
Synthesis of high molar mass all-(meth)acrylic thermoplastic elastomers by photo-iniferter RAFT polymerisation†
Izabela
Kurowska
 a,
Maksym
Odnoroh
b,
Oleksandr
Ivanchenko
b,
Marc
Guerre
b and
Mathias
Destarac
*b
a,
Maksym
Odnoroh
b,
Oleksandr
Ivanchenko
b,
Marc
Guerre
b and
Mathias
Destarac
*b
aFaculty of Chemistry, University of Bialystok, Ciolkowskiego 1k, 15-245 Bialystok, Poland
bLaboratoire SOFTMAT, CNRS UMR 5623, Université of Toulouse, Université Toulouse III-Paul Sabatier, 118 route de Narbonne, F-31062 Toulouse, France. E-mail: mathias.destarac@univ-tlse3.fr
First published on 18th November 2024
Abstract
The synthesis of PMMA-b-PnBA-b-PMMA triblock copolymers using photoiniferter reversible addition–fragmentation chain transfer (PI-RAFT) polymerisation is reported. By utilising a combination of green and blue LED irradiation, copolymers with high molar masses up to Mn ∼800 kg mol−1, low dispersities (Đ < 1.25), and thermoplastic elastomer behaviour were synthesised.
Introduction
Thermoplastic elastomers (TPEs) represent a class of materials that combine the elasticity of rubbers with the processability of thermoplastics, offering diversity in applications such as adhesives, automotive components, coatings, fibers, and medical devices.1–3 Typical examples of TPEs are ABA triblock copolymers, which consist of outer hard amorphous or semicrystalline blocks (A) with a high glass transition temperature (Tg) and an inner thermodynamically immiscible soft segment (B) with a Tg below ambient temperature. Styrenic block copolymers (SBCs) are among the most studied and widely used TPEs, with typical examples being polystyrene-b-polybutadiene-b-polystyrene (SBS) and polystyrene-b-polyisoprene-b-polystyrene (SIS), prepared by living anionic polymerisation. Despite their commercial availability, SBCs face limitations, including UV stability and oxidative resistance arising from unsaturated bonds in polydiene soft blocks.1,4 These drawbacks underscore the need for alternative materials. In this context, all-(meth)acrylic-based TPEs have emerged as promising alternatives, offering numerous advantages, including improved optical transparency, weatherability, printability, and resistance to oils and abrasion.1,5 Importantly, they are also integral to the shift toward a sustainable plastics economy due to the recyclability of thermoplastics and possible use of biosourced alcohol precursors for monomer supply.In the quest to develop high-performance TPEs with properties comparable to those achieved through living anionic polymerisation, our previous research focused on the synthesis of methacrylate–acrylate–methacrylate (hard–soft–hard) ABA triblock copolymers using reversible addition–fragmentation chain transfer (RAFT) polymerisation.6 Until recently, the RAFT method had shown serious limitations in producing well-defined (meth)acrylic ABA copolymers. The available symmetrical trithiocarbonates (TTCs) used as chain transfer agents (CTAs) exhibited moderate reactivity with methacrylates, resulting in poorly controlled polymers with relatively high dispersities (Đ = 1.4–1.8).7–10 Our group has addressed this issue by developing a symmetrical TTC, bis-(2-methylpropanenitrile)trithiocarbonate (TTC-bCP), with a 2-cyanopropan-2-yl leaving group that provides an optimal fragmentation rate for methacrylates as reflected through the high apparent transfer constant (Capptr = 13.8) determined for methyl methacrylate (MMA).11 As a result, TTC-bCP allowed the successful synthesis of ABA triblock copolymers with a relatively high molar mass (Mn = 135 kg mol−1) and remarkably low dispersity (Đ = 1.04).
Growing interest in more environmentally-friendly RAFT polymerisation has driven the exploration of alternative initiation routes.12 One particularly promising strategy is photoiniferter reversible addition–fragmentation chain transfer (PI-RAFT) polymerisation,13,14 where light directly triggers the dissociation of the CTA, thus initiating the polymerisation without an exogenous radical source. This approach offers several advantages over the traditional azo-initiated method, including mild reaction conditions, high spatial and temporal control, and tolerance to oxygen. The utilisation of light-emitting diodes (LEDs) as the light source further enhances the efficiency and versatility of PI-RAFT polymerisation. LEDs offer distinct advantages, including specific wavelength emission, low voltage operation, minimal heat generation, extended lifespan, and cost-effectiveness.15,16 Moreover, incorporating LED light as an external control in PI-RAFT allows for fine-tuning parameters such as light intensity, wavelength, and duration of irradiation.13,14 This capability enables precise control over polymerisation kinetics, facilitating the synthesis of polymers for diverse applications, including 3D printing,17,18 production of well-defined nanostructures,19 surface functionalization,20 and synthesis of multiblock copolymers.21 Furthermore, PI-RAFT facilitates the synthesis of well-defined high molar mass (HMM) polymers,22–24 which are highly desirable for rheology modifiers in liquid formulations,25,26 and for enhanced mechanical and thermal properties of 2D- and 3D-materials due to their decreased contribution of chain ends,27 enhanced intermolecular forces and increased chain entanglement density.28,29 Unlike traditional RAFT polymerisation, PI-RAFT eliminates the need for traditional low-molar mass radical initiators, thus potentially minimizing termination events.
