
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron-based non-fullerene small molecule acceptors via nitrogen substitution: a theoretical study†
Jie
Yang
,
Xun
Wu
,
Quan-Song
Li
 * and
Ze-Sheng
Li
*
* and
Ze-Sheng
Li
*
Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, 100081 Beijing, China. E-mail: liquansong@bit.edu.cn; zeshengli@bit.edu.cn
First published on 21st February 2022
Abstract
Acceptors play vital roles in absorbing sunlight and consequently producing charge in organic solar cells (OSCs). Boron-based non-fullerene acceptors have received particular attention due to their tunable electronic structure and high optoelectronic performance. Herein, a set of boron-based acceptors (M-BNP4P-1 and its nine derivatives) with double B ← N bridged bipyridine unit were theoretically studied by density functional theory (DFT) and time-dependent density functional theory (TD-DFT). The calculation results showed that nitrogen substitutions could precisely tune the energy levels of the frontier molecular orbitals in a wide range. The designed acceptors A1a, A1b, A2a, and A3a exhibit enhanced electron mobility of up to two orders of magnitude (about a few tenths of cm2 V−1 S−1) compared to the parent molecule M-BNBP4P-1 (A0). When nitrogen is substituted at the para position of the nitrogen atom of the pyridine ring of A0, the resulting acceptor A2d exhibits the maximum absorption wavelength of 727 nm with a considerably large oscillator strength. Moreover, the interfacial properties of PTB7-Th/A0 and PTB7-Th/A2d composed of A0 and A2d paired with the donor PTB7-Th have been systematically studied. Importantly, PTB7-Th/A2d exhibits charge transfer states as high as 67%, facilitating charge transfer via a direct excitation mechanism. Furthermore, the hot exciton mechanism and intermolecular electric field mechanism are more favourable in PTB7-Th/A2d. Our results provide a new design strategy to tune the optoelectronic properties of organic semiconductors and a valuable skeleton for the design of new and high-performance organoboron small molecular acceptors.
Introduction
Organic solar cells (OSCs) have received great attention due to their wide applications in energy conversion and storage. Since 1995, fullerenes and fullerene derivatives as electron acceptors in OSCs have been widely investigated due to their advantages of light weight, low cost, and high electron mobility.1–3 However, fullerene-based acceptors possess considerable limitations, such as weak visible light absorption, poor tuning of the energy levels, and unfavourable photo-stability in air.4 Meanwhile, non-fullerene acceptors (NFAs) have the merits of synthetic versatility, excellent electron-accepting properties, high absorption coefficients, facile synthesis, and simplified purification.5–7 As a result, enormous efforts have been devoted to the design of novel NFAs and their application in OSCs.8–13Recently, Liu and co-workers have performed a multitude of studies on boron-based acceptors for OSCs.14–24 For instance, Liu et al. synthesized a boron-based NFA P-BNBP-fBT with good crystallinity and high electron mobility. OSCs based on the donor PTB7-Th and acceptor P-BNBP-fBT blend showed a power conversion efficiency (PCE) as high as 6.26% at a remarkably low energy loss of 0.51 eV.25 Next, they reported the boron-based molecule PD-4F and a PCE of 6.45%.26 Among these, they developed a boron-based small molecule acceptor M-BNBP4P-1 with two unique strong absorption bands in the UV-vis region. The OSCs based on the PTB7-Th/M-BNBP4P-1 blend exhibited a PCE of 7.06% due to the wide photo-response of the acceptor.27 The boron–nitrogen coordination bond units can give rise to a significant change of the electronic structure, and a large downshift of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) energy levels,28–34 which have been successfully applied in high-performance OSCs.27,35–39 However, the development of n-type boron-based small molecular acceptors lags far behind that of their p-type polymer counterparts in material diversity and device performance.25,40–45
Doping or modification with heteroatoms is an attractive approach to enhance the acceptor character of conjugated organic materials.46 The nitrogen atom provides excess electrons due to its five valence electrons and an atomic size comparable to that of carbon, which results in a noticeable change in the optical properties and a significant improvement in electrical conductivity.47 The lone electron pair character of the nitrogen substituent significantly impacts intramolecular charge transfer and the π–π interactions.48 Nitrogen substitution has emerged as a powerful strategy to tune the properties and design materials.49–53 In 2011, Hashimoto and co-workers reported PDI-based acceptors containing fluorene (PF-PDI), carbazole (PC-PDI), and dibenzosilole (PDBS-PDI) to fine-tune the absorption and energy levels, where nitrogen-containing PC-PDI exhibited a better PCE of 2.23% as a blend with donor PT1.54 Huang and co-workers synthesized acceptor IDT-4CN, and got a PCE of 8.13% owing to multiple noncovalent conformational locks of N···S and C–H···N by introducing nitrogen atoms.55 In addition, Huang et al. found that OSCs based on acceptor IDTzCR, which has a better planar structure due to conformation locked IDT and thiazole units via S…N bonding, exhibited a higher PCE of 8.71% compared to those based on the inferior acceptor IDTCR (6.10%).56
In this work, we designed nine boron-based non-fullerene small acceptors (A1a, A1b, A2a, A2b, A2c, A2d, A3a, a3b, and A4) via nitrogen substitutions on M-BNBP4P-1 (A0). As shown in Fig. 1, the numbers (1, 2, 3, and 4) in the names of the newly designed acceptors indicate the number of added nitrogen atoms on A0, while the letters (a, b, c, and d) are used to discriminate the isomers. Our calculations showed that the newly designed acceptors possess different frontier molecular orbitals (FMOs), display distinct interfacial properties, and exhibit various types of optoelectronic behaviour. Notably, A2d shows the largest red-shifted absorbance. In the case of pairing with donor PTB7-Th, the constructed D/A interface PTB7-Th/A2d shows more robust charge transfer characteristics under direct excitation, hot exciton, and intermolecular electric field mechanisms. Our results not only provide a new strategy to tune the optoelectronic properties of organic semiconductors and a valuable skeleton for the design of new and better acceptors, but also demonstrate a deep insight into the effect of nitrogen substitution on boron-based non-fullerene small molecule acceptors.
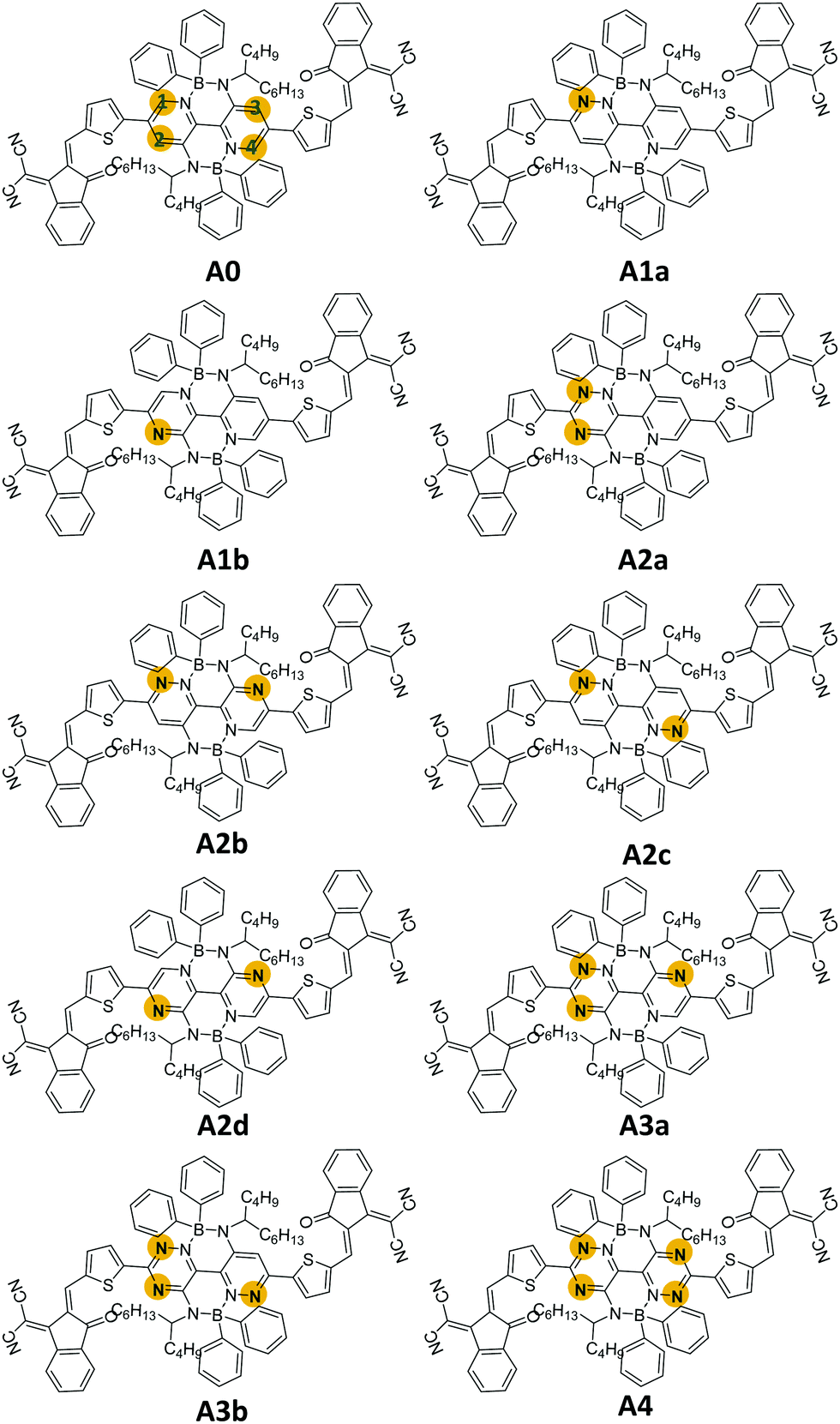 | ||
| Fig. 1 Molecular structures of the parent molecule M-BNBP4P-1 (A0) and newly designed acceptors. | ||
Computational methods
The geometries of the investigated molecules were relaxed to their potential energy minima without imaginary frequency using the DFT-D method at the B3LYP/6-31G(d,p) level.57,58 The TD-DFT method at the PBE38/6-31G(d,p) level was used to evaluate the properties related to excited states, which is in good agreement with the experimental value (calculated value 700 nm vs. experimental value 698 nm for A0).27 The empirical D3 dispersion corrections59 were included using the Becke–Johnson60 damping potential in DFT and TD-DFT calculations to describe the weak interactions accurately. The polarizable continuum model (PCM)61 was employed to consider the solvation effect in chlorobenzene.27 The above calculations were performed using the Gaussian 16 code.62 The crystal structures of the studied acceptors were predicted using the Materials Studio software,63 employing the Forcite module with the setting in fine. Both optical properties and electrostatic potentials (ESP) were analysed using Multiwfn software.64The electron transport behaviour in our work was described by using Marcus theory.65,66 The electron hopping rate (k) is expressed as67,68
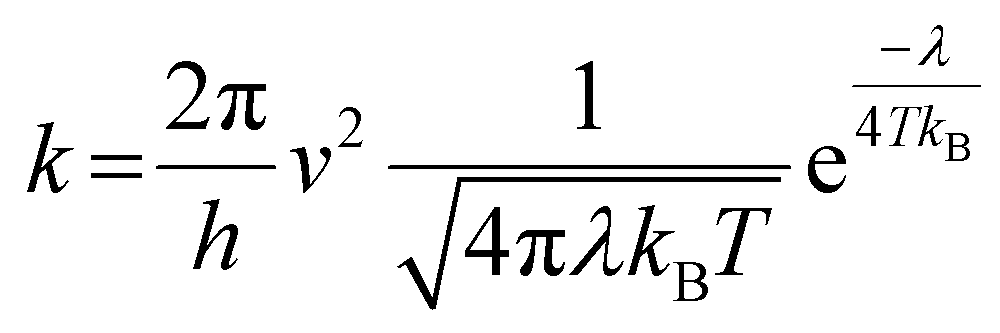
where
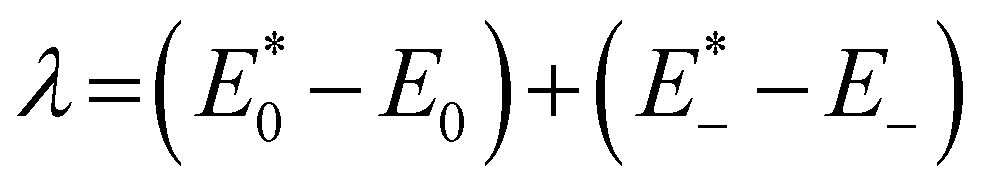
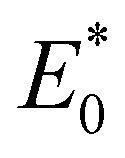 and E0 are the energies of the neutral species in the anionic and neutral geometries, respectively.
and E0 are the energies of the neutral species in the anionic and neutral geometries, respectively. 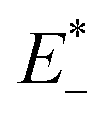 and E− represent the energies of the anionic species with the geometries of neutral and anionic molecules, respectively.
and E− represent the energies of the anionic species with the geometries of neutral and anionic molecules, respectively.
The transfer integral (v) was obtained by adopting a direct approach at the M06-2X/6-31G(d,p) level.70 In our work, v was expressed as71
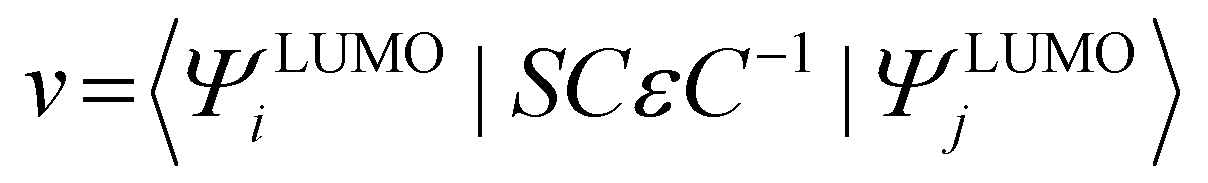
The electron mobility (μe) of the investigated molecules were calculated by the Einstein relation71,72
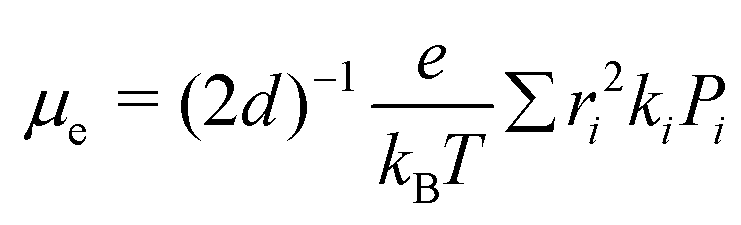
The hopping probability Pi is defined as
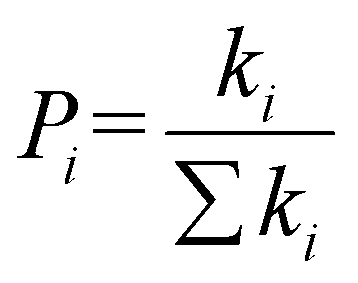
The net transferred electrons (Δq) from the donor (D) to the acceptor (A) were computed according to the following formula:
| Δq = QD,A − QA,D |
where wai and
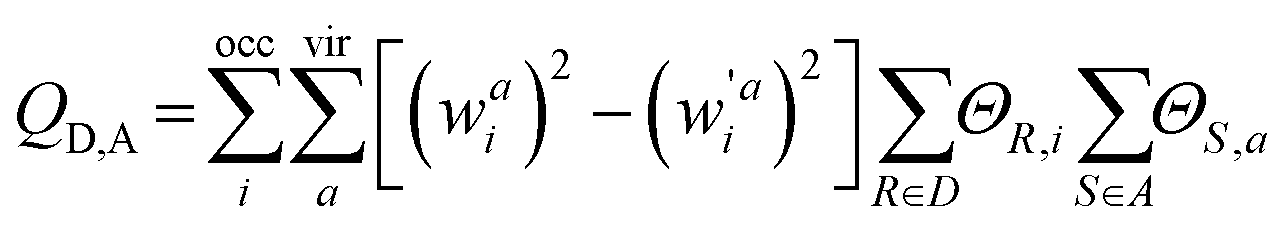
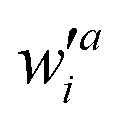 are the configuration coefficients of the excitation molecular orbital i to a and de-excitation molecular orbital a to i, respectively; ΘR,i (ΘS,a) is the contribution of atom R(S) to the molecular orbital i(a).
are the configuration coefficients of the excitation molecular orbital i to a and de-excitation molecular orbital a to i, respectively; ΘR,i (ΘS,a) is the contribution of atom R(S) to the molecular orbital i(a).
The D index is defined as the distance from the hole centroid to the electron centroid, which can be obtained from the following formula:64,73
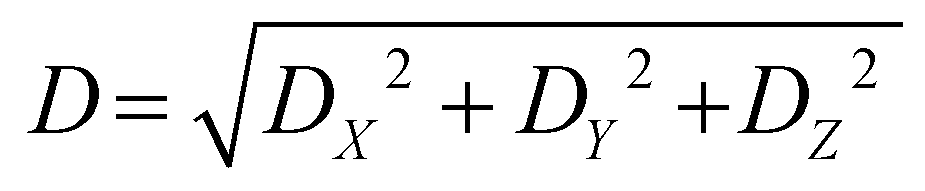
The charge transfer (CT) length in X/Y/Z can be measured by the centroid distances between the hole and the electron in the corresponding directions:
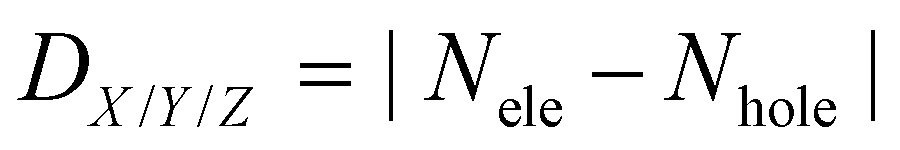
The electron centroid (Nele) or the hole centroid (Nhole) was computed as follows:
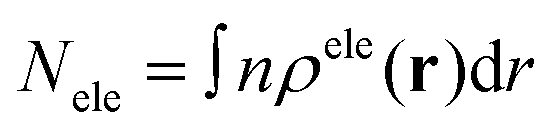
where n is the X (Y or Z) component of position vector r. ρele and ρhole present the spatial charge distribution.
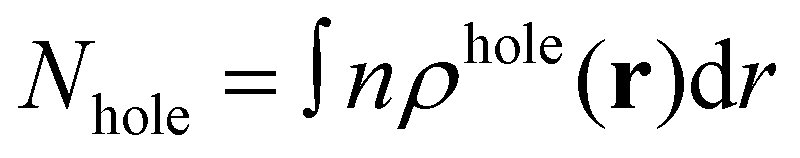
Results and discussion
Molecular structure and electronic properties
It is well known that material properties have some clues in the molecular structures. In this context, the structures of the acceptors are the precondition and foundation of their physicochemical properties. The key bond length and dihedral angle values are shown in Fig. 2 and Table S1 (ESI†). Obviously, the C–N bond lengths of the substituted nitrogen decrease by about 0.05 Å compared to the corresponding C–C bond lengths in unsubstituted ones because of the smaller radius (0.75 Å of N vs. 0.77 Å of C) and the lone-pair electron of the nitrogen atom. In contrast, the other bond lengths remain almost unchanged. The structural characteristics are also reflected in the Mulliken atomic charge distribution diagram in Fig. S1 (ESI†). We can see the electronegative nitrogen atoms in the newly designed acceptors pull the partial charges from the carbon atoms. Hence, the carbon atoms become more positive and the charge differences between C and N become more prominent, which is conducive to the charge flow on the skeleton. Another point that needs to be mentioned is about the NCCC dihedral angle (α) in the B-containing six-membered ring. As can be seen in Table S1 (ESI†), the α values in the acceptors (A0, A2b, A2c, A2d, and A4) with an equal number of nitrogen atoms on the left and right sides of the core are about 11°–12°, which are slightly fewer than those (13°–15°) in other acceptors (A1a, A1b, A2a, A3a, and A3b). Moreover, the dipole moments (see Fig. S2, ESI†) of central symmetric molecules, such as A0 (2.23 Debye), A2c (1.86 Debye), A2d (1.49 Debye), and A4 (1.01 Debye) are relatively smaller than those of other asymmetric ones (3.72, 5.71, 5.70, 4.68, and 4.55 Debye for A1a, A1b, A2a, A2b, A3a, and A3b, respectively).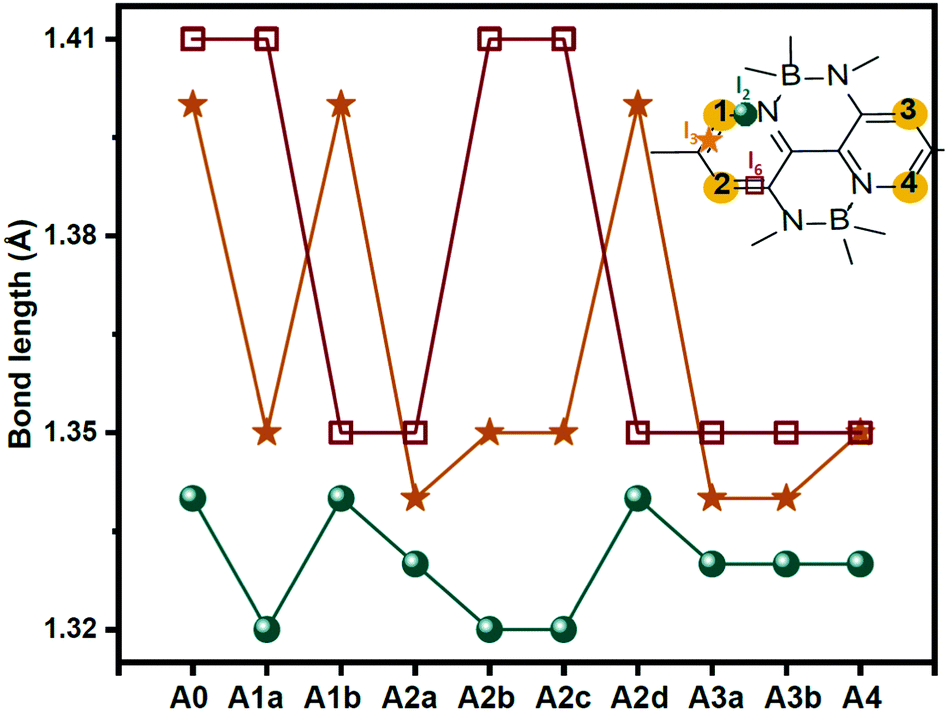 | ||
| Fig. 2 Key bond lengths of the studied molecules. | ||
Exciton binding energy and frontier molecular orbitals
Exciton generation and following diffusion are key factors affecting the PCE of OSCs.74 Coulombic attraction has to be overcome to guarantee effective charge separation, which is crucial for electron injection into the semiconductor.75 As a measure of Coulombic attraction, the exciton binding energy (Eb) can be expressed as the difference between the electric and optical band gaps. In this work, Eb is defined by the equation76,77| Eb = VIP − VEA − E1 |
| VIP = E0+ − E0 |
| VEA = E0 − E0− |
The LUMO offset between the donor and the acceptor is considered to be the energetic driving force for charge separation.78 The HOMO offsets are related to hole transfer (from acceptor to donor) and charge recombination.79,80 The difference between the energy level of the LUMO (ELUMO) of the acceptor and the energy level of the HOMO (EHOMO) of the donor is associated with the open-circuit voltage (Voc) of OSCs.81–85 Accordingly, tuning the ELUMO/EHOMO is very important. Fig. 3 displays the ELUMO and the EHOMO of the studied acceptors. What is striking about the data is that ELUMO/EHOMO can be precisely tuned in a wide range by substitution of nitrogen:
| ELUMO ≈ −3.43 − 0.05n − 0.10m (eV) |
| EHOMO ≈ −5.23 − 0.32n − 0.11m(eV) |
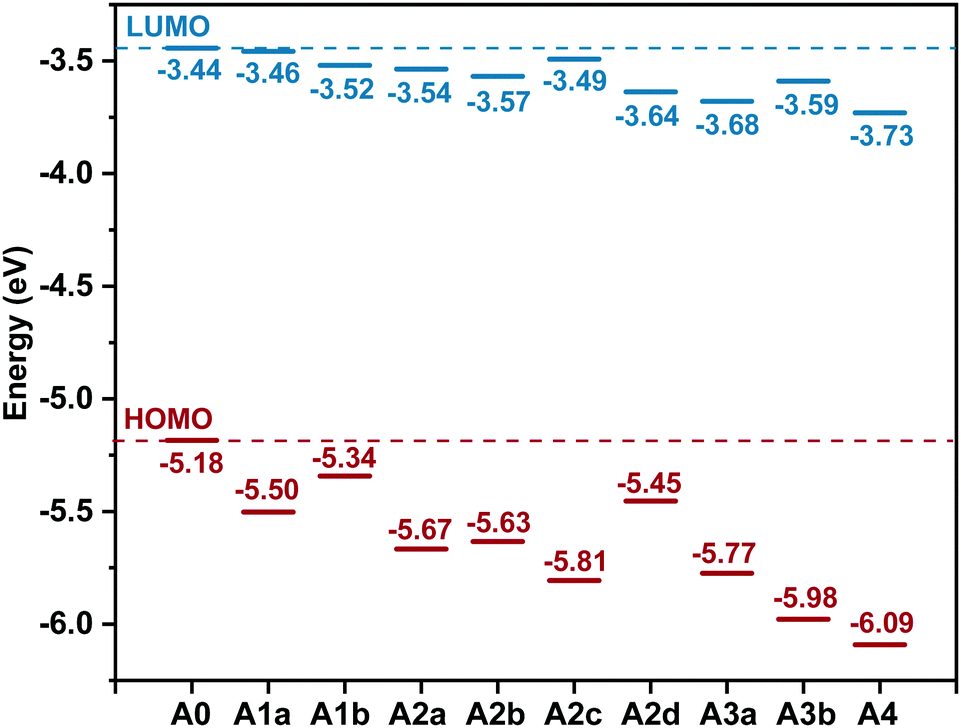 | ||
| Fig. 3 Energy levels of the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO) for the studied molecules. | ||
Optical properties
The optical characteristics of acceptors are of utmost importance because capturing sunlight and generating excitons are the first steps of photoelectric conversion.87,88 The absorption spectra of the studied boron-based acceptors were simulated by the TD-PBE38 method and are shown in Fig. 4. To verify the importance of the boron atoms for this type of acceptor, we also computed the absorption spectrum of A0–C, which was derived from A0 by changing the two boron atoms to carbon atoms. As shown in Fig. S4 (ESI†), the absorption spectrum of A0 is characterized by a wide absorption range with two peaks at around 700 nm and 457 nm, which covers most of the strong sunlight-irradiation region. This is in good agreement with previous experimental observation.27 Meanwhile, the absorption spectrum of A0–C mainly lies in the weak sunlight-irradiation region with a wide peak centred at about 1100 nm. Moreover, the EHOMO of A0–C is −3.76 eV, which is much higher than those (−4 to −5 eV) of most widely used donors,89 so the hole transfer channel will be blocked if A0–C is used as an acceptor for OSCs. Hence, the boron atoms in A0 are important and necessary in terms sunlight absorption and energy levels.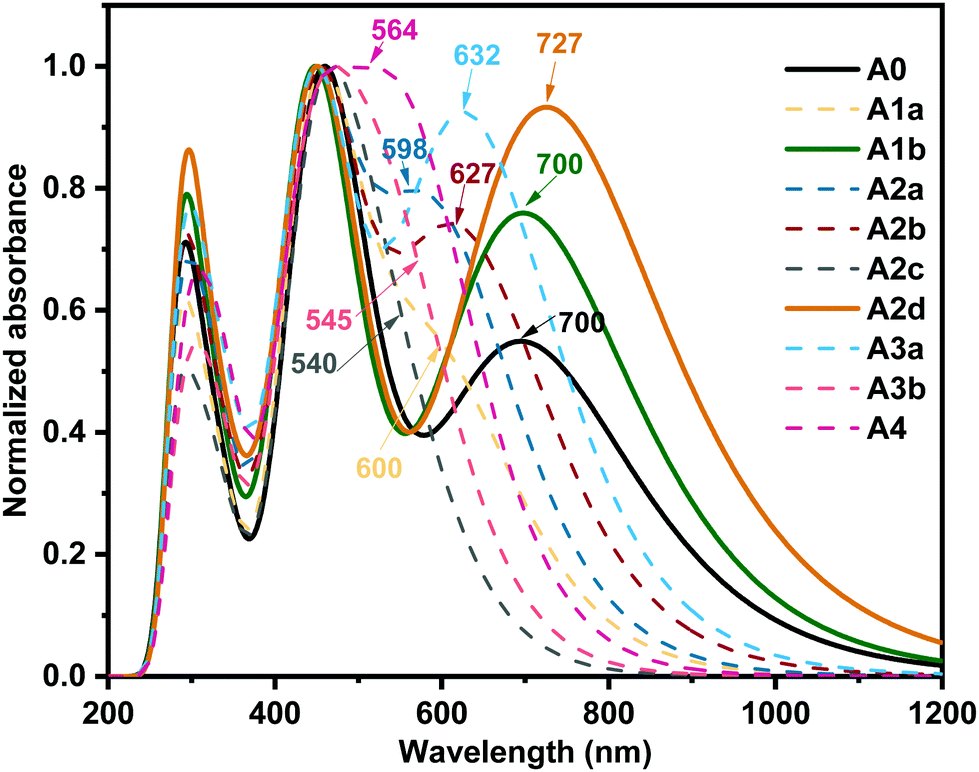 | ||
| Fig. 4 Calculated absorption spectra of the studied acceptors at the PBE38/6-31G(d,p) level (the values indicate the maximum absorption wavelength in nm). | ||
In Fig. 4, we can see the acceptors A1b and A2d exhibit unique wide absorption spectra with two strong absorption bands in the UV-vis region, indicating superior sunlight collection capacity as observed for A0 in the experiments.27 Moreover, compared with A0, A1b and A2d exhibit larger absorption coefficients in favour of a higher short-circuit current density (Jsc).90 In particular, A2d exhibits an evidently enhanced absorption from 600 nm to 1000 nm enabling a broad photon harvest to a high Jsc.73 The largest maximum absorption wavelength (727 nm) of A2d could be a result of the unique quinoid-enhancing character, which enhances the π-effect and results in a large bathochromic shift.91–95 On the whole, the absorption spectra of the studied boron-based acceptors can be well adjusted in a wide range (about 200 nm) by nitrogen substitution on the core, which complements the absorption of more kinds of donors to form high-performance OSCs.96,97
Electron mobility
Electron mobility (μe) is an essential indicator of the acceptors in OSCs.98 The calculated μe values and related parameters based on the Einstein equation71,72 and Marcus theory65,66 for the studied acceptors are shown in Table 1 and Fig. S5 (ESI†). In Table 1, we can see that the computed μe value of A0 is 2.45 × 10−3 cm2 V−1 s−1. For the newly designed acceptors, the μe values of A2b, A2c and A3b are of the same order of magnitude (10−3) as that of A0, while the μe values of other acceptors are one or two orders of magnitude larger (10−2–10−1). According to the formula of Marcus theory on the charge-transfer rate, the two key parameters governing the charge-transport rate and then the electron mobility are the reorganization energy (λ) and the transfer integral (v). As shown in Table 1, the λ values of the newly designed acceptors are in the range of 0.16–0.22 eV, which are either the same as or 0.03–0.06 eV larger than that (0.16 eV) of A0, while the vmax2 values have increased by up to two orders of magnitude after nitrogen substitutions. It is clear that the enhanced electron mobility of the newly designed acceptors mainly arise from the increased transfer integral, which strongly depends on the relative position of the interacting molecules.99| Molecules | λ (eV) | v max 2 (eV2) | μ e (cm2 V−1 s−1) |
|---|---|---|---|
| A0 | 0.16 | 1.51 × 10−6 | 2.45 × 10−3 |
| A1a | 0.16 | 1.17 × 10−4 | 2.02 × 10−1 |
| A1b | 0.19 | 9.98 × 10−5 | 1.64 × 10−1 |
| A2a | 0.20 | 7.31 × 10−4 | 8.41 × 10−1 |
| A2b | 0.20 | 2.14 × 10−6 | 2.36 × 10−3 |
| A2c | 0.16 | 3.81 × 10−6 | 4.94 × 10−3 |
| A2d | 0.21 | 2.93 × 10−5 | 3.47 × 10−2 |
| A3a | 0.21 | 2.73 × 10−4 | 3.45 × 10−1 |
| A3b | 0.21 | 3.46 × 10−5 | 9.15 × 10−3 |
| A4 | 0.22 | 1.33 × 10−4 | 3.20 × 10−2 |
In order to get more insights into the noncovalent interactions of the acceptors, the independent density gradient model (IGM)100 analysis was carried out on the most favoured hopping pathway of the studied acceptors. As shown in Fig. S6 (ESI†), the noncovalent interactions of A1a, A1b, A2a and A3a are strong and mainly distributed in the end groups of the interacting molecules. The large overlapping degree on molecular backbones, face-on stacking, and small distances (3.42–3.54 Å) between the two interacting molecules of A1a, A1b, A2a, and A3a, resulting in relatively large mobility μe of up to a few tenths of cm2 V−1 s−1.
Donor/acceptor interfacial properties
The donor/acceptor (D/A) interface is critical for the exciton dissociation, which to some extent determines the PCE of OSCs.101,102 The optimized geometries of D/A interfaces paired with the donor PTB7-Th are shown in Fig. S7 (ESI†). The centroid-to-centroid distances, the N⋯S distances, and intermolecular interactions of the studied interfaces are similar, indicating that A0 and its N-substitution counterparts have strong intermolecular stacking paired with PTB7-Th. Light-harvesting is a pivotal factor affecting the PCE of OSCs by dominating Jsc.90 As shown in Fig. S8, PTB7-Th/A1b and PTB7-Th/A2d exhibit significant red shifts because A1b and A2d have strong absorption in the UV-vis region.Charge transfer (CT) mechanisms play vital roles in determining the performance of bulk heterojunction OSCs and layer-by-layer OSCs.101–103 To better understand the CT mechanisms, the vertical excitation properties were investigated for the D/A interfaces of PTB7-Th/A0 and PTB7-Th/A2d. As shown in Fig. S9 and Table S3 (ESI†), the studied sixty lowest singlet excited states mainly lie in the visible light region (1.44–3.51 eV). The excited states in OSCs can be classified into two types, the Frenkel excitons (electron excitation localized on the donor or the acceptor) and CT states (the two singly occupied molecular orbitals separately locate on the donor and the acceptor).104 Importantly, among the first 60 low-energy excited states, there are 40 CT states, accounting for 67% in PTB7-Th/A2d, which is much more than that of 50% in PTB7-Th/A0. This suggests easier charge separation in PTB7-Th/A2d given that CT states are the precursors of the charge separation process.105
Fig. 5 displays the excitation energies and related electron transfer parameters for the six lowest excited states (S1–S6), since the low-lying excited states are essentially important in photochemical and photophysical processes.106 We can see that there are five CT states and one FE state in the S1–S6 states for both studied interfaces. Lying 1.71 eV above the ground state minimum, the S1 state of PTB7-Th/A0 is a charge transfer state (CT1) with a transferred charge of 0.78 |e| along an electron–hole distance of 2.37 Å and an oscillator strength of 0.04. As for the PTB7-Th/A2d interface, the S1 state is much lower in energy (1.44 eV) and also a charge transfer state (CT1), where the transferred charge is 0.89 |e| along a larger electron–hole distance of 2.88 Å with an enhanced oscillator strength of 0.05. Regarding the charge transfer mechanisms, the oscillators strengths (0.05, 0.82, 0.17, 0.05, and 0.09) of CT1–CT4 states for PTB7-Th/A2d are larger than those (0.04, 0.74, 0.06, 0.01, and 0.02) for PTB7-Th/A0, indicating a larger possibility of charge transfer via direct excitation mechanism. In addition, the energy gap between the FE1 state and its below CT state is 0.15 eV and 0.09 eV for PTB7-Th/A0 and PTB7-Th/A2d, implying that charge transfer via the hot exciton mechanism107 is more favourable for the latter interface with a relatively smaller FE-CT energy gap. As for the intermolecular electric field (IEF) mechanism, in which a lower energy FE state is converted to a CT state under the effect of the intermolecular electric field formed by the different electrostatic potentials (ESP) of the donor and the acceptor surfaces,108 the average ESP of A0 and A2d is equivalent (3 kcal mol−1, see Fig. S10, ESI†) while the energy gap between the FE1 state and its above CT state is 0.09 eV and 0.08 eV for PTB7-Th/A0 and PTB7-Th/A2d, slightly in favour of the latter interface.
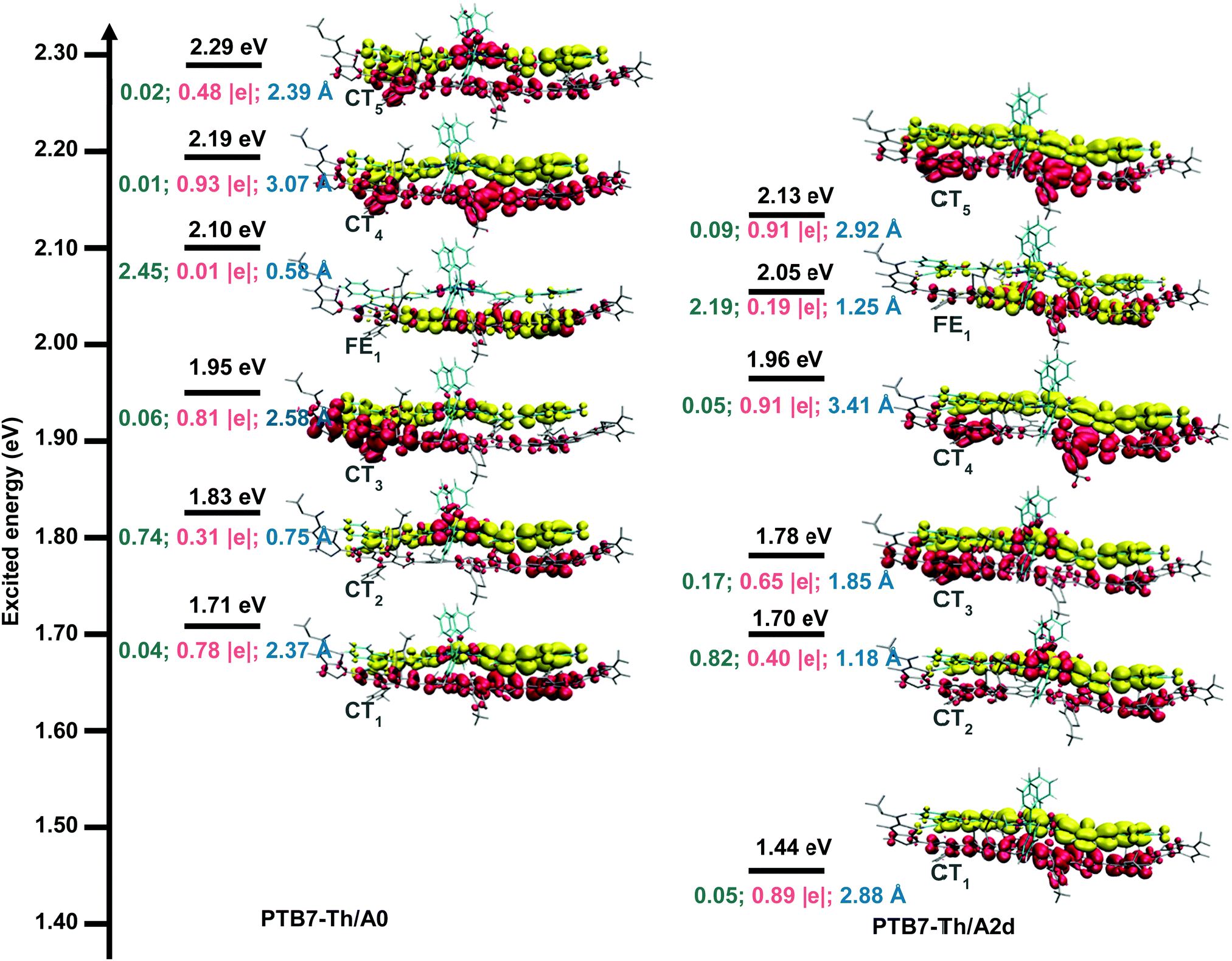 | ||
| Fig. 5 Excitation energies (black lines), oscillator strengths (green fonts), D index (blue fonts), charge density difference (CDD: the red region stands for the electron depletion zone, and the yellow region for electron accumulation) maps, and transferred charge (|Δq|, pink fonts) from the donor PTB7-Th to the acceptor of the six lowest excited states for the PTB7-Th/A0 and PTB7-Th/A2d interfaces. | ||
In short, multiple CT mechanisms including the direct excitation mechanism, the hot exciton mechanism, and the IEF mechanism, are feasible for the PTB7-Th/A0 and PTB7-Th/A2d interfaces. The three mechanisms are more favoured in the PTB7-Th/A2d system than in the PTB7-Th/A0 one.
Conclusions
In this work, we designed a series of boron-based non-fullerene small acceptors for OSCs based on DFT and TD-DFT calculations. Compared with the experimentally synthesized parent molecule A0, the newly designed acceptors exhibit improved photovoltaic properties, including a wider and red-shifted absorption spectrum, and larger electron mobility. Analysis of the D/A interfacial properties shows that there are up to 68% CT states in the first 60 excited states of the PTB7-Th/A2d interface, while the CT state proportion is 50% for the PTB7-Th/A0 interface. Furthermore, the three widely accepted CT mechanisms (direct excitation, hot exciton, and IEF mechanisms) were found to be more favoured in PTB7-Th/A2d than PTB7-Th/A0. Our results not only provide a set of promising boron-containing acceptors for OSCs, but also demonstrate that nitrogen substitution is an effective strategy to tune the optoelectronic properties of organic semiconductors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (22173008), and the Beijing Key Laboratory for Chemical Power Source and Green Catalysis (2013CX02031). In addition, we thank Professor Li-Jie Li for the code to calculate crystal structures.References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS
.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC
- B. C. Thompson and J. M. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed
- I. Fraga Domínguez, A. Distler and L. Lüer, Adv. Energy Mater., 2017, 7, 1601320 CrossRef
- A. A. F. Eftaiha, J. P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201–1213 RSC
- H. Sun, X. Song, J. Xie, P. Sun, P. Gu, C. Liu, F. Chen, Q. Zhang, Z. K. Chen and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 29924 CrossRef CAS PubMed
- J. D. Chen, T. Y. Jin, Y. Q. Li and J. X. Tang, Nanoscale, 2019, 11, 18517–18536 RSC
- D. Cai, J. Zhang, J. Y. Wang, Y. Ma, S. Wan, P. Wang, Z. Wei and Q. Zheng, J. Mater. Chem. A, 2020, 8, 24543–24552 RSC
- Z. Wang, H. Jiang, X. Liu, J. Liang, L. Zhang, L. Qing, Q. Wang, W. Zhang, Y. Cao and J. Chen, J. Mater. Chem. A, 2020, 8, 7765–7774 RSC
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee, T. Wang, H. Ade, T. Heumüller, C. Brabec and P. Meredith, Adv. Energy Mater., 2021, 11, 20003570 Search PubMed
- Y. C. Lin, C. H. Chen, N. Z. She, C. Y. Juan, B. Chang, M. H. Li, H. C. Wang, H. W. Cheng, A. Yabushita, Y. Yang and K. H. Wei, J. Mater. Chem. A, 2021, 9, 20510–20517 RSC
- Z. Wu, Y. Chen, L. Zhang, D. Yuan, R. Qiu, S. Deng, H. Liu, Z. Zhang and J. Chen, J. Mater. Chem. A, 2021, 9, 3314–3321 RSC
- J. Hai, S. Luo, H. Yu, H. Chen, Z. Lu, L. Li, Y. Zou and H. Yan, Mater. Adv., 2021, 2, 2132–2140 RSC
- J. Xu, B. Meng, J. Liu and L. Wang, Chem. Commun., 2020, 56, 5701 RSC
- J. Wang, Y. Gao, Y. Yu, R. Zhao, L. Zhang and J. Liu, Org. Electron., 2021, 92, 106134 CrossRef CAS
- Z. Ding, R. Zhao, Y. Yu and J. Liu, J. Mater. Chem. A, 2019, 7, 26533–26539 RSC
- R. Zhao, B. Lin, J. Feng, C. Dou, Z. Ding, W. Ma, J. Liu and L. Wang, Macromolecules, 2019, 52, 7081–7088 CrossRef CAS
- R. Y. Zhao, C. D. Dou, J. Liu and L. X. Wang, Chin. J. Polym. Sci., 2016, 35, 198–206 CrossRef
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed
- C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2017, 60, 450–459 CrossRef CAS
- L. Zhang, Z. Ding, R. Zhao, F. Jirui, W. Ma, J. Liu and L. Wang, J. Mater. Chem. C, 2020, 8, 5613–5619 RSC
- X. Long, Y. Gao, H. Tian, C. Dou, D. Yan, Y. Geng, J. Liu and L. Wang, Chem. Commun., 2017, 53, 1649–1652 RSC
- R. Zhao, Y. Min, C. Dou, J. Liu and L. Wang, Chem. – Eur. J., 2017, 23, 9486–9490 CrossRef CAS PubMed
- C. Dong, S. Deng, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 16184–16190 CrossRef CAS PubMed
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS PubMed
- N. Wang, S. Zhang, R. Zhao, J. Feng, Z. Ding, W. Ma, J. Hu and J. Liu, ACS Appl. Electron. Mater., 2020, 2, 2274–2281 CrossRef CAS
- F. Liu, Z. Ding, J. Liu and L. Wang, Chem. Commun., 2017, 53, 12213 RSC
- X. Shao, C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2019, 62, 1387–1392 CrossRef CAS
- Y. Min, C. Dou, H. Tian, J. Liu and L. Wang, Chem. Commun., 2019, 55, 3638–3641 RSC
- C. Dong, B. Meng, J. Liu and L. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 10428–10433 CrossRef CAS PubMed
- Y. Min, X. Cao, H. Tian, J. Liu and L. Wang, Chem. – Eur. J., 2021, 27, 2065–2071 CrossRef CAS PubMed
- Z. Zhang, Z. Ding, C. Dou, J. Liu and L. Wang, Polym. Chem., 2015, 6, 8029–8035 RSC
- J. Miao, H. Li, T. Wang, Y. Han, J. Liu and L. Wang, J. Mater. Chem. A, 2020, 8, 20998–21006 RSC
- T. Wang, C. Dou, J. Liu and L. Wang, Chem. – Eur. J., 2018, 24, 13043–13048 CrossRef CAS PubMed
- J. M. Farrell, C. Mutzel, D. Bialas, M. Rudolf, K. Menekse, A. M. Krause, M. Stolte and F. Wurthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed
- F. Liu, J. Liu and L. Wang, Org. Chem. Front., 2019, 6, 1996–2003 RSC
- J. Endres, I. Pelczer, B. P. Rand and A. Kahn, Chem. Mater., 2016, 28, 794–801 CrossRef CAS
- J. J. Chen, S. M. Conron, P. Erwin, M. Dimitriou, K. McAlahney and M. E. Thompson, ACS Appl. Mater. Interfaces, 2015, 7, 662 CrossRef CAS PubMed
- K. S. Schellhammer, T. Y. Li, O. Zeika, C. Körner, K. Leo, F. Ortmann and G. Cuniberti, Chem. Mater., 2017, 29, 5525–5536 CrossRef CAS
- R. Zhao, N. Wang, Y. Yu and J. Liu, Chem. Mater., 2020, 32, 1308–1314 CrossRef CAS
- Y. Li, H. Meng, T. Liu, Y. Xiao, Z. Tang, B. Pang, Y. Li, Y. Xiang, G. Zhang, X. Lu, G. Yu, H. Yan, C. Zhan, J. Huang and J. Yao, Adv. Mater., 2019, 31, e1904585 CrossRef PubMed
- R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557 CrossRef CAS PubMed
- X. Long, Z. Ding, C. Dou, J. Liu and L. Wang, Mater. Chem. Front., 2017, 1, 852–858 RSC
- X. Long, C. Dou, J. Liu and L. Wang, Chin. Chem. Lett., 2018, 29, 1343–1346 CrossRef CAS
- Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, Chem. Sci., 2016, 7, 6197–6202 RSC
- M. Stepien, E. Gonka, M. Zyla and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef CAS PubMed
- P. Ayala, R. Arenal, A. Loiseau, A. Rubio and T. Pichler, Rev. Mod. Phys., 2010, 82, 1843–1885 CrossRef CAS
- J. Wu, B. A. Wilson, D. W. Smith Jr and S. O. Nielsen, J. Mater. Chem. C, 2014, 2, 2591–2599 RSC
- J. Li and Q. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28049–28062 CrossRef CAS PubMed
- P. Y. Gu, Z. Wang, G. Liu, H. Yao, Z. Wang, Y. Li, J. Zhu, S. Li and Q. Zhang, Chem. Mater., 2017, 29, 4172–4175 CrossRef CAS
- R. Zhu, Q. S. Li and Z. S. Li, Nanoscale, 2018, 10, 17873–17883 RSC
- Z. Wang, Z. Wang, Y. Zhou, P. Gu, G. Liu, K. Zhao, L. Nie, Q. Zeng, J. Zhang, Y. Li, R. Ganguly, N. Aratani, L. Huang, Z. Liu, H. Yamada, W. Hu and Q. Zhang, J. Mater. Chem. C, 2018, 6, 3628–3633 RSC
- S. Li, C. Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 CrossRef CAS PubMed
- L. Yang, W. Gu, Y. Yang, L. Hong, X. Zhang, Y. Xiao, X. Wu, A. Peng and H. Huang, Small Methods, 2018, 2, 1700330 CrossRef
- P. Ye, Y. Chen, J. Wu, X. Wu, S. Yu, W. Xing, Q. Liu, X. Jia, A. Peng and H. Huang, J. Mater. Chem. C, 2017, 5, 12591–12596 RSC
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed
- A. McLean and G. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed
- B. Mennucci, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404 CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. W. Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016, vol. 3 Search PubMed
- One Molecular Simulation Software, https://www.accelrys.com.
