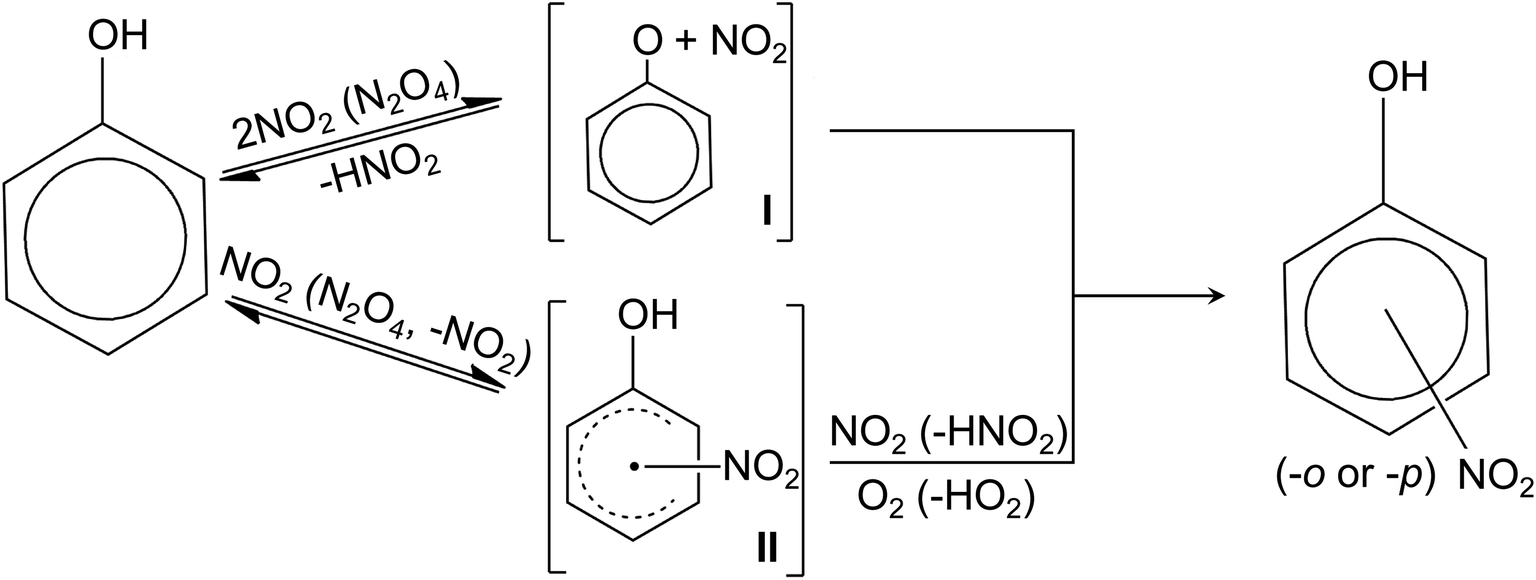 | ||
| Fig. 3 Proposed reaction mechanisms for phenol nitration.213,214 | ||
Nitration of phenols by NO2 is enhanced in the presence of OH scavengers such as 2-propanol.213 Scavengers inhibit the recombination of OH with NO2 to regenerate NO3− + H+,218 allowing more NO2 available for the nitration of phenol. The formation of nitrated phenols via nitrate photolysis was observed to decrease with increasing pH.213 At high pH, N2O4 can react with OH− to form NO3− and NO2−. At pH < 3, the formation of nitrophenols can be enhanced by thermal reactions (i.e., in the dark) involving HNO2.219,220 An example is the HNO2-catalyzed phenol nitration in which phenol directly reacts with N(III) (HNO2 or N2O3) in the dark.220 Although the reaction between phenol and HNO2 or N2O3 is a thermal process, nitrate irradiation is required to generate nitrite.
HOONO, an isomer of nitric acid (HNO3), is formed upon nitrate photoisomerization (R4). It is a powerful nitrating agent for both phenolic and non-phenolic aromatic substrates such as phenol, benzene, and naphthalene.201,221,222 Although the direct formation of HOONO upon nitrate irradiation requires wavelength that is not atmospherically relevant (<290 nm), it can also be generated from HNO2 and NO produced from nitrate photolysis via the reaction between NO and O2−.221 The irradiation of solid nitrate salts (NH4NO3, NaNO3) with benzene can also yield phenol and nitrobenzene, possibly due to the generation of OH and NO2.212,223 In the presence of hematite (α-Fe2O3),224 significant enhancement in nitrobenzene formation occurs likely due to the protonation of peroxynitrite (formed upon nitrate photoisomerization) to HOONO.225
6. Future directions
We have discussed the potential impacts of particulate nitrate photolysis in the atmosphere. Yet, many issues remain unresolved. Here, we propose the following questions to be addressed to better constrain the impacts of particulate nitrate photolysis.(1) How much can particulate nitrate photolysis promote multiphase oxidation for the secondary formation of inorganic and organic compounds in the particles, respectively? Earlier works have mainly studied the production of gaseous photoproducts of NO2 and HONO from nitrate photolysis (Table 3). Particular attention needs to be paid to quantifying the oxidation capacity of particulate nitrate photolysis in the particle phase. While gas phase chemistry is not the focus of this paper, particulate nitrate photolysis can affect gas phase chemistry by producing NOx. The NOx recycling from particulate nitrate photolysis can lead to enhancements in NOx, OH, and O3 concentrations in the atmosphere.34 It is also possible that photolysis of organic nitrates could influence the nitrogen cycle and O3 production, but there are very few studies on this topic.47
(2) What roles does particulate nitrate photolysis play in the formation and aging of brown carbon aerosols? Nitrate has been recognized as a nitrating agent in brown carbon formation.226 On the other hand, nitrate photolysis has the potential to accelerate the aging of brown carbon.175 Nonetheless, studies on both the formation and aging of brown carbon during particulate nitrate photolysis are scarce.
(3) What is the role of the surface/interfacial effects (Sections 3.1 and 3.6.1) in promoting nitrate photolysis in the particle phase? When nitrate anions are localized at the air/particle interface, they are not fully solvated. Nitrate photolysis in the incomplete solvent cage can proceed faster than in the complete solvent cage, which is one of the plausible reasons to differentiate nitrate photolysis in the particle phase from bulk solutions. However, whether nitrate anions are so surface-active to affect the rate constant in particles is still controversial. What makes nitrate photolysis in particles so different from in bulk solutions needs to be elucidated.
(4) What parameters best describe particulate nitrate photolysis in air quality models? The photolysis rate constants are one of the most practical parameters that can be used in air quality models to implement particulate nitrate photolysis. The majority of studies have measured photoproducts of gaseous species such as NO2 and HONO to estimate the rate constants. However, this method might potentially underestimate the constants (Section 4). Measurements of both gas and particle phase photoproducts generated from particulate nitrate photolysis are recommended to better constrain the rate constants. In addition to the photolysis rate constants, the branching ratio of photoproducts N(III) to NO2 is also an important parameter that affects the product yields during nitrate photolysis. A N(III):NO2 molar ratio of 0.33–0.67 was assumed in earlier modeling works.40,227 However, the product yields can be affected by many factors as discussed in Section 3. Systematic studies under more realistic complex aerosol systems such as nitrate particles internally mixed with black carbon, BrC, radical scavengers, surface-active species (e.g., halide ions), and heterogeneously mixed (e.g., liquid–liquid phase separated) particles are needed.
Author contributions
Masao Gen: conceptualization, organization, data analysis, writing–original draft, writing–review & editing. Zhancong Liang: writing–original draft, writing–review & editing. Ruifeng Zhang: writing–original draft, writing–review & editing. Brix Raphael Go: writing–original draft, writing–review & editing. Chak K. Chan: conceptualization, organization, data analysis, writing–original draft, writing–review & editingConflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge support from the National Natural Science Foundation of China (42075100 and 41875142), the Guangdong Basic and Applied Basic Research Foundation (2020B1515130003), the Japan Science and Technology Agency (JST) for Fusion Oriented Research for disruptive Science and Technology program (JPMJFR206V), the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Early-Career Scientists (21K17876).References
- X. Y. Boucher, O. Randall, D. Artaxo, P. Bretherton, C. Feingold, G. Forster, P. Kerminen, V. M. Kondo, Y. Liao, H. Lohmann, U. Rasch, P. Satheesh, S. K. Sherwood, S. Stevens and B. Zhang, Climate Change 2013: the Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, New York, 2013 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: from Air Pollution to Climate Change, Wiley, New York, 2006 Search PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- D. J. Donaldson, A. F. Tuck and V. Vaida, Chem. Rev., 2003, 103, 4717–4730 CrossRef CAS PubMed.
- P. S. Monks, Chem. Soc. Rev., 2005, 34, 376–395 RSC.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 1999 Search PubMed.
- P. S. Monks, A. T. Archibald, A. Colette, O. Cooper, M. Coyle, R. Derwent, D. Fowler, C. Granier, K. S. Law, G. E. Mills, D. S. Stevenson, O. Tarasova, V. Thouret, E. von Schneidemesser, R. Sommariva, O. Wild and M. L. Williams, Atmos. Chem. Phys., 2015, 15, 8889–8973 CrossRef CAS.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS PubMed.
- M. E. Monge, T. Rosenorn, O. Favez, M. Muller, G. Adler, A. Abo Riziq, Y. Rudich, H. Herrmann, C. George and B. D'Anna, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6840–6844 CrossRef CAS PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- M. Gen, R. Zhang, D. D. Huang, Y. Li and C. K. Chan, Environ. Sci. Technol., 2019, 53, 8757–8766 CrossRef CAS PubMed.
- M. Gen, R. Zhang, D. D. Huang, Y. Li and C. K. Chan, Environ. Sci. Technol. Lett., 2019, 6, 86–91 CrossRef CAS.
- H. Zheng, S. Song, G. Sarwar, M. Gen, S. Wang, D. Ding, X. Chang, S. Zhang, J. Xing, Y. Sun, D. Ji, C. K. Chan, J. Gao and M. B. McElroy, Environ. Sci. Technol. Lett., 2020, 7, 632–638 CrossRef CAS PubMed.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS PubMed.
- M. Tang, D. J. Cziczo and V. H. Grassian, Chem. Rev., 2016, 116, 4205–4259 CrossRef CAS PubMed.
- B. C. Faust and R. G. Zepp, Environ. Sci. Technol., 1993, 27, 2517–2522 CrossRef CAS.
- C. Weller, A. Tilgner, P. Bräuer and H. Herrmann, Environ. Sci. Technol., 2014, 48, 5652–5659 CrossRef CAS PubMed.
- J. Mack and J. R. Bolton, J. Photochem. Photobiol., A, 1999, 128, 1–13 CrossRef CAS.
- S. Goldstein and J. Rabani, J. Am. Chem. Soc., 2007, 129, 10597–10601 CrossRef CAS PubMed.
- R. Zellner, M. Exner and H. Herrmann, J. Atmos. Chem., 1990, 10, 411–425 CrossRef CAS.
- D. M. Bulman, S. P. Mezyk and C. K. Remucal, Environ. Sci. Technol., 2019, 53, 4450–4459 CrossRef CAS PubMed.
- S. Gligorovski and J. P. D. Abbatt, Science, 2018, 359, 632–633 CrossRef CAS PubMed.
- J. X. Warner, R. R. Dickerson, Z. Wei, L. L. Strow, Y. Wang and Q. Liang, Geophys. Res. Lett., 2017, 44, 2875–2884 CrossRef CAS PubMed.
- C. Li, C. McLinden, V. Fioletov, N. Krotkov, S. Carn, J. Joiner, D. Streets, H. He, X. Ren, Z. Li and R. R. Dickerson, Sci. Rep., 2017, 7, 14304 CrossRef PubMed.
- J. L. Hand, B. A. Schichtel, W. C. Malm and M. L. Pitchford, Atmos. Chem. Phys., 2012, 12, 10353–10365 CrossRef CAS.
- A. Franchin, D. L. Fibiger, L. Goldberger, E. E. McDuffie, A. Moravek, C. C. Womack, E. T. Crosman, K. S. Docherty, W. P. Dube, S. W. Hoch, B. H. Lee, R. Long, J. G. Murphy, J. A. Thornton, S. S. Brown, M. Baasandorj and A. M. Middlebrook, Atmos. Chem. Phys., 2018, 18, 17259–17276 CrossRef CAS.
