
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHorizontal lithium growth driven by surface dynamics on single crystal Cu(111) foil†
Min-Ho
Kim‡
a,
Dong Yeon
Kim‡
 bg,
Yunqing
Li
cd,
Juyoung
Kim
a,
Min Hyeok
Kim
ce,
Jeongwoo
Seo
a,
Benjamin V.
Cunning
c,
Taewon
Kim
a,
Sang-Wook
Park
b,
Rodney S.
Ruoff
*acde,
Dong-Hwa
Seo
*b,
Sunghwan
Jin
*f and
Hyun-Wook
Lee
*a
bg,
Yunqing
Li
cd,
Juyoung
Kim
a,
Min Hyeok
Kim
ce,
Jeongwoo
Seo
a,
Benjamin V.
Cunning
c,
Taewon
Kim
a,
Sang-Wook
Park
b,
Rodney S.
Ruoff
*acde,
Dong-Hwa
Seo
*b,
Sunghwan
Jin
*f and
Hyun-Wook
Lee
*a
aDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: rsruoff@ibs.re.kr; hyunwooklee@unist.ac.kr
bDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon, 34141, Republic of Korea. E-mail: dseo@kaist.ac.kr
cCenter for Multidimensional Carbon Materials (CMCM), Institute for Basic Science (IBS), 50 UNIST-gil, Ulsan, 44919, Republic of Korea
dSchool of Materials Science and Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan, 44919, Republic of Korea
eDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan, 44919, Republic of Korea
fDepartment of Materials Science and Engineering, Kangwon National University, 346 Jungang-ro, Samcheok-si, Gangwon-do, 25913, Republic of Korea. E-mail: shjin00@kangwon.ac.kr
gDepartment of Chemistry, Jeonbuk National University, 567 Baekje-daero, Jeonju, 54896, Republic of Korea
First published on 5th July 2024
Abstract
Anode-free lithium (Li) batteries that function via direct Li plating/stripping on metal current collectors have garnered significant interest in the field of metallic Li as an ideal negative electrode. However, dendritic Li growth creates unoccupied space in the battery, diminishing volumetric energy density. Our research reveals that Li adatoms can reposition on metal substrates via surface migration by interacting with individual grains at the atomic level after electro-adsorption of Li ions. By examining Li morphologies on diverse types of substrates, we find that the near-zero migration barrier of Li adatoms, especially on single crystal Cu(111) foils, is a key parameter that inhibits Li dendrites by initiating horizontal growth in an isotropically faceted nucleation shape, with 80% capacity retention even after 120 cycles (versus 29 cycles for polycrystalline Cu), paired with LiNi0.8Co0.1Mn0.1O2 (NCM811) cathodes with a high-loading level (5.3 mA h cm−2). Based on our findings, we highlight the importance of metal substrates that promote favorable surface dynamics of Li adatoms.
Broader contextLithium (Li) metal formation during electrochemical reactions has traditionally been characterized by dendritic growth. The common stages of chemical reactions—nucleation, particle growth, and growth cessation—are mirrored in Li metal deposition, which involves mass transport from the electrolyte, charge transfer at the collector surface, and dendrite formation. Previously, the role of Li adatom surface migration in this process was overlooked. Herein, we present a fundamental approach to mitigate dendritic Li growth by promoting the active surface migration of Li adatoms on the current collector. By employing single-crystal Cu(111) foil, we found that Li adatoms face an almost negligible migration barrier, which supports their lateral growth and coalescence, thereby minimizing dendrite formation. The migration of Li in the form of rhombic dodecahedra on copper foil plays a crucial role in suppressing dendritic Li growth during the plating process. We directly observed the surface migration of Li rhombic dodecahedra instead of Li dendrites during the plating process. Our findings suggest a paradigm shift in Li growth from vertical to horizontal, offering a promising pathway toward realizing commercial anode-free Li batteries. |
Introduction
Dendritic lithium (Li) refers to vertically grown metallic Li structures that create unoccupied spaces on metal current collectors and safety issues during Li plating, posing a critical challenge in advancing anode-free Li batteries.1,2 The extensive growth of dendritic Li across a large surface area has been identified as a primary factor contributing to low Coulombic efficiency (CE) due to side reactions with electrolytes and the subsequent formation of inactive Li.3–6 To date, the origin of such dendritic Li has been attributed to the limited mass transport of Li ions (Li+) in electrolytes.7 Given the sufficiently fast electro-adsorption of Li+ (Li+ + e− → Li0) near the electrodes, the relatively slow movement of Li+ within bulk electrolytes can trigger a steep concentration gradient of Li+ within the electrolytes, taking it out of equilibrium and creating uneven Li+ flux towards the current collector. Although many researchers have focused on mitigating the concentration gradient of Li+ and controlling Li morphology through lithiophilic agents,4,8,9 a few studies have definitively addressed the dendritic Li issue, and it sometimes misleads us about the ‘genuine meaning’ of lithiophilicity, where the scope has expanded to the realm of Li deposition behaviour (originally, the term was proposed to refer to the wettability of molten Li to hosts;10–12 this will be discussed later).Recently, a previous work13 suggested that the planar growth of Li can be achieved through lithiophobicity of an FeF3 thin film. This proposition is further supported by a theoretical model,14 which suggests that activating the self-rearrangement of metal atoms on anode surfaces could suppress the local electrodeposition of metal ions, conflicting with the conventional Sand theory where Li+ flux is critical in determining Li morphology.15,16 Although few papers have emphasized the importance of the rearrangement of the deposited Li, the ‘surface migration’ of Li adatoms remains ambiguous, and direct evidence for it has not been reported yet.
Herein, we demonstrate a fundamental approach to mitigate dendritic Li growth by merging the Cu grains of the as-received polycrystalline Cu foil into a monolith and by changing their crystallographic orientations into (111) orientation (Fig. 1(a)). By comparing diverse Cu foils (including as-received Cu with rolling marks, electropolished polycrystalline Cu, low-index single crystal Cu(111), and high-index single crystal Cu(410)) as current collectors for anode-free Li batteries, we observed that the active coalescence of Li adatoms and horizontal Li growth occurs only on single crystal Cu(111) foil, instead of Li dendrites. We reveal that this unique behaviour is attributed to a near-zero migration barrier for Li adatoms and particle-type seeds on single crystal Cu(111) foil. This has not been handled in previous studies, and distinguishes the Cu(111) foil, designed by us, from other foils. In addition, using twin-boundary-containing Cu foils, we first demonstrate that the grain boundary consistently induces non-uniform Li+ flux and Li plating even in various advanced electrolytes (including 0.6 M lithium difluoro(oxalate)borate (LiDFOB) and 0.6 M lithium tetrafluoroborate (LiBF4) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v fluoroethylene carbonate (FEC) and diethylene carbonate (DEC), 2 M lithium bis(fluorosulfonyl)imide (LiFSI) in dimethyl ether (DME) and 2 wt% lithium nitrate (LiNO3), 1 M LiFSI in 1:4 v/v DME and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE), and 4 M LiFSI in diethyl ether (DEE)), as well as conventional organic electrolytes (1.3 M lithium hexafluorophosphate (LiPF6) in 3:7 v/v ethylene carbonate (EC) and DEC + 10 wt% FEC).
:2 v/v fluoroethylene carbonate (FEC) and diethylene carbonate (DEC), 2 M lithium bis(fluorosulfonyl)imide (LiFSI) in dimethyl ether (DME) and 2 wt% lithium nitrate (LiNO3), 1 M LiFSI in 1:4 v/v DME and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE), and 4 M LiFSI in diethyl ether (DEE)), as well as conventional organic electrolytes (1.3 M lithium hexafluorophosphate (LiPF6) in 3:7 v/v ethylene carbonate (EC) and DEC + 10 wt% FEC).
 | ||
| Fig. 1 Design of a single crystal Cu(111) foil and Li growth on diverse Cu substrates. (a) Illustration of the preparation of a single crystal Cu(111) foil. Cu(111) foil is prepared via abnormal growth of grains with (111) orientation. (b) Characterization of polycrystalline Cu foil and single-crystal Cu(111) foil: EBSD inverse pole figure (IPF) map of the foil (left), XRD 2θ scan and WAXS results (right). The single-crystal foil has (111) surface orientation, as indicated by the uniform blue color in the IPF map, the {111} peaks in the XRD spectrum, and the triangle-shaped spots in the WAXS results, in contrast to polycrystalline Cu consisting of numerous grains with different orientations, respectively. (c) Li morphologies on four different Cu foils with areal capacity of 0.5 mA h cm−2: as-received Cu, electropolished polycrystalline Cu, low-index single crystal Cu(111), and high-index single crystal Cu(410). In contrast to other Cu foils, Cu(111) foil exhibits uniform Li growth in a planar manner. Li plating on each Cu substrate was conducted in an electrolyte system containing 1.3 M LiPF6 in EC/DEC = 3:7 + 10 wt% FEC. | ||
This unique Li deposition chemistry inspired us to coin the term “epihorizontaxy.” This compound word, derived from ‘epi’ (upon), ‘horizont’ (horizontal), and ‘axy’ (order), describes a process or phenomenon in which the movement or arrangement of a material (in this case, Li growth during deposition) may transform depending on the crystallographic orientation of a substrate or surface. The epihorizontaxy that we observed in our model system reflects a change in the Li growth model from vertically to horizontally on Cu(111) foil, unlike Cu(410) foil. In this context, we believe that this work establishes a better understandings of Li deposition chemistry on the current collector, and paves the way for the potential realization of commercial anode-free Li batteries.
