
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binding of exogenous cyanide reveals new active-site states in [FeFe] hydrogenases†
Maria Alessandra
Martini
 *a,
Konstantin
Bikbaev
b,
Yunjie
Pang
ac,
Christian
Lorent
d,
Charlotte
Wiemann
de,
Nina
Breuer
a,
Ingo
Zebger
d,
Serena
DeBeer
a,
Ingrid
Span
b,
Ragnar
Bjornsson
af,
James A.
Birrell
*ag and
Patricia
Rodríguez-Maciá
*h
*a,
Konstantin
Bikbaev
b,
Yunjie
Pang
ac,
Christian
Lorent
d,
Charlotte
Wiemann
de,
Nina
Breuer
a,
Ingo
Zebger
d,
Serena
DeBeer
a,
Ingrid
Span
b,
Ragnar
Bjornsson
af,
James A.
Birrell
*ag and
Patricia
Rodríguez-Maciá
*h
aDepartment of Inorganic Spectroscopy, Max Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: maria.martini@cec.mpg.de
bDepartment of Chemistry and Pharmacy, Friedrich Alexander University Erlangen-Nürnberg, Bioinorganic Chemistry, Erlangen, Germany
cCollege of Chemistry, Beijing Normal University, 100875, Beijing, China
dInstitut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany
eRuanda-Zentrum und Büro für Afrika-Kooperationen, Universität Koblenz-Landau, Universitätsstraße 1, 56070 Koblenz, Germany
fUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 17 Rue des Martyrs, F-38054 Grenoble, Cedex, France
gSchool of Life Sciences, University of Essex, Colchester, CO4 3SQ, UK. E-mail: james.birrell@essex.ac.uk
hDepartment of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: patricia.rodriguezmacia@chem.ox.ac.uk
First published on 8th February 2023
Abstract
[FeFe] hydrogenases are highly efficient metalloenyzmes for hydrogen conversion. Their active site cofactor (the H-cluster) is composed of a canonical [4Fe-4S] cluster ([4Fe-4S]H) linked to a unique organometallic di-iron subcluster ([2Fe]H). In [2Fe]H the two Fe ions are coordinated by a bridging 2-azapropane-1,3-dithiolate (ADT) ligand, three CO and two CN− ligands, leaving an open coordination site on one Fe where substrates (H2 and H+) as well as inhibitors (e.g. O2, CO, H2S) may bind. Here, we investigate two new active site states that accumulate in [FeFe] hydrogenase variants where the cysteine (Cys) in the proton transfer pathway is mutated to alanine (Ala). Our experimental data, including atomic resolution crystal structures and supported by calculations, suggest that in these two states a third CN− ligand is bound to the apical position of [2Fe]H. These states can be generated both by “cannibalization” of CN− from damaged [2Fe]H subclusters as well as by addition of exogenous CN−. This is the first detailed spectroscopic and computational characterisation of the interaction of exogenous CN− with [FeFe] hydrogenases. Similar CN−-bound states can also be generated in wild-type hydrogenases, but do not form as readily as with the Cys to Ala variants. These results highlight how the interaction between the first amino acid in the proton transfer pathway and the active site tunes ligand binding to the open coordination site and affects the electronic structure of the H-cluster.
Introduction
Hydrogenases are the most powerful natural catalyst for the production and utilization of molecular hydrogen.4,5 Depending on the metal content of the cofactor at their active site, hydrogenases are classified as [FeFe], [NiFe] or [Fe] hydrogenases.7 For the [FeFe] type, the cofactor at the active site is called the H-cluster and consists of a canonical [4Fe-4S] cluster ([4Fe-4S]H) linked through a cysteine thiolate to a unique organometallic diiron cluster ([2Fe]H) (Fig. 1A).8,9 In [2Fe]H, the two irons (distinguished as proximal, Fep, and distal, Fed, depending on their relative distance from [4Fe-4S]H) are bridged by a 2-azapropane-1,3-dithiolate (ADT) ligand and a CO ligand, while additional CO and CN− ligands are terminally bound to each Fe. The open coordination site at the apical position on Fed is where activation/formation of H2 occurs, but also where inhibitors including CO,10 H2S2,11 and O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 bind. Binding of O2 generally leads to the destruction of the H-cluster, making [FeFe] hydrogenases highly oxygen-sensitive.12–15
12 bind. Binding of O2 generally leads to the destruction of the H-cluster, making [FeFe] hydrogenases highly oxygen-sensitive.12–15
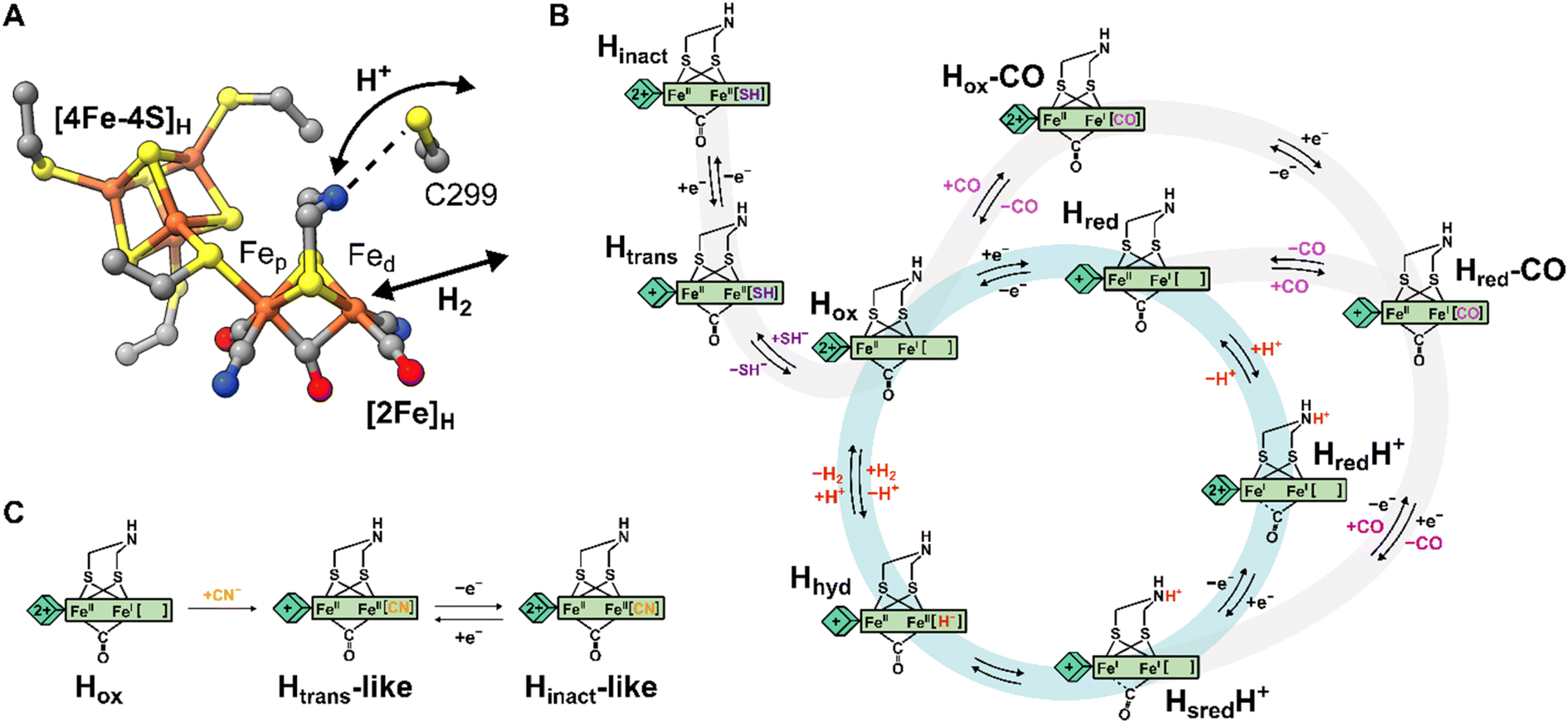 | ||
| Fig. 1 Structure of the H-cluster and proposed catalytic cycle. (A) Structure of the [2Fe]H and [4Fe-4S]H subclusters from Clostridium pasteurianum HydA1 (CpHydA1, PDB: 4XDC)1 with the Cys in the proton transfer pathway also shown (C299 in CpHydA1, C178 in DdHydAB, C169 in CrHydA1). (B) Proposed catalytic cycle also showing the pathways of reversible inactivation by H2S and CO, respectively. The pathway for Hinact formation is only applicable to DdHydAB as the Htrans state has not been identified for other [FeFe] hydrogenases. A different catalytic cycle has been proposed by some authors.6 (C) Schematic showing the proposed chemical structure of the Htrans-like and Hinact-like states identified in this study. | ||
Several states of the H-cluster differing in electron and proton distribution at the two subclusters and ligand binding to the open coordination site have been identified. However, the precise structure and the involvement of some of these states in the catalytic cycle (Fig. 1B) of [FeFe] hydrogenase is still a matter of debate.6,16 In [2Fe]H the strong-field CO and CN− ligands stabilize low-spin and low-oxidation states for the two Fe ions, which cycle between Fe(II) and Fe(I) during catalysis. For instance, the active oxidized state Hox has mixed valence Fep(II)Fed(I) in [2Fe]H.17 The one-electron reduced state Hred retains the Fep(II)Fed(I) valence in [2Fe]H but has a reduced [4Fe-4S]H. The reduced states HredH+ and HsredH+ are thought to have an Fep(I)Fed(I) configuration at [2Fe]H that is favored by concomitant protonation of the ADT ligand.18–20 The crucial two-electron reduced Hhyd state contains a terminal hydride on Fed and an overoxidized [2Fe]H with a formal Fep(II)Fed(II) configuration.21–25 Recently, states with a terminal hydride on Fed differing in the redox state of [4Fe-4S]H have been identified.26
