
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reducing hybrid ligand-based alane and chasing aluminium(I): dialane and unusual transient dialumene†
Xiaobai
Wang
 ,
Raphael F.
Ligorio
,
Franziska
Rüttger
,
David M. J.
Krengel
,
Nico
Graw
,
Regine
Herbst-Irmer
,
Anna
Krawczuk
and
Dietmar
Stalke
*
,
Raphael F.
Ligorio
,
Franziska
Rüttger
,
David M. J.
Krengel
,
Nico
Graw
,
Regine
Herbst-Irmer
,
Anna
Krawczuk
and
Dietmar
Stalke
*
Georg-August-Universität Göttingen, Institut für Anorganische Chemie, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: anna.krawczuk@uni-goettingen.de; dstalke@chemie.uni-goettingen.de
First published on 29th July 2024
Abstract
In this work, an alane, [DNIAlH2] (1) (DNI = 3,3-dimethyl-2-[2-methyl-2-(2,6-diisopropyl-aniline)ethenyl]-3H-indolenine), stabilised by a hybrid ligand was reduced by Jones's Mg(I) ([(MesBDIMg)2]) and Roesky's Al(I) ([DippBDIAl:]). The resulting dialane compound [{DNI(H)Al}2] (2) was characterised using NMR spectroscopy, mass spectrometry, DFT calculations and single-crystal XRD experiments. The reaction of aluminium dihydride [DNIAlH2] (1) with [DippBDIAl:] at high temperatures gives an intramolecular C(sp2)–H bond-activated compound 3. To study the monomeric hybrid ligand-based Al(I), characterisations and computational calculations were performed, which elucidate that compound 5, consisting of two inequivalent aluminium atoms in an Al2CN four-membered ring, resulting from the activation of a carbon–nitrogen bond in the reaction of [DNINa] with [(Cp*Al)4].
Introduction
Recent research on aluminium coordination compounds has continued to advance the development of aluminium applications in catalysis and organic synthesis, despite the fact that the most abundant metal in the Earth's crust was once historically very difficult to produce.1 The first milestone structure of low oxidation state aluminium was Al(II) in [{(TMS)2CH}2Al–Al{CH(TMS)2}2] (Scheme 1, A), published by Uhl in 1988,2,3 followed by Al(I) in [(Cp*Al)4] (Cp* = C5Me5) (B), published by Schnöckel et al. in 1991.4 Another landmark structure, [DippBDIAl:] (C), was revealed by Roesky and co-workers in 2000.5 This well-designed, bulky BDI compound has gained widespread interest from main-group chemists owing to its electronic structure and reactivity toward small molecules.6,7 In 2007, Jones and co-workers isolated the first magnesium(I) compound [(DippBDIMg)2] based on the BDI ligand.8 The unique properties of [(RBDIMg)2] (R = Mes, Dipp; Mes = 2,4,6-trimethylphenyl)9,10 and [DippBDIAl:]11 (C) as mild reducing agents opened up new options in low oxidation state aluminium chemistry. Their abilities to transfer electrons/hydrogen atoms efficiently or participate in oxidative addition reactions to form reactive Al(II) centers allow for many further investigations.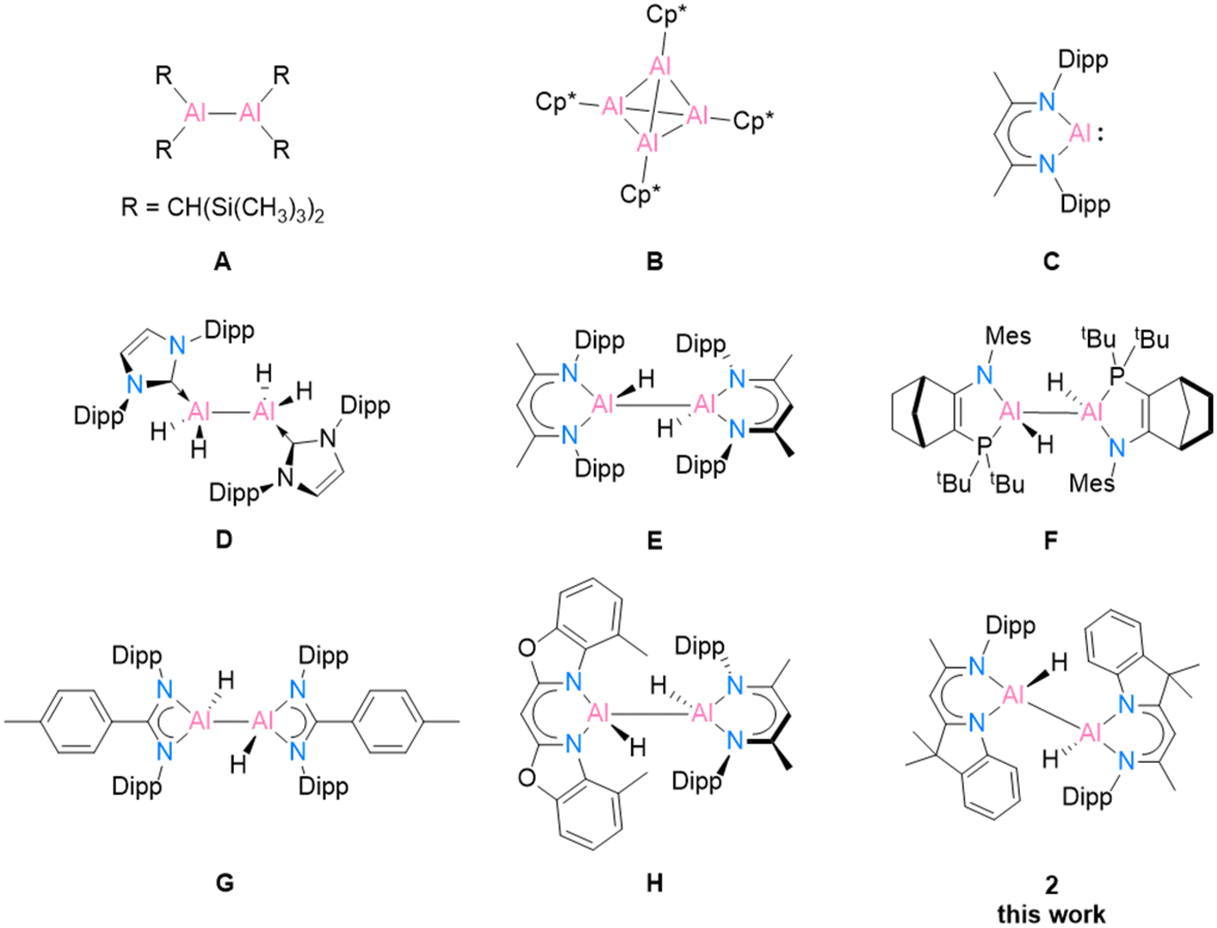 | ||
| Scheme 1 Selected examples of low oxidation state aluminium compounds A–C, and dialane complexes D–H obtained using [(DippBDIMg)2] or [DippBDIAl:] (C) as reducing agents. | ||
In 2010, the first neutral low oxidation state aluminium hydrides [{(IPr)(H)Al}2] (IPr = :C{(DippNCH)2}) (D) and [{(DippN)2}CRAlH2]2 (R = Me, p-tolyl (G), t-Bu, and Ni-Pr2) were isolated by Jones and co-workers by treatment with previously mentioned [(MesBDIMg)2] and the corresponding aluminium(III) hydride precursor.9 This remarkable work provides a mild way to prepare dialanes. In Nikonov and co-workers’ work, the reactivity of the aluminium(I) species [DippBDIAl:] (C) was demonstrated as nucleophiles, which can undergo oxidative addition reactions with electrophiles [DippBDIAlH2] to form the dihydrodialane complex [{DippBDI(H)Al}2] (E).11 A few years later, Cowley et al. synthesised the amidophosphine ligand [NR1/PR2] (R1 = Mes and Dipp; R2 = t-Bu2 and N2Si)-supported dihydroalane (F), which exhibits a reversible reductive elimination process involving the transfer of electrons. The optional breaking of the Al(II)–Al(II) bond leads to the formation of different diastereomers.12 Recently, Bakewell et al.13 reported a series of low oxidation state aluminium compounds using [DippBDIAl:] (C) as the reducing agent. Their work capitalises on Al(I), Al(II) and Al(III) to control the reaction equilibria and employs precise stoichiometry, resulting in the attainment of a wide scope of diverse reactions (G). In 2020, our group synthesised the first heteroleptic dihydroalane [4-MeBox(H)Al–Al(H)DippBDI] (H, 4-MeBox = [4-methyl-benzoxazol-2-yl]-methanide) by reducing an alane starting material with [DippBDIAl:] (C).14 In the current work, we present an innovative hybrid ligand system inspired by the fusion of two established successful ligand types, DippBDI and Box. Our objective is to explore the reduction reactions of a hybrid ligand-based aluminium compound and reveal its potential stabilisation ability for monomeric aluminium(I).
