
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale chirality generated in zinc(II) orthophosphate clusters: evidence by vibrational circular dichroism†
Sumio
Aisawa
 *a,
Nami
Horiguchi
a,
Chika
Chida
a,
Jing
Sang
a,
Hidetoshi
Hirahara
a,
Akihiko
Yamagishi
b and
Hisako
Sato
*c
*a,
Nami
Horiguchi
a,
Chika
Chida
a,
Jing
Sang
a,
Hidetoshi
Hirahara
a,
Akihiko
Yamagishi
b and
Hisako
Sato
*c
aFaculty of Science and Engineering, Iwate University, 4-3-5 Ueda, Morioka, Iwate, 020-8551, Japan. E-mail: aisawa@iwate-u.ac.jp
bFaculty of Medicine, Toho University, 5-21-16 Oomori-nishi, Ota-ku, Tokyo 143-8540, Japan
cFaculty of Science, Ehime University, 2-5 Bunkyo-cho, Matsuyama, Ehime, 790-8577, Japan. E-mail: sato.hisako.yq@ehime-u.ac.jp
First published on 17th October 2024
Abstract
Layered zinc(II) hydroxides (LZH) intercalating the deprotonated forms of R-(−) or S-(+)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (denoted as R- or S-BNDHPH, respectively) were prepared from Zn(NO3)2 at pH 5 and 60 °C by the mixing method. The obtained hybrid compounds (denoted as R- or S-BNDHP−/LZH, respectively) were heated from room temperature up to 800 °C under nitrogen atmosphere. According to the thermal gravimetric/differential thermal analysis measurements, hydroxyl groups were dehydrated at 270–400 °C, followed by the decomposition of organic components at 420–600 °C. X-ray diffraction patterns and scanning electron microscopy images indicated that the final products were a mixture of α-Zn3(PO4)2, ZnO crystals and non-crystalline zinc(II) orthophosphates. Vibrational circular dichroism (VCD) spectra were recorded before and after calcination. Before calcination, R- or S-BNDHP−/LZH exhibited VCD peaks assigned to intercalated R- or S-BNDHP−. The calcined products exhibited several VCD peaks in the range of 900–1200 cm−1, maintaining the mirror-image relationship between R-BNDHP−/LZH and S-BNDHP−/LZH used as starting materials. The observed peaks were assigned to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (symmetric), –POO−, and PO (asymmetric) stretching vibrations of the PO43− groups. According to theoretical simulations, the observed VCD activity can be rationalised in terms of vibrational coupling between two PO43− groups in a generated chiral zinc(II) orthophosphate cluster.
O (symmetric), –POO−, and PO (asymmetric) stretching vibrations of the PO43− groups. According to theoretical simulations, the observed VCD activity can be rationalised in terms of vibrational coupling between two PO43− groups in a generated chiral zinc(II) orthophosphate cluster.
Introduction
Chiral inorganic compounds containing no organic components are targeted as materials of both fundamental and practical value.1 The mechanism of generation of chirality in the network structures of inorganic oxides, hydroxides, or phosphates2 is in contrast with that of chiral organic compounds, in which chirality originates mostly from the asymmetric structures around carbon, nitrogen, or sulphur atoms or from the steric hindrance of bulky functional groups. From a practical viewpoint, chiral inorganic materials may possess characteristics that are hardly achieved by organic polymers, such as stability at high temperatures, resistance to either light illumination or chemical reagents, and transparency in the UV-vis region. These compounds are used in various functional applications, such as asymmetric catalysis, chiral sensors, and chromatographic adsorbents for optical resolution.Several studies have reported the generation of chirality in inorganic polymers.3–7 An example is the induction of chirality in the –O–Si–O– networks of silica. During synthesis, chirality is transferred from the organic adducts as a template or the asymmetric side chains in siloxanes during polymerisation. Chiral structures on the scale of a few nanometres, such as helically twisted ribbons, have been confirmed using scanning electric microscopy (SEM) or transmission electron microscopy (TEM).4
Most inorganic polymers are transparent in the UV-vis region. This property makes it difficult to apply chiroptical spectroscopy, such as electronic circular dichroism (ECD). To overcome this difficulty, vibrational circular dichroism (VCD) and Raman optical activity (ROA) have been applied to demonstrate nanoscale chirality.8–12 In particular, solid-state VCD (SD-VCD) is applicable for solid samples, in which the effects of linear interference are eliminated.13–23
As an example of two-dimensional nanomaterials, Zong et al. used layered zinc hydroxide (LZH) in which chirality was induced when the small molecules formed chiral supramolecular assemblies.24 The typical formula of LZH is expressed by Zn5(OH)8Ann−2/n·mH2O, where An is an intercalated anion such as Cl−, NO3−, SO42− and various organic anions.25 LZH basal layers consist of both octahedral and tetrahedral coordination and interlayer anions.26–30
In this study, LZH was prepared from Zn(NO3)2 using a mixing method. Deprotonated forms of R-(−) or S-(+)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate anions (denoted as R- or S-BNDHPH, respectively) were intercalated to replace –OH groups in LZH or electrostatic interactions between LZH and BNDHP−. The intercalated compounds were calcined at 800 °C to remove the organic components. The R- or S-BNDHP− anion was expected to act as a template to induce chirality. The produced inorganic materials (probably zinc(II) orthophosphate clusters) were analysed using SD-VCD. Consequently, evidence was obtained confirming that chirality was induced near the phosphate groups.
Results and discussion
Organic/inorganic hybrids as a precursor
The organic and inorganic hybrids of LZH and R- or S-BNDHP− were prepared by the mixing method, as described in the Experimental section. The products are denoted as R- or S-BNDHP−/LZH.Fig. 1(A) shows the powder X-ray diffraction (XRD) patterns of R-BNDHP−/LZH and S-BNDHP−/LZH at pH 5.0 and 60 °C. Fig. S1(A)† shows the XRD patterns of R-BNDHP−/LZH and S-BNDHP−/LZH, which were prepared at two different temperatures (40 °C and 60 °C) and pH value 3.8. Well-crystallised BNDHP−/LZH was difficult to prepare using this mixing method. As depicted in the inset of Fig. 1(A), a broad diffraction peak was observed at 2θ = 5.4°, indicative of BNDHP− intercalation within the LZH interlayer. Consequently, the resulting organic–inorganic hybrids exhibited a layered structure with a basal spacing of 1.63 nm. Among the samples, the diffraction peak was the sharpest for compounds prepared at 60 °C and pH 5.0, indicating the highest crystallinity under these conditions. The simultaneous formation of the LZH basal layer and the intercalation of BNDHP− may lead to the random orientation of BNDHP− anions in the interlayer space of LZH.
 | ||
| Fig. 1 (A) XRD patterns and (B) FT-IR spectra of BNDHP−/LZH, respectively: (a) R-BNDHP−/LZH (pH 5.0, 60 °C), and (b) S-BNDHP−/LZH (pH 5.0, 60 °C). The inset of (A) shows an expanded view of XRD patterns in the low-angle region (2θ = 0–10°). | ||
Fig. 1(B) shows the Fourier transform infrared (FT-IR) spectra of the hybrid products at pH 5.0 and 60 °C. Other data obtained under different conditions are shown in Fig. S1(B).† The broad absorption band in the wavenumber region of 3600–3000 cm−1 was assigned to the O–H stretching vibration of the hydroxide basal layer. Several weak absorption bands in the region 1590–1465 cm−1 were assigned to the ring-stretching vibration of the binaphthyl group. Strong and moderately strong absorption bands of the PO (symmetric), –POO−, and PO (asymmetric) stretching vibrations in R- or S-BNDHP− were observed in the region 1240–965 cm−1. The absorption band at 1380 cm−1 was assigned to the N–O stretching vibration of co-intercalated NO3− ions. The band near 1010 cm−1 was assigned to the OH bending mode of layer hydroxyl groups (OH–Zn).
XRD and IR results confirmed that the products were intercalated compounds of LZH with R- or S-BNDHP−. Because deprotonated R- or S-BNDHP− behaved as a mono-negative anion, the product was expressed as Zn5(OH)8(R- or S-BNDHP−)2−x(NO3)x·yEtOH.31 Further spectroscopic results are presented in the following section together with the calcined samples.
Calcination of R- or S-BNDHP−/LZH
R- or S-BNDHP−/LZH were used as precursors for the synthesis of chiral inorganic compounds. Fig. 2(a) and (b) show the thermal gravimetric/differential thermal analysis (TG/DTA) curves of BNDHP−/LZH (pH 5.0, 60 °C), respectively, when temperature was raised from 30 to 800 °C at a heating rate of 10 °C min−1. Fig. S2(a)–(d)† present the additional TG/DTA curves of BNDHP−/LZH, which were synthesized under various conditions.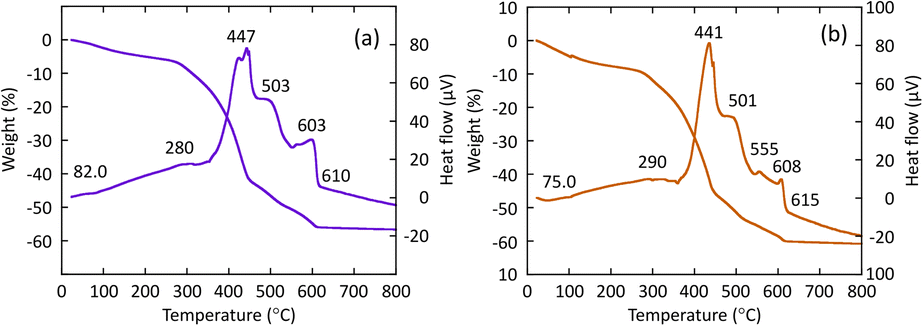 | ||
| Fig. 2 TG/DTA curves of BNDHP−/LZH, respectively: (a) R-BNDHP−/LZH (pH 5.0, 60 °C) and (b) S-BNDHP−/LZH (pH 5.0, 60 °C). | ||
The first weight loss and weak endothermic events observed at 70–82 °C were attributed to the desorption of ethanol molecules adsorbed on or intercalated between the LZH layers. The second major weight loss occurred at 270–450 °C due to the decomposition and combustion of the intercalated BNDHP−. This change was accompanied by an exothermic event, which was ascribed to the dehydration of the –OH groups in the LZH basal layer. A network of zinc(II) oxides may form during this step. The third slow weight loss took place at 450–600 °C. The fourth small weight loss with a medium exothermic event took place over the range 600–650 °C and was attributed to the crystallisation processes to form α-Zn3(PO4)2 and ZnO crystals, as indicated in the XRD patterns below.
Fig. 3(A) shows the XRD patterns of the final products when R- or S-BNDHP−/LZH formed at pH 5.0 and 60 °C were calcined at 500–800 °C. The diffraction peak of BNDHP−/LZH completely disappeared at 500 °C. The residual product might experience further crystallisation above 600 °C.
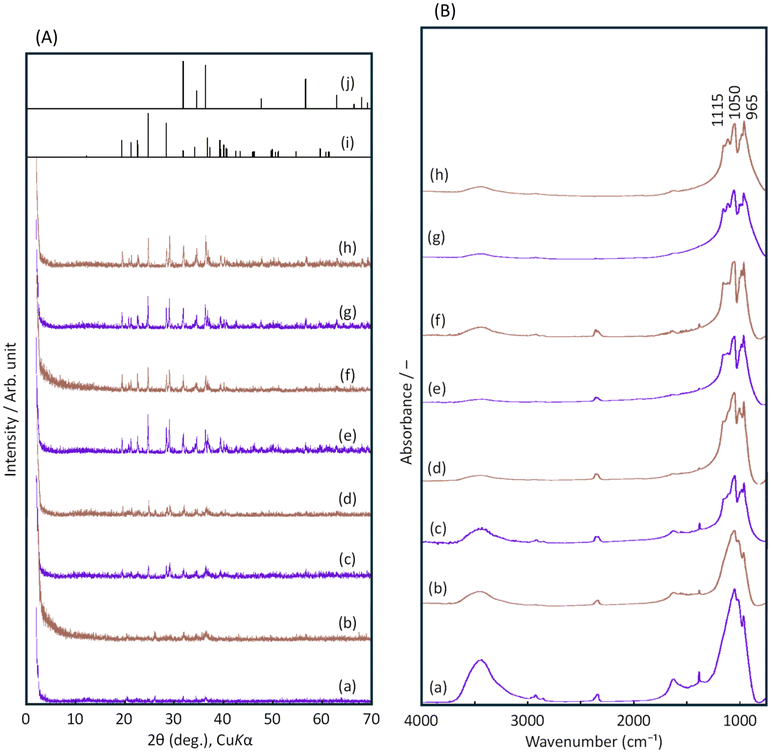 | ||
| Fig. 3 (A) XRD patters and (B) FT-IR spectra of calcined BNDHP−/LZH (pH 5.0, 60 °C), respectively: (a, c, e, and f) R-BNDHP−/LZH, (b, d, f, and h) S-BNDHP−/LZH. Calcination temperature (°C): (a and b) 500 °C; (c and d) 600 °C; (e and f) 700 °C; (g and h) 800 °C. (i) and (j) assigned to α-Zn3(PO4)2 (JCPDS #29-1390) and ZnO (JCPDS #36-1451), respectively. | ||
As a result, the final products exhibited small diffraction peaks in the range of 2θ = 20–40°. These peaks were attributed to α-Zn3(PO4)2 in comparison with the reported diffraction data of inorganic crystals.32,33 Additional diffraction peaks were attributed to zinc oxide.
Fig. 3(B) shows the FT-IR spectra of the calcined products. The band assigned to the stretching vibrations of the –OH groups almost disappeared. This reflected the dehydration of the OH groups. Three broad peaks at 1115, 1064, and 956 cm−1 were assigned to the asymmetric and symmetric stretching vibrations of the PO43− and the bending vibration of the P–O bonds, respectively, indicating the presence of PO43− groups. Summarising the results, the calcined final products were concluded to be a mixture of α-Zn3(PO4)2 (JCPDS #29-1390) and ZnO (JCPDS #36-1451) crystals and non-crystalline zinc orthophosphates.
X-ray photoelectron (XPS) measurements were performed before and after R-BNDHP−/LZH was calcinated at 800 °C. The results are shown in Fig. S3 and S4,† respectively. The C 1s, O 1s, Zn 2p3 and P 2p XPS core-level spectra are presented, along with the deconvoluted spectra of O 1s and Zn 2p3. The appearance of the C 1s signal in the region of 282–294 eV is consistent with the intercalation of BNDHP− and ethanol into the LZH interlayer space before calcination. After calcination, the signal intensity was significantly reduced because of the decomposition of BNDHP. The O 1s spectra exhibited a shift from 532.1 eV to 531.8 eV after calcination, indicating the formation of inorganic phosphate from the decomposition of the organic phosphate of BNDHP−.34 Deconvolution of the O 1s spectrum before calcination showed the presence of Zn–O and Zn–O–P (531.0 eV), Zn–OH and P–OH (532.1 eV), and P–O–C and ethanol (533.3 eV) for BNDHP−/LZH before calcination.35 After calcination, the deconvolution of the O 1s spectrum revealed various bonding environments, including Zn–O (529.6 eV), Zn–OH and Zn3(PO4)2 (531.4 eV), and H2O (533.6 eV). The results indicated the formation of α-Zn3(PO4)2 crystals. The increase in the Zn 2p3 peak for the calcined sample was attributed to the formation of zinc phosphate through the thermal decomposition of BNDHP−/LZH. The deconvolution of the Zn 2p3 spectra revealed the presence of two peaks of Zn–O–P (1020.9 eV) and Zn–OH and Zn–O–Zn (1022.5 eV), suggesting that the phosphate groups of BNDHP− were directly bound to the tetrahedral LZH basal layer.36 The P 2p core-level XPS spectrum exhibited a shift from 134.5 eV to 133.4 eV after calcination, indicating the formation of inorganic phosphates as a result of the thermal decomposition of the intercalated BNDHP−.
The above XPS results indicated that the decomposition of organic phosphates in BNDHP−/LZH led to the formation of inorganic phosphates, such as crystalline α-Zn3(PO4)2. The Zn/P molar ratios were calculated to be 1.6 and 1.5 before and after calcination, respectively. The values were close to those calculated for R- and S-BNDHP− (1.6) and α-Zn3(PO4)2 (1.5), respectively. The results suggested that phosphate groups were cleaved and left behind in the solids when intercalated R- or S-BNDHP− anions were decomposed at 420–600 °C. Thereafter, PO43− coordinates with a Zn(II) atom to form zinc(II) orthophosphate.
The scanning electron microscopy (SEM) images of BNDHP−/LZH before and after calcination at 800 °C are shown in Fig. S5.† The original LZH sample exhibits a plate-like morphology. The particles of BNDHP−/LZH had a granular shape and a size of approximately 100 nm. The calcined samples showed that the BNDHP−/LZH particles were sintered and changed into a lump shape upon thermal treatment. The small particles in the SEM images suggest the formation of ZnO.
Chirality aspects as studied by solid-state VCD
SD-VCD measurements were performed on the powder samples before and after calcination. The details of the SD-VCD measurements are described in the Experimental section.Fig. 4(a) shows the IR and VCD spectra of R- and S-BNDHP−/LZH prepared at 60 °C and pH 5.0. The mirror-image relationship was maintained in the VCD spectra between R-BNDHP−/LZH and S-BNDHP−/LZH. These anions preserved the chiral structure of the intercalated states.
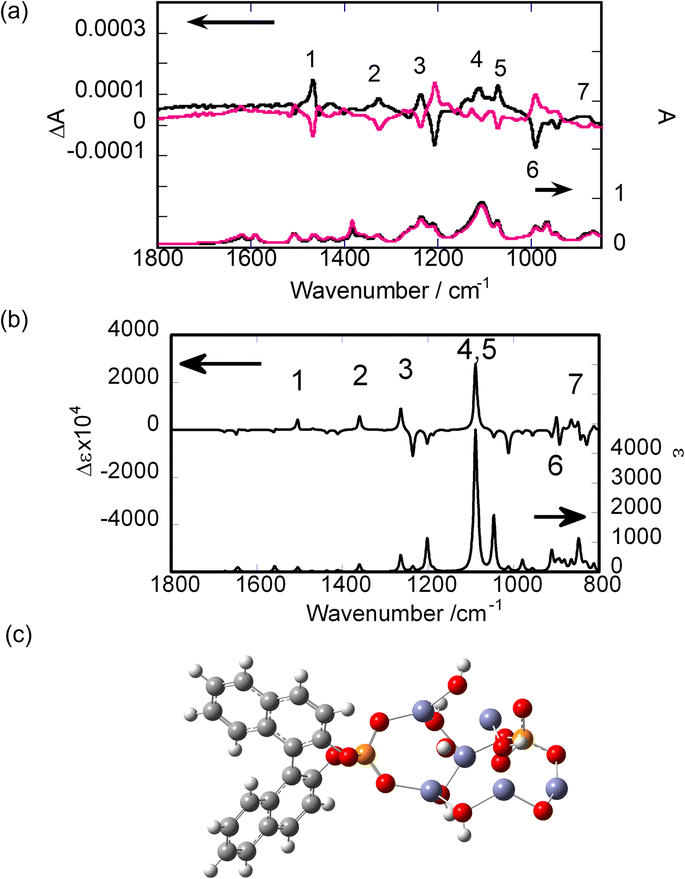 | ||
| Fig. 4 (a) Experimental IR (lower) and VCD (upper) spectra: black and red are R-BNDHP−/LZH and S-BNDHP−/LZH, respectively. (b) Calculated IR (lower) and VCD (upper) spectra for the assumed R-BNDHP−/LZH model. The number of each peak shows the correspondence between the experimental and theoretical VCD peaks. (c) The cluster model of R-BNDHP−/LZH employed for calculation. Number shows the correspondence between the experimental and theoretical VCD peaks. | ||
To assign the observed peaks, theoretical IR and VCD spectra were calculated. As a model structure, a cluster was truncated from an LZH crystal. A cluster with an elemental composition of Zn6O12H10 was selected. Deprotonated R-BNDHP− was attached to the cluster through the coordination of a phosphate group with two Zn(II) atoms. The IR and VCD spectra were calculated theoretically, as shown in Fig. 4(b). The optimal structure was obtained by minimising the total energy of formation, as shown in Fig. 4(c). The peaks were numbered for both the experimental and theoretical spectra. A satisfactory agreement was obtained between the positions and signs of the corresponding peaks. The IR and VCD spectra of KBr pellets for R- and S-BNDHPH are shown in Fig. S6.† The vibrations were assigned as reported for R- or S-BNDHPH.37
The vibrations assigned to the phosphate group were VCD-active in the range of 1200–1000 cm−1. Because the group itself is achiral, the observed activity might have been caused by cooperative vibrations between the phosphate group and the binaphthyl moiety. In fact, animation of the vibrational motion showed that both the phosphate group and binaphthyl moiety vibrated in a coherent fashion.
The calculation was made for the model system on a larger scale (Fig. S7†). As a result, the calculated VCD spectrum exhibited the peaks assigned to the OH vibrations in the inorganic layer. Such induction of chirality demonstrated the strong interaction between a guest molecule (R-BNDHP−) and a host (truncated LZH).
Next, SD-VCD measurements were performed on the calcinated samples. The lower part of Fig. 5(a) shows the IR spectra of the final samples. Sharp peaks assigned to R- or S-BNDHP− disappeared, leaving broad bands with overlapping small peaks in the region of 1200–900 cm−1. This reflects the elimination of organic components, as concluded from the TG/DTA results. The upper part of Fig. 5(a) shows the VCD spectra of the same samples. In contrast to the IR spectra, clear VCD signals are observed. Two couplet peaks were present around 1150 cm−1 and 1050 cm−1, respectively, numbered as 1, 2 and 3, 4, respectively. The order of +/− signs was the same for both couplets. Small peaks and broad bands were observed around 1100 cm−1 and 950 cm−1, respectively. All VCD peaks maintained a mirror-image relationship between the samples starting from either R-BNDHP− or S-BNDHP−/LZH as a precursor.
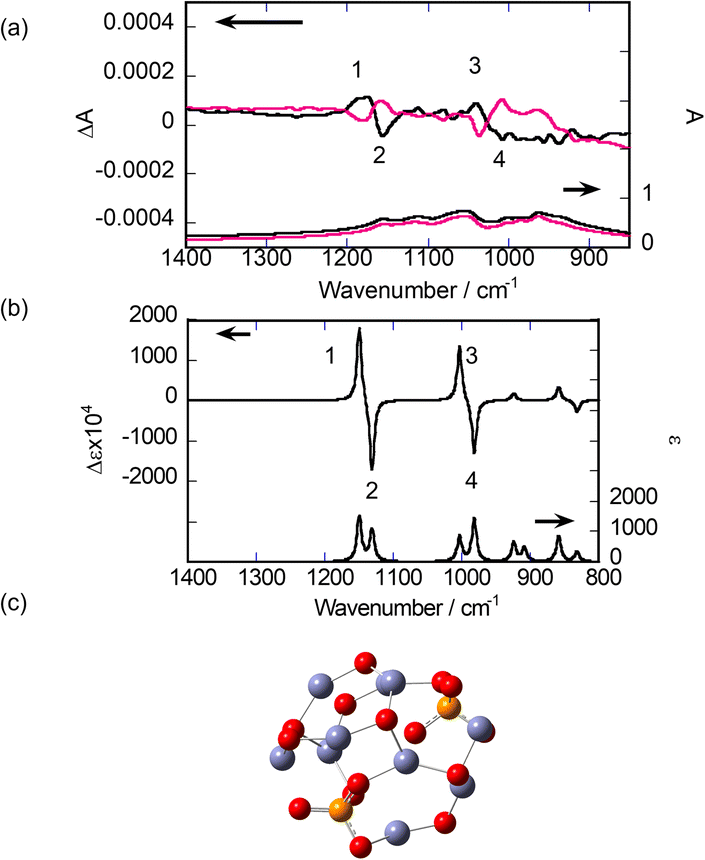 | ||
| Fig. 5 (a) Experimental IR (lower) and VCD (upper) spectra after calcination at 800 °C: black and red are the solid samples formed from R-BNDHP−/LZH or S-BNDHP−/LZH. (b) Calculated IR (lower) and VCD (upper) spectra for an assumed cluster model. (c) The most successful cluster model of Zn3(PO4)2(ZnO)7. Number shows the correspondence between the experimental and theoretical VCD peaks. | ||
The above peaks are most probably assigned to the stretching vibrations of P–O bonds in the PO43− groups. This is because the vibrational peaks assigned to the network of zinc(II) oxide (or O–Zn–O) are expected to appear below 800 cm−1, according to the reported IR data for ZnO.38
To analyse the observed results, the theoretical calculation of the VCD spectrum was performed for a model cluster containing Zn(II), PO43−, and bridging oxygen (–O–). In the simulation, the focus was on how to reproduce the two couplets (or 1, 2 and 3, 4) as observed experimentally. As described in the preceding section, the XRD patterns indicate the formation of α-Zn3(PO4)2 in the final product (Fig. 3). According to the literature, α-Zn3(PO4)2 crystals belong to the space group of P21/c.32,38 As the first trial, a cluster was truncated from an α-Zn3(PO4)2 crystal. After optimising the structure by minimising the formation energy, IR and VCD spectra were calculated. Seven clusters were chosen, as shown Fig. S8 in the ESI.† However, none of them predicted two couplets in the VCD spectra, as observed experimentally (or 1, 2 and 3, 4). The two neighbouring PO43− groups were bridged by one unit of –O–Zn–O– in these models. This distance was thought to be too short to correctly provide couplets in the VCD spectra.
As the next trial, a model structure was constructed by combining two components, α-Zn3(PO4)2 and ZnO, at different ratios, while aiming to place two PO43− groups at more remote positions. Several examples are shown in Fig. S9 in the ESI.† After optimising the structures by minimising the formation energy, the VCD spectra were theoretically calculated. Among the models, the most satisfactory result was obtained, as shown in Fig. 5(b). Its structure comprised one α-Zn3(PO4)2 and seven Zn–O pairs (Fig. 5(c)). According to the vibrational animations, the couplets around 1150 cm−1 (numbered 1, 2) and 1000 cm−1 (numbered 3, 4) represent the cooperative vibrations of P–O and PO bonds in the two attached PO43− groups, respectively. The orders of the +/− signs of the peaks were identical to those of the calcinated sample of R-BNDHP−/LZH.
The main reason for the success of the theoretical reproduction of the VCD couplets may lie in choosing the proper distance between the two PO4 groups, as represented by –P–O–Zn–O–Zn–O–P–. As a further attempt, the dimer comprising two α-Zn3(PO4)2 and fourteen Zn–O pairs was constructed. IR and VCD spectra were obtained (Fig. S10†). In addition to these two couplets, the dimer model predicts small peaks between them. Presently, the monomeric and dimeric models proposed above are considered the most plausible for rationalising the VCD activity in the calcined products.
Proposal of calcination mechanisms
Based on the above experimental and theoretical results, the following reactions are proposed to occur successively during the calcination of R- or S-BNDHP−/LZH.(i) 270–400 °C: the –OH groups were dehydrated around the Zn(II) ions, forming –Zn–O– networks. Chirality may be transferred from intercalated R- or S-BNDHP− to the networks.
(ii) 420–600 °C: R- or S-BNDHP− was thermally decomposed, cleaving the PO43− group. The PO43− groups remained coordinated with the Zn(II) ions in the –Zn–O– network. During this process, chiral clusters containing Zn(II) and PO43− were formed.
(iii) 620–800 °C: the crystal growth of α-Zn3(PO4)2 and ZnO occurred. Chiral clusters containing Zn(II) and PO43− groups constituted amorphous parts.
In cases where the above mechanism is correct, the present results are the first examples showing the induction of chirality around a P atom in purely inorganic compounds.39
Experimental part
General procedure for synthesis of compounds BNDHP−/LZH and Zn3(PO4)2
R- and S-BNDHPH were obtained from Tokyo Chemical Industry Co., Ltd. Zn(NO3)2·6H2O, NaOH, and 99.5% EtOH were purchased from FUJIFILM Wako Pure Chemical Corporation and used without further purification. The reagents used for the synthesis of BNDHP−/LZH were Zn(NO3)2, NaOH, and 99.5% EtOH as solvent. BNDHP−/LZH was synthesized using a mixing method, namely, 20.0 mL of 0.10 M Zn(NO3)2 solution was added to 200 mL of 10.0 mM BNDHP solution with a BNDHP/Zn molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by stirring for 30 minutes. The mixture was then adjusted to pH 3.8–5.0 using NaOH. The precipitate was collected via centrifugation and washed once with 99.5% EtOH. The solid product was dried at 40 °C for 24 h in a decompression oven. The BNDHP−/LZH was calcined at temperatures of 500, 600, 700 and 800 °C for 3 h under air atmosphere using an electric furnace.
:1, followed by stirring for 30 minutes. The mixture was then adjusted to pH 3.8–5.0 using NaOH. The precipitate was collected via centrifugation and washed once with 99.5% EtOH. The solid product was dried at 40 °C for 24 h in a decompression oven. The BNDHP−/LZH was calcined at temperatures of 500, 600, 700 and 800 °C for 3 h under air atmosphere using an electric furnace.
XRD measurements
Powder X-ray diffraction (XRD) measurements were performed on a Bruker D8 Advance diffractometer using CuKα radiation at 40 mA and 40 kV in the 2θ range from 2° to 70°, with the sampling step and counting time of 0.01° and 1.0 s, respectively.TG/DTA measurements
Thermogravimetry and differential thermal analysis (TG/DTA) were recorded using a Rigaku Thermo plus EVO2 TG-DTA8120 analyser in the range of 30–800 °C under an N2 atmosphere at a heating rate of 10 °C min−1.FT-IR measurements
Fourier transform infrared (FT-IR) spectra were recorded using a JASCO FT/IR-4600AC spectrophotometer in the wavenumber range of 4000–650 cm−1 by the standard KBr disk method.XPS measurement
X-ray photoelectron (XPS) spectra of BNDHP−/LZH and its calcined products were obtained using an ULVAC-PHI PHI 5000 V Versa Probe analyser. The X-ray source was AlKα radiation, and the electron takeoff angle was θ = 45°. The C 1s peak energy of 284.8 eV was used as the calibrated energy standard.SEM measurements
Scanning electron microscopy (SEM) images of BNDHP−/LZH before and after calcination were obtained using an ultralow accelerating voltage scanning electron microscope (ULV-SEM) on a JEOL JSM-7800 PRIME instrument. The sample was coated with osmium at a thickness of 2–6 nm, and measurements were made at an accelerating voltage of 5.0 kV.VCD measurements
VCD spectra were measured using a spectrometer developed in-house with the cooperation of the JASCO Corporation, Japan (MultiD-MIRAI-2020).20 The machine was a concurrent system combined with a quantum cascade laser (QCL-VCD) covering a wavenumber range of 1500–1740 cm−1 and FT-VCD covering 800–2000 cm−1. In this study, the samples were measured in FT-VCD mode using a normal cell holder under normal positioning. The wavenumber resolution was 4 cm−1. The sample was prepared by mixing an intercalation compound with KBr (0.90 or 1.0%) and pressing it onto a disc with a diameter of 10 mm. Signals were accumulated more than 10000 times. No baseline correction was performed for any of the samples.
Computational details
The theoretical IR and VCD spectra of the complexes were calculated using the Gaussian 16 program (C.01).40 The truncated cluster model was created on the basis of the X-ray structure of LZH, α-Zn3(PO4)2 or γ-Zn3(PO4)2.31,32 Geometry optimisation was performed at the DFT level, in which the B3LYP functional with LanL2DZ was employed for Zn(II) and 6-31G(d,p) for the other atoms. The VCD intensities were determined from the vibrational rotational strength and magnetic dipole moments, which were calculated using the magnetic field perturbation theory formulated using magnetic-field gauge-invariant atomic orbitals. The calculated intensities were converted to Lorentzian bands with a half-width of 4 cm−1 at half-height. The observed spectra were assigned based on animations of the molecular vibration with Gaussview 6.0 (Gaussian Inc.).Conclusions
A layered zinc(II) hydroxide intercalating the deprotonated anion of R-(−) or S-(+)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate was prepared. The compound was heated to 800 °C to remove organic components. The calcined material was a mixture of α-Zn3(PO4)2, ZnO crystals, and amorphous zinc(II) orthophosphates. With the help of theoretical simulations, the VCD spectra suggested that the two PO43− groups vibrated coherently when they were present in the same chiral cluster of zinc orthophosphate. The VCD method demonstrated its utility for analysing nanoscale chirality in noncrystalline inorganic materials.Author contributions
Sumio Aisawa: conceptualisation, methodology, investigation and writing the original draft; Nami Horiguchi: methodology, formal analysis, investigation, data curation and visualisation; Chika Chida: methodology, formal analysis, investigation, data curation and visualisation; Jing Sang: conceptualisation, methodology, investigation and XPS methodology; Hidetoshi Hirahara: conceptualisation, methodology, investigation and XPS methodology; Akihiko Yamagishi: conceptualisation, investigation, discussion and writing the original draft; Hisako Sato: conceptualisation, investigation, VCD methodology, DFT calculation and writing of the original draft.Data availability
The data supporting this article have been included as part of the ESI† and the digital data of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI (grant number: JP 20K21090, JP22K05263, JP22H02033 and JP23K23301). The computations were performed in collaboration with the Research Center for Computational Science, Okazaki, Japan (Project24-IMS-C162).References
- N. H. Cho, A. Guerrero-Martínez, J. Ma, S. Bals, N. A. Kotov, L. M. Liz-Marzán and K. T. Nam, Nat. Rev. Bioeng., 2023, 1, 88–106 CrossRef.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 12, 8041–8093 CrossRef PubMed.
- Y. Duan and S. Che, Adv. Mater., 2023, 35, 2205088 CrossRef CAS PubMed.
- S. Che, Z. Liu, T. Ohsuna, K. Sakamoto, O. Terasaki and T. Tatsumi, Nature, 2004, 429, 281–284 CrossRef CAS PubMed.
- H. Jin, Z. Liu, T. Ohsuna, O. Terasaki, Y. Inoue, K. Sakamoto, T. Nakanishi, K. Ariga and S. Che, Adv. Mater., 2006, 18, 593–596 CrossRef CAS.
- Q. Huibin and C. Shunai, Adv. Mater., 2023, 35, 2205088 CrossRef PubMed.
- J. Xie, Y. Duan and S. Che, Adv. Funct. Mater., 2012, 22, 3784–3792 CrossRef CAS.
- M. Krupová, J. Kessler and P. Bouř, ChemPlusChem, 2020, 85, 561–575 CrossRef PubMed.
- E. Er, T. H. Chow, L. M. Liz-Marzán and N. A. Kotov, ACS Nano, 2024, 18, 12589–12597 CrossRef CAS PubMed.
- J. Kwon, K. H. Park, W. J. Choi, N. A. Kotov and J. Yeom, Acc. Chem. Res., 2023, 56, 1359–1372 CrossRef CAS PubMed.
- P. R. Lassen, L. Guy, I. Karame, T. Roisnel, N. Vanthuyne, C. Roussel, X. Cao, R. Lombardi, J. Crassous, T. B. Freedman and L. A. Nafie, Inorg. Chem., 2006, 45, 10230–10239 CrossRef CAS PubMed.
- H. Sato, K. Tamura, K. Takimoto and A. Yamagishi, Phys. Chem. Chem. Phys., 2018, 20, 3141–3147 RSC.
- J. E. Rode, K. Łyczko, D. Kaczorek, R. Kawęcki and J. C. Dobrowolski, Spectrochim. Acta, Part A, 2024, 310, 123939 CrossRef CAS PubMed.
- J. E. Rode, J. Wasilczenko and M. Górecki, Spectrochim. Acta, Part A, 2024, 310, 123851 CrossRef CAS PubMed.
- S. Jähnigen, Angew. Chem., Int. Ed., 2023, 62, e202303595 CrossRef PubMed.
- M. Krupová, P. Leszczenko, E. Sierka, S. E. Hamplová, B. Klepetářová, R. Pelc and V. Andrushchenko, Spectrochim. Acta, Part A, 2024, 319, 124381 CrossRef PubMed.
- Y. Okazaki, T. Buffeteau, E. Siurdyban, D. Talaga, N. Ryu, R. Yagi, E. Pouget, M. Takafuji, H. Ihara and R. Oda, Nano Lett., 2016, 16, 6411–6415 CrossRef CAS PubMed.
- Y. Kaneko, H. Toyodome and H. Sato, J. Mater. Chem., 2011, 21, 16638–16641 RSC.
- H. Sato, Phys. Chem. Chem. Phys., 2020, 22, 7671–7679 RSC.
- H. Sato, J. Yoshida and A. Yamagishi, Multi-dimensional Vibrational Circular Dichroism, Springer Nature Singapore Ltd, Singapore, 2024 Search PubMed.
- H. Sato, K. Takimoto, A. Yamagishi, J. Yoshida and M. Hara, Appl. Clay Sci., 2022, 228, 106621 CrossRef CAS.
- H. Sato, K. Takimoto, I. Kawamura and S. Aisawa, Bull. Chem. Soc. Jpn., 2019, 92, 1779–1784 CrossRef CAS.
- S. Aisawa, C. Chida, H. Ida, J. Sang, H. Hirahara and H. Sato, Appl. Clay Sci., 2023, 244, 107108 CrossRef CAS.
- Y. Zong, S.-M. Xu, W. Shi and C. Lu, ACS Nano, 2023, 17, 3838–3846 CrossRef CAS PubMed.
- W. Stählin and H. R. Oswald, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 860–863 CrossRef.
- W. Ma, R. Ma, J. Liang, C. Wang, X. Liu, K. Zhou and T. Sasaki, Nanoscale, 2014, 6, 13870–13875 RSC.
- A. H. M. Leung, S. D. Pike, A. J. Clancy, H. C. Yau, W. J. Lee, K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Sci., 2018, 9, 2135–2146 RSC.
- N. Gorodylova, S. Cousy, P. Šulcová and L. Svoboda, J. Therm. Anal. Calorim., 2017, 127, 675–683 CrossRef CAS.
- D. A. Almasri, R. Essehli, Y. Tong and J. Lawler, RSC Adv., 2021, 11, 30172–30182 RSC.
- J.-R. Zhang, Y. Mo, W. Fang, Y.-X. Yuan, J.-L. Yao and J. H. Wu, Inorg. Chem., 2024, 63, 3692–3701 CrossRef CAS PubMed.
- T. Shinagawa, M. Watanabe, T. Mori, J. I. Tani, M. Chigane and M. Izaki, Inorg. Chem., 2018, 57, 13137–13149 CrossRef CAS PubMed.
- C. Calvo, Can. J. Chem., 1965, 43, 436–445 CrossRef CAS.
- B. Boonchom, R. Baitahe, S. Kongtaweelert and N. Vittayakorn, Ind. Eng. Chem. Res., 2010, 49, 3571–3576 CrossRef CAS.
- A. M. Kuchkaev, A. M. Kuchkaev, A. V. Sukhov, S. V. Saparina, O. I. Gnezdilov, A. E. Klimovitskii, S. A. Ziganshina, I. R. Nizameev, I. P. Asanov, K. A. Brylev, O. G. Sinyashin and D. G. Yakhvarov, Int. J. Mol. Sci., 2023, 24, 3095 CrossRef CAS PubMed.
- P. I. Hernández-Velázquez, J. A. Gutiérrez-Ortega, G. G. Carbajal-Arizaga, R. Manríquez-González, W. D. Cruz-Hernández and S. Gómez-Salazar, J. Mex. Chem. Soc., 2019, 63, 130–152 Search PubMed.
- N. S. Hassan, A. A. Jalil, L. Y. Twu, N. A. A. Fatah, H. U. Hambali, I. Hussain and M. L. Firmansyah, Int. J. Hydrogen Energy, 2023, 48, 20525–20537 CrossRef CAS.
- V. Setnička, M. Urbanová, P. Bouř, V. Král and K. Volka, J. Phys. Chem. A, 2001, 105, 8931–8938 CrossRef.
- G. Hitkari, S. Singh and G. Pandey, Nano-Struct. Nano-Objects, 2017, 12, 1–9 CrossRef CAS.
- L. V. Shvanskaya, O. S. Volkova and A. N. Vasiliev, J. Alloys Compd., 2020, 835, 155028 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2019.
Footnote |
| † Electronic supplementary information (ESI) available: XRD and FT-IR spectra under various conditions of the mixing method; TG/DTA curves under various conditions of the mixing method; XPS spectra of R-or S-BNDHP−/LZH before and after calcination at 800 °C; the deconvoluted XPS spectra of R-BNDHP−/LZH before and after calcination at 800 °C; SEM images of R- or S-BNDHP−/LZH before and after calcination at 800 °C; experimental solid-state IR and VCD spectra of R- and S-BNDHPH; IR and VCD spectra calculated for the intercalation model; IR and VCD spectra calculated for the model clusters truncated from α- or γ-Zn3(PO4)2 crystals; IR and VCD spectra calculated for the model clusters composed of α-Zn3(PO4)2 and ZnO at various ratios; IR and VCD spectra calculated for the dimer model of the zinc(II) orthophosphate cluster. See DOI: https://doi.org/10.1039/d4nr03809f |
| This journal is © The Royal Society of Chemistry 2024 |