Results and discussion
Fabrication and characterization of single crystal Cu(111) foil
Polycrystalline Cu foil (pCu), commonly employed as a metal current collector for anode-free batteries, comprises numerous grains with different crystallographic orientations. Since each grain and its grain boundaries (GBs) possess varying energies based on their atomic arrangements,17,18 interactions with Li atoms, including adsorption and surface migration, can differ across the foil, resulting in high localized current densities and heterogeneous Li plating on the Cu current collector (Fig. S1, ESI†). Given this, we hypothesized that a foil with a single grain could facilitate planar and homogenous Li plating throughout the entire foil.As a model system, we prepared a large-area single-crystal Cu(111) foil (approximately 80 mm × 190 mm; less than 0.05 mm thick) using contact-free annealing (CFA) near the melting temperature of Cu from a commercially available polycrystalline Cu foil (Fig. 1 and Fig. S2, ESI†). During the CFA process,19 abnormal growth of the grain with the (111) surface (having the lowest surface energy in the face-centred cubic (FCC) structure) occurs to minimize the surface energy of the entire foil, if the surface energy of the metal is the major driving force for grain growth without any additional interfacial energy, other than the contact region between the quartz rod and Cu foil (Fig. S2b, ESI†). Especially, the employed CFA process yields a ‘perfect’ single crystal Cu(111) foil (sCu(111)) with no defects, and its mass-production capability ensures suitability for practical use in both academic and industrial field (see Note S1 and Fig. S2c, d, ESI†).
Li nucleation and growth on single crystal Cu(111)
To examine the Li morphology on our sCu(111) foil, Cu|Li coin cells were assembled using commercially available organic electrolytes (1.3 M LiPF6 in EC/DEC = 3:7 v/v + 10 wt% FEC). Employing the galvanostatic deposition of small amounts of Li (with an areal capacity of 0.1 mA h cm−2) at 0.5 mA cm−2, we observed the emergence of numerous Li particles on sCu(111), which appeared to undergo coalescence, forming larger islands (Fig. 2(a)). Surprisingly, these particles possessed a rhombic dodecahedron shape with an average size of 0.54 ± 0.13 μm, known to be the thermodynamically most stable polymorph of the body-centred cubic (BCC) structure (Fig. 2(b) and Fig. S3, ESI†).13,20–22 Recent studies22,23 have reported that this particular faceted morphology appears under conditions where neither the solid electrolyte interphase (SEI) layer nor the electrolyte chemistry affects the deposition morphology, such as at exceptionally high current densities (>1000 mA cm−2). By contrast, we observed this Li polymorph on a bare Cu surface of a current collector at a realistic current density under battery operation. Notably, the inherent morphology of particles, particularly when strongly adhered to a substrate, is significantly influenced by the substrate itself. Given that rhombic dodecahedral shapes are composed exclusively of {110} planes having the lowest surface energy of a BCC structure,20,21 the rise of this Li polymorph on the sCu(111) foil represents that Li growth is solely dictated by the surface energy of the deposited Li itself, implying that Li adatoms are loosely attached on the surface.
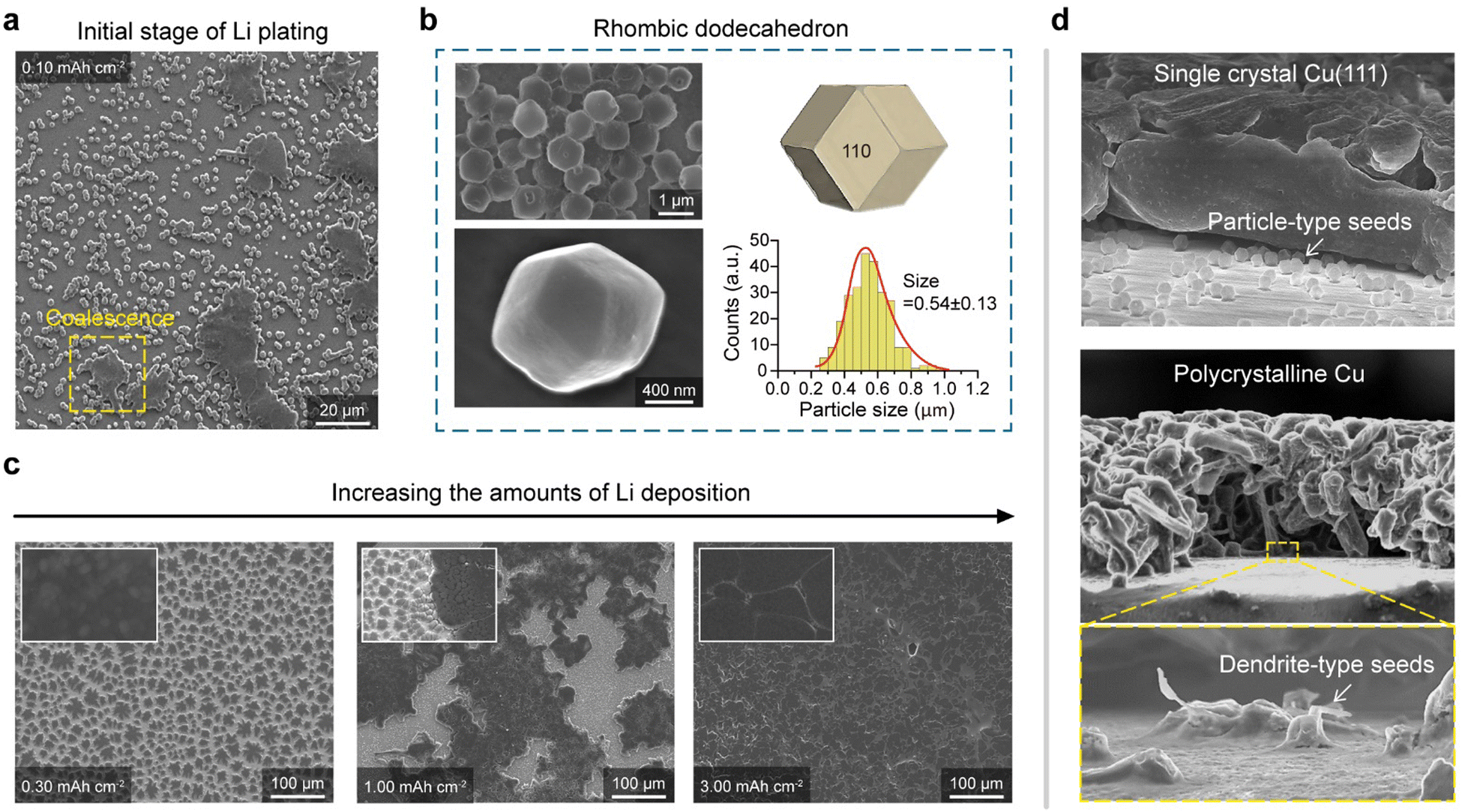 | ||
| Fig. 2 The formation of Li rhombic dodecahedra followed by horizontal Li growth. (a) SEM image of electrodeposited Li on a Cu(111) foil and polycrystalline Cu foils with an areal capacity of 0.10 mA h cm−2, showing the coalescence of Li particles. (b) SEM images of rhombic dodecahedral Li particles on a Cu(111) foil and their size distributions (mean = 54 ± 13 nm). (c) SEM images of electrodeposited Li on Cu(111) foils with capacities of 0.35, 0.50, and 1.00 mA h cm−2. The insets are enlarged images, clarifying the surface morphology of Li deposits. (d) Tilted SEM images of Li deposits on a Cu(111) and polycrystalline Cu foil at an areal capacity of 3.5 mA h cm−2. Although dense and uniform Li plating on Cu(111) foils is initiated by particle-type Li seeds, porous and nonuniform Li plating on polycrystalline Cu is initiated by dendrite-type Li seeds. All of the sampling was conducted in an electrolyte system containing 1.3 M LiPF6 in EC/DEC = 3:7 + 10 wt% FEC. | ||
In light of this, we speculated that Li adatoms behave like unimpeded particles on sCu(111), and this behaviour significantly affects the Li morphology in subsequent depositions. Consequently, the deposition process onto the sCu(111), with areal capacity ranging from 0.3 mA h cm−2 to 3.0 mA h cm−2, involves the coalescence of particle-type Li seeds into 2D Li islands and the subsequent horizontal expansion of these islands (Fig. S4, ESI† and Fig. 2(c)). While vertical Li growth is initiated from dendrite-type seeds on pCu, horizontal Li growth on sCu(111) results in the formation of densely packed Li plates (Fig. 2(d)), which is close to the ideal Li plating model to mitigate the volume expansion and stress evolution within the cell.
Optical observation of facet-dependent Li dynamics
To determine whether Li adatoms actively migrate on the sCu(111) surface in practice, we devised a hand-made optical cell, and conducted operando optical analysis. As widely accepted, Li grew vertically towards a dendritic form from its initial nucleation site on the pCu foil (Video S1, ESI†). However, upon deposition on sCu(111), we observed a significant crowding and waggling of Li particles (Video S2, ESI†). In stark contrast to the behaviour of Li nucleation on polycrystalline Cu, these Li particles displayed continuous movement instead of remaining stationary. Moreover, we confirmed that the motion of a single Li particle lying in open space without any disturbance consistently followed a straight trajectory (Fig. 3(a) and Video S3, ESI†). The rate of migration of the particle exhibited a logarithmic decrease, starting from an initial velocity of approximately 52.2 μm s−1 and diminishing to 2.1 μm s−1 within approximately 6 s (Fig. 3(b) and (c)). After roughly 10 s, the particle's motion came to a complete halt. This declining trend closely corresponds to the graph depicting changes in contrast intensity over time (Fig. 3(c), cyan), leading us to hypothesize that when the size of the Li nuclei exceeds a critical threshold, their surface migration may be impeded, at least when viewed optically.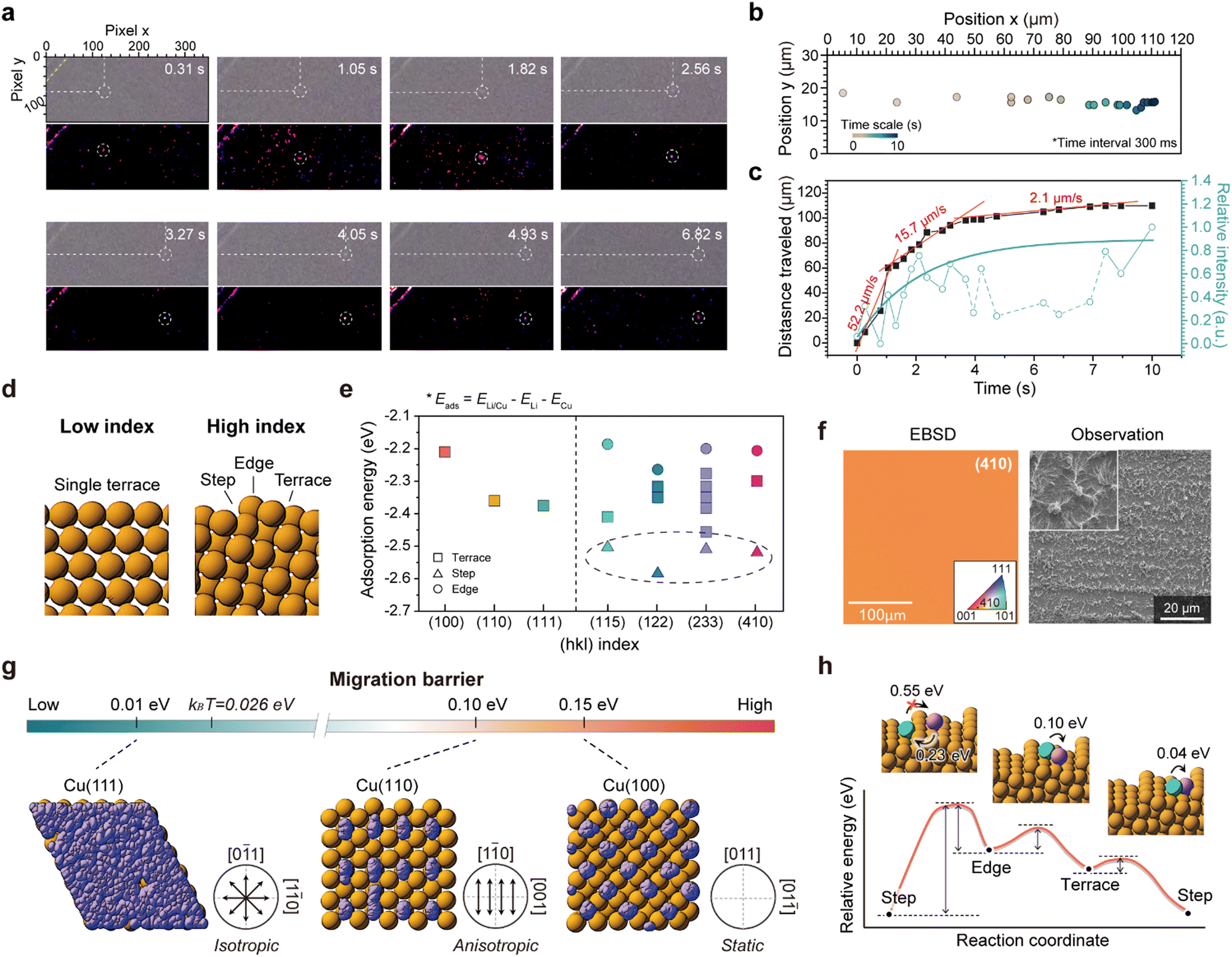 | ||
| Fig. 3 Surface migration of Li adatoms on a single-crystal Cu(111). (a)–(c) Optical images of the surface migration of a Li adatom on a Cu(111) upon Li plating for roughly 10 s. (a) and its trajectory (b). A Li adatom initially migrates quickly at 52.2 μm s−1 and then gradually stops (c). (d) Model surfaces of low-index Cu and high-index Cu. Low-index surfaces have a flat and single surface, whereas high-index surfaces consist of terrace, edge, and step sites, which can prevent uniform Li plating on their surfaces. (e) Calculated Li adsorption energies on diverse Cu facets with low index (e.g., (100), (110), and (111)) and high index (e.g., (115), (122), (233), and (410)). In contrast to Cu facets with low index, Cu facets with high index have multiple adsorption energies. (f) EBSD IPF map of the single crystal Cu(410) foil (left) and Li morphology on it, with an areal capacity of 0.5 mA h cm−2, showing dendritic Li growth. (g) Li migration barriers for low-index surfaces and the motion of Li adatoms on these surfaces. On the Cu(111) surface, the Li migration barrier is lower than the thermal energy at room temperature (kBT = 0.026 eV); thus, Li adatoms migrate along the Cu surface rather than forming Li dendrites. All the trajectories of Li on each facet, computed using ab initio molecular dynamics (AIMD) simulation for 10 ps with a timestep of 2 fs at 500 K, were collected in single snapshots (Cu: light brown and Li: purple). (h) Calculated reaction energy profiles for the migration of Li adatoms on the Cu(410) surface. | ||
To gain a deeper understanding of the distinct Li dynamics on sCu(111) and pCu foils, we conducted first-principles calculations of the Li adsorption energy (Eads) and migration activation energy (Emig) across various Cu model surfaces: low-index facets (e.g., (100), (110), and (111) surfaces) with a single terrace and high-index facets (e.g., (115), (410), and (122)) with various terraces, steps, and edges (Fig. 3(d) and Fig. S5, ESI†). In the case of low-index facets with a single terrace, each having one equivalent position, Li atoms tend to adsorb most easily to the (111) facet, indicating a higher Li affinity of the (111) surface compared to the (100) and (110) surfaces (see Fig. 3(e) and Fig. S6, ESI† for details). On the contrary, the high-index facets with different atomic arrangements from position to position possess varying adsorption energies, leading to potential non-uniform Li adsorption on these surfaces. Notably, Li adsorption on high-index facets is most thermodynamically favourable, compared to any other low-index Cu facets, particularly at step sites (Fig. 3(e) and Fig. S7, ESI†). In other words, high-index Cu facets exhibit a highly lithiophilic feature, with a predilection for Li atoms to adhere at the step sites.
To date, it has been widely accepted that lithiophilicity is critical in mitigating dendritic Li growth.4 Contrary to the conventional wisdom, upon Li deposition on a lithiophilic sCu(410) foil, we observed a unique needle-type Li morphology where numerous Li ‘whiskers’ are interconnected by Li ‘chunks’ (Fig. 3(f) and Fig. S8, ESI†), revealing the hidden side of lithiophilicity. In light of this, we propose that lithiophilicity should be subdivided into two categories: (1) the high affinity of Li to the surface, and (2) the preferential reaction of Li with alloying agents. Previous studies on lithiophilicity typically utilize lithiophilic agents, such as Al,24 Au,4,25 Si,26,27 Sn,28 and Ag.29,30 This alloying reaction enables spatial control of Li deposition, forming an alloy phase.31 By contrast, high affinity itself may lead to a steep concentration gradient in the vicinity of the electrode, resulting in a heterogeneous flux and resultant vertical growth (Fig. S9, ESI†). In this regard, we once again emphasize the importance of the rearrangement of the deposited Li on the substrate during the growth stage.32
To illustrate the Li dynamics after Li adsorption on Cu facets, we calculated the surface migration barriers of Li adatoms across various Cu facets using climbing-image nudged elastic band (CI-NEB) calculations,33 and computed the surface migration behaviours of Li adatoms on these facets through ab initio molecular dynamics (AIMD) simulations.34 Considering the surface symmetry of the low-index Cu facets (corresponding to the atomic arrangement of terraces of the high-index Cu facets, as well) (Fig. S5, ESI†), it is reasonable to assume that the migration pathways for Cu(100) and Cu(111) are isotropic, whereas that for Cu(110) is anisotropic, favouring the [1−10] and [001] directions, as indicated by the migration barriers for each facet (0.55 eV for Cu(110) to the [001] direction), 0.15 eV for Cu(100), 0.10 eV for Cu(110) to the [1−10] direction, and 0.01 eV for Cu(111) (see Fig. S10, ESI†).
Our simulation results clarify stark differences in the Li dynamics on each Cu facet. As illustrated by the Li trajectories in Fig. 3(g), the Li adatoms on Cu(100) are isolated onto equivalent sites near octahedral holes. On Cu(110), they migrate only along the [1−10] direction (Videos S4 and S5, ESI†). Interestingly, the migration barrier (0.01 eV) of Li adatoms on Cu(111) is significantly lower than that on other facets and even lower than the thermal vibration energy at room temperature (kBT: 0.026 eV, where kB represents Boltzmann's constant and T represents temperature). Consequently, Li adatoms on Cu(111) with a negligible migration barrier can migrate isotropically without any disturbance and uniformly cover the Cu(111) surface (Video S6, ESI†). This behaviour not only encourages horizontal Li growth, but also strongly validates our optical analyses and the corresponding Li morphologies on sCu(111) foil demonstrated earlier. In addition, we postulate that despite the uniform Li adsorption onto Cu(100) and Cu(110) facets, their subsequent Li growth modes are predominantly vertical (Fig. S11, ESI†), aligning with the trend exhibited on the high-index Cu(410) facet.
To interpret the experimental results on sCu(410) that we showed, we performed identical simulations with a Cu(410) model system having a stepped surface (Fig. 3(h) and Fig. S12, ESI†). As mentioned, the adsorption energy of Li atoms at the ‘step’ site is much lower than that of other sites, meaning that Li tends to preferentially adsorb onto the step sites on Cu(410). We also found that the activation barrier for Li migration on the Cu(410) surface is significantly lower for migration towards the steps than out of the steps or within an equivalent position (Fig. S12b, ESI†). This indicates that during Li plating, pre-deposited Li tends to accumulate at step sites (Video S7, ESI†), with incoming Li adsorbing directly onto these sites, elucidating the formation of the unique dendritic Li structure.
The influence of grain boundaries on Li dynamics
In addition to studying facet-dependent Li dynamics, we investigated the influence of GBs between different Cu facets. For this study, we utilized a twin-boundary-containing Cu(111) foil with some grains having a (115) surface orientation (denoted as tCu(111)/(115)), easily distinguishable using SEM and OM analyses (Fig. 4(a) and Fig. S13a, ESI†). The Cu(115) surface consists of high-index facets, comprising multiple (001) terraces and steps (Fig. S13b, ESI†). After deposition of a small quantity of Li (0.10 mA h cm−2) on the tCu(111)/(115) foil, Li was found to be primarily deposited at the boundary between the (115) and (111) grains (Fig. 4(b) and Fig. S14, ESI†). The amount of Li deposited at this GB was much higher than inside the grains, and the (115) grains exhibited more deposited Li (per area) than the (111) grains (This was also confirmed in Video S8, ESI†). Notably, different morphologies of Li were observed on each grain: particle-type Li deposits (rhombic dodecahedra, as described above) on the (111) grain (blue dashed area) and dendrite-type Li deposits on the (115) grain (red dashed area). These observations allowed us to discern which factor (‘adsorption’ or ‘surface migration’) predominates in determining Li growth behaviours (that is, this highlights the importance of surface migration for uniform Li plating, even if adsorption affects the uniformity of the Li ionic flux, as well as the initial Li plating).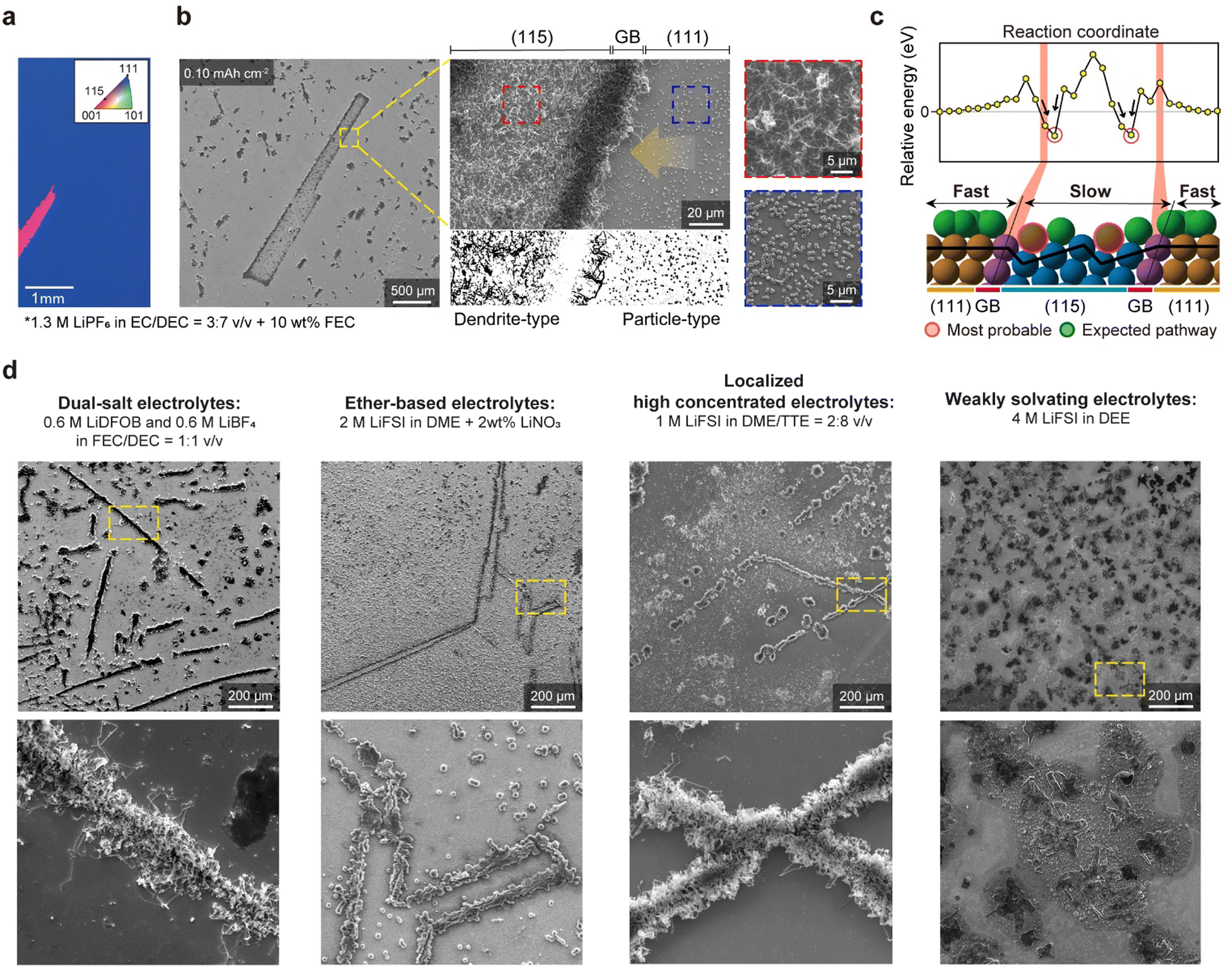 | ||
| Fig. 4 Facet-dependent Li deposition behaviours. (a) Inverse pole figure (IPF) map of the electron backscatter diffraction (EBSD) of twin boundary-containing Cu(111) foil (tCu(111)). The EBSD clarifies the presence of a (115) twin grain embedded in the tCu(111) foil. (b) SEM images of Li deposits on tCu(111). Two distinct morphologies of Li deposits can be distinguished through Image J software: particle-like Li on (111) grains and needle-like Li on (115) grains, and their boundaries. Primarily, Li tends to accumulate along the GBs. All the samplings were conducted in an electrolyte system containing 1.3 M LiPF6 in EC/DEC = 3:7 + 10 wt% FEC. (c) Computational model of the Cu surface composed of (111) and (115) grains and their boundaries; relative energies of the Li adatoms (top) and Li pathway on this model surface (bottom). The red circles indicate the thermodynamically most stable Li adsorption sites, which are ‘steps’ within (115) grains. (d) SEM images (top) and their magnified images (bottom) of Li morphologies on identical tCu(111) foils at 0.5 mA cm−2 for 30 min, as a function of diverse electrolyte formulations. | ||
As shown in Fig. S13b, ESI,† the adsorption energy of Li onto the Cu surface is lower for the step sites (−2.50 eV) on the Cu(115) surface than for any other sites on the model surface of tCu(111)/(115). The system is the most stabilized when Li atoms are trapped on the step sites, representing the thermodynamically preferred Li adsorption to the step site of the Cu(115) surface. The relative energies of Li adatoms at various positions on the model surface with reference to the Li adatom on the (111) surface, far from the boundary, also show that the relative energy begins to change as it approaches GBs. The relative energy drops significantly when the Li adatom is located on the step sites of the Cu(115) surface near GBs, while the energies on the (111) surface remain almost constant (Fig. 4(c)). However, from a kinetic perspective, this clearly demonstrates that the GBs between the Cu(111) and Cu(115) can be the most preferred sites for Li growth due to the high migration barrier, supporting our SEM data. Our AIMD simulation results also indicate that the Li adatoms move freely and faster on the (111) surface, but their motions are restricted and slower on the Cu(115) surface due to its stepped structure (Video S9, ESI†). As a result, Li is primarily concentrated on the GBs, as the Li adatoms that migrate on the Cu surface eventually become stuck near the boundary.
Over the past decade, researchers have explored a multitude of liquid electrolytes, focusing on advanced formulations, such as high concentrated electrolytes (HCE),35 localized high concentrated electrolytes (LHCE),36,37 and weakly solvating electrolytes (WSE).38,39 Numerous studies have reported the efficacy of these advanced electrolytes in achieving a more uniform Li+ flux and mitigating Li dendrites. Here, we used tCu(111)/(115) substrates as an indicator of effective electrolytes in homogenizing Li plating and suppressing dendritic Li growth. Four different electrolytes were selected for our study: dual-salt organic electrolytes, LiNO3-containing ether-based electrolytes, LHCE, and WSE. Obviously, rhombic dodecahedral Li seeds were formed on the sCu(111) foil, regardless of the electrolyte formulations (Fig. S15, ESI†). Surprisingly, however, when we deposited a small amount of Li (0.1 mA h cm−2) at 0.5 mA cm−2, they consistently exhibited Li deposition behaviours along the GBs (Fig. 4(d)), conflicting with previous works. This clarifies that recent electrolyte modifications may be still insufficient to resolve non-uniform Li plating, as well as resultant localized high current densities, due to the interaction between Li atoms and Cu grains, suggesting that the substrates (and their crystallographic orientations) have dominant roles in controlling the ‘Li coverage’ on themselves during Li plating. Interestingly, we discovered that in the case of LiNO3-containing ether-based electrolytes and WSE systems, the Li morphology near GBs is far from the dendrite-type. In particular, WSE electrolytes appear to somewhat ignore the intimate interactions between Cu and Li, showing unclear Li plating near grain boundaries. This indicates that the nucleation stage and growth stage are separate concerning dendritic Li growth, and we additionally suggest that an understanding of the LiNO3 additive and WSE electrolyte chemistry40 may be key solutions in transforming the Li plating model from the vertical to horizontal direction under any circumstance.
Electrochemistry of single crystal Cu(111)
To prove the superiority of sCu(111) on the electrochemical evaluations, we compared Cu|Li cells using the conventional organic electrolytes. When we measured the averaged CE (CEavg) over 10 cycles at a current density of 0.5 mA cm−2, the CEavg of the cell with sCu(111) is 97.2%, which is significantly higher than that (94.9%) of the cell with pCu (Fig. 5(a)). This finding indicates that side reactions with the electrolytes can be effectively alleviated by the surface area reduction of Li deposits through epihorizontaxial plating on sCu(111) foil. This result coincides with the cyclability of the cells. During galvanostatic tests, the cells using the sCu(111) foil showed stable cycling over 200 cycles at 0.50 mA cm−2 and over 150 cycles at 1.00 mA cm−2. Conversely, the cells using pCu foil had much shorter cell lifetimes with severe overpotential fluctuations (Fig. 5(b)). Consistent outcomes were observed not only in the organic electrolyte system, but also in the ether-based electrolyte system (Fig. 5(c)). Both cells utilizing sCu(111) and pCu demonstrated significantly enhanced cyclability compared to the organic system, which was attributed to their enhanced chemical compatibility with bare Li. However, cells with pCu still exhibited much poorer cyclability, leading to early short circuits compared to those with sCu(111). This suggests that even under mild conditions, excluding severe reactions between electrolytes and Li, sCu(111) has been proved to exhibit excellent electrochemical reversibility, accompanying the morphological merits of deposited Li, in terms of mitigated dead Li and SEI layer formation (Fig. S16–S19, ESI†).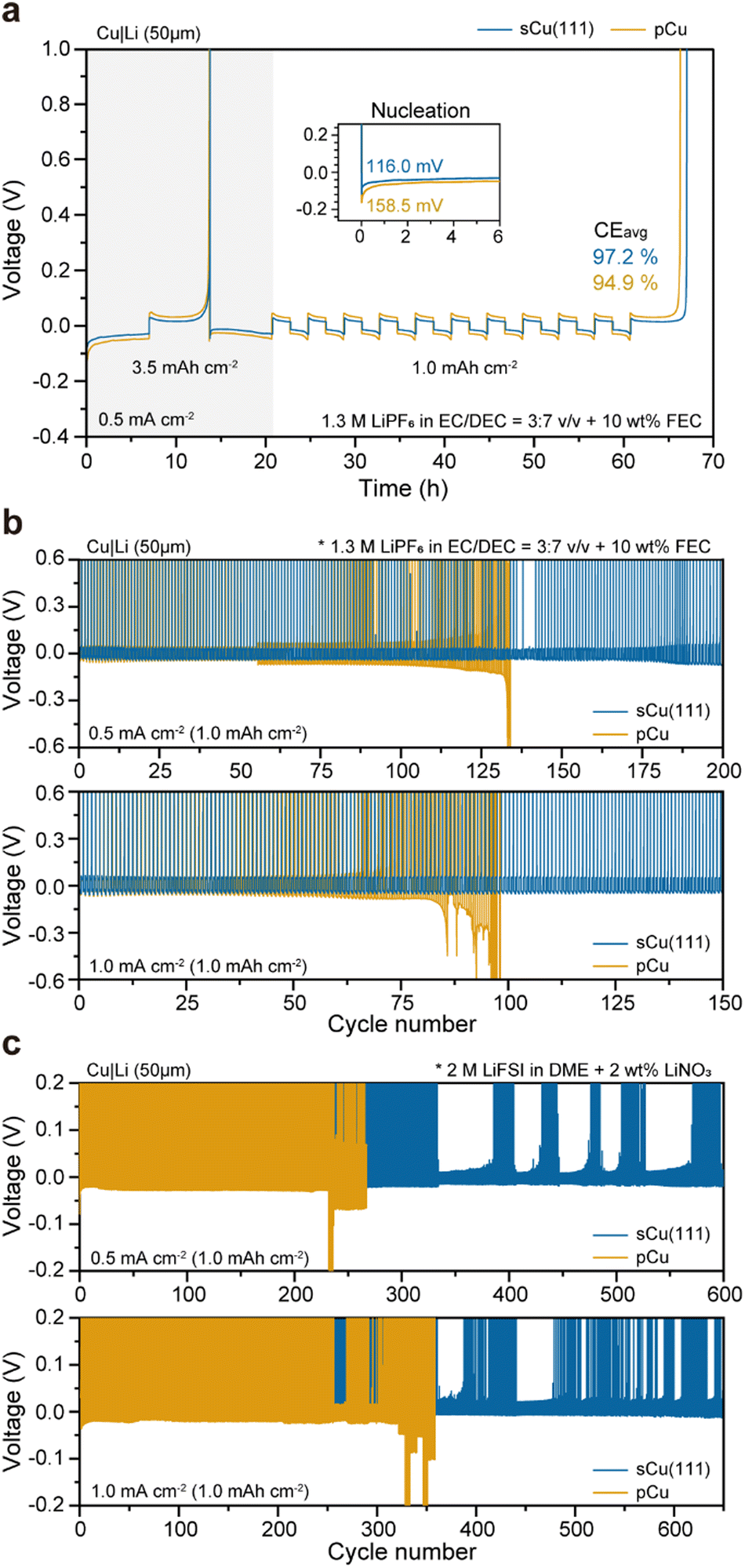 | ||
| Fig. 5 Electrochemical performances of Cu|Li half cells. (a) Measurement of the averaged Coulombic efficiencies (CEavg) using sCu(111) and pCu foil at 0.5 mA cm−2, in a conventional organic electrolyte system. CEavg was calculated using the Aurbach method described in the Methods section. (b) and (c) Galvanostatic Li plating/stripping voltage profiles of Cu|Li cells operated at 0.5 mA cm−2 (top) and 1.0 mA cm−2 (bottom) with a cycling capacity of 1.0 mA h cm−2 in a carbonate-based electrolyte (b) and ether-based electrolyte (c). | ||
To further test the feasibility of our single crystal foils for practical anode-free Li batteries, we assembled Cu|LiNi0.8Co0.1Mn0.1O2 (NCM811) and measured its electrochemical performances (Fig. 6). Fig. 6(a) displays the first-cycle voltage profiles of Cu|NCM811 anode-free cells, operated at 0.1C-rate (0.5 mA cm−2; 1C-rate is defined as 5.0 mA h cm−2) between 3.0 V and 4.4 V. When charging up to the upper cut-off voltage (4.4 V), both exhibit 247.2 mA h g−1, corresponding to 5.3 mA h cm−2 areal capacity. As the cells are discharged back to 3.0 V at 0.2C charging (0.2C) and 0.5 D discharging (0.5 D), the cell with sCu(111) has a comparatively higher initial CE (ICE) of 93.7% (vs. 93.2% for that with pCu), which is consistent with previous CE measurement of Cu|Li cells. However, as illustrated in Fig. 6(b), the Cu|NCM811 cells experience severe decay in conventional organic electrolyte systems during repeated cycles. This renders them unsuitable as electrolytes for anode-free Li full-cells without excess Li. Consequently, dual-salt organic electrolytes were chosen as alternatives to address the electrochemical instability, which were designed by the Dahn group.41,42 Despite the identical operating conditions, both cells exhibit significantly prolonged cycle life features with electrolyte modification. This indicates effective suppression of Li galvanic corrosion43 and electrolyte decomposition. In contrast with the cell using pCu, which shows rapid degradation after 60 cycles, the cell utilizing sCu(111) maintains high CEs of more than 97% over 100 cycles (Fig. 6(b)), accompanying relatively superior capacity retention (Fig. 6(c)).
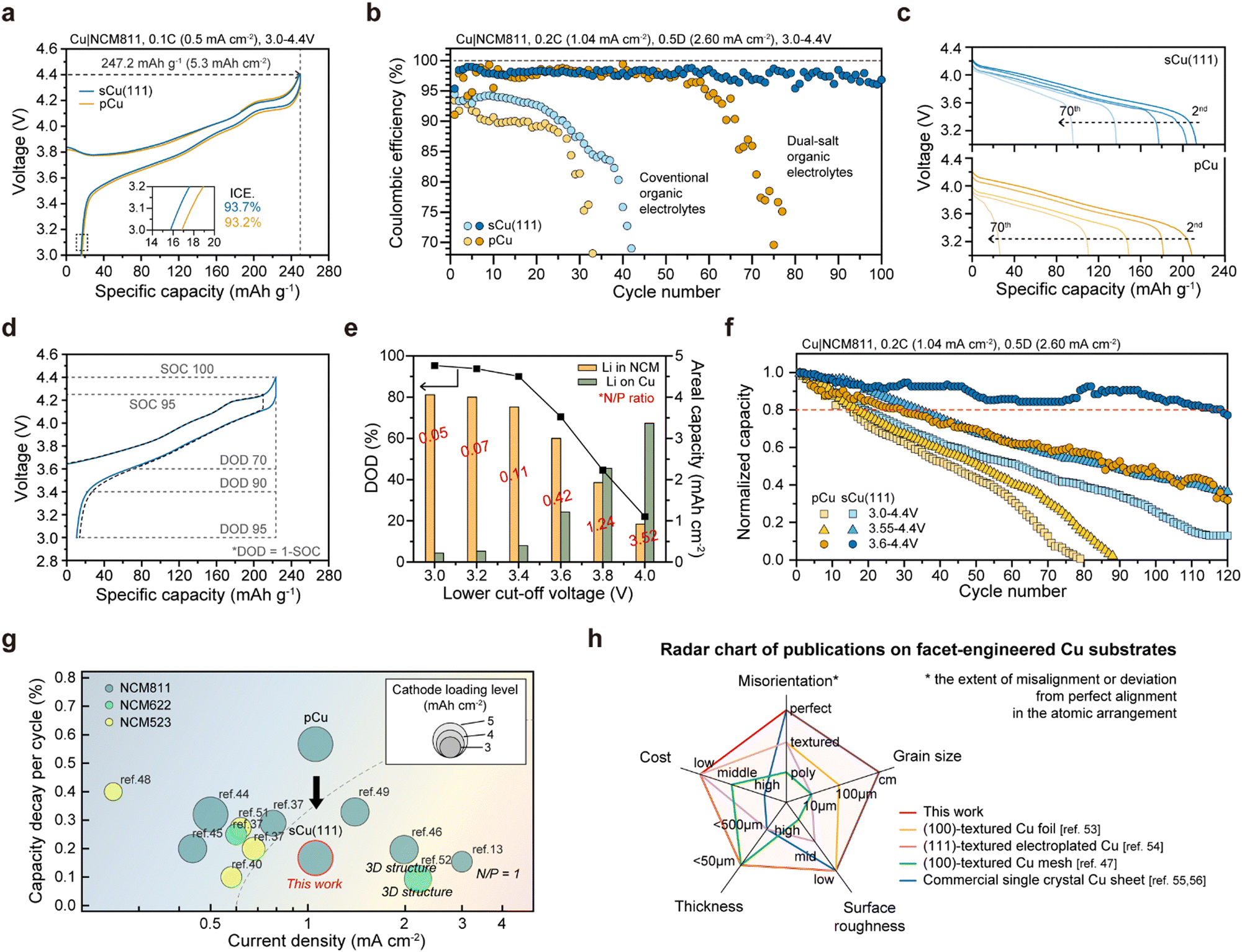 | ||
| Fig. 6 Electrochemical performances of anode-free full-cells with NCM811 cathodes. Areal capacity of the NCM811 electrode is 5.3 mA h cm−2. (a) First-cycle voltage profiles of anode-free cells using pCu (yellow) and sCu(111) (blue) measured at a 0.1C-rate (0.5 mA cm−2) between 4.4 and 3.0 V. The inset shows enlarged curves for the dotted box and initial Coulombic efficiencies (ICE.). (b) CE versus cycle number of Cu|NCM811, operated at 0.2C (1.04 mA cm−2) on charge and 0.5 D (2.60 mA cm−2) on discharge in 3.0–4.4 V, in conventional organic electrolyte (1.3 M LiPF6 in EC/DEC = 3:7 v/v + 10 wt% FEC) and dual-salt organic electrolyte system (0.6 M LiDFOB and 0.6 M LiBF4 in FEC/DEC = 1:2 v/v). Discharge curves in dual-salt organic electrolytes are expressed in (c). (d) Second-cycle voltage profiles of sCu|NCM811 cells at 0.2C and 0.5 D in 3.0–4.4 V (blue) and 3.0–4.3 V (black dash). SOC and DOD refer to the state of charge and the depth of discharge, respectively. (e) Variation of DOD and cycling areal capacity as a function of the lower cut-off voltage. N/P ratio refers to the ratio of the amount of remaining Li on Cu foil and the amount of Li present in the NCM cathodes. (f) Normalized capacity versus cycle number of Cu|NCM811 cells at 0.2C and 0.5 D, with different operating voltages (3.0–4.4 V, 3.55–4.4 V, and 3.6–4.4 V, corresponding to DOD95, DOD78, and DOD60, respectively). (d)–(f) the electrochemical data were obtained in a dual-salt organic electrolyte system. (g) Capacity decays per cycle and applied current densities in published studies. Circle size represents the cathode loading level, and circle colour represents the cathode type (NCM523, NCM622, and NCM811). (h) Radar chart of publications on facet-engineered Cu substrates, in terms of the misorientation, grain size, surface roughness, thickness, and cost, which are critical parameters for the substrate in anode-free batteries. | ||
Meanwhile, it is recognized that the state-of-charge (SOC) and depth-of-discharge (DOD), reflecting the charge status, are critical parameters in determining the cell lifetime.44 Particularly, it has been demonstrated that DOD significantly influences the cyclability of anode-free batteries, underscoring the importance of optimizing the operating conditions to maximize the battery lifespan.45Fig. 6(d) represents the second-cycle voltage profile of sCu(111)|NCM811 cells at 0.2C and 0.5 D, following the formation cycle. When comparing two different higher cut-off voltages (4.3 V vs. 4.4 V), their CEs are nearly the same (95.3% for 4.4 V and 93.5% for 4.3 V). This emphasizes that the control of DOD should take precedence over SOC. When modifying DOD by adjusting the lower cut-off voltage, we can utilize the Li residue left on the Cu surface during charging to 4.4 V again, as illustrated in Fig. 6(d) and (e). It is reasonable to expect that a higher amount of Li residue (i.e., lower DOD) will extend the cell lifetime, and this is likely influenced by the morphology and surface area of the Li deposited on Cu substrates.
We evaluated the cells by employing diverse DODs (DOD95 for 3.0 V, DOD78 for 3.55 V, and DOD60 for 3.6 V; corresponding to 213.0, 173.3, and 156.5 mA h g−1, respectively) (Fig. 6(f)). Among them, limiting the DOD to 60% resulted in dramatic changes for both pCu and sCu(111). Particularly for sCu(111)|NCM811 cell, its capacity retention reaches 80% at the 118th cycle, significantly outlasting the pCu|NCM811 cell, which reaches 80% capacity retention at the 29th cycle. The above results, obtained by us, are strongly correlated with the Li plating/stripping behaviours on each Cu foil under the same cell configuration and operating conditions. As depicted in Fig. 2(d) and Fig. S11, ESI,† the Li deposition layer with over 3 mA h cm−2 appears porous and non-uniform on the pCu foil, in stark contrast to that on the sCu(111) foil, which exhibits a highly dense and uniform morphology. We believe that this morphological merit of Li on sCu(111) may be definitely prerequisite for minimizing side reactions with the electrolytes and enhancing the cycling CEs.
Previous published papers13,38,42,46–54 on anode-free Li batteries mostly have primarily demonstrated their academic superiorities by employing low current densities, low cathode loading levels, thick 3D porous structures (resulting in a decrease in volumetric capacity), or a high N/P ratio (i.e., no anode-free Li batteries) (Fig. 6(g)). In terms of the capacity decay per cycle, we achieved remarkable outcomes simply by modulating the facet and grain boundary of the Cu substrate coupled with the cathode at a commercial level.
To date, a few previous reports51,55–58 have demonstrated that the Li plating behaviour is dependent on the crystallographic orientation of single crystal Cu substrates (Fig. 6(h) and Table S1, ESI†). It is noteworthy that they employed imperfect Cu substrates containing relatively small grains with different orientations and grain boundaries (strictly speaking, not single crystal Cu), leading to some technical errors in their conclusions. Furthermore, they often conducted insufficient control experiments using only commercially available single crystal Cu sheets, which are of no practical use in battery applications due to their dimension, thickness, and expense. In this regard, our work is totally different in terms of our discoveries and approach, and it is of great interest to both scientific research and industrial R&D communities studying and developing high-energy-density Li metal and anode-free batteries.
Conclusions
In summary, we have described the novel epihorizontaxial behaviour of Li adatoms on Cu substrates during plating. Particularly on low-index sCu(111) facet, the active surface migration of Li adatoms transforms the Li growth model from the vertical to horizontal direction, in stark contrast to high-index Cu(410) and (115) facets, and pCu containing numerous grains and grain boundaries. Based on our observations and modelling, we propose certain conditions that are necessary to induce epihorizontaxial Li plating on a substrate. First, the substrate should have not only an atomically ‘flat’ surface, but also a near-zero migration barrier. This activates the coalescence of Li atoms and particles, and induces the horizontal growth of Li islands. Second, the substrate should possess comparatively few defects. The presence of grains with different orientations and their boundaries on the substrate leads to distinct Li plating behaviours that inhibit the development of epihorizontaxial Li plating. The resultant heterogeneous Li plating can happen with as-received Cu foils that have surface roughness at the micrometre scale due to the rolling marks formed during their production, as well as with electropolished pCu foils. Given the design rules outline for the substrate in anode-free Li batteries that we propose here, our findings of a near-zero migration barrier for Li adatom migration on the metal current collector provide novel scientific insight and indicate the potential for commercial realization of high-energy lithium batteries, including both anode-free and solid-state batteries.Methods
Materials
The thickness of the Cu foils is 50 μm. Li metal foil (50 μm thick) was purchased from Wellcos. Lithium bisfluorosulfonylimido (LIFSI), lithium tetrafluoroborate (LiBF4), lithium difluoro(oxalate)borate (LiDFOB), dimethyl ether (DME) (99.5%), and LiNO3 were purchased from Sigma Aldrich. 2,2-Tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether (TTE) and 1,2-diethoxyethane (DEE) were purchased from TCI. The electrolyte (1.3 M LiPF6 in ethylene carbonate (EC)/diethyl carbonate (DEC) + 10 wt% fluoroethylene carbonate (FEC)) was purchased from Wellcos.Sample preparation
We used the contact-free annealing (CFA) process for the preparation of single-crystal Cu(111) and Cu(410) foils. We suspended commercial polycrystalline Cu foil on the quartz holder, and annealed near the melting temperature of Cu (at 1323 K) for 12 h in an atmosphere of flowing H2 and Ar, in order to induce the colossal growth of the (111) grain. Flowing H2 gas is required to prevent the oxidation of Cu foil. During annealing, few micro-scale {111} grains grew abnormally to the centimetre-scale, with {111} 〈112〉 orientation. Since the initial textures of the polycrystalline Cu foil is critical in determining the final crystal orientation, we can obtain diverse single crystal Cu foils with different orientations by changing the as-received Cu foils. To remove the rolling mark of the as-received polycrystalline Cu foil, we electropolished the as-received Cu foil using a simple electropolishing setup with a metal tweezer to clip the foil and flow the current through (Fig. S23, ESI†). This electropolishing was conducted under a constant voltage of 2 V for 10 min in the electrolyte made by mixing 750 ml H3PO4, 250 ml ethanol, 50 ml IPA, and 5 g urea.Materials characterization
Scanning electron microscopy (SEM) and electron backscatter diffraction (EBSD) results were obtained using a Quanta 3D FEG and Verios 460 (FEI company) with an EBSD accessory system (Digiview V from Ametek). To obtain SEM images of the Li deposits on the Cu foil, charged Cu|Li cells were disassembled in an argon-filled glove box, and Cu foils were then gently washed with dimethyl carbonate (DMC) to eliminate residual electrolyte salts. Synchrotron X-ray diffraction (XRD) and wide-angle X-ray scattering data were collected on the 6D beamline at the Pohang Light Source (PLS-II). The operating voltage of the accelerator was 3.5 GeV and the current was 400 mA. Operando Optical microscopy (Olympus, BX53M) measurements were carried out using a home-made optical cell and a side-by-side-type cell (EL-CELL, ECC-Opto-SBS). For this, a Cu foil as a working electrode, a Li metal foil as a counter electrode, and 1.3 M LiPF6 in EC/DEC + 10 wt% FEC as the electrolyte, were assembled in an argon-filled glove box.Electrochemical evaluation
For fabrication of Cu|Li and Cu|NCM811 cells, 16pi Cu foil, 14pi Li metal (and cathode electrode), and 10–20 μL of the electrolytes were used. The thickness of the Li metal is fixed at 50 μm. NCM811 electrodes were prepared by mixing 94 wt% active material, 3 wt% Super P carbon, and 3 wt% PVDF binder using a Thinky mixer. The electrodes were then dried at 110 °C for 6 h in a vacuum oven and roll-pressed. The average CEs with a current density of 0.5 mA cm−2 were calculated using the following equation (Aurbach method):| CEavg = (nQc + Qs)/(nQc + QT) |
Computational details
The simulations of the atomic-scale behaviour of Li on the various Cu slabs were performed using a spin-polarized density functional theory (DFT) calculation with the Perdew–Burke–Ernzerhof (PBE) functional and the projected augmented-wave pseudopotential. The kinetic energy cutoff of 500 eV for the plane-wave-basis set, energy convergence of 10−5 eV per atom, and force criteria of 0.03 eV Å−1 were used with a specified Monkhorst–Pack grid of k-point (5 × 5 × 1 for single facet slabs and 6 × 3 × 1 for slab constituting two grains). All theoretical calculations reported in this work were performed using the Vienna ab initio simulation package (VASP). The slab surface of the (hkl) plane is denoted as Cu(hkl). The Cu(100) and Cu(111) slabs were built using a 5 × 5 supercell with five layers, and Cu(110) was built using a 4 × 5 supercell with five layers. The upper two layers were relaxed, and the bottom three were fixed to give the bulk properties. The Cu(115), Cu(410), and Cu(223) slabs were built large enough to avoid the undesirable neighbouring interactions. To construct a huge Cu slab model constituting the grain boundaries of Cu(111) and Cu(115), the coherent atoms of each slab surface were integrated as a mirror plane. All simulation boxes contained a vacuum space of ∼15 Å. The Li adsorption energy (Eads) was calculated as follows:| Eads = ELi@sur – Esur − ELi, |
Author contributions
M.-H. K., S. J., and H.-W. L. conceived the idea and designed the experiments. R. S. R., S. J., and H.-W. L. supervised the project. M.-H. K. performed the characterization, carried out the electrochemical measurements and analyzed the data. Y. L., M. H. K., and S. J. prepared the Cu foils. D. Y. K., S.-W. P. and D.-H. S. performed the DFT and MD simulations. J. S. and T. K. assisted with the characterization. J. K. performed the in situ OM. M.-H. K., D. Y. K., R. S. R., D.-H. S., S. J., and H.-W. L. wrote the manuscript. All coauthors revised and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank UCRF (UNIST Central Research Facilities) for support of using the equipment. The WAXS experiment was performed at Beamline 6D of the Pohang Accelerator Laboratory. This work was supported by 2024 Research Fund (1.240005.01) of UNIST, National Research Foundation of Korea (RS-2024-00428511, RS-2023-00208929, 2021R1C1C2003118, and 2021M3A7C2089743), National Supercomputing Center of Korea Institute of Science and Technology Information (KSC-2023-CRE-0392), Korea Institute of Energy Technology Evaluation and Planning (20221B1010003B), National Research Council of Science & Technology (NST) grant by the Korea government (MSIT) (No. GTL24011-000), and the Institute for Basic Science (IBS-R-019-D1).Notes and references
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed
.
- G. Zheng, S. W. Lee, Z. Liang, H.-W. W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 CrossRef CAS
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS
- F. Liu, R. Xu, Y. Wu, D. T. Boyle, A. Yang, J. Xu, Y. Zhu, Y. Ye, Z. Yu, Z. Zhang, X. Xiao, W. Huang, H. Wang, H. Chen and Y. Cui, Nature, 2021, 600, 659–663 CrossRef CAS PubMed
- M. J. Zachman, Z. Tu, S. Choudhury, L. A. Archer and L. F. Kourkoutis, Nature, 2018, 560, 345–349 CrossRef CAS PubMed
- J. Xiao, Science, 2019, 366, 426–427 CrossRef CAS PubMed
- R. Zhang, X. R. X. X.-R. Chen, X. R. X. X.-R. Chen, X. B. Cheng, X.-Q. Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7764–7768 CrossRef CAS PubMed
- X. Chen, X. R. Chen, T. Z. Hou, B. Q. Li, X. B. Cheng, R. Zhang and Q. Zhang, Sci. Adv., 2019, 5, eaau7728 CrossRef CAS PubMed
- Z. Liang, D. Lin, J. Zhao, Z. Lu, Y. Liu, C. Liu, Y. Lu, H. Wang, K. Yan, X. Tao and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2862–2867 CrossRef CAS PubMed
- D. Lin, Y. Liu, Z. Liang, H.-W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef PubMed
- W. Go, M. H. Kim, J. Park, C. H. Lim, S. H. Joo, Y. Kim and H. W. Lee, Nano Lett., 2019, 19, 1504–1511 CrossRef PubMed
- Z. Wu, C. Wang, Z. Hui, H. Liu, S. Wang, S. Yu, X. Xing, J. Holoubek, Q. Miao, H. L. Xin and P. Liu, Nat. Energy, 2023, 8, 340–350 CrossRef
- F. Hao, A. Verma and P. P. Mukherjee, Energy Storage Mater., 2019, 20, 1–6 CrossRef
- Y. Lee, B. Ma and P. Bai, Energy Environ. Sci., 2020, 13, 3504–3513 RSC
- R. Akolkar, J. Power Sources, 2013, 232, 23–28 CrossRef
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef
- Y. N. Wen and J. M. Zhang, Solid State Commun., 2007, 144, 163–167 CrossRef
- S. Jin, M. Huang, Y. Kwon, L. Zhang, B.-W. Li, S. Oh, J. Dong, D. Luo, M. Biswal, B. V. Cunning, P. V. Bakharev, I. Moon, W. J. Yoo, D. C. Camacho-Mojica, Y.-J. Kim, S. H. Lee, B. Wang, W. K. Seong, M. Saxena, F. Ding, H.-J. Shin and R. S. Ruoff, Science, 2018, 362, 1021–1025 CrossRef PubMed
- X. Zhang, C. Dong, J. Zapien, S. Ismathullakhan, Z. Kang, J. Jie, X. Zhang, J. Chang, C.-S. Lee and S.-T. Lee, Angew. Chem., Int. Ed., 2009, 121, 9285–9287 CrossRef
- A. Hagopian, M.-L. Doublet and J.-S. Filhol, Energy Environ. Sci., 2020, 13, 5186–5197 RSC
- X. Yuan, B. Liu, M. Mecklenburg and Y. Li, Nature, 2023, 620, 86–91 CrossRef CAS PubMed
- D. T. Boyle, Y. Li, A. Pei, R. A. Vilá, Z. Zhang, P. Sayavong, M. S. Kim, W. Huang, H. Wang, Y. Liu, R. Xu, R. Sinclair, J. Qin, Z. Bao and Y. Cui, Nano Lett., 2022, 22, 8224–8232 CrossRef CAS PubMed
- H. Li, T. Yamaguchi, S. Matsumoto, H. Hoshikawa, T. Kumagai, N. L. Okamoto and T. Ichitsubo, Nat. Commun., 2020, 11, 1584 CrossRef PubMed
- H. Wang, Y. Li, Y. Li, Y. Liu, D. Lin, C. Zhu, G. Chen, A. Yang, K. Yan, H. Chen, Y. Zhu, J. Li, J. Xie, J. Xu, Z. Zhang, R. Vilá, A. Pei, K. Wang and Y. Cui, Nano Lett., 2019, 19, 1326–1335 CrossRef CAS PubMed
- H. Chen, Y. Yang, D. T. Boyle, Y. K. Jeong, R. Xu, L. S. de Vasconcelos, Z. Huang, H. Wang, H. Wang, W. Huang, H. Li, J. Wang, H. Gu, R. Matsumoto, K. Motohashi, Y. Nakayama, K. Zhao and Y. Cui, Nat. Energy, 2021, 6, 790–798 CrossRef CAS
- J. Sung, S. Y. Kim, A. Harutyunyan, M. Amirmaleki, Y. Lee, Y. Son and J. Li, Adv. Mater., 2023, 35, 2210835 CrossRef CAS PubMed
- M. Wan, S. Kang, L. Wang, H. W. Lee, G. W. Zheng, Y. Cui and Y. Sun, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed
- Y. G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D. S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS
- Y. Jeon, J. Kim, H. Jang, J. Lee, M. G. Kim, N. Liu and H. K. Song, J. Mater. Chem. A, 2022, 10, 1768–1779 RSC
- Y. Ye, H. Xie, Y. Yang, Y. Xie, Y. Lu, J. Wang, X. Kong, S. Jin and H. Ji, J. Am. Chem. Soc., 2023, 145, 24775–24784 Search PubMed
- Q. Yun, Y. B. He, W. Lv, Y. Zhao, B. Li, F. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 6932–6939 CrossRef PubMed
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed
- X. Ren, P. Gao, L. Zou, S. Jiao, X. Cao, X. Zhang, H. Jia, M. H. Engelhard, B. E. Matthews, H. Wu, H. Lee, C. Niu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J. G. Zhang and W. Xu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28603–28613 CrossRef CAS PubMed
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS
- Y. Yao, X. Chen, C. Yan, X. Zhang, W. Cai, J. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS PubMed
- Z. Wang and B. Zhang, Energy Mater. Devices, 2023, 1, 9370003 CrossRef
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. DeGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Nat. Energy, 2020, 5, 693–702 CrossRef CAS
- J. Ding, R. Xu, Y. Xiao, S. Zhang, T. Song, C. Yan and J. Huang, Adv. Energy Mater., 2023, 2204305, 1–8 Search PubMed
- B. Xu, A. Oudalov, A. Ulbig, G. Andersson and D. S. Kirschen, IEEE Trans. Smart Grid, 2018, 9, 1131–1140 Search PubMed
- A. J. Louli, M. Coon, M. Genovese, J. deGooyer, A. Eldesoky and J. R. Dahn, J. Electrochem. Soc., 2021, 168, 020515 CrossRef CAS
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Y. Yang, Z. Yu, S. T. Oyakhire, W. Zhang, L. C. Greenburg, M. S. Kim, D. T. Boyle, P. Sayavong, Y. Ye, J. Qin, Z. Bao and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef
- H. Chen, A. Pei, J. Wan, D. Lin, R. Vilá, H. Wang, D. Mackanic, H.-G. Steinrück, W. Huang, Y. Li, A. Yang, J. Xie, Y. Wu, H. Wang and Y. Cui, Joule, 2020, 4, 938–952 CrossRef
- L. Lin, L. Suo, Y. Hu, H. Li, X. Huang and L. Chen, Adv. Energy Mater., 2021, 11, 2003709 CrossRef
- M. Mao, X. Ji, Q. Wang, Z. Lin, M. Li, T. Liu, C. Wang, Y. Hu, H. Li, X. Huang, L. Chen and L. Suo, Nat. Commun., 2023, 14, 1082 CrossRef
- H. Kwon, J.-H. Lee, Y. Roh, J. Baek, D. J. Shin, J. K. Yoon, H. J. Ha, J. Y. Kim and H.-T. Kim, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed
- Y. Gu, H. Xu, X. Zhang, W. Wang, J. He, S. Tang, J. Yan, D. Wu, M. Zheng, Q. Dong and B. Mao, Angew. Chem., Int. Ed., 2019, 58, 3092–3096 CrossRef PubMed
- S. T. Oyakhire, W. Zhang, A. Shin, R. Xu, D. T. Boyle, Z. Yu, Y. Ye, Y. Yang, J. A. Raiford, W. Huang, J. R. Schneider, Y. Cui and S. F. Bent, Nat. Commun., 2022, 13, 3986 CrossRef PubMed
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J. G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef
- T. T. Hagos, B. Thirumalraj, C. J. Huang, L. H. Abrha, T. M. Hagos, G. B. Berhe, H. K. Bezabh, J. Cherng, S. F. Chiu, W. N. Su and B. J. Hwang, ACS Appl. Mater. Interfaces, 2019, 11, 9955–9963 CrossRef PubMed
- Y.-J. Kim, S. H. Kwon, H. Noh, S. Yuk, H. Lee, H. Soo Jin, J. Lee, J.-G. Zhang, S. G. Lee, H. Guim and H.-T. Kim, Energy Storage Mater., 2019, 19, 154–162 CrossRef
- S.-T. Liu, H.-Y. Ku, C.-L. Huang and C.-C. Hu, Electrochim. Acta, 2022, 430, 141011 CrossRef CAS
- K. Ishikawa, S. Harada, M. Tagawa and T. Ujihara, ACS Appl. Mater. Interfaces, 2020, 12, 9341–9346 CrossRef CAS
- C. Shen, J. Gu, N. Li, Z. Peng and K. Xie, J. Power Sources, 2021, 501, 229969 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee01766h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |