
Cationic vacancies and interface engineering on crystalline–amorphous gamma-phase Ni–Co oxyhydroxides achieve ultrahigh mass/areal/volumetric energy density flexible all-solid-state asymmetric supercapacitor†
Xue
Ren‡
 ,
Menggang
Li‡
,
Longyu
Qiu‡
,
Xin
Guo
,
Fenyang
Tian
,
Guanghui
Han
,
Weiwei
Yang
* and
Yongsheng
Yu
*
,
Menggang
Li‡
,
Longyu
Qiu‡
,
Xin
Guo
,
Fenyang
Tian
,
Guanghui
Han
,
Weiwei
Yang
* and
Yongsheng
Yu
*
State Key Laboratory of Urban Water Resource and Environment, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin, Heilongjiang 150001, China. E-mail: yangww@hit.edu.cn; ysyu@hit.edu.cn
First published on 14th February 2023
Abstract
Construction of gamma-phase transition metal oxyhydroxides for electrode materials is an effective strategy for improving electrochemical properties. However, the preparation of gamma-phase transition metal oxyhydroxides with various defects remains an arduous challenge. Herein, gamma-phase Ni–Co oxyhydroxides with cationic vacancy defects and crystalline–amorphous interfaces are synthesized successfully via electrochemical reconstruction to achieve an ultrahigh mass/areal/volumetric energy density, flexible all-solid-state asymmetric supercapacitor (ASC). The Ni–Co oxyhydroxides consist of γ-NiOOH, γ-CoOOH and NiOOH phases; the γ-NiOOH and γ-CoOOH phases can effectively improve electronic conductivity and theoretical capacitance due to the abundant Ni4+/Co4+. The cationic vacancy defects and crystalline–amorphous interfaces not only endow the gamma-phase Ni–Co oxyhydroxides with abundant electrochemical active sites, but also enable fast electron transfer between the electrode and electrolyte. Therefore, the as-obtained gamma-phase Ni–Co oxyhydroxides display a high capacitance of 20.9 F cm−2 at 4 mA cm−2 and 3483 F g−1 at 0.67 A g−1, as well as excellent rate characteristics (90.5% capacitance retention at a high current density of 240 mA cm−2 and 40 A g−1). Density functional theory calculations reveal that Ni–Co oxyhydroxides with cationic vacancies can increase the adsorption energy of H2O, which is beneficial for the capture of H2O to occur for subsequent charge storage reactions. Furthermore, the assembled Ni–Co oxyhydroxides//active carbon all-solid-state ASC shows ultrahigh mass/areal/volumetric energy density of 92.6 W h kg−1/3.3 mW h cm−2/19.5 mW h cm−3 (at 1156 W kg−1/34.6 mW cm−2/204.1 mW cm−3) and possesses excellent electrochemical stability with 91% retention after 7000 cycles.
1 Introduction
Nowadays, the increasing demand for portable electronic devices has presented a new challenge in the area of novel energy storage devices, among which all-solid-state asymmetric supercapacitors (ASCs), which exhibit excellent portability and fast charge–discharge characteristics, have been demonstrated as efficient energy storage devices.1–5 However, all-solid-state ASC devices usually exhibit low energy density, limiting their large-scale applications. Moreover, producing high-energy-density all-solid-state ASCs typically requires an electrode material with abundant redox active sites and developed electrical percolative networks. The abundant redox active sites can ensure the high capacitance of the all-solid-state ASC device, and the developed electrical percolative network speeds the e− or OH− transport and improves the rate capability of the ASC.6–8 Transition metal oxyhydroxides, especially gamma-phase nickel/cobalt oxyhydroxides (γ-NiOOH and γ-CoOOH), have emerged as new electrode materials for ASCs because of their high theoretical capacitance, abundant redox active sites, good electronic conductivity and high operational potential window.9–11 However, only a few works have reported the use of single-phase γ-NiOOH or γ-CoOOH as electrode materials for supercapacitors. In particular, there is a lack of reports on binary γ-phase Ni–Co oxyhydroxides.The past decade has witnessed rapid development in the design and preparation of nanostructured transition-metal-based electrode materials with varied compositions/structures and defects.12–16 Electrode materials with cationic vacancies or crystalline–amorphous interfaces can provide better electrochemical performance on account of their abundant defects/distortions, electrochemical redox active sites and fast electron transfer, thus becoming an effective strategy for improving electrochemical properties.17,18 In addition, metal cationic vacancy defects can be constructed by dissolving the corresponding anions.19 The loss or leakage of cation clusters such as [MO]x+ and [MOH]x+ can also occur in addition to simple M vacancy (VM, M = Ni or Co), that is, there are also vacancy defects of [MO]x+ vacancy (VMO) and [MOH]x+ vacancy (VMOH) during the electrochemical reconstruction processes.20 Based on the above considerations, via the introduction of cationic vacancy defects and crystalline–amorphous interfaces, γ-phase Ni–Co oxyhydroxides have a promising future as high-performance electrode materials for high-energy-density all-solid-state ASCs.
In this work, we report novel γ-phase Ni–Co oxyhydroxide nanoplate networks with cationic vacancy defects and crystalline–amorphous interfaces generated in situ on Ni foam for ultrahigh mass/areal/volumetric energy density, flexible all-solid-state ASCs. Physical characterizations confirmed that the Ni–Co oxyhydroxides consist of γ-NiOOH, γ-CoOOH and NiOOH phases. The γ-NiOOH and γ-CoOOH phases can significantly improve the electrical conductivity and theoretical capacitance due to the presence of Ni4+/Co4+ cations. Furthermore, the crystalline–amorphous interfaces greatly increase the amount of charge storage sites to improve the specific capacitance; additionally, the cationic vacancy defects provide more electrochemical active sites and enhance the adsorption energy of H2O. The idea of “dissolving out” the anions within the nanosized Ni–Co–P@CoNi2S4 to create defects originated from the fact that many solids in the nano-regime can flexibly undergo anion- or cation-exchange processes. As a consequence, the γ-phase Ni–Co oxyhydroxides showed an ultrahigh capacitance of 20.9 F cm−2 at 4 mA cm−2 (3483 F g−1 at 0.67 A g−1), as well as an excellent rate capability with 90.5% capacitance retention at a high current density of 240 mA cm−2 and 40 A g−1. In addition, a flexible Ni–Co oxyhydroxides//AC all-solid-state ASC was fabricated and exhibited an ultrahigh mass/areal/volumetric energy density of 92.6 W h kg−1/3.3 mW h cm−2/19.5 mW h cm−3 (at 1156 W kg−1/34.6 mW cm−2/204.1 mW cm−3). We also confirmed that a watchband with three Ni–Co oxyhydroxides//AC all-solid-state ASCs in series can power a smartwatch for more than three minutes.
2 Results and discussion
Ni–Co oxyhydroxides supported on Ni foam were successfully constructed using an electrochemical reconstruction strategy (Scheme 1). Specifically, NiOOH nanosheets with a thickness of ∼20 nm were synthesized on Ni foam during an improved Fast-Repeated-Charging/Discharging (FRCD) electro-oxidation process (Fig. S1 and S2†). After sulfur doping, NiOOH@Ni3S2 nanosheets with a thickness of 20–30 nm can be observed, with similar morphology to NiOOH (Fig. S3†). Furthermore, the vertically standing and cross-linked NiCo-LDH@CoNi2S4 nanoplates (6–8 μm in lateral dimensions and a thickness of 30–50 nm) can be uniformly transformed from NiOOH@Ni3S2 by a hydrothermal method (Fig. S4†). This indicates that the cobalt ion exchange plays a vital role in the fabrication of NiCo-LDH@CoNi2S4 nanoplate networks. Phosphorization causes the NiCo-LDH@CoNi2S4 nanoplates to transform into Ni–Co–P@CoNi2S4 nanoplates with a greater thickness of 100–150 nm and a rougher surface (Fig. S5†). To finally obtain the Ni–Co oxyhydroxides, we attempted to leach out the elements P and S from Ni–Co–P@CoNi2S4via electrochemical reconstruction methods, realized by cyclic voltammetry (CV) curves (Fig. S6†). From the CVs, we can observe that the current density gradually increases from reconstruction cycles 1 to 20 and that both the oxidation and reduction peaks move to both sides, revealing the formation of the Ni–Co oxyhydroxides with potential electrochemical performance. | ||
| Scheme 1 Schematic illustration of the preparation of γ-phase Ni–Co oxyhydroxides. | ||
The scanning electron microscopy (SEM) images of the Ni–Co oxyhydroxides display three-dimensional (3D) vertically standing and cross-linked nanoplate arrays consisting of two-dimensional (2D) hexagonal nanosheets, whose formation is mainly due to the electrochemical reconstruction (Fig. 1a and S7†). The nanoplates have a lateral size of 6–8 μm and a thickness of 400–600 nm, and the attached hexagonal nanosheets show a lateral size of 200–300 nm and a thickness of 20–30 nm. The transmission electron microscopy (TEM) image shows that the edge zone of the nanoplate is relatively thin and layered (Fig. 1b). The X-ray diffraction (XRD) pattern demonstrates that the as-formed Ni–Co oxyhydroxides show a typical mixed phase consisting of γ-NiOOH, γ-CoOOH and NiOOH phases (Fig. S8†), indicating the successful preparation of γ-phase Ni–Co oxyhydroxides. The corresponding selected-area electron diffraction (SAED) pattern further confirms the polycrystalline structure of the Ni–Co oxyhydroxides (Fig. 1c). From the HRTEM image of Ni–Co–P@CoNi2S4 (Fig. S5c†), we can observe the existence of crystalline–amorphous interfaces in the Ni–Co–P@CoNi2S4 precursor; this provides good evidence for the preparation of crystalline–amorphous Ni–Co oxyhydroxides via electrochemical reconstruction. A large number of crystalline and amorphous regions are scattered over the nanoplate, as confirmed by the high-resolution TEM (HRTEM) image (Fig. 1d). The densely heterogeneous crystalline–amorphous interfaces are marked by purple dashed lines. The abundant defects in the crystalline and amorphous regions reveal the presence of ionic vacancies. Moreover, the interplanar spacing was determined to be 0.236 and 0.209 nm, corresponding to the (102) and (105) planes of γ-NiOOH, and the lattice spacing of 0.242 and 0.220 nm corresponded to the (101) and (006) crystal planes of γ-CoOOH, further indicating the presence of γ-phase Ni–Co oxyhydroxides. In addition, the lattice spacing of 0.249 nm corresponds to the (110) plane of NiOOH. Energy dispersive spectroscopy (EDS) elemental mapping images reveal that the elements Ni, Co, P and S are homogeneously distributed throughout the Ni–Co–P@CoNi2S4 nanoplates, while the lack of an O signal confirms that Ni and Co were incorporated in the phosphide and sulfide forms (Fig. 1e). However, the lack of P and S signals and appearance of O signals after the electrochemical reconstruction process confirm that the Ni–Co–P@CoNi2S4 was successfully converted into Ni–Co oxyhydroxides (Fig. 1f).
 | ||
| Fig. 1 Morphology and composition characterization of the Ni–Co oxyhydroxides. (a) SEM image, (b) TEM image, (c) corresponding SAED pattern and (d) HRTEM image of Ni–Co oxyhydroxides. EDS elemental mapping of (e) Ni–Co–P@CoNi2S4 and (f) Ni–Co oxyhydroxides. | ||
To clarify the surface elemental compositions and change in the chemical valence states caused by the electrochemical reconstruction, high-resolution X-ray photoelectron spectroscopy (XPS) was conducted (Fig. 2a, b and S9†). The survey spectra show that the elements Ni, Co, P, S and O were present in Ni–Co–P@CoNi2S4, while the elements Ni, Co and O were found in the Ni–Co oxyhydroxides, further indicating that the Ni–Co oxyhydroxides were successfully prepared from Ni–Co–P@CoNi2S4 (Fig. S9a†). The Ni 2p spectrum of Ni–Co–P@CoNi2S4 contains two spin–orbit peaks (2p3/2 and 2p1/2) around 852.3/869.5 eV corresponding to Ni2+ and 855.5/873.3 eV corresponding to Ni3+ (Fig. 2a). The Ni 2p spectrum for the Ni–Co oxyhydroxides was fitted to two spin–orbit peaks (2p3/2 and 2p1/2) around 855.4/873.1 for Ni3+ and 857.4/875.8 eV for Ni4+, resulting in an average Ni valence of 3.3, in line with γ-NiOOH.21 Compared with that in Ni–Co–P@CoNi2S4, the increased proportion of Ni3+/Ni4+ in the Ni–Co oxyhydroxides indicates the generation of Ni3+/4+, [NiO]x+ and [NiOH]x+ cationic vacancies, also further confirming that the defects observed in the high-resolution TEM image are cationic vacancies.22 With regard to the Co 2p spectrum for Ni–Co–P@CoNi2S4, the peaks at 781.7/797.6 eV correspond to Co 2p3/2 and Co 2p1/2 of Co2+,23,24 and the two peaks at 779.9 and 795.2 eV are attributed to the Co 2p3/2 and Co 2p1/2 of Co3+ (Fig. 2b). The peaks at 780.3/795.6 eV and 783.7/798.5 eV correspond to Co3+/Co4+ in the high-resolution Co 2p spectrum of the Ni–Co oxyhydroxides, revealing an average cobalt valence of 3.27, which is associated with γ-CoOOH. Compared with that of Ni–Co–P@CoNi2S4, the increase in the cobalt oxidation state will lead to a decrease in the cobalt coordination number and give rise to Co-based cationic vacancies. Furthermore, the P 2p, S 2p, and O 1s spectra further prove the successful preparation of Ni–Co oxyhydroxides (Fig. S9b–d†).
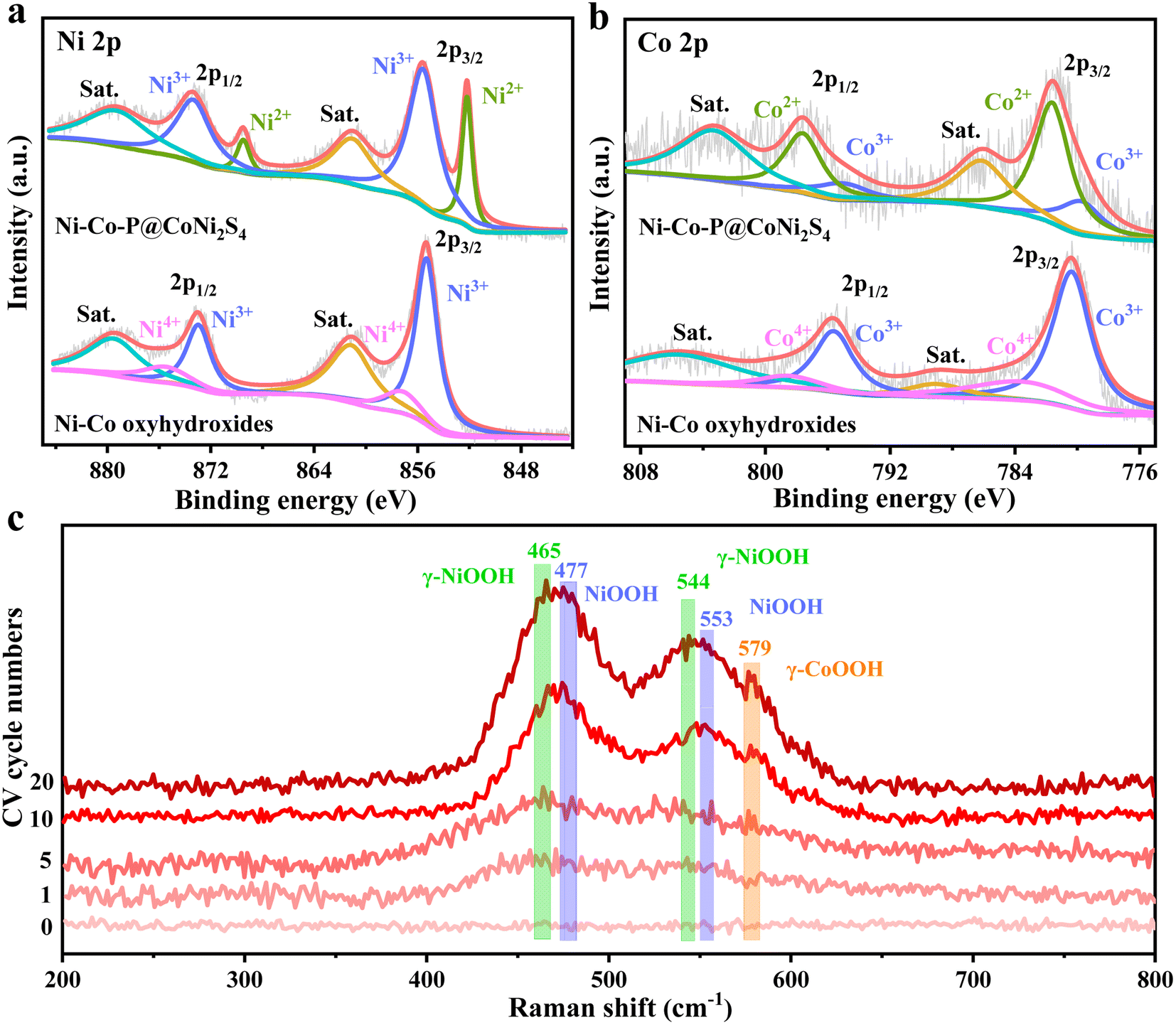 | ||
| Fig. 2 Electronic and crystallographic structure characterization before and after electrochemical reconstruction. High-resolution (a) Ni 2p and (b) Co 2p XPS spectra of Ni–Co–P@CoNi2S4 and Ni–Co oxyhydroxides. (c) In situ Raman spectroscopy during the electrochemical reconstruction process from Ni–Co–P@CoNi2S4 to Ni–Co oxyhydroxides. | ||
To further confirm the formation of γ-phase Ni–Co oxyhydroxides, in situ Raman spectra were obtained during electrochemical reconstruction (Fig. 2c). The pristine Ni–Co–P@CoNi2S4 does not show any visible peaks, indicating that are no Ni–Co oxyhydroxides before electrochemical reconstruction. As the electrochemical reconstruction progresses, the peaks of γ-NiOOH (465 cm−1 and 544 cm−1), γ-CoOOH (579 cm−1) and NiOOH (477 cm−1 and 553 cm−1) begin to form, revealing the initial formation of γ-NiOOH, γ-CoOOH and NiOOH. Electrochemical reconstruction is continued until the peak intensity and positions are constant; the peak positions become more obvious and the peak intensity becomes larger, further indicating the complete formation of the γ-NiOOH, γ-CoOOH and NiOOH phases. The above-mentioned results suggest that the Ni–Co oxyhydroxides were successfully prepared from Ni–Co–P@CoNi2S4via electrochemical reconstruction.
The electrochemical performances of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4 and Ni–Co oxyhydroxides were investigated in a traditional three-electrode system. The CV curves of the Ni–Co oxyhydroxides exhibit two pairs of redox peaks derived from the abundant redox reactions of Ni–Co oxyhydroxides during the electrochemical charge/discharge processes (Fig. 3a). The shift in the position of the oxidation and reduction peaks with increased scanning rate was due to the existence of polarization. The charge/discharge platforms in the galvanostatic charge–discharge (GCD) curves for the Ni–Co oxyhydroxides verify the battery-type nature (Fig. 3b and S10†). The good symmetry and obvious pseudocapacitance charge/discharge plateaus imply the high coulombic efficiency and abundant faradaic reactions of the Ni–Co oxyhydroxides. In addition, the CV curve of the Ni–Co oxyhydroxides has the largest integral area with broader and stronger redox peaks than that of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4 and Ni–Co–P@CoNi2S4 (Fig. 3c and S11†). This reveals that Ni–Co oxyhydroxides can store the most energy under the same conditions, which is mainly due to the abundant cationic vacancy defects, crystalline–amorphous interfaces, presence of Ni4+/Co4+ and unique 2D–3D/nano–micron hierarchical structure. The GCD curves at 4 mA cm−2 show that the Ni–Co oxyhydroxides have the longest discharge time, and the areal capacitance of the Ni–Co oxyhydroxides was derived to be 20.9 F cm−2, which is much higher than that of NiOOH (3.4 F cm−2), NiOOH@Ni3S2 (4.1 F cm−2), NiCo-LDH@CoNi2S4 (6.2 F cm−2) or Ni–Co–P@CoNi2S4 (11.2 F cm−2) (Fig. 3d). Additionally, the areal capacitance (Fig. 3e) and specific capacitance (Fig. 3f) of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4 and the Ni–Co oxyhydroxides at different current densities indicate that the Ni–Co oxyhydroxides have the highest areal/specific capacitance and excellent rate performance. The Ni–Co oxyhydroxides deliver an ultrahigh areal specific capacitance of 20.9, 19.8, 19.3, 19.2, 19.1 and 18.9 F cm−2 at 4, 8, 20, 80, 160 and 240 mA cm−2, respectively, and show a superhigh capacitance retention rate of 90.5% even when the current density is increased 60-fold. Additionally, the Ni–Co oxyhydroxides display a super-high specific capacitance of 3483 F g−1 at a current density of 0.67 A g−1 (90.5% capacitance retention from 0.67 to 40 A g−1), which is much higher than that of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4 or Ni–Co–P@CoNi2S4 (Fig. S12 and S13†).
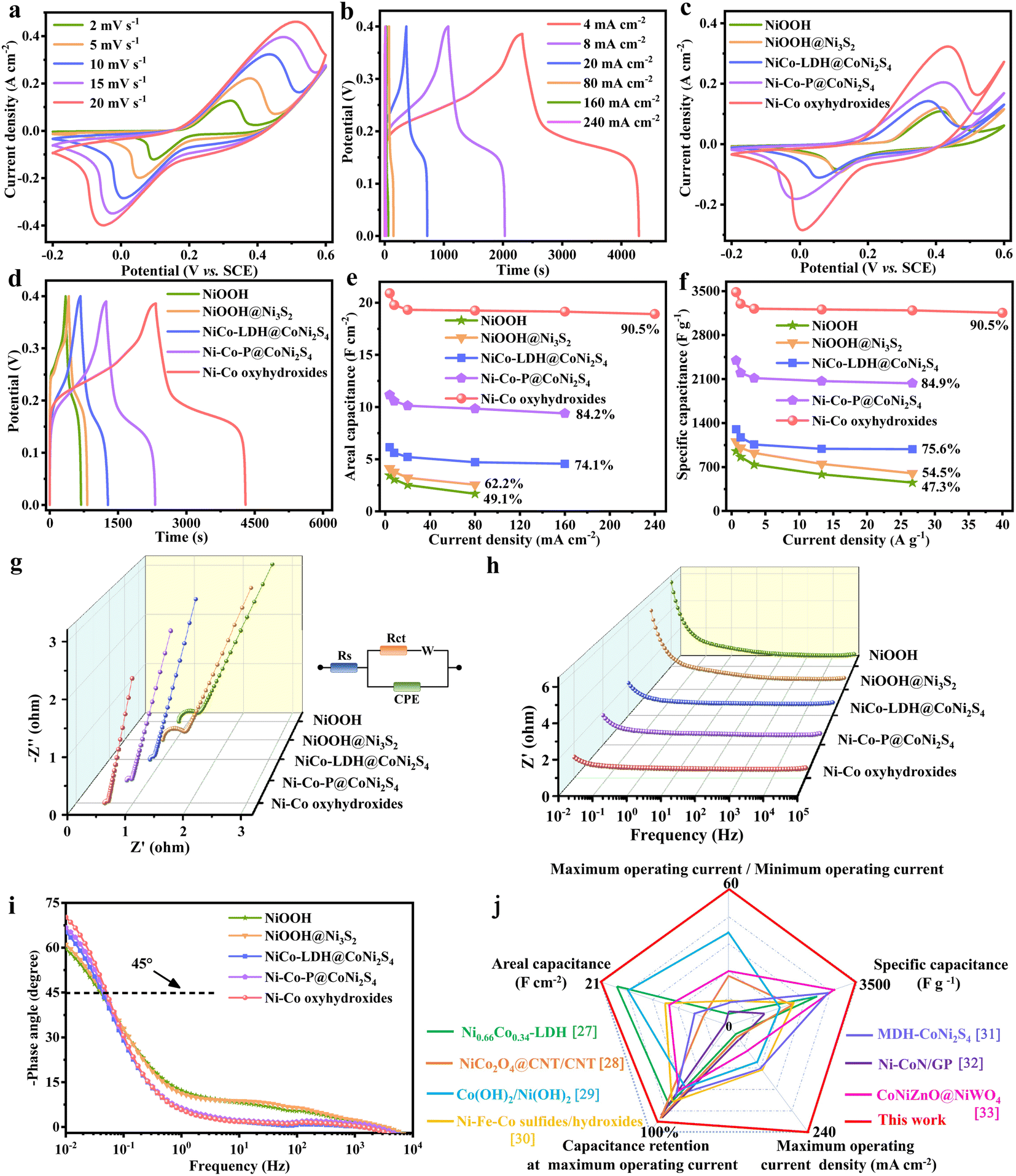 | ||
| Fig. 3 Electrochemical performances of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4 and Ni–Co oxyhydroxides in a three-electrode system. (a) CV curves at different scan rates and (b) GCD curves at various current densities of Ni–Co oxyhydroxides. Comparison of (c) CV curves at 10 mV s−1, (d) GCD curves at 4 mA cm−2, (e) areal capacitance at 4 to 240 mA cm−2, (f) specific capacitance at 0.67 to 40 A g−1, (g) Nyquist plots, (h) Bode modulus plots and (i) Bode phase plots of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4 and Ni–Co oxyhydroxides. (j) Comparison of the electrochemical performance of Ni–Co oxyhydroxides with other recent works in five dimensions. | ||
Subsequently, electrochemical impedance spectroscopy (EIS) tests were used to investigate the ion diffusion and migration kinetics of the electrolyte for these electrode materials (Fig. 3g–i). The Nyquist plot of the Ni–Co oxyhydroxides shows a smaller equivalent series resistance (0.503 Ω), charge-transfer resistance (0.063 Ω) and internal diffusion/transfer resistance (the angle of the straight line at low frequencies is about 79°) compared with those of NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4 and Ni–Co–P@CoNi2S4 (Fig. 3g). In addition, the electrolyte ions usually move near the holes of the electrode material at high frequencies and then migrate to the holes at lower frequency; hence, the penetration of the electrolyte decreases with increasing frequency.25 A horizontal line is observed at high frequencies, which represents an advantage in terms of electron transport. Additionally, the line in the low-frequency region indicates the accumulation of ions in the electrolyte. The Ni–Co oxyhydroxides display much smaller total impedance accumulation at low frequencies than the other materials, revealing the excellent ion diffusion achieved by the 2D–3D/nano–micron hierarchical structure (Fig. 3h). Compared with those of the other materials, the phase angle (at low frequency) of the Ni–Co oxyhydroxides (70.2°) is closer to 90°, indicating an ideal capacitive nature (Fig. 3i). The smallest phase angle of NiOOH is related to the higher pseudocapacitance contribution. The relaxation time constant (T0) calculated from the phase angle of 45° for NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4 and the Ni–Co oxyhydroxides are approximately 25.4, 23.2, 23.5, 21.3 and 19.7 s. The lowest relaxation time constant of the Ni–Co oxyhydroxides further suggests highly efficient ion diffusion within the 2D–3D/nano–micron hierarchical nanoplate networks, consistent with excellent rate capability performance.26 In order to comprehensively and objectively evaluate the electrochemical performance of the Ni–Co oxyhydroxides, the electrochemical performance of the Ni–Co oxyhydroxides in five dimensions was compared with that of recent state-of-the-art electrode materials (Fig. 3j).27–33 Obviously, the graph composed of five dimensional lines corresponding to the Ni–Co oxyhydroxides is close to a regular pentagon and has the largest area, further revealing their preeminent and comprehensive electrochemical performance for energy storage. The Ni–Co oxyhydroxides show super cycling stability with 86% capacitive retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (Fig. S14†), and their coulombic efficiency remains at nearly 99% throughout the cycles of the charge/discharge tests, indicating remarkable charge storage capability.
000 cycles (Fig. S14†), and their coulombic efficiency remains at nearly 99% throughout the cycles of the charge/discharge tests, indicating remarkable charge storage capability.
To better understand the role of the cationic vacancies for enhancing the electrochemical performance, the nine types of typical cationic vacancies that are formed by the leaching of the M3+/4+, [MO]x+ and [MOH]x+ (M = Ni or Co) cations (defined as γ-VNi, γ-VNiO, γ-VNiOH, γ-VCo, γ-VCoO, γ-VCoOH, VNi, VNiO, and VNiOH), were evaluated using density functional theory (DFT) calculations (Fig. S15–S17†). The charge density of the pristine and cationic vacancy surfaces of γ-NiOOH (102), γ-CoOOH (101) and NiOOH (110) show that metal atoms (Ni and Co) are electron donors, while the adjacent O atoms are electron acceptors (Fig. 4a). The electron cloud density of the O atoms in the cationic vacancies of defective Ni–Co oxyhydroxides for absorbing H2O molecules is more complex and larger than that of the pristine Ni–Co oxyhydroxides. This indicates that more electrons are transferred from Co/Ni to O and that the electron interaction between Ni/Co–O is enhanced, making the adsorption of H2O molecules stronger. To further investigate the redox reaction kinetics of the Ni–Co oxyhydroxides with cationic vacancy defects, the adsorption capacity of H2O was calculated on the optimized atomic structure models (Ni or Co sites with the lowest adsorption energy were selected as adsorption sites). The water adsorption energies of the pristine γ-NiOOH (102), γ-CoOOH (101) and NiOOH (110) surfaces are −0.9133, −0.3450 and −0.5823 eV, respectively, indicating that they all have good hydrophilicity (Fig. 4b–d). Obviously, the Ni–Co oxyhydroxides with cationic vacancy defects show enhanced adsorption strength towards H2O compared with the pristine Ni–Co oxyhydroxides, regardless of their vacancy configurations. Moreover, the charge storage can be ascribed to the redox reactions in eqn (1) and (2).34 This indicates that the Ni–Co oxyhydroxides with various cationic vacancy defects have good adsorption toward H2O, which is beneficial to capture H2O in order for the subsequent charge storage reactions (redox reactions) to occur. As a result, the electrochemical properties of the Ni–Co oxyhydroxides are greatly enhanced.
| NiOOH + H2O + e− ⇔ Ni2+ + 3OH− | (1) |
| CoOOH + H2O + e− ⇔ Co2+ + 3OH− | (2) |
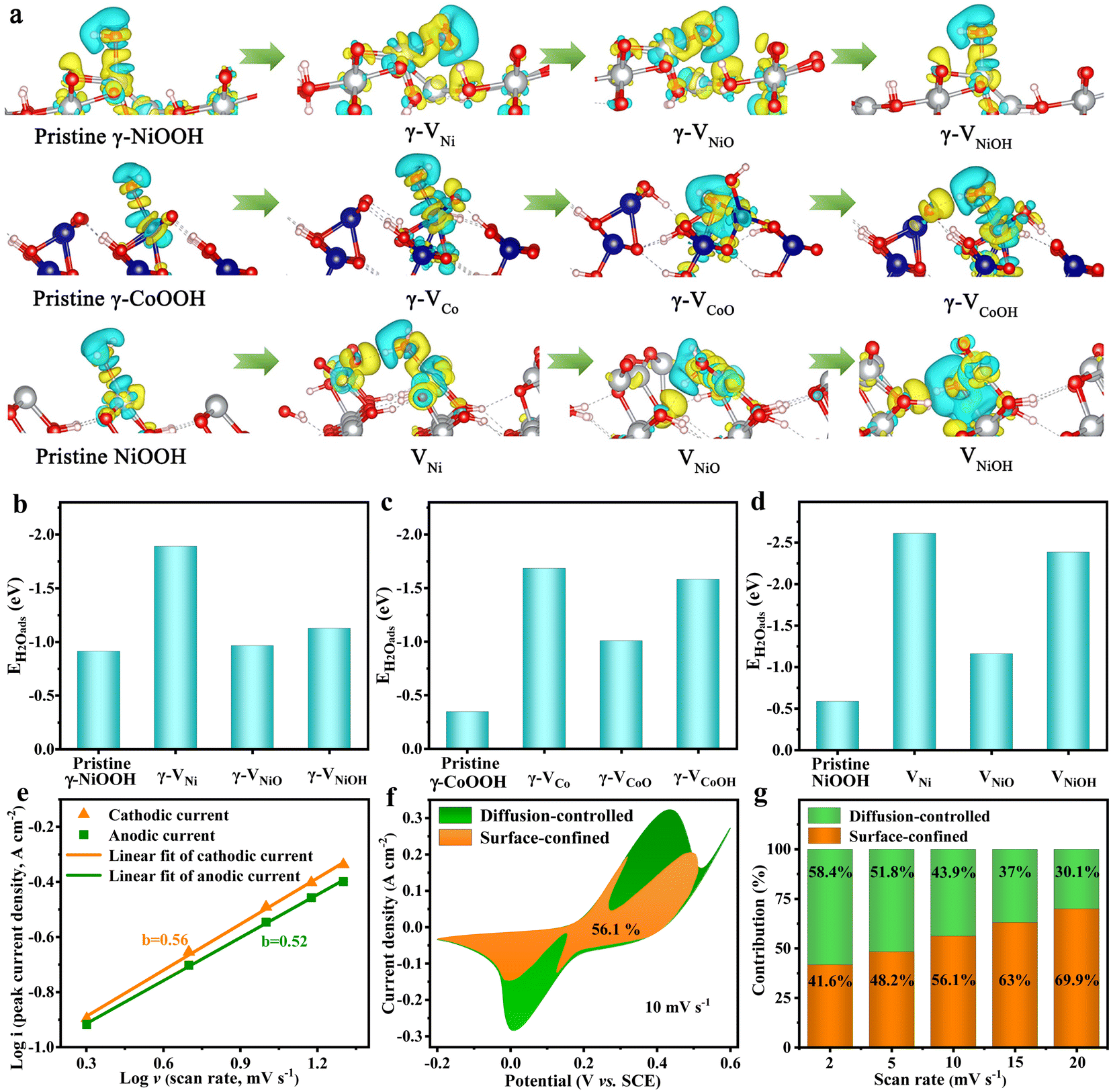 | ||
| Fig. 4 DFT calculations and charge storage contribution analysis. (a) Charge density difference between the material surface and water molecule. (b) Adsorption energies of H2O for pristine γ-NiOOH, γ-VNi, γ-VNiO and γ-VNiOH. (c) Adsorption energies of H2O for pristine γ-CoOOH, γ-VCo, γ-VCoO and γ-VCoOH. (d) Adsorption energies of H2O for pristine NiOOH, VNi, VNiO and VNiOH. (e) Relationship between log(i) and log(ν) for Ni–Co oxyhydroxides. (f) Surface-confined and diffusion-controlled contribution to the charge storage of Ni–Co oxyhydroxides at 10 mV s−1. (g) Surface-confined and diffusion-controlled contribution ratios of Ni–Co oxyhydroxides at various scan rates. | ||
Density of states (DOS) maps of the pristine γ-NiOOH, γ-CoOOH, NiOOH and the nine types of typical cationic vacancies show that the peaks of Ni/Co in pristine γ-NiOOH, γ-CoOOH and NiOOH are relatively sharper, indicating that the chemical bond between Ni/Co and O exhibits ionic bond characteristics (Fig. S18–S20†). The peaks of Ni/Co after the formation of cationic vacancies are gradual, indicating enhanced covalence of the M–O bonds. Additionally, the conduction band electronic states increase to a certain extent after the generation of cationic vacancies. This system exhibits an electron-deficient state, which is consistent with the XPS results. The increase of vacant orbitals is conducive to interaction between the vacant orbitals at the M site on the surface and the lone-pair electrons of O in water, thus enhancing the adsorption of O species such as H2O molecules. In addition, the pristine γ-NiOOH, γ-CoOOH and NiOOH show high electronic states at the adsorption site of Ni/Co near the Fermi level, which is mainly attributed to the 3d orbital of the transition metal (Fig. S21–S23†). The electrons near the Fermi level are active, and the adsorptions are unstable, resulting in a low adsorption energy on the original surface. However, the DOS peaks of the Co/Ni atoms around the Fermi level after the formation of cationic vacancies change from narrow/sharp (in the pristine model) to short/wide (in the vacancy model). Additionally, the coincidence of the DOS peaks between O and Ni/Co is better, indicating stronger electronic interaction between adsorption sites and water molecules after the formation of cationic vacancies, further confirming the calculation results for the adsorption energy of H2O.
The corresponding relationships of the peak current (i) and square root of the scan rate (ν1/2) of the as-prepared electrode materials further confirm their battery-type characteristic (Fig. S24 and S25†). Furthermore, the b value can be estimated by plotting log(ip) against log(ν); the slope of the plot is the value of b.35 The b values of the anodic and cathodic current for NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4 and Ni–Co oxyhydroxides are close to 0.5, indicating that both surface-confined and diffusion-controlled processes are existent (Fig. 4e and S26†). Fig. 4f displays the CV curve for the Ni–Co oxyhydroxides at 10 mV s−1, along with the surface-confined and diffusion-controlled contributions; the surface-confined contribution occupies an area of 56.1%. In addition, the diffusion-controlled contribution ratio decreases from 58.4% to 30.1% as the scan rate is changed from 2 to 20 mV s−1, revealing the consequence of restricted ion transfer at high scan rate (Fig. 4g and S27†). Furthermore, the species formed were analysed during charge–discharge processes using in situ Raman tests (Fig. S28†). The in situ Raman results show that γ-NiOOH (465 cm−1 and 545 cm−1), γ-CoOOH (579 cm−1), and NiOOH (478 cm−1 and 556 cm−1) species are formed by the charging process, and α-Ni(OH)2 (453 cm−1 and 484 cm−1), α-Co(OH)2 (496 cm−1), Ni(OH)2 (460 cm−1) species are formed by the discharging process.
In order to better explore the practical application of Ni–Co oxyhydroxides, active carbon (AC) was used as a negative electrode, the Ni–Co oxyhydroxides as a positive electrode and polyvinyl alcohol/potassium hydroxide (PVA/KOH) gel as an electrolyte/separator to assemble flexible Ni–Co oxyhydroxides//AC all-solid-state ASC (Fig. 5a). The CV curves for the Ni–Co oxyhydroxides//AC all-solid-state ASC show a similar shape without distortion, revealing capacitive behavior from both electric double-layer capacitance and pseudocapacitance (Fig. 5b). This indicates fast charge storage kinetics and excellent rate characteristics. Furthermore, the good symmetry of the GCD curves for the Ni–Co oxyhydroxides//AC all-solid-state ASC shows outstanding charge/discharge reversibility and high coulombic efficiency (Fig. 5c and S29†). In addition, the Ni–Co oxyhydroxides//AC all-solid-state ASC displays a super-high areal capacitance of 5.7 F cm−2 (at 30 mA cm−2) and retains a capacitance of 4.3 F cm−2, showing a high capacitance retention rate of 75.4% (at 300 mA cm−2) as well as high coulombic efficiency, revealing superior rate characteristics (Fig. 5d). Additionally, the Ni–Co oxyhydroxides//AC all-solid-state ASC delivers a high specific capacitance of 168 F g−1 at 1 A g−1 and exhibits a specific capacitance of 121 F g−1 at 10 A g−1, demonstrating a capacitance retention of 72% (Fig. S30†). Moreover, the Ni–Co oxyhydroxides//AC all-solid-state ASC shows an excellent electrochemical stability with 91% capacitance retention after 7000 charge/discharge cycles, and the coulombic efficiency remains at nearly 99% throughout the cycles of the charge/discharge tests, indicating remarkable charge storage capability (Fig. 5e). The Nyquist plot of the Ni–Co oxyhydroxides//AC all-solid-state ASC displays low equivalent series resistance (2.36 Ω cm−2), small charge-transfer resistance (∼1 Ω cm−2) and low ion diffusion resistance, further revealing its excellent rate capability performance (Fig. S31†). Ragone plots of the energy and power density of the Ni–Co oxyhydroxides//AC all-solid-state ASC and other reported devices reveal that the Ni–Co oxyhydroxides//AC all-solid-state ASC delivers an ultra-high mass/areal/volumetric energy density of 92.6 W h kg−1/3.3 mW h cm−2/19.5 mW h cm−3 (at 1156 W kg−1/34.6 mW cm−2/204.1 mW cm−3) (Fig. 5f–h). These values of mass/areal/volumetric energy density are much higher than those reported in recent studies.36–59
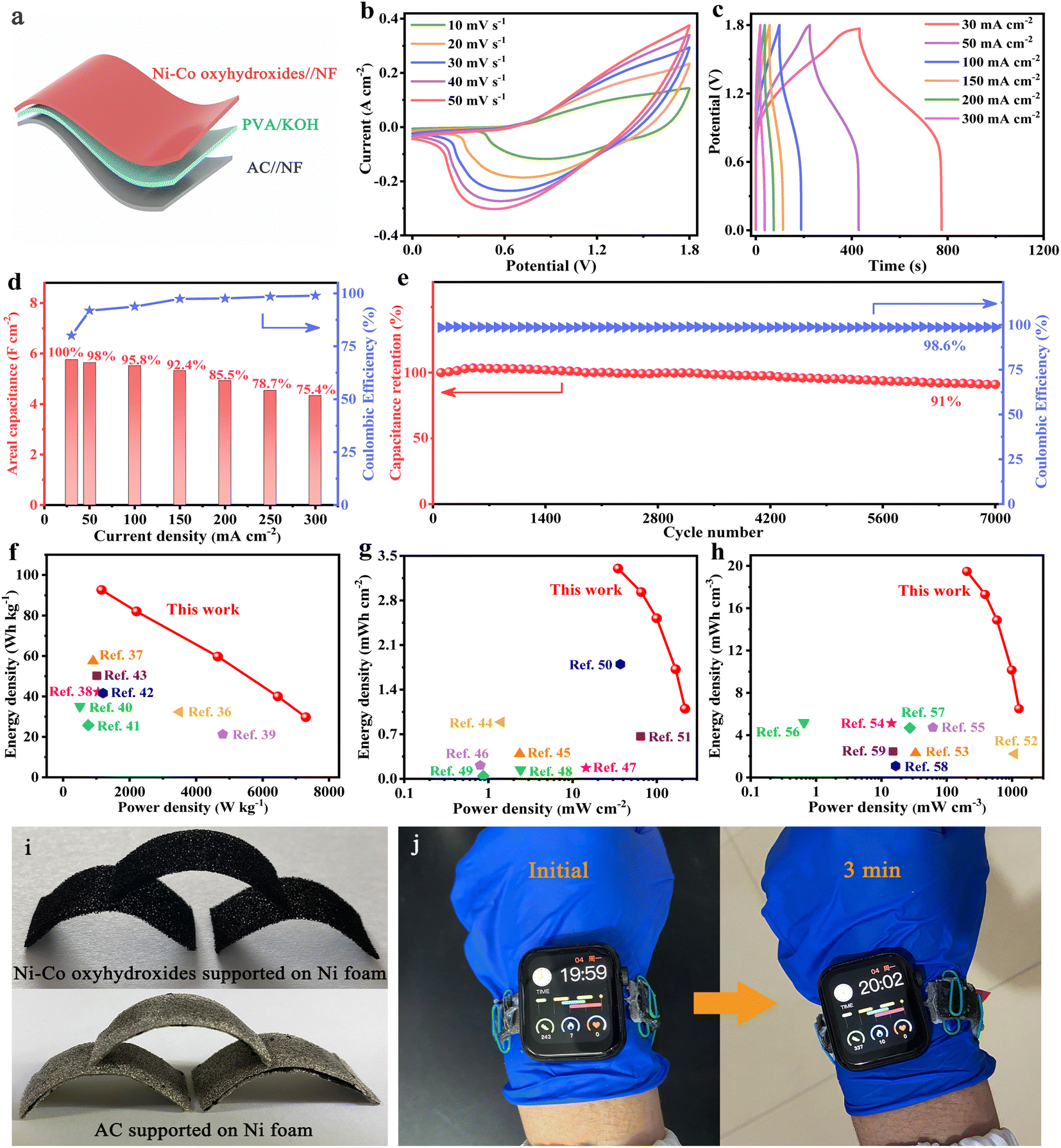 | ||
| Fig. 5 Electrochemical performance of wearable Ni–Co oxyhydroxides//AC all-solid-state ASC. (a) Schematic illustrations of assembled ASC. (b) CV curves at various scan rates. (c) GCD curves at different current densities. (d) Areal capacitance and coulombic efficiency at 30–300 mA cm−2. (e) Prolonged cycling performance and coulombic efficiency at 200 mA cm−2 over 7000 continuous charge/discharge cycles. Ragone plots for (f) mass energy/power densities, (g) areal energy/power densities and (h) volumetric energy/power densities compared with other reported devices. (i) Ni–Co oxyhydroxides and AC supported on Ni foam curved into arches. (j) Three wearable Ni–Co oxyhydroxides//AC all-solid-state ASC in series can power a smart watch. | ||
Furthermore, the flexibility of the Ni–Co oxyhydroxides supported on Ni foam and AC supported on Ni foam were utilized to achieve shape-tailored arched electrodes (Fig. 5i). Subsequently, an Ni–Co oxyhydroxides//AC all-solid-state ASC was assembled from the shape-tailored arched Ni–Co oxyhydroxides and AC. A watchband consisting of three flexible Ni–Co oxyhydroxides//AC all-solid-state ASCs in series can power a smartwatch for more than three minutes, further proving excellent mechanical flexibility and energy storage properties, which broaden its practical application in the field of energy storage (Fig. 5j).
3 Conclusions
In summary, we have developed unique 2D–3D/nano–micron hierarchical γ-phase Ni–Co oxyhydroxides with cationic vacancy defects and crystalline–amorphous interfaces via electrochemical reconstruction. The 2D–3D/nano–micron hierarchical nanoplates arrays can offer fast electron transport and preeminent mechanical stability. The γ-NiOOH and γ-CoOOH phases show superior electrochemical performance due to the presence of abundant Ni4+/Co4+, significantly enhancing the electrical conductivity and theoretical capacitance. Abundant crystalline–amorphous interfaces and cationic vacancy defects greatly increase the charge storage sites for improved specific capacitance. As a result, the Ni–Co oxyhydroxides show an ultrahigh capacitance of 20.9 F cm−2 at 4 mA cm−2 (3483 F g−1 at 0.67 A g−1), as well as excellent rate capability with 90.5% capacitance retention even when the current density was increased 60-fold. In addition, DFT calculations indicated that the cationic vacancy defects can enhance the strong affinity toward H2O, further facilitating the charge storage reactions. Under the optimal conditions, the fabricated flexible Ni–Co oxyhydroxides//AC all-solid-state ASC device exhibits an ultrahigh mass/areal/volumetric energy density of 92.6 W h kg−1/3.3 mW h cm−2/19.5 mW h cm−3 (at 1156 W kg−1/34.6 mW cm−2/204.1 mW cm−3) and a high capacitance retention ratio of 91% after 7000 cycles. The research provides a new approach for the design of the high-performance energy storage materials for flexible/wearable devices.4 Experimental
4.1 Synthesis of NiOOH
The Ni foam was immersed in 3.0 M HCl under ultrasonic treatment for 10 min and then repeatedly cleaned using ethanol and deionized water. Subsequently, it was dried for 5 h at 60 °C in a vacuum oven. NiOOH supported on Ni foam was prepared using an improved Fast-Repeated-Charging/Discharging (FRCD) electro-oxidation process.60 In a typical experiment, the anode was prepared using a 1 × 2.5 cm2 Ni foam, and the cathode contained an approximately 1 × 1 cm2 platinum sheet. The anode and cathode were then immersed in 45 mL of 2.0 M KOH. The cathode and anode were repeatedly charged and discharged at a current of 400 mA (∼2.8 V) for 4 hours using a CT3001A battery testing system. Finally, the laminar NiOOH was synthesized in situ on the Ni foam.4.2 Synthesis of NiOOH@Ni3S2
First, the NiOOH was immersed in 30 mL of 27 per mmol L sodium sulfide aqueous solution. The mixture was then transferred into a water bath kettle and heated at 80 °C for 1 h to obtain NiOOH@Ni3S2.4.3 Synthesis of NiCo-LDH@CoNi2S4
Typically, the as-obtained NiOOH@Ni3S2 was added to 35 mL of 13 mmol per L cobalt nitrate aqueous solution. After 15 min of stirring, the mixture was transferred to a Teflon-lined stainless-steel autoclave and heated at 150 °C for 7 h. The NiCo-LDH@CoNi2S4 was obtained through washing with deionized water several times and dried at 60 °C for 12 h in a vacuum oven.4.4 Synthesis of Ni–Co–P@CoNi2S4
The as-obtained NiCo-LDH@CoNi2S4 and NaH2PO2·H2O were placed at two separate positions in a porcelain boat with 1.5 g NaH2PO2·H2O at the upstream side of the furnace. The porcelain boat was placed at the center of a tube furnace at 300 °C for 1 h under an argon gas flow. After being allowed to cool to room temperature naturally, the Ni–Co–P@CoNi2S4 was obtained, washed with deionized water and then dried at 60 °C for 12 h in a vacuum oven.4.5 Synthesis of gamma-phase Ni–Co oxyhydroxides
The Ni–Co–P@CoNi2S4 was further electrochemical reconstructed in 2.0 M KOH aqueous electrolyte with cyclic voltammetry (CV) test between −0.2 and 0.6 V (at a scan rate of 5 mV s−1) in three-electrode system. After 20 cycles, the Ni–Co oxyhydroxides were synthesized via P and S leaching-induced strategy, and the Ni–Co oxyhydroxides were obtained through washed with deionized water and ethanol, then dried at 40 °C for 12 h in a vacuum oven (high mass loading of 6 mg cm−2).4.6 Electrochemical measurements
The working electrodes were as-prepared self-supporting NiOOH, NiOOH@Ni3S2, NiCo-LDH@CoNi2S4, Ni–Co–P@CoNi2S4, or Ni–Co oxyhydroxides electrodes. The standard calomel electrode (SCE) and a platinum electrode (Pt sheet) were used as the reference electrode and counter electrode in a 2.0 M KOH solution, respectively. Subsequently, the electrochemical tests were carried out by connecting the traditional three-electrode system to an electrochemical workstation (CHI 760E). The areal capacitance of the as-prepared self-supporting electrode was obtained from the charge/discharge curves using the equation below: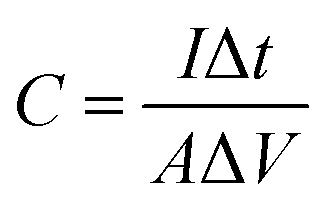 | (3) |
In addition, the charge storage contribution (the presence of diffusion/surface-controlled kinetics) can be investigated using the following equation:
| i = avb | (4) |
The capacitive contributions can be quantified for the overall charge storage using the following equation:
| iV = k1v + k2v1/2 | (5) |
This equation can be rewritten as:
| iV/v1/2 = k1v1/2 + k2 | (6) |
4.7 Assembly of flexible all-solid-state asymmetric supercapacitor (ASC)
Commercial active carbon (AC), acetylene black (conductive agent) and polytetrafluoroethylene (PTFE, binder) were mixed at a mass ratio of 8:1:1 on Ni foam as the negative electrode. Subsequently, a flexible all-solid-state ASC was assembled, in which the Ni–Co oxyhydroxides served as the positive electrode, AC served as the negative electrode and polyvinyl alcohol/potassium hydroxide (PVA/KOH) served as the solid electrolyte and separator. The Ni–Co oxyhydroxides and AC were closely assembled in parallel onto a PVA/KOH gel film. The mass loading balancing of the Ni–Co oxyhydroxides and AC is expressed according to the balance of charge on both electrodes (q+ = q−) using the following equations:| q+ = C+V+m+ | (7) |
| q− = C−V−m− | (8) |
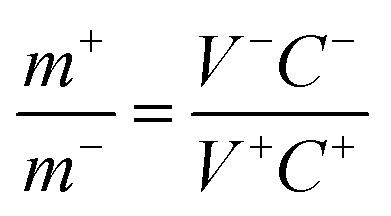 | (9) |
The calculations of the energy density (E) and power density (P) were based on the total weight of the two electrodes in the all-solid-state ASCs according to the following equations:
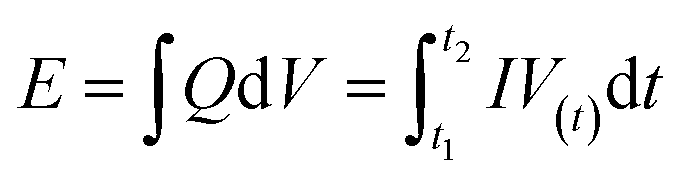 | (10) |
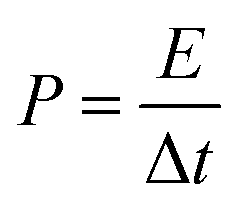 | (11) |
4.8 Computational methods
Density functional theory (DFT) calculations were carried out using the Vienna Ab initio Simulation Package (VASP) and the Perdew–Burke–Ernzerhof (PBE) exchange–correlation function. The interaction between valence electrons and the ionic core was described using the Projector-Augmented-Wave (PAW) method. The cut-off kinetic energies in the plane waves were set to 400 eV for all the calculations. Ionic relaxations were carried out under conventional energy (10–5 eV). A gamma-point centered 3 × 4 × 1 k-mesh was employed for structural optimizations. All atoms except the bottom three layers in the model were allowed to adjust until the magnitude of all residual forces was less than 0.02 eV Å−1. Spin polarization was also considered in all the calculations. Moreover, Grimme's DFT-D3 scheme was used to perform dispersion correction and describe the van der Waals (vdW) interactions in the models.Author contributions
Xue Ren: investigation, writing – original draft, visualization. Menggang Li: conceptualization, formal analysis. Longyu Qiu: software, formal analysis. Xin Guo: conceptualization. Fenyang Tian: visualization. Guanghui Han: validation. Weiwei Yang: supervision, writing – review & editing. Yongsheng Yu: conceptualization, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51871078 and 52071119), the Fundamental Research Funds for the Central Universities (Grant No. HIT.OCEF. 2021025), State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. 2022TS38) and the Heilongjiang Science Foundation (No. LH2020B006). The calculations were performed on TianHe-2 at Shanxi Supercomputing Center of China.Notes and references
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, eaan8285 CrossRef CAS PubMed
.
- J. Huang, J. Wei, Y. Xiao, Y. Xu, Y. Xiao, Y. Wang, L. Tan, K. Yuan and Y. Chen, ACS Nano, 2018, 12, 3030–3041 CrossRef CAS PubMed
- S. K. Hussain, B. N. V. Krishna and J. S. Yu, J. Mater. Chem. A, 2022, 10, 23352 RSC
- J. Acharya, G. P. Ojha, B. Pantab and M. Park, J. Mater. Chem. A, 2021, 9, 23977 RSC
- M. Li, W. Yang, Y. Huang and Y. Yu, Sci. China Mater., 2018, 61, 1167–1176 CrossRef CAS
- Z. Ma, F. Jing, Y. Fan, L. Hou, L. Su, L. Fan and G. Shao, Small, 2019, 15, 1900862 CrossRef PubMed
- D. P. Chatterjee and A. K. Nandi, J. Mater. Chem. A, 2021, 9, 15880 RSC
- P. Wu, S. Cheng, M. Yao, L. Yang, Y. Zhu, P. Liu, O. Xing, J. Zhou, M. Wang, H. Luo and M. Liu, Adv. Funct. Mater., 2017, 27, 1702160 CrossRef
- B. Tian, H. Shin, S. Liu, M. Fei, Z. Mu, C. Liu, Y. Pan, Y. Sun, W. A. Goddard III and M. Ding, Angew. Chem., Int. Ed., 2021, 60, 16448–16456 CrossRef CAS PubMed
- L. Aguilera, P. C. M. Aguiar, Y. L. Ruiz, A. Almeida, J. A. Moreira, R. R. Passos and L. A. Pocrifka, J. Mater. Sci.: Mater. Electron., 2020, 31, 3084–3091 CrossRef CAS
- Y. Sato, S. Takeuchi and K. Kobayakawa, J. Power Sources, 2001, 93, 20–24 CrossRef CAS
- G. Han, M. Li, H. Liu, W. Zhang, L. He, F. Tian, Y. Liu, Y. Yu, W. Yang and S. Guo, Adv. Mater., 2022, 34, 2202943 CrossRef CAS PubMed
- H. Liu, Q. Lei, R. Miao, M. Sun, C. Qin, L. Zhang, G. Ye, Y. Yao, B. Huang and Z. Ma, Adv. Funct. Mater., 2022, 2207408 CrossRef CAS
- X. Zhang, M. Gao, L. Qiu, W. Yang and Y. Yu, J. Energy Chem., 2023, 79, 64–71 CrossRef CAS
- X. Guo, M. Li, L. Qiu, F. Tian, L. He, S. Geng, Y. Liu, Y. Song, W. Yang and Y. Yu, Chem. Eng. J., 2023, 453, 139796 CrossRef CAS
- M. Gao, F. Tian, Z. Guo, X. Zhang, Z. Li, J. Zhou, X. Zhou, Y. Yu and W. Yang, Chem. Eng. J., 2022, 446, 137127 CrossRef
- P. Gao, Z. Chen, Y. Gong, R. Zhang and H. Liu, Adv. Energy Mater., 2020, 10, 1903780 CrossRef CAS
- B. Huang, J. Yuan, Y. Lu, Y. Zhao, X. Qian, H. Xu, G. He and H. Chen, Chem. Eng. J., 2022, 436, 135231 CrossRef CAS
- J. Abed, S. Ahmadi, L. Laverdure, A. Abdellah, C. P. O'Brien, K. Cole, P. Sobrinho, D. Sinton, D. Higgins, N. J. Mosey, S. J. Thorpe and E. H. Sargent, Adv. Mater., 2021, 2103812 CrossRef CAS PubMed
- Y. Wu, J. Yang, T. Tu, W. Li, P. Zhang, Y. Zhou, J. Li, J. Li and S.-G. Sun, Angew. Chem., Int. Ed., 2021, 60, 26829–26836 CrossRef CAS PubMed
- L. Huang, H. Shin, W. A. Goddard III and J. Wang, Nano Energy, 2020, 75, 104885 CrossRef CAS
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 2200434 CrossRef CAS
- X. Guo, M. Li, L. He, S. Geng, F. Tian, Y. Song, W. Yang and Y. Yu, Nanoscale, 2021, 13, 14179–14185 RSC
- I. Hussain, S. Iqbal, T. Hussain, Y. Chen, M. Ahmad, M. S. Javed, A. AlFantazibe and K. Zhang, J. Mater. Chem. A, 2021, 9, 17790 RSC
- H.-L. Hsu, W.-C. Chiu, C.-C. Yang, L.-C. Chen, C.-L. Lin, C.-M. Huang and S.-S. Hou, Mater. Today Chem., 2022, 23, 100644 CrossRef CAS
- H. Peng, B. Yao, X. Wei, T. Liu, T. Kou, P. Xiao, Y. Zhang and Y. Li, Adv. Energy Mater., 2019, 9, 1803665 CrossRef
- H. Wu, X. Zhang, H. Zhang, W. Zhu and S. Li, J. Solid State Chem., 2020, 282, 121073 CrossRef CAS
- P. Wu, S. Cheng, M. Yao, L. Yang, Y. Zhu, P. Liu, O. Xing, J. Zhou, M. Wang, H. Luo and M. Liu, Adv. Funct. Mater., 2017, 27, 1702160 CrossRef
- J.-J. Zhou, W. Ji, L. Xu, Y. Yang, W. Wang, H. Ding, X. Xu, W. Wang, P. Zhang, Z. Hua and L. Chen, Chem. Eng. J., 2022, 428, 132123 CrossRef CAS
- X. Ren, Y. Zhou, Y. Du, Y. Jiang, Y. Chen, J. Wan and F. Ma, Appl. Surf. Sci., 2020, 514, 145951 CrossRef CAS
- L. Mei, T. Yang, C. Xu, M. Zhang, L. Chen, Q. Lin and T. Wang, Nano Energy, 2014, 3, 36–45 CrossRef CAS
- F. Liu, L. Zeng, Y. Chen, R. Zhang, R. Yang, J. Pang, L. Ding, H. Liu and W. Zhou, Nano Energy, 2019, 61, 18–26 CrossRef CAS
- S. Kumar, G. Saeed, N. H. Kim and J. H. Lee, Composites, Part B, 2019, 176, 107223 CrossRef CAS
- C. Shang, S. Dong, S. Wang, D. Xiao, P. Han, X. Wang, L. Gu and G. Cui, ACS Nano, 2013, 7, 5430–5436 CrossRef CAS PubMed
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed
- N. Zhang, Y. Li, J. Xu, J. Li, B. Wei, Y. Ding, I. Amorim, R. Thomas, S. M. Thalluri, Y. Liu, G. Yu and L. Liu, ACS Nano, 2019, 13, 10612–10621 CrossRef CAS PubMed
- J. Zhao, C. Ge, Z. Zhao, Q. Wu, M. Liu, M. Yan, L. Yang, X. Wang and Z. Hu, Nano Energy, 2020, 76, 105026 CrossRef CAS
- B. Huang, H. Wang, S. Liang, H. Qin, Y. Li, Z. Luo, C. Zhao, L. Xie and L. Chen, Energy Storage Mater., 2020, 32, 105–114 CrossRef
- H. Luo, B. Wang, T. Liu, F. Jin, R. Liu, C. Xu, C. Wang, K. Ji, Y. Zhou, D. Wang and S. Dou, Energy Storage Mater., 2019, 19, 370–378 CrossRef
- Z. Jia, R. Ding, W. Yu, Y. Li, A. Wang, M. Liu, F. Yang, X. Sun and E. Liu, Adv. Funct. Mater., 2022, 32, 2107674 CrossRef CAS
- J. Ning, M. Xia, D. Wang, X. Feng, H. Zhou, J. Zhang and Y. Hao, Nano-Micro Lett., 2021, 13, 2 CrossRef PubMed
- N. Parveen, M. Hilal and J. I. Han, Nano-Micro Lett., 2020, 12, 25 CrossRef CAS PubMed
- S. He, F. Guo, Q. Yang, H. Mi, J. Li, N. Yang and J. Qiu, Small, 2021, 17, 2100353 CrossRef CAS PubMed
- L. Yu, W. Li, C. Wei, Q. Yang, Y. Shao and J. Sun, Nano-Micro Lett., 2020, 12, 143 CrossRef CAS
- G. Nagaraju, S. C. Sekhar, L. K. Bharat and J. S. Yu, ACS Nano, 2017, 11, 10860–10874 CrossRef CAS PubMed
- K. Li, B. Zhao, J. Bai, H. Ma, Z. Fang, X. Zhu and Y. Sun, Small, 2020, 16, 2001974 CrossRef CAS PubMed
- Y. Wang, X. Hong, Y. Guo, Y. Zhao, X. Liao, X. Liu, Q. Li, L. He and L. Mai, Small, 2020, 16, 2000293 CrossRef CAS PubMed
- J. Gong, W. Luo, Y. Zhao, M. Xie, J. Wang, J. Yang and Y. Dai, Chem. Eng. J., 2022, 434, 134640 CrossRef CAS
- H. Zhang, Y. Lv, X. Wu, J. Guo and D. Jia, Chem. Eng. J., 2022, 431, 133233 CrossRef CAS
- Y. Zhou, X. Tong, N. Pang, Y. Deng, C. Yan, D. Wu, S. Xu, D. Xiong, L. Wang and P. K. Chu, ACS Appl. Mater. Interfaces, 2021, 13, 34292–34300 CrossRef CAS PubMed
- N. Jayababu, S. Jo, Y. Kim and D. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 19938–19949 CrossRef CAS PubMed
- R. Li, X. Ba, H. Zhang, P. Xu, Y. Li, C. Cheng and J. Liu, Adv. Funct. Mater., 2018, 28, 1800497 CrossRef
- D. Kong, Y. Wang, S. Huang, J. Hu, Y. V. Lim, B. Liu, S. Fan, Y. Shi and H. Y. Yang, Energy Storage Mater., 2019, 23, 653–663 CrossRef
- J. Li, S. Luo, B. Zhang, J. Lu, W. Liu, Q. Zeng, J. Wan, X. Han and C. Hu, Nano Energy, 2021, 79, 105410 CrossRef CAS
- W. Zuo, C. Xie, P. Xu, Y. Li and J. Liu, Adv. Mater., 2017, 29, 1703463 CrossRef PubMed
- C. Guan, W. Zhao, Y. Hu, Q. Ke, X. Li, H. Zhang and J. Wang, Adv. Energy Mater., 2016, 6, 1601034 CrossRef
- H. Yuan, G. Wang, Y. Zhao, Y. Liu, Y. Wu and Y. Zhang, Nano Res., 2020, 13, 1686–1692 CrossRef
- J. Wen, B. Xu, J. Zhou, J. Xu and Y. Chen, Small, 2019, 15, 1901313 CrossRef PubMed
- X. Zhou, X. Li, D. Chen, D. Zhao and X. Huang, J. Mater. Chem. A, 2018, 6, 24603–24613 RSC
- X. Ren, Y. Du, M. Song, Y. Zhou, Y. Chen, F. Ma and J. Wan, Chem. Eng. J., 2019, 375, 122063 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09035j |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2023 |