Results and discussion
While the [DippBDIAlH2] has been known for quite some time,15 the starting material [DNIAlH2] (1) was synthesised according to our recent work.16 We initially started to react 1 with Jones’ Mg(I) complex [(MesBDIMg)2] according to the literature method.9 The reaction was monitored by 1H NMR spectroscopy and extended to 3 days to isolate compound 2 in crystalline form from a mixed solvent (benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-hexane = 1:1) in 23% yield (Scheme 2).
:n-hexane = 1:1) in 23% yield (Scheme 2).
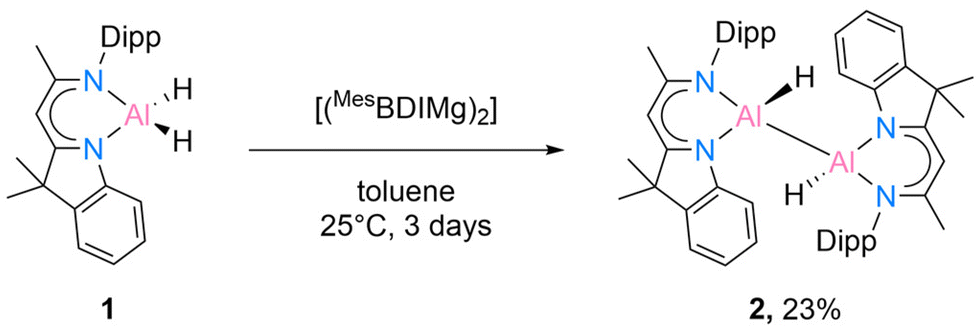 | ||
| Scheme 2 Reduction of [DNIAlH2] (1) with [(MesBDIMg)2] to yield dialane [{DNI(H)Al}2] (2). | ||
Compound 2 is crystallised in the space group P21/c with one molecule in the asymmetric unit. The molecular structure is depicted in Fig. 1. Compound 2 is a homoleptic dimer with a noticeable Al–Al bond. Each aluminium atom is coordinated with one hydrogen atom and one hybrid ligand. The Al–Al bond of 2.6007(6) Å is in the range (2.467 Å–2.751 Å) reported for complexes with Al–Al bonds.2,12 The hybrid ligand induces significant deviations in the dislocation patterns of the aluminium atoms relative to the ligand C3N2 plane: d(Al1⋯C3N2) = 0.0793(15) Å and d(Al2⋯C3N2) = 0.7368(14) Å, respectively. The 1H NMR experiment showed the chemical shift of the backbone proton at 5.01 ppm and a broad peak was observed at 5.15–4.65 ppm, which was assigned to alane hydrides. Further analyses such as mass spectrometry (LIFDI [+], toluene) and elemental analysis confirmed the successful synthesis of compound 2 (see ESI†).
 | ||
| Fig. 1 Molecular structure of [{DNI(H)Al}2] (2). The anisotropic displacement parameters are depicted at 50% probability level. All ligand-based hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Al(1)–Al(2) 2.6007(6), Al(1)–H(1A) 1.519(12), Al(2)–H(2A) 1.528(13), Al(1)–N(1) 1.9235(11), Al(1)–N(2) 1.9407(11), Al(2)–N(3) 1.9257(11), Al(2)–N(4) 1.9391(11); N(1)–Al(1)–N(2) 93.27(5), N(3)–Al(2)–N(4) 92.60(5), H(1A)–Al(1)–Al(2) 107.6(6), H(2A)–Al(2)–Al(1) 125.0(6), C(10)–C(11)–C(12) 125.64(12), C(35)–C(36)–C(37) 125.58(11). | ||
The inspection of the steric maps of buried volumes (VB) reveals the used ligand (50.9%) and the two deprotonated ligands (49.5%, 51.5%) in 2 to exhibit only minimal alterations (Fig. 2). The DNI ligand has significantly smaller buried volumes than those of DippBDI (61.9%), tBu, DippBDI (66.1%) and DIPePBDI (65.2%, DIPeP = 2,6-diisopentylphenyl).17 The UV-vis spectrum of 2 shows two absorption maxima at 447 nm and 352 nm (calculated values = 488 nm and 347 nm, Fig. S43–S45†), where the former one is responsible for the red colour of the compound. When analysing the frontier molecular orbitals of 2 (Fig. S59†), two features stand out.
 | ||
| Fig. 2 Calculated buried volumes VB for the free DNI ligand (left)18 and that at Al1 (mid) and Al2 in 2 (right) (3.5 Å sphere around Al, the right side of each map is the Dipp group, Dipp = 2,6-diisopropylphenyl). | ||
While the lowest unoccupied molecular orbital (LUMO, Fig. S59a†) is predominantly extended across the ligands and have a π* character, the highest occupied molecular orbitals (HOMO and HOMO−2, Fig. S59b and c†) correspond to Al–Al σ-bonding orbitals. The HOMO–LUMO gap is 3.44 eV. It is also interesting to take a look at the nature of the Al–Al bond using Bader's Quantum Theory of Atoms in Molecules (QTAIM).19
The graphical representation of the second derivative of the electron density, Laplacian ∇2ρ(r), in the Al–Al region is shown in Fig. 3. The first immediate element that attracts attention is the presence of two nuclear attractors (Al atoms) connected by two (3,–1) bond critical points (BCPs, green spheres) and one (3,–3) point (magenta sphere) that is not related to the nuclear position. A non-nuclear attractor (NNA) is found. This rather unique arrangement has been already observed in several metal complexes with close interatomic distances and is considered to be an ‘electronic glue’ binding together the metal ions.20 The value of electron density at the NNA is relatively low (0.059 e− Bohr−3); however, it corresponds well to previously reported metal–metal bonds with NNA present.21 The electron densities associated with the two BCPs are smaller than the one for NNA, however the difference is less than 0.001 e− Bohr−3. The Laplacian operator of the electron density at the NNA is negative, indicating the local concentration of electron density.
