
Synthesis of atomically precise Ag16 nanoclusters and investigating solvent-dependent ultrafast relaxation dynamics†
Sikta
Chakraborty
 a,
Sarita
Kolay
a and
Amitava
Patra
*ab
a,
Sarita
Kolay
a and
Amitava
Patra
*ab
aSchool of Materials Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, India. E-mail: msap@iacs.res.in; Fax: +91-33-2473-2805; Tel: +91-33-2473-4971
bInstitute of Nano Science and Technology, Knowledge City, Sector 81, Mohali 140306, India
First published on 16th August 2024
Abstract
In this article, the main focus is to employ a new synthetic strategy to prepare atomically precise Ag nanoclusters (NCs) and unveil the critical role played by the solvents in the excited state dynamics of Ag NCs. The compositional analysis confirms the formula of the nanoclusters as Ag16(PDT)8(PPh3)4 (Ag-PDT NCs). These NCs showed a sharp absorption band at 525 nm and a comparatively broad absorption band at 633 nm. The emission maximum was 630 nm with a quantum yield (QY) of 0.23%. Three-component relaxation dynamics was retrieved from global analysis and described as core relaxation (664 fs), core-to-surface state relaxation (500 ps), and ground state relaxation (>1 ns) for Ag NCs in the DCM solvent. The time constants are slightly higher at 1.25 ps, 624.25 ps, and >1 ns for Ag NCs in the DMF solvent because of the less effective charge separation. The high QY in DMF follows this low charge separation (0.23% vs. 0.63%). The straight-chain dithiol capping agent (with lower electron density than an electron-rich aromatic ring) is mainly responsible for this less effective charge separation. Finding the pivotal role of the solvent in NC chemistry will help to characterize it thoroughly and produce a strategy for precise applications in various fields.
Introduction
The synthesis of atomically precise luminescent metal nanoclusters (MNCs) has attracted significant interest due to the potential applications in sensing, hydrogen generation, light harvesting, and energy conversion.1–11 The discrete electronic transitions in absorption are due to the quantum effect in MNCs when the size approaches the Fermi wavelength of electrons.12–16 Their unique photophysical properties, such as significant Stokes shift, tunable photoluminescence (PL), and long excited-state lifetime, have potential for light-energy conversion.17–27 The excited state relaxation dynamics and electron–phonon coupling of NCs are tailored by modulating several atoms, surface staple motifs, nature of ligands, etc.28–35 Various spectroscopic methods have been used to understand the complex relaxation processes of NCs, such as internal conversion, charge transfer, and electron–hole recombination on different time scales.18,36–38 Recently, the origin of visible and NIR emission has been well explored by Zhou et al., who reported that the visible emission of Au25(SR)18 NCs arises from the surface state, and the Au13 core state is responsible for NIR emission.39 Jin et al. investigated the effects of structural isomerism on the ultrafast relaxation dynamics of two isomers of Au38. Au38Q showed a rapid decay (1.5 ps) followed by a nanosecond relaxation, whereas the other structural isomer Au38T displayed similar relaxation, yet the rapid decay was accelerated by 50% (1 ps).40 Our previous study reported the influence of single-atom doping on the excited state relaxation dynamics of a series of MAg24n− (M = Ag, Au, Pd, and Pt) NCs. We have described there how the electron affinity and rigidity of the framework influence the ultrafast electron dynamics.41 Recently, we demonstrated that the electronegativity of surface ligands and polarization can control the electron–phonon interaction, which eventually modifies the relaxation from the core state to the surface state of Ag44.42 Although the structure, doping of metal atoms or effects from the surface ligand, etc. are the critical parameters in determining the relaxation dynamics, the variable nature of NCs in the solvent helps in understanding the origin of the transitions.43,44 Recently Antoine's group demonstrated solvent-dependent two-photon excited photoluminescence (2PEPL) for platinum doped Ag29.45 Knappenberger Jr. recently analyzed the interband excitation (400 nm) of the Au25(PET)18 moiety followed by rapid internal conversion and picosecond relaxation depending on both the ligand structure and the dielectric of the dispersing medium. However, the intraband excitation (800 nm) showed solvent and ligand-independent relaxation dynamics.46 In another recent study, we demonstrated that the strong electron–phonon interaction is due to the combined effect of the metal core and surface of Ag21(CHT)5(DPPB) NCs that influence the excited state relaxation dynamics.47 Primary emphasis has been paid to the influence of core atoms, ligands, and electron–phonon interactions and less attention has been paid to solvent-dependent behaviors such as relaxation pathways, charge separation, dipole–dipole interactions, etc.48,49 In molecular systems, the solvent plays a pivotal role in excited state dynamics because of the dielectric constant. However, there has been little detailed study of the excited state dynamics behavior of Ag NCs with solvents.Here, we have used a new synthetic strategy for atomically precise Ag16 NCs using pentane-1,5-dithiol in DCM solvent and investigate their excited state dynamics with varying solvents using ultrafast spectroscopy. The atomically precise Ag16(PDT)8(PPh3)4 NCs were characterized using MALDI-MS, ESI-MS, and TGA analysis with FTIR and NMR studies. The distinct absorption bands at around 525 nm and 633 nm suggest a molecular nature. The red-emitting Ag NCs showed emission at 630 nm (excitation at 420 nm) with a QY of 0.23%. The excited state dynamics of the Ag-PDT NCs was investigated using transient absorption spectroscopy, and three components were retrieved from global analysis. Relatively less polar aliphatic thiol (compared to the electron-rich aromatic group) capped Ag NCs relaxed faster in DCM solvent as the relaxation is favorable in this solvent having a low dielectric constant. Meanwhile, the ultrafast relaxation of Ag-PDT NCs in the DMF solvent required more relaxation time. The less effective charge separation of the less polar aliphatic thiol-capped Ag-PDT system does not favor fast relaxation in a solvent with a higher dielectric constant. The ultrafast decay dynamics of the as-synthesized Ag NCs and analysis of decay dynamics in different solvents can be used to explore the field of several applications.
Results and discussion
A bottom-up synthetic procedure was followed using a small chain thiol for Ag NCs synthesis. AgNO3 was taken in a round bottom flask with 2 mL of MeOH and sonicated to dissolve the metal precursor. Then DCM was added under ice-cold conditions, followed by the pentane dithiol (PDT) ligand, triphenylphosphine, and sodium borohydride (details are given in the Experimental section in the ESI†). The feeding ratio of the metal and thiolate ligand is maintained at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Fig. 1 represents a synthetic illustration of the Ag-PDT NC system.
:5. Fig. 1 represents a synthetic illustration of the Ag-PDT NC system.
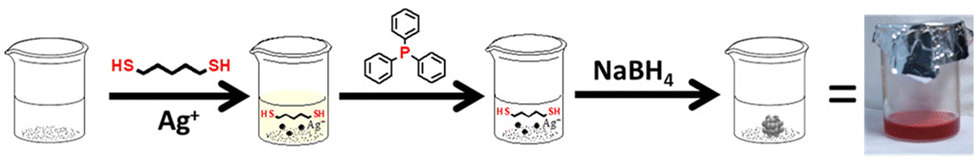 | ||
| Fig. 1 Schematic representation of the synthetic procedure of Ag-PDT NCs in DCM. | ||
The TEM image of NCs depicts the size of the Ag NCs (Fig. S1A and B†). The Ag-PDT NCs have an average size of around 1.8 ± 0.1 nm (<2 nm). In the FTIR spectra of the PDT ligand, a peak appears at around 2554 cm−1 due to the stretching of the S–H bond (Fig. S2A†). The disappearance of this peak in the case of Ag-PDT NCs indicates the formation of metal–sulfur bonds (Fig. S2A†). Another peak at around 500–600 cm−1 is the Ag–S bond in Ag-PDT NCs (Fig. S2B†).50 In 1H NMR spectra, the PDT ligand shows a characteristic proton peak, also observed in Ag NCs (Fig. S3 and S4†). The peak position shifts slightly due to the metal surface near the ligands.51,52 The δ value for hydrogen in the 2 position changes greatly (from 2.51 to 2.70), which is closest to the metal surface compared to the 3 and 4 positions of the ligand in Ag-PDT NCs (Fig. S3† insets). The 31P NMR spectra of PPh3 and Ag-PDT NCs are demonstrated in Fig. S5 and S6.† A peak was observed at around −5.41 ppm for the pure PPh3 ligand. The characteristic peak at 29 ppm was observed for Ag-PDT NCs. This indicates the presence of PPh3 in the NC system. The 1H NMR spectra of PPh3 and Ag-PDT NCs refurbished this idea as the characteristic proton peak of PPh3 (from 7–8 ppm) was also observed in the case of NCs (Fig. S7†).
The composition of Ag NCs was determined using the MALDI-MS and ESI-MS analysis. A broad peak at around m/z 3847 was obtained for Ag-PDT NCs, and no peak was obtained in the higher range MALDI-MS spectrum (Fig. 2A asterisk). This ensured that this is the molecular ion peak of the NCs. Almost the same value was obtained in the ESI-MS experiment, confirming the molecular ion peak assignment (devoid of some hydrogens in the Fig. 2A insets). The formula was calculated as [Ag16(PDT)8(PPh3)4] i.e., at m/z 3849.16. The other high-intensity peak (∼m/z 3705) corresponds to the decomposition of one PDT ligand and 4H, i.e., [Ag16(PDT)7(PPh3)4 − 4H+]. The TGA graph further supports this analysis, where 56% mass loss is obtained for NCs (Fig. 2B).
 | ||
| Fig. 2 (A) MALDI mass experiment with the DCTB matrix (inset represents the ESI-MS data) and (B) TGA spectra of Ag-PDT NCs. | ||
A sharp peak at around 525 nm and a small hump at around 633 nm were observed in the absorption spectra. The UV-vis-NIR absorption spectra ruled out the presence of all near or far-infrared absorption bands. The dominant peak at 525 nm is found to have the same signature as other reported Ag16 NCs from our observation.53 The emission maximum was obtained at 630 nm with 420 nm excitation (Fig. 3A). This significant Stokes shift (around 140 nm) and multiple absorption features confirmed the molecular nature of the system. A difference in absorption and PL excitation was observed due to the different origins of the absorption and emission centers. The emission at 630 nm was observed due to the involvement of surface staples (relating to the ligands) and not only involving the metal core, whereas the absorption solely depends on the metal core.39,47 Very few Ag NCs were found in the literature that give emission and are helpful in various applications. However, these Ag NCs showed emissions at a smaller size (Ag16) compared to other reported systems.54 The QY was calculated using an ethanol solution of rhodamine B (QY = 0.97) as a reference and found to be 0.23%.55 All possible control experiments (without PDT, PPh3, etc.) were performed, and the corresponding UV-vis absorption spectra are depicted in Fig. S8† to confirm the utility of the substances. The characteristic peaks (at around 525 or 633 nm) were not observed for other cases. The UV-vis and photoluminescence spectra displayed a distinct difference between the PDT-capped and pentanethiol (PT) capped Ag NCs (Fig. S9†). In the absorption spectrum, the prominent peak at around 525 nm was broadened, and the photoluminescence was not observed in the case of the Ag–PT system. Here, pentanedithiol provides significant rigidity to the system, enabling a highly structured system with less vibration and significant luminescence.56 The time-resolved luminescence decay curve of the Ag-PDT NCs is sufficiently fitted with a tri-exponential function. The average lifetime of these Ag-PDT NCs was found to be 2.19 ± 0.05 ns, having three components of 0.38 ± 0.01 ns (40%), 2.21 ± 0.04 ns (40%), and 5.78 ± 0.02 ns (20%) (Fig. 3B). This short lifetime in ns confirms that the emission mainly arises from fluorescence involving the surface states. In addition, the excitation was changed from 375 nm to 500 nm, and a tiny shift in emission spectra was noticed (Fig. S10A†). The PL decay lifetime was calculated at different excitation wavelengths, and no such change was observed (Fig. S10B†). This spectroscopic analysis confirmed the homogeneous nature of the emissive states in Ag-PDT NCs.57 The optical gap for this Ag-PDT NCs was found to be 1.75 eV using the Tauc plot (Fig. 3A insets).
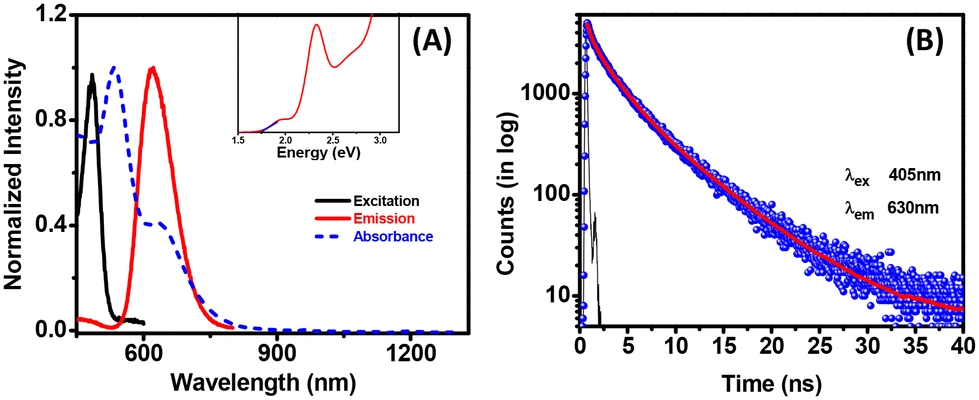 | ||
| Fig. 3 (A) Absorption, emission, and excitation spectra of Ag-PDT NCs (inset represents the Tauc plot) and (B) the PL decay curve with 405 nm excitation. | ||
The XPS of Ag-PDT NCs was analyzed to shed light on the valence state behavior of the NCs. The survey spectrum depicted in Fig. S11A† confirms the presence of constituent elements like Ag, S, O, C, etc. A peak at 368 eV is observed for Ag 3d5/2, and another is observed at 374 eV for Ag 3d3/2. The binding energy of Ag 3d5/2 lies between 367.9 eV (bulk Ag(0)) and 368.87 eV (Ag(I)), confirming that both Ag(0) and Ag(I) are present in Ag-PDT NCs (Fig. S11B†).58 In the XPS spectra of S, the S 2p3/2 and S 2p1/2 peak positions are spotted at 161.8 and 162.9 eV, respectively (Fig. S12†). The S 2p3/2 peak for unbound thiol is at around 163–164 eV. As for the S 2p3/2 peak position for Ag-PDT NCs, it is found at a slightly lower range (161.8 eV), confirming the attachment of sulfur to a silver metal surface as a thiolate species.59
The fs-transient absorption (TA) spectroscopy was analyzed at time scales of up to 8 nanoseconds (ns) to reveal the excited state dynamics of Ag-PDT NCs (in DCM). The sample was measured with 400 nm excitation at a 120 femtoseconds (fs) time resolution. The TA spectra depict prominent ground state bleaching (GSB) at around 525 nm, corresponding to the ground state absorption peak. A broad excited state absorption (ESA) was observed from 560 nm to 680 nm, and both the GSB and the ESA almost spread over the visible range. Fig. 4(A) depicts the heat map of the TA of Ag-PDT NCs. The amplitude of the GSB at 525 nm is enhanced with a delay time of up to 50 ps and subsequently decreases with time. The signal does not recover within the time window of the instrument setup. The ESA of around 610 nm is red-shifted gradually with time. In contrast, the peak position of the GSB remains constant in the total time window, as observed from the time evolution of the TA spectra in Fig. 4(B). Global fitting of the data with a sequential model ensures that three components are sufficient to fit the whole spectra in this case. The three extracted components are 664 fs, 500 ps, and >1 ns. These three evolution-associated difference spectra are described as EADS 1, EADS 2, and EADS 3, respectively, in Fig. 4(C). The time traces at the selected wavelengths are depicted in Fig. 4(D), and the population profile, which corresponds to the goodness of fit, is described in Fig. S13.† Analysing these results, we have proposed a model for the relaxation dynamics where the fast 664 fs component corresponds to the core state relaxation. In contrast, the picosecond component (500 ps) is ascribed to the core to surface state relaxation. Finally, the slowest decay greater than 1 ns corresponds to the relaxation to the ground state from the surface-related state (Fig. 5).
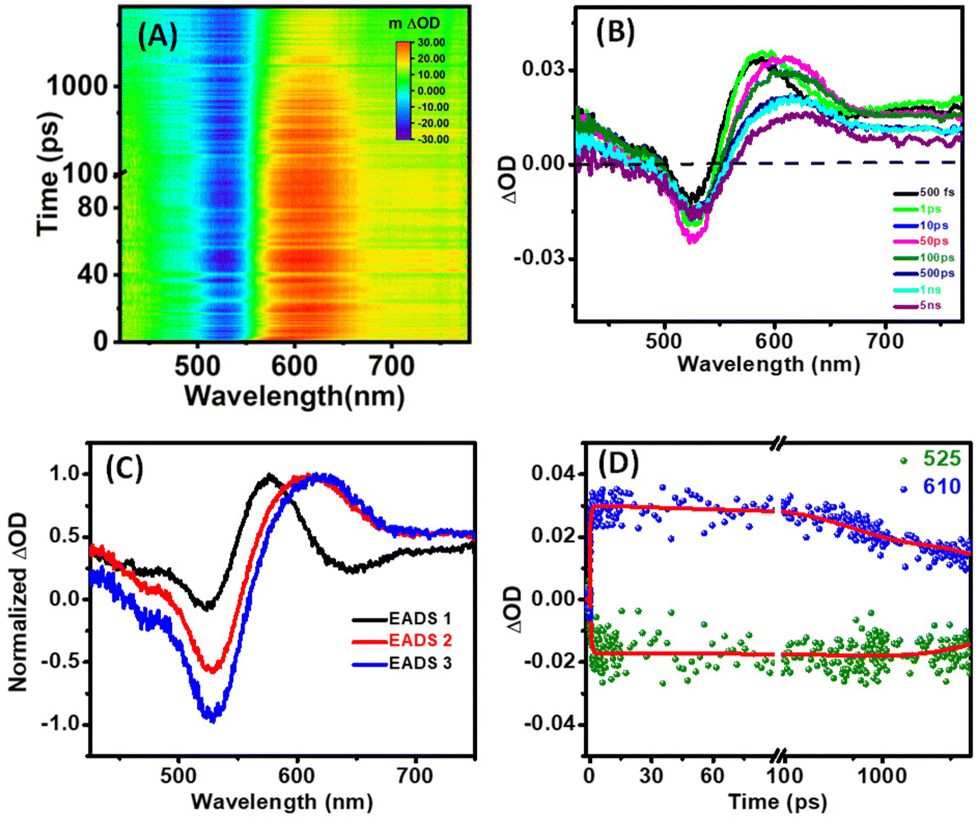 | ||
| Fig. 4 (A) 2D false color map of fs-TA data, (B) TA spectra at different time delays, (C) evolution-associated decay spectra, and (D) kinetic traces at a selected wavelength of Ag-PDT NCs in DCM. | ||
 | ||
| Fig. 5 Plausible relaxation dynamics of Ag-PDT NCS. | ||
Due to the instrument's limitations, the exact lifetime of the long-lived state is calculated from the TCSPC measurement as 2.19 ± 0.05 ns. Thyrhaug et al. studied the ultrafast dynamics of the Ag16 (DNA stabilized Ag16) system using TA and two-dimensional electronic spectroscopy (2DES). This NIR emissive system showed different ultrafast dynamics associated with a nanosecond time scale to a microsecond lifetime state.53 The tangled DNA structure may be responsible for the complex nature of the ultrafast dynamics. Hakkinen et al. theoretically analyzed the similar absorption band at around 525 nm for DNA2–Ag16Cl2 which corresponds to the simple HOMO-to-LUMO molecular transition.60
Solvent dependence on excited state dynamics
Herein, we extracted the Ag-PDT NCs in two different solvents, and we obtained the core relaxation and core-to-ligand-based surface state relaxation that are affected by the solvents. We use 400 nm excitation in fs-TA analysis for both cases, considering the interband excitation involving both the core and motif-related states simultaneously.46 As mentioned, a three-state relaxation was observed for Ag-PDT NCs in DCM with components of 664 fs (τ1), 500 ps (τ2), and >1 ns (τ3), i.e., core relaxation, core to surface state relaxation, and finally the ground state decay. The ultrafast relaxation of Ag-PDT NCs was also investigated in the DMF solvent, and the data are fitted satisfactorily in three components (Fig. S14†). However, the values are significantly different, and the three components of Ag-PDT NCs in DMF are 1.25 ps, 624.25 ps, and >1 ns, summarised in Table S1.† The kinetic traces at a selected wavelength (here 530 nm) were compared for both DCM and DMF-dissolved Ag-PDT NCs (Fig. S15†). In a previous study, we observed that in the case of Au25(PET)18, the time (τ2) decreased with increasing dielectric constant of the solvent and did not affect the core relaxation process (τ1).46 Generally, τ2 arises due to the electron relaxation within the semi-ring units (core–shell structure of MNCs), which appreciably depends on the ligand. DMF (dielectric constant 37) is a more polar solvent than DCM (dielectric constant 8.93). Here, we observed that the core and ligand-based states in DMF take more time to relax than DCM for Ag-PDT NCs. To compare the steady state behavior, the UV-vis-NIR spectra of Ag-PDT NCs in both solvents are depicted in Fig. S16.† In both cases, the distinct absorption peak at 525 nm did not shift, confirming that the structural features remain similar. However, the peak was broadened due to the dipolar aprotic nature of DMF.46,61 The QY was measured for both cases and found to be 0.23% and 0.62% for DCM and DMF solutions of Ag-PDT NCs, respectively (Fig. S17†). To eliminate the valence state involvement in the excited state behavior, we analyzed the binding energy of Ag in Ag-PDT NCs in both DCM and DMF, and there is no deviation in the 3d peak of Ag (Fig. S18†). In addition, we have analyzed previously how solvents with different dielectric constants can affect the visible emission channel as the emissive states are significantly associated with the ligand-centered charge transfer process.41 In this study, aliphatic thiol was used as the primary ligand, which is supposed to provide less polarity to the energy states than aromatic thiol-capped NCs. In general, the individual absorbing PDT-capped Ag NCs possess individual dipole and interact with the inherent dipole of the solvent.46,61 Therefore, solvents reorganize around these excited NCs dipoles, affecting the excited state dynamics. Effective charge separation was observed in the molecular system, and effective dipole–dipole interaction occurred between excited state species and solvent dipoles. This effective charge separation is more favorable in a polar solvent with a high dielectric constant, and relaxation becomes very fast compared to a less polar solvent. In this case, the capping ligand used is a non-polar type (compared to other electron-rich aromatic thiols); therefore, theoretically, we assume that the charge separation is less effective and results in dissimilar vibrational cooling of higher excited states. Hence, the relaxation requires more time in a more polar solvent as the interaction between the dipole of the excited state species and the solvent dipole is not so effective. As a result, the time required for both components (τ1 and τ2) is increased in the case of DMF solvent. The higher QY of Ag-PDT in DMF than DCM (0.63% vs. 0.23%) is also consistent with this less effective charge separation.62 This phenomenon also affects the core relaxation (τ1) and the overall lifetime obtained from the TCSPC decay curve. The average lifetime is 2.19 ± 0.05 ns in DCM, whereas it is 2.64 ± 0.05 ns in DMF solvents for Ag-PDT NCs. For the DMF solution, the luminescence of Ag-PDT NCs was stable at RT for a longer time (Fig. S19†). Only DCM and DMF solutions of Ag-PDT NCs have been studied, considering the stability of the Ag16 NCs.Conclusions
Here, a new strategy to prepare atomically precise Ag16(PDT)8(PPh3)4 NCs has been highlighted with an emission maximum at 630 nm and a QY of 0.23%. The ultrafast relaxation is favorable in the solvent with a lower dielectric constant (DCM) than the DMF solvent due to less effective charge separation. Here, the solvent effect on the excited state dynamics is demonstrated, as the solvent polarity with the ligand effects is reflected in their ultrafast behavior. This new synthetic strategy and the solvent-dependent excited state pathway investigations are essential to determine the structure–property correlation. With appropriate characterization and a complete understanding of different properties, these precise MNCs genuinely benefit various potential applications.Author contributions
A. P.: conceptualization, project administration, and supervision. S. C.: investigation, data curation, and writing – original draft. S. K.: conceptualization, formal analysis, and writing – review & editing.Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
DST-TRC is gratefully acknowledged for its research support. SC acknowledges IACS for providing fellowship. SK thanks UGC for awarding the fellowship. The authors thank IACS for its support throughout their experiment.References
- Rashi, D. Bain, A. Devi, S. Chakraborty and A. Patra, ACS Sustainable Chem. Eng., 2023, 11, 1995–2004 CrossRef CAS.
- D. Bain, S. Maity, B. Paramanik and A. Patra, ACS Sustainable Chem. Eng., 2018, 6, 2334–2343 CrossRef CAS.
- L. Ai, Z. Liu, D. Zhou, J. Liu, H. Zou, Z. Wu, Y. Liu, H. Zhang and B. Yang, Nanoscale, 2017, 9, 18845–18854 RSC.
- S. Li, J. Wei, Q. Yao, X. Song, J. Xie and H. Yang, Chem. Soc. Rev., 2023, 52, 1672–1696 RSC.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- S. Chakraborty, S. Kolay, S. Maity and A. Patra, Langmuir, 2024, 40, 317–324 CrossRef CAS.
- M. Ghosal Chowdhury, L. Sahoo, S. Maity, D. Bain, U. K. Gautam and A. Patra, ACS Appl. Nano Mater., 2022, 5, 7132–7141 CrossRef CAS.
- D. Bain, B. Paramanik, S. Sadhu and A. Patra, Nanoscale, 2015, 7, 20697–20708 RSC.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, ACS Nano, 2017, 11, 6904–6910 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- A. K. Das, S. Biswas, S. S. Manna, B. Pathak and S. Mandal, Chem. Sci., 2022, 13, 8355–8364 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- K. Kwak, V. D. Thanthirige, K. Pyo, D. Lee and G. Ramakrishna, J. Phys. Chem. Lett., 2017, 8, 4898–4905 CrossRef PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691–5695 CrossRef.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed.
- S. Kolay, S. Maity, D. Bain, S. Chakraborty and A. Patra, Nanoscale Adv., 2021, 3, 5570–5575 RSC.
- K. G. Stamplecoskie and A. Swint, J. Mater. Chem. A, 2016, 4, 2075–2081 RSC.
- Z. Liu, M. Zhou, L. Luo, Y. Wang, E. Kahng and R. Jin, J. Am. Chem. Soc., 2023, 145, 19969–19981 CrossRef CAS.
- S. Maity, D. Bain and A. Patra, Nanoscale, 2019, 11, 22685–22723 RSC.
- D. Bain, S. Maity and A. Patra, Phys. Chem. Chem. Phys., 2019, 21, 5863–5881 RSC.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS.
- S. Kolay, D. Bain, S. Maity, A. Devi, A. Patra and R. Antoine, Nanomaterials, 2022, 12, 544 CrossRef CAS.
- T.-Q. Yang, B. Peng, B.-Q. Shan, Y.-X. Zong, J.-G. Jiang, P. Wu and K. Zhang, Nanomaterials, 2020, 10, 261 CrossRef CAS PubMed.
- S. Chakraborty, D. Bain, S. Maity, S. Kolay and A. Patra, J. Phys. Chem. C, 2022, 126, 2896–2904 CrossRef CAS.
- D. Bain, S. Maity and A. Patra, Chem. Commun., 2020, 56, 9292–9295 RSC.
- M. Zhou, C. Yao, M. Y. Sfeir, T. Higaki, Z. Wu and R. Jin, J. Phys. Chem. C, 2018, 122, 13435–13442 CrossRef CAS.
- Y. Song, K. Lambright, M. Zhou, K. Kirschbaum, J. Xiang, A. Xia, M. Zhu and R. Jin, ACS Nano, 2018, 12, 9318–9325 CrossRef CAS.
- M. Zhou, C. Zeng, Y. Song, J. W. Padelford, G. Wang, M. Y. Sfeir, T. Higaki and R. Jin, Angew. Chem., Int. Ed., 2017, 56, 16257–16261 CrossRef.
- P. J. Herbert, C. J. Ackerson and K. L. Knappenberger, Jr., J. Phys. Chem. Lett., 2021, 12, 7531–7536 CrossRef PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef.
- H. Qian, M. Y. Sfeir and R. Jin, J. Phys. Chem. C, 2010, 114, 19935–19940 CrossRef.
- S. Maity, D. Bain, K. Bhattacharyya, S. Das, R. Bera, B. Jana, B. Paramanik, A. Datta and A. Patra, J. Phys. Chem. C, 2018, 122, 13354–13362 CrossRef CAS.
- W.-T. Chen, Y.-J. Hsu and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 2493–2499 CrossRef CAS PubMed.
- M. Zhou, R. Jin, M. Y. Sfeir, Y. Chen, Y. Song and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4697–E4705 CrossRef CAS.
- M. Zhou and Y. Song, J. Phys. Chem. Lett., 2021, 12, 1514–1519 CrossRef CAS.
- M. Zhou, S. Tian, C. Zeng, M. Y. Sfeir, Z. Wu and R. Jin, J. Phys. Chem. C, 2017, 121, 10686–10693 CrossRef CAS.
- S. Maity, S. Kolay, S. Ghosh, S. Chakraborty, D. Bain and A. Patra, J. Phys. Chem. Lett., 2022, 13, 5581–5588 CrossRef CAS.
- S. Kolay, S. Maity, S. Chakraborty, S. Ghosh and A. Patra, J. Phys. Chem. C, 2023, 127, 3769–3777 CrossRef.
- V. D. Thanthirige, E. Sinn, G. P. Wiederrecht and G. Ramakrishna, J. Phys. Chem. C, 2017, 121, 3530–3539 CrossRef.
- M. Zhou, S. Long, X. Wan, Y. Li, Y. Niu, Q. Guo, Q.-M. Wang and A. Xia, Phys. Chem. Chem. Phys., 2014, 16, 18288–18293 RSC.
- H. Fakhouri, E. Salmon, X. Wei, S. Joly, C. Moulin, I. Russier-Antoine, P.-F. Brevet, X. Kang, M. Zhu and R. Antoine, J. Phys. Chem. C, 2022, 126, 21094–21100 CrossRef.
- C. Yi, H. Zheng, P. J. Herbert, Y. Chen, R. Jin and K. L. Knappenberger, Jr., J. Phys. Chem. C, 2017, 121, 24894–24902 CrossRef.
- S. Kolay, S. Chakraborty, S. Pramanik and A. Patra, J. Phys. Chem. C, 2024, 128, 7643–7651 CrossRef.
- H. Yang, Y. Wang and N. Zheng, Nanoscale, 2013, 5, 2674–2677 RSC.
- L. Gell, L. Lehtovaara and H. Häkkinen, J. Phys. Chem. A, 2014, 118, 8351–8355 CrossRef PubMed.
- R. Zamiri, H. Abbastabar Ahangar, A. Zakaria, G. Zamiri, M. Shabani, B. Singh and J. M. F. Ferreira, Chem. Cent. J., 2015, 9, 28 CrossRef PubMed.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CrossRef CAS.
- Z. Wu, C. Gayathri, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2009, 131, 6535–6542 CrossRef CAS PubMed.
- J. Chen, A. Kumar, C. Cerretani, T. Vosch, D. Zigmantas and E. Thyrhaug, J. Phys. Chem. Lett., 2023, 14, 4078–4083 CrossRef CAS.
- C. Zeng, Y. Chen, G. Li and R. Jin, Chem. Mater., 2014, 26, 2635–2641 CrossRef CAS.
- Y. Song, Y. Li, M. Zhou, X. Liu, H. Li, H. Wang, Y. Shen, M. Zhu and R. Jin, Sci. Adv., 2021, 7, eabd2091 CrossRef CAS PubMed.
- M. Bodiuzzaman, E. Khatun, K. S. Sugi, G. Paramasivam, W. A. Dar, S. Antharjanam and T. Pradeep, J. Phys. Chem. C, 2020, 124, 23426–23432 CrossRef CAS.
- S. Maity, D. Bain, S. Chakraborty, S. Kolay and A. Patra, ACS Sustainable Chem. Eng., 2020, 8, 18335–18344 CrossRef CAS.
- X. Zou, Y. Lv, X. Kang, H. Yu, S. Jin and M. Zhu, Inorg. Chem., 2021, 60, 14803–14809 CrossRef CAS.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- S. Malola and H. Häkkinen, Chem. Commun., 2024, 60, 3315–3318 RSC.
- L. G. AbdulHalim, M. S. Bootharaju, Q. Tang, S. Del Gobbo, R. G. AbdulHalim, M. Eddaoudi, D.-e. Jiang and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11970–11975 CrossRef CAS.
- M. Pelton, Y. Tang, O. M. Bakr and F. Stellacci, J. Am. Chem. Soc., 2012, 134, 11856–11859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02392g |
| This journal is © The Royal Society of Chemistry 2024 |