A similar overoxidized [2Fe]H can be found in two inactive states called Htrans and Hinact.11,27–30 These states form upon reversible inactivation of [FeFe] hydrogenases by sulfide, which binds to the H-cluster under oxidizing conditions in some enzymes,2,11 or by binding of a nearby cysteine thiolate in others.31–33 In Hinact, an RS− ligand (where R can be H or the rest of the cysteine amino acid) is thus bound in the apical position to Fed, [2Fe]H is in the overoxidized state and the [4Fe-4S]H subcluster is oxidized ([4Fe-4S]H2+–[Fep(II)Fed(II)-SR]H). Notably, the Hinact state is stable under air as the RS− ligand prevents O2 from binding to Fed. Reversible one-electron reduction of Hinact, in which SH− is bound, yields the Htrans state, which has a reduced [4Fe-4S]H+ ([4Fe-4S]H+–[Fep(II)Fed(II)-SH]H). Conversion of Htrans to the active hydrogenase appears to require an additional reduction step.29 However, the exact mechanism of conversion is not clear, but several theories have been proposed.2,11,34 So far, Htrans has not been identified in enzymes that bind a cysteine thiolate in Hinact.33
Efficient exchange of protons between the solvent and the active site is crucial during H2 conversion and is facilitated by a proton channel (also called the proton transfer pathway, PTP). This pathway is formed by a series of largely conserved (at least in prototypical hydrogenases)5 amino acids and water molecules that form a network of hydrogen bonds connecting the protein surface with the H-cluster.35–37 Site-directed mutagenesis of amino acids along the proton transfer pathway can impair or even completely abolish catalytic activity, as a consequence of the slower proton exchange with the H-cluster.36,38–40 Additionally, it was observed that some H-cluster states accumulate differently in wild-type enzymes and in variants with deficient proton transfer. Closest to the H-cluster is a cysteine residue (C299 in Clostridium pasteurianum HydA1, C178 in Desulfovibrio desulfuricans HydAB, DdHydAB, and C169 in Chlamydomonas reinhardtii HydA1) whose thiol is within hydrogen-bond distance of the bridgehead amine of the ADT ligand (Fig. 1A). When this Cys was mutated to alanine (Ala) or serine (Ser) in Chlamydomonas reinhardtii HydA1 (CrHydA1 C169A and C169S) and to Ser in Clostridium pasteurianum HydA1 (CpHydA1 C299S), these hydrogenases formed readily the Hhyd state.21–23,39,41,42 In addition, the CrHydA1 C169S variant was shown, using electron paramagnetic resonance (EPR) spectroscopy, to accumulate a state similar to Htrans, but the precise nature of this state remains unknown.21,22,43 Finally, the C169A variant of CrHydA1 has been reported to react with oxygen to form an Hox–O2 state (so far observed only in this particular mutant), which has been suggested to have a superoxide bound to Fed and an oxidized [4Fe-4S] cluster, yielding a [4Fe-4S]H2+–[Fep(I)Fed(III)-O2−]H electronic configuration.44 However, the infrared (IR) spectrum of this state is very similar to the Hinact state and so a formal Fep(II)Fed(II) valence would seem more likely.
In this study, we investigated the effects of replacing the Cys in the proton transfer pathway with alanine in the hydrogenases DdHydAB and CrHydA1. DdHydAB is an exceptionally active bidirectional hydrogenase that contains two additional [4Fe-4S] clusters (F-clusters) for electron transfer between the H-cluster and the protein surface. The mutation of amino acids along the proton transfer pathway of DdHydAB has not been investigated before. The C169A variant of CrHydA1 has already been studied in particular in relation to the Hhyd and Hox–O2 states,23,41,44 but here we report two new active site states in CrHydA1 C169A never identified before. In both DdHydAB C178A and CrHydA1 C169A, we observed formation of unprecedented H-cluster states similar to Htrans and Hinact. By combining their spectroscopic and structural characterization, we demonstrated that these Htrans-like and Hinact-like states form upon binding of CN− to the H-cluster (Fig. 1C). These CN−-bound states form also in wild-type (WT) hydrogenases, but are stabilized in the Cys to Ala mutants. This study highlights how the interaction between the Cys in the proton transfer pathway and the H-cluster (specifically the bridgehead amine in [2Fe]H) tunes the electronic structure of the H-cluster and regulates ligand binding to the apical position of Fed.
Results
DdHydAB C178A is isolated in an Htrans-like state
The DdHydAB C178A mutant was recombinantly expressed in E. coli as an “apo”-hydrogenase (i.e. containing the [4Fe-4S]H subcluster and all the accessory F-clusters but lacking [2Fe]H) and artificially maturated in vitro, as routinely performed with the WT enzyme.45 The WT DdHydAB is commonly isolated after maturation (under 2% H2 and 98% N2) as a mixture of states, mainly Hox, Hox–CO, and HredH+.45 As shown in the IR spectra in Fig. 2A, after artificial maturation the C178A mutant was, surprisingly, isolated in an almost pure unprecedented state that greatly differs from the states normally observed in freshly maturated WT DdHydAB (Table S1†). In particular, the spectrum of the as isolated C178A mutant exhibits a broad absorption at 1853 cm−1 attributed to the bridging CO ligand, a single broad band at 1989 cm−1 attributed to the terminal CO ligands (potentially resulting from the two overlapping CO bands, exhibiting a shoulder at ∼2002 cm−1), and three bands at 2116, 2100 and 2087 cm−1 attributed to CN− ligands (instead of the expected two bands). Relative to the IR bands observed for the Hox state in WT DdHydAB, the IR bands of the C178A variant are shifted to higher energies (blue-shifted) suggesting decreased electron density on [2Fe]H. The frequencies of the IR bands are very similar to those of the Htrans state in WT DdHydAB (Table S1†),2,29 therefore, we hypothesized that this new state could be an Htrans-like state (i.e. with the same electronic configuration [4Fe-4S]H+–[Fep(II)Fed(II)]H). An Hinact-like state ([4Fe-4S]H2+–[Fep(II)Fed(II)]H) was formed both by oxidation of the as-isolated C178A variant under anaerobic conditions (using hexaammineruthenium(III) chloride, HAR) or by exposure to atmospheric oxygen (Fig. 2A, green bands), yielding identical IR spectra in each case. Exposure of the as-isolated sample to air did not result in any significant decrease in IR signal intensity, suggesting that all active site species present in the Htrans-like state transform into an Hinact-like state. Notably, this Hinact-like state is another example of an air-stable state in [FeFe] hydrogenases. Therefore, in addition to having the same electronic configuration of Htrans and Hinact, respectively, we hypothesized that the new states might have an additional ligand (likely different from SH−) bound at the open coordination site.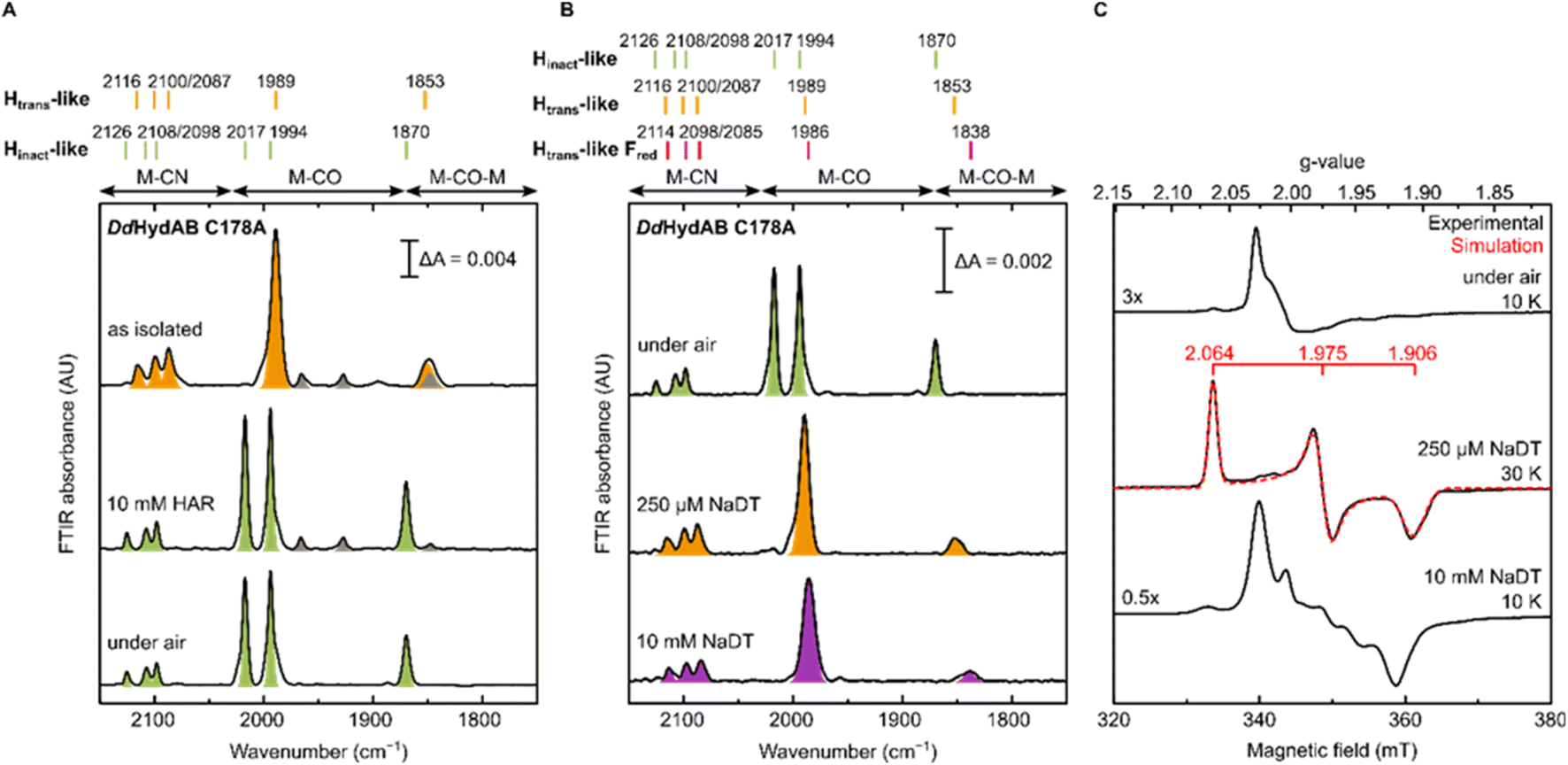 | ||
| Fig. 2 IR and EPR spectra of DdHydAB C178A exhibit new Htrans-like and Hinact-like states. (A) IR spectra of freshly maturated DdHydAB C178A (in 25 mM Tris pH 8.0, 25 mM KCl): as isolated; oxidized under anaerobic conditions (with 10 mM HAR); oxidized under air. Bands from the Htrans-like and Hinact-like states are colored in orange and green, respectively. (B and C) Samples of DdHydAB C178A were prepared under different conditions. One aliquot was used for room temperature IR measurements (B), while the remaining sample was used for CW X-band EPR measurements (C). In (A and B), bands are color coded as follows: green for Hinact-like state, orange for Htrans-like state, purple for Htrans-like Fred state. The gray bands correspond to traces of an unidentified state that potentially lacks the third CN− ligand (which protects the H-cluster from O2 attack), as this state disappears after exposure to air. In (C), experimental spectra are in black, overlaid in one case with spectral simulations (dashed red line). EPR experimental conditions: microwave frequency = 9.64 GHz; microwave power = 1 mW for the first two conditions (under air, 250 μM NaDT), 0.1 mW for the bottom one (10 mM NaDT); temperature is specified in the figure. | ||
To confirm our assignment of Htrans-like and Hinact-like states, we measured EPR spectra of DdHydAB C178A poised in different states as confirmed by IR spectroscopy (Fig. 2C). The Hinact-like state is EPR silent, like the Hinact state in WT DdHydAB.30 Only a signal from a [3Fe-4S] cluster could be detected, probably due to oxidative damage to the F-cluster located in vicinity of the protein surface. Addition of one equivalent of reducing agent (sodium dithionite, NaDT) to the DdHydAB C178A sample, which had partially converted to the Hinact-like state during storage (Fig. S1A†) reverts it to the Htrans-like state (Fig. 2B). The Htrans-like state exhibits a rhombic EPR signal (g = 2.06, 1.98, 1.91) similar to the one of Htrans in WT DdHydAB (Fig. S2†),30 confirming our initial assignment of DdHydAB C178A being isolated in an Htrans-like state. Addition of excess NaDT results in only minor shifts to lower energies (red-shift) of all IR bands and gives rise to a complex EPR interaction spectrum. We interpreted this behaviour with the H-cluster remaining in an EPR-active Htrans-like state while the accessory F-clusters are being reduced to EPR-active states by the excess of NaDT, resulting in strong dipolar spin-coupling (Htrans-like Fred state) as observed previously for the WT enzyme in the Hox or Hox–CO state with reduced F-clusters.46 Samples in the as-isolated state and purged with CO showed no change to the IR spectrum (Fig. S1B†) indicating that this state was unable to bind CO, most likely due to an already occupied coordination site.
Evidence for an additional CN− ligand at the H-cluster in DdHydAB C178A
For all the states we could observe in DdHydAB C178A (Htrans-like, Hinact-like and Htrans-like Fred), the IR spectra always exhibit three bands in the CN− region. To test if all the bands derive from CN− vibrations associated with the H-cluster, we performed the artificial maturation of the C178A mutant with a precursor of the [2Fe]H cluster with both CN− ligands labelled with 13C. As shown in Fig. 3A, we observed an isotope shift (46–44 cm−1) of all three CN− absorptions in the IR spectrum of the Hinact-like state. We could interpret these results in three ways: (i) three CN− ligands are present at the H-cluster; (ii) two CN− vibrations couple in an unusual way giving rise to three IR bands; (iii) spectra represent two very similar states with one strongly overlapping CN− band, while the other CN− band in each state is distinct. The second hypothesis is not likely as the CN− ligands are on different Fe ions and such a structure is unlikely to give significant quadratic coupling.29,47 Previous isotope editing experiments on the CO ligands in WT enzyme showed very little perturbation in the vibrational frequency of the pCO ligand (the terminal CO on Fep), when dCO (the terminal CO on Fed) or μCO (the bridging CO) were exchanged with 13CO.29,47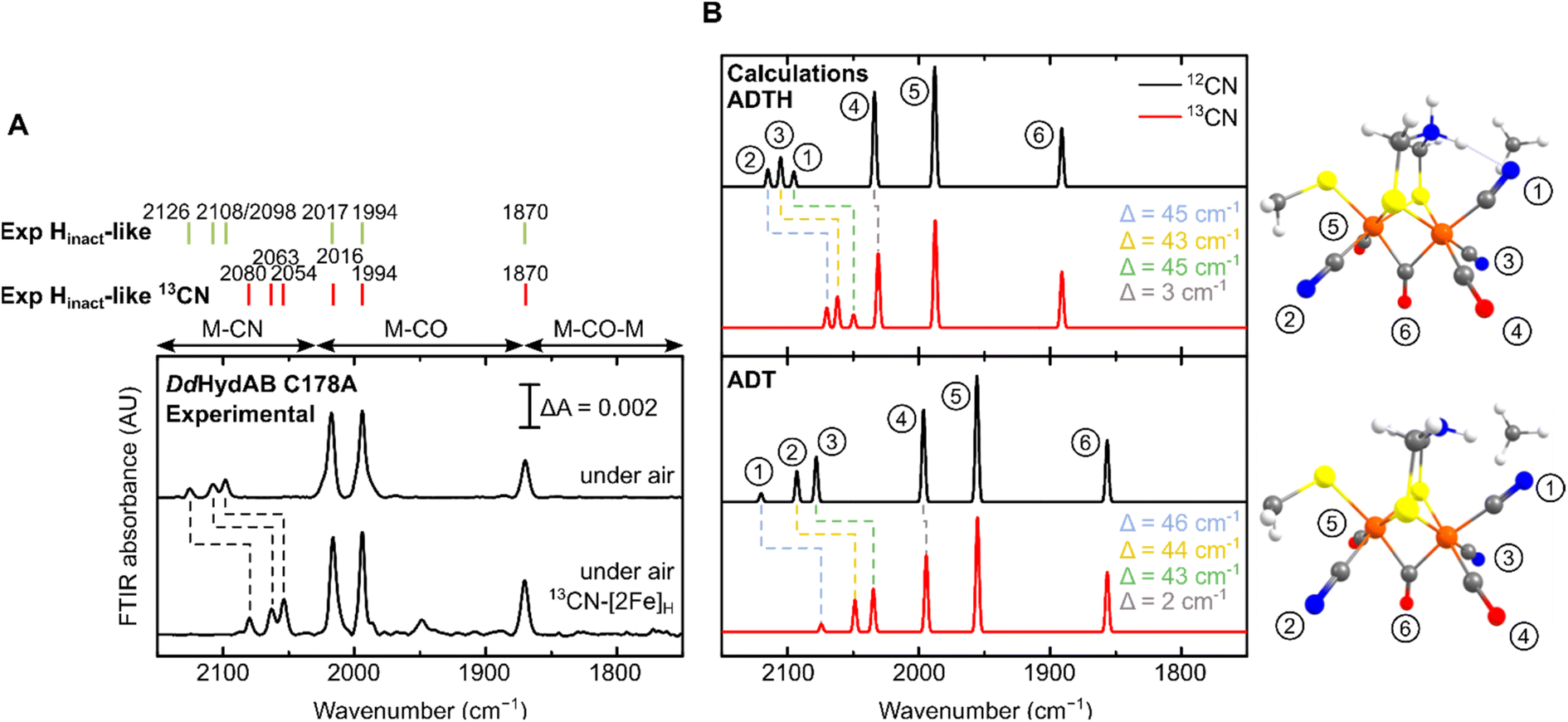 | ||
| Fig. 3 (A) Experimental IR spectra of DdHydAB C178A maturated with natural abundance (top) and 13CN−-labeled [2Fe]H precursor (bottom), both exposed to air to form the Hinact-like state. The small additional feature in the 13CN spectrum may represent a small amount of an unknown degradation product. (B) Calculated IR spectra of the H-cluster model with either a singly protonated (ADT) or doubly protonated (ADTH) ADT ligand. Insets are [2Fe]H structures with chemical groups associated with modes labelled. A scaling factor of 0.964 was used. | ||
The isotope shift could be reproduced by quantum mechanics/molecular mechanics (QM/MM) calculations of an H-cluster model (Fe(II)Fe(II) redox state) of the C178A variant of DdHydAB, with CN− as the exogenous ligand on Fed, as shown in Fig. 3B. Two protonation states of the amine in the ADT ligand were calculated: singly protonated (ADT) and doubly protonated (ADTH). The experimental 13CN isotope shifts of 43–46 cm−1 could be satisfactorily reproduced with both models: 43–46 cm−1 (ADT) and 43–45 cm−1 (ADTH). The absolute experimental frequencies are reasonably well reproduced by scaled harmonic frequencies, though with some differences between ADT and ADTH models. The terminal CO modes were somewhat better predicted by the ADTH model while the CN− modes and bridging CO modes were better predicted with the ADT model. We note that the CO frequencies are quite dependent on the quality of the model, density functional and scaling factor while the CN− frequencies are less so (Fig. S3 and Materials and methods in the ESI†). The calculated relative intensities of the three CN− modes, however, differ more strongly between models. The ADT model predicts an increase in CN− mode intensity with decreasing energy, consistent with the experimental intensities, while the ADTH model does not. The reason is that the order of the assigned CN− modes differs between ADT/ADTH models; the exogenous CN− mode is the highest-energy CN− mode for the ADT model but it is the lowest for the ADTH. These differences can be explained by a stronger exogenous CN−-binding in the ADTH model (aided by stronger H-bonding to the doubly protonated amine). Other conformers of the ADT and ADTH models were explored (Fig. S4†) but were found to be energetically unfavorable. Overall the calculations suggest the Hinact-like state as best described by an [Fep(II)Fed(II)]H model featuring an exogenous CN− ligand in the apical position with a singly protonated bridgehead amine of the ADT ligand.