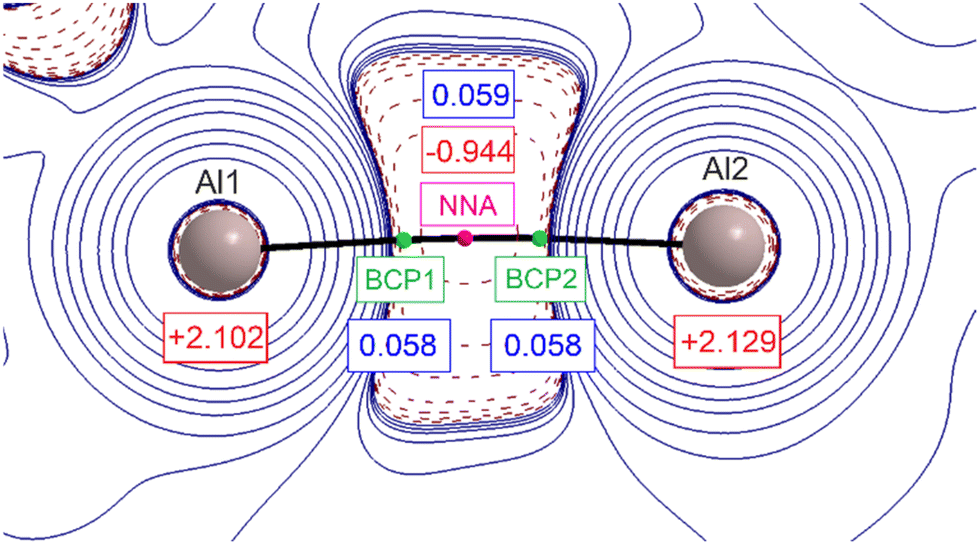 | ||
| Fig. 3 Laplacian operator of the electron density in a plane containing both Al nuclei of [{DNI(H)Al}2] (2). Contours are drawn in a logarithmic scale: positive as solid blue lines and negative ones as dashed red lines. The non-nuclear attractor (NNA) is depicted in a magenta box, bond critical points (BPCs) are given in green box, and AIM charges are given in red boxes for both Al ions and charge density magnitude at both bond critical points and the NNA are given in blue boxes (e− Bohr−3). | ||
The large negative region of ∇2ρ(r) around the Al center not only points at the covalent character of the Al–Al chemical bonding but also suggests the σ-orbital origin, exactly as the frontier molecular orbitals (FMOs), discussed earlier. The summary taken from DFT single-molecule calculations is shown in Table S7.†
Recently, Bakewell13 has reported a series of low oxidation state aluminium compounds through the reductive elimination reaction of the Al–H bond in aluminium dihydride compounds using the aluminium(I) species [DippBDIAl:] (C). To obtain further information on this reactivity, compounds [DNIAlH2] (1) and C were reacted in a 1:1 ratio (Scheme 3).
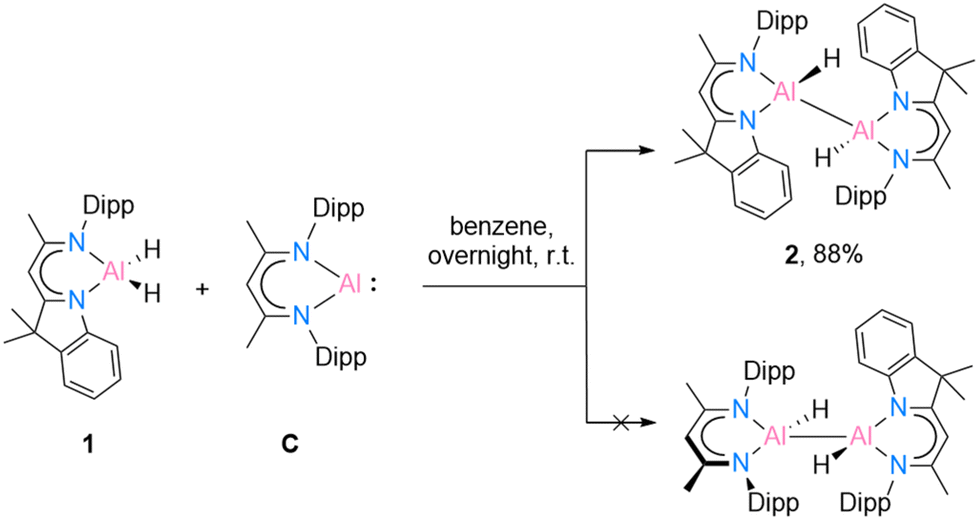 | ||
| Scheme 3 Reduction reaction of [{DNI(H)Al}2] (2) through oxidative addition reaction of [DippBDIAl:] (C). | ||
In benzene at 25 °C, it led to an immediate colour change of the mixture, indicating the formation of an envisaged new dialane [{DNI(H)Al}–{Al(H)DippBDI}]. The reaction was monitored by 1H NMR spectroscopy, and after 24 hours, most of the solvent was removed under reduced pressure. This way, after leaving the saturated mixture in benzene overnight, identical red crystals of dialane 2 were obtained in very good yield (88%). It is highly likely, therefore, that dialane 2 is the most stable compound in the equilibrium of all possible formed homo- and heteroleptic Al(I)/Al(II) species. To find out whether the transient dialumene equilibrium [Al(I) = Al(I)] is present during the reaction, the aluminium dihydride compound 1 was heated with excess [DippBDIAl:] (C) and a rare example of C(sp2)–H cleavage by the transient dialumene was observed in the reaction (Scheme 4). Notably, DFT calculations (see Theoretical section) indicate an endergonic reaction, ΔG = 12.8 kcal mol−1, hence justifying the formation of 3 occurring above room temperature. In respect to the first step, the formation of the heteroleptic dimer suggests an endergonic reaction, ΔG = 12.8 kcal mol−1, with a subsequent exergonic disproportionation of the dimer, ΔG = −11.7 kcal mol−1. The formation of 3* indicates an unfavourable reaction, ΔG = 31.1 kcal mol−1, while the last step occurs at a favorable intramolecular C–H activation, producing 3 (ΔG = −19.4 kcal mol−1).
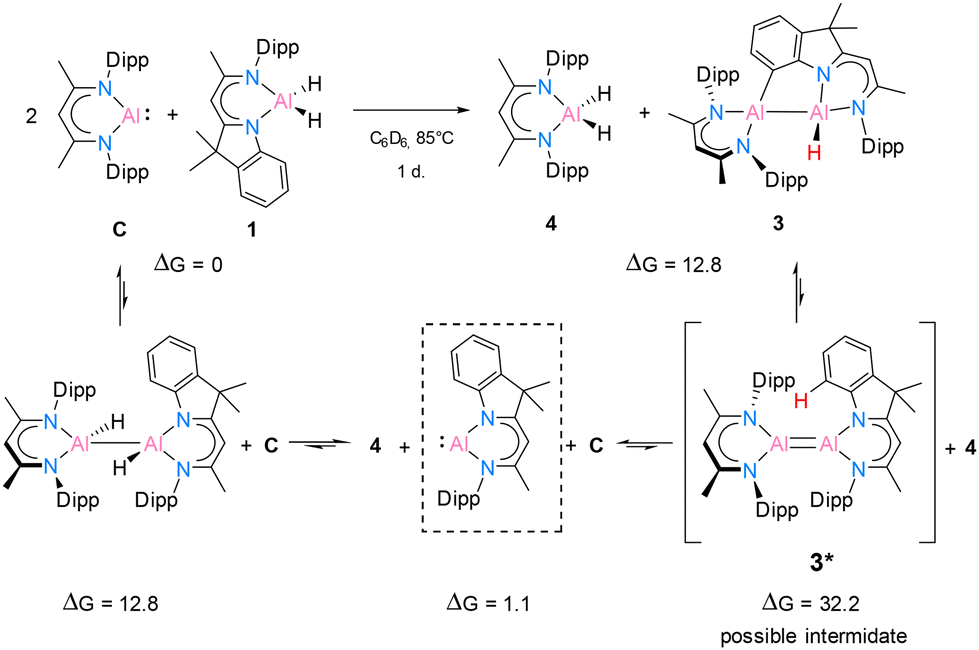 | ||
| Scheme 4 Possible dialumene intermediate 3* for the reaction of 1 with excess [DippBDIAl:] (C). Intramolecular C–H activation forms compound 3 at 85 °C after 1 day. Free energies (ΔG), given in kcal mol−1, are relative to the initial reactants, [DippBDIAl:] + [DNIAlH2] (C + 1). | ||
A deep red colour was observed immediately by solving both substances in benzene-d6. The reaction was monitored by 1H NMR spectroscopy and after one day it showed complete consumption of C (see ESI†). Crystallization from n-hexane yielded colourless crystals first, which were identified by 1H NMR and SC-XRD experiments to be [DippBDIAlH2] (4). The remaining red solution was completely dried and the residue was recrystallised from benzene. The crystals of 3 suitable for SC-XRD experiments were obtained from a saturated mixture at room temperature after one day. However, the formation of colourless crystals of 4 still occurred as well. The NMR spectroscopic data in C6D6 confirms the formation of both 4 and 3 in a ratio of roughly 1/0.6. Here, 4 shows a 1H backbone chemical shift of 4.87 ppm, while both backbone signals of 3 are shifted to lower fields (5.02 ppm for H28 and 5.07 ppm for H11, for numbering see Fig. 4). One carbon signal in the 13C NMR spectrum at 141.6 ppm is significantly broadened and very likely belongs to the C–H bond-activated aromatic carbon C2 of the DNI ligand.
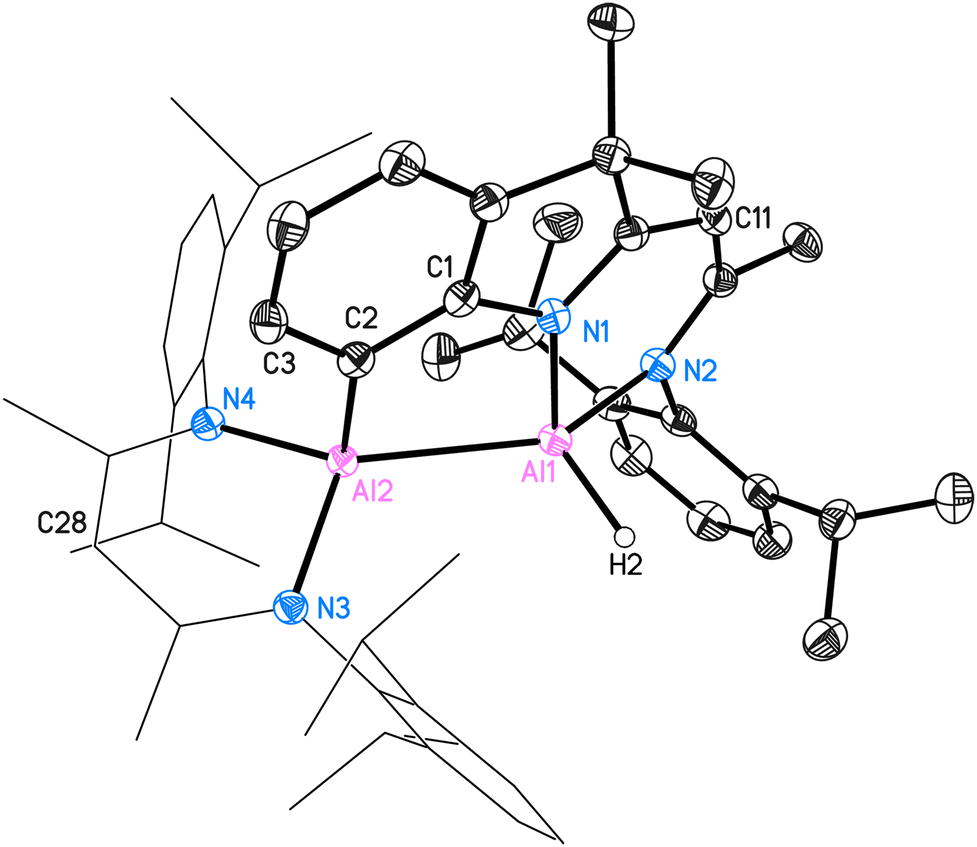 | ||
| Fig. 4 Molecular structure of intramolecular C–H bond-activated (C2–H2) compound 3. The anisotropic displacement parameters are depicted at the 50% probability level. All ligand-based hydrogen atoms and disorders are omitted for clarity. Selected bond lengths (Å) and angles (°): Al(1)–Al(2) 2.6044(6), Al(2)–C(2) 2.0172(12), C(1)–C(2) 1.3947(17), C2)–C(3) 1.4103(17), C(1)–N(1) 1.4220(15), Al(1)–N(1) 1.9046(10), Al(1)–N(2) 1.9422(11), Al(2)–N(3) 1.9303(11), Al(2)–N(4) 1.9149(11), Al(1)–H(2) 1.538(15); C(2)–Al(2)–Al(1) 88.65(4), C(1)–C(2)–C(3) 112.77(11), C(1)–C(2)–Al(2) 115.52(8), C(1)–N(1)–Al(1) 122.31(8), N(1)–Al(1)–Al(2) 86.09(3), N(1)–Al(1)–N(2) 91.31(5), N(3)–Al(2)–N(4) 95.58(5), H(2)–Al(1)–Al(2) 112.6(6). | ||
The crystal structure of 3 is shown in Fig. 4. This C–H bond-activated compound crystallises in the monoclinic space group P21/c with one molecule in the asymmetric unit. The Al–Al bond distance is 2.6044(6) Å, which is very close to that in 2 (2.6007(6) Å). Thus, the reduced steric impact of the dimethylindole moiety is evident, given that the DippBDI ligand is notably bulkier than DNI. The hybrid ligand system creates space for intramolecular reactions within the possible intermediate 3* (see Scheme 4) and is believed to cleave the CAr(sp2)–H bond, rather than undergoing addition to the aromatic solvent as the Birch-type reductions (e.g. benzene or toluene).22 The bond distance d(Al2–C2) = 2.0172(12) Å shows good agreement with the most published Al−CAr(sp2) bond lengths by Power,23 Tokitoh,24 Bakewell,13 Braunschweig25 and Roesky.26 The distances d(C1–C2) = 1.3947(17) Å and d(C1–N1) = 1.4220(15) Å are marginally elongated due to the formation of a five-membered ring (Al1–N1–C1–C2–Al2). On the contrary, the bond length of Al1–N1 decreases (1.9046(10) Å), compared with 2 (1.9235(11) Å). It seems worthy to note that we found approximately 6% dialuminoxane in the solid-state aligning, which was possibly caused by heating the mixture in glassware. The 2D NMR experiments and LIFDI ([+], toluene) proved the successful synthesis of 3. Bond lengths, angles and disorder are shown in the ESI.†
Further attempted reductions of dihydrodialane 2 with Na or K by using Jones’ Na/NaCl and K/KI10 or deprotonation of the backbone with [KCH(SiMe3)2] or [KN(SiMe3)2],7 however, demonstrated either a certain inertness of 2 or led to total decomposition. In order to investigate whether monomeric low oxidation state aluminium can be stabilised in the presence of only a unilateral bulky protecting group, while increasing the reaction space of the aluminium ion at the other side, the reaction of [DNINa]16 with [(Cp*Al)4] (B) was tested according to the synthetic protocols of Schnöckel27 and Kretschmer28 In anticipation of the successful stabilization of monomeric low oxidation state aluminium, an interesting structure that results from the reaction of an aluminium(I) complex intermediate and the sodiated ligand has been determined serendipitously.
The treatment of the solvent-free sodium compound [DNINa] with 1.0 eq. [(Cp*Al)4] (B) in toluene gave compound 5. The mixture was stirred at 90 °C for 1 hour. The undissolved [(Cp*Al)4] (B) was continuously dissolved into the mixture and it turned brown rapidly. Formed [Cp*Na] was filtered and the filtrate was re-dissolved in n-hexane (Scheme 5). In the absence of the monomeric aluminium(I) compound, the red crystals of 5 were found after leaving the saturated n-hexane solution at −30 °C overnight.
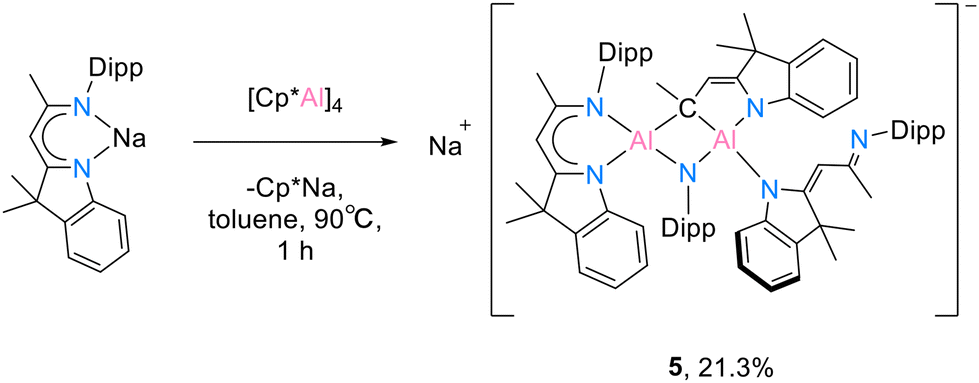 | ||
| Scheme 5 Reaction of solvent-free [DNINa] with [(Cp*Al)4] (B). | ||
Two molecules in the asymmetric unit crystallise in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with four n-hexane molecules. The structure of 5 is shown in Fig. 5. The two inequivalent aluminium atoms in 5 are four-coordinated by three nitrogen atoms (N1–N5) and one bridging carbon atom (C38). They both form distorted tetrahedra. The whole molecule comprises two entire deprotonated DNI ligands (N1, N2 chelating to Al1; N5 pending to Al2) and one activated fragmented ligand (around N3 and N4). This ligand, which originally formed the C3N2 residue with N3–C38–C40–C41–N4, was activated and the N3–C38 bond was cleaved to form a Dipp-amide.
with four n-hexane molecules. The structure of 5 is shown in Fig. 5. The two inequivalent aluminium atoms in 5 are four-coordinated by three nitrogen atoms (N1–N5) and one bridging carbon atom (C38). They both form distorted tetrahedra. The whole molecule comprises two entire deprotonated DNI ligands (N1, N2 chelating to Al1; N5 pending to Al2) and one activated fragmented ligand (around N3 and N4). This ligand, which originally formed the C3N2 residue with N3–C38–C40–C41–N4, was activated and the N3–C38 bond was cleaved to form a Dipp-amide.
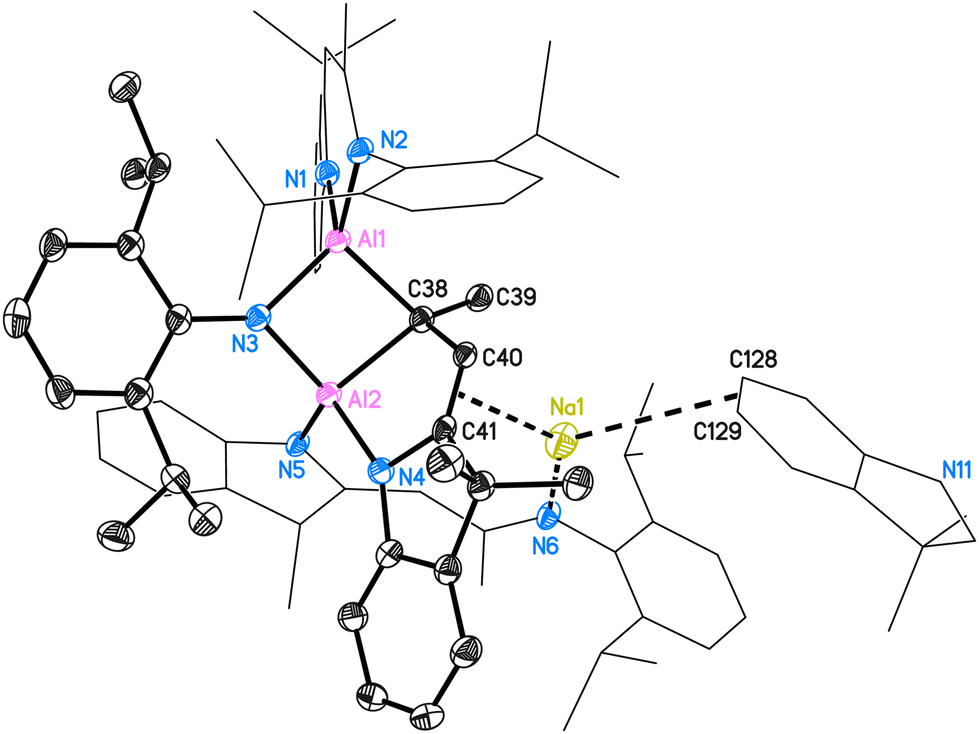 | ||
| Fig. 5 Molecule 1 of structure 5. The anisotropic displacement parameters are depicted at the 50% probability level. All ligand-based hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): N(1)–Al(1) 1.941(2), N(2)–Al(1) 1.927(2), N(3)–Al(1) 1.871(2), Al(1)–C(38) 1.968(2), N(3)–Al(2) 1.871(2), Al(2)–C(38) 2.026(3), C(38)–C(40) 1.521(3), C(40)–C(41) 1.351(3), N(4)–C(41) 1.414(3), Al(2)–N(4) 1.876(2), Al(2)–N(5) 1.917(2), Na(1)–C(40) 2.588(3), Na(1)–C(41) 2.718(3), Na(1)–N(6) 2.343(2), Na(1)–C(129) 3.028(3); N(1)–Al(1)–N(2) 93.45(9), Al(1)–N(3)–Al(2) 89.59(9), Al(1)–C(38)–Al(2) 82.60(9), N(3)–Al(2)–C(38) 91.98(9), N(4)–Al(2)–N(5) 108.51(9), C(39)–C(38)–C(40) 111.24(19), C(38)–C(40)–C(41) 121.0(2), N(6)–Na(1)–C(40) 135.30(8), N(6)–Na(1)–C(41) 123.12(8). | ||
The bridging nitrogen atom N3 shows the same bond lengths of 1.871(2) Å to Al1 and Al2 (for molecule 2: d(Al3–N9) = 1.870(2) Å, d(Al4–N9) = 1.866(2) Å), while the Al2–N4 and Al2–N5 distances are different (for molecule 1: 1.876(2) Å and 1.917(2) Å; for molecule 2: 1.873(2) Å and 1.930(2) Å, respectively). The intact part of the C3N2 fraction forms with the second aluminium atom a new five-membered C3NAl ring. The aluminium–carbon bond Al1–C38 (1.968(2) Å, 1.970(2) Å) is shorter than the Al2–C38 distance (2.026(3) Å, 2.030(2) Å). The solid-state structure of 5 also reveals the C38–C40, C40–C41 and C41–N4 distances of 1.521(3) Å, 1.351(3) Å and 1.414(3) Å, hence the second significantly shorter than the first, which proves C40–C41 to be the double bond (for molecule 2 and further details, please see ESI†). The deprotonated pendent ligand around N5 and N6 only coordinates once to Al2 while the second nitrogen coordinates the sodium atom. Due to the complexity of the structure posing challenges for characterisation, all other analyses were performed using isolated crystals of compound 5.
The 1H NMR spectrum of 5 is very complex (see ESI†). Many signals overlap, particularly in the aromatic and methyl region, preventing unambiguous signal assignment. The signals of all three variations of the backbone proton can be identified from the 1H, 15N HMBC spectrum at 6.12 ppm, 5.22 ppm and 4.68 ppm, where the first two show a downfield shift with respect to the ligand backbone (∼1.1 ppm and 0.2 ppm, respectively). The 27Al NMR spectrum shows two very broad signals, but no reliable assignment is possible. Despite the difficulty of analysing the product by NMR, both elemental analysis and mass spectrometry (LIFDI [+], toluene) confirm the existence of product 5.
Based on the above-mentioned discussion and experimental steps leading to 5, we were challenged to define a mechanism standing behind the formation of this rather unusual product. The free energy diagram of a possible mechanism route is provided in Fig. 6. A detailed description of the reaction path is given in the ESI† file (see Computational details section).
 | ||
| Fig. 6 Reaction path for obtaining compound 5 and corresponding free energies (kcal mol−1) of the reactants in the gas phase. Blue lines represent the gas phase route, whereas the orange lines depict the reaction in the toluene solvated medium. Transition state 2 could not be reliably defined. The meaning of A, B, C and D paths is described in the ESI† file (see the Computational details section). | ||
Conclusions
We successfully synthesised and characterised a hybrid ligand-based dialane [{DNI(H)Al}2] (2) featuring less steric strain by reducing of the precursor alane [DNIAlH2] (1) with [(MesBDIMg)2] and [DippBDIAl:] (C), respectively. Ongoing research continues to investigate deeper into the properties and reactivities of [DNIAl(I)]. Treatment of alane 1 with [DippBDIAl:] C at high temperature, yields an aromatic C(sp2)–H bond activation and the heteroleptic dialumene [DippBDIAl![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) AlDNI] acted as a key intermediate. The existence and reactivity of the transient monomeric [DNIAl:] was proved by computational calculations. Further reactivity is outlined in the ESI.† We are now investigating deeper into the reactivity of this promising dialane [{DNI(H)Al}2].
AlDNI] acted as a key intermediate. The existence and reactivity of the transient monomeric [DNIAl:] was proved by computational calculations. Further reactivity is outlined in the ESI.† We are now investigating deeper into the reactivity of this promising dialane [{DNI(H)Al}2].
Experimental section
All manipulations that require processing under inert conditions were carried out in an argon atmosphere using standard Schlenk techniques or an MBraun Glovebox in an argon atmosphere. Toluene was distilled from sodium. Toluene-d8, benzene and benzene-d6 were distilled from potassium. n-Hexane was distilled from the Na/K alloy. Solvents were all routinely degassed three times by the freeze–pump–thaw method and stored in a glove box over molecular sieves. [DNIAlH2],16 [(MesBDIMg)2],10 [DippBDIAl:],5 [(Cp*Al)4],29 and [DNINa]16 were prepared according to literature methods. Elemental analysis was performed using an Elementar Vario EL3 by the Analytisches Labor, Institut für Anorganische Chemie, Universität Göttingen. Mass spectra were recorded by the Zentrale Analytik within the Faculty of Chemistry, Universität Göttingen applying a Liquid Injection Field Desorption Ionisation-technique using a JEOL accuTOF instrument with an inert-sample application setup in an argon atmosphere. The 1H, 13C, 27Al and 2D NMR spectroscopic data were recorded using a Bruker Avance III 300 MHz, a Bruker Avance III HD 400 MHz, a Bruker Avance III HD 500 MHz and a Bruker Avance NEO 600 MHz instrument. The measurements were carried out at room temperature in solutions of deuterated solvents. For solubility and comparison reasons, the most used deuterated solvent for the 1H-NMR spectra is toluene-d8. The chemical shifts δ are given in ppm and the coupling constants J in Hz. The observed multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Young-type Teflon-valve borosilicate NMR tubes and standard NMR tubes were used throughout this work. The crystals were selected under cold protective conditions using an X-Temp2 device.30 The diffraction data were acquired using a Bruker Photon II detector with Mo Kα radiation. The data were integrated with SAINT.31 A multi-scan absorption correction was performed using SADABS.32 The structures were solved using SHELXT33 and refined on F2 using SHELXL34 in the graphical user interface ShelXle.35 The crystallographic information files (CIFs) can be obtained free of charge from the Cambridge Crystallographic Data Centre (CCDC 2313962 (2), 2313964 (3), 2313966 (4), and 2313965 (5)).†Preparation of 2
Route 1: In an argon atmosphere glove box, alane [DNIAlH2] 1 (453 mg, 1.17 mmol, 1.00 eq.) and [(MesBDIMg)2] (425.5 mg, 0.595 mmol, 0.51 eq.) were dissolved and stirred in about 15 mL dry and degassed toluene at room temperature for 3 days. The resulting mixture was filtered, dried and extracted with cold n-hexane (−30 °C, 1 mL each, several times). The n-hexane solution was evaporated to dryness and the first crop of deep red crystals of 2 (176 mg, 0.227 mmol) was obtained in a mixed solvent (benzene/n-hexane from the liquid–liquid diffusion). The supernatant was decanted and the crystals were washed with cold n-hexane. All remaining solutions were collected and dried to completeness. Benzene was added and left standing overnight. The second crop of crystal of 2 (40 mg, 0.052 mmol) was washed with cold n-hexane and dried under vacuum. Yield: 216.0 mg, 0.279 mmol, 23.90%. Route 2: In an argon glove box, [DippBDIAl:] (C) (20 mg, 0.045 mmol, 1.00 eq.) was dissolved in 5 mL benzene. Alane 1 (17.5 mmol, 0.045 mmol, 1.00 eq.) was added and a red clear mixture was stirred overnight at ambient temperature. The red mixture was concentrated in vacuo to approximately 0.05 mL and left standing overnight. Deep red crystals of 2 were formed and washed with cold n-hexane (2 × 0.1 mL). Yield: 15.4 mg, 0.02 mmol, 88%. 1H NMR (298 K, tol-d8, 300 MHz, solvent peak = 2.09 ppm): 7.13–6.90 (m, 10H, Ar–H), 5.01 (s, 2H, 11-H), 5.15–4.65 (br., 2H, Al–H(H1A, H2A)), 3.36–3.25 (m, 4H, 20-H, 23-H, 45-H, 48H, J = 7.0 Hz), 1.64 (s, 6H, 13-H, 38-H), 1.36 (s, 6H, 8,9,33,34-H), 1.20–1.13 (d, J = 6.6 Hz, 18H, iPr–H, 1.13 = 8,9,33,34-H), 1.05 (d, 6H, iPr–H, J = 6.9 Hz), 0.79 (br., 6H, iPr–H). 13C{1H} NMR (298 K, tol-d8, 75 MHz, solvent peak = 20.40 ppm): 180.23 (10-C), 171.59 (12-C), 150.13 (1-C), 144.96 (14-C), 141.28 (6-C), 124.33 (16-C, 18-C), 122.26 (4-C), 121.23 (5-C), 114.93 (2-C), 88.73 (11-C), 49.76 (7-C), 28.15 (20-C, 23-C), 25.38 (8-C, 9-C), 24.74 (21-C, 24-C), 24.61 (22-C, 25-C), 23.76 (13-C). 27Al NMR: not observed. Elemental analysis: C50H64Al2N4 calculated C 77.49, H 8.32, N 7.23, found C 76.51, H 8.46, N 7.33. LIFDI-MS ([+], toluene): 389.9(4) [(DNI)AlH2 + H]+, 774.5(100) [2]+.Preparation of 3
Compound 1 [DNIAlH2] (15.29 mg, 0.039 mmol, 1.00 eq.) and [DippBDIAl:] (C) (21 mg, 0.047 mmol, 1.20 eq.) were dissolved in a J. Young NMR tube with 0.6 mL C6D6. The mixture was heated to 85 °C and monitored by 1H NMR spectroscopy. After 1 day, the 1H NMR spectrum showed the completion of the reaction. The mixture was filtered and transferred into a glass vial. The volatiles were removed in vacuo and n-hexane was added. [DippBDIAlH2] (4) was obtained as colourless/pale yellow crystals and the red residue was isolated and dried. Then, 1–2 drops of benzene were added and the red crystals of 3 formed at room temperature after 24 h, which was still co-crystallised with [DippBDIAlH2] (4). The isolation of 3 from 4 seems impossible. The red crystal of 3 suitable for SC-XRD experiments should be carefully fragmentised from 4 under a microscope. NMR experiments could be carried out without this purification process. Due to the co-crystallisation with 4, the yield and element analysis could not reliably be determined. 1H NMR (298 K, C6D6, 500 MHz, solvent peak = 7.16 ppm): 8.03 (dd, 1H, 3-H, J = 6.78, 1.51 Hz), 6.95–7.25 (m, 11H, Ar–H), 5.07 (s, 1H, 11-H), 5.02 (s, 1H, 1-H), 3.15–3.4 (m, 6H, iPr–H), 2.67 (hept, 1H, iPr–H, J = 6.92 Hz), 1.65 (s, 3H, 30-H/26-H), 1.59 (s, 3H, 13-H), 1.49 (s, 3H, 26-H/30-H), 1.32 (s, 3H, 8-H/9-H), 1.29 (d, 3H, iPr–Me–H, J = 6.88 Hz), 1.26 (d, 3H, iPr–Me–H, J = 6.67 Hz), 1.24 (s, 3H, 8-H/9-H), 1.11–1.13 (m, 6H, iPr–Me–H), 1.10 (d, 3H, iPr–Me–H, J = 6.88 Hz), 1.07 (d, 3H, iPr–Me–H, J = 6.78 Hz), 1.03 (d, 3H, iPr–Me–H, J = 6.88 Hz), 0.98 (d, 3H, iPr–Me–H, J = 6.88 Hz), 0.92 (d, 3H, iPr–Me–H, J = 6.88 Hz), 0.60 (d, 3H, iPr–Me–H, J = 6.67 Hz), 0.43 (d, 3H, iPr–Me–H, J = 6.56 Hz), 0.34 (d, 3H, iPr–Me–H, J = 6.88 Hz). 13C NMR (298 K, C6D6, 125 MHz, solvent peak = 128.06 ppm): 179.47 (1C, 10-C), 171.41 (1C, 27-C/29-C), 169.43 (1C, 27-C/29-C), 169.38 (1C, 12-C), 160.49 (1C, 1-C), 145.75 (1C, Dipp-C), 145.13 (1C, Dipp-C), 145.05 (1C, Dipp-C), 143.69 (1C, Dipp-C), 143.60 (1C, Dipp-C), 142.61 (1C, Dipp-C), 142.55 (1C, Dipp-C), 142.23 (1C, Dipp-C), 141.90 (1C, Dipp-C), 141.58 (1C, 2-C), 138.52 (1C, 6-C), 136.11 (1C, 3-C), 127.15 (1C, Dipp-C), 126.96 (1C, Dipp-C), 126.43 (1C, Dipp-C), 125.14 (1C, Dipp-C), 124.69 (1C, Dipp-C), 124.45 (1C, Dipp-C), 124.33 (1C, Dipp-C), 123.94 (1C, Dipp-C), 123.72 (1C, Dipp-C), 122.06 (1C, 4-C), 120.61 (1C, 5-C), 98.41 (1C, 28-C), 89.04 (1C, 11-C), 50.20 (1C, 7-C), 29.33 (1C, iPr–C), 28.97 (1C, iPr–C), 28.53 (1C, iPr–C), 28.37 (1C, iPr–C), 28.36 (1C, iPr–C), 27.89 (1C, iPr–C), 26.37 (1C, Me–C), 26.15 (1C, 8-C/9-C), 26.02 (1C, 8-C/9-C), 25.32 (1C, Me–C), 25.13 (1C, Me–C), 24.81 (2C, Me–C), 24.68 (1C, Me–C), 24.58 (1C, Me–C), 24.52 (1C, Me–C), 24.39 (1C, Me–C), 24.35 (1C, Me–C), 24.07 (1C, 26-C/30-C), 23.86 (1C, 13-C), 23.69 (1C, Me–C), 23.67 (1C, Me–C), 23.52 (1C, Me–C), 23.35 (1C, 26-C/30-C). 15N NMR (298 K, C6D6, 50.7 MHz, relative to CH3NO2): −189.03 (1N, 1-N), −198.49 (1N, 3-N/4-N), −213.30 (1N, 2-N), −215.48 (1N, 3-N/4-N). LIFDI-MS ([+], toluene): 389.9 (2) [1 + H]+, 447.3 (3) [4 + H]+, 774.3 (42) [2]+, 830.4 (100) [3]+, 846.4 (3) [3 + O]+.Preparation of 5
To a solution of THF, free [DNINa] (471.64 mg, 1.23 mmol, 1.00 eq.) in dry, degassed toluene (15 mL) [(Cp*Al)4] (C) (200 mg, 0.31 mmol, 0.25 eq.) was added and the mixture was stirred rapidly at 90 °C for 1 hour. Over the course of heating, undissolved (Cp*Al)4 was continuously consumed and the mixture turned brown quickly. As the reaction proceeded, a large amount of insoluble solid-state material was formed. The mixture was cooled to ambient temperature and the insoluble solid was filtered using a syringe equipped with a glass fibre filter. The volatiles were removed under reduced pressure and the dark-brown residue was dissolved in n-hexane and filtered again. Crystals of 5 suitable for SC XRD were grown in a concentrated n-hexane solution at −30 °C. Yield: 99.9 mg, 0.086 mmol, 21.3%. The 1H and 13C{1H} NMR signals cannot be assigned unambiguously to the structure. The signal intensity of 13C{1H} NMR is weak due to the poor solubility of the crystal in most deuterated solvents. 27Al NMR (104 MHz, tol-d8): 73.91, −5.75. Elemental analysis: C75H92Al2N6Na calculated C 78.02, H 8.03, N 7.28; observed C 77.57, H 8.04, N 7.09. LIFDI-MS ([+], toluene): 1132.6(100) [5 − Na + H]+, 1155.5(100) [5 + H]+.Theoretical section
Electronic structure, topological analysis of electron density and theoretical UV-VIS spectrum
Gas-phase DFT calculations were performed to get access to the electronic structure of compound 2 and time-dependent TD-DFT was employed to model theoretical UV-vis spectra. For both methods, the PBE0 functional and the def2-TZVP basis sets were used in the Gaussian 16 software.36 Grimme's empirical dispersion correction37 with Becke–Johnson damping (GD3BJ),38 as well as an ultrafine integration grid, was used. The input coordinates for each calculation were generated using crystallographic coordinates. Full optimisation of the structures led to bond lengths and angles to be reproduced well with good agreement to the experimentally determined values. The final geometries were confirmed to be minimum at the potential energy surface (PES) via calculation of the associated vibrational frequencies, which turned to be all positive. The DFT-obtained wavefunction was further used to perform full topological analysis of the electron density employing the AIMAll software.39Transition state and reaction path
In recent investigations, the benchmarking of several density functional theory (DFT) functionals by Mats and Brinck aimed to elucidate the accurate prediction of the transition state in Diels–Alder reactions involving small organic molecules.40 The study unveiled that both the hybrid B3LYP (h-GGA) and the long-range corrected wB97X (lc-GGA) functionals exhibited a systematic overestimation of approximately 5 kcal mol−1 in the activation energy when compared to the highly accurate CCSD(T) reference method. However, the incorporation of dispersion corrections within the wB97X functional (wB97XD) yielded remarkable improvements, closely aligning the results with those obtained using the meta-hybrid M06-2x (hm-GGA) functional, showcasing deviations of approximately 2 kcal mol−1 relative to the reference. In terms of the basis set choice, even the inclusion of diffuse functions within small basis sets, such as 6-31+G(d), demonstrated substantial reductions in the basis set superposition error, leading to enhanced precision in the calculations. In our study, despite exhaustive efforts invested in performing calculations using the hm-GGA and lc-GGA functionals with a diffuse basis set, challenges in achieving convergence were encountered, particularly during the final stages of the reaction. In order to bypass these limitations, geometry minimisations and energy calculations were performed using the Gaussian 16 software,36 at the B3LYP/6-31G(d) level of theory, which aligns with recent studies investigating transition states governed by a similar mechanism.7,28 Geometries were initially minimised in gas phase with further optimization employing the periodic continuum medium (PCM) model, mimicking a toluene solvated environment. After optimising the transition state in toluene, a substantially different geometry was obtained, resulting in the loss of the unique imaginary mode. Nonetheless, a single-point calculation was conducted using the geometry obtained from the gas phase optimisation. The analysis revealed the presence of a dissociative mode with a wave number close to −38 cm−1, accompanied by a secondary negative frequency of very low intensity. Despite the presence of two imaginary modes, we found that the free energy (ΔG) associated with the solvation of the reactants and intermediate (−12 kcal mol−1) remained preserved for the non-optimised transition state in toluene. Additionally, the geometries were highly similar in both optimisations for products and reactants. Hence, we considered to have a reasonable approximation for the energy of the transition state within the solvated medium.Oxidative addition reaction of aluminium(I)
To maintain uniformity with the quantum mechanical approach selected for determining transition state geometries and energies, we utilized the same theoretical level to compute the Gibbs energy for the reaction depicted in Scheme 5. Here, however, the implicit solvent simulated is coherent with the Experimental section, thus gave benzene. Noteworthily, compounds 3 and 4 were initially subjected to gas-phase minimisation, followed by a subsequent single-point calculation using the PCM method, due to their difficult energy/geometry convergence employing the PCM method. Notably, all positive frequencies were retained for compound 4, indicating its stability at the minimum energy structure with and without the simulated solvent. However, in the case of compound 3, a negative wavenumber of −5 cm−1 was detected.Data availability
The data are available on request from the author.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This paper is dedicated to Prof. V. Chandrashekar on the occasion of his 65th birthday, all the best Chandra! AK and RFL gratefully acknowledge the Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within the computational grant no. PLG/2022/015866. This project has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 405832858.References
- D. Rabinovich, Nat. Chem., 2013, 5, 76 CrossRef CAS PubMed.
- W. Uhl, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 1113–1118 CrossRef CAS.
- W. Uhl, R. Graupner, S. Pohl, W. Saak, W. Hiller and M. Neumayer, Z. Anorg. Allg. Chem., 1997, 623, 883–891 CrossRef CAS.
- (a) C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564–565 CrossRef; (b) S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., 1993, 105, 1828–1830 CrossRef CAS.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- (a) C. Cui, S. Köpke, R. Herbst-Irmer, H. W. Roesky, M. Noltemeyer, H. G. Schmidt and B. Wrackmeyer, J. Am. Chem. Soc., 2001, 123, 9091–9098 CrossRef CAS PubMed; (b) Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., 2004, 116, 3525–3527 CrossRef; (c) Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS; (d) S. Nagendran and H. W. Roesky, Organometallics, 2008, 27, 457–492 CrossRef CAS; (e) H. W. Roesky, Inorg. Chem., 2004, 43, 7284–7293 CrossRef CAS PubMed.
- S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 15982–15986 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865–869 CrossRef CAS PubMed.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 CrossRef CAS.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- R. L. Falconer, G. S. Nichol, I. V. Smolyar, S. L. Cockroft and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 2047–2052 CrossRef CAS PubMed.
- C. Bakewell, K. Hobson and C. J. Carmalt, Angew. Chem., Int. Ed., 2022, 61, e202205901 CrossRef CAS PubMed.
- J. Kretsch, A. Kreyenschmidt, T. Schillmöller, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2020, 59, 13690–13699 CrossRef CAS PubMed.
- (a) C. Cui, H. W. Roesky, H. Hao, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2000, 39, 1815–1817 CrossRef CAS; (b) M. D. Anker, A. L. Colebatch, K. J. Iversen, D. J. D. Wilson, J. L. Dutton, L. García, M. S. Hill, D. J. Liptrot and M. F. Mahon, Organometallics, 2017, 36, 1173–1178 CrossRef CAS.
- X. Wang, F. Rüttger, A. Krawczuk, R. Herbst-Irmer and D. Stalke, Eur. J. Inorg. Chem., 2023, e202300629 Search PubMed.
- S. Grams, J. Mai, J. Langer and S. Harder, Organometallics, 2022, 41, 2862–2867 CrossRef CAS.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed; (c) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS; (d) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766 CrossRef; (e) I. Koehne, N. Graw, T. Teuteberg, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2017, 56, 14968–14978 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in molecules. A quantum theory, Clarendon Press; Oxford University Press, Oxford England, New York, 1994, vol. 22 Search PubMed.
- (a) W. L. Cao, C. Gatti, P. J. MacDougall and R. Bader, Chem. Phys. Lett., 1987, 141, 380–385 CrossRef CAS; (b) J. T. Boronski, L. R. Thomas-Hargreaves, M. A. Ellwanger, A. E. Crumpton, J. Hicks, D. F. Bekiş, S. Aldridge and M. R. Buchner, J. Am. Chem. Soc., 2023, 145, 4408–4413 CrossRef CAS PubMed; (c) W. Haider, D. M. Andrada, I.-A. Bischoff, V. Huch and A. Schäfer, Dalton Trans., 2019, 48, 14953–14957 RSC; (d) K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2021, 143, 18172–18180 CrossRef CAS PubMed.
- (a) J. A. Platts, J. Overgaard, C. Jones, B. B. Iversen and A. Stasch, J. Phys. Chem. A, 2011, 115, 194–200 CrossRef CAS PubMed; (b) G. K. H. Madsen, P. Blaha and K. Schwarz, J. Chem. Phys., 2002, 117, 8030–8035 CrossRef CAS; (c) M. Michalski, A. J. Gordon and S. Berski, J. Mol. Model, 2019, 25, 211 CrossRef CAS PubMed.
- P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed.
- (a) R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS PubMed; (b) J. D. Queen and P. P. Power, Chem. Commun., 2022, 59, 43–46 RSC.
- T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818–10821 CrossRef CAS PubMed.
- D. Dhara, A. Jayaraman, M. Härterich, R. D. Dewhurst and H. Braunschweig, Chem. Sci., 2022, 13, 5631–5638 RSC.
- A. Kumar, K. Yadav, N. Graw, M. K. Pandey, R. Herbst-Irmer, U. Lourderaj, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2023, 29, e202300546 CrossRef CAS PubMed.
- H. Sitzmann, M. F. Lappert, C. Dohmeier, C. Üffing and H. Schnöckel, J. Organomet. Chem., 1998, 561, 203–208 CrossRef CAS.
- O. Kysliak, H. Görls and R. Kretschmer, Dalton Trans., 2020, 49, 6377–6383 RSC.
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Chem. Commun., 2013, 49, 2858–2860 RSC.
- D. Stalke, Chem. Soc. Rev., 1998, 27, 171 RSC.
- Bruker AXS Inc., SAINT v8.40B, Madison (WI), USA, 2019 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- AIMAII (Version 19.10.12), Todd A. Keith, Overland Park KS, USA, 2019, https://aim.tkgristmill.com Search PubMed.
- M. Linder and T. Brinck, Phys. Chem. Chem. Phys., 2013, 15, 5108–5114 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2313962, 2313964–2313966. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01798f |
| This journal is © The Royal Society of Chemistry 2024 |