- T. Lu and F. J. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- L. Tender, M. T. Carter and R. W. Murray, Anal. Chem., 1994, 66, 3173–3181 CrossRef CAS
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS
- J. Cornil, J. L. Brédas, J. Zaumseil and H. Sirringhaus, Adv. Mater., 2007, 19, 1791–1799 CrossRef CAS
- H. Y. Chen and I. Chao, Chem. Phys. Lett., 2005, 401, 539–545 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- X. Yang, Q. Li and Z. Shuai, Nanotechnology, 2007, 18, 424029 CrossRef PubMed
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed
- C. Yao, Y. Yang, L. Li, M. Bo, J. Zhang, C. Peng, Z. Huang and J. Wang, J. Phys. Chem. C, 2020, 124, 23059–23068 CrossRef CAS
- O. V. Mikhnenko, P. W. M. Blom and T. Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC
- S. M. Menke and R. J. Holmes, Energy Environ. Sci., 2014, 7, 499 RSC
- S. Sun, Z. Fan, Y. Wang and J. Haliburton, J. Mater. Chem., 2005, 40, 1429–1443 CAS
- J. L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC
- Y. Li, D. Qian, L. Zhong, J. D. Lin, Z. Q. Jiang, Z. G. Zhang, Z. Zhang, Y. Li, L. S. Liao and F. Zhang, Nano Energy, 2016, 27, 430–438 CrossRef CAS
- S. Li, L. Zhan, C. Sun, H. Zhu, G. Zhou, W. Yang, M. Shi, C. Z. Li, J. Hou, Y. Li and H. Chen, J. Am. Chem. Soc., 2019, 141, 3073–3082 CrossRef CAS PubMed
- P. Peumans and S. R. Forrest, Chem. Phys. Lett., 2004, 398, 27 CrossRef CAS
- L. Huo, T. Liu, B. Fan, Z. Zhao, X. Sun, D. Wei, M. Yu, Y. Liu and Y. Sun, Adv. Mater., 2015, 27, 6969–6975 CrossRef CAS PubMed
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS
- L. Huo, J. Hou, S. Zhang, H. Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 CrossRef CAS PubMed
- L. Zhu, M. Zhang, W. Zhong, S. Leng, G. Zhou, Y. Zou, X. Su, H. Ding, P. Gu, F. Liu and Y. Zhang, Energy Environ. Sci., 2021, 14, 4341–4357 RSC
- R. Heuvel, F. J. M. Colberts, J. Li, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2018, 6, 20904–20915 RSC
- S. Dey, Small, 2019, 15, e1900134 CrossRef PubMed
- K. Wang, Z. Gao, W. Zhang, Y. Yan, H. Song, X. Lin, Z. Zhou, H. Meng, A. Xia, J. Yao and Y. S. Zhao, Sci. Adv., 2019, 5, eaaw2953 CrossRef CAS PubMed
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Nat. Commun., 2017, 8, 15171 CrossRef CAS PubMed
- K. Zhang, L. Ying, H. L. Yip, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2020, 12, 39937–39947 CrossRef CAS PubMed
- C. Chen, S. Zheng and H. Song, Chem. Soc. Rev., 2021, 50, 7250–7329 RSC
- D. Yuan, L. Liu, X. Jiao, Y. Zou, C. R. McNeill, W. Xu, X. Zhu and D. Zhu, Adv. Sci., 2018, 5, 1800947 CrossRef PubMed
- N. Kleinhenz, L. Yang, H. Zhou, S. C. Price and W. You, Macromolecules, 2011, 44, 872–877 CrossRef CAS
- H. Lissau, R. Frisenda, S. T. Olsen, M. Jevric, C. R. Parker, A. Kadziola, T. Hansen, H. S. van der Zant, M. Brondsted Nielsen and K. V. Mikkelsen, Nat. Commun., 2015, 6, 10233 CrossRef PubMed
- F. Yang, C. Li, Y. Wei, N. Yan, X. Wang, F. Liu, S. You, J. Wang, W. Ma and W. Li, Macromol. Rapid Commun., 2018, 39, e1800546 CrossRef PubMed
- P. Rietsch, S. Sobottka, K. Hoffmann, A. A. Popov, P. Hildebrandt, B. Sarkar, U. Resch Genger and S. Eigler, Chem. – Eur. J., 2020, 26, 17361–17365 CrossRef CAS PubMed
- R. Xue, J. Zhang, Y. Li and Y. Li, Small, 2018, 14, e1801793 CrossRef PubMed
- Z. Wang, L. Zhu, Z. Shuai and Z. Wei, Macromol. Rapid Commun., 2017, 38, 1700470 CrossRef PubMed
- L. J. A. Koster, E. C. P. Smits, V. D. Mihailetchi and P. W. M. Blom, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085205 CrossRef
- G. Han, Y. Guo, L. Ning and Y. Yi, Sol. RRL, 2019, 3, 1800251 CrossRef
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras Garcia and E. Henon, Phys. Chem. Chem. Phys., 2017, 19, 17928 RSC
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736 CrossRef CAS PubMed
- J. L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef CAS PubMed
- W. Xu, X. Ma, J. H. Son, S. Y. Jeong, L. Niu, C. Xu, S. Zhang, Z. Zhou, J. Gao, H. Y. Woo, J. Zhang, J. Wang and F. Zhang, Small, 2022, 18, e2104215 CrossRef PubMed
- Z. W. Zhao, Q. Q. Pan, Y. Geng, Y. Wu, L. Zhao, M. Zhang and Z. M. Su, ACS Sustainable Chem. Eng., 2019, 7, 19699–19707 CrossRef CAS
- J. L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef CAS PubMed
- K. Vandewal, S. Albrecht, E. T. Hoke, K. R. Graham, J. Widmer, J. D. Douglas, M. Schubert, W. R. Mateker, J. T. Bloking, G. F. Burkhard, A. Sellinger, J. M. Frechet, A. Amassian, M. K. Riede, M. D. McGehee, D. Neher and A. Salleo, Nat. Mater., 2014, 13, 63–68 CrossRef CAS PubMed
- Y. J. Liang, Z. W. Zhao, Y. Geng, Q. Q. Pan, H. Y. Gu, L. Zhao, M. Zhang, S. X. Wu and Z. M. Su, New J. Chem., 2020, 44, 9767–9774 RSC
- H. Yao, D. Qian, H. Zhang, Y. Qin, B. Xu, Y. Cui, R. Yu, F. Gao and J. Hou, Chin. J. Chem., 2018, 36, 491–494 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: The Mulliken charge, dipole moments, frontier molecular orbitals, the packing information of the studied acceptors, donor/acceptor interfaces, and excitation properties. See DOI: 10.1039/d1ma01010g |
| This journal is © The Royal Society of Chemistry 2022 |