In this work, we investigated the synthesis of high molar mass methacrylate–acrylate–methacrylate (hard–soft–hard) ABA triblock copolymers via PI-RAFT polymerisation with TTC-bCP as a chain transfer agent (Scheme 1). By proposing an energy-saving initiation mode using LED light, we seek to contribute to the development of more environmentally friendly strategies for TPE material synthesis. We demonstrate our approach using PMMA as the hard block (A) and PnBA as the soft block (B).6
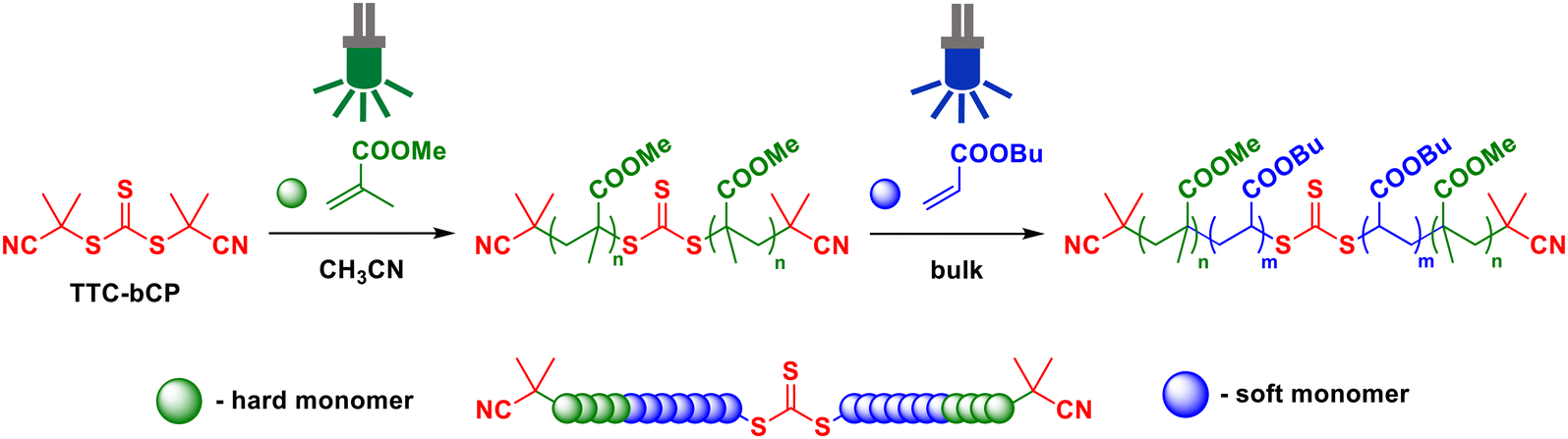 | ||
| Scheme 1 General scheme for the synthesis of ABA triblock copolymers using PI-RAFT. | ||
Synthesis of a hard block: PMMA
The UV-VIS absorption spectrum of TTC-bCP was investigated to understand its behaviour during PI-RAFT polymerisation (Fig. S1†). TTC-bCP absorbs light in the UV region (265–365 nm) due to the spin allowed π → π* electronic transition of the thiocarbonyl group, and the absorption in the visible light region (385–545 nm) is caused by the spin forbidden n → π* electronic transition. Although the band related to the spin forbidden n → π* transition is much weaker, it facilitates efficient excitation and photolysis required to start polymerisation.13,30 Consequently, TTC-bCP can be excited by either blue or green light (Fig. S1†).Initially, two light wavelengths, 460 nm (blue) and 530 nm (green), were employed to drive MMA polymerization targeting Mn,th = 50 kg mol−1. Polymerizations were conducted in a 50% w/w acetonitrile (CH3CN) solution ([MMA]0 = 4.28 mol L−1) using handmade photoreactors equipped with 60 LEDs, with light intensities of 3.33 ± 0.43 mW cm−2 for the blue light and 2.35 ± 0.17 mW cm−2 for the green light (Fig. S2, and Table S1†). Due to the cylindrical form of our reactor and the fact that the detector can absorb only a small fraction of the light and only from one side, the accuracy of the intensity measurements is questionable. The reported values should be considered more qualitatively than quantitatively, only tending to show that more LEDs give higher intensity as well as blue LEDs have higher intensity compared to green ones. After 16 h of irradiation, blue light resulted in a higher conversion of 81%, compared to 61% with green light. This difference can be attributed to the higher energy of blue light, which promotes faster radical generation and thus enhances the polymerisation rate. However, SEC analysis revealed significant differences in polymerisation control between these samples. Specifically, the molar mass of PMMA synthesized under green light showed close agreement between Mn obtained from SEC (Mn,SEC = 28 kg mol−1, Đ = 1.22) and the theoretical value (Mn,th = 31 kg mol−1). Conversely, PMMA synthesized with blue light exhibited a lower Mn,SEC = 30 kg mol−1 than the expected value (Mn,th = 41 kg mol−1) and was characterized by higher dispersity (Đ = 1.62). This was corroborated by SEC-RI chromatograms, indicating a broader non-monomodal profile for blue light-initiated polymerisation (Fig. S3a†). Moreover, TTC mid-chain fragments were detected in green light-initiated polymerisation, evidenced by strong UV absorbance at 290 nm. In contrast, no signal was observed for polymerisation with blue LEDs, indicating the photodegradation of TTC fragments and a lack of control over the process (Fig. S3b†). These findings indicated that green light enables a more controlled polymerisation of MMA, attributed to the lower energy provided for the fragmentation of TTC-bCP and subsequent PMMA-TTC-bCP. Consequently, it was chosen for further investigations. The selected conditions (green LED light, 50% w/w MMA in CH3CN) were tested for a targeted Mn of 200 kg mol−1. Polymerisation proceeded for 32 hours, achieving a final conversion of 85%. However, the molar masses determined by SEC were notably lower than theoretical ones (Fig. 1 and Table S2†). In order to increase the rate of polymerisation, a higher concentration of MMA in acetonitrile 70% w/w ([MMA]0 = 6.21 mol L−1) was tested. This adjustment resulted in a closer alignment between the molar masses measured by SEC and the theoretical values. Nevertheless, longer irradiation times resulted in bimodal chromatograms (Fig. S4†) coupled with broad dispersity (Đ ∼ 1.5), presumably because of photodegradation of the TTC sulfanyl radical as reported by several groups.23,31,32 To address this issue, we decided to decrease the radical concentration by reducing the number of LEDs in the photoreactor.33
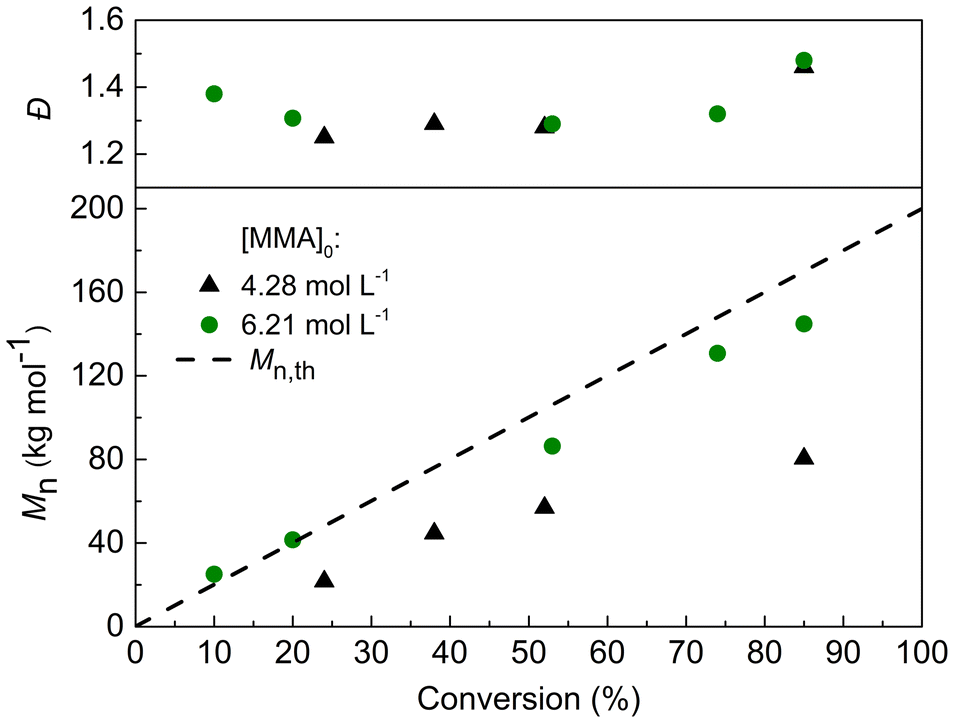 | ||
| Fig. 1 Evolution of Mn and Đ during PI-RAFT polymerisation at ambient temperature with different concentrations of MMA in acetonitrile. | ||
To further investigate PI-RAFT polymerisation of MMA, two additional initial concentrations of TTC-bCP (12.49 and 1.55 mmol L−1) corresponding to theoretical Mn of 50 and 400 kg mol−1 were selected. The resulting polymers demonstrated a linear increase in Mn,SEC with monomer conversion, aligning with theoretical values and exhibiting dispersities below 1.5 (Fig. 2 and Table S3†). SEC traces were monomodal throughout polymerisation and shifted toward a lower elution time with increasing monomer conversion (Fig. S5†). The polymerisation rate increased with increasing initial concentration of the CTA, which is a typical pattern in PI-RAFT approach. After 32 hours of irradiation, the conversion reached 94% for [TTC-bCP]0 = 12.49 mmol L−1. In contrast, for [TTC-bCP]0 = 1.55 mmol L−1, the monomer conversion was lower (58%) after the same time. Also, dispersities for polymers of [TTC-bCP]0 = 12.49 mmol L−1 (Đ = 1.27–1.33) were lower than those of [TTC-bCP]0 = 1.55 mmol L−1 (Đ = 1.29–1.40).
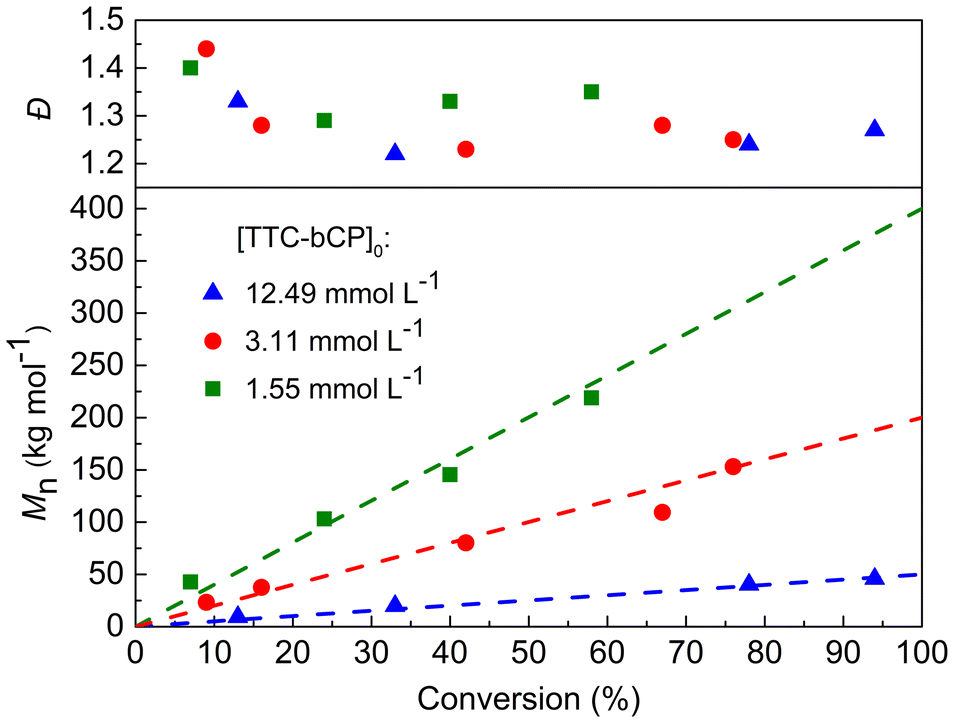 | ||
| Fig. 2 Evolution of Mn and Đ during PI-RAFT polymerisation of MMA for different targeted molar masses. Dashed lines represent the theoretical molar masses. | ||
It is also worth mentioning that attempts were made to polymerise MMA in bulk. For example, for a Mn,th of 400 kg mol−1, the conversion reached 52% after 16 hours of irradiation. The molar mass determined by SEC (Mn, SEC = 192 kg mol−1) was close to the theoretical assumption (Mn,th = 208 kg mol−1). The dispersity was also low (Đ = 1.24), and the chromatogram exhibited a monomodal distribution (Fig. S6†). However, this method was subsequently abandoned because of the difficulties in solubilizing the polymer samples for SEC or NMR analysis, and for the purification of PMMA-TTC-bCP prior to triblock synthesis due to the excessively high viscosity of the reaction medium.
Synthesis of a soft block: PnBA
PI-RAFT with green light activation was then tested for the polymerisation of n-butyl acrylate (nBA). As our previous study showed the presence of chain transfer to solvent using toluene,6 this reaction was performed in bulk. However, no evidence of polymerisation was observed even after 24 h of irradiation. Thus, the choice of the light wavelength depends not only on the type of CTA but also on the reactivity of the monomer considered. Upon cleavage of TTC-bCP, acrylates form secondary radicals, while methacrylates produce more stable tertiary radicals. The C–S bond dissociation energy increases with the insertion of an acrylate monomer that provides less radical stabilisation but results in a more stable acrylate-TTC adduct. Therefore, the lower energy green light was insufficient in the polymerisation of nBA. In the case of PMMA-TTC-bCP, which has a weaker C–S bond dissociation energy, blue light was too powerful, as indicated by the lack of control over the polymerisation. Based on these findings, the reaction conditions were changed, and blue light was tested. The polymerisations were conducted in bulk with a photoreactor equipped with 60 LEDs targeting molar masses of 200, 400, 800, and 1500 kg mol−1 ([TTC-bCP]0 = 4.45, 2.23, 1.11, 0.59 mmol L−1, respectively). All polymerisations showed high efficiency. Analysis of the polymers via SEC revealed a linear correlation between their molar masses and monomer conversion, aligning with theoretical predictions (Fig. 3, and Table S4†). Moreover, all polymers were characterised by remarkably low dispersities. However, in the early stages of all polymerisations, higher dispersities were observed, which subsequently diminished as monomer conversion progressed. As with any RAFT polymerization, the average number of nBA units added to propagating chains between two reversible transfer events decreases with monomer concentration, leading to a decrease in dispersities over the course of polymerisation.6 Once approximately 30% of monomer conversion was attained, dispersities stabilised at a lower level of <1.07. In line with previous findings on PI-RAFT, the highest conversion achieved tended to decrease with an increase in Mn,th, which corresponds to a decrease in the concentration of TTC-bCP as the only source of radicals. For instance, final conversions ranged from a minimum of 45% for [TTC-bCP]0 = 0.59 mmol L−1 to a maximum of 88% for [TTC-bCP]0 = 4.45 mmol L−1 (Fig. 3). SEC-RI traces also confirmed the controlled character of the polymerisation (Fig. S7†). SEC chromatograms were symmetrical and monomodal in all cases and shifted toward a lower elution time with increasing monomer conversion. Importantly, our approach significantly enhanced the accessibility of high molar mass PnBA with remarkably low dispersity (Mn = 718 kg mol−1, Đ = 1.02) compared to other RDRP methods reported in the literature. For example, PnBA of high molar mass (Mn = 612 kg mol−1) with Đ = 1.25 was prepared by ATRP under a high-pressure condition (5 kbar).34 PnBA with Mn > 500 kg mol−1 was also synthesized by TERP, but polymers were characterized by higher dispersity (∼1.4).35 However, in the case of methyl acrylate, photoiniferter-mediated polymerisation enabled the synthesis of ultrahigh molar mass polymers with Mn up to 5520 kg mol−1 and low dispersity (Đ = 1.10).23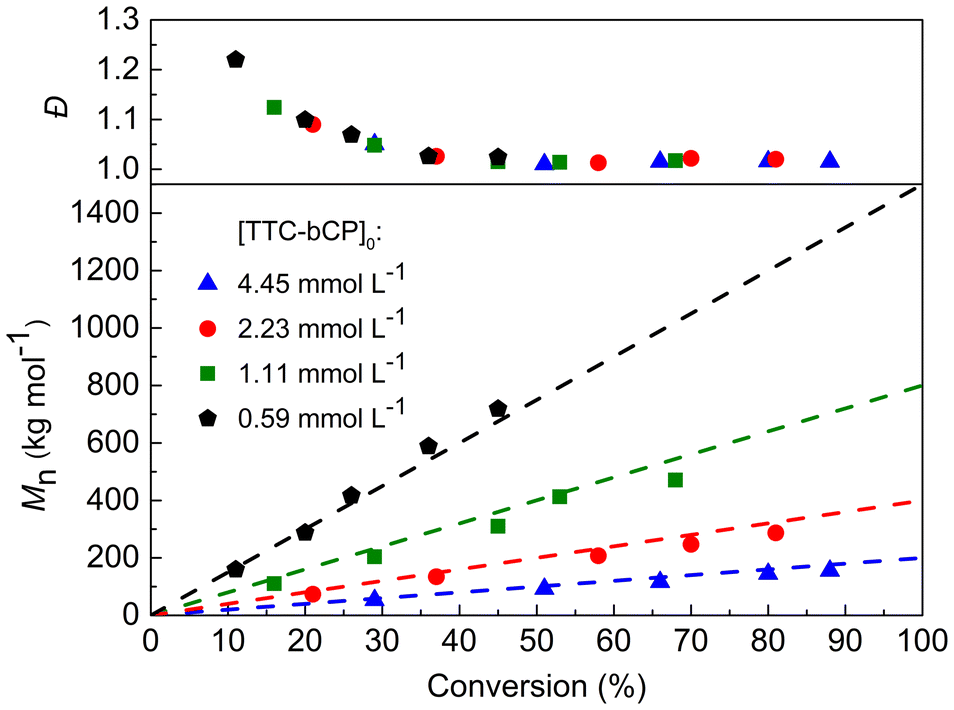 | ||
| Fig. 3 Evolution of Mn and Đ during PI-RAFT bulk polymerisation of nBA for different targeted molar masses. Dashed lines represent the theoreotical molar masses. | ||
Synthesis of the ABA triblock copolymer
The possibility of using PI-RAFT to synthesise methacrylate–acrylate–methacrylate triblock copolymers was then examined. The key to achieving the desired properties of these copolymers, such as high elongation, flexibility, UV, and scratch resistance, lies in the precise combination of methacrylate and acrylate blocks.The performance of these materials as TPEs depends on the ratio of hard to soft blocks, with optimal properties observed when the hard phase accounts for about 15 to 30% of the composition.5,36–39 The first block was synthesised using a PI-RAFT polymerisation method optimized earlier in this study (green light, 30 LEDs, [MMA]0 = 6.21 mol L−1 in CH3CN) with a [TTC-bCP]/[MMA] ratio of 1/4000, reaching 40% conversion after 24 hours of irradiation (Fig. 4). After purification by precipitation in toluene, PMMA with an Mn of 154 kg mol−1 and a dispersity of 1.25 was used for chain extension with n-butyl acrylate, targeting a Mn of the soft block of 800 kg mol−1. The polymerisation was carried out in bulk using blue LEDs, conditions which were previously identified as suitable for nBA. During the first 5 hours of polymerisation, the dispersity remained similar as for starting macro-CTA (1.25–1.24), while the lowest dispersity (1.07) was achieved after 16 hours with a 68% conversion (Fig. S8, and Table S5†). An additional 8 hours of reaction increased this value to 1.11 at 80% conversion. The Mn values of the obtained triblock copolymers were in good agreement with theoretical values, indicating high polymerisation control throughout the reaction (Fig. S8†). SEC chromatograms of high-conversion samples exhibited a small shoulder in the lower molar mass region, meaning that some starting PMMA remained unreacted (Fig. S9†). It was hypothesised that the extended irradiation time (24 hours) during the synthesis of PMMA led to the degradation of a fraction of the thiocarbonylthio group (vide supra), reducing the livingness of the macro-CTA.23,31,32,40 After precipitation of the 16 h sample, the PMMA77k-b-PnBA531k-b-PMMA77k triblock copolymer was obtained (Fig. 4).
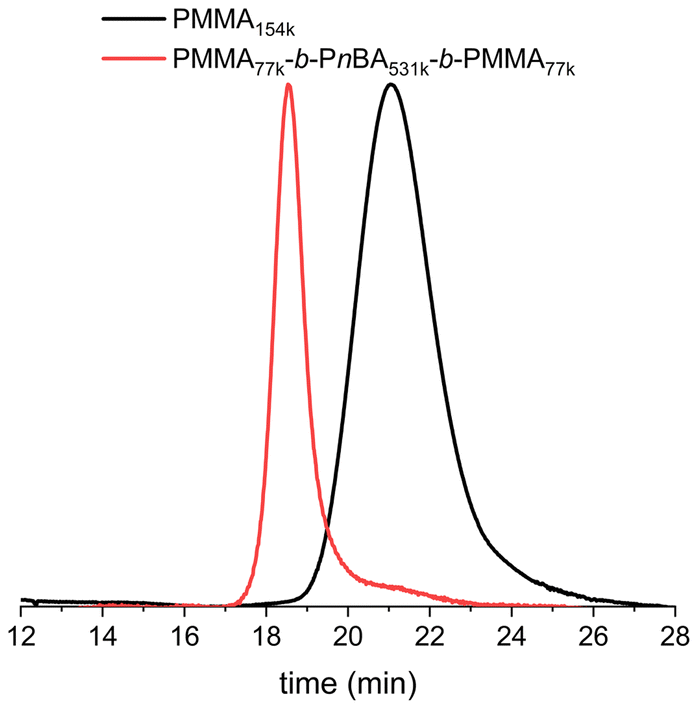 | ||
| Fig. 4 SEC-RI chromatograms of PMMA154k and the corresponding PMMA77k-b-PnBA531k-b-PMMA77k triblock copolymer. | ||
Thermal analysis
The Tg value of PMMA77k-b-PnBA531k-b-PMMA77k was measured by differential scanning calorimetry (DSC) (Fig. S10†). While the Tg of the soft block was found to be −50 °C, which is in accordance with the established value for PnBA, the Tg of PMMA was not visible during the analysis.This is possibly due to the relatively low weight fraction of the hard block (22.4%), which was previously observed for a similar triblock copolymer synthesized by our group.11 The thermal stability of PMMA77k-b-PnBA531k-b-PMMA77k was then tested using thermogravimetric analysis (TGA) under air and inert atmosphere (Fig. S11†). The 5% weight loss reached at 259 and 326 °C in air and in an inert atmosphere, respectively, which shows good potential for applications as TPE materials. For the similar triblock copolymer synthesised by living anionic polymerisation by Kuraray, 5% loss at 276 °C and 316 °C in air and in an inert atmosphere, respectively, was obtained.41 This shows the competitiveness of the PI-RAFT process in relation to more stringent anionic polymerisation.
Rheology
We then evaluated the thermomechanical properties of the PMMA77k-b-PnBA531k-b-PMMA77k triblock copolymer using rheology measurements over a temperature range from 20 to 180 °C. Fig. 5 shows the storage modulus (G′) for this elastomeric material, which is in the order of 1 MPa, similar to what has been reported for similar block copolymers obtained with an azo initiation.6,11 The glass transition observed in Fig. 5 at around 120 °C confirms the tendency of PMMA blocks to exhibit higher Tg values in (meth)acrylic block copolymers than for homopolymer. These results suggest that light irradiation, despite forming PMMAs of slightly higher dispersity with a small fraction of dead chains, does not significantly alter the macroscopic properties of the all-(meth)acrylic triblock copolymer.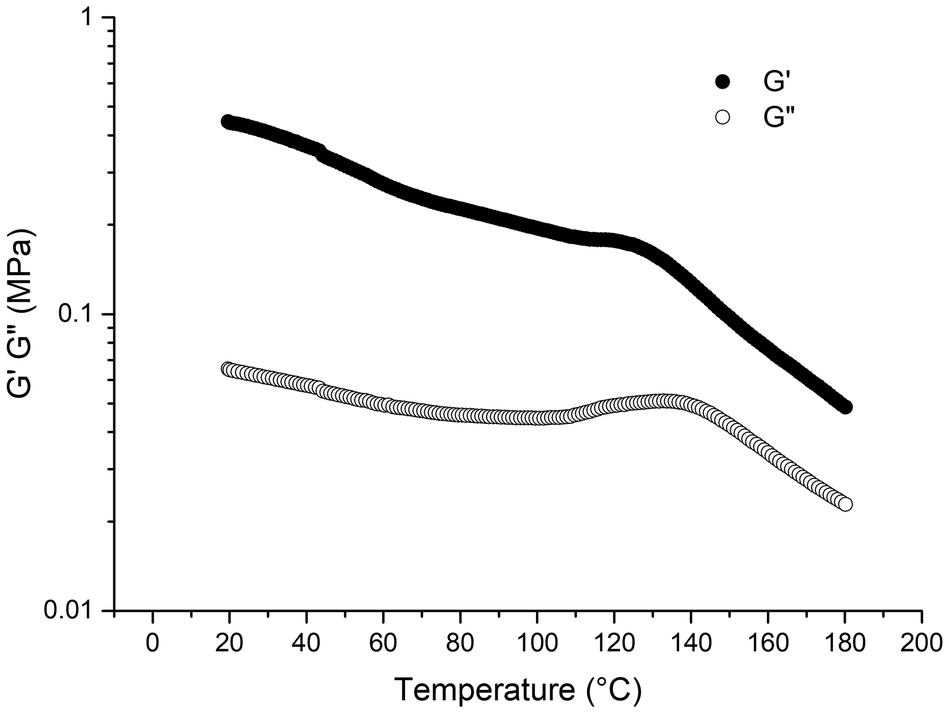 | ||
| Fig. 5 Temperature dependence of G′ and G′′ of PMMA77k-b-PnBA531k-b-PMMA77k triblock copolymer from 20 to 180 °C. | ||
Conclusions
This study demonstrated the synthesis of high molar mass methacrylate–acrylate–methacrylate (hard–soft–hard) ABA triblock copolymers using photoiniferter reversible addition–fragmentation chain transfer (PI-RAFT) polymerisation with TTC-bCP as a chain transfer agent. The use of varied light wavelengths, monomer concentration, and light intensities allowed the optimization of the PI-RAFT process and successful two-step synthesis of PMMA-b-PnBA-b-PMMA with high molar masses (up to Mn ∼ 800 kg mol−1) and low dispersities (Đ < 1.25). This work underscores the potential of initiator-free, light-mediated polymerisation in creating environmentally friendly synthetic routes for ABA all-(meth)acrylic triblock copolymers with application as TPEs. A more detailed study on material properties and phase separation will be presented in a forthcoming article, which will focus on the interplay between dispersity and material properties, phase separation, and long-range ordering.Author contributions
Conceptualization: M.D.; formal analysis: I.K., M.O., M.G.; experimentation: I.K., M.O.; supervision: M.D., I.K.; funding acquisition: M.D.; investigation: I.K., I.O., M.O., M.G.; validation: M.D. I.K., M.O.; project administration: M.D; visualization: I.K.; writing – original draft: I.K., M.O.; writing & review & editing: I.K, M.O, O.I, M.G, M.D.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
I. K. thanks the French Ministry of Foreign and European Affairs for the Eiffel scholarship.References
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N.-G. Kang, K. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- N. De Alwis Watuthanthrige, J. A. Reeves, M. T. Dolan, S. Valloppilly, M. B. Zanjani, Z. Ye and D. Konkolewicz, Macromolecules, 2020, 53, 5199–5207 CrossRef CAS.
- T. J. Neal, R. D. Bradley, M. W. Murray, N. S. J. Williams, S. N. Emmett, A. J. Ryan, S. G. Spain and O. O. Mykhaylyk, Macromolecules, 2022, 55, 9726–9739 CrossRef CAS PubMed.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- O. Ivanchenko, M. Odnoroh, S. Mallet-Ladeira, M. Guerre, S. Mazières and M. Destarac, J. Am. Chem. Soc., 2021, 143, 20585–20590 CrossRef CAS PubMed.
- R. L. Atkinson, O. R. Monaghan, M. T. Elsmore, P. D. Topham, D. T. W. Toolan, M. J. Derry, V. Taresco, R. A. Stockman, D. S. A. D. Focatiis, D. J. Irvine and S. M. Howdle, Polym. Chem., 2021, 12, 3177–3189 RSC.
- L. G. Williams, K. Sullivan and J. Tsanaktsidis, RAFT Agents for Making Well-Defined Functionalized Polymers, https://www.sigmaaldrich.com/PL/pl/technical-documents/technical-article/materials-science-and-engineering/polymer-synthesis/raft-agents-for-functionalized-polymers, (accessed January 14, 2024).
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- J. Ma and H. Zhang, Macromol. Res., 2015, 23, 67–73 CrossRef CAS.
- O. Ivanchenko, M. Odnoroh, F. Rolle, A. A. Kroeger, S. Mallet-Ladeira, S. Mazières, M. Guerre, M. L. Coote and M. Destarac, Macromol. Rapid Commun., 2024, 45, 2400317 CrossRef CAS.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2021, 222, 2000311 CrossRef CAS.
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS.
- R. W. Hughes, M. E. Lott, R. A. Olson S and B. S. Sumerlin, Prog. Polym. Sci., 2024, 101871 CrossRef CAS.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef CAS.
- V. Bellotti and R. Simonutti, Polymers, 2021, 13, 1119 CrossRef CAS PubMed.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, Polym. Chem., 2020, 11, 641–647 RSC.
- B. Zhao, J. Li, Z. Li, X. Lin, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 7181–7192 CrossRef CAS.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS.
- P. Akarsu, S. Reinicke, A.-C. Lehnen, M. Bekir, A. Böker, M. Hartlieb and M. Reifarth, Small, 2023, 19, 2301761 Search PubMed.
- A.-C. Lehnen, J. A. M. Kurki and M. Hartlieb, Polym. Chem., 2022, 13, 1537–1546 RSC.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS.
- S. Lian, S. P. Armes and Z. An, CCS Chem., 2024, 1–24 Search PubMed.
- K. Sparnacci, T. Frison, E. Podda, D. Antonioli, M. Laus, M. Notari, G. Assanelli, M. Atzeni, G. Merlini and R. Pó, ACS Appl. Polym. Mater., 2022, 4, 8722–8730 CrossRef CAS.
- L. Despax, J. Fitremann, M. Destarac and S. Harrisson, Polym. Chem., 2016, 7, 3375–3377 RSC.
- G. A. Adam, J. N. Hay, I. W. Parsons and R. N. Haward, Polymer, 1976, 17, 51–57 CrossRef CAS.
- J.-Y. Choi, S.-W. Jin, D.-M. Kim, I.-H. Song, K.-N. Nam, H.-J. Park and C.-M. Chung, Polymers, 2019, 11, 477 CrossRef.
- G. Vaganov, M. Simonova, M. Romasheva, A. Didenko, E. Popova, E. Ivan'kova, A. Kamalov, V. Elokhovskiy, V. Vaganov, A. Filippov and V. Yudin, Polymers, 2023, 15, 2922 CrossRef CAS PubMed.
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 14–19 CrossRef CAS.
- M. A. Beres, J. Y. Rho, A. Kerr, T. Smith and S. Perrier, Polym. Chem., 2024, 15, 522–533 RSC.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Polym. Chem., 2015, 6, 5362–5368 RSC.
- I. Kurowska, A. Dupre-Demorsy, S. Balayssac, M. Hennetier, A. Ric, V. Bourdon, T. Ando, H. Ajiro, O. Coutelier and M. Destarac, Macromol. Rapid Commun., 2023, 44, 2200729 CrossRef CAS.
- Y. Wang, H. Schroeder, J. Morick, M. Buback and K. Matyjaszewski, Macromol. Rapid Commun., 2013, 34, 604–609 CrossRef PubMed.
- Y. Jiang, W. Fan, M. Tosaka, M. F. Cunningham and S. Yamago, Macromolecules, 2021, 54, 10691–10699 CrossRef CAS.
- Z. Huo, S. Arora, V. A. Kong, B. J. Myrga, A. Statt and J. E. Laaser, Macromolecules, 2023, 56, 1845–1854 CrossRef CAS.
- G. Moineau, M. Minet, P. Teyssié and R. Jérôme, Macromol. Chem. Phys., 2000, 201, 1108–1114 CrossRef CAS.
- W. Lu, A. Goodwin, Y. Wang, P. Yin, W. Wang, J. Zhu, T. Wu, X. Lu, B. Hu, K. Hong, N.-G. Kang and J. Mays, Polym. Chem., 2017, 9, 160–168 RSC.
- A.-V. Ruzette, S. Tencé-Girault, L. Leibler, F. Chauvin, D. Bertin, O. Guerret and P. Gérard, Macromolecules, 2006, 39, 5804–5814 CrossRef CAS.
- A.-C. Lehnen, J. Gurke, A. M. Bapolisi, M. Reifarth, M. Bekir and M. Hartlieb, Chem. Sci., 2023, 14, 593–603 RSC.
- KURARITYTM Acrylic Block Copolymer Technical Information, (accessed February 3, 2023).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01123f |
| This journal is © The Royal Society of Chemistry 2024 |