
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiposomal formulation of a gold(III) metalloantibiotic: a promising strategy against antimicrobial resistance†
Alejandro
Llamedo
ab,
Pablo
Rodríguez
a,
Yaiza
Gabasa
cd,
Raquel G.
Soengas
 *b,
Humberto
Rodríguez-Solla
b,
David
Elorriaga
b,
Francisco J.
García-Alonso
*b and
Sara M.
Soto
*cd
*b,
Humberto
Rodríguez-Solla
b,
David
Elorriaga
b,
Francisco J.
García-Alonso
*b and
Sara M.
Soto
*cd
aNanovex Biotechnologies S.L., Parque Tecnológico de Asturias Edificio CEEI, 33428 Llanera, Spain
bDepartamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles”, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: rsoengas@uniovi.es
cISGlobal, Hospital Clínic - Universitat de Barcelona, Barcelona, Spain
dCIBER Enfermedades Infecciosas (CIBERINFEC), Instituto de Salud Carlos III, Madrid, Spain
First published on 28th August 2024
Abstract
A novel lipoformulation was developed by encapsulating cationic (S^C)-cyclometallated gold(III) complex [Au(dppta)(N2Py-PZ-dtc)]+ (AuPyPZ) in liposomes. The liposomal form of compound AuPyPZ has a bactericidal action similar to that of the free drug without any appreciable effect on the viability of mammalian cells. Furthermore, the nanoformulation reduces metalloantibiotic-induced inhibition of hERG and the inhibition of cytochromes, significantly decreasing the potential liabilities of the metallodrug. The obtained metalloantibiotic liposomal formulation shows high stability and suitable properties for drug delivery, representing an effective strategy to fight against drug-resistant bacteria.
Introduction
Antimicrobial resistance (AMR) is a global health disaster that is already killing 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people a year, and it is predicted to cause 10 million deaths per year by 2050 if the current situation is not improved.1,2 Antibiotics, which allowed us to treat deadly bacterial infections and save many lives, are now becoming less and less effective.3,4 When antibiotics fail to work, it undermines the foundations of modern healthcare, from joint replacements to chemotherapy, threatens to reverse declines in mortality and morbidity from infectious diseases, and poses potentially crippling financial effects.5,6
000 people a year, and it is predicted to cause 10 million deaths per year by 2050 if the current situation is not improved.1,2 Antibiotics, which allowed us to treat deadly bacterial infections and save many lives, are now becoming less and less effective.3,4 When antibiotics fail to work, it undermines the foundations of modern healthcare, from joint replacements to chemotherapy, threatens to reverse declines in mortality and morbidity from infectious diseases, and poses potentially crippling financial effects.5,6
Despite the clear need for more antimicrobial agents, such drugs have not been forthcoming.7,8 There are currently only 42 antibiotic drugs in clinical development, which is clearly not enough considering the current rate of AMR.9 Furthermore, a closer analysis of their structures reveals an even more pressing problem: most of these compounds are simply variations of well-known antibiotic families, meaning that bacteria will quickly develop resistance to these antibiotics.10 The only long-term solution is to develop new chemical classes of antibiotic drugs to replace the increasingly ineffective ones.11 Since these new generations of antibiotics would have completely new structures, they would be insensitive to existing resistance mechanisms and effective against the most dangerous forms of antibiotic-resistant bacteria.12
Most of the lead compounds in preclinical and clinical antibiotic development share a common structural feature: they are purely organic compounds. However, a radically new approach that has emerged in recent years has been to explore the antimicrobial properties of metal complexes, the so-called metalloantibiotics.13,14 Although yet to be advanced to clinical trial stage, metalloantibiotics have several advantages over purely organic drug candidates.15 The great diversity of ligand types and geometries makes metal-based complexes very useful to access a very little explored chemical space for drug development, especially relevant for the design of new antimicrobials.16 Furthermore, metal-based complexes can provide unique modes of action: ligand exchange or release, redox activation and catalytic generation of toxic species (reactive oxygen species, ROS), as well as depletion of essential substrates, making them capable of abolishing enzyme activities, disrupt membrane function or damage DNA.17 As recently reported, metallodrugs display a 10-fold higher hit-rate towards resistant pathogens compared to purely organic molecules.18 Among the metal-containing species, gold complexes have attracted considerable attention for exploration as metalloantibiotics.19,20 This interest was sparked by the potent antibacterial activity discovered in the FDA-approved drug auranofin (Aur, Ridaura™), a gold(I) complex in clinical use since 1985 for the treatment of severe rheumatoid arthritis (Fig. 1).21 Since then, a wide variety of gold(I) and gold(III) complexes have reported for their antibiotic potential.22 However, despite the potential of gold complexes for antimicrobial drug development, the clinical application of metallodrugs poses some major challenges,23 including limited aqueous solubility and short in vivo half-lives, resulting in inadequate bioavailability and low accumulation at the therapeutic site.24 In addition, several metallodrugs exerted systemic toxicity, mainly related to hepatic damage and cardiotoxic effects.25–27 A general strategy to overcome these limitations focuses on the encapsulation of metallodrugs in nanotechnology-based formulations.28,29 Encapsulation systems limit the interaction of the drug with healthy cells and create a protective home for the drug, dramatically improving toxicity as well as the efficacy of the treatment.30,31
 | ||
| Fig. 1 Gold metalloantibiotics. | ||
Among the synthetic nanocarriers used to encapsulate metallodrugs, liposomes are by far the most successful. Liposomes have a morphology similar to that of cellular membranes: they are spherical vesicles with an aqueous inner core surrounded by lipid bilayers.32 Liposomes are one of the first nanosized vehicles used in drug delivery applications, as they can encapsulate hydrophilic drugs in an aqueous core and hydrophobic derivatives in the lipidic bilayer.33,34 Liposomes are therefore ideal vehicles for the delivery of metallodrugs, and several studies has been published so far, mainly dedicated to anticancer platinum metallodrugs. In this regard, a liposomal formulation of cisplatin (Lipoplatin®) has reached Phase III clinical trials.35–37 Liposomal formulations for anticancer metallodrugs of ruthenium,38,39 iridium40,41 and gold42 have also received attention.
To address the global shortage of effective antibiotics, our research group is dedicated to the development of novel antibiotic classes based on Au(III) complexes. It was recently reported the synthesis of a series of cationic (dppta)Au(III) complexes (dppta = N,N-diisopropyl-P,P-diphenylphosphinothioic amide-κ2C,S) with κ2S,S’-dithiocarbamate (dtc) auxiliary ligands (AuDTC) displaying potent antibacterial activity against methicillin-resistant S. aureus (MRSA).43
These promising results indicate that the gold(III) (C^S, S^S)-cyclometallated center could be used as a scaffold to develop new classes of antibiotics. However, these metalloantibiotics present a very low aqueous solubility, considerable cytotoxicity in hepatic cell lines and potential cardiotoxicity, which have hampered its clinical application.
Herein we report the synthesis and chemical and biological characterization of a (C^S, S^S)-cycloaurated gold(III) dithiocarbamate complex with improved properties. Furthermore, the encapsulation of this gold(III) complex in liposomes was investigated. The obtained liposomal formulations were physico-chemically characterized and subsequently analyzed for their antibacterial activity, toxicity and relevant pharmacological properties.
Results and discussion
Synthesis and chemical characterization of [Au(dppta)(N2Py-PZ-dtc)]PF6 complex
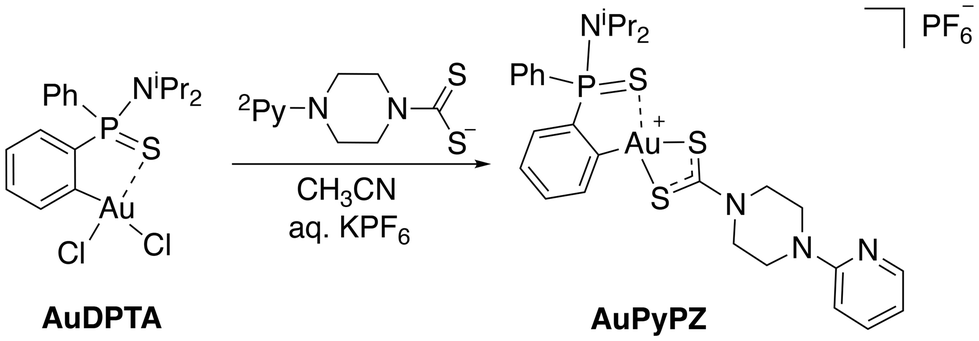
In the search for structures with improved features for liposomal encapsulation, we focused our attention on dithiocarbamate ligands with a pendant 2-pyridine ring. The target (C^S)-cyclometallated gold(III) complex was synthesized using established procedures with minor modifications.43 Thus, overnight reaction of Au(dppta)Cl2 complex (AuDPTA) with 4-(pyridin-2-yl)piperazine-1-dithiocarbamate sodium salt in methanol for 12 hours at room temperature and subsequent addition of aqueous KPF6 afforded the corresponding [Au(dppta)(N2Py-PZ-dtc)]PF6 complex (AuPyPZ) in 74% yield (Scheme 1). | ||
| Scheme 1 Synthesis of [Au(dppta)(N2Py-PZ-dtc)]PF6 complex (AuPyPZ). | ||
The complex was characterized by 1H, 13C{1H} and 31P{1H} NMR, IR, X-ray and HRMS. Complex formation with the dithiocarbamate ligand was confirmed by the upfield shift in the characteristic signal of the H ortho relative to Au (from δH 8.47 to 7.84 ppm) in the 1H NMR. On the other hand, the 31P NMR spectra of AuPyPZ display a significant downfield shift from δP 68.2 to 74.6 ppm. The 13C NMR resonance for the NCS2 carbon appeared at δC 195.0 ppm, the signals corresponding to the pyridine ring at δC 107.0, 113.7, 138.7, 149.5 and 157.8 ppm and the quaternary carbon directly attached to the Au(III) center in the dppta ligand at δC 144.7 ppm (1JPC 27.9 Hz). The band in the IR spectra at 1527 cm−1 is attributed to the delocalized dithiocarbamate NCS2 system, while the band around 840 cm−1 is due to the PF6− anion. The formation of the desired complex is further supported by the ion [M]+ observed in mass spectrometry (calculated for C28H35AuN4PS3+: 751.1421; found: 751.1414). The lack of fragmentation indicates appreciable stability, and is consistent with other gold(III) complexes containing dithiocarbamate ligands.
Slow vapour diffusion of hexane into a concentrated chloroform solution of AuPyPZ gold(III) complex gave crystals suitable for X-ray diffraction. The molecular structure is shown in Fig. 2, and the most significant bond lengths and bond angles are given in Table 1. The unit cell contains the two enantiomers of chiral phosphorous-containing complex AuPyPZ; one of the enantiomers is omitted in Fig. 2 for clarity. Diffraction studies revealed a mononuclear compound where the dithio ligand is κ2-S,S′ coordinated to the gold centre; however, the S2–Au1 and S3–Au1 involved distances are 2.377 Å and 2.235 Å, respectively, and this difference is attributed to the stronger trans influence of the C6H4 aromatic ring compared to that of the S = P moiety. The bite angle of the dithiocarbamate ligand is 74.63°, which is in agreement with the values reported in the literature for related dithiocarbamate complexes.44,45
| Bond lengths | |||
|---|---|---|---|
| Au1–S1 | 2.3229(13) | Au1–S2 | 2.3772(13) |
| Au1–S3 | 2.3347(11) | Au1–C13 | 2.0625(5) |
| Bond angles | |||
| S1–Au1–C18 | 91.10(1) | C13–Au1–S2 | 173.02(1) |
| S2–Au1–S1 | 95.89(5) | C13–Au1–S3 | 98.50(14) |
| S2–Au1–S3 | 74.63(5) | S1–Au1–S3 | 169.84(5) |
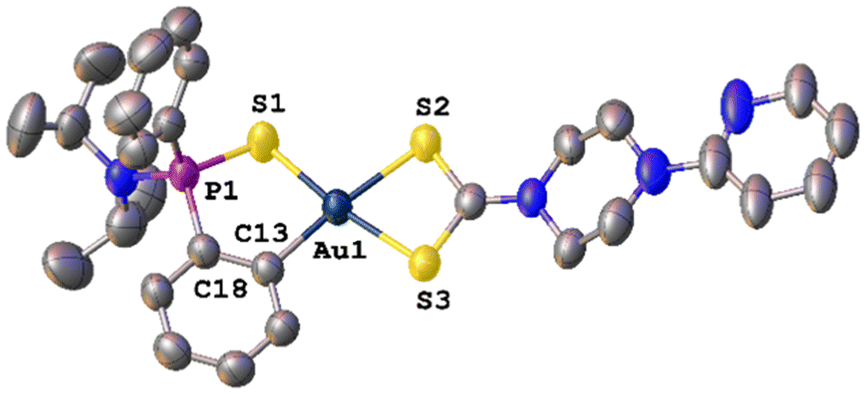 | ||
| Fig. 2 Molecular structure of complex AuPyPZ. Ellipsoids are shown at 50% probability. Hydrogen atoms, chloroform molecule and PF6 ion are omitted for clarity. | ||
The metallacycle is slightly displaced from planarity, with the phosphorus atom pointing out the plane with a torsion angle S1–P1–C18–C13 of 26.23°. Still, the value of the four-coordinate geometry index (τ4 = 0.10) is close to the ideal square planar geometry (τ4 = 0).46
In a biomedical context, a truthful chemical characterization requires not only an unambiguous description of the structure but also of the purity. Regardless of development stage, purity assessment is critical whenever chemistry is linked with biological activity and therapeutic application. Impurity analysis in a drug should include organic impurities and inorganic impurities.47 Organic impurities for AuPyPZ complex were analyzed by quantitative NMR (qNMR) using 1,3,5-trimethoxybenzene as internal reference,48 indicating high purity (≥99.5%) and an impurity level below the accepted limits.49 On other hand, inorganic impurities were assessed by ICP-MS, considering the most relevant class 1 elemental impurities (Cd, Hg, Pb) and also Sn, as the preparation of the starting complex involves a Sn–Au transmetallation step. The concentration of elemental impurities is in all cases below the accepted limits for any route of administration, as shown by the results compiled in Table 2.50
Chemical and biological stability of gold(III) complex AuPyPZ
The presence of a phosphorous atoms in the (C^S)-cyclometallated ligand allows rapid and non-destructive assessment of the stability of the gold(III) metallo-complex by 31P{1H} NMR. To investigate the stability of AuPyPZ under physiological conditions, a solution of the complex in DMSO-d6 was treated with phosphate buffer saline (PBS), heated to 37 °C and monitored by 31P{1H} NMR over 48 h. No significant deviation of the chemical shift of the phosphorous signal was observed over time, indicating that both dppta and dtc ligands are suitable for stabilizing gold(III) under physiological conditions (Fig. 3). | ||
| Fig. 3
31P{1H} NMR spectra of AuPyPZ in 0.5 mL of (CD3)2SO/PBS (1:1) (a) at t = 0; (b) after heating at 37 °C for 24 h; (c) after heating at 37 °C for 48 h. | ||
Despite the promising biological profile of gold(III) complexes, their application in pharmacology has been limited by their ability to oxidize biologically relevant molecules. If the metallic centre is not adequately stabilized, Au(III) complexes can be easily reduced to Au(I)/Au(0) in the presence of thiol containing amino acids. This reaction not only results in the loss of the chemical integrity of the complex, but also in the oxidation of sulfur-containing proteins. In fact, the oxidation of thiol containing amino acids by metal ions has been studied for decades because of their important role in both protein structure and function.51 One of the most abundant thiol-containing peptide in biological systems is the tetrapeptide glutathione (GSH, L-γ-Glu-Cys-Gly), present at mM concentrations in mammalian cells. To determine the redox stability of AuPyPZ complex in the presence of biological reductants, the behaviour of the complex in the presence of GSH was studied. A solution of complex AuPyPZ in DMSO-d6 and PBS was treated with an excess of GSH. The 31P NMR spectra of the resulting solution showed that the metallo-complex maintained its chemical integrity (Fig. 4).
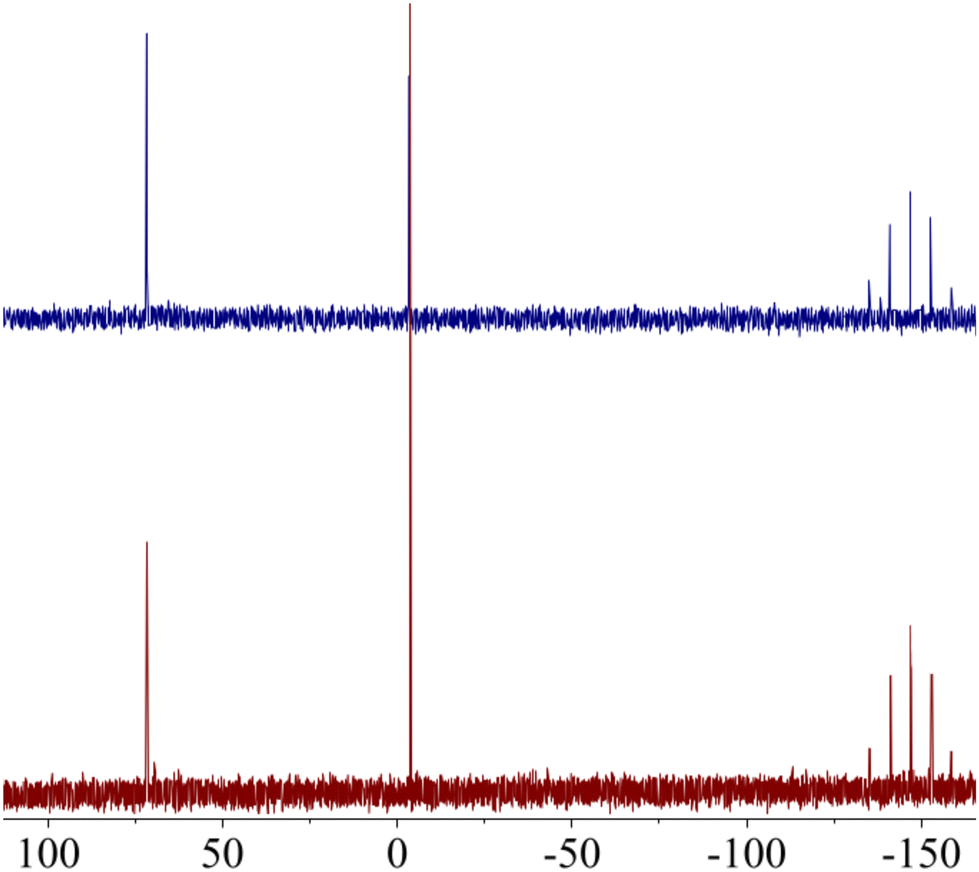 | ||
| Fig. 4
31P{1H} NMR NMR spectra of AuPyPZ in 0.5 mL of (CD3)2SO/PBS (1:1) (a) at t = 0; (b) after addition of aq. GSH (5 equiv.). | ||
In vitro biological evaluation
In vitro antibacterial activity of gold(III) complex AuPyPZ was screened using a panel of 21 strains belonging to six different bacterial species representing clinically important microorganisms included in the WHO critical pathogens list (Table 3 and ESI Table S1†). These bacterial strains have been selected among the most representative Gram-positive and Gram-negative bacteria causing important clinical infections and taking into account their resistance profile. AuPyPZ displayed the highest selective activity towards Gram-positive pathogens, especially against multidrug-resistant strains of S. aureus (MRSA), with a minimum inhibitory concentration (MIC) value of 0.5 mg L−1. For all S. aureus and S. epidermidis strains tested, complex AuPyPZ showed higher inhibitory efficacy than clinical antibiotics ciprofloxacin (CIP) or tobramycin (TOB). Such preference for Gram-positive strains has already been described for gold complexes, including those belonging to this chemical class. Most Gram-negative pathogens showed moderate MIC values (4 mg L−1 for S. maltophilia, 16 mg L−1 for P. aeruginosa, 8 mg L−1 for E. coli and K. pneumoniae). The highest efficacy compared to reference antibiotics was found for multidrug-resistant A. baumannii (MRAB), with MIC values of 2 mg L−1, which are lower than those of CIP and TOB. Particularly relevant is the activity shown against A. baumannii AbCr17, a pan-resistant strain that shows resistance even to last resort antibiotics such as colistin (ESI, Table S1†).| Entry | Bacterial speciesa | No. tested strains | MIC (mg L−1) | ||
|---|---|---|---|---|---|
| AuPyPZ | CIP | TOB | |||
| a MDR, multidrug-resistant bacteria; ATCC, American Type Culture Collection. | |||||
| 1 | MRSA (MDR clinical) | 4 | 0.5 | 32– > 128 | 2– > 128 |
| 2 | MSSA (ATCC 25923) | 1 | 1 | 0.5 | 0.5 |
| 3 | S. epidermidis (MDR clinical) | 3 | 1 | 128 | 0.25–32 |
| 4 | P. aeruginosa (MDR clinical) | 2 | 16 | 0.5– > 128 | 1– > 128 |
| 5 | P. aeruginosa (ATCC 27853) | 1 | 16 | 1 | 1 |
| 6 | S. malthophilia (MDR clinical) | 2 | 4 | 4–64 | 4– > 128 |
| 7 | E. coli (MDR clinical) | 1 | 8–16 | >128 | >128 |
| 8 | E. coli (ATCC 25922) | 1 | 8 | 0.25 | 0.25 |
| 9 | A. baumanii (MDR clinical) | 3 | 2 | >128 | 64– > 128 |
| 10 | A. baumanii (ATCC 19606) | 1 | 2 | 0.5 | 1 |
Considering the potential of AuPyPZ as a therapeutic antimicrobial agent, we evaluated the in vitro toxicity profile on tumor and non-tumor eukaryotic cell lines from liver tissue. The in vitro cytotoxicity results show an IC50 value of 1.48 μM (1.33 mg L−1) on the tumor cell line Hep G2, whereas the toxicity profile using the non-tumor cell line THLE-2 was slightly less favorable with an IC50 value of 1.12 μM (1.00 mg L−1). Overall, these results point to an acceptable but narrow therapeutic window, with a Therapeutic Index (TI) in the range of 2–4 for S. aureus and S. epidermidis.
Early assessment of absorption, distribution, metabolism and excretion (ADME) properties and safety is critical, not only to guide the initial selection of a drug lead and avoid failures in later stages of development, but also to establish benchmarks against which subsequent drug optimizations can be evaluated. Considering the previous information from other metallodrugs, we decided to evaluate three critical parameters: water solubility, potential cardiotoxicity and cytochrome inhibition.
Oral ingestion is the most convenient route of drug delivery due to its ease of administration, cost effectiveness, and patient comfort.52 However, a major challenge in the design of oral dosage forms of metallodrugs lies in their low bioavailability, mainly related to low solubility. This is the case for many gold metallodrugs, whose poor water solubility has hampered their pharmaceutical use.53 The water solubility determination of AuPyPZ was performed by turbidimetric measurement, showing intermediate to low solubility (11–33 μM).
One of the main reasons for drug withdrawal or drug label revision is drug-induced sudden cardiac death associated with QT prolongation in the electrocardiogram. Inhibition of the hERG (human ether-a-go-go related gene) potassium channel is the most common mechanism responsible for drug-induced QT interval prolongation in humans; therefore, testing the interaction of a compound with the hERG potassium channel is crucial to assess the safety of a drug. Several gold-based drugs showed significant inhibition of the hERG potassium channel, indicating a potential risk for cardiac problems.54AuPyPZ was tested as an hERG potassium channel inhibitor, displaying an IC50 value of 6.2 μM. As drugs with an IC50 < 10 μM are classified as potentially cardiotoxic, AuPyPZ is below safe limits, indicating a potential risk for sudden cardiac death.
Another relevant parameter to determine the safety of a drug compound is the inhibition of cytochrome P450 (CYP450) enzymes, as this is the most common mechanism leading to drug–drug interaction. For early assessment, the inhibition of cytochrome CYP3A4 is used as a reference. In this regard complex AuPyPZ shows a moderate inhibition of CYP3A4, with an IC50 value of 3.57 μM (3.20 mg L−1).
Synthesis of Au(III)-loaded liposomes
Despite achieving positive results in the antibacterial efficacy of the gold complex, its practical application is constrained by its limited bioavailability and safety concerns. In this regard, encapsulation proved to be a suitable strategy to improve bioavailability and cardiotoxicity of gold metallodrugs.55,56 Among all nanotechnological solutions for drug delivery, liposomal encapsulation is the most widely used strategy to improve the bioavailability of water-insoluble drugs and mitigate drug-induced inhibition of the cardiac IKR channel and QTc prolongation.57 Consequently, we decided to investigate the encapsulation of the gold(III) metallodrug AuPyPZ in liposomes and its impact on biological properties.For the preparation of the liposomal formulations loaded with the AuPyPZ metalloantibiotic, the optimal conditions were carefully investigated, selecting the best components to preserve the structure of the encapsulated metallodrug and achieve suitable nanoparticle size, drug loading and release properties. In this regard, liposomes were prepared using thin film hydration method.58 Phosphatidylcholine, 1-hexadecanol and oleic acid in a molar ratio of 64/28/8 were dissolved in ethanol for empty liposomes. In the case of loaded liposomes, AuPyPZ metalloantibiotic was added to the solution. The organic solvent was removed using a rotary evaporator at 50 °C and decreasing the pressure for 45 minutes. Then, the residue was hydrated with 3 mL of ultrapure water and sonicated in an ultrasound bath for 3 minutes. The solution was then homogenised for 5 minutes at 11.000 rpm using a Micron homogenizer, obtaining a yellow solution of liposomes of bilayer structure.
Characterization of the loaded and unloaded liposomes
Particle size, zeta potential, polydispersity index (PDI), and encapsulation efficiency parameters of the investigated liposomal systems are presented in Tables 4 and S2 (ESI).†| Diameter (nm) | PDI | Zeta potential (mV) | EE (%) | |
|---|---|---|---|---|
| Unloaded | 185.0 nm ± 19.1 | 0.322 ± 0.070 | −30.9 mV ± 0.8 | — |
| AuPyPZ loaded | 181.8 nm ± 25.5 | 0.246 ± 0.023 | 9.54 mV ± 1.22 | 94% ± 2 |
Particle size was determined by dynamic light scattering (DLS), confirming a similar size between unloaded liposomes and gold complex-loaded liposomes. Unloaded liposomes have an average diameter of 185.0 nm ± 19.1 while loaded liposomes had an average diameter of 181.8 nm ± 25.5. The polydispersity index (PDI) is below 0.35, indicating that the size distribution of liposomes is monodisperse.
Zeta potential analysis was performed by combining laser Doppler velocimetry (LDV) and phase analysis light scattering (PALS).59 LDV measurements are performed using a procedure called “mixed mode measurement” (M3).60 Unloaded liposomes exhibit a high negative value of −30.9 mV ± 0.8, whereas loaded liposomes exhibit a positive zeta potential of 9.54 mV ± 1.22. The negative charge of unloaded liposomes can be explained by the presence of oleic acid in the liposome membrane. The gold complex in the liposome membrane modifies this charge from negative to positive.
The efficacy of encapsulation (EE%) of AuPyPZ was determined by high-performance liquid chromatography with ultraviolet/visible light detection (HPLC-UV/VIS). The amount of free metallodrug was analyzed and the EE% was calculated by comparison with the total concentration. The high value found for the encapsulation efficiency (94%) shows that the designed experimental procedure succeeded in incorporating the drug almost completely in the lipoformulation.
Another important feature of liposomes is their stability over time. Stability studies performed over a week (Fig. 5) demonstrated that the liposomal formulations remained stable with no observable clumps or precipitations. Measurements show that there are minimal differences in size, PDI and z-potential over the days measured.
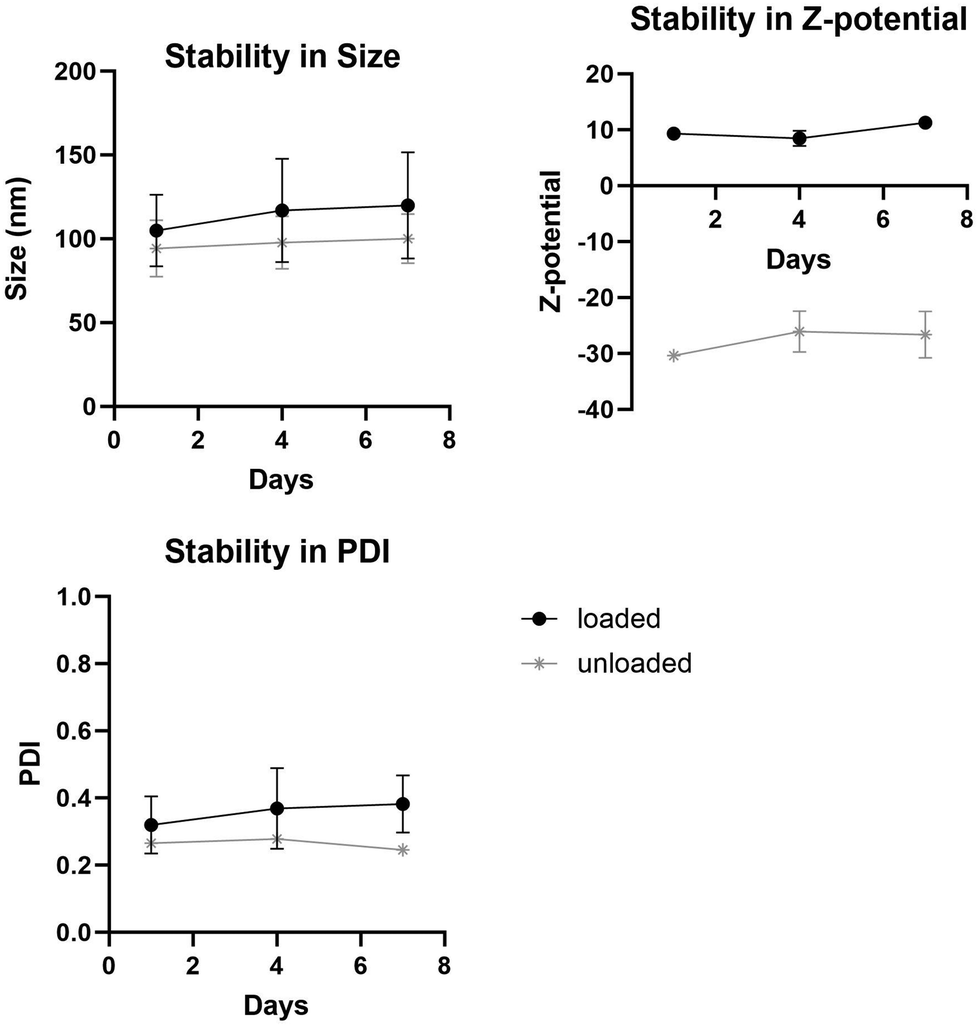 | ||
| Fig. 5 Stability in (a) size, (b) z-potential and (c) PDI of loaded and unloaded liposomes over a week. | ||
Biological characterization and pharmacological profile of the Au(III)-loaded liposomes
The antibacterial activity of the liposomal formulation was studied in comparison with free gold(III) complex and unloaded liposomes against bacterial strains of the Gram positive species of interest, S. aureus (MRSA and MSSA) and S. epidermidis. As depicted in Table 5, unloaded liposomes had no observable effects on bacterial cell viability. While for S. aureus the liposomal formulation shows similar activity, for S. epidermidis the MIC decreased 1–2 fold.| Entry | Bacterial species | No. tested strains | IC50 (mg L−1) | ||
|---|---|---|---|---|---|
| Free AuPyPZ | Liposomal AuPyPZ | Unloaded liposomes | |||
| 1 | MRSA (MDR clinical) | 4 | 0.5 | 0.5–1 | >50 |
| 2 | MSSA (ATCC 25923) | 1 | 1 | 1 | >50 |
| 3 | S. epidermidis (MDR clinical) | 3 | 1 | 0.25–0.5 | >50 |
In view of the excellent antibacterial activity of the metalloantibiotic-loaded liposomes, in vitro toxicity was next investigated. Considering that S. aureus and S. epidermidis are responsible for most of the diagnosed resistant skin infections and that liposomal formulations are especially useful for topical use, cell viability for both skin and liver cells was investigated.
As depicted in Table 6, unloaded liposomes have no appreciable effect on cell viability for any of the cell lines studied. Surprisingly, the liposomal metalloantibiotic formulation also has no effect on cell viability, in contrast to the relatively low IC50 values obtained for the free metallodrug for all liver and skin eukaryotic cell lines studied. Even though the antibacterial activity is similar to that of the free drug in most cases, the steep decrease in cytotoxicity results in an optimal Therapeutic Index for the metalloantibiotic liposomal formulation.
| Entry | Cell line | IC50 (mg L−1) | ||
|---|---|---|---|---|
| Free AuPyPZ | Liposomal AuPyPZ | Unloaded liposomes | ||
| 1 | Hep G2 (tumoral, liver) | 1.33 | >50 | >50 |
| 2 | THLE-2 (non-tumoral, liver) | 1.00 | >50 | >50 |
| 3 | CCD-1064-sk (tumoral, skin) | 0.86 | >50 | >50 |
Furthermore, liposomes ameliorate metalloantibiotic-induced inhibition of hERG, significantly decreasing the cardiac liability of the metallodrug. Thus, AuPyPZ-loaded liposomes display a 26 ± 2% inhibition of hERG at a maximum concentration of 1.12 μM. Cytochrome inhibition is also improved by encapsulation, as the metalloantibiotic-loaded liposomes show low CYP3A4 inhibition (27 ± 5% inhibition at a maximum concentration of 1.12 μM).
Conclusions
A novel lipoformulation was developed by encapsulating cationic (S^C)-cyclometallated gold(III) complex [Au(dppta)(N2Py-PZ-dtc)]+ (AuPyPZ) in liposomes. Under the investigated experimental conditions, liposomes with high loading efficiency (∼94%) of structurally preserved metallodrug were obtained. The gold(III)-loaded liposomes show high stability and particle size and zeta potential values suitable for drug delivery. While the encapsulation had no observable effects on bacterial activity compared to the free drug, a steep decrease in the cytotoxicity of the gold(III) metalloantibiotic was observed, resulting in an optimal Therapeutic Index for the liposomal formulation of the metalloantibiotic. Furthermore, the liposomes ameliorate the metalloantibiotic-induced inhibition of hERG and the inhibition of cytochromes, thereby significantly decreasing the potential liabilities of the metallodrug. The development of this novel metalloantibiotic liposomal formulation broadens the perspective of using gold(III) metallodrugs to combat bacterial resistance.Experimental section
General experimental procedures
All reagents were obtained from commercial suppliers and used without further purification. NMR spectra were recorded on Bruker Avance III HD 300 (1H 300.13 MHz; 13C 75.47 MHz; 31P 121.49 MHz). All chemical shifts (δ) are given in parts per million (ppm) and referenced to the residual solvent signal as internal standard. The following abbreviations are used to indicate the multiplicity of signal: s – singlet, d – doublet, t – triplet, q – quartet, hept – heptet. High Resolution Mass Spectra (HRMS) were recorded on an Agilent Technologies LC/MSD-TOF and HP 1100 MSD spectrometer using electrospray ionization. IR spectra were recorded in neat form on a Jasco FT/IR-4700 spectrophotometer and, and νmax values are given in cm−1 for the main absorption bands.Synthesis of [Au(dppta)(N2Py-PZ-dtc)]PF6 complex
To a solution of AuDPTA complex (117 mg, 0.20 mmol) in MeOH (6 mL), sodium 4-(pyridin-2-yl)piperazine-1-carbodithioate (0.20 mmol) was added. The reaction mixture was stirred at rt for 12 h and then saturated aqueous potassium hexafluorophosphate was added and the mixture was stirred for 15 min. After partial evaporation of the methanol, the resulting solid was filtered and the residue was washed with water and diethyl ether to afford desired AuPyPZ complex (133 mg, 74% yield) as a yellow solid. HRMS (ESI+) [M]+ calcd for C28H35AuN4PS3+, 751.1421; found: 751.1414. IR (KBr, ν cm−1): 555 (P = S), 840 (PF6), 1527 (NCS2). 1H NMR (300 MHz, CD3CN) δ 1.18 (d, 3JHH 6.8 Hz, 6H, H12), 1.28 (d, 3JHH 6.8 Hz, 6H, H12), 3.65–3.87 (m, 6H, H11, H13), 3.94–4.04 (m, 4H, H14), 7.342–7.47 (m, 1H, H5), 7.54–7.68 (m, 5H, H6, H, 9, H10, H18), 7.75–7.78 (m, 1H, H4), 7.81–7.88 (m, 1H, H3), 8.13–8.20 (m, 3H, H8, H16) ppm. 13C NMR (101 MHz, (CD3)2CO) δ 22.3 (d, 3JPC 3.0 Hz, C12), 23.5 (d, 3JPC 3.6 Hz, C12), 44.59 (C14), 44.6 (C14), 49.8 (C13), 50.0 (C13), 51.6 (d, 2JPC 3.3 Hz, C11), 108.4 (C19), 115.1 (C17), 127.6 (d, 1JPC 101.8 Hz, C7), 129.3 (d, 2JPC 12.3 Hz, C6), 130.5 (d, 3JPC 13.7 Hz, C9), 133.4 (d, 3JPC 16.9 Hz, C5), 134.6 (d, 3JPC 11.9 Hz, C3, C8), 135.7 (d, 4JPC 3.1 Hz, C4), 136.5 (d, 4JPC 3.7 Hz, C10), 138.7 (C18), 139.8 (d, 1JPC 124.2 Hz, C1), 144.7 (d, 2JPC 27.9 Hz, C2), 148.7 (C16), 159.0 (C15), 195.0 ppm. 31P NMR (121 MHz, CD3CN) δ −144.7 (sep, 1JPF 706.6 Hz), 74.6 ppm.Crystallography
A crystal of compound AuPyPz was mounted on a glass fibre. Data were collected on an Oxford Diffraction Xcalibur Nova single crystal diffractometer, at room temperature and using CuKα radiation. Images were collected at a fixed distance of 62 mm between the crystal and the detector using the oscillation method, with a oscillation of 1.0–1.2° and variable exposure time per image. The data collection strategy was calculated using CrysAlis Pro CCD software,61 and data reduction and cell refinement were performed using CrysAlis Pro RED software.61 An empirical absorption correction was applied using the SCALE3 ABSPACK algorithm as implemented in CrysAlis Pro RED software. Structures were solved using ShelXT,62 all structures were refined by full matrix least squares against F2 in ShelXL63,64 using Olex2.65 All non-hydrogen atoms were refined anisotropically, while all hydrogen atoms were geometrically localized and refined using a riding model. The structure shows some disordered fragments and PF6 molecules and the occupancies of the disordered group were refined with their sum set to equal 1 and subsequently fixed at the refined values. Restraints were applied to maintain sensible thermal and geometric parameters. The X-ray crystallographic coordinates for the structure reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2361824.†Liposome preparation
Liposomes were prepared using a standard method. Initially, all lipids were dissolved in methanol and combined in a round-bottom flask. Gold complexes were dissolved in DMSO at 22 mg ml−1. The solvent was evaporated using a rotary evaporator at 50 °C and decreasing the pressure for 45 minutes. The lipid film was hydrated by adding 3 mL of ultrapure water and gently mixing. The resulting liposomal suspension was homogenized for 5 minutes at 10000 rpm using a Micra homogenizer.
High performance liquid chromatography with ultraviolet/light detection (HPLC/UV-VIS)
Quantification of free gold complex was performed by high performance liquid chromatography (HPLC). Reversed phase HPLC analysis was performed with SHIMAZDU UFPLC equipped with a reversed-phase column. The mobile phase consisted of 30% citrate buffer 50 mM, pH 6 and 80% methanol at room temperature in an isocratic gradient at a flow 1 ml min−1. The injection volume was 10 μL and the total run time was set at 5 min. The Au(III) complex was detected at 220 nm using a UV/VIS detector. All experiments were performed in triplicate.Size and zeta-potential determination
Liposome sizes and zeta potentials were assessed at 25 °C using a ZetasizerNano-ZS (Malvern Instruments Ltd, Malvern, UK) equipped with a 633 nm He–Ne laser. Prior to measurement, all samples were diluted with distilled water at a ratio of 1:100 v/v for size and 3:100 v/v for z-potential. The polydispersity index (PDI) was determined using the instrument's built-in software.
Encapsulation efficiency
To determine the encapsulation efficacy (EE%), free gold complex was separated from liposomes using Vivaspin2 centrifugal concentrators with MWCO of 30.000 (Sartorius AG, Göttingen, Germany). One milliliter of the solution was added to the Vivaspin2 and centrifuged for 30 min at 3000g. The free Au(III) complex that passed through the filter was analyzed by HPLC.Liposome stability
To monitor liposome stability, changes in size, PDI, and zeta potential were tracked during storage at 4 °C over a 7-day period by measuring days 1, 4 and 7.Antibacterial activity
The antibacterial activity was tested against a panel of MDR strains of clinical origin. Methicillin-resistant Staphylococcus aureus (MRSA) strains 162065-705, 163501-000, 161071-210 and 162058-967 are respiratory isolates from cystic fibrosis patients at the Hospital Clinic of Barcelona (Barcelona, Spain). Staphylococcus epidermidis a FG22014, FG03015 and FG14013 are wound isolates from the Hospital Clinic of Barcelona (Barcelona, Spain). S. maltophilia strains 9510-524 and 895 are respiratory isolates from cystic fibrosis patients at the Hospital Clinic of Barcelona (Barcelona, Spain). P. aeruginosa strains 166097-953 and 30302995-242 are respiratory isolates from cystic fibrosis patients at the Hospital Clinic of Barcelona (Barcelona, Spain). E. coli strains E4, E8 and E11 were isolated from the blood of patients in the Manhiça District Hospital (Manhiça, Mozambique). Acinetobacter baumannii strains Ab177 and Ab210 were originally identified in 43 hospitals in Spain during February–March 2010; they produce OXA-24 and 58-like β-lactamases and are highly resistant to several clinical antibiotics. Acinetobacter baumannii Abcr17 is a cerebrospinal fluid isolate from the Virgen del Rocío Universiy Hospital (Seville, Spain). This is a pan-drug resistant strain, meaning that is nonsusceptibility to all agents in all antimicrobial categories, including carbapenems. Laboratory reference strains of S. aureus ATCC 29213, P. aeruginosa ATCC 27853, E. coli ATCC 25922, A. baumannii ATCC 19606 were used as the control wild-type strains in this study. Identification of bacterial species was confirmed by Matrix-Assisted Laser Desorption Ionization-Time Of Flight (MALDI-TOF) mass spectrometry. Isolates were cultured on Columbia sheep blood agar (Becton Dickinson, Heidelberg, Germany) and incubated at 37 °C for 18 h.The minimum inhibitory concentrations (MICs) of AuPyPZ complex were determined in triplicate by the broth microdilution method recommended by the Clinical and Laboratory Standards Institute (CLSI) in 96-well round-bottom microtiter plates. Auranofin, a reference Au(I) metalloantibiotic, was included for comparison. The assays were performed in ISO-Sensitest broth (Oxoid, Madrid, Spain). The plates were incubated at 37 °C and read after 18 h to check the absence of turbidity. The same procedure was performed to determine the MICs of unloaded and AuPyPZ-loaded liposomes. MIC values were defined as the lowest concentration of the compound that inhibited visible growth.
Citotoxicity
In vitro prediction of the hepatic safety of compounds was carried out on liver-derived tumor and non-tumor lines and skin-derived non-tumor lines. Human hepatic cancer cell line HepG2 was obtained from the American Type Cultures Collection (Ref: HB-8065, ATCC, Manassas, VA, USA) by the cell bank of Fundación Medina (Granada, Spain). Human immortalized liver cell line THLE-2 was obtained from the American Type Cultures Collection (Ref: CRL-2076, ATCC, Manassas, VA, USA) by the cell bank of Fundación Medina (Granada, Spain). Human dermal fibroblast cell line CCD-1064Sk was obtained from the American Type Cultures Collection (Ref: CRL-2076, ATCC, Manassas, VA, USA) by the cell bank of Fundación Medina (Granada, Spain).Cytotoxicity was evaluated using the MTT metabolic test. Cells were seeded at a density of 10.000 per well in a 96-wells plate and were incubated in a humidified atmosphere at 37 °C with 5% CO2 for 24 h. Solutions of AuPyPZ and AuPyPZ-loaded liposomes were prepared at 25 mM in 100% DMSO and analyzed in an increasing dose curve from 100 to 0.2 μM. After 72 h of treatment, plates were treated with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, ACROS Organics) at 5 μg mL−1 in Minimum Essential Medium Eagle (MEM) for 2 h. Then, DMSO was added to the plates to solubilizing the formazan crystals formed in viable cells and plates were stirred for 5 minutes to homogenize the solution. Absorbance was measured at 570 nm by Envision Multiplate Reader (PerkinElmer), and AC50 calculations were done using GeneData Screener Software.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received financial support from the Instituto de Salud Carlos III (PI1900478, PI22/00148), Agaur Producte (2021 PROD 00047), Proyectos de Desarrollo Tecnológico en Salud (DTS21/00004) and MINECO (PID2020-115204RB-I00). P.P.R. thanks NextGenerationEU for a predoctoral contract (Programa Investigo, AYUD/2022/9313). ISGlobal is a CERCA center from the Generalitat of Catalunya and a Severo Ochoa Center (Spanish Ministry of Science, Innovations, and Universities).References
- M. Irfan, A. Almotiri and Z. A. AlZeyadi, Antibiotics, 2022, 11, 1362 CrossRef CAS PubMed.
- E. M. Darby, E. Trampari, P. Siasat, M. S. Gaya, I. Alav, M. A. Webber and J. M. A. Blair, Nat. Rev. Microbiol., 2023, 21, 280 CrossRef CAS PubMed.
- K. W. Chin, H. L. M. Tiong, V. Luang-In and N. L. Ma, Environ. Adv., 2023, 11, 100331 CrossRef CAS.
- G. Muteeb, M. T. Rehman, M. Shahwan and M. Aatif, Pharmaceuticals, 2023, 16, 1615 CrossRef CAS PubMed.
- N. D. Friedman, E. Temkin and Y. Carmeli, Clin. Microbiol. Infect., 2016, 22, 416–422 CrossRef CAS PubMed.
- P. Dadgostar, Infect. Drug Resist., 2019, 12, 3903 CrossRef CAS PubMed.
- J. H. Powers, Clin. Microbiol. Infect., 2004, 10, 23 CrossRef PubMed.
- V. Gigante, H. Sati and P. Beyer, ADMET DMPK, 2022, 10, 147 CrossRef PubMed.
- M. S. Butler, I. R. Henderson, R. J. Capon and M. A. T. Blaskovich, J. Antibiot., 2023, 76, 431 CrossRef CAS PubMed.
- M. Bergkessel, B. Forte and I. H. Gilbert, ACS Infect. Dis., 2023, 11, 2062 CrossRef PubMed.
- S. Walesch, J. Birkelbach, G. Jézéquel, F. P. J. Haeckl, J. D. Hegemann, T. Hesterkamp, A. K. H. Hirsch, P. Hammann and R. Müller, EMBO Rep., 2022, 24, e56033 CrossRef PubMed.
- K. Gadar and R. R. McCarthy, Antimicrob. Resist., 2023, 1, 11 CrossRef PubMed.
- R. J. Turner, Microb. Biotechnol., 2017, 10, 1062 CrossRef PubMed.
- M. Claudel, Chemistry, 2020, 2, 849 CrossRef CAS.
- A. Frei, A. D. Verderoca, A. G. Eliott, J. Zuegg and M. K. T. Braskovich, Nat. Rev. Chem., 2023, 7, 202 CrossRef CAS PubMed.
- A. Frei, Antibiotics, 2020, 9, 90 CrossRef CAS PubMed.
- A. Pandey and E. Boros, Chem. – Eur. J., 2021, 27, 7340 CrossRef CAS PubMed.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627 RSC.
- C. I. Yeo, C. H. P. Goh, E. R. T. Tiekink and J. Chew, Coord. Chem. Rev., 2024, 500, 215429 CrossRef CAS.
- C. Ratia, R. G. Soengas and S. M. Soto, Front. Microbiol., 2022, 13, 846959 CrossRef PubMed.
- S. Thangamani, H. Mohammad, M. F. Abushahba, T. J. Sobreira, V. E. Hedrick, L. N. Paul and M. N. Seleem, Sci. Rep., 2016, 6, 22571 CrossRef CAS PubMed.
- B. Glišić and M. I. Djuran, Dalton Trans., 2014, 43, 5950 RSC.
- E. J. Anthony, O. W. L. Carter, E. M. Bolitho, H. E. Bridgewater, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F. Wang, W. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888 RSC.
- E. Kabir, M. R. O. K. Noyon and M. A. Hossain, Results Chem., 2023, 5, 100935 CrossRef CAS.
- S. S. Bradford and J. A. Cowan, Metallodrugs, 2014, 1, 10 Search PubMed.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071 CrossRef CAS.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645 RSC.
- M. Poursharifi, M. T. Wlodarczyk and A. J. Mieszawska, Inorganics, 2019, 7, 2 CrossRef CAS.
- Y. Jia, Y. Jiang, Y. He, W. Zhang, J. Zou, K. T. Magar, H. Boucetta, C. Teng and W. He, Pharmaceutics, 2023, 15, 774 CrossRef CAS PubMed.
- I. Iacobucci, S. La Manna, I. Cipollone, V. Monaco, L. Canè and F. Cozzolino, Pharmaceutics, 2023, 15, 1997 CrossRef CAS PubMed.
- D. Ezhilarasan and K. S. Harini, Int. J. Drug Deliv. Technol., 2023, 86, 104731 CrossRef CAS.
- A. Sharma and U. S. Sharma, Int. J. Pharm., 1997, 154, 123 CrossRef CAS.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef PubMed.
- M. Dymek and E. Sikora, Adv. Colloid Interface Sci., 2022, 309, 102757 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202 RSC.
- J. Kim, S. Pramanick, D. Lee, H. Par and W. J. Kim, Biomater. Sci., 2015, 3, 1002 RSC.
- M. Poursharifi, M. T. Wlodarczyk and A. J. Mieszawska, Inorganics, 2019, 7, 1 RSC.
- J. Shen, H. C. Kim, J. Wolfram, C. Mu, W. Zhang, H. Liu, Y. Xie, J. Mai, H. Zhang, Z. Li, M. Guevara, Z. Mao and H. Shen, Nano Lett., 2017, 17, 2913 CrossRef CAS PubMed.
- F. P. Rodrigues, Z. A. Carneiro, P. K. Mascharak, C. Curti and R. S. da Silva, Coord. Chem. Rev., 2016, 306, 701 CrossRef CAS.
- Y. Chen, Y. Gu, H. Hu, H. Liu, W. Li, C. Huang, J. Chen, L. Liang and Y. Liu, J. Inorg. Biochem., 2023, 241, 112134 CrossRef CAS PubMed.
- S. Gadre, M. Manikandan, G. Chakraborty, A. Rayrikar, S. Paul, C. Patra and M. Patra, J. Med. Chem., 2023, 66, 13481 CrossRef CAS PubMed.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., Int. Ed., 2023, 62, e202218000 CrossRef PubMed.
- S. Sueiro, C. Ratia, R. G. Soengas, M. J. Iglesias, F. López-Ortiz and S. M. Soto, Antibiotics, 2022, 11, 1728 CrossRef PubMed.
- M. Altaf, A. A. Isab, J. Vanco, Z. Dvorak, Z. Travnicek and H. Stoeckli-Evans, RSC Adv., 2015, 5, 81599 RSC.
- M. R. M. Williams, B. Bertrand, D. L. Hughes, Z. A. E. Waller, C. Schmidt, I. Ott, M. O'Connell, M. Searcey and M. Bochmann, Metallomics, 2018, 10, 1655 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- A. B. Bhoi, M. Dalwadi and U. M. Upadhyay, Int. J. Pharm. Res. Appl., 2020, 5, 477 Search PubMed.
- G. F. Pauli, S. Chen, C. Simmler, D. C. Lankin, T. Gödecke, B. U. Jaki, J. B. Friesen, J. B. McAlpine and J. G. Napolitano, J. Med. Chem., 2014, 57, 9220 CrossRef CAS PubMed.
- ICH guidelines for impurities in new drug substances: https://www.ema.europa.eu/en/ich-q3a-r2-impurities-new-drug-substances-scientific-guideline.
- ICH guidelines for elemental impurities: https://www.ema.europa.eu/en/ich-q3d-elemental-impurities-scientific-guideline.
- L. A. Ba, T. Burkholz, T. Schneider and C. Jacob, Role of Cysteine in Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Hughes, Wiley-VCH, Heidelberg, 2012, vol 5, ch. 10, pp. 361–394 Search PubMed.
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, ISRN Pharm., 2012, 2012, 195727 Search PubMed.
- R. C. Blodgett Jr, Am. J. Med., 1983, 75, 86 CrossRef CAS PubMed.
- G. D. Hoke, R. A. Macia, P. C. Meunier, P. J. Bugelski, C. K. Mirabelli, G. F. Rush and W. D. Matthews, Toxicol. Appl. Pharmacol., 1989, 100, 293 CrossRef CAS PubMed.
- M. F. Tomasello, C. Nardon, V. Lanza, G. Di Natale, N. Pettenuzzo, S. Salmaso, D. Milardi, P. Caliceti, G. Pappalardo and D. Fregona, Eur. J. Med. Chem., 2017, 138, 115 CrossRef CAS PubMed.
- D. Cirri, I. Landini, L. Massai, E. Mini, F. Maestrelli and L. Messori, Pharmaceutics, 2021, 13, 727 CrossRef CAS PubMed.
- G. M. Shopp, L. Helson, A. Bouchard, D. Salvail and M. Majeed, Anticancer Res., 2014, 34, 4733 CAS.
- H. Zhang, Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation, in Liposomes. Methods in Molecular Biology, ed. G. D'Souza, Humana Press, New York, NY, 2017, vol. 1522 Search PubMed.
- W. W. Tscharnuter, Appl. Opt., 2001, 40, 3995 CrossRef CAS PubMed.
- M. T. Connah, M. Kaszuba and A. Morfesis, J. Dispersion Sci. Technol., 2002, 23, 663 CrossRef CAS.
- O.D Rigaku, CrysAlis PRO, Rigaku Oxford Diffraction, Yarnton, England, 2023 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2361824. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01867b |
| This journal is © The Royal Society of Chemistry 2024 |