To further investigate the properties of the H-cluster in the C178A variant of DdHydAB, we solved crystal structures of the enzyme in the Hinact-like and the Htrans-like states using X-ray crystallography (Fig. 4 and S5–S9†). After exposure to air to form the Hinact-like state, the protein was crystallized under aerobic conditions (Fig. 4A and S7†) as performed previously for the SH-bound Hinact state in the WT enzyme.11 IR and resonance Raman (RR) measurements on crystals48 prepared under the same conditions confirmed that the enzyme was indeed in the Hinact-like state (Fig. S5†). However, small shifts in the band positions compared with solution measurements were observed and are likely related to temperature-dependent changes or crystal packing effects. Such effects have been observed previously for [FeFe]11,49 and [NiFe] hydrogenases.48,50,51 We also solved a structure of the Htrans-like state of the enzyme from crystals grown under anaerobic conditions (2% H2, 98% N2) (Fig. 4B). DdHydAB C178A crystallized in an orthorhombic space group P212121 as observed for the previously reported WT DdHydAB in the Hinact state.11 The crystals diffracted up to 1.0 Å and the structures were solved at high resolution, 1.04 Å for the Hinact-like state and 1.01 Å for the Htrans-like state. The overall structure of the C178A variant in the Hinact-like state and the WT enzyme in the Hinact state is virtually identical with an RMSD of 0.237 Å (calculated for all Cα atoms of all residues, Table S2) (Fig. S7 and S8†). The structure at atomic resolution clearly shows a diatomic ligand in the apical position of Fed and the Ala residue that replaced the Cys at position 178. Moreover, we identified two additional well-defined water molecules appearing near the Ala178. Interestingly, in the crystal structure of the C299A variant from CpHydA1 reported by Duan et al.,36 the space of the missing thiol group was replaced by an additional H2O molecule (Wat962), which was located at 3.4–3.7 Å from the NH group of the ADT ligand and at 3.6–3.7 Å from the Wat826 molecule of the PTP.36 This new water molecule was hypothesised to rescue proton transfer activity in the absence of the thiol; however, the authors could not measure any significant catalytic activity.
 | ||
| Fig. 4 Crystal structures of the DdHydAB C178A mutant in two different states. (A) DdHydAB in the Hinact-like state (PDB ID 8BJ7) is shown as cartoon and colored in gray. (B) DdHydAB in the Htrans-like state (PDB ID 8BJ8) is shown as cartoon and colored in magenta. Close-up view of the active site showing the H-cluster, the Cys ligating the cofactor, the side chain of Ala178 and the well-defined water molecules with a distance <4.0 Å from Ala178. The protein backbone is shown as cartoon, the amino acid side chains and the H-cluster including the bound CN− are shown as stick model, and water molecules are shown as spheres. The cofactors and amino acid side chains are colored according to the element-specific color code. A 2Fo–Fc electron density map (blue mesh, contoured at 1.0σ) is shown for the H-cluster including the CN− ligand, the side chain of Ala178, and the water molecules. | ||
The overall architecture of the Hinact-like and the Htrans-like states are also virtually identical with an RMSD of 0.053 Å (calculated for all Cα atoms of all residues, Table S2) (Fig. S8†). While for the structure of WT DdHydAB in the Hinact state a reduced occupancy of the [2Fe]H subcluster led to better agreement between modelled and experimental data,11 here we observed no negative difference density when refining the structural models with an occupancy of 100% for the [2Fe]H subcluster. This could be evidence for a better incorporation of the [2Fe]H subcluster during artificial maturation or higher stability of the H-cluster during crystallization in the C178A mutant compared to the WT protein.
The crystal structure clearly shows a well-defined, relatively symmetric bridging CO (Fig. S9†), with roughly equal Fep—Cb and Fed—Cb bond distances. A similar observation was made for the Hinact state in wild type DdHydAB,11 whereas other [FeFe] hydrogenase structures show slight lengthening of the Fep—Cb bond and shortening of the Fed—Cb bond.8,9,52 The differences here are attributed to the oxidation states and coordination environment of Fep and Fed. In our structure with cyanide bound and the previously published Hinact state11 both Fe ions were Fe(II) and hexacoordinate. Meanwhile for structures obtained of the active enzyme,8,9,52 the Fe ions are more reduced (for Hox Fed is reduced to Fe(I) and for HredH+ both Fe ions are reduced to Fe(I)) and Fed is pentacoordinate. These effects lead to shortening of the Fed—Cb bond giving a semi-bridging CO.
On the basis of these results and the fact that the IR spectra of all Hinact-like and Htrans-like states in DdHydAB C178A exhibit three CN− bands, we suggest that in these states a third CN− is present at the H-cluster, bound at the apical position on Fed. Therefore, in the structure we modelled a CN− ligand coordinated to the distal iron ion through its carbon atom, with a Fe–C distance of 1.90 Å. To confirm the assignment of the ligands, we calculated an omit map in the absence of the [2Fe]H subsite and the additional ligand at the apical position on Fed. The omit map (Fig. S9†) supports the positioning of the atoms in the electron density. In addition, we are able to distinguish between the N and O atoms of the terminal and bridging ligands when increasing the contouring level to σ = 2.8 Å (Fig. S9C†). This result suggests that the exogenous CN− ligand remains in the apical position, which agrees with the QM/MM calculations as well as the illumination experiments on CrHydA1 C169A (see below).
A similar Htrans-like state in CrHydA1 C169A
To gain further understanding of these CN− bound states and on the role of the Cys in the proton transfer pathway in their formation, we decided to re-investigate the C169A variant of CrHydA1. As mentioned in the introduction, this particular mutation in CrHydA1 has already been studied and showed accumulation of the Hhyd and Hox–O2 states.23,41,44 In contrast with what we observed with DdHydAB C178A, and consistent with the previous reports on CrHydA1 C169A, this mutant is isolated after artificial maturation without an additional CN− ligand on the H-cluster. Indeed, CrHydA1 C169A is initially isolated under 2% H2 in a mixture of the Hhyd state (which can be enriched upon addition of NaDT) and another state that has been previously assigned as Hox on the basis of the position of the IR bands (Fig. 5A).23,53 However, the EPR spectrum of the as-isolated enzyme lacks the characteristic rhombic signal for the Hox state and shows only the Hhyd state as major component (75%) (Fig. 5B). The EPR spectrum of the Hhyd state in CrHydA1 C169A (g = 2.075, 1.942, 1.884) is nearly identical to the one reported for the same state in the C169S mutant (g = 2.07, 1.94, 1.88).22 Therefore, we suggest that the state originally labelled as Hox may instead be an EPR silent state with a similar electronic structure to the Hred state (Hred-like, light blue IR bands in Fig. 5A), which has a reduced [4Fe-4S]H ([4Fe-4S]H+–[Fep(II)Fed(I)]H). Oxidation of this state with one equivalent of oxidizing agent (HAR) under anaerobic conditions formed a new state with its most intense IR band at 1948 cm−1, with an EPR spectrum (g = 2.104, 2.046, 2.000) similar to that observed for the Hox state in WT CrHydA1 (dark blue bands in Fig. 5A). Interestingly, these Hred-like and Hox-like states have similar FTIR spectra to those observed for the recently characterised HoxH and H′redH states,53–55 with bands shifted to higher energy compared with the Hred and Hox states in WT CrHydA1.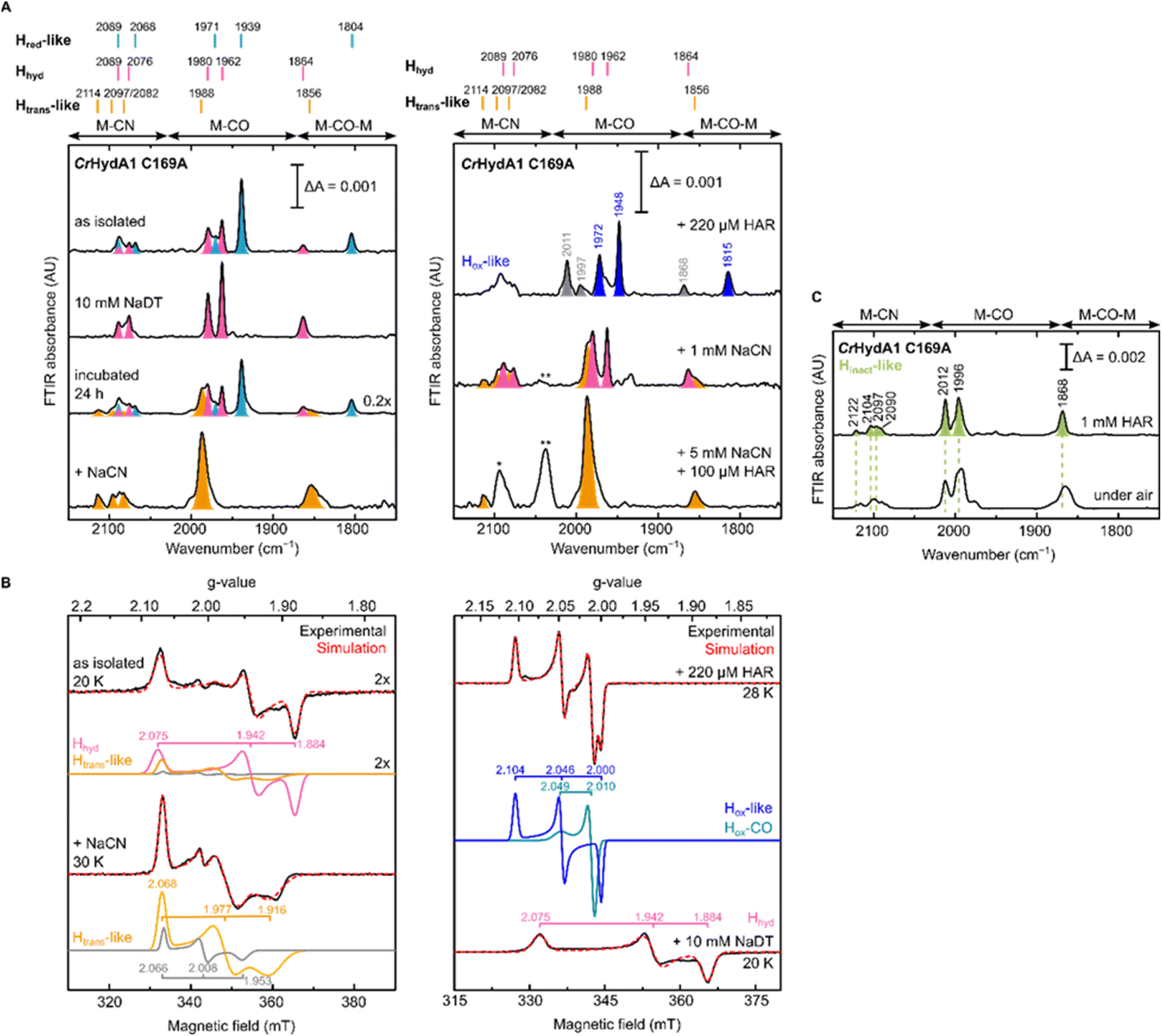 | ||
| Fig. 5 IR and EPR spectra suggest formation of a CN−-dependent Htrans-like state also in CrHydA1 C169A. (A) Room temperature IR spectra of CrHydA1 C169A under different conditions: as isolated; with NaDT; after 24 h incubation under 2% H2; after addition of 5 mM NaCN, 100 μM HAR and then buffer exchanged (“+NaCN”); oxidized with 220 μM (1.1 eq.) HAR; after addition of 1 mM NaCN; after addition of 5 mM NaCN and 100 μM (0.5 eq.) HAR. Bands are color-coded as follows: light blue for Hred-like, pink for Hhyd, orange for the Htrans-like, and blue for the Hox-like state. Bands in gray correspond to traces of the Hinact-like and Hox–CO states. For the Hox-like state the bands in the complex CN− region could not be assigned. In the Hhyd state, CN− binding is likely disfavored as a hydride is bound to Fed. The single asterisk marks the band of HCN (2093 cm−1), while the double asterisk marks the band of [Fe(CN)6]4− (2037 cm−1),3 which suggests partial cofactor degradation upon CN− addition. Clean spectra for the Htrans-like state where obtained after buffer-exchanging the protein to eliminate HCN, free CN− and degradation products. (B) CW X-band EPR spectra for some of the conditions shown in (A). Experimental spectra are shown in black and are overlaid with spectral simulations (dashed red line) with component spectra underneath. The pink component corresponds to the Hhyd state. The orange component is likely the Htrans-like state, while the gray component may represent an alternative, as yet unidentified, state. Presence of the Htrans-like components in the EPR spectrum of as isolated enzyme suggests that a small amount of Htrans-like state has already formed in freshly maturated CrHydA1 C169A (the small shoulder at 1988 cm−1 in the IR spectrum of the same sample is also consistent with the presence of traces of the Htrans-like state). The blue trace corresponds to the Hox-like state, while the dark cyan trace corresponds to the Hox–CO state. EPR experimental conditions: microwave frequency = 9.64 GHz; microwave power = 1 mW; temperature is specified in the figure. (C) IR spectra of the Hinact-like states in CrHydA1 C169A. From the Htrans-like state, two similar but slightly different Hinact-like states form by oxidation of the enzyme with HAR under anaerobic (top) or by oxidation by atmospheric oxygen (bottom). | ||
Despite being initially isolated in states lacking an additional CN− ligand (Hred-like, Hhyd), incubation of CrHydA1 C169A (pH 8) in the glovebox (2% H2, 98% N2) for 24 h at room temperature led to the appearance in the IR spectrum of an Htrans-like state similar to the one observed in DdHydAB C178A, including a third CN− band appearing at high energy (2114 cm−1, Fig. 5A). This Htrans-like state could also be enriched upon addition of exogenous CN− to freshly maturated enzyme (Fig. 5A), which is initially isolated as a mixture of Hred-like and Hhyd (Fig. 5A). CN− binding to the Hhyd state seems less favored, consistent with the presence of a terminal hydride bound to Fed in Hhyd (Fig. 5A). Therefore, complete conversion of the as-isolated enzyme to the Htrans-like state required addition of half an equivalent of oxidizing agent (HAR) to first oxidize the Hhyd state (Fig. 5A). Addition of excess CN− induces partial degradation of the H-cluster, as demonstrated by the appearance in the IR spectra of a broad band around 2037 cm−1, which suggests formation of [Fe(CN)6]4− (Fig. 5A),3 as already reported for other Fe-containing metalloenzymes like CODH upon treatment with CN−.56 Therefore, after the formation of the Htrans-like state, samples were buffer exchanged to remove degradation products as well as the excess of free CN− to give cleaner IR spectra as in Fig. 5 and 6.
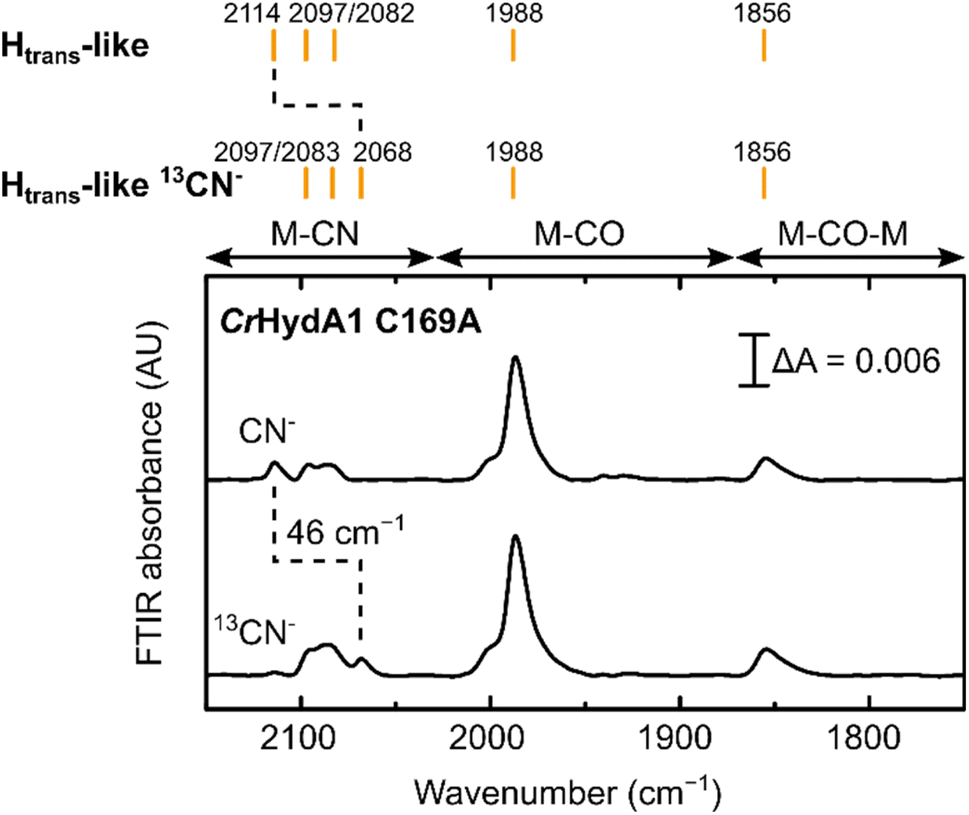 | ||
| Fig. 6 IR spectra of CrHydA1 C169A prepared in the Htrans-like state using natural abundance and 13C labelled CN− exhibit an expected isotope shift of one of the CN− bands. Small peaks in the region between 1930 and 1940 cm−1 are likely due to small contributions from states without CN− bound. | ||
The EPR spectrum of the Htrans-like state in CrHydA1 C169A could be simulated with two components having similar rhombic signals (Fig. 5B). The first component (g = 2.068, 1.977, 1.916), accounting for ca. 87% of the signal, resembles the EPR spectrum of the Htrans-like state in DdHydAB C178A as well as a similar Htrans-like state previously observed in CrHydA1 C169S (g = 2.065, 1.969, 1.906), which was never assigned to a particular structure of the H-cluster.21,22,43 The second rhombic component (g = 2.066, 2.008, 1.935) could potentially relate to a different protein or H-cluster conformation. As observed for DdHydAB C178A, oxidation of CrHydA1 C169A in the Htrans-like state yields an Hinact-like state. However, two similar Hinact-like states were formed depending on whether the oxidation was performed under anaerobic conditions by HAR or by atmospheric oxygen (Fig. 5B). A reason for this difference could be damage to the [4Fe-4S]H during the different oxidative treatments. Notably, illumination of the air-oxidized CrHydA1 C169A at cryogenic temperature (Fig. S10†) did not reveal any photosensitivity, confirming again the assignment of a terminally bound CN− for the Htrans-like and Hinact-like states, since a CO species in the apical position would likely be photolyzed.29,30
Treatment of freshly-maturated CrHydA1 C169A with 13CN− yielded an Htrans-like state which exhibited an isotope shift (46 cm−1) of one of the CN− band in the IR spectrum (Fig. 6). This result unambiguously confirms that a third CN− ligand is bound at the H-cluster in the Htrans-like and Hinact-like states. Additionally, this result allows us to assign the exogenous CN− ligand on Fed to the band with the highest vibrational frequency (in samples prepared with 12CN−) among the three CN− bands. In line with this observation, resonance Raman (RR) measurements on CrHydA1 C169A in the Hinact-like state (Fig. S11A†) revealed 1–3 cm−1 downshifts in the region characteristic for metal–ligand vibrations with contributions from the cyanide ligands (390–600 cm−1), when comparing samples prepared with 12CN− and 13CN−. This is consistent with an expected lower vibrational frequency in the presence of a heavier atom. Calculated Raman spectra of the apical CN−-bound H-cluster models in a [Fep(II)Fed(II)]H redox state (note: using the DdHydAB QM/MM model) reproduce the experimental RR spectra and 13C isotope shifts fairly well (Fig. S11†). An observed mode at 603 cm−1 (experimental), assigned as a bridging CO bending mode, could only be reproduced (calculated 605 cm−1) with the ADT model, suggesting the ADT ligand to be singly protonated.
Addition of CN− to WT hydrogenases
Can these CN−-dependent states only be formed in [FeFe] hydrogenases with a disrupted proton transfer pathway? IR spectra of artificially maturated WT DdHydAB quite often show a band at 1987 cm−1,45 which has previously been difficult to assign to any known state of the H-cluster (Fig. 7A). However, we note that the vibrational frequency of this band is similar to the one of the terminal CO ligands in the CN−-dependent Htrans-like state in DdHydAB C178A (1989 cm−1) (Fig. 2). Indeed, addition of exogenous CN− to WT DdHydAB caused an increase in intensity of this band, together with the appearance of other bands characteristic of the CN−-dependent Htrans-like state (Fig. 7B).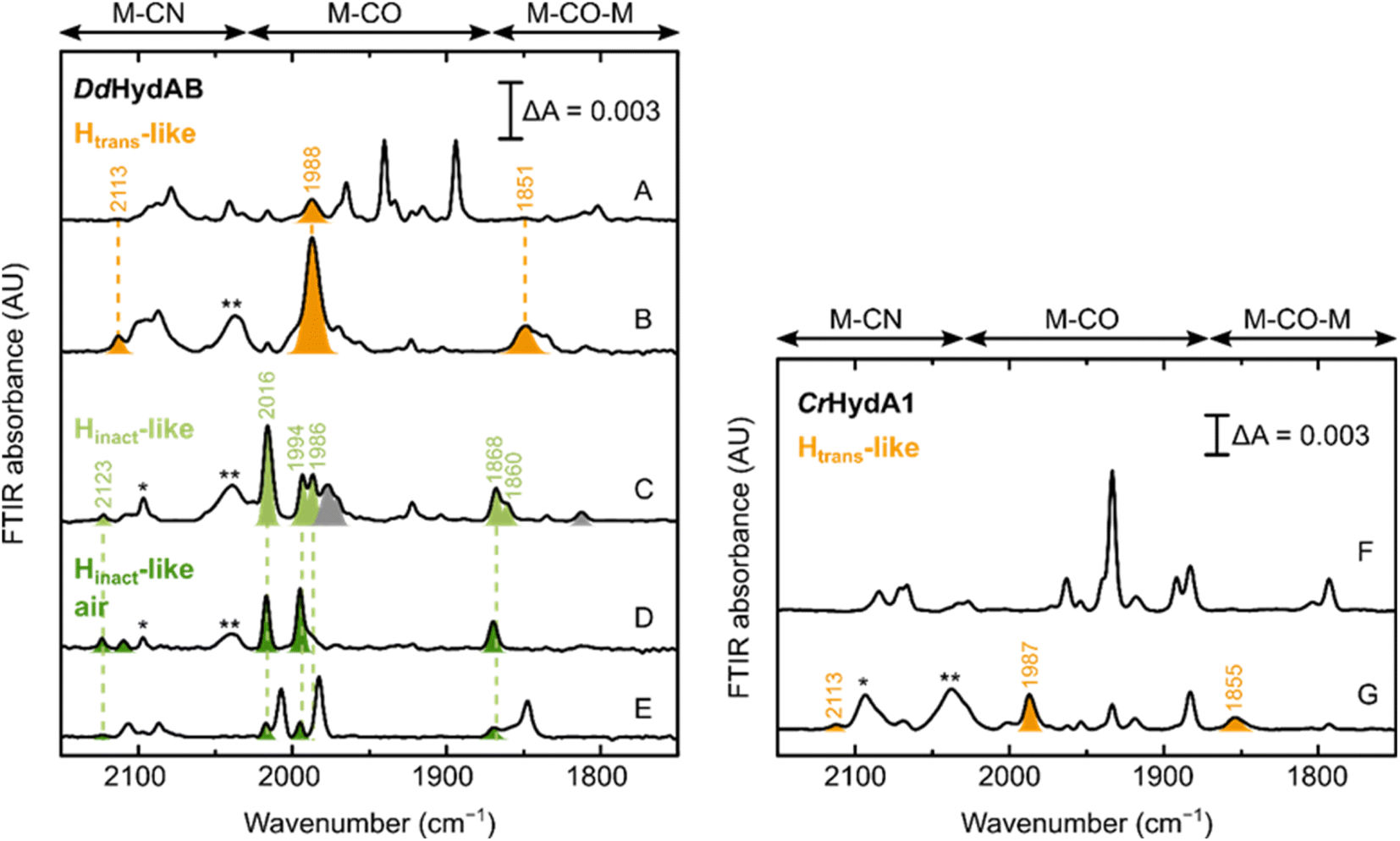 | ||
| Fig. 7 Formation of the Htrans-like and Hinact-like states in WT [FeFe] hydrogenase. IR spectra of WT DdHydAB and WT CrHydA1 under several conditions. Left panel – DdHydAB: as isolated (A); with 10 mM NaCN (B); with 10 mM NaCN and 10 mM HAR (C); with 10 mM NaCN, 10 mM HAR and after exposure to air (D); after preparation of the Hinact state with Na2S as described in ref. 2. Right panel – CrHydA1: as isolated (F) and with 10 mM NaCN. Bands from the Htrans-like state are colored in orange, those from the Hinact-like state obtained under anaerobic conditions in light green and those from the Hinact-like state under air in dark green. The single and double asterisk indicate HCN and [Fe(CN)6]4− respectively. In (A), the additional bands (not colored) correspond to the Hox and HredH+ states, with small contributions from the Hox–CO and Hred states. A small proportion of the Htrans-like state is often present even in freshly-maturated DdHydAB. In (C), the gray bands belong to the Hox–CO state. In (D), the small blue-shift observed when the enzyme is exposed to oxygen after addition of NaCN and HAR (Hinact-like air) might be caused by oxidation of the F-clusters not already oxidized by HAR. As shown in (E), the same “Hinact-like air” state is routinely present as a minor component when the Hinact state (not colored) is prepared by addition of Na2S and HAR,2 as a result of the oxidation of the Htrans-like state formed during artificial maturation (shown in A). In (F), the additional bands belong to the Hred state, with smaller contributions from the HredH+ and HsredH+ states. The Hred and HsredH+ states are present also in (G), after addition of CN− to CrHydA1. Addition of CN− to CrHydA1 seems to induce a lot more H-cluster degradation, as demonstrated by the loss in intensity in the IR spectra (while the protein concentration was similar, 420 μM in (F) and 500 μM in (G)) and the presence of an intense band for [Fe(CN)6]4−. | ||
Oxidation with HAR under anaerobic conditions gives rise to multiple bands in the IR spectrum, some belonging to the Hox–CO state and others similar to the Hinact-like state of the C178A mutant. Subsequent exposure of the enzyme to oxygen, yields a simpler IR spectrum with only an Hinact-like state present (slightly blue-shifted compared to the analogous state observed under anaerobic conditions, potentially reflecting a difference in the oxidation state of F-clusters, fully oxidized under aerobic conditions) (Fig. 7D). This same CN−-dependent Hinact-like state is present in small amounts when WT DdHydAB is prepared in the Hinact state by addition of sulfide,2 and probably derives from the small amount of the CN−-dependent Htrans-like state that forms during artificial maturation of this enzyme. Addition of exogenous CN− to WT CrHydA1 also induces formation of a CN−-dependent Htrans-like state (Fig. 7F and G). However, we noted that in this case addition of excess CN− caused substantial degradation of the H-cluster (more than with the CrHydA1 C169A mutant or WT DdHydAB). Therefore, we did not investigate the formation of the Hinact-like state in WT CrHydA1 in more detail.
Discussion
In this work, we identified two new H-cluster redox states with electronic structures similar to those of the Htrans and Hinact states.2,11,29 These states have been characterized in detail via a combination of spectroscopic, crystallographic and computational techniques. We revealed that the Htrans-like and Hinact-like states form upon reaction of [FeFe] hydrogenases with CN−. Isotope labelling experiments and X-ray crystallography, supported by computational calculations, suggest that CN− binds at the open coordination site of the H-cluster and, therefore, protects it from O2 binding, as it blocks the vacant site. Unlike typical [FeFe] hydrogenase inhibitors such as CO and H2S, which bind to the H-cluster reversibly,2,57 CN− binding appears to be irreversible, at least under the conditions studied here (pH 8). As noted earlier, purging the DdHydAB C178A variant with CO had no effect (Fig. S1B†) and all samples are initially prepared under an atmosphere of 2% H2, suggesting that neither CO nor H2 can effectively compete off CN−. Although CN− binding to the H-cluster confers air-stability to [FeFe] hydrogenases, the irreversible nature of CN− binding does not make the formation of the Hinact-like state a suitable strategy to protect [FeFe] hydrogenases during aerobic handling, in contrast to the reversible formation of the H2S-dependent Hinact state.2,58For the Cys-to-Ala variants and, to a smaller extent, also for the artificially maturated WT DdHydAB, the Htrans-like state could form even in the absence of exogenous CN−. We suggest that the source of CN− in this case derives from the degradation of the [2Fe]H synthetic cofactor during artificial maturation. Degradation of the [2Fe]H cofactor leads to the dissociation of the CO and CN− ligands, which can in turn bind to the ‘intact’ H-clusters. This process of cofactor “cannibalization” is a well-known source of the Hox–CO state in WT enzymes,29 but this is the first time that CN− binding is also observed. The reason why DdHydAB enzymes (mutant and WT) form more of the Htrans-like state compared with CrHydA1 is because this enzyme requires longer maturation times with a large excess of cofactor and higher temperature (see Materials and methods in the ESI†), therefore, promoting degradation of the [2Fe]H cofactor and allowing the accumulation of more free CN− in solution. In contrast, artificial maturation of CrHydA1 can be achieved in one hour with only a small excess of the [2Fe]H cofactor. Nevertheless, DdHydAB shows an unusually high affinity towards strong-field ligands, i.e. CO and CN−, with a tendency to stabilize them in the apical position of the distal Fe ion, as is well known for the CO-inhibited state Hox–CO in native DdHydAB. For example, Goldet et al. showed that DdHydAB had a 25-fold higher KI (inhibition constant for CO during H2 oxidation) than CrHydA1, and a 330-fold higher KI than HydA1 [FeFe] hydrogenase from Clostridium acetobutylicum (CaHydA).57 In addition, the C169A variant of CrHydA1,23,41,44 the C299A variant of CpHydA1 36 and the C298A variant of CaHydA59 have been produced and studied before. In none of these cases was spontaneous formation of CN−-bound states observed during artificial maturation, further highlighting that the C178A variant of DdHydAB had especially high affinity for CN−. The Hinact-like crystal structure presented in this work (Fig. 4) at atomic resolution, shows a diatomic ligand at the apical position on Fed, which we have modelled as CN−. The reason for this high affinity of DdHydAB towards strong-field ligands is not well understood and needs to be investigated further. Very recently, Duan et al. showed binding of CN− to WT CpHydA1 and CrHydA1 using X-ray crystallography and IR spectroscopy.60 Their structures show an identical binding mode for the CN− ligand, but the authors propose additional hydrogen bonding interactions from the ADT ligand and nearby cysteine to the nitrogen of the CN− ligand. While their IR spectra of CN−-bound WT CpHydA1 appear to be analogous to those from the Hinact-like states of DdHydAB C178A and CrHydA1 C169A reported here, their IR spectrum of CN−-bound CrHydA1 appears to be analogous to our spectrum of CrHydA1 C169A in the Htrans-like state.
Another example that illustrates the exceptional ligand binding properties of DdHydAB is the formation of the H2S-dependent Hinact state. Previous work on WT DdHydAB showed that anaerobic oxidation in the presence of sulfide results in binding of SH2 to the open coordination site, forming the Hinact state, for which the crystal structure revealed a SH− ligand on the apical position of Fed. Sulfide binding to the active site also requires an overoxidized [2Fe]H subcluster (i.e. Fep(II)Fed(II)).2,11 This has been shown to occur in both DdHydAB and CrHydA1,2 but much less effectively in CpHydA1, CaHydA and MeHydA from Megasphaera elsdenii.34 Interestingly, during electrochemistry DdHydAB inactivates with a slightly more negative potential (Eswitch) than CrHydA1 suggesting that H2S binding is faster and H2S release is slower for DdHydAB compared with CrHydA1.2
In both enzymes, DdHydAB and CrHydA1, the Cys-to-Ala mutation in the proton transfer pathway favors the formation of the CN− bound states. We hypothesize that this may be related to the fact that the H-cluster in the Cys-to-Ala variants seems to be electron-deficient compared to the WT enzymes, as suggested by the blue-shifted bands observed in the IR spectra. This is also in line with previous reports on the altered Hox/Hred thermodynamics in CrHydA1 C169S.22 In contrast, in the HydA1 [FeFe] hydrogenase from Clostridium acetobutylicum, the Cys mutation in the PTP to the ionizable residue aspartic acid seems to favor formation of Hox over Hred.40
Why does CN− binding favor Htrans-/Hinact-like states? While CN− is generally considered a good π-acceptor, recent experimental and theoretical studies have shown that it is dominated by σ-donating properties with only weak π-accepting properties.61,62 Therefore, CN− might stabilize higher oxidation states (e.g. the overoxidized [2Fe]H subcluster in the Htrans-like and Hinact-like states [Fep(II)Fed(II)]H) relative for example to CO, which is a better π-acceptor than CN−. According to the IR spectra, the Cys-to-Ala variants appear more electron-deficient and, therefore, they have higher affinity for CN− compared to CO. Although we note that the interaction between the cysteine in the PTP and the H-cluster is crucial to modulate the electronic structure at the active site, the detailed understanding of how the Cys-to-Ala mutation affects the electronic distribution at the H-cluster is beyond the scope of this work. However, we suggest that this mutation affects the hydrogen-bonding network surrounding the H-cluster, in particular concerning the ADT amine which has been shown to be involved in electronic delocalization from the two Fe ions in [2Fe]H model compounds.63 The reduced steric hindrance in the Cys-to-Ala mutants might also provide easier access to the open coordination site for the additional CN− ligand.
Interestingly, during the synthesis of the [2Fe]H precursor, only two of the CO ligands in Fe2[(SCH2)2NH](CO)6 can be substituted with CN−.64 This shows how the protein scaffold plays a crucial role in stabilizing an additional CN− bound to [2Fe]H. One reason could be that within the H-cluster, electron transfer from [2Fe]H to [4Fe-4S]H allows formation of an overoxidized binuclear site, [Fep(II)Fed(II)]H, which is necessary for CN− binding.
Our report of CN− binding to [FeFe] hydrogenases also sheds light on the nature of previously uncharacterized active-site states. We have demonstrated that the unknown states present as impurities in artificially maturated samples of WT DdHydAB are indeed CN− bound states caused by the long artificial maturation of this enzyme. Previous EPR studies on CrHydA1 C169S have shown the formation of an unidentified Htrans-like state, exhibiting almost identical g-values to the one studied here.21,22,43 Thus, it is also plausible that the previously observed Htrans-like state is caused by binding of CN− to the H-cluster, favored by the Cys-to-Ser mutation. A recent study reported the accumulation over a long time-scale (24 h) of this very similar Htrans-like state for both the C169S variant and WT CrHydA1 artificially maturated inside E. coli cells, leading to the inhibition of H2 production by the culture.43 We hypothesize that this Htrans-like state is also CN−-dependent as the one described here.
Cyanide binding to metals in biology is well known, with the most classic example being cytochrome c oxidase of the mitochondrial respiratory chain, where CN− binds between heme a3 and the CuB site.65,66 Interestingly, in the reduced structure, with Fe(II) the Fe–C distance is 2.4 Å (ref. 65) and shortens to 2.0 Å in the oxidized structure, with Fe(III),66 suggesting a shorter stronger bond. Cyanide has also been reported to bind ferric heme-proteins with a very high affinity, e.g. myoglobin67–69 and hemoglobin.70,71 In [NiFe] hydrogenase, CN− is thought to bind transiently to the Ni(II) in the Ni–SIa state, promoting oxidation to Ni(III) and formation of the Ni–B state.72 In CODH, CN− binds again to a Ni(II) ion with a 1.8 Å Ni–C bond,73 and inhibits CO oxidation rather than CO2 reduction suggesting that it also binds favorably to a more oxidized active site. Overall, our results are consistent with literature observations that CN− binds preferentially to more oxidized active sites, or alternatively that binding of CN− favors metal oxidation.
Conclusions
Here, we have reported for the first time, a detailed spectroscopic and computational characterisation of the binding of CN− to the active site of [FeFe] hydrogenases. CN− binding is clearly favored in the Cys-to-Ala mutants, exemplifying the crucial role of the second coordination sphere of the H-cluster in preventing CN− binding, and reflecting the electronic structure adaptations of the H-cluster environment to facilitate stabilization of a terminal Fe(II)-hydride species during catalysis. Overall, our studies showed how the interaction between the Cys in the PTP and the ADT in [2Fe]H tunes the electronic structure of the active site, controlling ligand binding at the open coordination site.Data availability
Data supporting the findings of this study are available in the article and the associated ESI files. Structural data for DdHydAB C178A have been deposited into the Protein Data Bank (PDB) under the following accession codes: 8BJ7 for DdHydAB C178A in the Hinact-like state and 8BJ8 for DdHydAB C178A in the Htrans-like state.†Author contributions
Conceptualization: M. A. M., J. A. B. and P. R.-M.; methodology: M. A. M., K. B., Y. P., C. L., N. B., I. S., R. B.; investigation: M. A. M., K. B., Y. P., C. L., C. W.; writing – original draft: M. A. M., J. A. B and P. R.-M; writing & editing: all authors; supervision: M. A. M., I. S., R. B., I. Z., S. D., J. A. B and P. R.-M.; funding acquisition: J. A. B, P. R.-M., S. D., I. S., I. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Inge Heise and Tabea Mussfeld, for synthesizing the diiron cofactors. We also thank Melissa Jansing, Institut für Physikalische Biologie at Heinreich Heine University Düsseldorf for preliminary data. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. M. A. M., Y. P., N. B., S. D., R. B. and J. A. B. would like to thank the Max Planck Society for funding. J. A. B., M. A. M., I. S., K. B., S. D., C. L., C. W. and I. Z. also acknowledge the Deutsche Forschungsgemeinschaft (DFG) Priority Programme “Iron–Sulfur for Life: Cooperative Function of Iron–Sulfur Centers in Assembly, Biosynthesis, Catalysis and Disease” (SPP 1927) Projects BI 2198/1-1 (J. A. B. and M. A. M.), IS 1476/4-1 (I. S. and K. B.), DE 1877/1-2 (S. D.), and ZE 510/2-2/311062227 (C. L., C. W. and I. Z.). P. R.-M. thanks the University of Oxford for a Glasstone Research Fellowship and Linacre College Oxford for a Junior Research Fellowship. Y. P. thanks the China Scholarship Council for a visiting scholar fellowship.References
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- P. Rodriguez-Macia, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rüdiger and J. A. Birrell, J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef CAS PubMed.
- S. Yoshikawa, D. H. O'Keeffe and W. S. Caughey, J. Biol. Chem., 1985, 260, 3518–3528 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- M. Haumann and S. T. Stripp, Acc. Chem. Res., 2018, 51, 1755–1763 CrossRef CAS PubMed.
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- B. J. Lemon and J. W. Peters, Biochemistry, 1999, 38, 12969–12973 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, L. M. Galle, R. Bjornsson, C. Lorent, I. Zebger, Y. Yoda, S. P. Cramer, S. DeBeer, I. Span and J. A. Birrell, Angew. Chem., Int. Ed., 2020, 59, 16786–16794 CrossRef PubMed.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 17331–17336 CrossRef CAS PubMed.
- C. Lambertz, N. Leidel, K. G. V. Havelius, J. Noth, P. Chernev, M. Winkler, T. Happe and M. Haumann, J. Biol. Chem., 2011, 286, 40614–40623 CrossRef CAS PubMed.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS PubMed.
- J. Esselborn, L. Kertess, U.-P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS PubMed.
- J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, Coord. Chem. Rev., 2021, 449, 214191 CrossRef CAS.
- E. J. Reijerse, V. Pelmenschikov, J. A. Birrell, C. P. Richers, M. Kaupp, T. B. Rauchfuss, S. P. Cramer and W. Lubitz, J. Phys. Chem. Lett., 2019, 10, 6794–6799 CrossRef CAS PubMed.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS PubMed.
- S. Katz, J. Noth, M. Horch, H. S. Shafaat, T. Happe, P. Hildebrandt and I. Zebger, Chem. Sci., 2016, 7, 6746–6752 RSC.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 16894–16902 CrossRef CAS PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- A. J. Pierik, W. R. Hagen, J. S. Redeker, R. B. Wolbert, M. Boersma, M. F. Verhagen, H. J. Grande, C. Veeger, P. H. Mutsaers and R. H. Sands, et al. , Eur. J. Biochem., 1992, 209, 63–72 CrossRef CAS PubMed.
- D. S. Patil, J. J. Moura, S. H. He, M. Teixeira, B. C. Prickril, D. V. DerVartanian, H. D. Peck Jr, J. LeGall and B. H. Huynh, J. Biol. Chem., 1988, 263, 18732–18738 CrossRef CAS PubMed.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, JBIC, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- S. P. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS PubMed.
- P. S. Corrigan, J. L. Tirsch and A. Silakov, J. Am. Chem. Soc., 2020, 142, 12409–12419 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U.-P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Léger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- C. Felbek, F. Arrigoni, D. de Sancho, A. Jacq-Bailly, R. B. Best, V. Fourmond, L. Bertini and C. Léger, ACS Catal., 2021, 11, 15162–15176 CrossRef CAS.
- G. Hong, A. J. Cornish, E. L. Hegg and R. Pachter, Biochim. Biophys. Acta Bioenerg., 2011, 1807, 510–517 CrossRef CAS PubMed.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef PubMed.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS PubMed.
- A. J. Cornish, K. Gärtner, H. Yang, J. W. Peters and E. L. Hegg, J. Biol. Chem., 2011, 286, 38341–38347 CrossRef CAS PubMed.
- P. Knorzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- S. Morra, A. Giraudo, G. Di Nardo, P. W. King, G. Gilardi and F. Valetti, PLoS One, 2012, 7, e48400 CrossRef CAS PubMed.
- S. Rumpel, C. Sommer, E. Reijerse, C. Fares and W. Lubitz, J. Am. Chem. Soc., 2018, 140, 3863–3866 CrossRef CAS PubMed.
- C. C. Pham, D. W. Mulder, V. Pelmenschikov, P. W. King, M. W. Ratzloff, H. Wang, N. Mishra, E. E. Alp, J. Zhao, M. Y. Hu, K. Tamasaku, Y. Yoda and S. P. Cramer, Angew. Chem., Int. Ed., 2018, 57, 10605–10609 CrossRef CAS PubMed.
- M. Lorenzi, P. Ceccaldi, P. Rodriguez-Macia, H. J. Redman, A. Zamader, J. A. Birrell, L. S. Meszaros and G. Berggren, J. Biol. Inorg Chem., 2022, 27, 345–355 CrossRef CAS PubMed.
- S. Mebs, R. Kositzki, J. Duan, L. Kertess, M. Senger, F. Wittkamp, U. P. Apfel, T. Happe, S. T. Stripp, M. Winkler and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 28–41 CrossRef CAS PubMed.
- J. A. Birrell, K. Wrede, K. Pawlak, P. Rodriguez-Maciá, O. Rüdiger, E. J. Reijerse and W. Lubitz, Isr. J. Chem., 2016, 56, 852–863 CrossRef CAS.
- P. Rodríguez-Maciá, K. Pawlak, O. Rüdiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, J. Am. Chem. Soc., 2017, 139, 15122–15134 CrossRef PubMed.
- M. Senger, S. Mebs, J. Duan, F. Wittkamp, U.-P. Apfel, J. Heberle, M. Haumann and T. Stripp Sven, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, 8454–8459 CrossRef CAS PubMed.
- C. Lorent, V. Pelmenschikov, S. Frielingsdorf, J. Schoknecht, G. Caserta, Y. Yoda, H. Wang, K. Tamasaku, O. Lenz, S. P. Cramer, M. Horch, L. Lauterbach and I. Zebger, Angew. Chem., Int. Ed., 2021, 60, 15854–15862 CrossRef CAS PubMed.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS PubMed.
- M.-E. Pandelia, H. Ogata, L. J. Currell, M. Flores and W. Lubitz, JBIC, J. Biol. Inorg. Chem., 2009, 14, 1227–1241 CrossRef CAS PubMed.
- G. Caserta, V. Pelmenschikov, C. Lorent, A. F. Tadjoung Waffo, S. Katz, L. Lauterbach, J. Schoknecht, H. Wang, Y. Yoda, K. Tamasaku, M. Kaupp, P. Hildebrandt, O. Lenz, S. P. Cramer and I. Zebger, Chem. Sci., 2021, 12, 2189–2197 RSC.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- M. A. Martini, O. Rüdiger, N. Breuer, B. Nöring, S. DeBeer, P. Rodríguez-Maciá and J. A. Birrell, J. Am. Chem. Soc., 2021, 143, 18159–18171 CrossRef CAS PubMed.
- A. Ciaccafava, D. Tombolelli, L. Domnik, J. Fesseler, J.-H. Jeoung, H. Dobbek, M. A. Mroginski, I. Zebger and P. Hildebrandt, Chem. Sci., 2016, 7, 3162–3171 RSC.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- A. A. Oughli, S. Hardt, O. Rüdiger, J. A. Birrell and N. Plumeré, Chem. Commun., 2020, 56, 9958–9961 RSC.
- T. Lautier, P. Ezanno, C. Baffert, V. Fourmond, L. Cournac, J. C. Fontecilla-Camps, P. Soucaille, P. Bertrand, I. Meynial-Salles and C. Léger, Faraday Discuss., 2011, 148, 385–407 RSC.
- J. Duan, A. Hemschemeier, D. J. Burr, S. T. Stripp, E. Hofmann and T. Happe, Angew. Chem., Int. Ed., 2023, 62(7), e202216903 CrossRef CAS PubMed.
- N. Levin, S. Peredkov, T. Weyhermüller, O. Rüdiger, N. B. Pereira, D. Grötzsch, A. Kalinko and S. DeBeer, Inorg. Chem., 2020, 59, 8272–8283 CrossRef CAS PubMed.
- S. K. Singh, J. Eng, M. Atanasov and F. Neese, Coord. Chem. Rev., 2017, 344, 2–25 CrossRef CAS.
- Ö. F. Erdem, M. Stein, S. Kaur-Ghumaan, E. J. Reijerse, S. Ott and W. Lubitz, Chem.–Eur. J., 2013, 19, 14566–14572 CrossRef PubMed.
- M. Schmidt, S. M. Contakes and T. B. Rauchfuss, J. Am. Chem. Soc., 1999, 121, 9736–9737 CrossRef CAS.
- K. Muramoto, K. Ohta, K. Shinzawa-Itoh, K. Kanda, M. Taniguchi, H. Nabekura, E. Yamashita, T. Tsukihara and S. Yoshikawa, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7740–7745 CrossRef CAS PubMed.
- N. Yano, K. Muramoto, M. Mochizuki, K. Shinzawa-Itoh, E. Yamashita, S. Yoshikawa and T. Tsukihara, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2015, 71, 726–730 CrossRef CAS PubMed.
- Y. Dou, J. S. Olson, A. J. Wilkinson and M. Ikeda-Saito, Biochemistry, 1996, 35, 7107–7113 CrossRef CAS PubMed.
- M. Bolognesi, C. Rosano, R. Losso, A. Borassi, M. Rizzi, J. B. Wittenberg, A. Boffi and P. Ascenzi, Biophys. J., 1999, 77, 1093–1099 CrossRef CAS PubMed.
- P. Ascenzi, A. di Masi, F. Gullotta, M. Mattu, C. Ciaccio and M. Coletta, Biochem. Biophys. Res. Commun., 2010, 393, 196–200 CrossRef CAS PubMed.
- M. Milani, Y. Ouellet, H. Ouellet, M. Guertin, A. Boffi, G. Antonini, A. Bocedi, M. Mattu, M. Bolognesi and P. Ascenzi, Biochemistry, 2004, 43, 5213–5221 CrossRef CAS PubMed.
- A. Boffi, E. Chiancone, E. S. Peterson, D. L. Rousseau and J. M. Friedman, Biochemistry, 1997, 36, 4510–4514 CrossRef CAS PubMed.
- S. V. Hexter, M.-W. Chung, K. A. Vincent and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 10470–10477 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, J. Am. Chem. Soc., 2009, 131, 9922–9923 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06098a |
| This journal is © The Royal Society of Chemistry 2023 |