- J.-P. Putaud, F. Raes, R. Van Dingenen, E. Brüggemann, M.-C. Facchini, S. Decesari, S. Fuzzi, R. Gehrig, C. Hüglin, P. Laj, G. Lorbeer, W. Maenhaut, N. Mihalopoulos, K. Müller, X. Querol, S. Rodriguez, J. Schneider, G. Spindler, H. ten Brink, K. Tørseth and A. Wiedensohler, Atmos. Environ., 2004, 38, 2579–2595 CrossRef CAS.
- G. Spindler, K. Müller, E. Brüggemann, T. Gnauk and H. Herrmann, Atmos. Environ., 2004, 38, 5333–5347 CrossRef CAS.
- I. Uno, Z. Wang, S. Itahashi, K. Yumimoto, Y. Yamamura, A. Yoshino, A. Takami, M. Hayasaki and B.-G. Kim, Sci. Rep., 2020, 10, 6450 CrossRef CAS PubMed.
- Y.-J. Jo, H.-J. Lee, H.-Y. Jo, J.-H. Woo, Y. Kim, T. Lee, G. Heo, S.-M. Park, D. Jung, J. Park and C.-H. Kim, Atmos. Res., 2020, 240, 104948 CrossRef CAS.
- Q. Xu, S. Wang, J. Jiang, N. Bhattarai, X. Li, X. Chang, X. Qiu, M. Zheng, Y. Hua and J. Hao, Sci. Total Environ., 2019, 689, 1293–1303 CrossRef CAS PubMed.
- X. Fu, T. Wang, J. Gao, P. Wang, Y. Liu, S. Wang, B. Zhao and L. Xue, Environ. Sci. Technol., 2020, 54, 3881–3889 CrossRef CAS PubMed.
- G. Mark, H. G. Korth, H. P. Schuchmann and C. Von Sonntag, J. Photochem. Photobiol., A, 1996, 101, 89–103 CrossRef CAS.
- P. Kasibhatla, T. Sherwen, M. J. Evans, L. J. Carpenter, C. Reed, B. Alexander, Q. Chen, M. P. Sulprizio, J. D. Lee, K. A. Read, W. Bloss, L. R. Crilley, W. C. Keene, A. A. P. Pszenny and A. Hodzic, Atmos. Chem. Phys., 2018, 18, 11185–11203 CrossRef CAS.
- Y. Wu, L. Bu, X. Duan, S. Zhu, M. Kong, N. Zhu and S. Zhou, J. Clean. Prod., 2020, 273, 123065 CrossRef CAS.
- R. G. Zepp, J. Holgne and H. Bader, Environ. Sci. Technol., 1987, 21, 443–450 CrossRef CAS PubMed.
- R. Zhang, M. Gen, D. Huang, Y. Li and C. K. Chan, Environ. Sci. Technol., 2020, 54, 3831–3839 CrossRef CAS PubMed.
- C. Ye, N. Zhang, H. Gao and X. Zhou, Environ. Sci. Technol., 2017, 51, 6849–6856 CrossRef CAS PubMed.
- Q. Shi, Y. Tao, J. E. Krechmer, C. L. Heald, J. G. Murphy, J. H. Kroll and Q. Ye, Environ. Sci. Technol., 2021, 55, 854–861 CrossRef CAS PubMed.
- C. Ye, X. Zhou, D. Pu, J. Stutz, J. Festa, M. Spolaor, C. Tsai, C. Cantrell, R. L. Mauldin, T. Campos, A. Weinheimer, R. S. Hornbrook, E. C. Apel, A. Guenther, L. Kaser, B. Yuan, T. Karl, J. Haggerty, S. Hall, K. Ullmann, J. N. Smith, J. Ortega and C. Knote, Nature, 2016, 532, 489–491 CrossRef CAS PubMed.
- P. Nissenson, D. Dabdub, R. Das, V. Maurino, C. Minero and D. Vione, Atmos. Environ., 2010, 44, 4859–4866 CrossRef CAS.
- N. K. Scharko, A. E. Berke and J. D. Raff, Environ. Sci. Technol., 2014, 48, 11991–12001 CrossRef CAS PubMed.
- L. M. Wingen, A. C. Moskun, S. N. Johnson, J. L. Thomas, M. Roeselová, D. J. Tobias, M. T. Kleinman and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2008, 10, 5668–5677 RSC.
- S. S. Brown and J. Stutz, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- A. E. Perring, S. E. Pusede and R. C. Cohen, Chem. Rev., 2013, 113, 5848–5870 CrossRef CAS PubMed.
- J.-F. Müller, J. Peeters and T. Stavrakou, Atmos. Chem. Phys., 2014, 14, 2497–2508 CrossRef.
- T. Nah, J. Sanchez, C. M. Boyd and N. L. Ng, Environ. Sci. Technol., 2016, 50, 222–231 CrossRef CAS PubMed.
- M. Daniels, R. V. Meyers and E. V. Belardo, J. Phys. Chem., 1968, 72, 389–399 CrossRef CAS.
- A. M. Grannas, A. E. Jones, J. Dibb, M. Ammann, C. Anastasio, H. J. Beine, M. Bergin, J. Bottenheim, C. S. Boxe, G. Carver, G. Chen, J. H. Crawford, F. Dominé, M. M. Frey, M. I. Guzmán, D. E. Heard, D. Helmig, M. R. Hoffmann, R. E. Honrath, L. G. Huey, M. Hutterli, H. W. Jacobi, P. Klán, B. Lefer, J. McConnell, J. Plane, R. Sander, J. Savarino, P. B. Shepson, W. R. Simpson, J. R. Sodeau, R. Von Glasow, R. Weller, E. W. Wolff and T. Zhu, Atmos. Chem. Phys., 2007, 7, 4329–4373 CrossRef CAS.
- K. B. Benedict, A. S. McFall and C. Anastasio, Environ. Sci. Technol., 2017, 51, 4387–4395 CrossRef CAS PubMed.
- P. Warneck and C. Wurzinger, J. Phys. Chem., 1988, 92, 6278–6283 CrossRef CAS.
- T. Arakaki, C. Anastasio, Y. Kuroki, H. Nakajima, K. Okada, Y. Kotani, D. Handa, S. Azechi, T. Kimura, A. Tsuhako and Y. Miyagi, Environ. Sci. Technol., 2013, 47, 8196–8203 CrossRef CAS PubMed.
- G. Chen, S. Hanukovich, M. Chebeir, P. Christopher and H. Liu, Environ. Sci. Technol., 2019, 53, 316–324 CrossRef CAS PubMed.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS.
- S. Goldstein and G. Czapski, Free Radic. Biol. Med., 1995, 19, 505–510 CrossRef CAS PubMed.
- S. V Lymar and J. K. Hurst, J. Am. Chem. Soc., 1995, 117, 8867–8868 CrossRef.
- O. Svoboda, L. Kubelová and P. Slavíček, J. Phys. Chem. A, 2013, 117, 12868–12877 CrossRef CAS PubMed.
- K. R. Wilson, A. M. Prophet, G. Rovelli, M. D. Willis, R. J. Rapf and M. I. Jacobs, Chem. Sci., 2020, 11, 8533–8545 RSC.
- D. E. Otten, P. B. Petersen and R. J. Saykally, Chem. Phys. Lett., 2007, 449, 261–265 CrossRef CAS.
- M. A. Brown, B. Winter, M. Faubel and J. C. Hemminger, J. Am. Chem. Soc., 2009, 131, 8354–8355 CrossRef CAS PubMed.
- C. Jie, C. D. Vecitis, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. B, 2006, 110, 25598–25602 CrossRef PubMed.
- M. Xu, R. Spinney and H. C. Allen, J. Phys. Chem. B, 2009, 113, 4102–4110 CrossRef CAS PubMed.
- C. Tian, S. J. Byrnes, H. L. Han and Y. R. Shen, J. Phys. Chem. Lett., 2011, 2, 1946–1949 CrossRef CAS.
- S. N. Wren and D. J. Donaldson, Chem. Phys. Lett., 2012, 522, 1–10 CrossRef CAS.
- L. M. Pegram and M. T. Record, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14278–14281 CrossRef CAS PubMed.
- J. L. Thomas, M. Roeselová, L. X. Dang and D. J. Tobias, J. Phys. Chem. A, 2007, 111, 3091–3098 CrossRef CAS PubMed.
- B. Minofar, R. Vácha, A. Wahab, S. Mahiuddin, W. Kunz and P. Jungwirth, J. Phys. Chem. B, 2006, 110, 15939–15944 CrossRef CAS PubMed.
- H. O. T. Pye, A. Nenes, B. Alexander, A. P. Ault, M. C. Barth, S. L. Clegg, J. L. Collett, K. M. Fahey, C. J. Hennigan, H. Herrmann, M. Kanakidou, J. T. Kelly, I. T. Ku, V. Faye McNeill, N. Riemer, T. Schaefer, G. Shi, A. Tilgner, J. T. Walker, T. Wang, R. Weber, J. Xing, R. A. Zaveri and A. Zuend, Atmos. Chem. Phys., 2020, 20, 4809–4888 CrossRef CAS PubMed.
- Q. Zhang, J. L. Jimenez, D. R. Worsnop and M. Canagaratna, Environ. Sci. Technol., 2007, 41, 3213–3219 CrossRef CAS PubMed.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7365–7369 CrossRef CAS PubMed.
- N. K. Richards and B. J. Finlayson-Pitts, Environ. Sci. Technol., 2012, 46, 10447–10454 CrossRef CAS PubMed.
- G. A. Poskrebyshev, P. Neta and R. E. Huie, J. Phys. Chem. A, 2002, 106, 11488–11491 CrossRef CAS.
- C. Anastasio and C. Liang, Environ. Sci. Technol., 2009, 43, 1108–1114 CrossRef CAS PubMed.
- E. Riordan, N. Minogue, D. Healy, P. O'Driscoll and J. R. Sodeau, J. Phys. Chem. A, 2005, 109, 779–786 CrossRef CAS PubMed.
- T. C. Vandenboer, C. J. Young, R. K. Talukdar, M. Z. Markovic, S. S. Brown, J. M. Roberts and J. G. Murphy, Nat. Geosci., 2015, 8, 55–60 CrossRef CAS.
- H. Su, Y. Cheng, R. Oswald, T. Behrendt, I. Trebs, F. X. Meixner, M. O. Andreae, P. Cheng, Y. Zhang and U. Pöschl, Science, 2011, 333, 1616–1618 CrossRef CAS PubMed.
- J. Y. Park and Y. N. Lee, J. Phys. Chem., 1988, 92, 6294–6302 CrossRef CAS.
- C. K. Chan, R. C. Flagan and J. H. Seinfeld, Atmos. Environ., Part A, 1992, 26, 1661–1673 CrossRef.
- M. C. Yeung and C. K. Chan, Aerosol Sci. Technol., 2010, 44, 269–280 CrossRef CAS.
- M. Roca, J. Zahardis, J. Bone, M. El-Maazawi and V. H. Grassian, J. Phys. Chem. A, 2008, 112, 13275–13281 CrossRef CAS PubMed.
- P. K. Hudson, J. Schwarz, J. Baltrusaitis, E. R. Gibson and V. H. Grassian, J. Phys. Chem. A, 2007, 111, 544–548 CrossRef CAS PubMed.
- S. A. Asher, D. D. Tuschel, T. A. Vargson, L. Wang and S. J. Geib, J. Phys. Chem. A, 2011, 115, 4279–4287 CrossRef CAS PubMed.
- N. K. Richards-Henderson, C. Anderson, C. Anastasio and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2015, 17, 32211–32218 RSC.
- W. Yang, C. Han, H. Yang and X. Xue, Environ. Pollut., 2018, 243, 679–686 CrossRef CAS PubMed.
- S. Romakkaniemi, K. Hämeri, M. Väkevä and A. Laaksonen, J. Phys. Chem. A, 2001, 105, 8183–8188 CrossRef CAS.
- L. Chu and C. Anastasio, J. Phys. Chem. A, 2003, 107, 9594–9602 CrossRef CAS.
- K. B. Benedict and C. Anastasio, J. Phys. Chem. A, 2017, 121, 8474–8483 CrossRef CAS PubMed.
- A. S. McFall, K. C. Edwards and C. Anastasio, Environ. Sci. Technol., 2018, 52, 5710–5717 CrossRef CAS PubMed.
- Y. Dubowski, A. J. Colussi, C. Boxe and M. R. Hoffmann, J. Phys. Chem. A, 2002, 106, 6967–6971 CrossRef CAS.
- M. Zatko, L. Geng, B. Alexander, E. Sofen and K. Klein, Atmos. Chem. Phys., 2016, 16, 2819–2842 CrossRef CAS.
- F. Dominé and P. B. Shepson, Science, 2002, 297, 1506–1510 CrossRef PubMed.
- G. Marcotte, P. Marchand, S. Pronovost, P. Ayotte, C. Laffon and P. Parent, J. Phys. Chem. A, 2015, 119, 1996–2005 CrossRef CAS PubMed.
- C. S. Blaszczak-Boxe and A. Saiz-Lopez, Atmos. Environ., 2018, 193, 224–241 CrossRef CAS.
- F. Domine, J. Bock, D. Voisin and D. J. Donaldson, J. Phys. Chem. A, 2013, 117, 4733–4749 CrossRef CAS PubMed.
- C. Meusinger, T. A. Berhanu, J. Erbland, J. Savarino and M. S. Johnson, J. Chem. Phys., 2014, 140, 244305 CrossRef PubMed.
- S. N. Wren and D. J. Donaldson, J. Phys. Chem. Lett., 2011, 2, 1967–1971 CrossRef CAS.
- K. J. Morenz, Q. Shi, J. G. Murphy and D. J. Donaldson, J. Phys. Chem. A, 2016, 120, 7902–7908 CrossRef CAS PubMed.
- C. L. Weagle, G. Snider, C. Li, A. Van Donkelaar, S. Philip, P. Bissonnette, J. Burke, J. Jackson, R. Latimer, E. Stone, I. Abboud, C. Akoshile, N. X. Anh, J. R. Brook, A. Cohen, J. Dong, M. D. Gibson, D. Griffith, K. B. He, B. N. Holben, R. Kahn, C. A. Keller, J. S. Kim, N. Lagrosas, P. Lestari, Y. L. Khian, Y. Liu, E. A. Marais, J. V. Martins, A. Misra, U. Muliane, R. Pratiwi, E. J. Quel, A. Salam, L. Segev, S. N. Tripathi, C. Wang, Q. Zhang, M. Brauer, Y. Rudich and R. V. Martin, Environ. Sci. Technol., 2018, 52, 11670–11681 CAS.
- C. K. Chan, Z. Liang, J. Zheng, S. L. Clegg and P. Brimblecombe, Aerosol Sci. Technol., 1997, 27, 324–344 CrossRef CAS.
- N. K. Richards-Henderson, K. M. Callahan, P. Nissenson, N. Nishino, D. J. Tobias and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2013, 15, 17636–17646 RSC.
- N. K. Richards, L. M. Wingen, K. M. Callahan, N. Nishino, M. T. Kleinman, D. J. Tobias and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2011, 115, 5810–5821 CrossRef CAS PubMed.
- L. Piatkowski, Z. Zhang, E. H. G. Backus, H. J. Bakker and M. Bonn, Nat. Commun., 2014, 5, 4083 CrossRef CAS PubMed.
- P. Jungwirth and D. J. Tobias, J. Phys. Chem. B, 2001, 105, 10468–10472 CrossRef CAS.
- S. Ghosal, J. C. Hemminger, H. Bluhm, B. S. Mun, E. L. D. Hebenstreit, G. Ketteler, D. F. Ogletree, F. G. Requejo and M. Salmeron, Science, 2005, 307, 563–566 CrossRef CAS PubMed.
- D. C. Blanchard, J. Geophys. Res. Ocean., 1985, 90, 961–963 CrossRef.
- W. R. Simpson, S. S. Brown, A. Saiz-Lopez, J. A. Thornton and R. Von Glasow, Chem. Rev., 2015, 115, 4035–4062 CrossRef CAS PubMed.
- X. Fu, T. Wang, S. Wang, L. Zhang, S. Cai, J. Xing and J. Hao, Environ. Sci. Technol., 2018, 52, 1644–1654 CrossRef CAS PubMed.
- H. W. Xiao, H. Y. Xiao, L. Luo, C. Y. Shen, A. M. Long, L. Chen, Z. H. Long and D. N. Li, Atmos. Chem. Phys., 2017, 17, 3199–3214 CrossRef CAS.
- B. Wang, Y. Chen, S. Zhou, H. Li, F. Wang and T. Yang, Sci. Total Environ., 2019, 649, 652–660 CrossRef CAS PubMed.
- H. W. Xiao, H. Y. Xiao, C. Y. Shen, Z. Y. Zhang and A. M. Long, Atmosphere, 2018, 9, 298 CrossRef.
- R. Kumar, A. Elizabeth and A. G. Gawane, Aerosol Sci. Technol., 2006, 40, 477–489 CrossRef.
- R. Rengarajan, A. K. Sudheer and M. M. Sarin, Atmos. Res., 2011, 102, 420–431 CrossRef CAS.
- L. Zhao, L. Wang, J. Tan, J. Duan, X. Ma, C. Zhang, S. Ji, M. Qi, X. H. Lu, Y. Wang, Q. Wang and R. Xu, Atmos. Environ., 2019, 206, 119–131 CrossRef CAS.
- H. Wang, J. Ding, J. Xu, J. Wen, J. Han, K. Wang, G. Shi, Y. Feng, C. E. Ivey, Y. Wang, A. Nenes, Q. Zhao and A. G. Russell, Sci. Total Environ., 2019, 646, 564–572 CrossRef CAS PubMed.
- D. Atzei, P. Fermo, R. Vecchi, M. Fantauzzi, V. Comite, G. Valli, F. Cocco and A. Rossi, Environ. Pollut., 2019, 246, 294–302 CrossRef CAS PubMed.
- D. K. Deshmukh, M. K. Deb, Y. I. Tsai and S. L. Mkoma, Aerosol Air Qual. Res., 2011, 11, 696–708 CrossRef CAS.
- P. Pipalatkar, V. V. Khaparde, D. G. Gajghate and M. A. Bawase, Aerosol Air Qual. Res., 2014, 14, 1089–1099 CrossRef CAS.
- B. J. Finlayson-Pitts, Chem. Rev., 2003, 103, 4801–4822 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts, Anal. Chem., 2010, 82, 770–776 CrossRef CAS PubMed.
- J. L. Moyers and R. A. Duce, J. Geophys. Res., 1972, 77, 5229–5238 CrossRef CAS.
- Y.-H. Zhang and C. K. Chan, J. Phys. Chem. A, 2000, 104, 9191–9196 CrossRef CAS.
- P. D. Pedersen, K. V. Mikkelsen and M. S. Johnson, Phys. Chem. Chem. Phys., 2020, 22, 11678–11685 RSC.
- C. Ye, N. Zhang, H. Gao and X. Zhou, Sci. Rep., 2019, 9, 4351 CrossRef PubMed.
- C. George, R. S. Strekowski, J. Kleffmann, K. Stemmler and M. Ammann, Faraday Discuss., 2005, 130, 195–210 RSC.
- J. Liu, S. Li, M. Mekic, H. Jiang, W. Zhou, G. Loisel, W. Song, X. Wang and S. Gligorovski, Environ. Sci. Technol. Lett., 2019, 6, 413–417 CrossRef CAS.
- C. Han, W. Yang, Q. Wu, H. Yang and X. Xue, Environ. Sci. Technol., 2016, 50, 5017–5023 CrossRef CAS PubMed.
- K. Stemmler, M. Ammann, C. Donders, J. Kleffmann and C. George, Nature, 2006, 440, 195–198 CrossRef CAS PubMed.
- X. Wang, E. Z. Dalton, Z. C. Payne, S. Perrier, M. Riva, J. D. Raff and C. George, Environ. Sci. Technol. Lett., 2021, 8, 53–58 CrossRef CAS.
- Z. Liang, R. Zhang, M. Gen, Y. Chu and C. K. Chan, J. Phys. Chem. A, 2021, 125, 3739–3747 CrossRef CAS PubMed.
- B. Mayer and S. Madronich, Atmos. Chem. Phys., 2004, 4, 2241–2250 CrossRef CAS.
- P. Nissenson, C. J. H. Knox, B. J. Finlayson-Pitts, L. F. Phillips and D. Dabdub, Phys. Chem. Chem. Phys., 2006, 8, 4700–4710 RSC.
- C. K. Chan, R. C. Flagan and J. H. Seinfeld, Appl. Opt., 1991, 30, 459–467 CrossRef CAS PubMed.
- B. Mayer, M. Schröder, R. Preusker and L. Schüller, Atmos. Chem. Phys., 2004, 4, 1255–1263 CrossRef CAS.
- F. J. Dentener, G. R. Carmichael, Y. Zhang, J. Lelieveld and P. J. Crutzen, J. Geophys. Res. Atmos., 1996, 101, 22869–22889 CrossRef CAS.
- B. R. G. Mabato, Y. Lyu, Y. Ji, Y. J. Li, D. D. Huang, X. Li, T. Nah, C. H. Lam and C. K. Chan, Atmos. Chem. Phys., 2022, 22, 273–293 CrossRef.
- J. Baltrusaitis, J. Schuttlefield, J. H. Jensen and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 4970–4980 RSC.
- G. Rubasinghege and V. H. Grassian, J. Phys. Chem. A, 2009, 113, 7818–7825 CrossRef CAS PubMed.
- D. M. B. Lesko, E. M. Coddens, H. D. Swomley, R. M. Welch, J. Borgatta and J. G. Navea, Phys. Chem. Chem. Phys., 2015, 17, 20775–20785 RSC.
- J. Du and L. Zhu, Chem. Phys. Lett., 2011, 511, 213–218 CrossRef CAS.
- C. Zhu, B. Xiang, L. Zhu and R. Cole, Chem. Phys. Lett., 2008, 458, 373–377 CrossRef CAS.
- C. R. Usher, A. E. Michel and V. H. Grassian, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS PubMed.
- A. Gankanda and V. H. Grassian, J. Phys. Chem. C, 2014, 118, 29117–29125 CrossRef CAS.
- A. Gankanda and V. H. Grassian, J. Phys. Chem. A, 2013, 117, 2205–2212 CrossRef CAS PubMed.
- J. J. Jankowski, D. J. Kieber, K. Mopper and P. J. Neale, Photochem. Photobiol., 2004, 71, 431–440 CrossRef.
- F. Bao, M. Li, Y. Zhang, C. Chen and J. Zhao, Environ. Sci. Technol., 2018, 52, 6309–6316 CrossRef CAS PubMed.
- P. S. Romer, P. J. Wooldridge, J. D. Crounse, M. J. Kim, P. O. Wennberg, J. E. Dibb, E. Scheuer, D. R. Blake, S. Meinardi, A. L. Brosius, A. B. Thames, D. O. Miller, W. H. Brune, S. R. Hall, T. B. Ryerson and R. C. Cohen, Environ. Sci. Technol., 2018, 52, 13738–13746 CrossRef CAS PubMed.
- C. Ye, H. Gao, N. Zhang and X. Zhou, Environ. Sci. Technol., 2016, 50, 3530–3536 CrossRef CAS PubMed.
- A. M. Baergen and D. J. Donaldson, Environ. Sci. Technol., 2013, 47, 815–820 CrossRef CAS PubMed.
- S. Laufs and J. Kleffmann, Phys. Chem. Chem. Phys., 2016, 18, 9616–9625 RSC.
- M. N. Sullivan, L. T. Chu and L. Zhu, Phys. Chem. Chem. Phys., 2018, 20, 30537–30539 RSC.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- T. C. Bond and R. W. Bergstrom, Aerosol Sci. Technol., 2006, 40, 27–67 CrossRef CAS.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chem. Rev., 2015, 115, 4335–4382 CrossRef CAS PubMed.
- M. Shiraiwa, Y. Kondo, N. Moteki, N. Takegawa, Y. Miyazaki and D. R. Blake, Geophys. Res. Lett., 2007, 34, L16803 CrossRef.
- D. A. Lack, J. M. Langridge, R. Bahreini, C. D. Cappa, A. M. Middlebrook and J. P. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14802–14807 CrossRef CAS PubMed.
- T. Moise, J. M. Flores and Y. Rudich, Chem. Rev., 2015, 115, 4400–4439 CrossRef CAS PubMed.
- R. F. Hems, E. G. Schnitzler, C. Liu-Kang, C. D. Cappa and J. P. D. Abbatt, ACS Earth Space Chem., 2021, 5, 722–748 CrossRef CAS.
- Y. Wang, D. D. Huang, W. Huang, B. Liu, Q. Chen, R. Huang, M. Gen, B. R. G. Mabato, C. K. Chan, X. Li, T. Hao, Y. Tan, K. I. Hoi, K. M. Mok and Y. J. Li, Environ. Sci. Technol., 2021, 55, 15694–15704 CrossRef CAS PubMed.
- M. Gen, R. Zhang and C. K. Chan, Environ. Sci. Technol., 2021, 55, 15715–15723 CrossRef CAS PubMed.
- A. Alif and P. Boule, J. Photochem. Photobiol., A, 1991, 59, 357–367 CrossRef CAS.
- A. Bianco, M. Passananti, H. Perroux, G. Voyard, C. Mouchel-Vallon, N. Chaumerliac, G. Mailhot, L. Deguillaume and M. Brigante, Atmos. Chem. Phys., 2015, 15, 9191–9202 CrossRef CAS.
- H. Herrmann, Chem. Rev., 2003, 103, 4691–4716 CrossRef CAS PubMed.
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Bräuer and A. Tilgner, ChemPhysChem, 2010, 11, 3796–3822 CrossRef CAS PubMed.
- H. Herrmann, T. Schaefer, A. Tilgner, S. A. Styler, C. Weller, M. Teich and T. Otto, Chem. Rev., 2015, 115, 4259–4334 CrossRef CAS PubMed.
- B. Ervens, A. Sorooshian, Y. B. Lim and B. J. Turpin, J. Geophys. Res., 2014, 119, 3997–4016 CAS.
- Y. Cheng, G. Zheng, C. Wei, Q. Mu, B. Zheng, Z. Wang, M. Gao, Q. Zhang, K. He, G. Carmichael, U. Pöschl and H. Su, Sci. Adv., 2016, 2, e1601530 CrossRef PubMed.
- A. C. Hong, S. N. Wren and D. J. Donaldson, J. Phys. Chem. Lett., 2013, 4, 2994–2998 CrossRef CAS PubMed.
- B. Zheng, Q. Zhang, Y. Zhang, K. B. He, K. Wang, G. J. Zheng, F. K. Duan, Y. L. Ma and T. Kimoto, Atmos. Chem. Phys., 2015, 15, 2031–2049 CrossRef CAS.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- A. G. Carlton, B. J. Turpin, H. J. Lim, K. E. Altieri and S. Seitzinger, Geophys. Res. Lett., 2006, 33, L06822 CrossRef.
- Y. Liu, I. El Haddad, M. Scarfogliero, L. Nieto-Gligorovski, B. Temime-Roussel, E. Quivet, N. Marchand, B. Picquet-Varrault and A. Monod, Atmos. Chem. Phys., 2009, 9, 5093–5105 CrossRef CAS.
- L. Poulain, Y. Katrib, E. Isikli, Y. Liu, H. Wortham, P. Mirabel, S. Le Calvé and A. Monod, Chemosphere, 2010, 81, 312–320 CrossRef CAS PubMed.
- X. Zhang, Z. M. Chen and Y. Zhao, Atmos. Chem. Phys., 2010, 10, 9551–9561 CrossRef CAS.
- A. K. Y. Lee, P. Herckes, W. R. Leaitch, A. M. MacDonald and J. P. D. Abbatt, Geophys. Res. Lett., 2011, 38, L11805 Search PubMed.
- D. D. Huang, Q. Zhang, H. H. Y. Cheung, L. Yu, S. Zhou, C. Anastasio, J. D. Smith and C. K. Chan, Environ. Sci. Technol., 2018, 52, 9215–9224 CrossRef CAS PubMed.
- R. Zhang, M. Gen, T.-M. Fu and C. K. Chan, Environ. Sci. Technol., 2021, 55, 5711–5720 CrossRef CAS PubMed.
- B. R. G. Mabato, M. Gen, Y. Chu and C. K. Chan, ACS Earth Space Chem., 2019, 3, 150–157 CrossRef CAS.
- M. Gen, D. D. Huang and C. K. Chan, Environ. Sci. Technol., 2018, 52, 6903–6911 CrossRef CAS PubMed.
- R. Zhang, M. Gen, Z. Liang, Y. J. Li and C. K. Chan, Environ. Sci. Technol., 2022 DOI:10.1021/acs.est.1c07211.
- M. Z. Jacobson, J. Geophys. Res. Atmos., 1999, 104, 3527–3542 CrossRef CAS.
- B. R. G. Mabato, Y. Lyu, Y. Ji, D. D. Huang, X. Li, T. Nah, C. H. Lam and C. K. Chan, Atmos. Chem. Phys. Discuss., 2021, 2021, 1–35 Search PubMed.
- J. Yang, W. C. Au, H. Law, C. H. Lam and T. Nah, Atmos. Environ., 2021, 254, 118401 CrossRef CAS.
- A. Kahnt, S. Behrouzi, R. Vermeylen, M. Safi Shalamzari, J. Vercauteren, E. Roekens, M. Claeys and W. Maenhaut, Atmos. Environ., 2013, 81, 561–568 CrossRef CAS.
- C. Mohr, F. D. Lopez-Hilfiker, P. Zotter, A. S. H. Prévoît, L. Xu, N. L. Ng, S. C. Herndon, L. R. Williams, J. P. Franklin, M. S. Zahniser, D. R. Worsnop, W. B. Knighton, A. C. Aiken, K. J. Gorkowski, M. K. Dubey, J. D. Allan and J. A. Thornton, Environ. Sci. Technol., 2013, 47, 6316–6324 CrossRef CAS PubMed.
- M. Teich, D. Van Pinxteren, M. Wang, S. Kecorius, Z. Wang, T. Müller, G. Močnik and H. Herrmann, Atmos. Chem. Phys., 2017, 17, 1653–1672 CrossRef CAS.
- F. Li, S. Tang, N. T. Tsona and L. Du, Atmos. Environ., 2020, 237, 117650 CrossRef CAS.
- Z. Kitanovski, A. Čusak, I. Grgić and M. Claeys, Atmos. Meas. Tech., 2014, 7, 2457–2470 CrossRef.
- A. L. Klodt, D. E. Romonosky, P. Lin, J. Laskin, A. Laskin and S. A. Nizkorodov, ACS Earth Space Chem., 2019, 3, 2736–2746 CrossRef CAS.
- H. Pang, Q. Zhang, X. Lu, K. Li, H. Chen, J. Chen, X. Yang, Y. Ma, J. Ma and C. Huang, Environ. Sci. Technol., 2019, 53, 14253–14263 CrossRef CAS PubMed.
- G. Loisel, M. Mekic, S. Liu, W. Song, B. Jiang, Y. Wang, H. Deng and S. Gligorovski, Atmos. Environ., 2021, 246, 118140 CrossRef CAS.
- W. Walcher, T. Franze, M. G. Weller, U. Pöschl and C. G. Huber, J. Proteome Res., 2003, 2, 534–542 CrossRef CAS PubMed.
- Y. K. Gruijthuijsen, I. Grieshuber, A. Stöcklinger, U. Tischler, T. Fehrenbach, M. G. Weller, L. Vogel, S. Vieths, U. Pöschl and A. Duschl, Int. Arch. Allergy Immunol., 2006, 141, 265–275 CrossRef CAS PubMed.
- Y. Zhang, H. Yang and U. Pöschl, Anal. Bioanal. Chem., 2011, 399, 459–471 CrossRef CAS PubMed.
- M. Shiraiwa, K. Selzle and U. Pöschl, Free Radic. Res., 2012, 46, 927–939 CrossRef CAS PubMed.
- M. Shiraiwa, K. Selzle, H. Yang, Y. Sosedova, M. Ammann and U. Pöschl, Environ. Sci. Technol., 2012, 46, 6672–6680 CrossRef CAS PubMed.
- A. Ghiani, M. Bruschi, S. Citterio, E. Bolzacchini, L. Ferrero, G. Sangiorgi, R. Asero and M. G. Perrone, Sci. Total Environ., 2016, 573, 1589–1597 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. J. Harrison, R. I. Olariu and C. Arsenec, Chem. Soc. Rev., 2006, 35, 441–453 CAS.
- R. Niessen, D. Lenoir and P. Boule, Chemosphere, 1988, 17, 1977–1984 CrossRef CAS.
- D. Guillaume, J. Morvan and G. Martin, Environ. Technol. Lett., 1989, 10, 491–500 CrossRef CAS.
- F. Machado and P. Boule, J. Photochem. Photobiol., A, 1995, 86, 73–80 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, M. Lucchiari and E. Pelizzetti, Chemosphere, 2004, 56, 1049–1059 CrossRef CAS PubMed.
- P. Lin, J. Liu, J. E. Shilling, S. M. Kathmann, J. Laskin and A. Laskin, Phys. Chem. Chem. Phys., 2015, 17, 23312–23325 RSC.
- Z. Finewax, J. A. De Gouw and P. J. Ziemann, Environ. Sci. Technol., 2018, 52, 1981–1989 CrossRef CAS PubMed.
- J. L. Chang and J. E. Thompson, Atmos. Environ., 2010, 44, 541–551 CrossRef CAS.
- J. D. Smith, H. Kinney and C. Anastasio, Atmos. Environ., 2016, 126, 36–44 CrossRef CAS.
- P. Lin, N. Bluvshtein, Y. Rudich, S. A. Nizkorodov, J. Laskin and A. Laskin, Environ. Sci. Technol., 2017, 51, 11561–11570 CrossRef CAS PubMed.
- M. Xie, X. Chen, M. D. Hays and A. L. Holder, Atmos. Chem. Phys., 2019, 19, 2899–2915 CrossRef CAS PubMed.
- K. Vidović, A. Kroflič, M. Šala and I. Grgić, Atmosphere, 2020, 11, 131 CrossRef.
- S. Morville, A. Scheyer, P. Mirabel and M. Millet, J. Environ. Monit., 2004, 6, 963–966 RSC.
- Y. Iinuma, O. Böge and H. Herrmann, Environ. Sci. Technol., 2010, 44, 8453–8459 CrossRef CAS PubMed.
- A. Roger, S. M. Aschmann and A. Janet, Environ. Sci. Technol., 1992, 26, 1397–1403 CrossRef.
- M. A. J. Harrison, S. Barra, D. Borghesi, D. Vione, C. Arsene and R. Iulian Olariu, Atmos. Environ., 2005, 39, 231–248 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, M. Vincenti and E. Pelizzetti, Chemosphere, 2001, 44, 237–248 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero and E. Pelizzetti, Chemosphere, 2001, 45, 893–902 CrossRef CAS PubMed.
- R. G. Coombes, A. W. Diggle and S. P. Kempsell, Tetrahedron Lett., 1994, 35, 6373–6376 CrossRef CAS.
- J. Dzengel, J. Theurich and D. W. Bahnemann, Environ. Sci. Technol., 1999, 33, 294–300 CrossRef CAS.
- J. J. Cole, E.-C. Lin, C. R. Barry and H. O. Jacobs, Appl. Phys. Lett., 2009, 95, 113101 CrossRef.
- P. Warneck and C. Wurzinger, J. Phys. Chem., 1988, 92, 6278–6283 CrossRef CAS.
- M. Fischer and P. Warneck, J. Phys. Chem., 1996, 100, 18749–18756 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero and E. Pelizzetti, Environ. Sci. Technol., 2002, 36, 669–676 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero, D. Borghesi, M. Lucchiari and E. Pelizzetti, Environ. Sci. Technol., 2003, 37, 4635–4641 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero and E. Pelizzetti, Environ. Sci. Technol., 2005, 39, 1101–1110 CrossRef CAS PubMed.
- D. Borghesi, D. Vione, V. Maurino and C. Minero, J. Atmos. Chem., 2005, 52, 259–281 CrossRef CAS.
- R. Arimoto, W. Balsam and C. Schloesslin, Atmos. Environ., 2002, 36, 89–96 CrossRef CAS.
- C. Minero, F. Bono, F. Rubertelli, D. Pavino, V. Maurino, E. Pelizzetti and D. Vione, Chemosphere, 2007, 66, 650–656 CrossRef CAS PubMed.
- Y. Wang, M. Mekic, P. Li, H. Deng, S. Liu, B. Jiang, B. Jin, D. Vione and S. Gligorovski, Environ. Sci. Technol., 2021, 55, 4553–4564 CrossRef CAS PubMed.
- C. Reed, M. J. Evans, L. R. Crilley, W. J. Bloss, T. Sherwen, K. A. Read, J. D. Lee and L. J. Carpenter, Atmos. Chem. Phys., 2017, 17, 4081–4092 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |