DOI:
10.1039/D4NR02883J
(Review Article)
Nanoscale, 2024,
16, 18644-18665
Atomic-level design of biomimetic iron–sulfur clusters for biocatalysis
Received
11th July 2024
, Accepted 30th August 2024
First published on 5th September 2024
Abstract
Designing biomimetic materials with high activity and customized biological functions by mimicking the central structure of biomolecules has become an important avenue for the development of medical materials. As an essential electron carrier, the iron–sulfur (Fe–S) clusters have the advantages of simple structure and high electron transport capacity. To rationally design and accurately construct functional materials, it is crucial to clarify the electronic structure and conformational relationships of Fe–S clusters. However, due to the complex catalytic mechanism and synthetic process in vitro, it is hard to reveal the structure–activity relationship of Fe–S clusters accurately. This review introduces the main structural types of Fe–S clusters and their catalytic mechanisms first. Then, several typical structural design strategies of biomimetic Fe–S clusters are systematically introduced. Furthermore, the development of Fe–S clusters in the biocatalytic field is enumerated, including tumor treatment, antibacterial, virus inhibition and plant photoprotection. Finally, the problems and development directions of Fe–S clusters are summarized. This review aims to guide people to accurately understand and regulate the electronic structure of Fe–S at the atomic level, which is of great significance for designing biomimetic materials with specific functions and expanding their applications in biocatalysis.
 Sufei Zhou | Sufei Zhou is a Ph.D. student at the Academy of Medical Engineering and Translational Medicine, Tianjin University, under the supervision of Prof. Xiao-Dong Zhang. Her research interests focus on the design and synthesis of metal nanomaterials for biomedical applications. |
 Di Liu | Di Liu is a Ph.D. student at the Academy of Medical Engineering and Translational Medicine, Tianjin University, under the supervision of Prof. Xiao-Dong Zhang. Her current research interests are focused on the design and synthesis of nanozymes for biomedical applications. |
 Kelong Fan | Kelong Fan is a full professor at Chinese Academy of Sciences. He is interested in exploring the novel functions and applications of nanozymes in biomedicine, with top priority to design functional nanozymes by learning from nature and to develop novel strategies for disease theranostics. |
 Haile Liu | Haile Liu, received her Ph.D. degree from Tianjin University under the supervision of Professor Xiaodong Zhang, then joined the School of Chemistry and Chemical Engineering of Northwest Normal University as an associate professor. Her research interests focus on the controllable design of ultra-small metal clusters and their application in disease diagnosis and treatment. |
 Xiao-Dong Zhang | Xiao-Dong Zhang is a full professor at Tianjin University. His primary research interests are the design of smart medical materials and their application in neurological diseases, and the application of NIR-II fluorescence imaging in brain diseases. |
1. Introduction
Iron–sulfur (Fe–S) clusters, one of the oldest proteins, play a vital role in biological processes. Back in the 1960s, it was first discovered that an unknown protein could act as an electron transport medium and was soon confirmed that it contained Fe.1 Around 1965, the proteins were revealed to be a complex of Fe, S atoms, and cysteine (Cys), called Fe–S clusters. The structures of natural Fe–S clusters with minor elements are relatively simple.2,3 And the delocalization of electron density on S and Fe atoms makes Fe–S clusters well-suited for bio-mediated electron transport.4–6 Moreover, the diverse and stable ligands enable Fe–S clusters to achieve strong performance by meeting different environmental requirements.7 For example, the Cys ligands of Fe–S clusters interact with the H atoms of peptides or water via S atoms, affecting the redox process of the clusters in the organism and achieving electron transfer.7 Recently, more and more discoveries about the biological functions of Fe–S clusters have been reported, such as electron transport,8–11 catalysis,12,13 cluster assembly,11,14–17 biosensing,18–23 metabolic pathway regulation,24,25etc. It has been shown that Fe–S clusters are being developed to be used in the biomedical field.16,18,26–28 However, due to the complex physiological environment, it is difficult to accurately reveal the relationship between the structure and functions of Fe–S clusters. Therefore, the structure–activity relationship needs to be investigated in vitro and appropriately modified to adapt to various application scenarios.
In general, there are two ways to obtain Fe–S clusters, including biological extraction and chemical synthesis, but the synthesis of Fe–S clusters in biological systems presents several challenges. Firstly, the process requires precise environmental conditions, such as optimal pH, temperature, and ion concentrations,29,30 which are difficult to control within a living organism. Additionally, the formation of Fe–S clusters involves the coordinated action of various enzymes and ligands,31 making the biosynthetic pathways and regulatory mechanisms highly complex and finely tuned. The stability of Fe–S clusters is another major issue, as they are susceptible to oxidation and reduction reactions.32–34 There is still limited understanding of the synthesis and assembly of Fe–S clusters, complicating their reconstruction in vitro. Consequently, the direct chemical synthesis method was well received. Holm prepared the first artificial [Fe4S4] cluster in the 1970s,35 and since then, many Fe–S clusters based on Fe–S structure have emerged. For example, the Fe6RHH-cluster ([Fe6S9(SEt)2]4−), the Mo-cluster ([(η5-pentamethylcy-clopentadienyl)MoFe5S9(SH)]3−),36 [Fe7S8] nanoflower37 and [MoFe3S4] cluster38 are adapted from the Fe–S clusters. There are antituberculosis drugs based on the Rieske-like structure [Fe2S2] cluster.39 In addition to the basic skeleton, the researchers have also examined the arrangement of ligands around Fe–S and found that the use of 3-Cys and 1-His attached to the [Fe2S2] cluster helps regulate the stability of the cluster.40,41 Based on functional modifications, Xie et al. incorporated FeS into bovine serum albumin (FeS@BSA) to generate reactive oxygen species (ROS) for tumor therapy.42 Using the same mechanisms of ROS, our group has developed a series of artificial nanoclusters that can inhibit ROS for the treatment of brain injury as well as inflammation in the body, including atom-precise Au25 clusterzymes,43,44 carbon nanozymes,45 trimetallic nanozyme,46 hollow PtPdRh nanocubes,47 Pt or Cr doped CeO2 nanozymes, etc.48,49 The above work gives us a clue to accurately understand the relationship between the structure and biological function of Fe–S clusters. However, these synthetic Fe–S cluster analogs are limited in terms of function and are not universal in interpreting their structure–activity relationships. Until now, the catalytic mechanism, structure–activity relationship and applications of Fe–S clusters have been poorly understood. The systematic presentation of Fe–S clusters is also scarce, which poses the challenge of precisely regulating the structure of the Fe–S clusters to realize future functionalization. In this review, the basic structure and catalysis of Fe–S clusters formation are discussed. Various strategies for the design of biomimetic Fe–S clusters, including configuration design, element doping and ligand regulation are presented. And the effect of structure on catalytic performance is clearly explained from the perspective of electron transfer. What's more, the progress of Fe–S clusters in biocatalytic fields, involving tumor therapy, antibacterial, viral suppression, and plant photoprotection is also listed. Finally, the problems and development direction of the Fe–S cluster are summarized. This review attempts to elucidate the connection between the structure and function of Fe–S clusters at the atom level, provide more theoretical design ideas for the precise synthesis of Fe–S clusters in the future, and achieve functional selective design.
2. The main structural types of Fe–S clusters
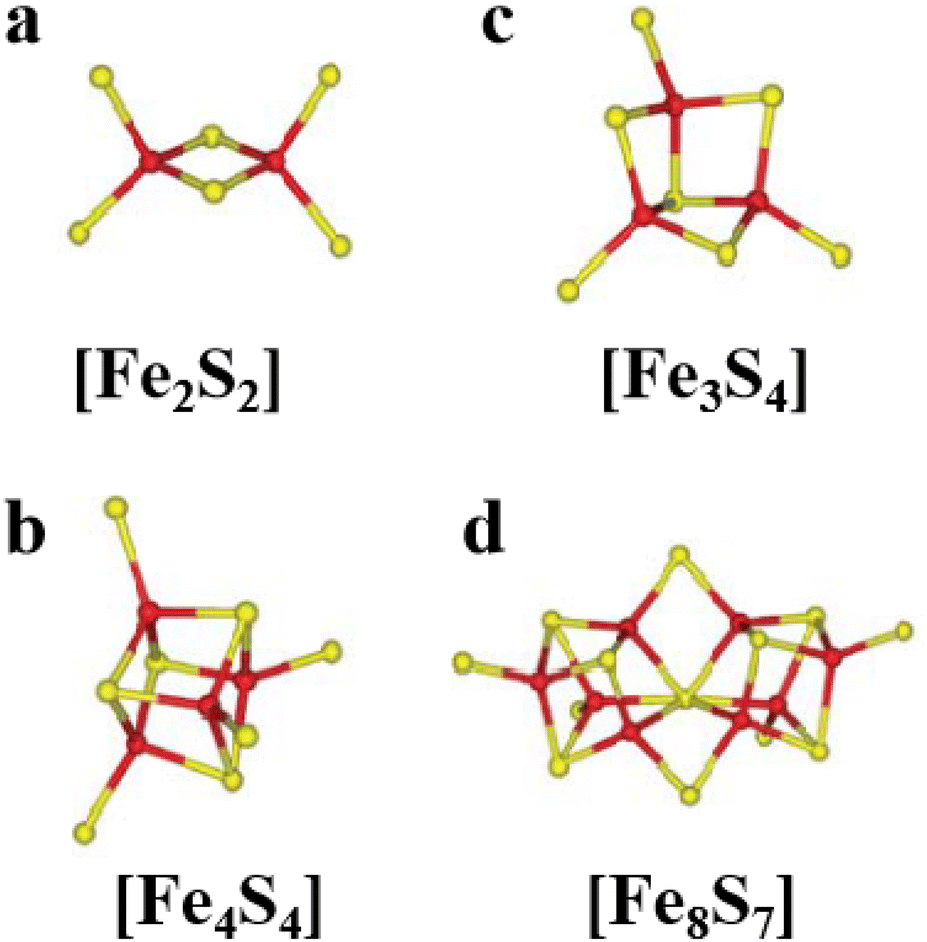
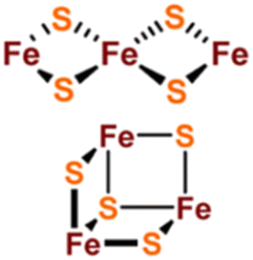
[Fe2S2], [Fe3S4] and [Fe4S4] clusters are common Fe–S clusters. The ligands in these clusters present in a typical homocysteine form,1,50 where the individual Cys can be removed or replaced by histidine (His), thereby deviating from the homocysteine form.5,28 For instance, the [Fe2S2] cluster behaves as the simplest polymetallic system among the Fe–S clusters. It consists of two Fe ions bridging two S ions to form a core part (Fig. 1a), which is blocked by four Cys ligands.2 Other common ligand forms include two Cys and two His, as well as three Cys and one His coordination. [Fe2S2] clusters exist in two oxidation states, [Fe2S2]2+ and [Fe2S2]+. Additionally, the [Fe4S4] cluster with four Fe ions and four S ions is the most widely used polymetallic system of Fe–S clusters,51 which is also the most classical cubic cluster (Fig. 1b). Others are linear structures containing four or three Cys ligands.50 The oxidation states of [Fe4S4] clusters add the all-iron state ([Fe4S4]0) and the highly oxidized state ([Fe4S4]3+) compared to [Fe2S2] cluster. [Fe3S4] cluster can be simply considered as [Fe4S4] cluster with one less iron in the core (Fig. 1c).50 The oxidation states of [Fe3S4] clusters include [Fe3S4]+, [Fe3S4]0, [Fe3S4]− and [Fe3S4].3,52 In addition, [Fe4S4] clusters may be part of P ([Fe8S7]) -clusters (Fig. 1d), which are the more complex Fe–S clusters in present-day organisms. The oxidized states contain both [Fe8S7]3+ and [Fe8S7]2+.53,54 Generally, the simple Fe–S clusters with various oxidation states have different electronic and magnetic properties, which contribute to the functional diversity and biological behavior of Fe–S clusters (Table 1).
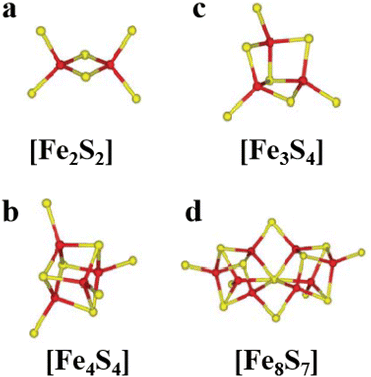 |
| | Fig. 1 Structure of common natural Fe–S clusters. The structure of (a) [Fe2S2], (b) [Fe3S4], (c) [Fe4S4] and (d) [Fe8S7] clusters. Reproduced with permission from ref. 14. Copyright 2005, Annual Reviews. | |
Table 1 The main structure types and physicochemical properties of Fe–S clusters
| Fe–S clusters |
Structure |
Oxidation state |
Spin state |
Function |
Example |
Ref. |
| [Fe2S2] |

|
[Fe2S2]2+, [Fe2S2]+ |
S = 0 S = 1/2 or S = 9/2 |
Electron transfer/coupled electron/regulation of gene expression |
Ferredoxins; redox enzymes/Rieske protein/SoxR; IscR |
29 and 143 |
| [Fe3S4] |
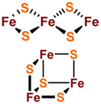
|
[Fe3S4]+, [Fe3S4]0, [Fe3S4]−, [Fe3S4]2− |
S = 1/2, S = 2, S = 5/2 |
Electron transfer |
Ferredoxins; redox enzymes |
143
|
| [Fe4S4] |
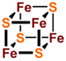
|
[Fe4S4]3+, [Fe4S4]2+, [Fe4S4]+, [Fe4S4]0 |
S = 1/2, S = 0, S = 1/2 or S = 3/2, S = 4 |
Electron transfer/substrate binding and activation/structural/regulation of gene expression |
Ferredoxins; redox enzymes/(de)Hydratases/Endonuclease III/FNR |
1
|
| [Fe8S7] |
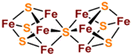
|
[Fe8S7]3+, [Fe8S7]2+ |
S = 1/2 or S = 5/2, S = 0 |
Coupled electron/proton transfer |
Nitrogenase |
15 and 144 |
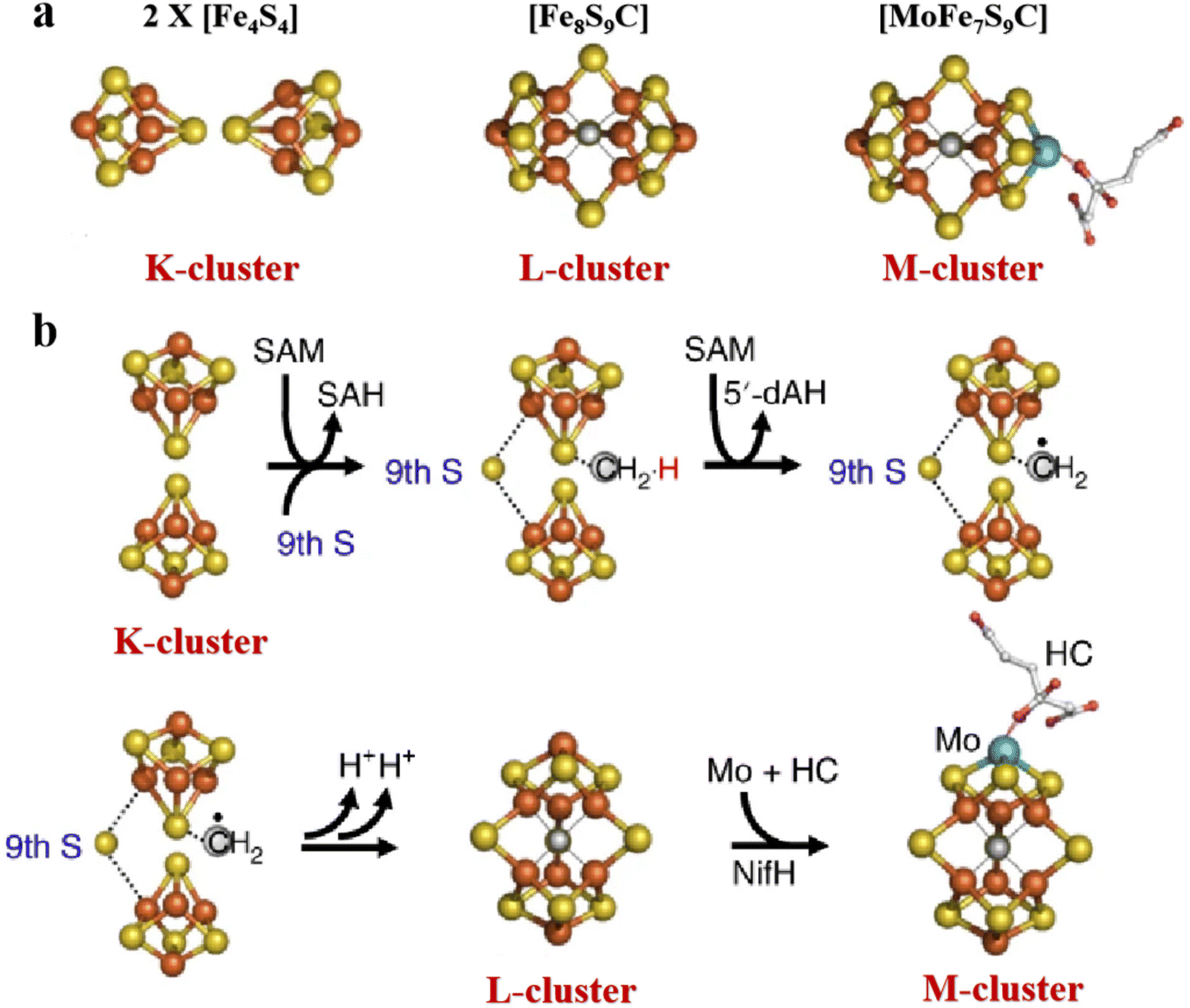
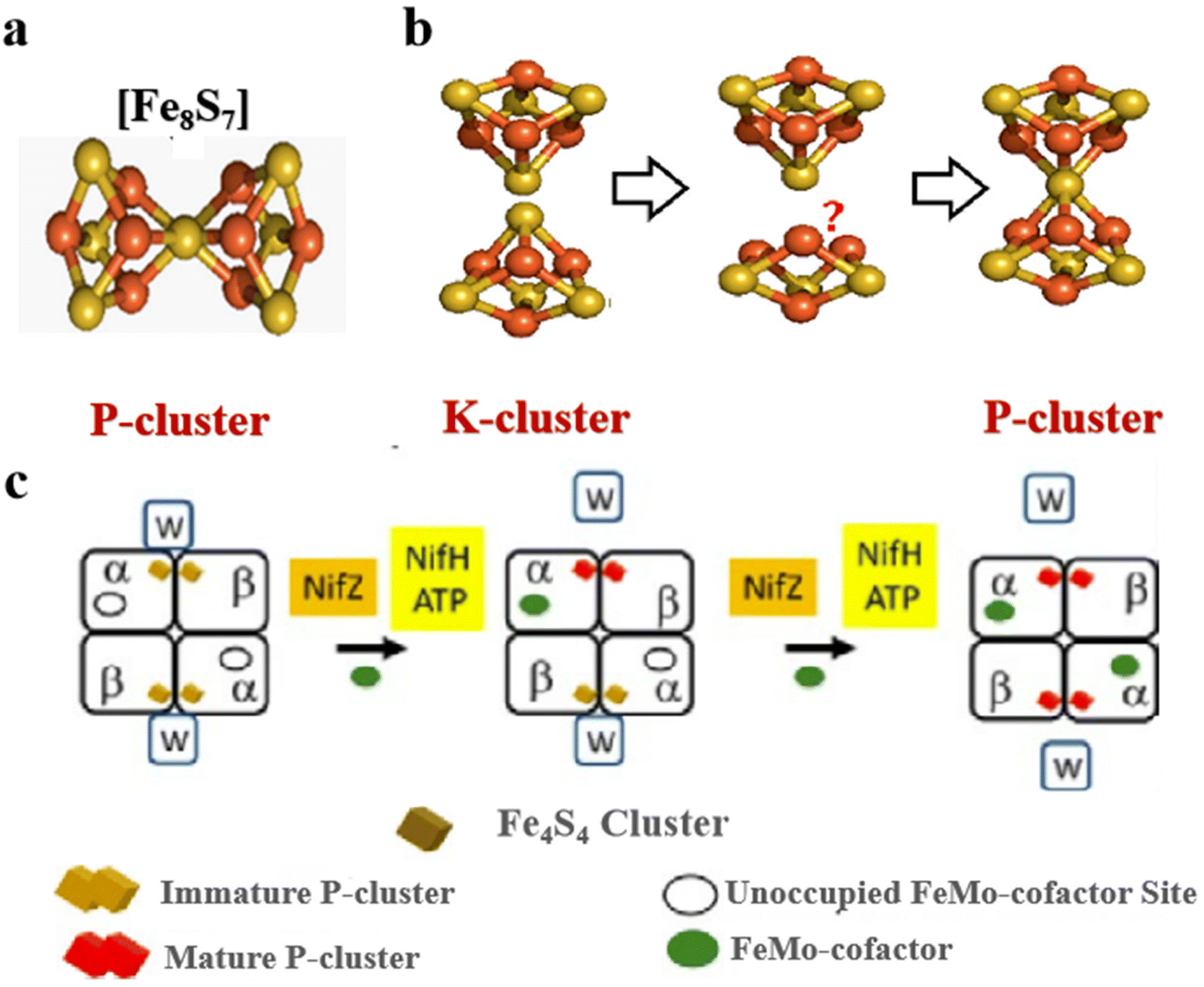
The most complex Fe–S clusters are almost present in nitrogenase in living organisms. With a deeper exploration of nitrogenase, an interesting phenomenon has attracted attention. The complex Fe–S clusters in nitrogenase can be assembled from the [Fe4S4] cluster. Tanifuji and co-workers researched the whole process of [Fe4S4] cluster assembly to form the M ([(homocitrate) MoFe7S9C]) -cluster, one of the significant catalytic cofactors in nitrogenase.16,28 And explained the origin and insertion order of the ninth “S”.14 Specifically, K-cluster (2 × [Fe4S4]) uses the C source provided by S-adenosyl-l-methionine (SAM)-cluster to link Fe in the two [Fe4S4] clusters, and SAM was further converted to 5′-deoxyadenosyl radical (5′-dAH). Subsequently, sulfite was inserted into the K-cluster containing C as the ninth “S” source to produce the L (Fe8S9C) -cluster.15 An apical Fe of the L-cluster in the assembly protein will be replaced by Mo, which then connects the homocitrate to become a mature M-cluster transferred to the assembly protein binding site (Fig. 2a and b).55,56 In addition to the M-cluster, which can be formed by the assembly of the [Fe4S4] cluster in the catalytic cofactor nitrogenase, another important catalytic P-cluster can be viewed as the coupling of two [Fe4S4] clusters (Fig. 3a and b). It is reported that P-cluster assembly occurs at the α–β interface of the assembly protein.57,58 NifW, NifZ and NifH assembly factors convert two immature P-clusters into mature P-clusters. NifW binds to the immature P-cluster in this process and subsequently dissociates from the mature P-cluster. Notably, the maturation of either the first or second P-cluster is equivalent (Fig. 3c).59 However, the formation process of the two P-clusters was different. The first P-cluster formed in 5 min, and the other one formed slowly over 2 h from a pair of [Fe4S4] clusters. In the process, a precursor [Fe4S4]+ is first converted into a [Fe4S3–4] cluster. Then, an antimagnetic cluster with the possible structure [Fe8S7–8]β is formed. The final transformation into mature P-clusters.17,60–63 It is worth mentioning that M-cluster and P-cluster are essential in catalyzing N2 reduction.64 Among them, the M-cluster provides the N2 catalytic binding site, while the P-cluster mainly mediates the intermolecular electron transfer to the M-cluster and thus realizes the reduction process. The biological processes of Fe–S proteins involve their synthesis, assembly, and function in the cell.65,66 First, the biosynthesis of Fe–S proteins begins with the synthesis of their precursors and the assembly of Fe–S clusters into proteins via specific biosynthetic pathways.67,68 Next, Fe–S proteins perform key functions within the cell, including participation in electron transfer, catalyzing reactions, and regulating cellular metabolism.69 These proteins typically function in the mitochondria, plastids, and cytoplasm of the cell.70,71 The function of Fe–S proteins is dependent on the structural stability and electron-transferring ability of their iron-sulfur clusters, and the assembly and repair of these clusters are tightly regulated.
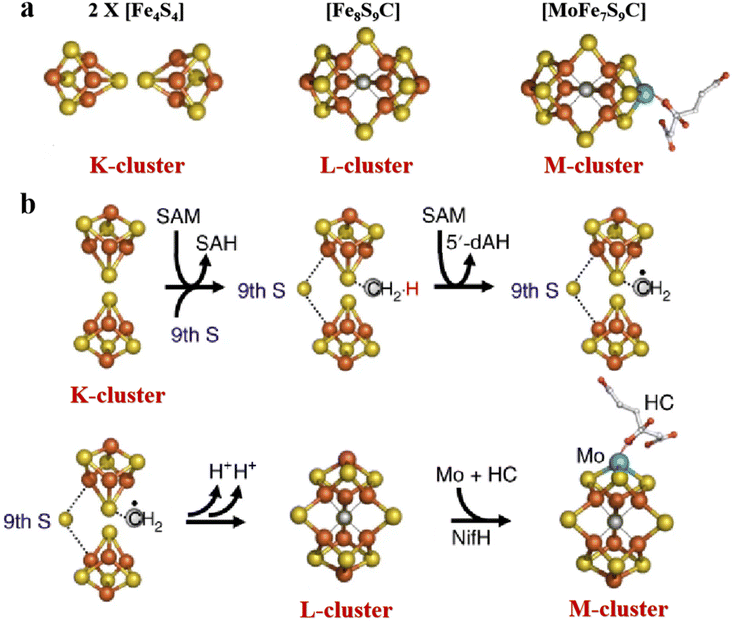 |
| | Fig. 2 Structure of complex natural Fe–S clusters and assembly of M-clusters. (a) Structures of K-, L- and M-clusters. Fe, orange; S, yellow; Mo, cyan; C, gray; N, blue; O, red. (b) M-cluster assembly process and its corresponding structure. Reproduced with permission from ref. 15. Copyright 2018, Springer Nature. | |
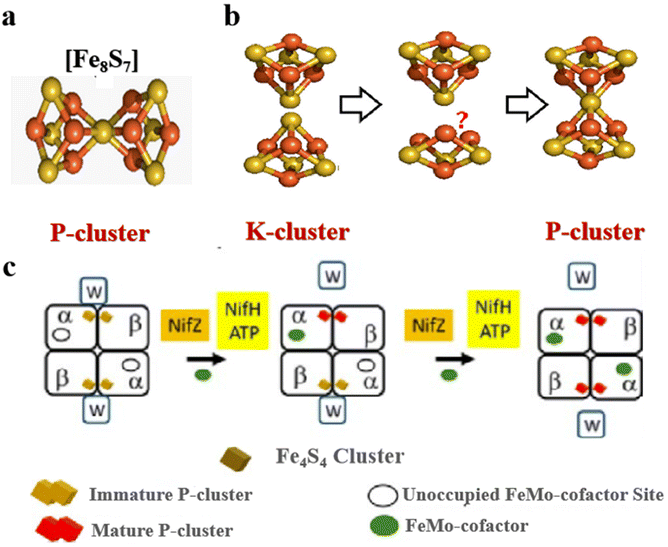 |
| | Fig. 3 Structure of complex natural Fe–S clusters and assembly of P-clusters. (a) Structures of P-clusters. Fe, orange; S, yellow; Mo, cyan; C, gray; N, blue; O, red. Reproduced with permission from ref. 61. Copyright 2018, American Chemical Society. (b and c) P-cluster assembly process and its corresponding structure in the organism. Reproduced with permission from ref. 59. Copyright 2019, ScienceDirect. | |
3. Catalytic mechanisms of Fe–S clusters
The excellent redox properties of Fe–S clusters make them indispensable in most biological processes.72 By examining the catalytic mechanism and the Fe–S clusters pathway, we can further regulate the structure of Fe–S clusters to construct stable Fe–S cluster systems for biomedical applications. Here, the mechanism of Fe–S clusters in plants, viruses and other organisms is introduced. The catalytic mechanisms and pathways of the Fe–S clusters for dipeptide biosynthesis and carbon dioxide (CO2) reduction are presented, and the general mechanisms of electron transfer in the Fe–S cluster redox processes are summarized. Fe–S clusters are widely present in functional proteins of organisms. When inhibitors are present (such as nitric oxide (NO), strong light, etc.), the Fe–S clusters in functional proteins of organisms undergo electron transfer with specific inhibitors, thus affecting the function of the organism. Dph2 is one of the critical enzymes for diphthamide synthesis. The reaction pathway and energy distribution of the Fe–S clusters in Dph2 were calculated by molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) calculations. The introduction of the elongation factor 2 (EF2) histidine residue led to the cleavage of the reductive S–C(γ) in the Fe–S clusters ligand to generate the 3-amino-3-carboxypropyl (ACP) radical species (Fig. 4aI). The structure of ACP is shown in Fig. 4b. Further, EF2 histidine residues bound to ACP via C–C coupling to form the ACP-His radical (Fig. 4aII). In the third step, the histidine proton is transferred to the residue and the electron is transferred to the Fe–S clusters, producing the dipeptide product (Fig. 4aIII). Additionally, the Fe atom attached to the ligand may be called Fe4, and the remaining three Fe atoms are numbered Fe1, Fe2 and Fe3. Feng found that the transition of S–C(γ) components strongly depends on the electron spin structure of the Fe–S class, and the electron transfer via the Fe1–Fe2–Fe3–Fe4 model to the Fe4 to C(γ) transfer. The entire Fe1–Fe2–Fe3 process acts as an electron reservoir. Fe4 regions are responsible for substrate coordination (Fig. 4c).73,74
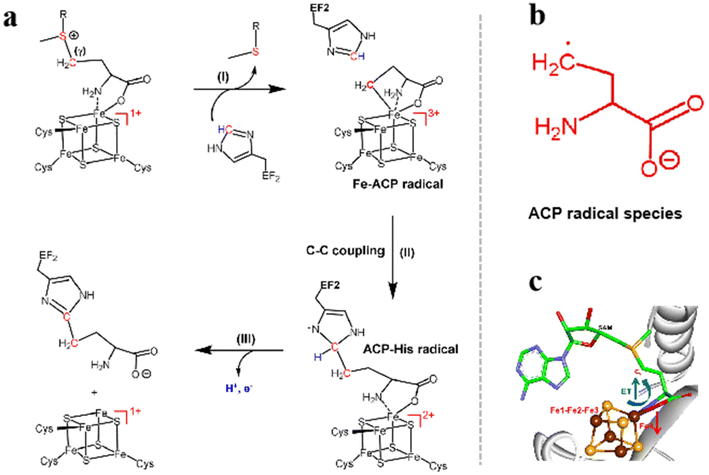 |
| | Fig. 4 The catalytic mechanism of Fe–S clusters catalyzing dipeptide synthesis. (a) Mechanism of synthesis of diphthamide by Dph2. (b) The structure of 3-amino-3-carboxypropyl (ACP) radical. (c) Spin-regulated electron transfer in Fe–S clusters-mediated redox reactions. Reproduced with permission from ref. 73. Copyright 2021 Wiley-VCH GmbH. | |
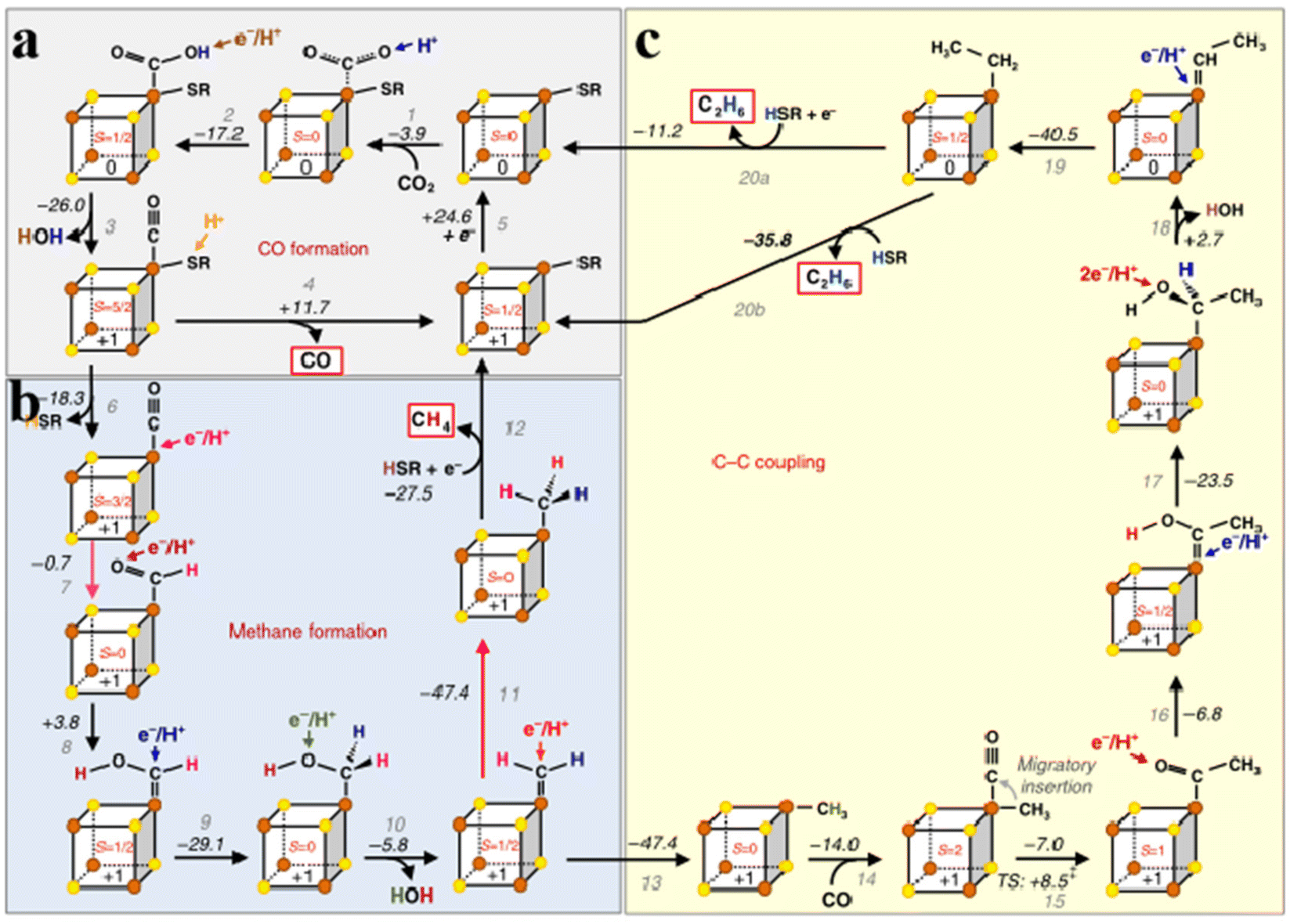
In a typical CO2 reduction process, Stiebritz proposes a possible way to reduce CO2 that can be divided into three parts (Fig. 5a).26,75 During CO2 reduction, CO2 binds to the [Fe4S4] cluster and produces a small amount of heat (Fig. 5a step 1). Electron transfer removes a water molecule and forms a [Fe4S4]1+ cluster in the form of connected CO (Fig. 5a, steps 2 and 3). After the dissociation of CO, [Fe4S4]1+ cluster is reduced to [Fe4S4]0 (Fig. 5a, steps 4 and 5). The reduction and proton-induced dissociation branches from step 3 depend on the thiol ligands at the same Fe center (Fig. 5a, step 6). Second, the proton-coupled electron is transferred to the bound CO site to form an aldehyde-like species, followed by a series of proton transfers and dissociation of water molecules, finally resulting in the binding of [Fe4S4]+ to CH3 (Fig. 5b, step 7–11). The free thiol molecules donate a proton to obtain CH4, and the thiol ligand rebinds to [Fe4S4]+ (Fig. 5b, step 12). C–C coupling relies on the activation of C1. When C1 binds to the [Fe4S4]+ cluster, the CO attached to the Fe center bound to C1 to form a Fe acetyl intermediate (Fig. 5c, steps 13–15). After a series of proton transfers, exothermic dehydration and Fe-bound ethyl species are combined (Fig. 5c, steps 16–19). Apparently, the dissociation of water results in the cluster conversion from the reduced state [Fe4S4]1+ to the all-ferrous state [Fe4S4]0. C2H6 is removed using a new thiocluster similar to C1. Whether to add an extra electron can be divided into two steps (Fig. 5c, steps 20a, 20b). Furthermore, the C–C bond has a synthetic pathway similar to that of CH3 and requires protonation competition between the two pathways, and protonation can occur at any stage. The CH4 formation process is shorter and CH4 reaction intermediates are more accessible, thus promoting CH4 production. Therefore, the yield of CH4 is higher than that of C2H6. These findings demonstrate the reliability of this approach.76–83 Overall, Fe–S clusters utilize electron spin and exchange to regulate redox in both biosynthesis and CO2 reduction, which is universal for understanding the redox mechanisms of various metalloenzymes.73 Understanding the origin of catalytic activity and structural design of FeS nanozymes is instructive for the rational design of mimetic Fe–S clusters. Fan et al. constructed pyrite (FeS2) nanozymes for tumor therapy.84 FeS2 possesses stronger peroxidase (POD)-like activity compared to Fe3S4 nanozymes. Density functional theory (DFT) calculations show that the POD-like activity of FeS2 mainly originates from the combination of Fe(II) on the surface of FeS2 with hydrogen peroxide to form an intraorbital complex, which leads to a smaller energy barrier for the base-like decomposition of hydrogen peroxide (H2O2).85 In addition, the inherent glutathione (GSH)-oxidase (OXD) activity of FeS2 nanozymes can oxidize GSH to oxidized glutathione (GSSH) and H2O2, which would produce abundant H2O2 and depleted GSH, thus causing tumor cell death.86 In their work, FeS2 nanozymes were further modified with -polyethylene glycol (PEG)-folic acid (FA), which increased water solubility while enabling the nanozymes to target tumor cells. Based on the ultra-high antibacterial properties of metal inorganic sulfides, Gao et al. used a nanotransformation strategy to convert organic sulfur compounds into inorganic nano-iron sulfur compounds (nFeS). The antibacterial properties of this inorganic compound were 500 times higher than those of natural sulfides in garlic, and the activity originated from the hydrogen polysulfanes released during the oxidation of nFeS to iron oxide. Not only that, the addition of H2O2 enhances the release of hydrogen polysulfanes.87 These studies provide new ideas for the design and application of bionic Fe–S clusters.
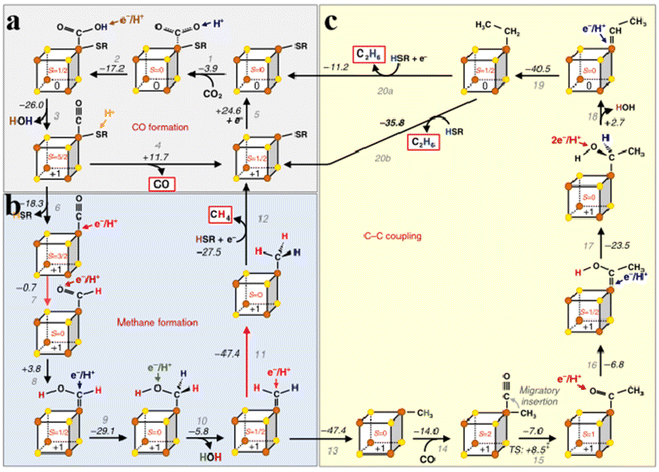 |
| | Fig. 5 Mechanism of CO2 reduction catalyzed by Fe–S clusters. (a) Fe–S clusters catalyze CO formation. (b) Fe–S clusters catalyze methane formation. (c) Fe–S clusters catalyze C–C coupling. Reproduced with permission from ref. 26. Copyright 2018, Springer Nature. | |
4. Bionic structure design strategy of Fe–S cluster
4.1 Artificial Fe–S clusters based on configuration design
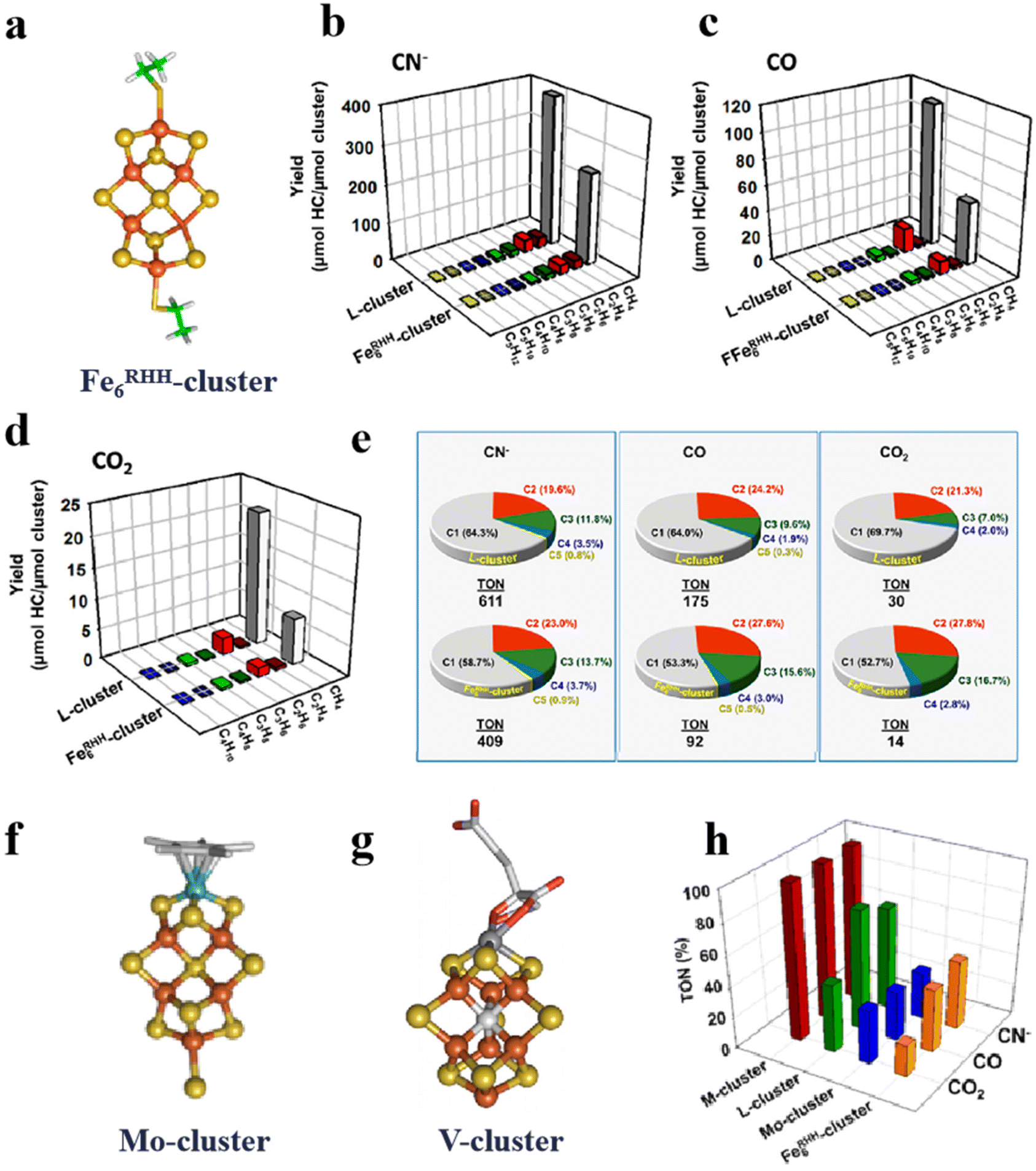
The artificial Fe–S clusters have the advantage of controlling the molecular structure. The performance of the Fe–S clusters by structural design is the major method in their development. As we all know, nitrogenase can fix nitrogen,38,51,88 and L-cluster and M-cluster can also reduce CO2 and CO. Due to the limitations of their synthesis, several Fe–S-like clusters have been designed and coordinated according to their active centers.89,90 Sickerman et al. constructed the Fe6RHH-cluster, which is homologous to the L-cluster and has a similar topological structure to the M-cluster (Fig. 6a).36 They suggest that both the L-cluster and M-cluster have three pairs of Fe atoms bonded to three S, which are considered to be catalytic for the L-cluster.91 Depending on the site structure of the L-cluster, the Fe6RHH-cluster with two pairs of S-bridged Fe atoms is constructed. And S in the center is similar to the single C atom in the L-cluster. Such a structure allows the introduction of the Fe6RHH-cluster as a catalytic component to the nitrogenase. As shown in Fig. 6b–d, comparing the catalytic activity of Fe6RHH-cluster with those of natural L-cluster, Fe6RHH-cluster also catalyzes the reduction of cyanide ions (CN−), CO and CO2 to C1–C5 hydrocarbons,36,92 especially CH4. In addition, Fig. 6e depicts the product occupancy and turnover numbers (TONs; calculated based on μmol total C in hydrocarbons/μmol cluster) for the hydrocarbon-forming of the L- and Fe6RHH-clusters. Fe6RHH-cluster appears to be more conducive to the formation of a higher proportion of long-chain hydrocarbons than L-cluster. Another Mo-dependent cofactor is a Mo (homocitrate MoFe5S10)-cluster, which is a complex of Mo bound to a homocitrate ligand (Fig. 6f). It has the same core configuration and catalytic activity as the Fe6RHH-cluster.92 In another example, the V (VFe7S9C)-cluster mimics the overall structure of the M-cluster (Fig. 6g).36 The catalytic reduction capacity of M-, L-, Mo-, and Fe6RHH-clusters for CN−, CO, and CO2 was compared by calculating the total TON (Fig. 6h).36 Unsurprisingly, the M-cluster exhibits strong catalytic activities for the reduction of CN−, CO and CO2.38 It illustrates that homocitrate and Mo addition are necessary and selected by the evolutionary process. The L-, Mo-, and Fe6RHH-clusters can selectively reduce substrates. The L-cluster tends to reduce CO and the Mo-cluster has an overall TON distribution of about 20%. Although the Mo-cluster contains Mo atoms, the Mo atoms are surrounded by larger citrates and Fe, which may affect the behavior of the substrate for Mo-active centers.92 However, the TON for the reduction of CN− by Fe6RHH-cluster is higher than that of CO and CO2, up to 30%, even higher than that of Mo-cluster. It suggests the catalytic reduction reaction of the Fe–S clusters independent of the center C.
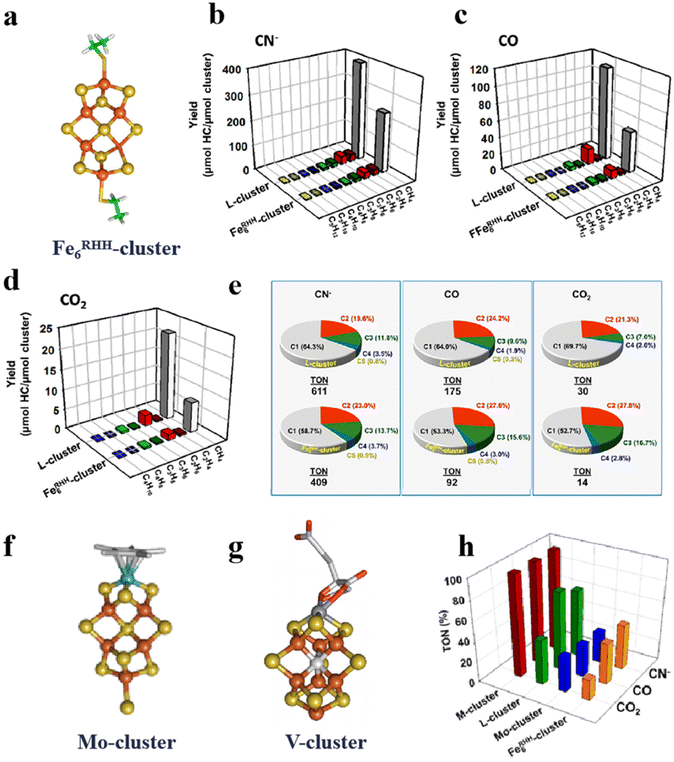 |
| | Fig. 6 Artificial Fe–S clusters based on configuration design to improve catalytic performance. (a) Structure of Fe6RHH-cluster. Types and yields of L- and Fe6RHH-clusters for the reduction of (b) CN−, (c) CO and (d) CO2 to hydrocarbons. (e) L- and Fe6RHH-clusters percentage of products forming hydrocarbons. (f) Structure of Mo-cluster. (g) Structure of V-cluster. (h) TON of M-, L-, Mo- and Fe6RHH-clusters for the reduction of CN−, CO and CO2. Reproduced with permission from ref. 36. Copyright 2017, American Chemical Society. | |
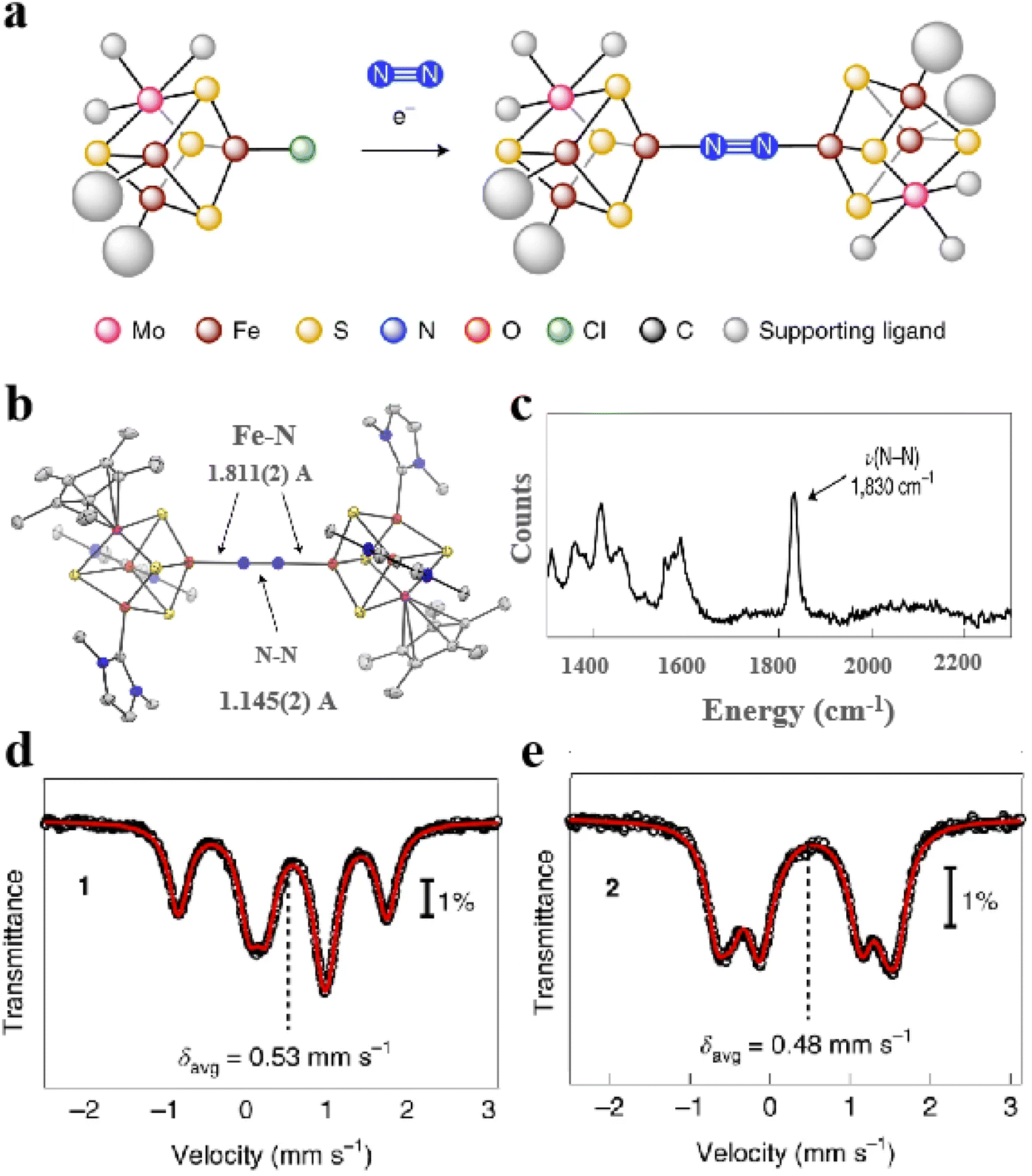
Moreover, artificial Fe–S clusters designed by conformational design can simulate not only the catalytic activity of natural Fe–S clusters but also some artificial Fe–S cluster reactions used in nitrogenase research.93 For example, a [MoFe3S4] cluster similar to the [Fe4S4] configuration can form definite coordination with N2 (Fig. 7a). Through X-ray crystal structure analysis, the N2 ligand is located in the inverted center of the crystal and binds to the Fe sites of two [(η5pentamethylcyclopentadienyl)-MoFe3S4(IPr)2] cubic (Fig. 7b). Raman spectrum was also observed for the N–N stretching vibrations (1830 cm−1) (Fig. 7c). Mössbauer spectra show that the average Fe–S distances decrease modestly after the [MoFe3S4] cluster binds N2. The covalent interaction of N2 with Fe–S clusters indicates the fixation process of N2 (Fig. 7d and e).38 Overall, the design of Fe–S clusters with high efficiency, specificity and biocompatibility based on the conformation is instructive for advancing the field of biomaterials science and biocatalytic applications.
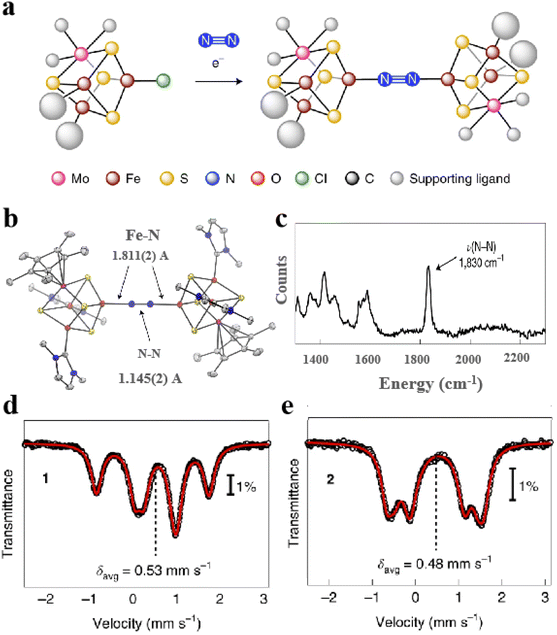 |
| | Fig. 7 Artificial Fe–S clusters based on configuration design are used to study the mechanism. (a) Synthesis of [MoFe3S4]2-N2 cluster. (b) X-ray crystal structure of [MoFe3S4]2-N2 cluster. (c) Raman spectra of [MoFe3S4]2-N2 cluster. (d) 80 K Mössbauer spectra of MoFe3S4Cl cluster and (e) [MoFe3S4]2-N2 cluster. Reproduced with permission from ref. 38. Copyright 2021, Springer Nature. | |
4.2 Artificial Fe–S clusters based on element doping
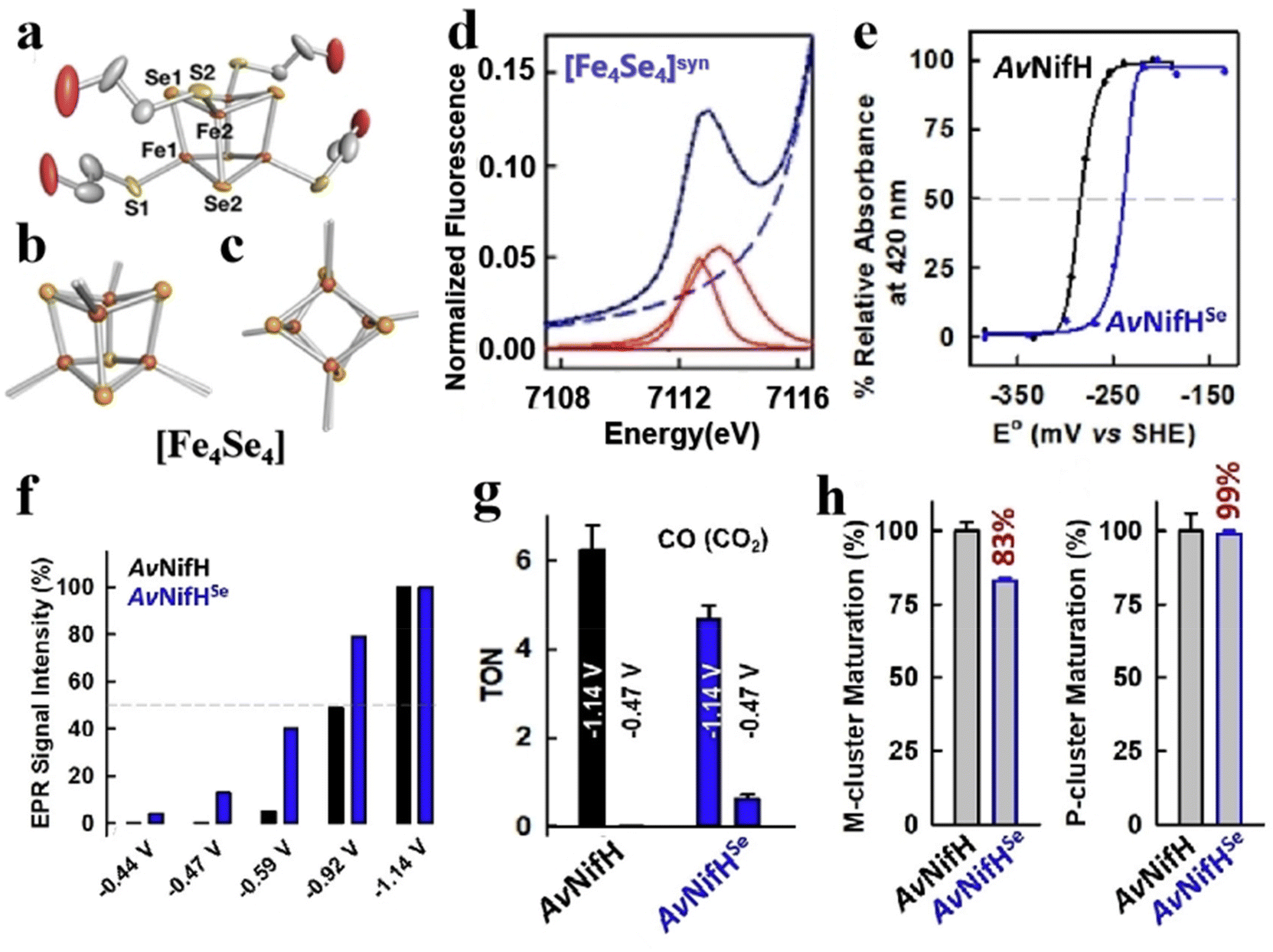
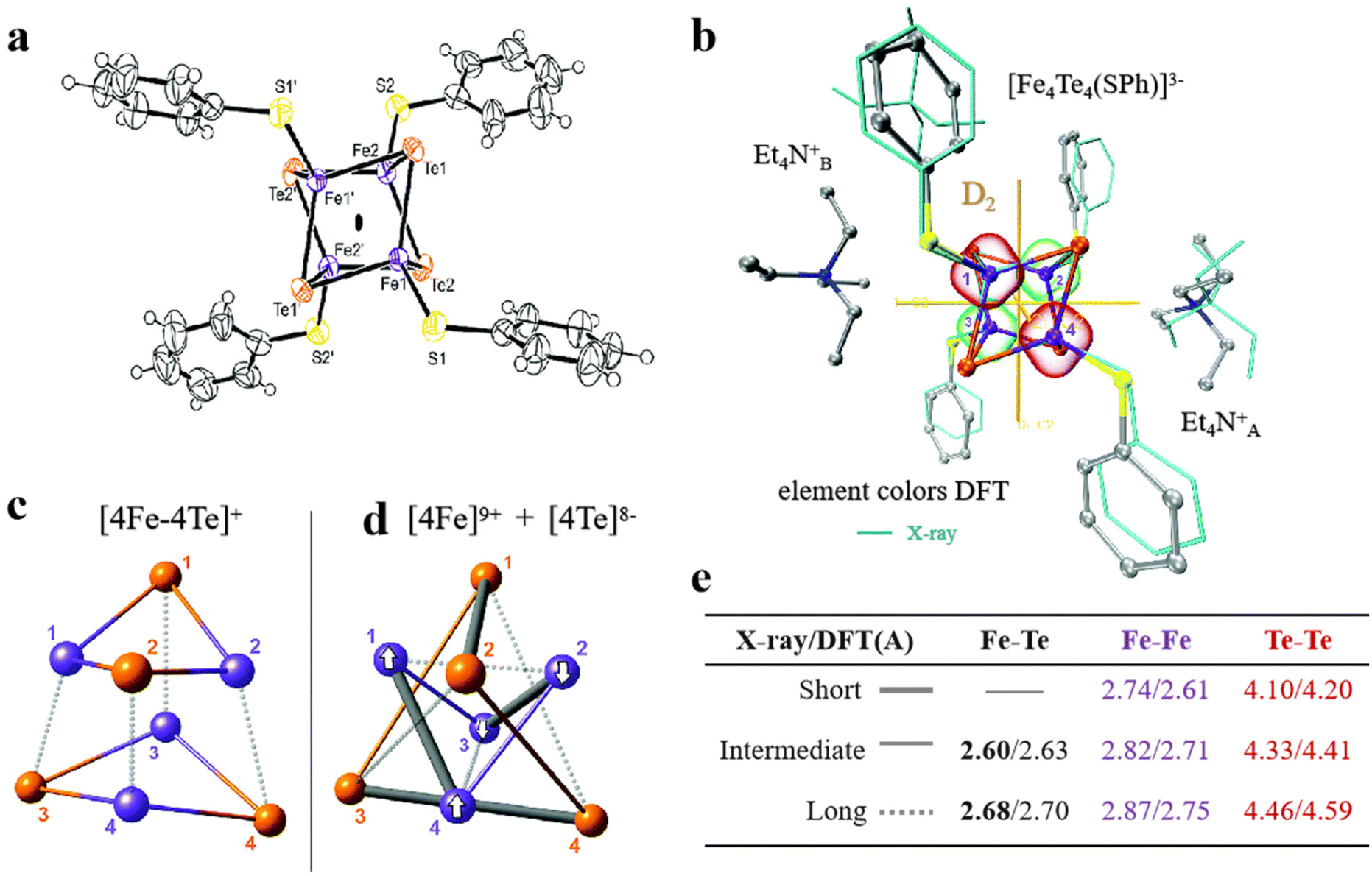
Selenium and tellurium are sulfur congeners, so the idea of replacing sulfur with selenium or tellurium is predictable. Solomon et al. successfully synthesized a water-soluble [Fe4Se4] cluster, whose structures were contrasted to the overlapping [Fe4S4] cluster as depicted in Fig. 8a–c. There is not much difference in terms of general structure or core portion, it still shows a cubic core structure. Compared with the Fe K-edge X-ray absorption spectroscopy (XAS) of the [Fe4S4] cluster, the deviation of the metal center from the symmetry center in the [Fe4Se4] cluster leads to an increase of 4.5 units in the three-dimensional jump intensity of the region below the leading edge (Fig. 8d).2 From the various structures and properties, it is clear that the Se substitution does not cause much change in the structure of the [Fe4S4] cluster, and the [Fe4Se4] cluster still has three redox states. However, the data were slightly different and their reaction properties varied significantly, which can be verified by measuring the midpoints between the three redox states of [Fe4S4] and [Fe4Se4], respectively. Midpoint potentials of [Fe4S4]1+/2+ and [Fe4Se4]1+/2+ were determined to be −240 mV and −285 mV by redox titration (Fig. 8e). Fig. 8f utilizes the electron paramagnetic resonance (EPR) data to represent the midpoint potentials of [Fe4S4]0/1+ and [Fe4Se4]0/1+. Since it is difficult to find the reduction potential where the EPR signal of the all-iron state (g = 16.4) reaches half of the maximum value, it is considered to be a midpoint potential.82,94,95 The [Fe4Se4] cluster exhibits lower reducing potential and higher activity than the [Fe4S4] cluster (Fig. 8g). When the reduction potential is −0.49 V (low potential), the [Fe4Se4] cluster forms an all-iron state [Fe4S4]0, showing a high CO2 reduction capacity.96,97 It indicates that the [Fe4Se4] cluster can form the reduced all-iron state [Fe4Se4]0 under the action of mild reductants, avoiding the effect of strong reductants on other components. Meanwhile, the effect of doping on the assembly process of the [Fe4S4] cluster was analyzed. It was found that the addition of selenium does not alter the inherent assembly capacity of [Fe4S4] (Fig. 8h).5 Additionally, Wittkamp et al. synthesized Te-doped [Fe4S4] and used isotope labels of Fe and Te (Fig. 9a). When combined with DFT calculations and nuclear resonance vibration spectroscopy (NRVS), the [Fe4Te4] cluster contains two Et4N+ ions and D2d symmetry at its core (Fig. 9b). Fig. 9c–e show the core structure and bond length information of the [Fe4Te4] clusters. Moreover, the [Fe4Te4] cluster was used as a probe for the electron transport process.98 Such doping of [Fe4S4] with other non-metal or electron donors provides an effective way to modify Fe–S clusters. The miraculous in-body conversion process of Fe–S clusters has also inspired a large number of researchers to explore, and they are trying to find a faster and more efficient way to design Fe–S clusters with high catalytic properties.
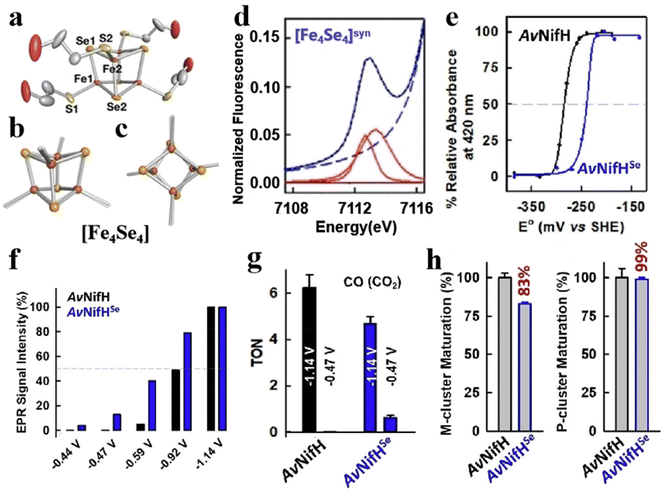 |
| | Fig. 8 Artificial Fe–S clusters based on Se element doping. (a) Crystal structure of [Fe4Se4] cluster. [Fe4Se4] (solid) and [Fe4S4] (transparent) superimposed side view (b) and top view (c) of the core structure. (d) [Fe4Se4] cluster pre-edge region analysis. The experimental data (black dashed line), baseline (red dashed line), pre-edge peak component (red solid), residual (brown solid), and total fit (blue solid). (e) [Fe4Se4]1+/2+ midpoint potential of AvNifHSe (blue) and [Fe4S4]1+/2+ midpoint potential of AvNifH (black). (f) EPR signal strength (%) (g = 16.4) at different reduction points of AvNifHSe (blue) or AvNifH (black). (g) AvNifHSe (blue) or AvNifH (black) act as independent reductases to reduce CO2 to CO. (h) reduce C2H2, H+(Ar), and N2 to C2H4, H2, and NH3/H2, respectively, where AvNifHSe (blue) or AvNifH (black). Reproduced with permission from ref. 2. Copyright 2022, Wiley-VCH GmbH. | |
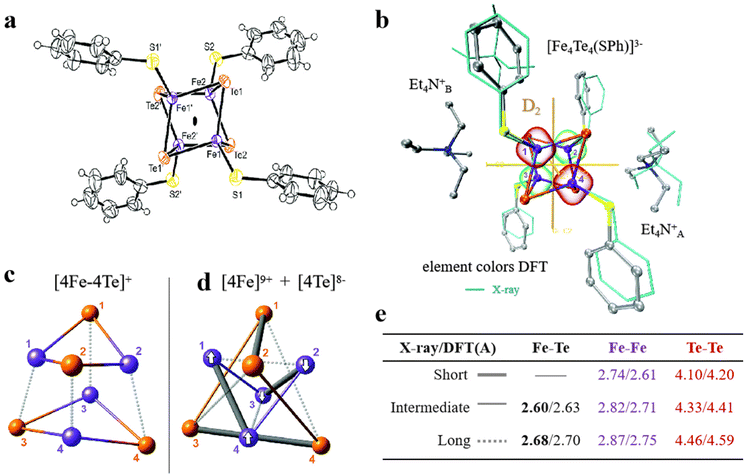 |
| | Fig. 9 Artificial Fe–S clusters based on Te element doping. (a) The structure of [Fe4Te4] cluster. (b) D2-symmetric [Fe4Te4] clusters obtained by X-ray and DFT optimization. (c and d) The bonding interactions and (e) bond length information of [Fe4Te4] cluster cores. Reproduced with permission from ref. 98. Copyright 2019, Royal Society of Chemistry. | |
4.3 Artificial Fe–S clusters based on ligand regulation
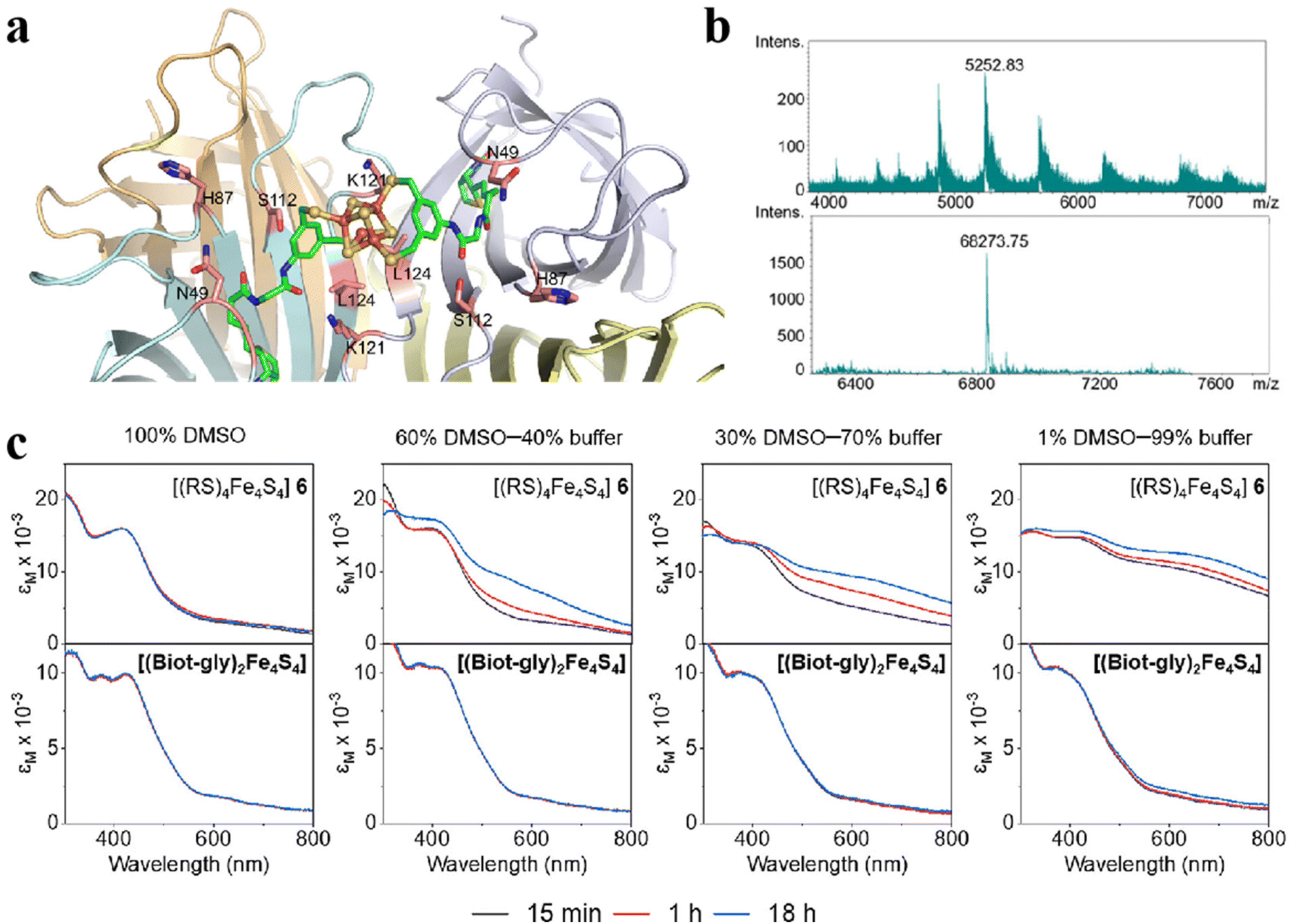
The properties and functions of clusters depend in part on the surface ligands. The type of ligands on the surface of the Fe–S clusters, weak interactions with the protein matrix and the degree of exposure to the medium affect the function of clusters. Therefore, the development of novel Fe–S clusters modified with ligands is an effective way to control the functionality of clusters. In a recent study, Waser and colleagues synthesized and characterized the bis-biotinylated clusters [(Biot-gly)2Fe4S4].99 Specifically, the [Fe4S4] center is tightly connected to the 3,5-dimercaptobenzene scaffold to reduce the distortion of the core. This structure also binds to glycine (Gly) as a blocking position and ultimately binds to biotin on the surface to achieve molecular orientation (Fig. 10a).100,101 Ultraviolet–visible spectroscopy (UV-Vis), high-resolution mass spectrometry (HRMS) and nuclear magnetic resonance (NMR) confirm the successful synthesis of Fe–S clusters (Fig. 10b). Results from the spectrophotometer show that the clusters have good stability in aqueous solution (Fig. 10c). The effect of the protein environment on the ligands was determined by cyclic voltammetry, demonstrating the outstanding accessibility of the bibiotin-substituted [Fe4S4] cluster. To improve the catalytic performance of Fe–S clusters, they reduced CO2 to hydrocarbons by chemo-genetic means, whose overall turnover is 14.102 What's more, the change of ligand can also regulate the redox level of clusters. An N-heterocyclic carbene ligand was designed in the outer layer of the [Fe4S4] core. When CO binds to [Fe4S4] core, the intramolecular valence disproportionation occurs, and the co-bound Fe site forms a low-priced Fe+ oxidation state, which can significantly activate the C–O bond.103 Modification of ligands can not only stabilize the structure, improve the water solubility of the material and adjust the level of redox but also improve its biocompatibility and enhance other properties (optical, electrical, etc.), which is indispensable for the design of artificial Fe–S clusters.
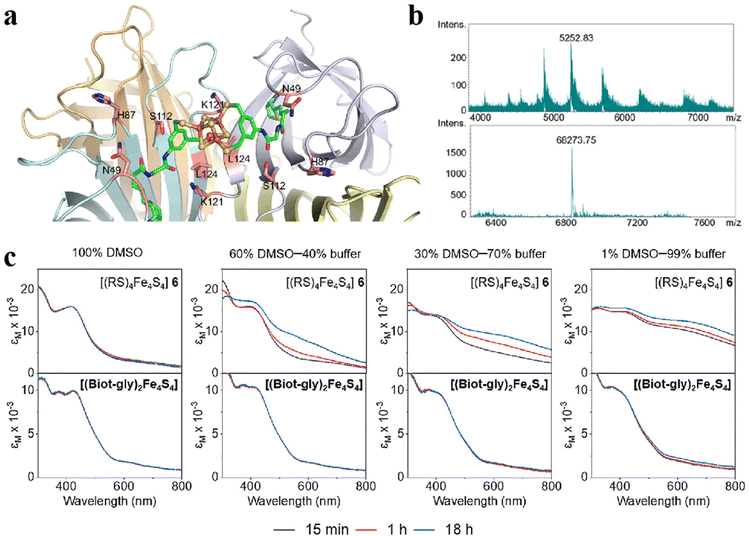 |
| | Fig. 10 Artificial Fe–S clusters based on ligand regulation. (a) The structure of Biot-gly ligand-protected Fe4S4 clusters. C: green, O: red, N: blue, Fe: orange, and S: yellow. (b) The mass spectrum of Biot-gly ligand-protected Fe4S4 clusters. (c) The stability of Fe4S4 protected by Biot-gly and RS was compared. Reproduced with permission from ref. 99. Copyright 2023, American Chemical Society. | |
5. Biocatalysis progress of Fe–S clusters
5.1 Fe–S clusters for cancer
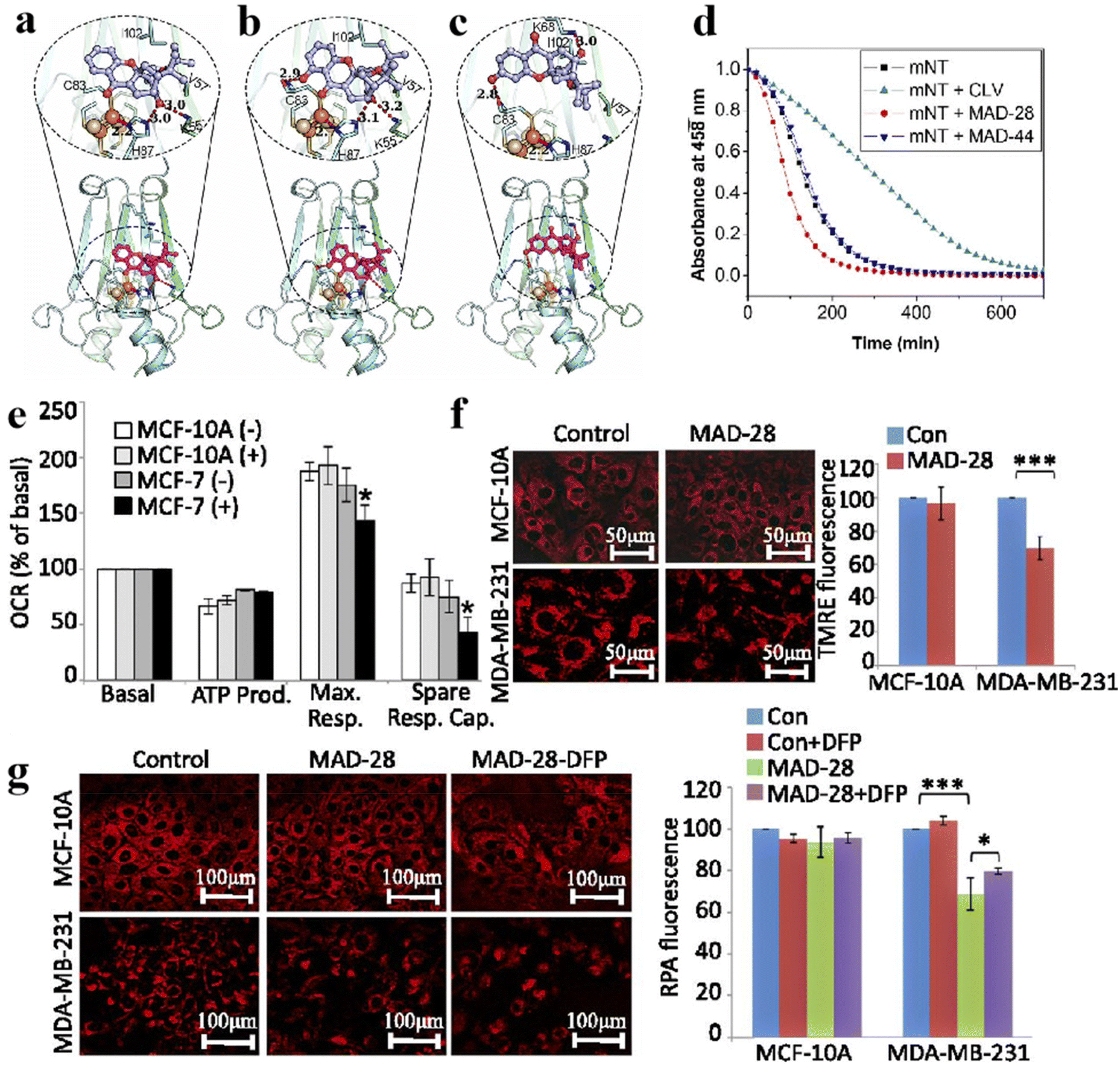
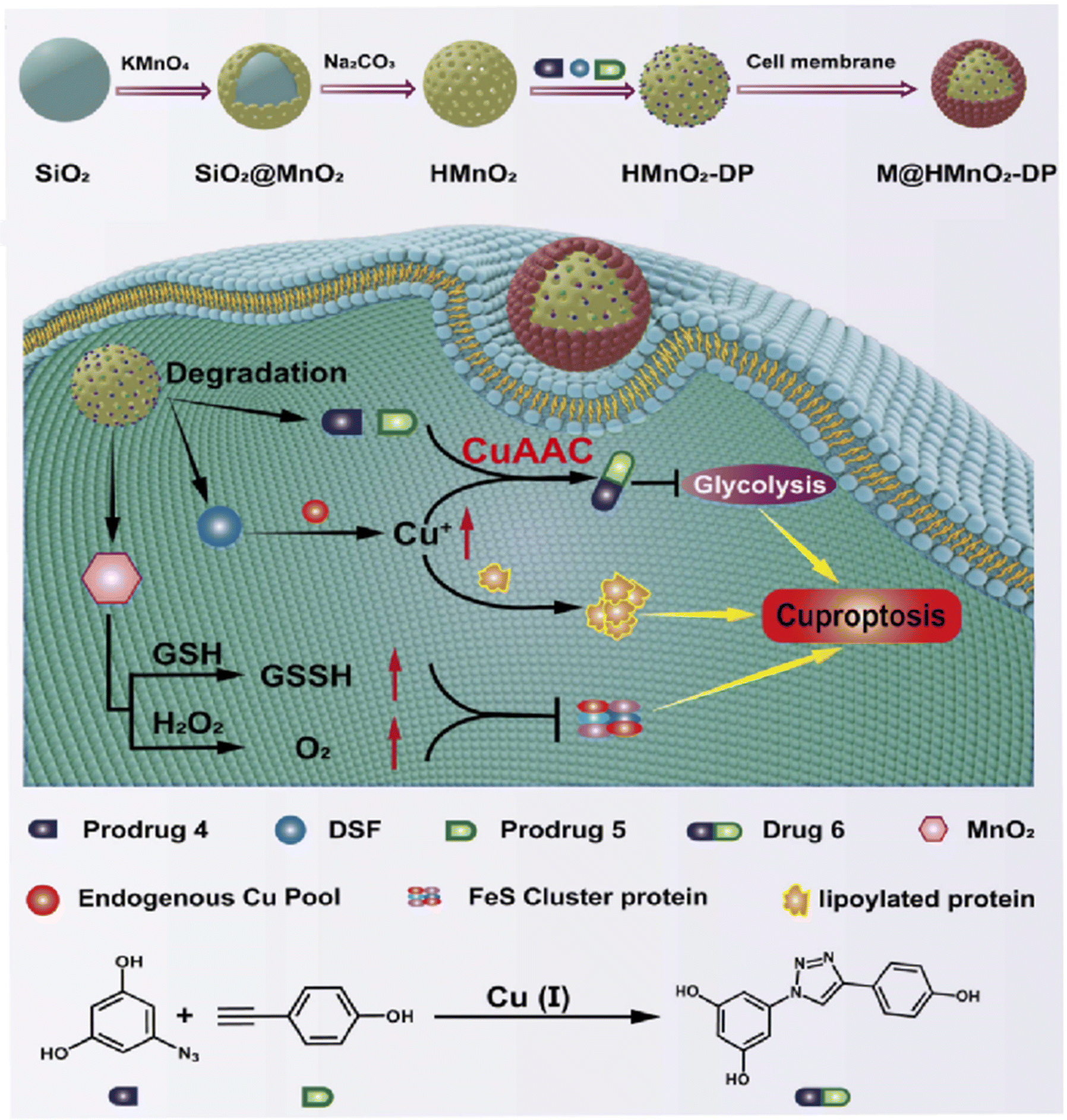
Cancer has been a leading cause of death worldwide for decades.104 Various anticancer strategies have been developed to increase tumor cell death while minimizing damage to normal tissues. Therefore, the discovery of new drug targets is significant. Mitochondria regulate cellular metabolism and produce cellular energy. There is no doubt that this will be an important target in cancer therapy. Based on this, Bai et al. compared three mitocan drugs (CLV, MAD-28, MAD-44) that target the Fe–S clusters in mito NEET (mNT) proteins (Fig. 11a–c) and found that the binding of MDA-28 to the Fe–S clusters breaks the bond between the mNT proteins and the Fe–S clusters to destabilize them (Fig. 11d). Notably, MAD-28 selectively targets cancer cell mitochondria, causing a decrease in the number of mito-primary cells and an increase in mito-primary Fe in breast cancer cells (Fig. 11e–g).105 For another example, Zhu et al. constructed a hollow HMnO2 drug carrier for loading prodrugs and disulfiram (DSF). They also loaded the cancer cell membranes on its surface to target the tumors while releasing the drug through membrane fusion (Fig. 12). In cancer cells, DSF reacts with intracellular Cu2+ to form Cu+. On the one hand, Cu+ leads to the acylation of proteolipids. On the other hand, Cu+ participates in the bioorthogonal reactions to achieve in situ synthesis of glycolysis inhibitors. In addition, the presence of Fe–S clusters enhances the resistance of cancer cells. Removal of excess intracellular GSH and H2O2 by MnO2 can effectively inhibit the biosynthesis of the Fe–S clusters, thus improving the effect of copper toxicity in the treatment of tumors.106,107 Ferroptosis as a novel mode of cell death provides new insights for tumor therapy.108,109 Liu et al. constructed iron-containing protein-mimic supramolecular iron delivery systems (SIDS) with ultra-high iron content and efficient iron delivery. In their work, SIDS promotes their accumulation in tumors through iron-containing protein-mimic structures and shuttle-like morphology.110,111 In tumor microenvironments containing high concentrations of GSH, SIDS inhibited glutathione peroxidase (GPx) activity by depleting GSH,112 accompanied by Fe2+ release, which triggered ferroptosis. In addition, some FeS nanozymes have been used in tumor therapy due to their unique catalytic activity photoelectric and photothermal effects.113,114 Fan et al. prepared FA-modified pyrite nanozymes with prominent POD-like activity and targeting properties.84 The intrinsic GSH-OXD activity of pyrite catalyzes the oxidation of GSH and the concomitant generation of H2O2, which is further reacted to hydroxyl radicals (˙OH), thereby inducing apoptosis and ferroptosis in tumor cells.115 In some typical examples, FeS nanozymes have been designed as multifunctional synergistic therapeutic nanoplatforms. For example, antiferromagnetic pyrite polyethylene glycol (FeS2-PEG) nanocubes were able to utilize excess peroxides in tumors to generate ˙OH, during which the Fe valence state on the surface of the nanozyme was markedly elevated thereby facilitating magnetic resonance imaging (MRI).116 Furthermore, the local heat generated by photothermal treatment can accelerate the Fenton reaction in tumors.117 Biologically amenable phototheranostic pyrite nanocrystals were developed for multispectral photoacoustic tomography (MSOT) and photothermal ablation, and near-infrared (NIR) light was utilized to enhance the Fenton reaction, triggering the dual death pathways of apoptosis and ferroptosis.118 Overall, whether by targeting mitochondria, optimizing drug delivery systems, and optimizing material properties to improve cancer therapy. Fe–S cluster mimetic materials and their derivatives have shown great potential in cancer therapy, and future research needs to optimize these strategies for clinical translation.
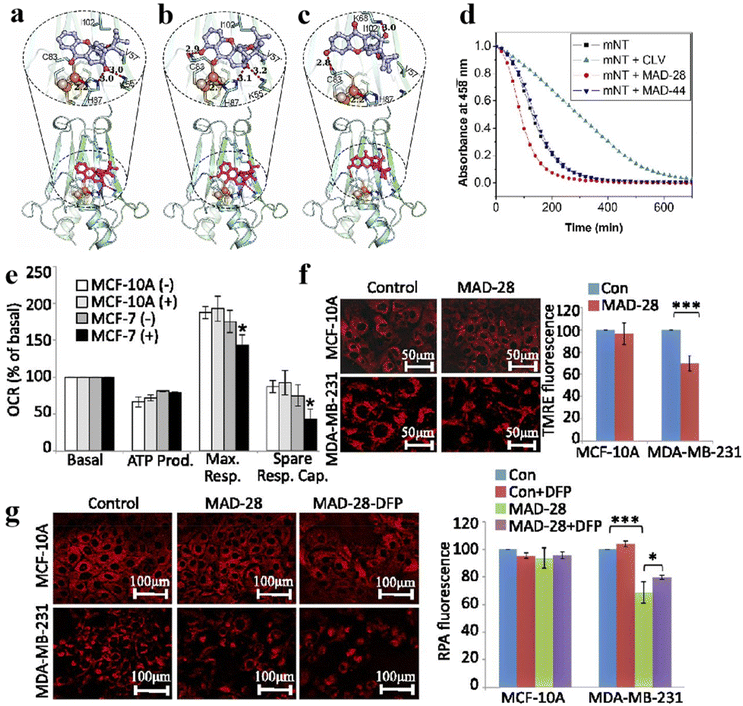 |
| | Fig. 11 Fe–S clusters as inhibitor targets for cancer treatment. Structural model of (a) CLV, (b) MAD-28, (c) MAD-44 binding Fe–S clusters. (d) Effect of three inhibitors on the stability of Fe–S clusters. (e–g) Effects of MAD-28 on mitochondrial function in breast cancer cells. Reproduced with permission from ref. 105. Copyright 2015, PNAS. | |
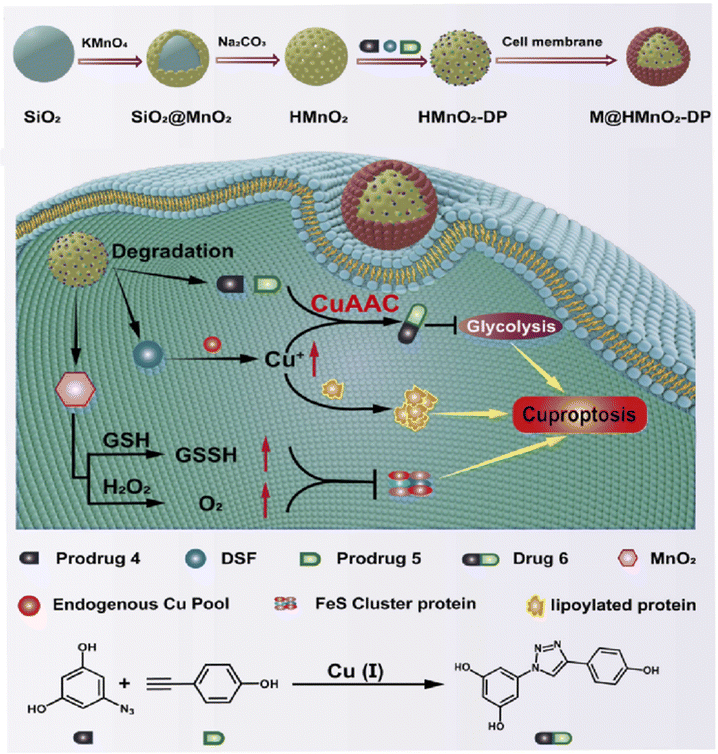 |
| | Fig. 12 Promote copper poisoning by destroying Fe–S clusters to treat cancer. Removal of excess intracellular GSH and H2O2 by MnO2 can effectively inhibit the biosynthesis of Fe–S clusters, thereby reducing the resistance of cancer cells. Reproduced with permission from ref. 106. Copyright 2024, ScienceDirect. | |
5.2 Fe–S clusters for antibacterial
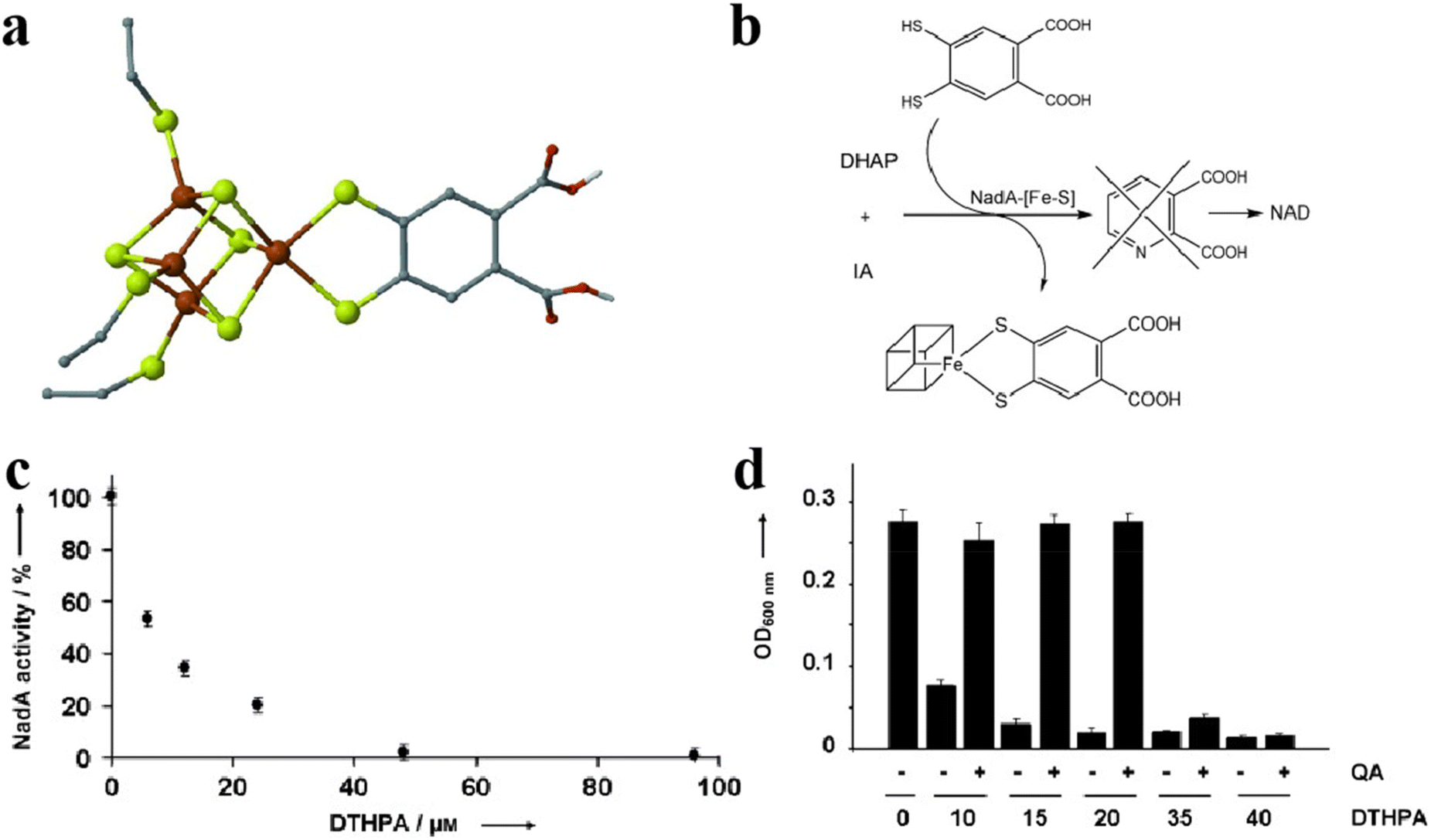
Fe–S clusters can bind to inhibitors to achieve antimicrobial effects.119,120 For example, binding of the Fe–S cluster to the inhibitor 4,5-dithiohydroxyphthalic acid (DTHPA) resulted in the inhibition of the synthesis of Nicotinamide adenine dinucleotide (NAD) (Fig. 13a).121 NAD is a cofactor involved in the majority of biological processes and plays a vital role in E. coli biosynthesis. Its synthesis involves the formation of quinolinic acid (QA), which requires the assistance of Fe–S clusters. However, the addition of inhibitors leads to the formation of new species of Fe–S clusters (Fig. 13b). The activity of L-aspartate oxidase (NadA) carrying Fe–S clusters decreased as the DTHPA concentration decreased (Fig. 13c). The inhibitory effect of DTHPA on bacteria can be mitigated by adding small but large amounts of QA, although QA DTHPA can cause the death of bacteria by inhibiting other metalloproteins (Fig. 13d). Wang and his colleagues designed a series of pyridine inhibitors to bind and disrupt the Fe–S clusters in IspH for the treatment of certain bacterial infections. Hyperfine sublevel correlation (HYSCORE) spectroscopy demonstrated that the pyridine inhibitors of IspH were directly linked to the Fe in the Fe–S clusters. IspH protein is involved in isoprenoid biosynthesis, and isoprenoid is associated with cell wall synthesis.122 These findings suggest that this may be an essential target for the development of new antibiotics.123 Gao et al. used Fe3S4 and Fe1−xS to convert organic sulfur compounds into inorganic nFeS, which has superior antibacterial activity, 500 times that of natural sulfide. nFeS releases hydrogen polysulfanes with strong antibacterial effects, which have significant inhibitory effects on Pseudomonas aeruginosa and Staphylococcus aureus. Topical application of nFeS can disrupt bacterial biofilm and accelerate the healing of infected wounds.87,114,124,125 They successfully prepared an antibacterial alternative. This work lays the foundation for the application of Fe–S clusters in the prevention and treatment of bacterial infections. Although fewer Fe–S clusters are currently used for antibacterial applications, these studies have demonstrated the promising application of mimetic Fe–S clusters in antimicrobial therapeutics and have provided new ideas for future antimicrobial drug development.
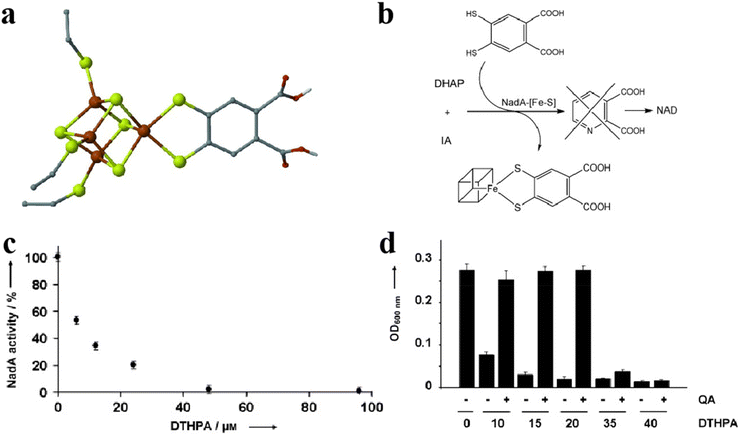 |
| | Fig. 13 Fe–S cluster binding inhibitors are used for antibacterial. (a) The structure of Fe–S cluster-binding inhibitors. (b) The pathways of Fe–S cluster-binding inhibitor. (c) Relationship between inhibitor content and Fe–S cluster activity. (d) The salvage ability of QA in the antibacterial process of inhibitors. Reproduced with permission from ref. 121. Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
5.3 The role of Fe–S clusters in viral suppression
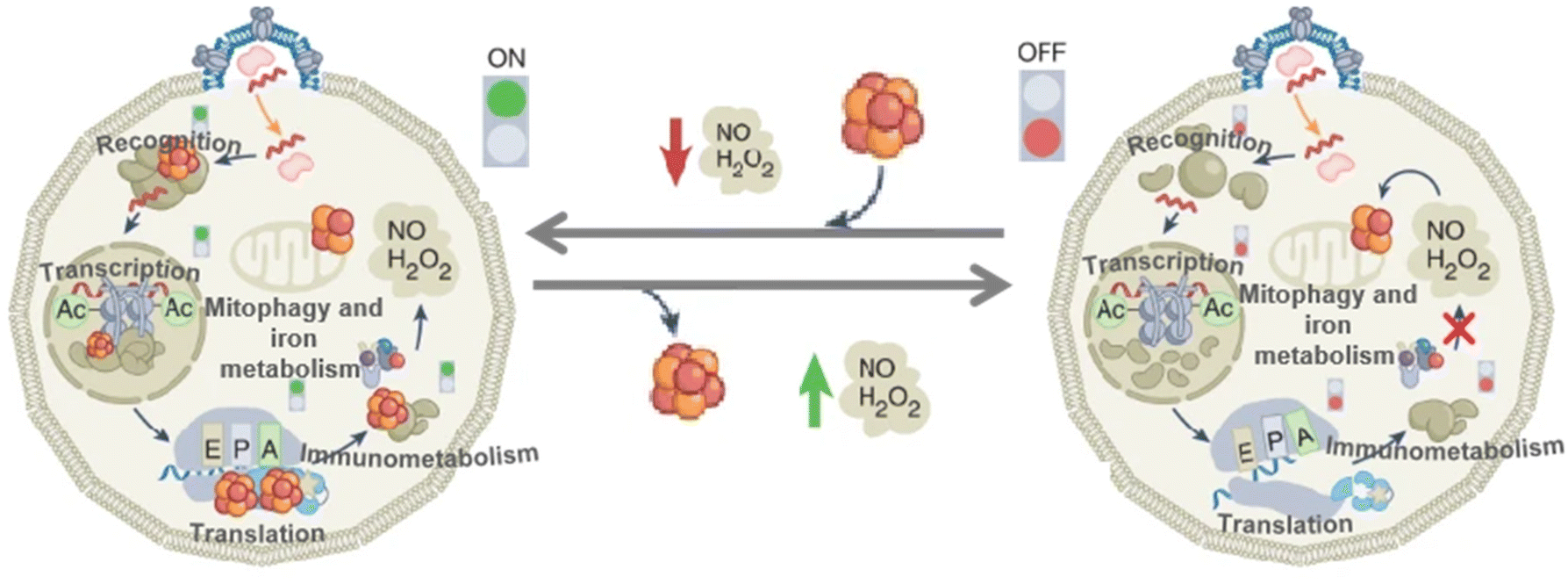
Virus infection has always been a global problem that plagues scholars from all walks of life, which has caused huge financial and human losses. The Fe–S clusters are involved in many biological regulatory processes.53,126 Ebrahimi et al. linked Fe–S clusters involved in immunity to viral replication, investigating how Fe–S clusters play a role in the immune process (Fig. 14).50 When the body responds to a viral infection, the Fe–S protein first recognizes the components of the invading virus and initiates the transcription of inflammatory factors such as type I and type II interferons (IFNs). Transcription of interferon stimulated genes (ISGs) is then induced by interferon stimulation. These Fe–S proteins have catalytic subunits-α (DNA Pol-α) and RNA polymerase (RNA Pol-III) of DNA polymerase.127–129 Translation occurs in ribosomes, and Fe–S proteins include the homodimers Dph2 h and ABCE1 in archaea.130 The primary role of Dph2 h is to translate and modify histidine residues in elongation factors; ABCE1 stimulates the degradation of circulating ribosomes. Stimulate the expression of ISGs proteins, which RSAD2 contains Fe–S clusters and can inhibit viral replication and regulate immune metabolism.131 Finally, mitochondrial autophagy and iron stabilization are regulated.132 The immunity response to viral infection leads to the expression of inflammatory factors. There is growing evidence that ROS are associated with the initiation, development and resolution of the inflammatory response. As a result, a large amount of ROS and reactive nitrogen species (RNS) will be produced in the body such as H2O2 and NO, which causes a decrease in oxygen. These ROS and RNS will inactivate the Fe–S clusters in Fe–S proteins involved in immune recognition, transcription, translation, and metabolism. To solve these problems, on the one hand, large amounts of H2O2 and NO caused by excessive stress can provide feedback to protect the body by preventing harmful substances. On the other hand, Fe–S proteins, which regulate Fe homeostasis and mitochondrial autophagy, cause mitochondrial autophagy by sensing excess H2O2 and NO in the environment. These include proteins such as IRP1, FBXL5, and mitoNEET.24,133,134
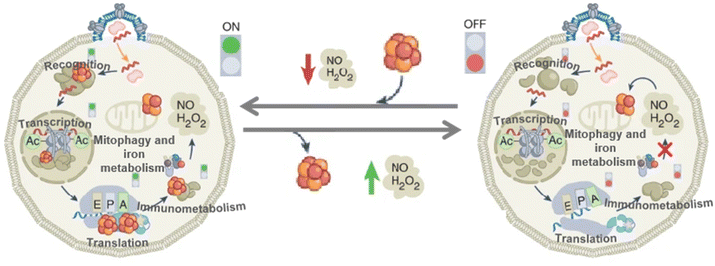 |
| | Fig. 14 Fe–S clusters for inhibition of viral replication. Schematic diagram of the process by which Fe–S clusters inhibit viral replication. Reproduced with permission from ref. 50. Copyright 2022, Springer Nature Limited. | |
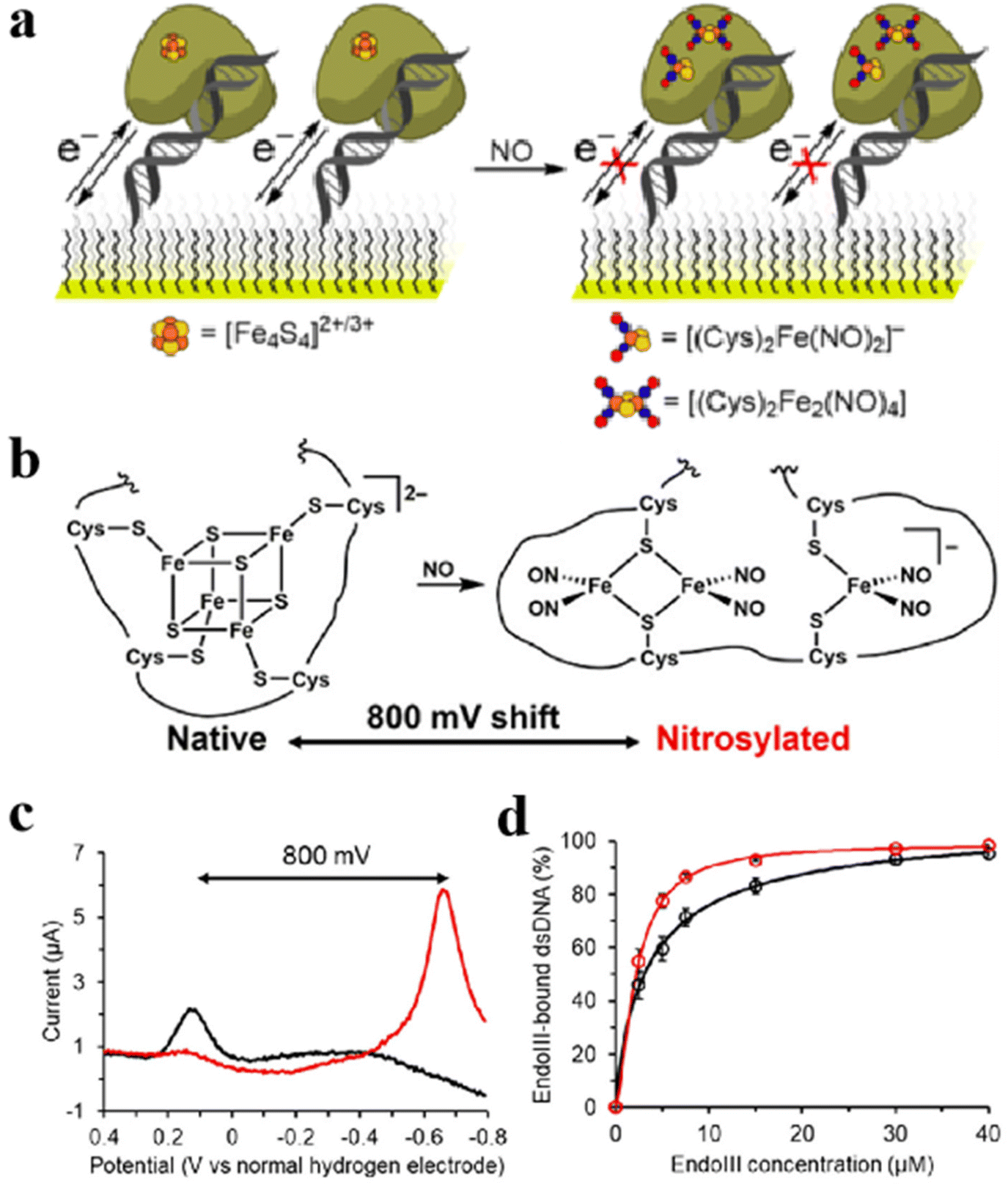
The most prominent achievement of Fe–S clusters in the immunization process is the formation of feedback by sensing H2O2, NO, etc.20 In addition to inhibiting viral replication through immunomodulation, Fe–S clusters can modulate the gene expression process of certain viruses to inhibit viruses.21,135 Ekanger et al. studied the migration of the [Fe4S4] cluster into the endonucleus after sensing environmental NO.16 After binding NO, the [Fe4S4] cluster produces nitrite products [(Cys)2Fe(NO)2]− and Roussin's red ester ([(μ-Cys)2Fe2(NO)4]). It causes [Fe4S4]2+/3+ midpoint potential to shift negatively by 800 mV. At the same time, the DNA binding potential is not affected (Fig. 15a and b).20,21 The electrochemical test was performed by graphite electrode with a wide potential window and the differential pulse voltammetry was drawn. The midpoint potential of redox after nitrosylation was observed to shift negatively by 800 mV compared to native endonucleases (Fig. 15c). Notably, affinity with DNA is also the main criterion for evaluating endonuclease activity. As displayed in Fig. 15d, electrophoretic mobility analysis and circular dichroic (CD) spectra showed that the affinity with DNA after nitrosylated endonuclease sensing and binding to NO was not reduced compared to the native endonucleases. It indicates that NO is inactive by negatively shifting the redox midpoint potential by the nitrosolated endonuclease [Fe4S4] cluster, inhibiting viral proliferation. The role of Fe–S clusters in immune responses and viral replication highlights their critical role in cellular defense and disease processes. Fe–S clusters are essential for the recognition of viral components and the activation of inflammatory pathways. Their role in the translation and modification of proteins that influence the immune response emphasizes their importance in the regulation of viral infection. In addition, Fe–S clusters modulate the immune response by altering the redox midpoint potential of endonucleases through the binding of NO, thereby effectively inhibiting viral replication. NO modifies Fe–S clusters by this mechanism, affecting their function, and demonstrating the complex balance in immune defense and the potential of Fe–S clusters in maintaining immune system function.
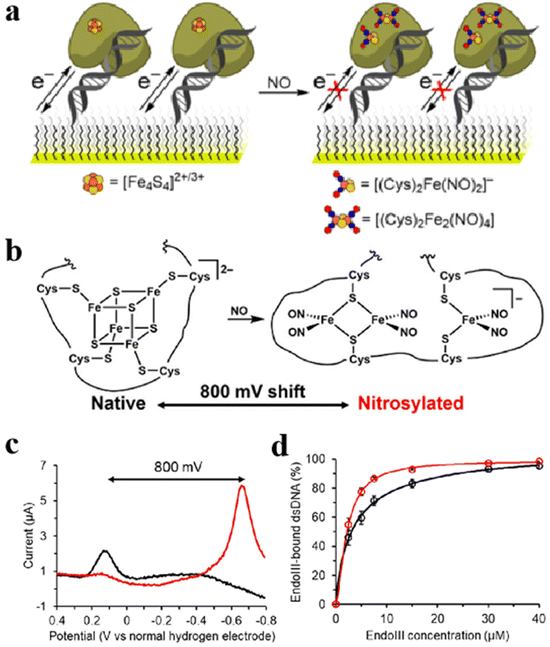 |
| | Fig. 15 Regulate the gene expression process of the virus to inhibit the virus. (a) Schematic diagram of nitrosylation process of natural endonuclease. (b) Structure of nitrosation products of natural endonuclease. (c) Differential pulse voltammograms of natural (black) and nitrosylated (red) nucleic acid endonucleases on edge-plane graphite electrodes. (d) CD chromatograms of the natural (black) and the nitrosylated (red) endonuclease. Reproduced with permission from ref. 16. Copyright 2018, American Chemical Society. | |
5.4 Role of Fe–S clusters in the photoprotection process
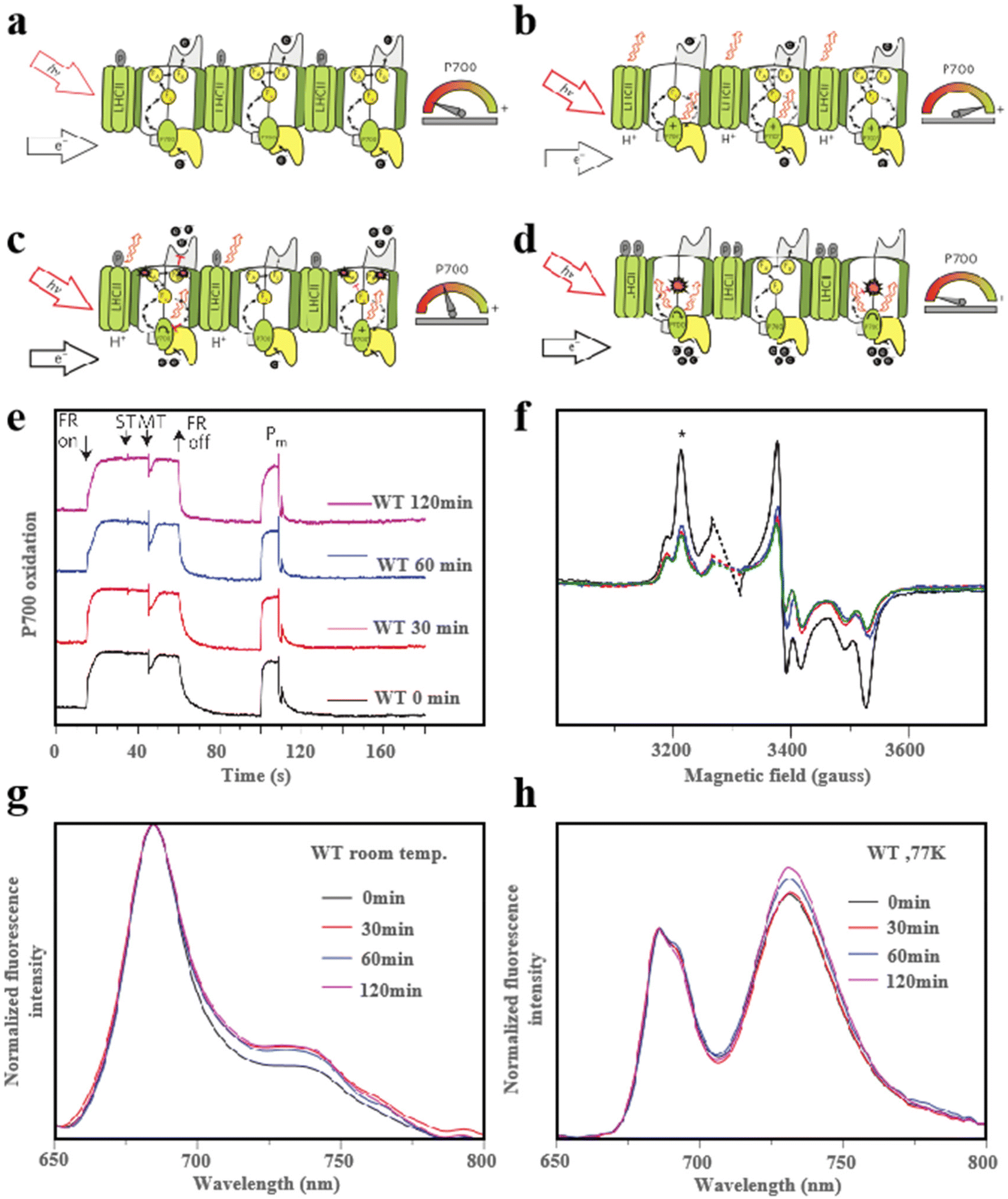
During photosynthesis, light is essential and hazardous to the photosystem, so protection from light plays an important role in this process. Currently, photosystem II (PSII) and light-harvesting complex II (LHCII) photoprotection mechanisms are the two major convergence points of research. The common photo-suppression reactions occur mainly in PSII. Photo suppression is when light energy exceeds the amount that the photosynthetic system can utilize, and the photosynthetic function decreases to protect itself. It reflects some defensive intensification of the excitation energy heat dissipation process. In contrast, the role of photosystem I (PSI) in the photoprotection process is rarely reported, especially the photoprotection caused by Fe–S cluster damage.136,137 PSI can reduce oxidized nicotinamide adenine dinucleotide (NADP+) to nicotinamide adenine dinucleotide phosphate (NADPH) using light energy and electrons produced by PSII. In addition, PSI can tolerate excess light but detect more electrons produced by PSII.138 During excitation and equilibrium electron flow at PSI, the photosynthetic process is related to the potential of the electron matrix. Simultaneously, the excitation energy reduces the electron transfer rate of PSI (Fig. 16a). Maintaining continuous oxidation of P700 is critical to protect PSI and the Fe–S clusters within it from photooxidative damage when highly excited and controlled electron flow to PSI (Fig. 16b). When the excitation energy is further increased and the electron flow cannot be controlled, P700 will be reduced by the uncontrolled electron flow of PSII. At this point, two kinds of Fe–S clusters, FA and FB, will disappear rapidly (Fig. 16c). When high excitation and unpaired electron flow enter PSI, both the P700 and PSI are over-reduction and the photooxidation ability of P700 disappears. In the meanwhile, the Fe–S cluster was destroyed, thereby the PSI receptor side restriction was reduced. The non-photochemical photoprotective energy dissipation of the damaged PSI center was turned on (Fig. 16d). To maximize the transformation of excitation energy to PSI, PSI dissipates excess excitation energy at the expense of the PSII excitation. In addition, the excess energy is consumed by the LHCII-dependent mechanisms.139 As shown in Fig. 16e, when the plants were transferred to high light for up to 120 minutes, the P700 redox kinetics were completely unaffected in typical plants, acting as an excess excitation energy dissipator. However, this process will cause some damage to the two forms of Fe–S clusters FA and FB after illumination (Fig. 16f). The fluorescence emission distribution also confirmed that PSI was a very good quencher after degradation (Fig. 16g) and confirmed that the quenching energy decreased as a result of lowering the temperature (Fig. 16h).27,72 In exploring the process of plant photoprotection, the role of Fe–S clusters in it cannot be ignored. Therefore, an in-depth study of the photoprotective mechanism of Fe–S clusters under different light intensities is of great significance for optimizing photosynthesis and improving plant stress tolerance.
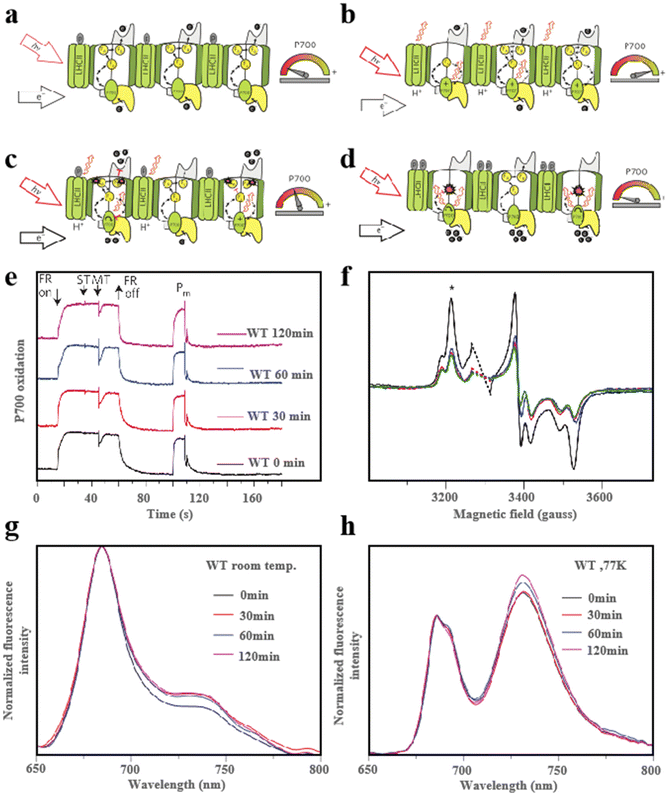 |
| | Fig. 16 Fe–S clusters for light protection. (a) Schematic diagram of the light protection process under low excitation and equilibrium electron flow. (b) Schematic diagram of light protection process under controlled electron flow state. (c and d) Schematic diagram of light protection process under uncontrolled electron flow state. (e) Redox kinetics of PSI. (f) FA and FB central EPR plots. Fluorescence emission spectra of WT plants. The excitation light used is 440 nm. At room temperature (g); (h) at 77K. Reproduced with permission from ref. 27. Copyright 2016, Macmillan Publishers Limited. | |
6. Conclusions
Over the past few decades, studies on the physicochemical properties and biotransformation of Fe–S proteins have elucidated the structure of Fe–S clusters and their transformation processes in vivo. There is no doubt that one of the factors affecting the properties of materials is structure. Fe–S clusters are naturally abundant in organic materials due to their flexibility and versatility, making them ideal candidates for the development of materials with specific functions. As we continue to update synthesis technology, it will become a trend to exploit its catalytic activity by mimicking the active sites of Fe–S clusters in vitro. It provides a platform for the formation of structure–activity relationships and expands the application of Fe–S clusters to other fields. Nowadays, synthetic Fe–S clusters offer a valuable model for the research of their structure, catalytic and sensing properties. However, the characteristics of many natural Fe–S clusters remain challenging to replicate in vitro. In particular, the rational design of materials that are stable and specific to satisfy the requirements of different directions is still an urgent problem that needs to be solved.
Firstly, at present, there are many types of artificial Fe–S clusters, but their performance is misty. Natural Fe–S clusters have powerful functions in vivo, but it is very difficult to synthesize stable clusters in vitro. It limits the use of Fe–S clusters in catalysis, disease treatment and other aspects. Therefore, it is of great importance for basic research to design a simple synthesis process for the Fe–S route that contains stable and high-yield products.
Secondly, structure–activity relationships are key to the whole of materials science. Previous reports have explored the relationship between the structures and functions of Fe–S clusters, revealing the influence of factors such as ligands, electron configuration and configuration on Fe–S clusters in organisms. So far, the creation of such relationship applications is still lacking. In particular, many specific factors and structural relationships have not been identified.
Thirdly, the biological functions of Fe–S clusters are limited to tumor therapy, antibacterial, etc. Investigating other biological functions of Fe–S clusters, such as redox balance in organisms and gene editing, is also one of the research directions.
Fourthly, although a large number of artificial Fe–S clusters have been developed, few are used in the clinic. Compared to other clusters, Fe–S clusters are more abundant in organisms and have the potential for clinical transformation. How to determine the clinical changes of the Fe–S cluster and how to fulfill the biological function of the Fe–S cluster is a question that we need to consider.140
Finally, the role of the Fe–S clusters in the innate immune system and the viral replication process is revealed. However, the development of drugs aimed at achieving better treatment and the creation of Fe–S models targeting other diseases (such as neurodegenerative diseases) to identify biological mechanisms between groups and diseases are still directions for future work.141,142
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2021YFF1200700), the National Natural Science Foundation of China (Grant No. 91859101, 81971744, U1932107, 82202324), Outstanding Youth Funds of Tianjin (2021FJ-0009), Natural Science Foundation of Tianjin (No. 23JCYBJC00710, 23YDTPJC00160), the Natural Science Foundation Project of Gansu Province (22JR5RA152), China Postdoctoral Science Foundation (2023M732601) and CAS Interdisciplinary Innovation Team (JCTD-2020-08).
References
- H. Beinert, R. Holm and E. Münck, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- J. B. Solomon, K. Tanifuji, C. C. Lee, A. J. Jasniewski, B. Hedman, K. O. Hodgson, Y. Hu and M. W. Ribbe, Angew. Chem., Int. Ed., 2022, 61, e202202271 CrossRef CAS PubMed.
- D. Rossetto, N. Cvjetan, P. Walde and S. S. Mansy, Acc. Chem. Res., 2024, 57, 2293–2302 CrossRef CAS PubMed.
- X. M. Xu and S. G. Møller, Antioxid. Redox Signal., 2011, 15, 271–307 CrossRef CAS PubMed.
- D. W. Bak and S. J. Elliott, Curr. Opin. Chem. Biol., 2014, 19, 50–58 CrossRef CAS PubMed.
- N. Rouhier, Nat. Chem. Biol., 2022, 19, 129–130 CrossRef PubMed.
- S. Ohta and Y. Ohki, Coord. Chem. Rev., 2017, 338, 207–225 CrossRef CAS.
- H. M. Dietrich and V. Müller, ACS Catal., 2023, 13, 12374–12382 CrossRef CAS.
- M.-E. Pandelia, W. Nitschke, P. Infossi, M.-T. Giudici-Orticoni, E. Bill and W. Lubitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6097–6102 CrossRef CAS PubMed.
- J. M. Schuller, J. A. Birrell, H. Tanaka, T. Konuma, H. Wulfhorst, N. Cox, S. K. Schuller, J. Thiemann, W. Lubitz, P. Sétif, T. Ikegami, B. D. Engel, G. Kurisu and M. M. Nowaczyk, Science, 2019, 363, 257–260 CrossRef CAS PubMed.
- L. A. Rettberg, J. Wilcoxen, A. J. Jasniewski, C. C. Lee, K. Tanifuji, Y. Hu, R. D. Britt and M. W. Ribbe, Nat. Commun., 2020, 11, 1757 CrossRef CAS PubMed.
- J. G. Rebelein, C. C. Lee, Y. Hu and M. W. Ribbe, Nat. Commun., 2016, 7, 13641 CrossRef CAS PubMed.
- C. C. Lee, Y. Hu and M. W. Ribbe, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6922–6926 CrossRef CAS PubMed.
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed.
- K. Tanifuji, C. C. Lee, N. S. Sickerman, K. Tatsumi, Y. Ohki, Y. Hu and M. W. Ribbe, Nat. Chem., 2018, 10, 568–572 CrossRef CAS PubMed.
- L. A. Ekanger, P. H. Oyala, A. Moradian, M. J. Sweredoski and J. K. Barton, J. Am. Chem. Soc., 2018, 140, 11800–11810 CrossRef CAS PubMed.
- B. Chica, J. Ruzicka, L. M. Pellows, H. Kallas, E. Kisgeropoulos, G. E. Vansuch, D. W. Mulder, K. A. Brown, D. Svedruzic, J. W. Peters, G. Dukovic, L. C. Seefeldt and P. W. King, J. Am. Chem. Soc., 2022, 144, 5708–5712 CrossRef CAS PubMed.
- J. C. Crack, J. Green, A. J. Thomson and N. E. L. Brun, Acc. Chem. Res., 2014, 47, 3196–3205 CrossRef CAS PubMed.
- M. T. Pellicer Martinez, A. B. Martinez, J. C. Crack, J. D. Holmes, D. A. Svistunenko, A. W. B. Johnston, M. R. Cheesman, J. D. Todd and N. E. Le Brun, Chem. Sci., 2017, 8, 8451–8463 RSC.
- A. Volbeda, E. L. Dodd, C. Darnault, J. C. Crack, O. Renoux, M. I. Hutchings, N. E. Le Brun and J. C. Fontecilla-Camps, Nat. Commun., 2017, 8, 15052 CrossRef CAS PubMed.
- R. Rohac, J. C. Crack, E. de Rosny, O. Gigarel, N. E. Le Brun, J. C. Fontecilla-Camps and A. Volbeda, Commun. Biol., 2022, 5, 769 CrossRef CAS PubMed.
- E. L. McCarthy and S. J. Booker, Science, 2017, 358, 373–377 CrossRef CAS PubMed.
- A. Volbeda, C. Darnault, O. Renoux, Y. Nicolet and J. C. Fontecilla-Camps, Sci. Adv., 2015, 1, 1–5 Search PubMed.
- H. Wang, H. Shi, M. Rajan, E. R. Canarie, S. Hong, D. Simoneschi, M. Pagano, M. F. Bush, S. Stoll, E. A. Leibold and N. Zheng, Mol. Cell, 2020, 78, 31–41 CrossRef CAS PubMed.
- E. M. Terzi, V. O. Sviderskiy, S. W. Alvarez, G. C. Whiten and R. Possemato, Sci. Adv., 2021, 7, 2375–2548 Search PubMed.
- M. T. Stiebritz, C. J. Hiller, N. S. Sickerman, C. C. Lee, K. Tanifuji, Y. Ohki and Y. Hu, Nat. Catal., 2018, 1, 444–451 CrossRef CAS.
- A. Tiwari, F. Mamedov, M. Grieco, M. Suorsa, A. Jajoo, S. Styring, M. Tikkanen and E.-M. Aro, Nat. Plants, 2016, 2, 16035 CrossRef CAS PubMed.
- S. Sharma, K. Sivalingam, F. Neese and G. K.-L. Chan, Nat. Chem., 2014, 6, 927–933 CrossRef CAS PubMed.
- J. C. Crack and N. E. Le Brun, Coord. Chem. Rev., 2021, 448, 1–12 CrossRef.
- C. Bonfio, L. Valer, S. Scintilla, S. Shah, D. J. Evans, L. Jin, J. W. Szostak, D. D. Sasselov, J. D. Sutherland and S. S. Mansy, Nat. Chem., 2017, 9, 1229–1234 CrossRef CAS PubMed.
- P. La, V. Ghiaccio, J. Zhang and S. Rivella, Blood, 2018, 132, 1048 CrossRef.
- R. Shi, W. Hou, Z.-Q. Wang and X. Xu, Front. Cell Dev. Biol., 2021, 9, 735678 CrossRef PubMed.
- T. A. Rouault and W. H. Tong, Nat. Rev. Mol. Cell Biol., 2005, 6, 345–351 CrossRef CAS PubMed.
- A. D. Read, R. E. T. Bentley, S. L. Archer and K. J. Dunham-Snary, Redox Biol., 2021, 47, 102164 CrossRef CAS PubMed.
- R. Lill, Nature, 2009, 460, 831–838 CrossRef CAS PubMed.
- N. S. Sickerman, K. Tanifuji, C. C. Lee, Y. Ohki, K. Tatsumi, M. W. Ribbe and Y. Hu, J. Am. Chem. Soc., 2017, 139, 603–606 CrossRef CAS PubMed.
- X. Wang, W. Zhou, Z. Chang, Z. Zhou and S. Wu, Chin. J. Chem., 2013, 31, 983–986 CrossRef CAS.
- A. McSkimming and D. L. M. Suess, Nat. Chem., 2021, 13, 666–670 CrossRef CAS PubMed.
- X. Zhou, Y. Gao, W. Wang, X. Yang, X. Yang, F. Liu, Y. Tang, S. M. Lam, G. Shui, L. Yu, C. Tian, L. W. Guddat, Q. Wang, Z. Rao and H. Gong, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022308118 CrossRef CAS PubMed.
- Y. K. Levy, S. Tamir, J. A. Zuris, L. Agranat, C. H. Lipper, A. R. Conlan, D. Michaeli, Y. Harir, M. L. Paddock, R. Mittler, Z. I. Cabantchik, P. A. Jennings and R. Nechushtai, PLoS One, 2013, 8, e61202 CrossRef PubMed.
- J. A. Zuris, Y. Harir, A. R. Conlan, M. Shvartsman, D. Michaeli, S. Tamir, M. L. Paddock, J. N. Onuchic, R. Mittler, Z. I. Cabantchik, P. A. Jennings and R. Nechushtai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13047–13052 CrossRef CAS PubMed.
- C. Xie, D. Cen, Z. Ren, Y. Wang, Y. Wu, X. Li, G. Han and X. Cai, Adv. Sci., 2020, 7, 1903512 CrossRef CAS PubMed.
- S. Sun, H. Liu, K. Chen, H. Ma, S. Liu, X. Mu, W. Hao, S. Liu, Y. Gao, Y. Wang, J. Pei, R. Zhao, S. Zhang, X. Zhang, H. Wang, Y. Li and X.-D. Zhang, Nano Lett., 2021, 21, 2562–2571 CrossRef CAS PubMed.
- H. Liu, Y. Li, S. Sun, Q. Xin, S. Liu, X. Mu, X. Yuan, K. Chen, H. Wang, K. Varga, W. Mi, J. Yang and X.-D. Zhang, Nat. Commun., 2021, 12, 114 CrossRef CAS PubMed.
- X. Mu, H. He, J. Wang, W. Long, Q. Li, H. Liu, Y. Gao, L. Ouyang, Q. Ren, S. Sun, J. Wang, J. Yang, Q. Liu, Y. Sun, C. Liu, X.-D. Zhang and W. Hu, Nano Lett., 2019, 19, 4527–4534 CrossRef CAS PubMed.
- X. Mu, J. Wang, Y. Li, F. Xu, W. Long, L. Ouyang, H. Liu, Y. Jing, J. Wang, H. Dai, Q. Liu, Y. Sun, C. Liu and X.-D. Zhang, ACS Nano, 2019, 13, 1870–1884 CAS.
- J. Y. Wang, X. Mu, Y. Li, F. Xu, W. Long, J. Yang, P. Bian, J. Chen, L. Ouyang, H. Liu, Y. Jing, J. Wang, L. Liu, H. Dai, Y. Sun, C. Liu and X. D. Zhang, Small, 2018, 14, 1703736 CrossRef PubMed.
- S. Zhang, Y. Liu, S. Sun, J. Wang, Q. Li, R. Yan, Y. Gao, H. Liu, S. Liu, W. Hao, H. Dai, C. Liu, Y. Sun, W. Long, X. Mu and X.-D. Zhang, Theranostics, 2021, 11, 2806–2821 CrossRef CAS PubMed.
- R. Yan, S. Sun, J. Yang, W. Long, J. Wang, X. Mu, Q. Li, W. Hao, S. Zhang, H. Liu, Y. Gao, L. Ouyang, J. Chen, S. Liu, X.-D. Zhang and D. Ming, ACS Nano, 2019, 13, 11552–11560 CrossRef CAS PubMed.
- K. H. Ebrahimi, S. Ciofi-Baffoni, P.-L. Hagedoorn, Y. Nicolet, N. E. Le Brun, W. R. Hagen and F. A. Armstrong, Nat. Chem., 2022, 14, 253–266 CrossRef PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- A. C. Brown and D. L. M. Suess, J. Am. Chem. Soc., 2023, 145, 2075–2080 CrossRef CAS PubMed.
- A. E. Boncella, E. T. Sabo, R. M. Santore, J. Carter, J. Whalen, J. D. Hudspeth and C. N. Morrison, Coord. Chem. Rev., 2022, 453, 214229 CrossRef CAS.
- G. Moula, A. Nagasaki, T. Matsumoto, M. E. Miehlich, K. Meyer, R. E. Cramer and K. Tatsumi, Angew. Chem., Int. Ed., 2021, 60, 15792–15797 CrossRef CAS PubMed.
- J. Liedtke, C. C. Lee, K. Tanifuji, A. J. Jasniewski, M. W. Ribbe and Y. Hu, ChemBioChem, 2020, 22, 151–155 CrossRef PubMed.
- A. J. Jasniewski, C. C. Lee, M. W. Ribbe and Y. Hu, Chem. Rev., 2020, 120, 5107–5157 CrossRef CAS PubMed.
- H. L. Rutledge and F. A. Tezcan, Chem. Rev., 2020, 120, 5158–5193 CrossRef CAS PubMed.
- M. W. Ribbe, Y. Hu, K. O. Hodgson and B. Hedman, Chem. Rev., 2013, 114, 4063–4080 CrossRef PubMed.
- E. Jimenez-Vicente, Z.-Y. Yang, J. S. Martin del Campo, V. L. Cash, L. C. Seefeldt and D. R. Dean, J. Biol. Chem., 2019, 294, 6204–6213 CrossRef CAS PubMed.
- S. Burén, E. Jiménez-Vicente, C. Echavarri-Erasun and L. M. Rubio, Chem. Rev., 2020, 120, 4921–4968 CrossRef PubMed.
- K. Rupnik, C. C. Lee, Y. Hu, M. W. Ribbe and B. J. Hales, Inorg. Chem., 2018, 57, 4719–4725 CrossRef CAS PubMed.
- C. V. Stappen, E. Jiménez-Vicente, A. Pérez-González, Z.-Y. Yang, L. C. Seefeldt, S. DeBeer, D. R. Dean and L. Decamps, Chem. Sci., 2022, 13, 3489–3500 RSC.
- E. Jimenez-Vicente, Z.-Y. Yang, W. K. Ray, C. Echavarri-Erasun, V. L. Cash, L. M. Rubio, L. C. Seefeldt and D. R. Dean, J. Biol. Chem., 2018, 293, 9812–9823 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 191–604 CrossRef PubMed.
- I. Coric and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 7200–7211 CrossRef CAS PubMed.
- R. Lill and S.-A. Freibert, Annu. Rev. Biochem., 2020, 89, 471–499 CrossRef CAS PubMed.
- S. P. Bennett, J. C. Crack, R. Puglisi, A. Pastore and N. E. Le Brun, Chem. Sci., 2022, 14, 78–95 RSC.
- S. F. Jordan, I. Ioannou, H. Rammu, A. Halpern, L. K. Bogart, M. Ahn, R. Vasiliadou, J. Christodoulou, A. Marechal and N. Lane, Nat. Commun., 2021, 12, 5925 CrossRef CAS PubMed.
- A. V. Goldberg, S. Molik, A. D. Tsaousis, K. Neumann, G. Kuhnke, F. Delbac, C. P. Vivares, R. P. Hirt, R. Lill and T. M. Embley, Nature, 2008, 452, 624–627 CrossRef CAS PubMed.
- D. Brancaccio, A. Gallo, M. Mikolajczyk, K. Zovo, P. Palumaa, E. Novellino, M. Piccioli, S. Ciofi-Baffoni and L. Banci, J. Am. Chem. Soc., 2014, 136, 16240–16250 CrossRef CAS PubMed.
- K. Gari, A. M. L. Ortiz, V. Borel, H. Flynn, J. M. Skehel and S. J. Boulton, Science, 2012, 337, 243–245 CrossRef CAS PubMed.
- J. Zhang, Z. Bai, M. Ouyang, X. Xu, H. Xiong, Q. Wang, B. Grimm, J. D. Rochaix and L. Zhang, EMBO J., 2021, 40, e106742 CrossRef CAS PubMed.
- J. Feng, S. Shaik and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 20430–20436 CrossRef CAS PubMed.
- M. Dong, M. Horitani, B. Dzikovski, J. H. Freed, S. E. Ealick, B. M. Hoffinan and H. Lin, J. Am. Chem. Soc., 2017, 139, 5680–5683 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- Y. Jia, F. Li, K. Fan and L. Sun, Adv. Powder Technol., 2022, 1, 100012 Search PubMed.
- X. Cai, W. Hu, S. Xu, D. Yang, M. Chen, M. Shu, R. Si, W. Ding and Y. Zhu, J. Am. Chem. Soc., 2020, 142, 4141–4153 CrossRef CAS PubMed.
- Z. Yin, G. T. R. Palmore and S. Sun, Trends Chem., 2019, 1, 739–750 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- T. Wagner, U. Ermler and S. Shima, Science, 2016, 354, 114–117 CrossRef CAS PubMed.
- L. Noodleman, Nat. Catal., 2018, 1, 383–384 CrossRef CAS.
- C. C. Lee, M. T. Stiebritz and Y. Hu, Acc. Chem. Res., 2019, 52, 1168–1176 CrossRef CAS PubMed.
- K. A. Vincent, G. J. Tilley, N. C. Quammie, I. Streeter, B. K. Burgess, M. R. Cheesman and F. A. Armstrong, Chem. Commun., 2003, 20, 2590–2591 RSC.
- X. Meng, D. Li, L. Chen, H. He, Q. Wang, C. Hong, J. He, X. Gao, Y. Yang, B. Jiang, G. Nie, X. Yan, L. Gao and K. Fan, ACS Nano, 2021, 15, 5735–5751 CrossRef CAS PubMed.
- H. Cao, Y. Yuan, R. Zhao, W. Shi, J. Jiang, Y. Gao, L. Chen and L. Gao, ACS Appl. Mater. Interfaces, 2024, 16, 30958–30966 CrossRef CAS PubMed.
- X. Huang, F. Xia and Z. Nan, ACS Appl. Mater. Interfaces, 2020, 12, 46539–46548 CrossRef CAS PubMed.
- Z. Xu, Z. Qiu, Q. Liu, Y. Huang, D. Li, X. Shen, K. Fan, J. Xi, Y. Gu, Y. Tang, J. Jiang, J. Xu, J. He, X. Gao, Y. Liu, H. Koo, X. Yan and L. Gao, Nat. Commun., 2018, 9, 3713 CrossRef CAS PubMed.
- T. M. Buscagan and D. C. Rees, Joule, 2019, 3, 2662–2678 CrossRef CAS PubMed.
- C. C. Lee, Y. Hu, M. W. Ribbe and D. G. Capone, mBio, 2015, 6, e00307–e00315 CrossRef PubMed.
- D. E. DeRosha, V. G. Chilkuri, C. V. Stappen, E. Bill, B. Q. Mercado, S. DeBeer, F. Neese and P. L. Holland, Nat. Chem., 2019, 11, 1019–1025 CrossRef CAS PubMed.
- I. Čorić, B. Q. Mercado, E. Bill, D. J. Vinyard and P. L. Holland, Nature, 2015, 526, 96–99 CrossRef PubMed.
- N. S. Sickerman, K. Tanifuji, Y. Hu and M. W. Ribbe, Chem. – Eur. J., 2017, 23, 12425–12432 CrossRef CAS PubMed.
- A. T. Thorhallsson, B. Benediktsson and R. Bjornsson, Chem. Sci., 2019, 10, 11110–11124 RSC.
- I. Bertini, S. Ciurli, A. Dikiy and C. Luchinat, J. Am. Chem. Soc., 1993, 115, 11663–12230 CrossRef.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed.
- J. B. Solomon, M. F. Rasekh, C. J. Hiller, C. C. Lee, K. Tanifuji, M. W. Ribbe and Y. Hu, J. Am. Chem. Soc. Au, 2020, 1, 119–123 Search PubMed.
- L. A. Rettberg, M. T. Stiebritz, W. Kang, C. C. Lee, M. W. Ribbe and Y. Hu, Chem. – Eur. J., 2019, 25, 13078–13082 CrossRef CAS PubMed.
- F. Wittkamp, N. Mishra, H. Wang, H.-C. Wille, R. Steinbrügge, M. Kaupp, S. P. Cramer, U.-P. Apfel and V. Pelmenschikov, Chem. Sci., 2019, 10, 7535–7541 RSC.
- V. Waser, M. Mukherjee, N. V. Igareta, R. V. Tachibana and T. R. R. Ward, J. Am. Chem. Soc., 2023, 145, 14823–14830 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661 CrossRef CAS PubMed.
- W. Lo, P. Zhang, C.-C. Ling, S. Huang and R. H. Holm, Inorg. Chem., 2012, 51, 9883–9892 CrossRef CAS PubMed.
- L. Grunwald, M. Inoue, P. C. Carril, M. Wörle and V. Mougel, Chem, 2023, 10, 1–23 Search PubMed.
- A. C. Brown, N. B. Thompson and D. L. M. Suess, J. Am. Chem. Soc., 2022, 144, 9066–9073 CrossRef CAS PubMed.
- A. Jassim, E. P. Rahrmann, B. D. Simons and R. J. Gilbertson, Nat. Rev. Cancer, 2023, 23, 710–724 CrossRef CAS PubMed.
- F. Bai, F. Morcos, Y.-S. Sohn, M. Darash-Yahana, C. O. Rezende, C. H. Lipper, M. L. Paddock, L. Song, Y. Luo, S. H. Holt, S. Tamir, E. A. Theodorakis, P. A. Jennings, J. N. Onuchic, R. Mittler and R. Nechushtai, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3698–3703 CrossRef CAS PubMed.
- J. Zhu, Y. You, W. Zhang, W. Wang, M. Jiang, F. Pu, J. Ren and X. Qu, Nano Today, 2024, 55, 1–10 Search PubMed.
- D. Brancaccio, A. Gallo, M. Piccioli, E. Novellino, S. Ciofi-Baffoni and L. Banci, J. Am. Chem. Soc., 2017, 139, 719–730 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison III and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- W. S. Yang and B. R. Stockwell, Trends Cell Biol., 2016, 26, 165–176 CrossRef CAS PubMed.
- S. Liu, M. Zhang, H. Jin, Z. Wang, Y. Liu, S. Zhang and H. Zhang, J. Am. Chem. Soc., 2023, 145, 160–170 CrossRef CAS PubMed.
- J. Yang, W. Xiong, L. Huang, Z. Li, Q. Fan, F. Hu, X. Duan, J. Fan, B. Li, J. Feng, Y. Xu, X. Chen and Z. Shen, J. Nanobiotechnol., 2024, 22, 204 CrossRef CAS PubMed.
- X. Zhong, X. Wang, L. Cheng, Y. a. Tang, G. Zhan, F. Gong, R. Zhang, J. Hu, Z. Liu and X. Yang, Adv. Funct. Mater., 2020, 30, 1907954 CrossRef CAS.
- Z. Zeng, C. Zhang, J. Li, D. Cui, Y. Jiang and K. Pu, Adv. Mater., 2021, 33, 2007247 CrossRef CAS PubMed.
- W. Yin, J. Yu, F. Lv, L. Yan, L. R. Zheng, Z. Gu and Y. Zhao, ACS Nano, 2016, 10, 11000–11011 CrossRef CAS PubMed.
- Y. Wang, K. Huang, T. Wang, L. Liu, F. Yu, W. Sun, W. Yao, H. Xiong, X. Liu, H. Jiang and X. Wang, Small, 2024, 20, 2310300 CrossRef CAS PubMed.
- Z. Tang, H. Zhang, Y. Liu, D. Ni, H. Zhang, J. Zhang, Z. Yao, M. He, J. Shi and W. Bu, Adv. Mater., 2017, 29, 1701683 CrossRef PubMed.
- C. Pan, M. Ou, Q. Cheng, Y. Zhou, Y. Yu, Z. Li, F. Zhang, D. Xia, L. Mei and X. Ji, Adv. Funct. Mater., 2020, 30, 1906466 CrossRef CAS.
- C. Zhao, Z. Liu, C.-C. Chang, Y.-C. Chen, Q. Zhang, X.-D. Zhang, C. Andreou, J. Pang, Z.-X. Liu, D.-Y. Wang, M. F. Kircher and J. Yang, ACS Nano, 2023, 17, 4261–4278 CrossRef CAS PubMed.
- J. Middaugh, R. Hamel, G. Jean-Baptiste, R. Beriault, D. Chenier and V. D. Appanna, J. Biol. Chem., 2005, 280, 3159–3165 CrossRef CAS PubMed.
- M. P. Thorgersen and D. M. Downs, J. Bacteriol., 2007, 189, 7774–7781 CrossRef CAS PubMed.
- A. Chan, M. Clemancey, J.-M. Mouesca, P. Amara, O. Hamelin, J.-M. Latour and S. O. de Choudens, Angew. Chem., Int. Ed., 2012, 51, 7711–7714 CrossRef CAS PubMed.
- G. Rao and E. Oldfield, Biochemistry, 2016, 55, 4119–4129 CrossRef CAS PubMed.
- W. Wang, J. Li, K. Wang, T. I. Smirnova and E. Oldfield, J. Am. Chem. Soc., 2011, 133, 6525–6528 CrossRef CAS PubMed.
- H.-Y. Wang, X.-W. Hua, F.-G. Wu, B. Li, P. Liu, N. Gu, Z. Wang and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 7082–7092 CrossRef CAS PubMed.
- X. Yang, J. Li, T. Liang, C. Ma, Y. Zhang, H. Chen, N. Hanagata, H. Su and M. Xu, Nanoscale, 2014, 6, 10126–10133 RSC.
- M. T. Pellicer Martinez, J. C. Crack, M. Y. Y. Stewart, J. M. Bradley, D. A. Svistunenko, A. W. B. Johnston, M. R. Cheesman, J. D. Todd and N. E. Le Brun, eLife, 2019, 8, 1–25 CrossRef PubMed.
- Q. Wang, S. Li, F. Wan, Y. Xu, Z. Wu, M. Cao, P. Lan, M. Lei and J. Wu, Nat. Struct. Mol. Biol., 2021, 28, 220–227 CrossRef CAS PubMed.
- C. Lancey, M. Tehseen, V.-S. Raducanu, F. Rashid, N. Merino, T. J. Ragan, C. G. Savva, M. S. Zaher, A. Shirbini, F. J. Blanco, S. M. Hamdan and A. De Biasio, Nat. Commun., 2020, 11, 1109 CrossRef CAS PubMed.
- J. t. Beek, V. Parkash, G. O. Bylund, P. Osterman, A. E. Sauer-Eriksson and E. Johansson, Nucleic Acids Res., 2019, 47, 5712–5722 CrossRef PubMed.
- X. Su, Z. Lin and H. Lin, Crit. Rev. Biochem. Mol. Biol., 2013, 48, 515–521 CrossRef CAS PubMed.
- K. H. Ebrahimi, J. Vowles, C. Browne, J. McCullagh and W. S. James, FEBS Lett., 2020, 594, 1631–1644 CrossRef PubMed.
- J. D. Wofford and P. A. Lindahl, J. Biol. Chem., 2015, 290, 26968–26977 CrossRef CAS PubMed.
- C. P. Anderson, M. Shen, R. S. Eisenstein and E. A. Leibold, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 1468–1483 CrossRef CAS PubMed.
- X. Li, Y. Wang, G. Tan, J. Lyu and H. Ding, Free Radical Biol. Med., 2018, 121, 98–104 CrossRef CAS PubMed.
- J. C. Crack, J. Munnoch, E. L. Dodd, F. Knowles, M. M. Al Bassam, S. Kamali, A. A. Holland, S. P. Cramer, C. J. Hamilton, M. K. Johnson, A. J. Thomson, M. I. Hutchings and N. E. Le Brun, J. Biol. Chem., 2015, 290, 12689–12704 CrossRef CAS PubMed.
- M. K. Terashima, Plant Cell Physiol., 2016, 57, 1405–1414 Search PubMed.
- K. Sonoike, Physiol. Plant., 2010, 142, 56–64 CrossRef PubMed.
- M. Kono, K. Noguchi and I. Terashima, Plant Cell Physiol., 2014, 55, 990–1004 CrossRef CAS PubMed.
- R. Barbato, L. Tadini, R. Cannata, C. Peracchio, N. Jeran, A. Alboresi, T. Morosinotto, A. A. Bajwa, V. Paakkarinen, M. Suorsa, E.-M. Aro and P. Pesaresi, Sci. Rep., 2020, 10, 6770 CrossRef CAS PubMed.
- H. Ma, L. Liu, H. He, S. Sun, S. Yang, Y. Huang, Y. Li, K. Tan, H. Liu, S. Liu, Y. Li, C. Liu, P. Liu, H. Wang and X.-D. Zhang, Matter, 2024, 7, 1660–1676 CrossRef CAS.
- M. T. Heneka, M. P. Kummer and E. Latz, Nat. Rev. Immunol., 2014, 14, 463–477 CrossRef CAS PubMed.
- F. Prischi, P. V. Konarev, C. Iannuzzi, C. Pastore, S. Adinolfi, S. R. Martin, D. I. Svergun and A. Pastore, Nat. Commun., 2010, 1, 1–10 Search PubMed.
- J. C. Crack, J. Green, A. J. Thomson and N. E. Le Brun, Acc. Chem. Res., 2014, 47, 3196–3205 CrossRef CAS PubMed.
- R. H. Holm and W. Lo, Chem. Rev., 2016, 116, 13685–13713 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Haile
Liu
*c and
Xiao-Dong
Zhang
b,
Haile
Liu
*c and
Xiao-Dong
Zhang