
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly sensitive nanochannel device for the detection of SUMO1 peptides†
Yue
Qin‡
ab,
Xiaoyu
Zhang‡
 b,
Yanling
Song
a,
Bowen
Zhong
b,
Lu
Liu
b,
Dongdong
Wang
b,
Yahui
Zhang
b,
Wenqi
Lu
b,
Xinjia
Zhao
b,
Zhiqi
Jia
a,
Minmin
Li
b,
Lihua
Zhang
*b and
Guangyan
Qing
*bc
b,
Yanling
Song
a,
Bowen
Zhong
b,
Lu
Liu
b,
Dongdong
Wang
b,
Yahui
Zhang
b,
Wenqi
Lu
b,
Xinjia
Zhao
b,
Zhiqi
Jia
a,
Minmin
Li
b,
Lihua
Zhang
*b and
Guangyan
Qing
*bc
aCollege of Pharmaceutical and Biological Engineering, Shenyang University of Chemical Technology, No. 11 Street, Economic and Technological Development Zone, Shenyang 110142, P. R. China
bCAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: qinggy@dicp.ac.cn; lihuazhang@dicp.ac.cn
cCollege of Chemistry and Chemical Engineering, Wuhan Textile University, 1 Sunshine Road, Wuhan 430200, P. R. China
First published on 13th July 2023
Abstract
SUMOylation is an important and highly dynamic post-translational modification (PTM) process of protein, and its disequilibrium may cause various diseases, such as cancers and neurodegenerative disorders. SUMO proteins must be accurately detected to understand disease states and develop effective drugs. Reliable antibodies against SUMO2/3 are commercially available; however, efficient detectors are yet to be developed for SUMO1, which has only 50% homology with SUMO2 and SUMO3. Here, using phage display technology, we identified two cyclic peptide (CP) sequences that could specifically bind to the terminal dodecapeptide sequence of SUMO1. Then we combined the CPs and polyethylene terephthalate conical nanochannel films to fabricate a nanochannel device highly sensitive towards the SUMO1 terminal peptide and protein; sensitivity was achieved by ensuring marked variations in both transmembrane ionic current and Faraday current. The satisfactory SUMO1-sensing ability of this device makes it a promising tool for the time-point monitoring of the SENP1 enzyme-catalyzed de-SUMOylation reaction and cellular imaging. This study not only solves the challenge of SUMO1 precise recognition that could promote SUMO1 proteomics analysis, but also demonstrates the good potential of the nanochannel device in monitoring of enzymes and discovery of effective drugs.
Introduction
The covalent attachment of small ubiquitin-like modifier (SUMO) protein (size, ∼11 kD) to substrate proteins is an important and widely distributed post-translational modification (PTM).1,2 This modification is highly conserved in eukaryotic cells and has various cellular regulatory functions.3 SUMOylation is a highly dynamic process;4 the disequilibrium of SUMOylation may lead to the development of neurodegenerative diseases5 and cancers.6 In mammalian cells, the SUMOylation of proteins involves a cascade of enzymatic reactions (Fig. 1a),7 including the activation of SUMO by the E1 enzyme SAE1/SAE2 (also known as Aosl1/Uba2), the transfer of SUMO to ubiquitin carrier protein 9 (UBC9) through the E2 conjugating enzyme, and the ligation of SUMO to the target protein through E3 ligases. Only one type of E2 conjugating enzyme but a variety of E3 ligases can be found in mammalian cells.8 SUMO proteins can bind to one or more lysine residues of a target protein, or a chain of SUMO proteins may attach to a single lysine residue of the target protein.9 In mammals, SUMO-specific proteases (SENPs), which are the primary regulators of intracellular SUMO-related homeostasis,10 catalyze the removal of SUMO proteins from a modified target protein by breaking isopeptide bonds; thus SUMO proteins are removed from the target protein and can re-enter the SUMOylation cycle.11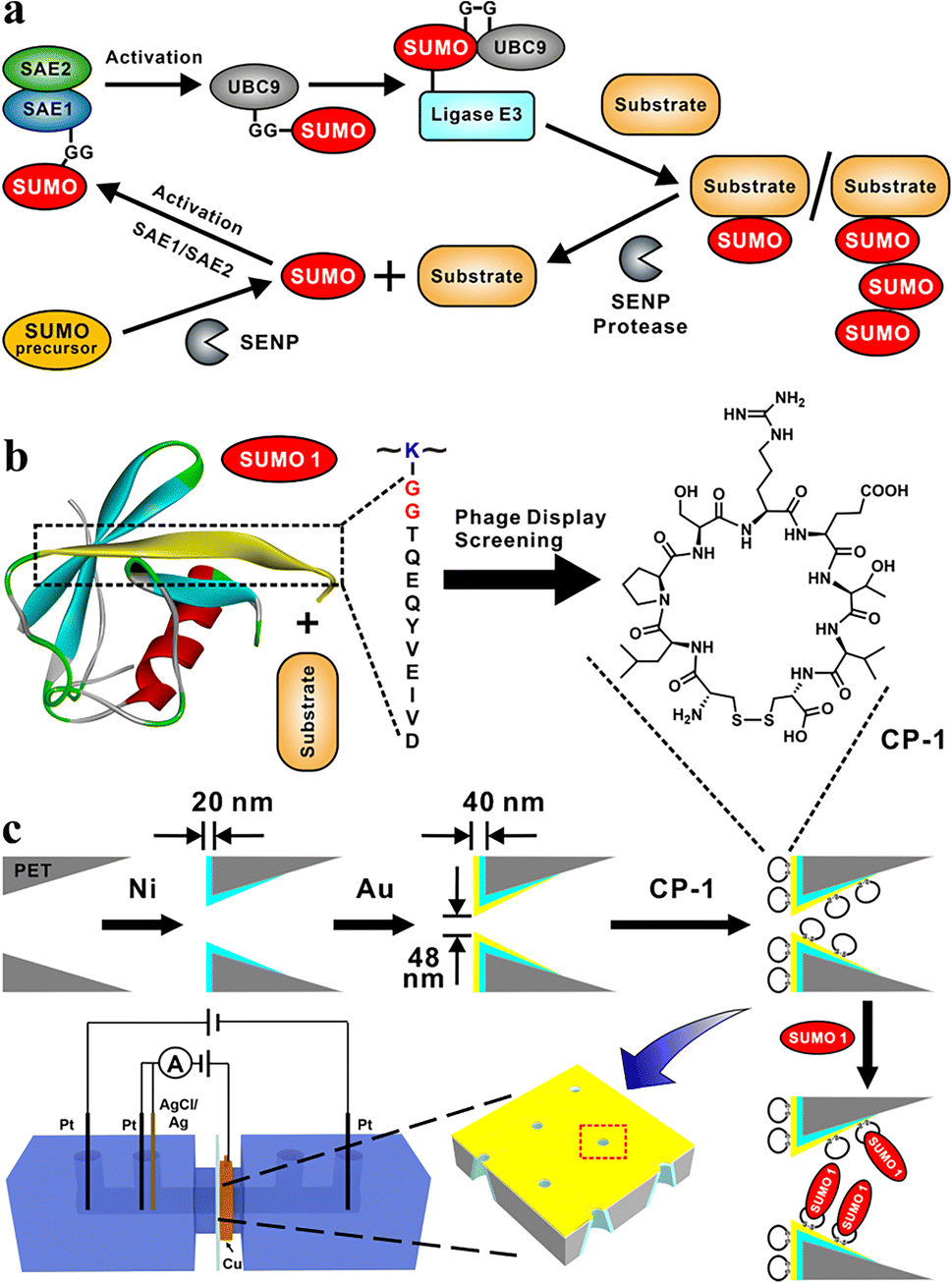 | ||
| Fig. 1 SUMOylation and the design of functional nanochannels for the detection of SUMO1. (a) Catalytic cycle of SUMOylation. (b) Phage display screening for CPs capable of binding DG12, a key region of SUMO1, leading to the identification of CP-1, which had strong affinity towards DG12. (c) Modifications, such as etching, nickel and gold plating, and CP-1 adsorption of nanochannel films to fabricate a highly sensitive SUMO1 detection device by using both transmembrane ionic current and Faraday current. | ||
Uncontrolled SUMOylation and de-SUMOylation may lead to homeostasis dysregulation in cells,12,13 and central to the monitoring of SUMOylation are SENPs, and the importance of SENP1 in clinical and molecular studies has been enumerated by Woźniak et al. and is shown in Table S1.†14,15 The silencing of cardiac-targeted PIASy through the de-SUMOylation of Cav-3 partially prevented the downregulation of the Nav 1.5 protein in the plasma membrane of cardiomyocytes during ischemia/reperfusion injury, thus reducing the incidence of ventricular arrhythmia in rats.16 TAK-981 selectively reduces SUMOylation, thereby preventing cell proliferation and causing mitotic failure and defective chromosome segregation in pancreatic cancer cells.17 For telomere maintenance, the disruption of the alternative lengthening of telomere-associated promyelocytic leukemia bodies is a potential target for cancer drugs; this disruption mainly involves SUMOylation.18 Considering the importance of de-SUMOylation, sensitive and specific detection methods must be developed for monitoring this process for facilitating drug development and imaging; for this, precise recognition and detection of SUMO proteins are crucial.
Mature forms of SUMO2 and SUMO3 exhibit 97% amino acid sequence homology and are indistinguishable; therefore, these two forms are often studied together. However, they exhibit only 50% homology with SUMO1, which remains to be studied extensively.8 SUMO1 varies considerably from SUMO2 and SUMO3 in terms of function and in vivo substrate proteins. Compared with other PTMs,19–21 SUMOylation is difficult to explore, primarily because of the transient nature of this process,4 low levels of SUMOylated proteins, and lack of reliable detection tools. Particularly, the available SUMO1 antibodies do not enable the effective detection of this protein, which hinders the discovery of the functions of SUMO1.22,23 These problems have been indicated as the major contributors to reproducibility crises.24
In the present study, we designed a sensitive nanochannel device25–27 for the detection of SUMO1. Using the phage display technology, we first identified a series of cyclic peptides (CPs) capable of specifically binding to the key region of SUMO1 (DG12: DVIEVYQEQTGG, Fig. 1b). Then, the CPs were immobilized on the gold-coated (thickness, 20 nm) tip side28 of nanochannels in etched conical polyethylene terephthalate (PET) films.29 High sensitivity and specificity30 of DG12 and SUMO1 detection were achieved using an electrochemical cell.31 This cell had two separate measurement systems, which enabled transmembrane ionic current and Faraday current to be simultaneously recorded32 (Fig. 1c and Schemes S1–S3 in the ESI†). This nanochannel device further realized the time-point monitoring of SENP1-catalyzed de-SUMOylation, which may facilitate the screening of effective drugs.
Results and discussion
Phage display biopanning helps screen peptides with high affinity towards target proteins of interest from a large library of candidate peptides (diversity, approximately 109). To identify SUMO1-specific peptides, we performed phage display screening. For this, we used a library constructed using peptide data from a filamentous phage displaying five copies of CPs on the phage surface; these CPs generally were cystine-linked cyclic heptapeptides (CX7C; X is any amino acid).33 The structures of CPs are more rigid than that of linear peptides of the same length; this reduces the conformational flexibility and increases the proteolytic stability of CPs.34 In this study, a series of SUMO1-binding CPs were identified through complete biopanning, which included one round of reverse biopanning against bovine serum albumin (BSA, which reduces the probability of non-specific binding) and four rounds of selection against DG12. After the complete biopanning procedure, we randomly selected DG12-binding phage clones and successfully obtained the DNA sequences of 7 of these clones. Fig. 2a depicts the corresponding peptide sequences. The DG12-binding affinity of the selected phages bearing CPs was confirmed through enzyme-linked immunosorbent assay; the results are presented in Fig. 2b. Finally, we selected CP-1 and CP-2, which exhibited relatively high affinities, as the candidate peptides for the subsequent experiments. Protein blast searches performed on the National Center for Biotechnology Information website (https://www.ncbi.nlm.nih.gov/BLAST/) by using CP-1 and CP-2 sequences revealed no homologous sequences in the database, indicating that the two CPs are novel peptides. The synthesized peptides (CP-1/2 and Rhodamine B-labelled CP-1) were characterized through high-performance liquid chromatography and mass spectrometry35,36 (Fig. S1–S3 in the ESI†).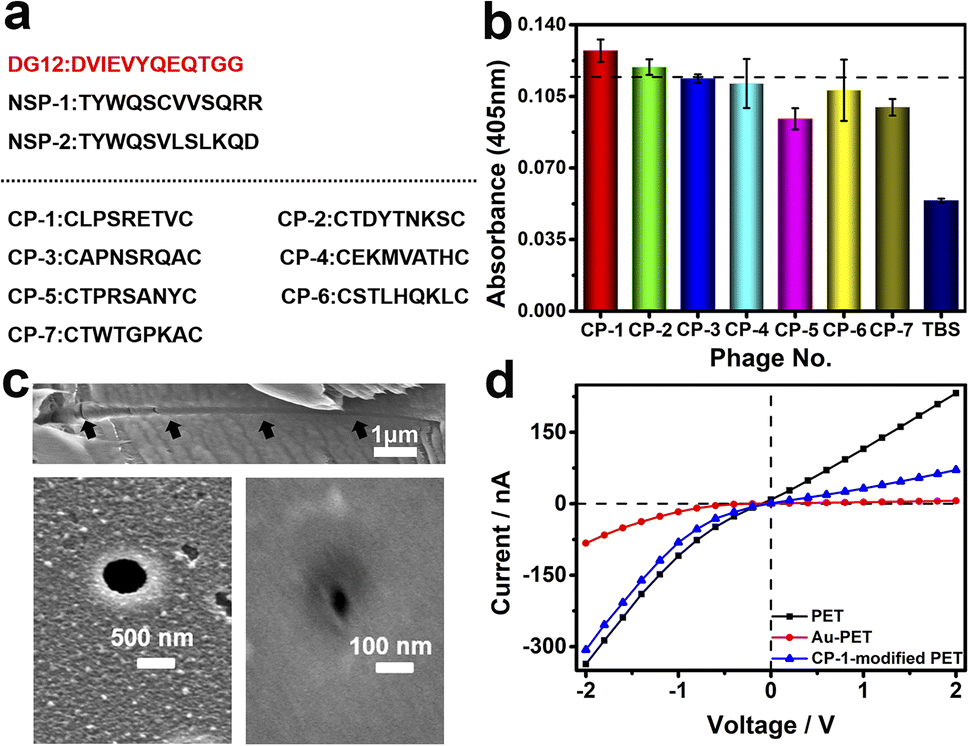 | ||
| Fig. 2 (a) Peptide sequences used in this study (NSP-1/2: non-SUMO peptide 1/2). (b) Results of the enzyme-linked immunosorbent assay performed to evaluate the DG12-binding affinity of seven CPs. Error bars represent the standard deviations from three measurements, n = 3. (c) Field emission scanning electron microscopy images of the cross-section (top), base side (bottom left), and tip side (bottom right) of PET conical nanochannels. (d) Current–voltage (I–V) curves of a bare PET film (black) and a gold-coated PET film before (red) and after (blue) CP-1-mediated modification (0.25 mg mL−1) at 25 °C. | ||
A PET film with conical nanochannels (1 × 107 pores per cm2) was prepared through asymmetric ion-track etching.37 The average diameters of the small (tip side) and large (base side) openings of the nanopores were 48 and 680 nm, respectively, and the length of the conical nanochannel was 12 μm (Fig. 2c). The distribution of surface charge on the tip side of nanopores strongly affects transmembrane ionic current;38 therefore, a thin layer of gold was plated on this side to improve the sensitivity of the nanochannel device. The deposition of a thin layer of Au on the surface of the tip side of the PET film has two roles; on the one hand, the nanopore becomes smaller, which will improve the sensitivity of the nanopore; on the other hand, Au has excellent conductivity, capable of building an electrode interface for the Faraday current measurement, which has been confirmed by Xia et al.39 The signals for both transmembrane ionic current and Faraday current could be recorded (Fig. 1c). Specifically, a layer of nickel was sputtered onto the tip side of the PET film by using a coater; then, a layer of gold was sputtered on the same side. The average thickness of both the nickel and gold layers was 20 nm (Fig. S4 in the ESI†); the deposition rate was 0.02 nm s−1.40 Subsequently, CP-1 was immobilized on the gold layer through gold–sulfur bonding.41 We used 2-mercaptoethanol to shield the residual active sites on the gold-coated surface and maintain the well-organized structure of the CP-1 monolayer.
The analysis of the X-ray photoelectron spectrum, quartz crystal microbalance with dissipation (QCM-D), and surface contact angle data of the gold-coated PET film indicated apparent changes after CP-1-mediated modification (Fig. S5–S7 in the ESI†). The film was tightly mounted between two Teflon modules in a current measurement apparatus made in our laboratory (Scheme S1 in the ESI†). Then, transmembrane ionic current was measured after injecting 5 mM Tris–HCl buffer solution (electrolyte, TBS) into the two modules. As shown in Fig. 2d, due to the deposition of gold, the diameter of the original tip side becomes smaller, resulting in a larger rectification ratio (ratio of 13). The transmembrane ionic current value at −2 V (a similar voltage was used in the subsequent experiment) increased from −337 to −83 nA. This value increased to −307 nA when the film was modified using CP-1 (Fig. 2d). The self-assembly of CP-1 on the gold layers deposited on the nanochannels further decreased the rectification ratio because CP-1 is a neutral biomolecule that changed the surface charge remarkably.42 These findings confirmed the successful modification of the PET film.
TBS containing different concentrations of DG12 was sequentially added to the electrolytic cell. For the CP-1-modified nanochannel device, when the concentration of DG12 was 1 × 10−12 M, the value of transmembrane ionic current increased from −307 to −278 nA, which further increased gradually with the increasing concentration of DG12 (Fig. 3a). Finally, the transmembrane ionic current increased to −48 nA in the presence of 1 × 10−6 M DG12, denoting a current increase ratio of approximately 84% compared with the value noted for a blank buffer solution. The complexation between CP-1 and DG12 caused the original monomolecular layer to become complex, impeding electrolyte transport in the narrow nanochannel space, thus leading to the change in the transmembrane ionic current. Furthermore, the differential pulse voltammetry43,44 spectra of Faraday current exhibited prominent variations (Fig. 3b). The variations in the current signal indicated a concentration (DG12)-dependent decreasing trend. The current value at −0.4 V increased from −3.3 to −2 μA in the presence of 1 × 10−6 M DG12 solution; the characteristic peak potential shifted from −0.4 to −0.6 V. Initially, only CP-1 is present on the gold electrode surface, and when DG12 is added, the complexation between CP-1 and DG12 contributes to a new characteristic peak, accompanied by a remarkable change in the Faraday current. Remarkable variations in both transmembrane ionic current and Faraday current indicated that the binding between CP-1 and DG12 influenced the ionic transport and charge transfer from the bulk solution to the gold electrode surface. We evaluated whether the concentration of TBS would impact the sensitivity of the device. As shown in Fig. S8 in the ESI,† the device displays remarkable response to DG12 in 10 mM TBS, and the variations are close to that detected in 5 mM TBS.
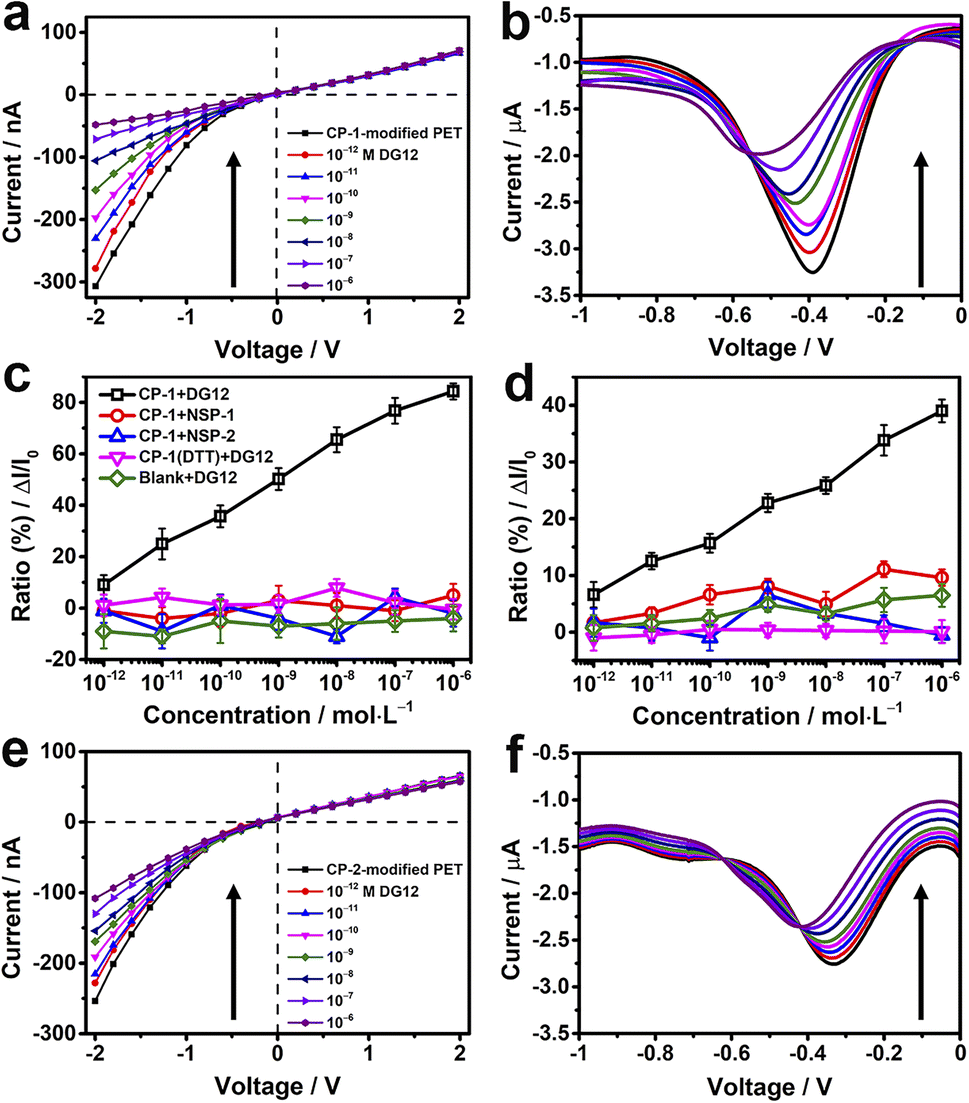 | ||
| Fig. 3 CP-modified nanochannels for the detection of DG12. (a, b, e and f) Transmembrane ionic current curves (a and e) and Faraday current curves (b and f) of CP-1-modified (a and b) and CP-2-modified (e and f) nanochannels in response to addition of different concentrations (10−12 to 10−6 M) of DG12 in TBS (5 mM and pH 8.0) at 25 °C. (c and d) Concentration-dependent transmembrane ionic current increase ratios (ΔI/I0) at −2 V (c) and Faraday current increase ratios (ΔI/I0) at −0.4 V (d) of CP-1-modified nanochannels in response to DG12 (black), two non-SUMO peptides NSP-1 (red) and NSP-2 (blue), or the ratios of the dithiothreitol (DTT)-treated CP-1-modified (purple) or bare gold-coated PET film (green) in response to DG12. n = 3. | ||
A linear relationship was identified between the concentration of DG12 and the variation in transmembrane ionic current or Faraday current (Fig. 3c and d; black lines), which was favorable for DG12 detection. To evaluate the specificity of the CP-1-modified nanochannel device for DG12, two non-SUMO peptides (NSPs) were used. No evident change was detected after the addition of NSP-1 or NSP-2. We further used BSA tryptic digest solution containing >100 types of peptides to mimic a relatively complex biological sample; notably, the CP-1-modified nanochannel device could detect DG12 even in this complex sample (Fig. S9 in the ESI†).45
We further investigated the role of the rigid cyclic structure of CP-1 in molecular recognition. When the sulfur–sulfur bond of CP-1 was broken using dithiothreitol (DTT), CP-1 transformed into a linear peptide, which was assembled onto the nanochannel device. Under these conditions, no considerable change was noted in the current signal (Fig. 3c and d; purple lines), highlighting the crucial role of the cyclic structure of CP-1 in DG12 detection. This presumption was further validated by a bare gold-coated PET film, which had negligible sensitivity toward DG12 (Fig. S10 in the ESI†). In addition to CP-1 that was screened from phage display, the CP-2-modified nanochannel device also exhibited considerable variations in the current signal upon interaction with DG12 (Fig. 3e and f), and the power of phage display screening technology speaks for itself. Finally, a control experiment was designed, in which CP-1 was immobilized on a bare PET film with conical nanochannels. The transmembrane ionic current of this device only decreased by 10% compared to the initial value when 1 μM DG12 was added (Fig. S11 in the ESI†). Therefore, we presumed that the detection sensitivity could be largely improved by plating a thin gold layer on the tip of the nanochannels.
To investigate the electrochemical behaviors of the surface of the nanochannel films, CP-1 was immobilized on a gold-coated electrode surface through gold–sulfur bonding,46 and electrochemical impedance spectroscopy was performed. The semicircle diameter representing resistance gradually increased with the increasing concentration of DG12 (Fig. 4a). Charge transfer resistance (Rct) exhibited a linear relationship with the DG12 concentration (Fig. 4b). Rct increased from 831 ± 138 to 2886 ± 481 Ω in the presence of 10 nM DG12, corresponding to a 277% Rct increase from the initial value. We assumed that complexation between CP-1 and DG12 impeded the transfer of Fe(CN)63−/4− from solution to the gold electrode surface, thus increasing Rct. Cyclic voltammetry further indicated the complex formation (Fig. S12 in the ESI†). Control experiments performed using NSP-1, the DTT-treated CP-1-modified electrode, and the bare electrode further confirmed the satisfactory specificity of CP-1 for DG12 (Fig. 4b and S13 in the ESI†).
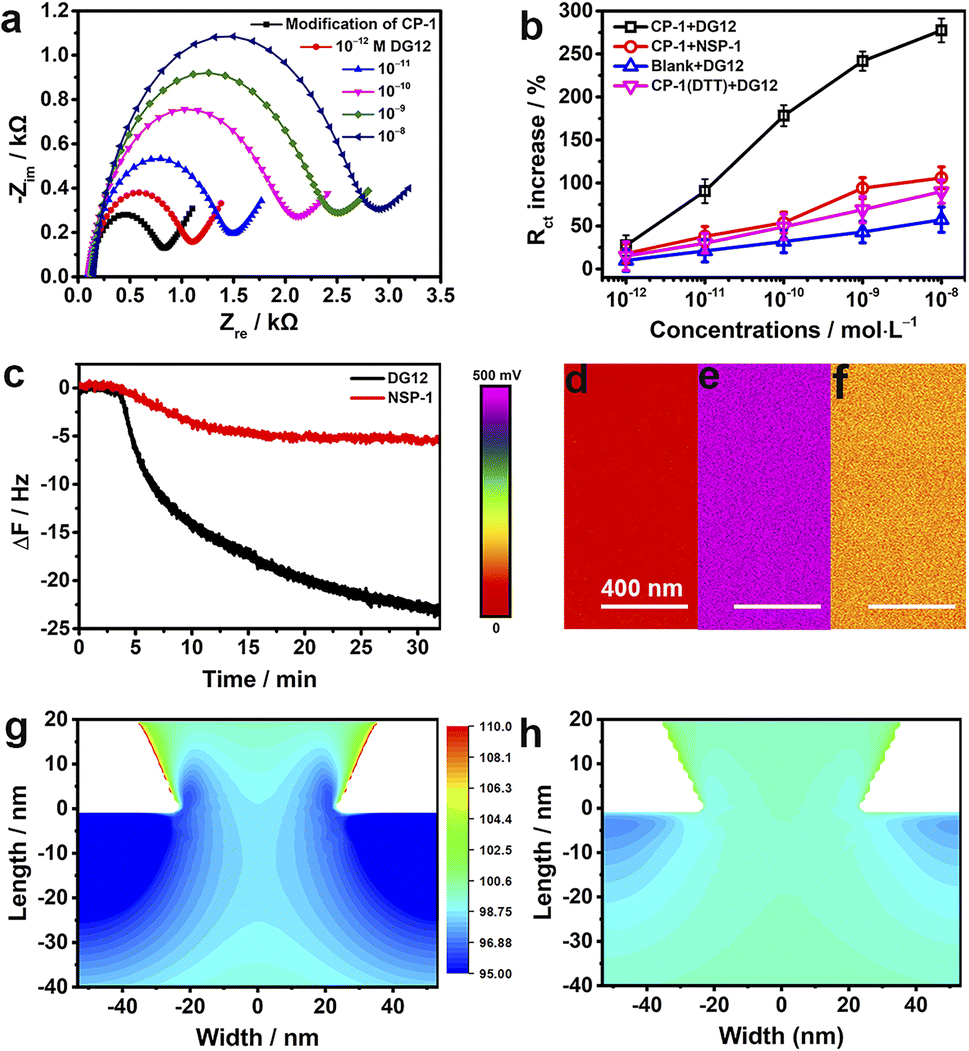 | ||
| Fig. 4 Mechanisms. (a) Nyquist plots (−Zimvs. −Zre) constructed using data obtained by measuring the electrochemical impedance of the CP-1-modified gold electrode in 0.1 M TBS containing 5 mM Fe(CN)63−/4− with different concentrations (10−12 to 10−6 M) of DG12. The solid lines indicate the data fitted to the equivalent circuit at 25 °C. (b) Concentration-dependent variations in the Rct of the CP-1-modified gold electrode in response to DG12 (blank) and NSP-1 (red) and of the DTT-treated CP-1-modified (purple) or bare gold electrode (blue) in response to DG12. n = 3. (c) ΔF on the CP-1-modified QCM-D sensor surface upon addition (1 mM) of DG12 (blank) or NSP-1 (red) in TBS at 25 °C and flow rate: 100 μL min−1. (d–f) Surface potential of the gold-coated PET film (d) and the CP-1-modified PET film before (e) and after (f) its immersion in DG12 solution (1 mM); measurements were performed using a Bruker Dimension Icon atomic force microscope in Kelvin probe force microscopy mode. COMSOL-based numerical simulation of the distribution of K+ on the tip side of the CP-1-modified nanochannel device (g) and the CP-1-nanochannel device after interaction with DG12 (h). | ||
The dynamic adsorption of DG12 on the CP-1-modified gold surface was monitored through QCM-D. CP-1 was immobilized on a quartz crystal sensor surface and then washed with ultrapure water and TBS (Fig. S14 in the ESI†). After equilibrium, DG12 buffer solution was added. As shown in Fig. 4c, the frequency of the sensor decreases gradually and plateaus at approximately 30 min, indicating a typical chemical absorption process. The final frequency variation (ΔF) was 24 Hz, and the adsorption amount of DG12 was 141.6 ng cm−2 according to the Sauerbrey equation. An upward curve of energy dissipation (D) was observed with a ΔD of 9 × 10−6 (Fig. S15 in the ESI†), which indicated that the adsorption of DG12 increased the viscoelasticity of the film. When using NSP-1, ΔF was only 6 Hz (Fig. 4c), which was substantially lower than that induced by DG12, suggesting a prominent difference between SUMO and NSP.
After confirming the strong interaction between CP-1 and DG12, we measured the surface potential47 of the PET films in different states by using a Bruker Dimension Icon atomic force microscope in Kelvin probe force microscope mode. As shown in Fig. 4d–f, the surface potential of the gold-coated PET film was 50 mV, which increased to 472 mV after CP-1-mediated modification. After the interaction with DG12, this value reduced to 137 mV.
We propose a plausible explanation for the remarkable variations in transmembrane ionic current from the perspective of surface charge density, which was calculated through an independently electroosmotic flow (EOF) experiment performed as described previously,48,49 which was independent of the surface potential. The EOF velocity was measured by recording the rate at which an electrically neutral probe molecule (e.g., phenol) passed through the nanochannels (Scheme S4 and Fig. S16 in the ESI†). As shown in Fig. S17,† the surface charge density of the bare gold-coated PET film was −0.008e nm2, which decreased to −0.029e nm2 after CP-1-mediated modification. After treatment with DG12, the surface charge density increased to −0.014e nm2, indicating that the negative charge of CP-1 was shielded by the binding between CP-1 and DG12. Changes in surface charge distribution may strongly influence transmembrane ionic current.50 To visualize the effect, we performed a steady-state finite simulation with the modified nanochannels according to Poisson and Nernst–Planck equations using COMSOL Multiphysics (version 5.4).51 The Au layer was considered to be about 20 nm in height inside the tip side, and the charge density change occurred on the Au-coated area. The simulation model was set only to contain the Au-coated area, where the size effect was strongest, and the detailed calculation is presented in Scheme S4 and Fig. S18 in the ESI.†Fig. 4g and h present the two-dimensional profile of the K+ concentration in the two types of nanochannel devices with the corresponding surface charge density values obtained. In the presence of DG12, the K+ concentration in the nanochannels was reduced. The ion reduction was due to the decreased electric double layer on the tip side of the negatively charged channel. DG12 entered the restricted CP-1 nanochannels and was then enriched in these nanochannels. The concentration gradient and the potential gradient induced the permeation of DG12. Finally, selective transport of DG12 was achieved.
Isothermal titration calorimetry was employed to investigate the binding affinity between CP-1 and DG12. Remarkable heat release was detected when CP-1 was titrated into DG12 solution (Fig. 5a). Curve non-linear fitting based on a sequential binding mode (Fig. 5b) gave a dissociation constant (KD) of 6.9 μM between CP-1 and DG12. By comparison, the titration of CP-1 into the NSP-1 solution only generated very weak heat variation, and under these conditions, a reliable KD could not be obtained through non-linear fitting (Fig. S19 in the ESI†). These results proved the satisfactory selectivity of CP-1 for DG12 in terms of binding affinity. Besides, the CP-1-modified sensor exhibited adequate binding to DG12 in biolayer interferometry and led to the generation of a steady-state fitted response curve, and a KD of 5.86 μM was obtained between CP-1 and DG12 (Fig. S20 in the ESI†), which is close to the KD value obtained by ITC titration.
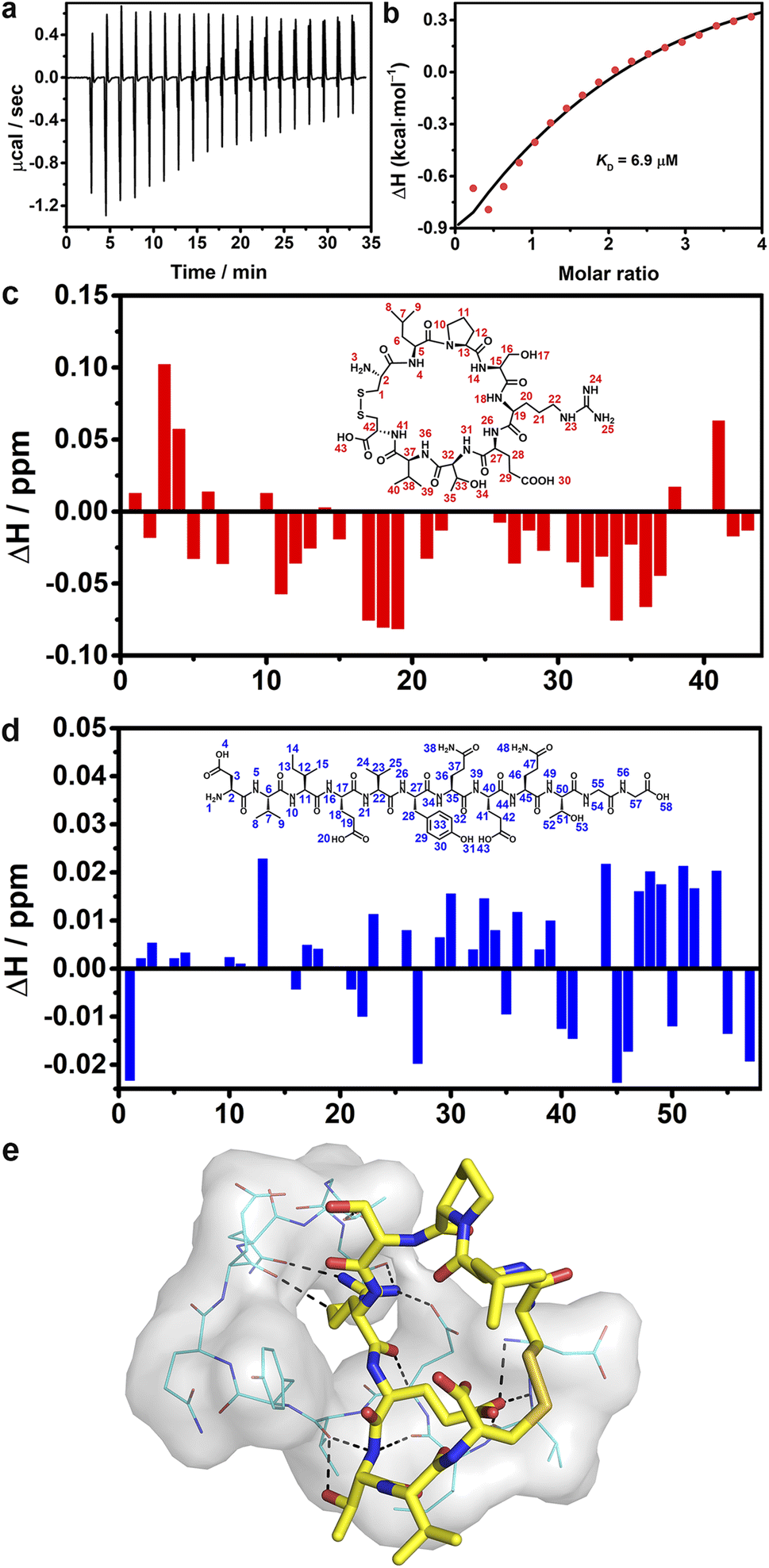 | ||
| Fig. 5 Binding site analysis. (a) ITC raw data of DG12 (0.25 mM) titrated with CP-1 (5 mM) at 25 °C and (b) corresponding fitting curve, and stirring speed: 1000 rpm. The number of titrations is 3. Complexation-induced 1H–1H COSY NMR chemical shift variations in CP-1 (c) and DG12 (d), and the peptide concentration was 20 mM. Data were obtained through an NMR titration experiment at 25 °C in d6-dimethylsulfoxide. (e) Possible binding between DG12 (shown as lines and surface) and CP-1 (shown as sticks), obtained through molecular docking. | ||
1H–1H nuclear magnetic resonance (NMR) spectra provided the details of binding between CP-1 and DG12.52,53 First, the chemical shift attribution of each H proton in CP-1 and DG12 was assigned according to the two-dimensional 1H–1H (correlation spectroscopy) COSY and 13C–1H (heteronuclear singular quantum correlation) HSQC NMR spectra (Fig. S21–S25 in the ESI†). Then, CP-1 was mixed with an equimolar amount of DG12. The chemical shift variation of these H protons was recorded (Fig. 5c and d). Acid–base interaction may cause prominent changes in terminal amines (e.g., H3 in CP-1 and H1 in DG12), which were absent here. The largest changes were observed for H18 (arginine; N–H), H19 (arginine; C–H), H34 (threonine; O–H), and H36 (valine; N–H) in CP-1, and H13 (isoleucine; C–H), H45 (glutamine; C–H), H51 (threonine; C–H), and H54 (glycine; N–H) in DG12. The data indicate that CP-1 mainly bound to the QTGG fragment of DG12, which is a key linkage unit between SUMO1 and the substrate protein. Considering the aforementioned binding site information, we performed molecular docking calculations. As shown in Fig. 5e, DG12 has a random coil conformation (Fig. S26 in the ESI;† circular dichroism spectrum and analysis), and CP-1 is inserted into the cavity of DG12, resulting in close complexation. Nine sets of hydrogen bonds form between CP-1 and DG12 (Fig. S27 in the ESI†). Specifically, the arginine of CP-1 binds to multiple amino acids of DG12 through hydrogen bonding. QTGG is a key binding region in DG12, and it may even be the main binding region. Fourier transform infrared spectroscopy further confirmed the strong binding between CP-1 and DG12 (Fig. S28 in the ESI†).
To evaluate the application potential of CP-1-modified nanochannels in the label free detection of de-SUMOylation reaction, we carried out experiments to monitor the de-SUMOylation reaction with our device. The enzymatic cleavage reaction is a process by which the SUMOylated RanGTPase-activating protein 1 (RanGAP1) is cleaved into RanGAP1 (the substrate protein) and SUMO1 by the action of the SENP1 enzyme (Fig. 6a). As the reaction proceeds, more SUMO1 is dispersed into the solution and recognized by CP-1 immobilized on the sensor surface, at which point the current changes. The more SUMO1 is recognized, the more pronounced the change in current. We used RanGAP1 which is the most abundant SUMO1-modified protein in mammalian cells.54 The SUMOylated RanGAP1 peptide (SUMO1 is located at the site K524 of RanGAP1) and SUMO1 peptide were introduced, and their peptide sequences are presented in Fig. 6a. First, the response of CP-1-modified nanochannels to these peptides was assessed. As shown in Fig. 6b and c, the addition of the SUMO1 caused considerable variations in transmembrane ionic current and Faraday current signals, which were reduced by 17% and 16%, respectively. By contrast, no apparent change was noted when using the RanGAP1 peptide or SUMOylated RanGAP1 (Fig. S29 in the ESI† and insets of Fig. 6b and c). Notably, the nanochannel device exhibited no response to SUMOylated RanGAP1; because the key QTGG fragment of SUMO1 was linked to RanGAP1, the steric hindrance might have hindered CP-1 recognition. This result meets the specificity requirement for SUMO1 detection, thus laying the foundation for the monitoring of the SENP-catalyzed de-SUMOylation reaction.
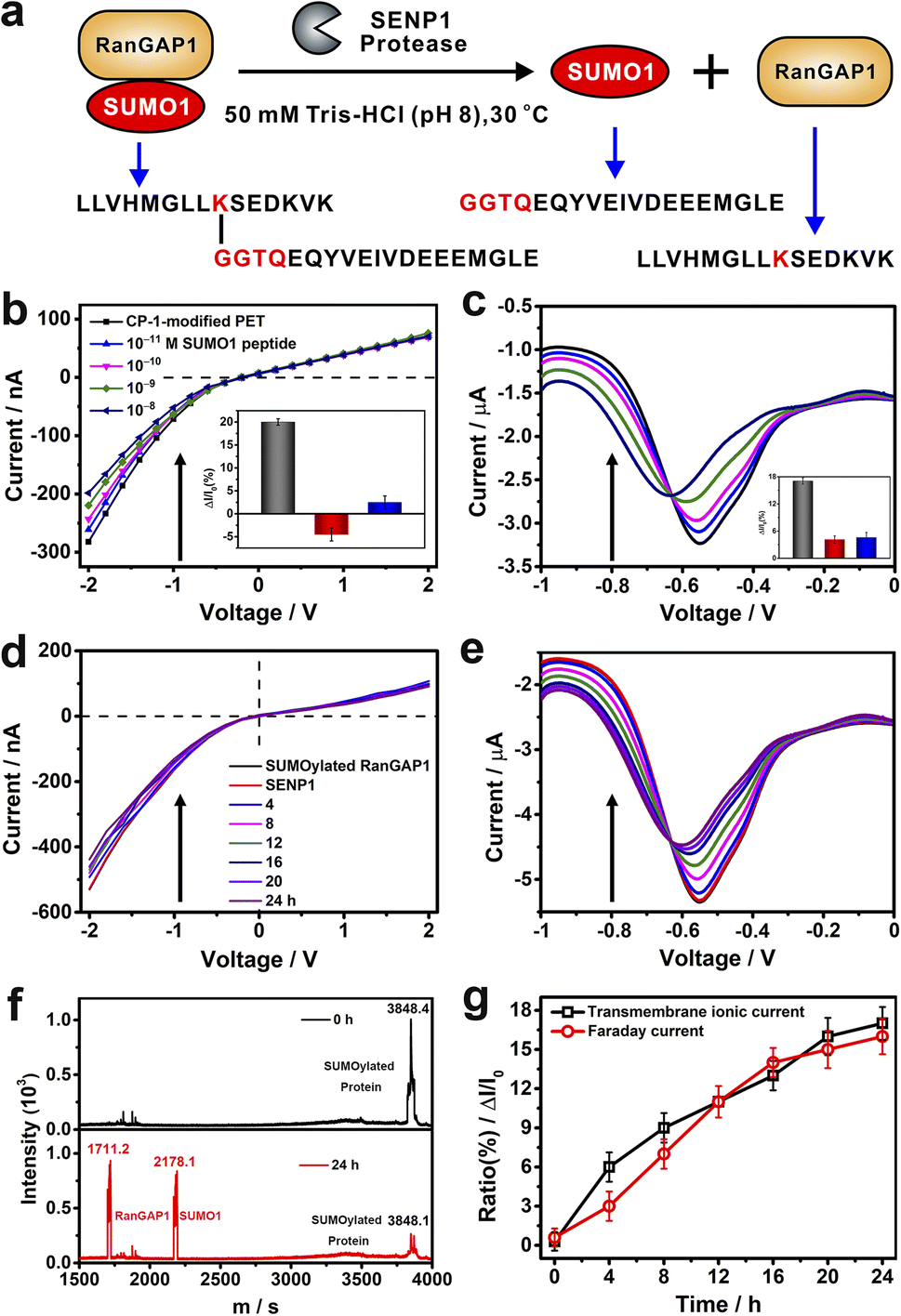 | ||
Fig. 6 (a) Schematic of de-SUMOylation. (b and c) Variations in transmembrane ionic current (b) and Faraday current (c) signals generated by the CP-1-modified nanochannel device in response to different concentrations of SUMO1 in TBS at 25 °C. The insets show the current increase ratios for SUMO1 (blank), RanGAP1 (red) and SUMOylated RanGAP1 (blue). Time-dependent transmembrane ionic current (d) and Faraday current (e) signals generated by CP-1-modified nanochannels for the time-point monitoring of the de-SUMOylation reaction at 30 °C (at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; m:m, protein:enzyme). (f) Matrix-assisted laser desorption/ionization-time of flight mass spectrometry spectra of the mixture of SUMOylated RanGAP1 and SENP1 before (top) and after 24 h (bottom) enzyme digestion at 25 °C. (g) Time-dependent current increase ratio ((I–I0)/I0); black: transmembrane ionic current variation at −2 V and red: Faraday current variation at −0.4 V. Error bars represent the standard deviation values calculated from three ionic current measurements. :1; m:m, protein:enzyme). (f) Matrix-assisted laser desorption/ionization-time of flight mass spectrometry spectra of the mixture of SUMOylated RanGAP1 and SENP1 before (top) and after 24 h (bottom) enzyme digestion at 25 °C. (g) Time-dependent current increase ratio ((I–I0)/I0); black: transmembrane ionic current variation at −2 V and red: Faraday current variation at −0.4 V. Error bars represent the standard deviation values calculated from three ionic current measurements. | ||
We further performed the de-SUMOylation reaction. SUMOylated RanGAP1 solution (1 μM) was dissolved in 50 mM TBS (pH 8.0) and was injected into the measurement apparatus mounted with a CP-1 modified nanochannel film. Then SENP1 was added to the SUMOylated RanGAP1 solution. According to the literature, and we recorded the enzymatic digestion reaction continuously for 24 hours.54 SUMOylated RanGAP1 was enzymatically cleaved by SENP1, resulting in the generation of two new peptides: RanGAP1 substrate peptide containing lysine modification sites and the SUMO1 peptide. Transmembanr ionic current was monitored continuously. The recorded transmembrane ionic current value increased gradually with the reaction time (Fig. 6d), from an initial value of −530 to −395 nA after 24 h. Simultaneously, the Faraday current value increased from −5.35 to −4.43 μA (Fig. 6e). De-SUMOylation was confirmed through matrix-assisted laser desorption/ionization-time of flight mass spectrometry.55 The signal peak intensity of SUMOylated RanGAP1 considerably reduced, and two new peaks corresponding to RanGAP1 and SUMO1 appeared (Fig. 6f). The time-dependent variations in transmembrane ionic current and Faraday current signals generated by the nanochannel device clearly display the de-SUMOylation process (Fig. 6g). Therefore, our device may be used for the time-point monitoring of this enzymatic reaction.
Studies on SUMOylation rely on the detection of endogenous SUMO family members using antibodies. Although approximately 100 monoclonal antibodies for SUMO are available commercially, a minority are cited and many lack complete or any validation by their vendors.56 Therefore, in this study, we aimed to demonstrate a proof of concept for the application of CP-1 in cellular imaging,57,58 which is of significant interest to researchers. We examined CP-1's capability to detect the subcellular localization of SUMO1 through immunofluorescence in cells. As shown in the right panels of Fig. 7 and S30 in the ESI,† Rhodamine B-labelled CP-1 exhibited predominant nucleus localization similar to the pattern of commercial SUMO1-specific antibodies, displaying a diffuse nuclear signal. Furthermore, Rhodamine B-labelled CP-1 was observed in the nucleolus (highlighted by the white arrow), which corresponds to the localization of SUMO1. As described in the literature, SUMO1 is mainly distributed in the nucleus,20 and the nucleolus is the site where SUMO1 modifications mainly occur in ribosomes and chromosomes. These data demonstrated that CP-1 could replace a specific antibody and efficiently indicate the localization of SUMO1 in cells. More broadly, our strategy shows potential for yielding various specific peptides for applications in both chemical and biological studies, laying the groundwork for the subsequent intracellular SUMO1 assays.
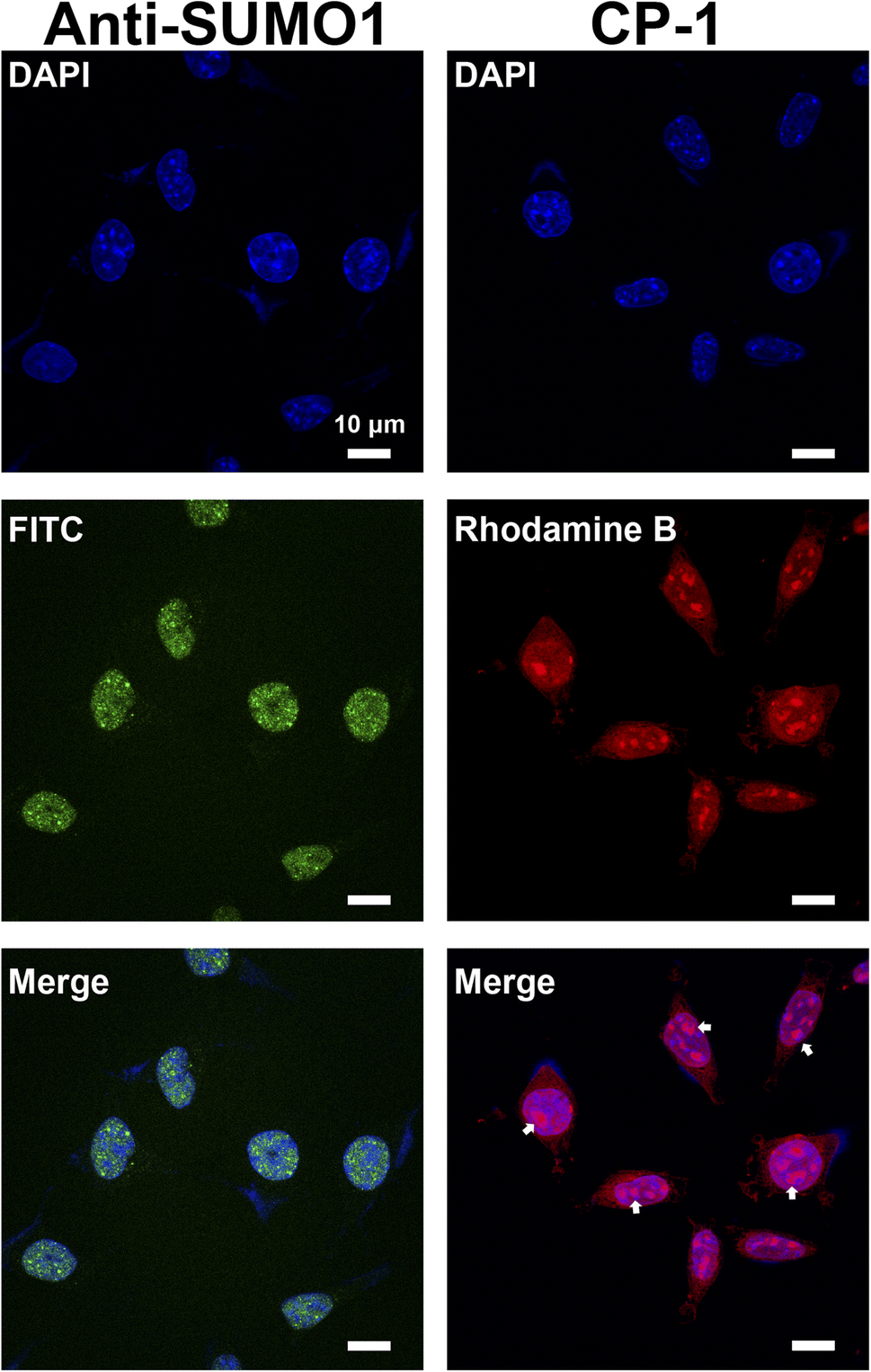 | ||
| Fig. 7 Imaging of SUMO1 with CP-1 in SH-SY5Y cells. 4′,6-Diamidino-2-phenylindole (DAPI) was used to display the localization of the nucleus. Anti-SUMO1 antibody was used as a positive control (left panels). Rhodamine B-labelled CP-1 was used for immunofluorescence imaging (right panels). Scale bars: 10 μm. | ||
Conclusions
In this study, we report the first small molecule ligand targeting SUMO1, and based on this we designed a nanochannel device for the detection of SUMO1 with satisfactory sensitivity and high specificity. This device was found to be useful for monitoring de-SUMOylation. The advantage of our device is that it does not require any labeling or radioactive/fluorescent reagent. Thus, this device can facilitate the development of drugs targeting SENP1. The present study considerably expands the scope of nanopore research to SUMOylation, and our strategy is also applicable to other important and rare PTM types, such as ubiquitination, glycosylation, and acetylation, proving efficient enrichment and analysis methods for biological studies. Notably, through phage display, we identified a series of CPs to improve the sensitivity of SUMO1 detection, thus resolving the bottleneck problem associated with the low efficiency of SUMO1 antibodies. The current work is just the beginning, and in the future, the identified CP sequences may be developed as an enrichment material for advancing SUMO1 proteomics, SUMO1 probes, SUMO1-targeting drug release systems, and drug design, which, in turn, would improve our understanding of the role of SUMO proteins, particularly SUMO1, in disease and physiology. This direction is currently being explored in our laboratory.Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
Y. Q. performed experiments and prepared the paper and ESI.† X. Z. and L. Z. conceived the concept and X. Z. conducted the phage display screening and cell imaging. Y. S. and D. W. performed the theoretical calculation. B. Z. and L. Z. provided the SUMO1 peptides and SENP1. L. L., Y. Z., and W. L. fabricated the nanochannel device, X. Z. performed the COMSOL calculation, Z. J. helped with the synthesis, and M. L. discussed the results and helped with the writing. G. Q. conceived and directed the project, contributed to the writing of the paper, discussed the results and commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2020YFC3400800), the National Natural Science Foundation of China (21922411 and 22174138), DICP Innovation Funding (DICP-I202008, I202243, and I202229), and the Liaoning Provincial Education Department General Research Project (LJ2020025). The authors thank Dr Yongjun Li from Bruker China BNSM for AFM measurements. The results presented here are based on a UMAT irradiation experiment, which was performed at the beamline X0 at the GSI Helmholtzzentrum fur Schwerionenforschung, Darmstadt (Germany) in the frame of FAIR-Phase 0.Notes and references
- I. A. Hendriks and A. C. O. Vertegaal, Nat. Rev. Mol. Cell Biol., 2016, 17, 581–595 CrossRef CAS PubMed
.
- R. J. Lumpkin, H. Gu, Y. Zhu, M. Leonard, A. S. Ahmad, K. R. Clauser, J. G. Meyer, E. J. Bennett and E. A. Komives, Nat. Commun., 2017, 8, 1171 CrossRef PubMed
- A. C. O. Vertegaal, Nat. Rev. Mol. Cell Biol., 2022, 23, 715–731 CrossRef CAS
- S. V. Barysch, N. Stankovic-Valentin, T. Miedema, S. Karaca, J. Doppel, T. N. Achour, A. Vasudeva, L. Wolf, C. Sticht, H. Urlaub and F. Melchior, EMBO Rep., 2021, 22, e49651 CrossRef CAS
- E. Knock, S. Matsuzaki, H. Takamura, K. Satoh, G. Rooke, K. Han, H. Zhang, A. Staniszewski, T. Katayama, O. Arancio and P. E. Fraser, Neurobiol. Dis., 2018, 110, 154–165 CrossRef CAS PubMed
- J. S. Seeler and A. Dejean, Nat. Rev. Cancer, 2017, 17, 184–197 CrossRef CAS PubMed
- L. Cappadocia, T. Kochańczyk and C. D. Lima, EMBO J., 2021, 40, e103787 CrossRef CAS PubMed
- R. Geiss-Friedlander and F. Melchior, Nat. Rev. Mol. Cell Biol., 2007, 8, 947–956 CrossRef CAS
- Y. Fan, X. Li, L. Zhang, Z. Zong, F. Wang, J. Huang, L. Zeng, C. Zhang, H. Yan, L. Zhang and F. Zhou, Adv. Sci., 2022, 9, 2104126 CrossRef
- K. M. Chauhan, Y. Chen, Y. Chen, A. T. Liu, X. X. Sun and M. S. Dai, J. Cell. Biochem., 2021, 122, 189–197 CrossRef CAS PubMed
- L. Shen, M. H. Tatham, C. Dong, A. Zagórska, J. H. Naismith and R. T. Hay, Nat. Struct. Mol. Biol., 2006, 13, 1069–1077 CrossRef CAS PubMed
- L. Du, W. Liu, G. Aldana-Masangkay, A. Pozhitkov, F. Pichiorri, Y. Chen and S. T. Rosen, J. Exp. Clin. Cancer Res., 2022, 41, 8 CrossRef CAS PubMed
- A. Nakamura, S. Grossman, K. Song, K. Xega, Y. Zhang, D. Cvet, A. Berger, G. Shapiro and D. Huszar, Blood, 2022, 139, 2770–2781 CrossRef CAS PubMed
- C. P. Cui, C. C. L. Wong, A. K. L. Kai, D. W. H. Ho, E. Y. T. Lau, Y. M. Tsui, L. K. Chan, T. T. Cheung, K. S. H. Chok, A. C. Y. Chan, R. C. L. Lo, J. M. F. Lee, T. K. W. Lee and I. O. L. Ng, Gut, 2017, 66, 2149–2159 CrossRef CAS
- P. Tokarz and K. Woźniak, Cancers, 2021, 13, 2059 CrossRef CAS PubMed
- C. C. Hu, X. Wei, J. M. Liu, L. L. Han, C. K. Xia, J. Wu, T. You, A. F. Zhu, S. L. Yao, S. Y. Yuan, H. D. Xu, Z. Y. Xia, T. T. Wang and W. K. Mao, Mil. Med. Res., 2022, 9, 58 CAS
- S. Kumar, M. J. A. Schoonderwoerd, J. S. Kroonen, I. J. de Graaf, M. Sluijter, D. Ruano, R. González-Prieto, M. Verlaan-de Vries, J. Rip, R. Arens, N. F. C. C. de Miranda, L. J. A. C. Hawinkels, T. van Hall and A. C. O. Vertegaal, Gut, 2022, 71, 2266–2283 CrossRef CAS PubMed
- J. Gao and H. A. Pickett, Nat. Rev. Cancer, 2022, 22, 515–532 CrossRef CAS PubMed
- I. Nir, D. Huttner and A. Meller, Biophys. J., 2015, 108, 2340–2349 CrossRef CAS PubMed
- Z. You, W. X. Jiang, L. Y. Qin, Z. Gong, W. Wan, J. Li, Y. Wang, H. Zhang, C. Peng, T. Zhou, C. Tang and W. Liu, Nat. Commun., 2019, 10, 5792 CrossRef CAS
- Y. Zhang, C. Fang, H. Bao, W. Yuan and H. Lu, Chin. J. Chem., 2021, 39, 550–558 CrossRef
- J. A. Daniel, B. H. Cooper, J. J. Palvimo, F. P. Zhang, N. Brose and M. Tirard, eLife, 2017, 6, e26338 CrossRef PubMed
- M. P. C. Mulder, R. Merkx, K. F. Witting, D. S. Hameed, D. E. Atmioui, L. Lelieveld, F. Liebelt, J. Neefjes, I. Berlin, A. C. O. Vertegaal and H. Ovaa, Angew. Chem., Int. Ed., 2018, 57, 8958–8962 CrossRef CAS PubMed
- S. L. Goodman, New Biotechnol., 2018, 45, 9–13 CrossRef CAS
- G. Pérez-Mitta, M. E. Toimil-Molares, C. Trautmann, W. A. Marmisollé and O. Azzaroni, Adv. Mater., 2019, 31, 1901483 CrossRef
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS
- D. Zhang, Y. Sun, Z. Wang, F. Liu and X. Zhang, Nat. Commun., 2023, 14, 1901 CrossRef CAS PubMed
- G. Laucirica, W. A. Marmisollé, M. E. Toimil-Molares, C. Trautmann and O. Azzaroni, ACS Appl. Mater. Interfaces, 2019, 11, 30001–30009 CrossRef CAS PubMed
- K. Lee, K. B. Park, H. J. Kim, J. S. Yu, H. Chae, H. M. Kim and K. B. Kim, Adv. Mater., 2018, 30, 1704680 CrossRef
- Y. Zeng, X. Qu, B. Nie, Z. Mu, C. Li and G. Li, Biosens. Bioelectron., 2022, 215, 114564 CrossRef CAS
- Q. Wen, N. Malik, Y. Addadi, M. Weissenfels, V. Singh, L. J. W. Shimon, M. Lahav and M. E. van der Boom, Angew. Chem., Int. Ed., 2023, 62, e202214041 CrossRef CAS PubMed
- D. Zhang and X. Zhang, Small, 2021, 17, 2100495 CrossRef CAS
- A. P. Mann, P. Scodeller, S. Hussain, G. B. Braun, T. Mölder, K. Toome, R. Ambasudhan, T. Teesalu, S. A. Lipton and E. Ruoslahti, Nat. Commun., 2017, 8, 1403 CrossRef PubMed
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed
- X. Zhang, M. Zhong, P. Zhao, X. Zhang, Y. Li, X. Wang, J. Sun, W. Lan, H. Sun, Z. Wang and H. Gao, Biomater. Sci., 2019, 7, 5197–5210 RSC
- X. Zhang, X. Zhang, M. Zhong, P. Zhao, C. Guo, Y. Li, T. Wang and H. Gao, ACS Chem. Neurosci., 2020, 11, 4240–4253 CrossRef CAS
- R. Fang, H. Zhang, L. Yang, H. Wang, Y. Tian, X. Zhang and L. Jiang, J. Am. Chem. Soc., 2016, 138, 16372–16379 CrossRef CAS
- S. Qian, S. W. Joo, Y. Ai, M. A. Cheney and W. Hou, J. Colloid Interface Sci., 2009, 329, 376–383 CrossRef CAS PubMed
- T. Liu, X. Wu, H. Xu, Q. Ma, Q. Du, Q. Yuan, P. Gao and F. Xia, Anal. Chem., 2021, 93, 13054–13062 CrossRef CAS PubMed
- X. Li, T. Zhai, P. Gao, H. Cheng, R. Hou, X. Lou and F. Xia, Nat. Commun., 2018, 9, 40 CrossRef PubMed
- S. Chen, L. Li, C. L. Boozer and S. Jiang, J. Phys. Chem. B, 2001, 105, 2975–2980 CrossRef CAS
- H. Yang and G. Qing, Chem. Phys. Rev., 2021, 2, 021306 CrossRef
- R. Hein, P. D. Beer and J. J. Davis, Chem. Rev., 2020, 120, 1888–1935 CrossRef CAS PubMed
- Y. Liu, Z. Liu and Y. Tian, Acc. Chem. Res., 2022, 55, 2821–2832 CrossRef CAS PubMed
- M. Li, Y. Xiong, W. Lu, X. Wang, Y. Liu, B. Na, H. Qin, M. Tang, H. Qin, M. Ye, X. Liang and G. Qing, J. Am. Chem. Soc., 2020, 142, 16324–16333 CrossRef CAS PubMed
- Q. Liang, Y. Huang, M. Wang, D. Kuang, J. Yang, Y. Yi, H. Shi, J. Li, J. Yang and G. Li, Chem. Eng. J., 2023, 452, 139646 CrossRef CAS PubMed
- S. Lin, L. Xu, A. C. Wang and Z. L. Wang, Nat. Commun., 2020, 11, 399 CrossRef CAS PubMed
- C. A. Amarasekara, U. S. Athapattu, C. Rathnayaka, J. Choi, S. Park and S. A. Soper, Electrophoresis, 2020, 41, 1627–1640 CrossRef CAS PubMed
- M. Li, Y. Cao, X. Zhang, D. Wang, S. Qian, G. Li, F. Zhang, Y. Xiong and G. Qing, Chem. Commun., 2021, 57, 7914–7917 RSC
- X. Huang, X. Y. Kong, L. Wen and L. Jiang, Adv. Funct. Mater., 2018, 28, 1801079 CrossRef
- S. Zhang, J. Zhou and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202204012 CAS
- X. Wang, C. Wang, H. Chu, H. Qin, D. Wang, F. Xu, X. Ai, C. Quan, G. Li and G. Qing, Chem. Sci., 2020, 11, 7369–7378 RSC
- C. Wang, Y. Yang, X. Zhang, Z. Shi, H. Gao, M. Zhong, Y. Fan, H. Zhang, B. Liu and G. Qing, Sci. Adv., 2023, 9, eade0293 CrossRef CAS
- Y. Li, M. Sun, Y. Hu, Y. Shan, Z. Liang, L. Zhang and Y. Zhang, Anal. Chim. Acta, 2021, 1154, 338324 CrossRef CAS PubMed
- Y. Liu, G. Li, T. J. Gu and L. Li, Anal. Chem., 2022, 94, 3774–3781 CrossRef CAS PubMed
- A. J. Garvin, A. J. Lanz and J. R. Morris, Sci. Rep., 2022, 12, 21343 CrossRef CAS PubMed
- H. Zhao, C. Liu, W. Ding, L. Tang, Y. Fang, Y. Chen, L. Hu, Y. Yuan, D. Fang and S. Lin, J. Am. Chem. Soc., 2022, 144, 6742–6748 CrossRef CAS PubMed
- L. Mao, Y. Han, Q. W. Zhang and Y. Tian, Nat. Commun., 2023, 14, 1419 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02140h |
| ‡ Y. Q. and X. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |