DOI:
10.1039/D4RA04686B
(Review Article)
RSC Adv., 2024,
14, 30647-30661
NSAIDs between past and present; a long journey towards an ideal COX-2 inhibitor lead
Received
27th June 2024
, Accepted 5th September 2024
First published on 25th September 2024
Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) are the most abundantly used classes among therapeutic agents in medicine. NSAIDs inhibit the enzyme cyclooxygenase (COX), which is responsible for the conversion of arachidonic acid to prostaglandins. Meanwhile, non-selective NSAIDs are considered as a double-edged weapon since inhibition of COX-1 can lead to gastrointestinal side effects and kidney damage, whereas selective COX-2 inhibition provides anti-inflammatory effects without gastrointestinal toxicity. The detection of COX-2 role in inflammation process launched a new era in its management. Several trials have been established to proceed towards selectivity of well-defined anti-inflammatory members. COX-2 selective inhibitors are evidently safer on the gastrointestinal tract than non-selective NSAIDs. Nevertheless, their unexpected cardiovascular risks cannot be ignored. This review article highlights the latest trials aimed at developing new compounds with promising selective COX-2 activity.
1. Introduction
Inflammation is a normal, essential immune system response through diverse interactions between soluble factors and cells. Inflammation occurs in any tissue during exposure to infections, toxins, or autoimmune disease processes.1 From a pathological point of view, inflammation can be categorized into acute and chronic inflammation. Acute inflammation has a fast onset and a short duration of time with several symptoms such as pain, redness, heat, swelling and loss of function in severe cases.2 On the other hand, chronic inflammation exhibits slower onset of symptoms while persisting for a longer duration of time.3,4 Normally, this process contributes to the recovery from infection and consequently healing. However, if selective degradation and aided reconstruction of the inflamed cells are inadequately phased out, this can lead to a persistent tissue damage by leukocytes, lymphocytes.5,6
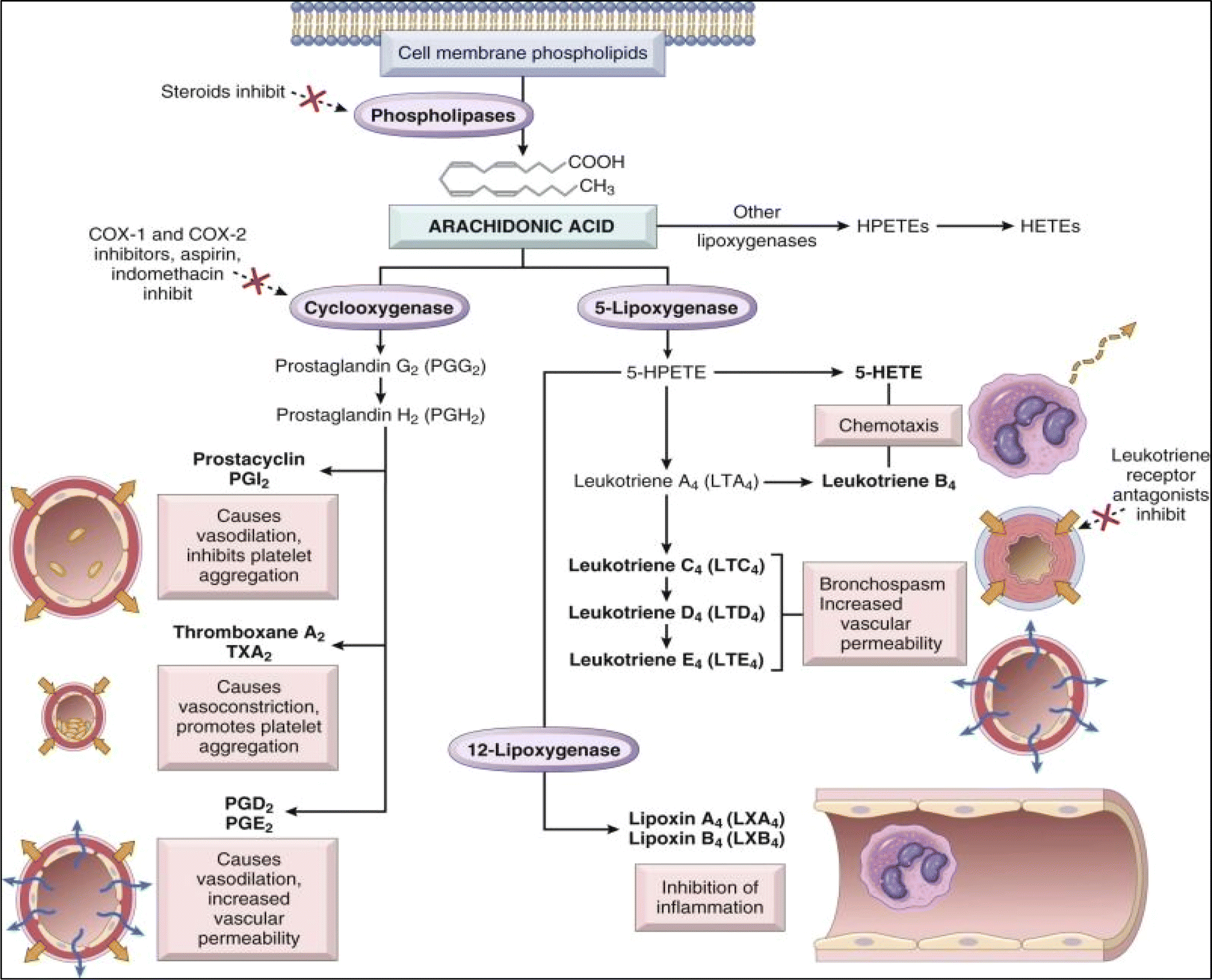
The normal pathway of inflammation starts with trauma, infection, or immune reactions. Such an incidence stimulates the cleavage of membrane's phospholipids by phospholipase A2 enzyme to produce arachidonic acid. In accordance, arachidonic acid is subjected to downstream modification with cyclooxygenase enzymes into prostaglandins G2 (PG2) and then converted to PGH2. PGH2 is a precursor that can be further converted into various prostaglandins, thromboxanes, and other eicosanoids (Fig. 1).6,8–10 Prostaglandins (PGs) are hormone-like bioactive substances that are directly implicated in many physiological and pathological processes.10 PGs are considered to be significant mediators involved in various therapeutic areas of concern, including inflammation, pain, pyrexia, cancer, glaucoma, male sexual dysfunction, osteoporosis.8 On the other hand, thromboxane A2 (TXA2) possesses vasoconstrictor and platelet aggregation effect.8 Meanwhile, 5-lipoxygenase enzyme (5-LOX) acts on arachidonic acid to produce leukotrienes (LTs). Leukotrienes (LTs) are responsible for anaphylaxis. Accordingly, therapies inhibiting inflammatory mediators are considered as an effective treatment for controlling inflammation.11–14
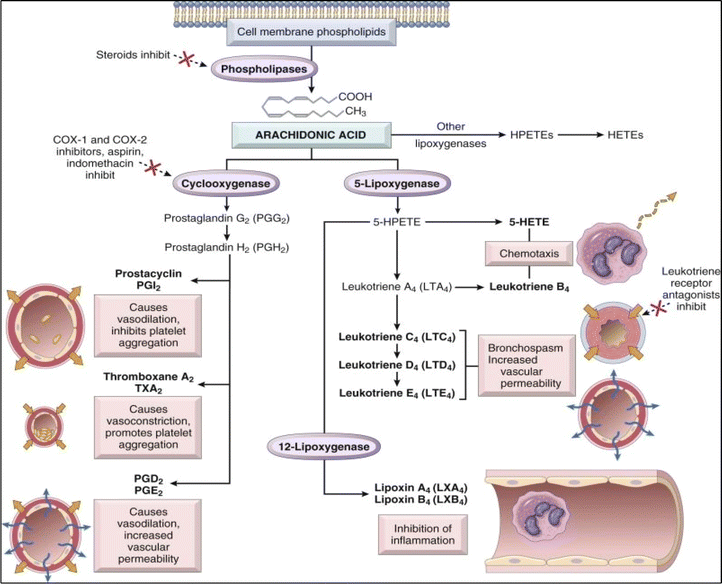 |
| | Fig. 1 Arachidonic acid metabolism products involved in the inflammatory process.7 | |
Depending on the enzyme targeted in the inflammation process, we can classify the anti-inflammatory agents into two major classes, steroidal anti-inflammatory agents, and non-steroidal anti-inflammatory ones.
1.1 Steroidal anti-inflammatory agents (glucocorticoids)
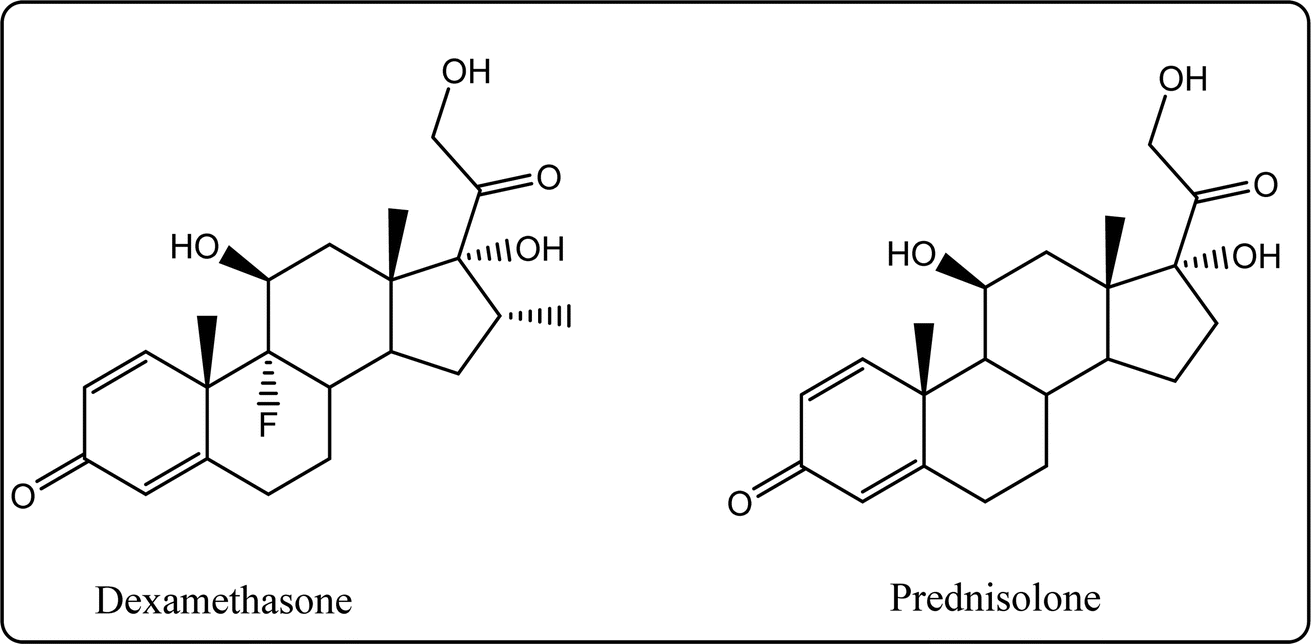
Glucocorticoids (GC) stop inflammation pathway by inhibiting phospholipase A2 (PLA2), which consequently reduces the production of arachidonic acid.15 The major use of such a class is mostly for patients suffering from diseases such as rheumatoid arthritis, osteoarthritis.16,17 Although GCs can be considered as a potent drug, their excessive use specifically in large doses for a long duration of time is associated with overwhelming dermatologic, musculoskeletal side effects. Suppression of the hypothalamic pituitary adrenal gland (HPA) axis or cushing syndrome are other expected possible drawbacks. Moreover, the gastrointestinal, ocular, cardiovascular, neuropsychiatric, and immunologic are serious side effects for glucocorticoids long-term use15,16,18 (Fig. 2).
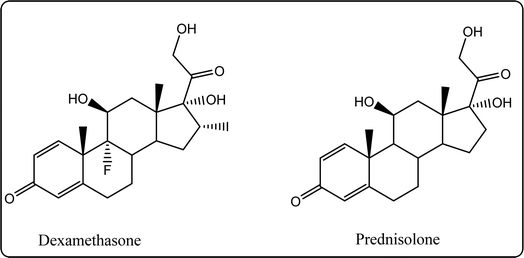 |
| | Fig. 2 Structures of the most famous corticosteroids. | |
1.2 Non-steroidal anti-inflammatory drugs (NSAIDs)
NSAIDs is a class of pharmaceutical agents most generally used to manage pain, fever, and numerous inflammatory forms. It is worth mentioning that there are two main COX enzymes possessing different functions, COX-1, and COX-2. COX-1 is indeed constitutively expressed and plays a role in maintaining homeostasis in the gastrointestinal tract, kidneys, and other organs. On the other hand, although COX-2 is primarily induced during inflammation, it also is constitutively expressed in some tissues and has roles in certain physiological processes such as renal function and healing.19,20 However, most of the NSAIDs cause significant gastrointestinal side effects due to COX-1 enzyme suppression20–22 while several studies proved that selective inhibition of COX-2 minimizes the GIT effects, and other undesired associated side effects.21
From the isoenzyme's selectivity point of view, NSAIDS can be categorized into non-selective and selective COX-2 inhibitors (Fig. 3).
 |
| | Fig. 3 Main classes of NSAIDs. | |
1.2.1 Non-selective COX inhibitors. This class lacks the selective inhibitory action on COX isoforms with fewer side effects in comparison with GCs class. Famous members for this class are ibuprofen, ketoprofen, naproxen and aspirin 23 (Fig. 4). Although this group is still recommended for the use as analgesic and anti-inflammatory agents in rheumatoid arthritis, degenerative joint disease, ankylosing spondylitis and periarticular diseases such as tendinitis,24 they cause serious gastrointestinal tract adverse effects. Gastrointestinal tract injury is attributed to two main reasons, the direct insult of the carboxylic acid moiety (–COOH) with GIT mucosal cells in combination with the decrease in PG production in tissues thus reducing the cytoprotective effect of PGs on GIT health.25,26
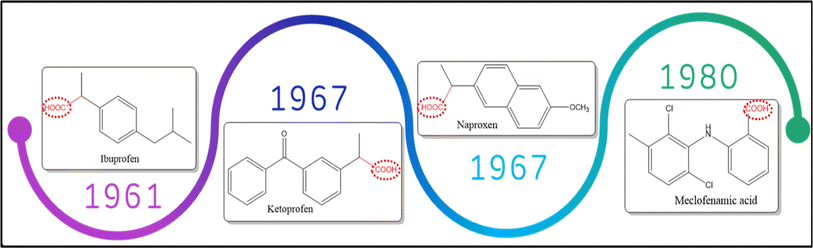 |
| | Fig. 4 Chronological order for the most famous non-selective COX inhibitors. | |
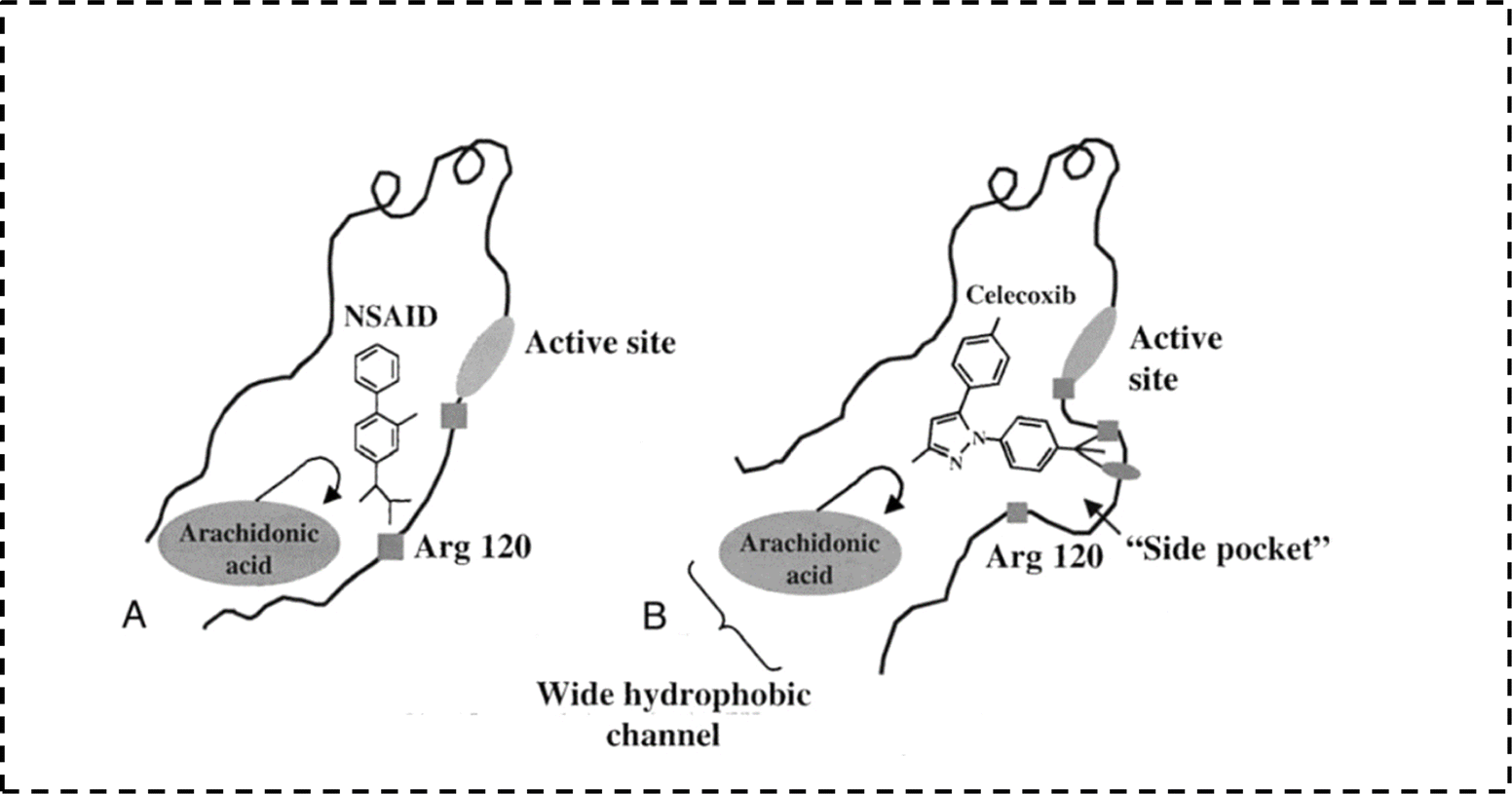
1.2.2 Selective cyclooxygenase-2 enzyme (COX-2) inhibitors (COXIBs). The search for ideality is one of the scientists' targets. Accordingly, researchers started to look for another alternatives aiming to avoid GIT side effects caused by non-selective NSAIDs.23,27 The first step in this journey was the discovery of COX isoforms. In early 1990s, Dan Simmons et al. raised the fact that COX does not exist as an only one enzyme. Actually, there are two isoenzymes of COX with structural and functional differences.28–31 From the structural point of view there is a very small but still a significant difference between COX-1 and COX-2 isoenzymes.32 Both enzymes have the same cavity to which arachidonic acid fits. On the other hand, the main difference is detected in the presence of a small additional pocket in COX-2 enzyme structure. So the key for increasing selectivity towards COX-2 enzyme is having an extension that specifically binds to COX-2 additional pocket (Fig. 5).33,35–37 Moreover, Table 1 highlights the most significant points of differences between the two isoenzymes.
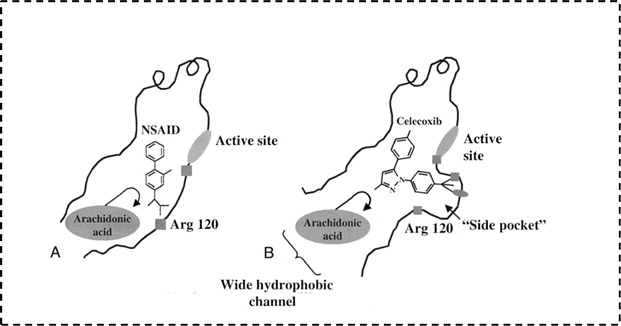 |
| | Fig. 5 Prominent features distinguishing the COX isoenzymes.33,34 | |
Table 1 The most significant points of difference between COX-1 and COX-2 (ref. 34 and 38–40)
| Points of comparison |
COX-1 |
COX-2 |
| Active site |
NSAIDs generally bind the top part of the COX channel located near Tyr385 and Arg120 which is present at the entrance of the COX channel34 |
| Significant amino acids |
(a) Isoleucine 523, (b) Isoleucine 434, Histidine 513 |
A smaller valine at amino acid residue at position 523, a valine amino acid also at position 434, and the secondary pocket which is accessible at the COX-2 active site |
| Volume of COX binding site |
316 Å (ref. 21) |
394 Å (ref. 21) |
COXIBs are selective COX-2 inhibitors characterized by the pharmacophore carboxylic or heterocyclic five-membered ring. Structure activity relationship studies proved that substituted sulfonyl groups are also considered as pharmacophores that recognize COX-2 active site pocket4,21,41,42 (Fig. 6).
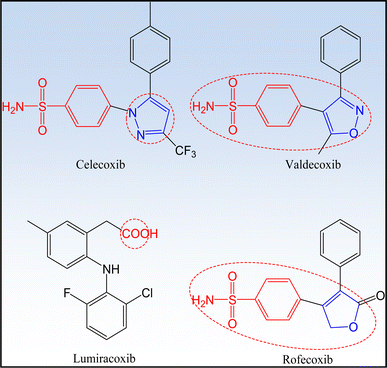 |
| | Fig. 6 Pharmacophores of selective COX-2 inhibitors. | |
They are used in the management of inflammatory pain, symptoms of osteoarthritis and rheumatoid arthritis with enhanced gastrointestinal safety profile compared to NSAIDs.43,44 Although, COXIBs cause lesser ulceration than traditional NSAIDs they can increase the risk of severe cardiovascular events such as heart attack, myocardial infarction (MI), and stroke.45–47 The safety of NSAIDs is influenced by multiple factors, not just their selectivity for COX-1 or COX-2. Both the COX-1 and COX-2 roles are inhibited during NSAID therapy.20,48
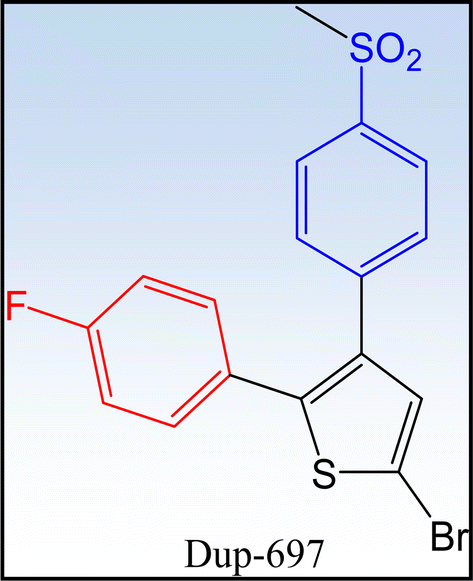
1.2.2.1 First generation COXIBs. Celecoxib® and rofecoxib® are two earliest and most widely recognized COX-2 inhibitors (Fig. 6). DuP-697 is one of the lead compounds that inspired the development of selective COX-2 inhibitors (Fig. 7). The comparison between the two drugs is illustrated in Table 2.55–57
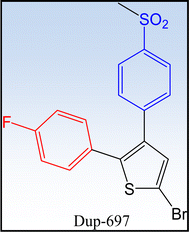 |
| | Fig. 7 DuP-697 is one of the lead compounds in the development of COX-2 inhibitors. | |
Table 2 Celecoxib and rofecoxib main differences4,29,49
| |
Celecoxib |
Rofecoxib |
| History |
The first approved selective COX-2 inhibitor. It was marketed by Pfizer |
A selective COX-2 inhibitor produced by Merck50,51 |
| Trade name |
Celebrex®, and elyxyb® |
Vioxx® (ref. 52) |
| Ratio of COX-2/COX-1 inhibition |
7.6-Fold |
35-Fold |
| Adverse side effects |
Dyspepsia, edema, moderate probability of CVS side effects53 |
| SAR |
Both members possess a diaryl substitution on a central heterocycle54 |
An acceptable explanation for the cardiovascular undesired side effects is the inhibition of endothelial PGI2 synthesis, which is mainly dependent on COX-2. PGI2 is the powerful inhibitor of platelet aggregation and thrombosis. Consequently, affecting PGI2 synthesis leads to triggering the acute coronary syndromes, myocardial infarction, thrombosis and atherosclerosis are expected.58,59
1.2.2.2 Second generation COXIBs. The most important members of this class are valdecoxib, parecoxib and etoricoxib (Fig. 8).30,47 A brief description for the characteristics of each member is illustrated in Table 3.
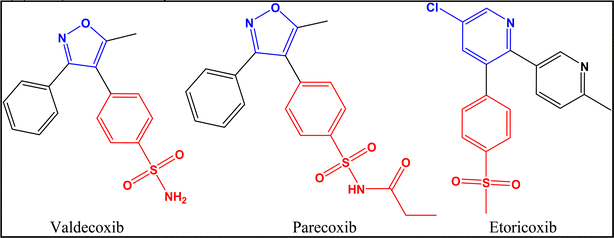 |
| | Fig. 8 Second generation of COXIBs. | |
Table 3 Characteristics of the members of COXIBs second generation4,29,47,60
| |
Valdecoxib |
Parecoxib |
Etoricoxib |
| History |
FDA-approved valdecoxib in 2001, shortly it was withdrawn by Pfizer at 2005 due to it’s cardiovascular and severe skin reaction risks |
Patency was in 1996 and FDA-approved in 2002, then produced by Pfizer |
Discovered and produced by Merck in 2002 and approved for medical use in 2007 |
| Trade names |
Bextra® |
Dynastat® |
Arcoxia® |
| COX-2/COX-1 inhibition ratio |
30-Fold |
Water soluble prodrug of valdecoxib |
106-fold |
| Adverse effects |
All showed high probability of cardiovascular side effect compared to non-selective NSAIDs45 |
| Central nucleus |
Oxazole ring |
Oxazole ring |
Pyridine ring |
| SAR |
Valdecoxib possesses a diaryl substitution on the central oxazole ring with a characteristic sulfonamide group on one of the aryl rings |
Acting as a prodrug, parecoxib possesses the same SAR of valdecoxib |
Etoricoxib possesses a diaryl substitution on the central pyridine ring with a characteristic methylsulfonyl moiety on one of the aryl rings |
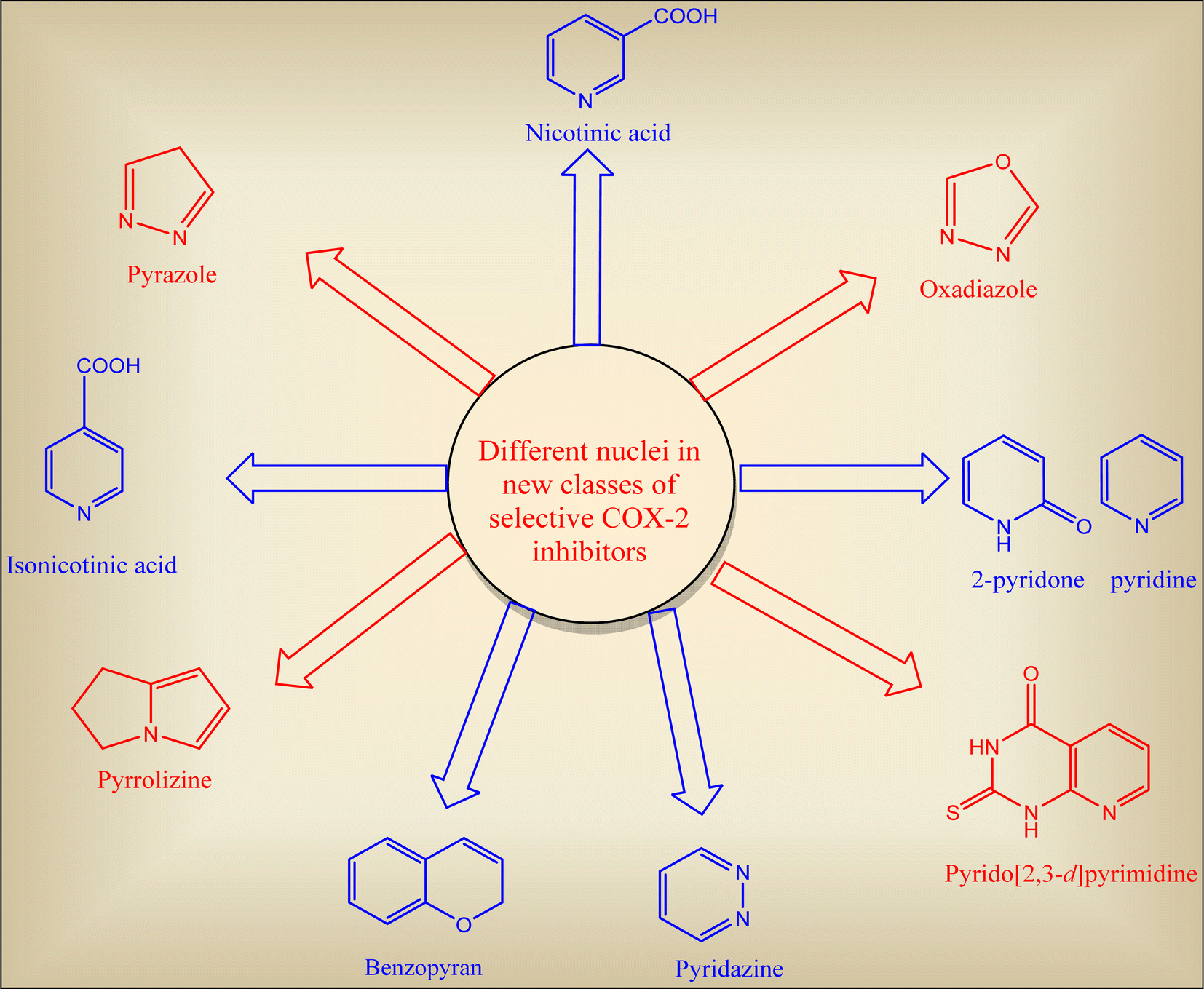
2. Promising approaches towards ideal COXIB lead
Tremendous efforts have been recorded to discover selective COX-2 inhibitors with lesser side effects. The synthesized are categorized according to their common nucleus in each class (Fig. 9).37,59,61,62
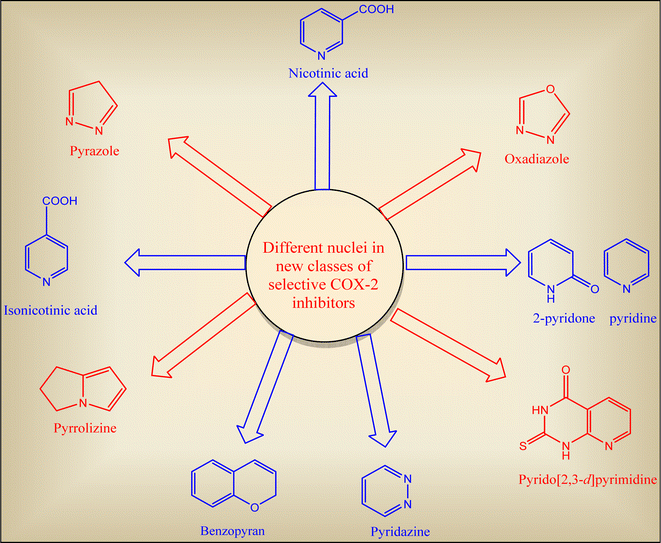 |
| | Fig. 9 Different heterocyclic nuclei incorporated in selective COX-2 inhibitors scaffold. | |
2.1 Nicotinic acid-containing compounds
Abouzid's team synthesized the 2-substituted phenyl derivatives of nicotinic acid 1 and 2a,b (Fig. 10) according to the reported procedure.12 These derivatives were biologically evaluated for their anti-inflammatory activity. Compounds 1, 2a,b (Fig. 10) showed unique analgesic and anti-inflammatory activities compared to mefenamic acid as a standard drug. Further investigation concerning the ulcerogenic effect of these compounds were performed.12 The screening of all the synthesized compounds revealed that compounds 2a,b exceeded mefenamic acid (54.5 ± 3.45%) regarding anti-inflammatory activity (64.5 ± 4.9%, 61.3 ± 5.6%, and 56.2 ± 5.7%, respectively) with a slightly improved ulcerogenic profile compared to mefenamic acid (ulcer indices = 0.9 ± 0.04%, 0.9 ± 0.03%, and 1 ± 0.08%, respectively c.f. 1.1 ± 0.03% for mefenamic acid) and acceptable analgesic activity.12 Their tumor necrosis factor TNF-α values were (3.25 ± 0.21%, 2.45 ± 0.18% and 2.67 ± 0.15%, sequentially) and interleukin IL-6 values were (9.2 ± 0.61, 14.3 ± 0.56, and 10.1 ± 0.82 ng mL−1).12
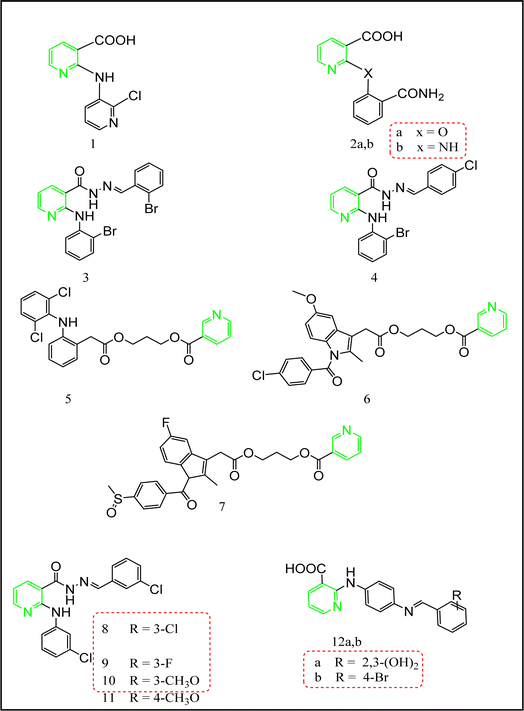 |
| | Fig. 10 The most promising nicotinic acid-containing compounds. | |
On the other hand, khalil's research team12 succeeded in synthesizing another series of nicotinic acid derivatives (compounds 3 and 4 Fig. 10). In accordance, this series was prepared via the straightforward nucleophilic substitution reaction of the respective primary aromatic amines with the 2-chloronicotinic acid. The activation of the carboxylic functionality was achieved through esterification followed by the reaction with hydrazine hydrate to afford the hydrazide derivatives. Finally, compounds 3 and 4 were synthesized through the condensation reaction of the respective aldehydes with hydrazide derivatives. Applying carrageenan induced rat paw edema,63 compounds 3 and 4 showed superior anti-inflammatory activity percentage of edema inhibition = 52.9 ± 3.8%, and 62.2 ± 5.4%, respectively c.f. mefenamic acid percentage of edema inhibition = 59.3 ± 4.21%), also they exhibited unique analgesic activities (61.7 ± 4.8%, and 61.7 ± 4.8%, successively c.f. mefenamic acid analgesic activity 72.4 ± 4.6), and an enhanced ulcerogenic profile (0.82 ± 0.05%, and 1.3 ± 0.09%, sequentially).64
Furthermore, new scaffolds of nicotinic acid derivatives 5, 6, and 7 (Fig. 11) were reported by Gund et al.65 The aim of this study was to combine a well-known NSAID with the nicotinic acid to benefit from the mutual prodrug strategy of different therapeutic agents in a single molecule by adopting the 1,3-propandiol ester linkage (Fig. 11).65,66 The 1,3-propandiol ester linkage was adopted for such an aim.
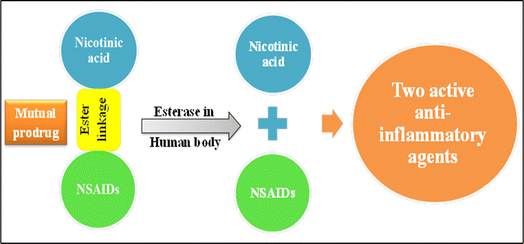 |
| | Fig. 11 Mutual prodrugs strategy. | |
These conjugates 5, 6 and 7 are privileged in the extended time of action, and as a result, decreasing the number of doses. Regarding their in vitro anti-inflammatory activity, the human TNF-α and IL-6 parameters were assessed. All the synthesized scaffolds have considerable in vitro potency when evaluated with the corresponding parent drug. Compound 5 as a hybrid of nicotinic acid and diclofenac showed IL-6 and TNF-α inhibition activities (44 ± 4.1%, and 48 ± 1.9%, respectively), on the other hand diclofenac itself showed IL-6 and TNF-α inhibition activities (35 ± 3.2%, and 42 ± 3.2%, respectively). Furthermore, compound 6 compared to indomethacin as a standard drug revealed IL-6 and TNF-α inhibition activities (37 ± 3.9%, and 40 ± 2.1%, sequentially), in contrast to indomethacin IL-6 and TNF-α inhibition activities (21 ± 3.5%, and 32 ± 7.2%, respectively). Compound 7 exceeded sulindac IL-6 and TNF-α inhibition activities (42 ± 3.1%, and 44 ± 7.1%, respectively), c.f. sulindac IL-6 and TNF-α inhibition activities (36 ± 2.1%, and 38 ± 8.8%, respectively).65
Navidpour et al. synthesized a new series of niflumic acid derivatives 8–11 according to the reported procedures.67–70 All the synthesized compounds were tested for their anti-inflammatory activity using carrageenan induced rat paw edema.63 Compound 8 was the most potent derivative (percentage of edema inhibition = 95.37 ± 4.45% at 30 min). Moreover, compound 8 was tested for its analgesic effect using rat writhing method,71 and showed percentage of inhibition = 49.43%. Compounds 9, 10 and 11 exhibited excellent anti-inflammatory activities (percentage of edema inhibition = 77.92 ± 4.04%, 79.07 ± 5.60%, 87.48 ± 5.04%, at 30 min., respectively) and considerable analgesic activities = 52.23%, 59.69%, and 97.27%, serially.68
In accordance, a new mefenamic acid analogues 12a, and 12b (Fig. 11) were successfully prepared through the nucleophilic attack of the 1,4-phenylenediamine on 2-chloronicotinic acid.72 Compounds 12a and 12b revealed unique analgesic and anti-inflammatory activities considering that celecoxib, indomethacin, diclofenac selectivity indices (SI) are 9.26, 0.39, and 1.19, successively as a standard reference. Furthermore, 12a and 12b showed the superior selective inhibition activity towards COX-2 enzyme. These compounds were also tested for their GIT ulcerogenic effect. The results for both COX-1 and COX-2 at 10 μM concentration were motivating towards their COX-2 enzyme selectivity. Compound 12a showed 66.9% of COX-1 inhibition and 77.2% of COX-2 inhibition, while compound 12b possessed 68.5% of COX-1 inhibition and 77.4% of COX-2 inhibition compared to celecoxib using 10 μM percentage of inhibition. Statistical analysis of the data was performed using one-way Anova followed by Tukey’ s Karmer post hoc test for multiple comparisons at probability levels of p < 0.05, results were considered statistically significant. Guided with the above promising results the gastric ulcerative effect was investigated and compared to the same three standard drugs. Upon recording the number of sores and their severity compound 12b was a competitor to the control with null sores while compound 12a exhibited 0.4 ± 0.01 sores with severity 0.74 ± 0.01%. On the other hand, celecoxib, diclofenac and indomethacin showed 3.3 ± 0.02, 4.1 ± 0.1 and 9.5 ± 0.4 sores with severity 7.1 ± 0.2, 8.6 ± 0.3 and 17.3 ± 0.5, respectively.72
2.2 1,3,4-Oxadiazole-containing compounds
Grover's team built up the scaffold 2,5-diaryl-1,3,4-oxadiazoles derivatives 13a–c (Fig. 12).73 The successful procedure started with the reaction of the appropriate benzaldehydes with the substituted hydrazides to yield the resultant acyl hydrazones followed by the reaction with N-bromosuccinimide (NBS) and triethylamine. The oxidation of thiomethyl group using oxone as an oxidizing agent in acetonitrile/water finally afforded the sulfone functionality.73 Series 13 revealed a significant COX-2 selectivity. Compound 13a exhibited COX-2 selectivity index = 132.83 c.f. celecoxib selectivity index 379.80, and IC50 of COX-2 (0.74 μM). Methylsulfonyl group was a distinguishing pharmacophore, since the compounds bearing methylsulfonyl group showed higher docking scores against COX-2 (46.27–55.53 kcal mol−1) in comparison to thiomethyl derivatives (10.52–53.14 kcal mol−1).73
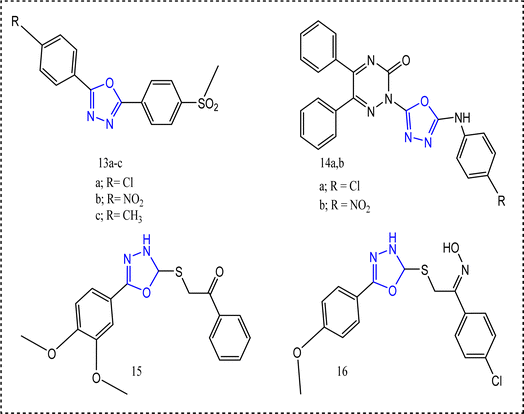 |
| | Fig. 12 The most promising oxadiazole-containing compounds. | |
Applying the hybrid molecular technique74 Banerjee's team conjoined five-membered heterocyclic rings with diphenyltriazine-3-ones 14a,b.75 5-Arylamino-1,3,4-oxadiazole compounds 14a, and 14b were prepared via the oxidative cyclization of thiosemicarbazides that results in elimination of H2S. Attaching electron-withdrawing group to 5-membered heterocyclic ring increased the activity significantly.75 Using carrageenan induced paw edema technique, it was obvious that compounds 14a,b had a quick onset (after 2 h) related to other compounds. In addition to the exceptional anti-inflammatory activities (60.97%, and 61.56%, respectively c.f. celecoxib 62.26%).75
In accordance, the 1,3,4-oxadiazole member 15 and the oxime core 16 exhibited noticeable anti-inflammatory activities displaying 72% and 83.33% inhibition of carrageenan induced edema,63 respectively. Nevertheless, both compounds 15 and 16 lacked a better selectivity profile towards COX-2 inhibition. The mono and dimethoxyphenyl substituted group seemed to increase the anti-inflammatory activity but not COX-2 selectivity. Compound 15 showed a noticeable inhibition of COX-1 IC50 = 1.10 ± 0.18 μM weighed against COX-2 IC50 = 2.30 ± 0.19 μM. On the other hand, 16 showed COX-1 IC50 0.94 ± 0.10 μM and COX-2 IC50 5.00 ± 0.61 μM76 (Fig. 13).
 |
| | Fig. 13 Pyridine-containing members. | |
2.3 Pyridine and 2-pyridone-containing compounds
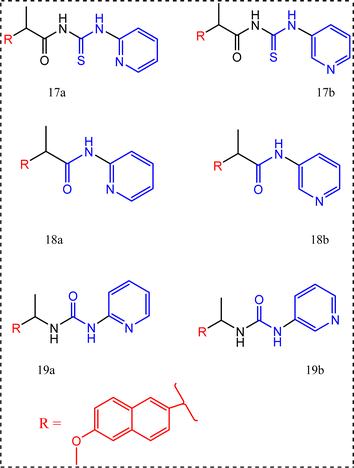
The pyridinamide functionality was a new attracting scaffold,77 the synthesis of these new scaffolds begins with reacting naproxen and thionyl chloride to prepare the corresponding acid chloride followed by reaction with 2-or 3-aminopyridine to yield the pyridinecarboxamides 17a,b. Moreover, compounds 18a,b were synthesized through reacting 2- or 3-aminopyridine with naproxenoyl isothiocyanate. Naproxen acid chloride was also converted to the urea derivatives 19a,b via the reaction with sodium azide, then addition of amino pyridines via the curtius rearrangement of the acid azide.77
All the synthesized compounds 17a,b, 18a,b, 19a,b were tested for their anti-inflammatory activity using carrageenan induced rat paw edema using naproxen with percentage of edema inhibition = 25.93% as a standard drug giving their percentage of edema inhibition (40.03%, 33.12%, 33.67%, 33.67%, 35.61%, and 33.12%, respectively). In another words their potency compared to potency of the standard drug naproxen after 5 h of induced inflammation were sequentially 1.54, 1.27, 1.29, 1.29, 1.37, and 1.27 times more than naproxen.77
Compounds 20 and 21 (Fig. 14) are examples for the 2-pyridone-containing compounds possessing anti-inflammatory activity.78 They exhibited percentage of edema inhibition (98.9%, and 90.8%) with 1.25 μg. Accordingly, from these results compound 20 exceeded indomethacin (98.2%) in its anti-inflammatory activity.79 Compound 20 was also identified as a selective COX-2 inhibitor, whereas 21 lacked COX-2 selectivity (Fig. 14).
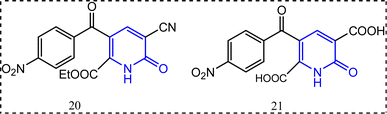 |
| | Fig. 14 2-Pyridone-containing compounds. | |
2.4 Tetrahydropyrido[2,3-d] pyrimidine-containing compounds
Abdelgawad et al. reported some compounds with pyrido[2,3-d:6,5-d]dipyrimidine-4,5-diones 22a–d, and the tetrahydropyrido[2,3-d]pyrimidine-6-carbonitrile derivatives 23a–d (Fig. 15).27 All compounds were tested for their in vitro anti-inflammatory activity. This was achieved by comparing the IC50 of an enzyme immunoassay (EIA) kit for ovine as the distinguishing parameter,80 and in vivo anti-inflammatory activity using carrageenan induced rat paw edema model and celecoxib was the standard drug.27,63 Compounds 22b,d and 23c showed selective COX-2 inhibition as shown by their selectivity indices (SI = 4.99, 6.43, and 17.08, respectively) c.f. celecoxib COX-2 selectivity index 6.61. Moreover, compound 23c exhibited an edema inhibition profile better than the standard drug celecoxib's result (74%). The gastrointestinal ulcerogenicity was also evaluated for the target molecules 22d and 23c with ulcer index 2.25, and 1.5, sequentially c.f. indomethacin ulcer index = 22.5.81,82
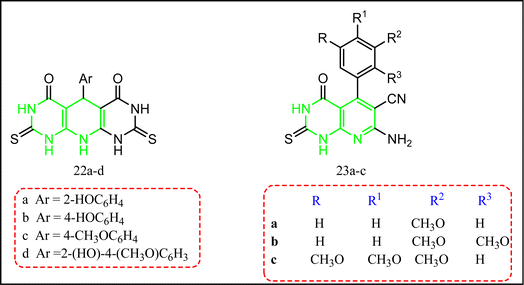 |
| | Fig. 15 Tetrahydropyrido[2,3-d] pyrimidine-containing compounds. | |
2.5 Pyridazinone-containing compounds
The synthesis of several novel pyridazinone-containing compounds as selective COX-2 inhibitors was reported.83 Biological evaluation revealed that compounds 24a,b, and 25a,b possess the most promising selective COX-2 inhibition. The COX-2 IC50 ranged between 15.56 and 19.77 nM. Moreover, their selectivity indices (SI) were 24, 38, 35, and 24, respectively. All synthesized compounds were 1.4 to 2.2 times more selective than celecoxib COX-2 IC50 17.79 ± 0.69 nM, and (SI) 17.18. Moreover, the in vivo results through the carrageenan induced rat paw edema technique and ulcerogenicity profile revealed that compounds 24b, 25a,b displayed greater anti-inflammatory activity in comparison with celecoxib, and no ulcerogenic effect was recognized by any of these compounds83 (Fig. 16).
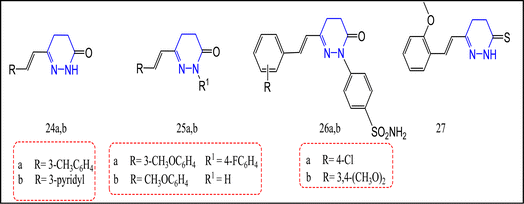 |
| | Fig. 16 Pyridazinone/thione-containing compounds. | |
Recently, the pyridazinone derivatives 26a,b, and pyridinethione one 27 were prepared and tested for their selective cyclooxygenase-2 inhibitory activities.84 Compounds 26a,b, and 27 exhibited significant influence towards COX-2 enzyme inhibition in comparison with celecoxib. COX-2 IC50 for these compounds were 67.23, 43.84, and 53.01 nM, respectively i.e., 1.1–1.7-fold more effective than celecoxib and indomethacin with IC50 = 73.53, 739.2 nM, successively. Compound 26b possessed a superior COX-2 selective inhibition with a SI = 11 equivalent to indomethacin and celecoxib. Furthermore, compound 26b exceeded both celecoxib and indomethacin in its ulcerogenic profile84 (Fig. 16).
2.6 Benzopyran-containing compounds
From its name, benzopyran is a bicyclic system composed of benzene ring fused to the six-membered hetero ring pyran. Chromene is the retained name of this class.85 Novel benzopyran derivatives were synthesized according to the reported procedure (Fig. 17).86 All the synthesized compounds were tested for their COX-1/COX-2 inhibitory ability. Compound 28 was considered the lead compound of this class. The selectivity index for 28 IC50 of COX-1/IC50 of COX-2 was 69 folds with excellent anti-inflammatory activity. The structure activity relationship study (SAR) revealed that substitution at position 7 is beneficial for both selectivity and enhanced activity.86 In accordance, compound 29 exhibited 16.45 folds selectivity towards COX-2 (IC50 = 0.062 μM).86
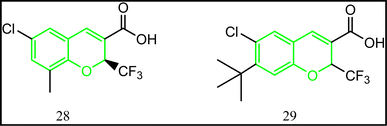 |
| | Fig. 17 Benzopyran-containing compounds. | |
2.7 Pyrrolizine-containing compounds
Recently, a series of pyrrolizine derivatives were synthesized achieving greater anti-inflammatory activity with minimal side effects.87 All the synthesized compounds were in vitro tested for their COX-1/COX-2 inhibitory ability using a COX colorimetric inhibitor screening assay kit.88 Also, their in vivo anti-inflammatory activities were applied using carrageenan induced rat paw edema.63,87 Compounds 30–34 (Fig. 18) revealed an exceptional anti-inflammatory activity and acceptable selectivity towards COX-2 inhibitory activity. Accordingly, compounds 30, 31, 32, 33 and 34 (Fig. 18) showed IC50 of COX-1/IC50 of COX-2 equal to 3.64, 3.48, 3.21, 3.17, and 2.89 folds, respectively c.f. 0.02 for indomethacin, while their percentages of inhibition of edema thickness after 3 hours were (44.79%, 52.31%, 16.37%, 24.91%, and 50.58%, sequentially) compared to ibuprofen 40.82%. Percentage of inhibition of ulcer formation was (66.38%, 68.18%, 66.38%, 80.66%, and 83.69%, respectively c.f. zero protection for ibuprofen).87
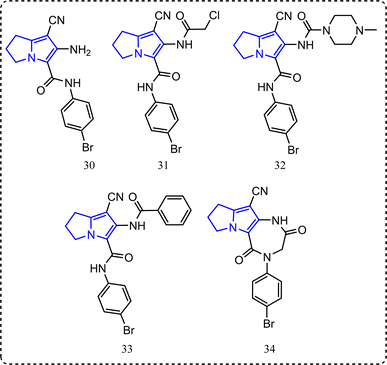 |
| | Fig. 18 Pyrrolizine-containing compounds. | |
2.8 Isonicotinic acid-containing compounds
New scaffolds of isonicotinic acid containing derivatives were reported by Zaheer et al.89 The anti-inflammatory activity was evaluated via chemiluminescence technique.90 The results revealed that compounds 35 and 36 (Fig. 19) showed superior anti-inflammatory activity, with percentage inhibition = 95.9%, and 67.3%, respectively. Compounds 35 and 36 showed COX-2 IC50 = 1.42 ± 0.1, and 8.6 ± 0.5 excelling IC50 of ibuprofen itself (11.2 ± 1.9) as a standard drug.
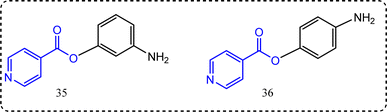 |
| | Fig. 19 Isonicotinic acid-containing compounds. | |
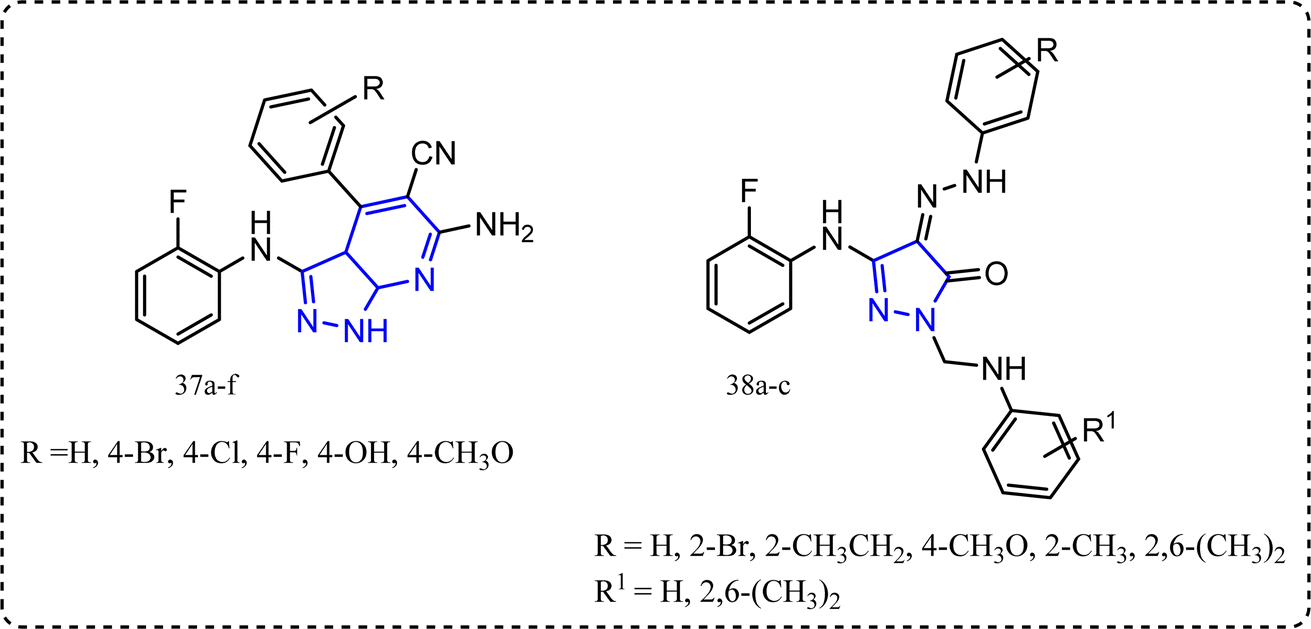
2.9 Pyrazoles and pyrazolo[3,4-b]pyridines-containing compounds
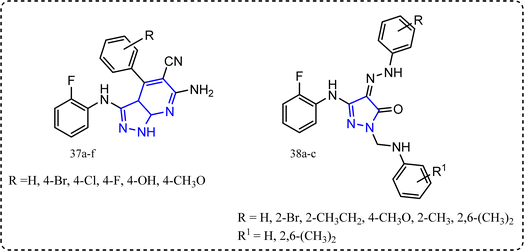
Mohamed et al. synthesized novel pyrazoles and pyrazolo[3,4-b]pyridines derivatives 37a–f and 38a–c (Fig. 20).91 All the target molecules were tested for their in vitro anti-inflammatory activity using cayman COX (ovine/human) inhibitor assay using diclofenac sodium,92 indomethacin and celecoxib as a standard drug. Pyridines derivatives 37a–f SI (IC50 COX-1/IC50 COX-2) ranged between 16.346 to 104.878 folds compared to celecoxib with SI 308.163, while compounds 38a–c exhibited SI = 258.333, 297.917, and 267.391, respectively. Furthermore, all the target molecules were evaluated for their in vivo anti-inflammatory activity using carrageenan induced rat paw edema. Compounds 38a–c exhibited in vivo anti-inflammatory activities 61%, 64%, and 62%, respectively c.f. 69% for celecoxib. Additionally, their ulcerogenic incidence was (20–30%).93,94
 |
| | Fig. 20 Pyrazoles and pyrazolo[3,4-b]pyridines-containing compounds. | |
3. Conclusion
Commencing these facts, we tried to track the most updated trials to attain novel compounds with promising anti-inflammatory, analgesic activities and if possible higher COX-2 enzyme selectivity. To the best of our knowledge no perfect balanced anti-inflammatory with minimal side effects was discovered. Consequently, the continuous and active trials for the development of new and selective coxibs with diminished side effects are still a hot research spot.
New scaffolds that may be eye-catching to explore could be built upon the promising findings from previous studies,95–97 future work should further explore the anti-inflammatory potential of 2-substituted arylaminonicotinic acid derivatives bearing an aminosulfonyl moiety. This approach combines the nicotinic acid pharmacophore with the biologically active sulfonamide moiety to develop compounds with improved anti-inflammatory properties. Specifically, it would be beneficial to investigate a wider range of substituents at positions 2 and 4 of the phenyl ring to optimize the anti-inflammatory efficacy. Additionally, replacing the carboxylic acid moiety with a 2-aminothiadiazole ring may be explored. The choice of the thiadiazole ring as a substitute for the carboxyl group is hypothesized to enhance anti-inflammatory activity while minimizing gastrointestinal toxicity.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
References
- W. Riedel, Nociception, pain, and antinociception: current concepts, Z. Rheumatol., 2001, 404–415 CrossRef CAS PubMed.
- N. A. A. Razzak, Design and synthesis of new mefenamic acid derivatives as anti-inflammatory agents, J. Al-Nahrain Univ., 2011, 14, 38–44 CrossRef.
- P. A. Todd and E. M. Sorkin, A reappraisal of its pharmacodynamic and phannacokinetic properties, and therapeutic efficacy diclofenac sodium, Drugs, 1988, 244–285 CrossRef CAS PubMed.
- R. W. McMurray and K. J. Hardy, Cox-2 inhibitors: today and tomorrow, Am. J. Med. Sci., 2002, 323, 181–189 CrossRef PubMed.
- A current view on inflammation, Nat. Immunol., 2017, 18, 825, DOI:10.1038/ni.3798.
- C. Nathan, Points of control in inflammation, Nature, 2002, 420, 846–852 CrossRef CAS PubMed.
- V. Kumar, A. K. Abbas, N. Fausto and J. C. Aster, Robbins and Cortan Pathologic Basis of Disease, Professional Edition, Elsevier health sciences, 9th edn, 2014, pp. 14–72 Search PubMed.
- J. Martel-Pelletier, D. Lajeunesse, P. Reboul and J. P. Pelletier, Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs, Ann. Rheum. Dis., 2003, 62, 501–509 CrossRef CAS PubMed.
- R. P. Choudhury, J. M. Lee and D. R. Greaves, Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis, Nat. Clin. Pract. Cardiovasc. Med., 2005, 2, 309–315 CrossRef CAS PubMed.
- P. N. P. Rao, E. E. Knaus, T. P. Road and L. Jolla, Evolution of nonsteroidal anti-inflammatory cyclooxygenase (COX) inhibition and beyond drugs (NSAIDs), J. Pharm. Pharm. Sci., 2008, 11, 81–110 Search PubMed.
- J. Pelletier, Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs, Ann. Rheum. Dis., 2003, 62, 501–509 CrossRef PubMed.
- K. A. M. Abouzid, N. A. Khalil, E. M. Ahmed and S. A. B. Zaitone, Synthesis and biological evaluation of new heteroaryl carboxylic acid derivatives as anti-inflammatory-analgesic agents, Chem. Pharm. Bull., 2013, 61, 222–228 CrossRef CAS PubMed.
- S. Bacchi, P. Palumbo, A. Sponta and M. F. Coppolino, Clinical pharmacology of non-steroidal anti-inflammatory drugs: a review. Anti-inflamm anti-allergy agents, Med. Chem., 2012, 11, 52–64 CAS.
- G. W. Schmid, Analysis of inflammation, Annu. Rev, 2006, 8, 93–151 Search PubMed.
- R. P. Schleimer, An overview of glucocorticoid anti-inflammatory actions, Eur. J. Clin. Pharmacol., 1993, 45, 3–7 CrossRef PubMed.
- J. W. J. Bijlsma, J. W. G. Jacobs and F. Buttgereit, Glucocorticoids in the treatment of rheumatoid arthritis, Clin. Exp. Rheumatol., 2015, 33, 34–36 Search PubMed.
- R. M. Stanbury and E. M. Graham, Systemic corticosteroid therapy – side effects and their management, Br. J. Ophthalmol., 1998, 82(6), 704–708 CrossRef CAS PubMed.
- V. J. Vardanyan and R. S. Hruby, Synthesis of Essential Drugs, Elsevier. 2006. pp. 19–55 Search PubMed.
- J. Braun, X. Baraliakos and T. Westhoff, Nonsteroidal anti-in fl ammatory drugs and cardiovascular risk À a matter of indication, Semin. Arthritis Rheum., 2019, 10–13, DOI:10.1016/j.semarthrit.2019.07.012.
- B. Monteiro and P. V. Steagall, Antiinflammatory drugs, Vet. Clin. North Am. Small Anim. Pract., 2019, 49, 993–1011 CrossRef PubMed.
- D. A. Al-Turki, L. A. Abou-Zeid, I. A. Shehata and M. A. Al-Omar, Therapeutic and toxic effects of new NSAIDS and related compounds: a review and prospective study, Int. J. Pharmacol., 2010, 6, 813–825 CAS.
- X. Zhang, P. T. Donnan, S. Bell and B. Guthrie, Non-steroidal anti-inflammatory drug induced acute kidney injury in the community dwelling general population and people with chronic kidney disease: systematic review and meta-analysis, BMC Nephrol., 2017, 18, 1–12 CrossRef PubMed.
- G. Faich, J. L. Goldstein, L. S. Simon, T. Pincus, A. Whelton and W. F. Stenson, et al., Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis, JAMA-Express, 2015, 284, 1247–1255 Search PubMed.
- R. N. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Sulindac: a review of its pharmacological properties and therapeutic efficacy in rheumatic diseases, Drugs, 1978, 16(2), 97–114 CrossRef CAS PubMed.
- C. J. Hawkey, New drug classes COX-2 inhibitors, Lancet, 1999, 353, 307–314 CrossRef CAS PubMed.
- S. Bindu, S. Mazumder and U. Bandyopadhyay, Non-steroidal anti-in fl ammatory drugs (NSAIDs ) and organ damage: a current perspective, Biochem. Pharmacol., 2020, 180, 114–147 CrossRef PubMed.
- M. A. Abdelgawad, R. B. Bakr and A. A. Azouz, Novel pyrimidine-pyridine hybrids: Synthesis, cyclooxygenase inhibition, anti-inflammatory activity and ulcerogenic liability, Bioorg. Chem., 2018, 77, 339–348 CrossRef CAS PubMed.
- D. L. Simmons, R. M. Botting and T. Hla, Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition, Pharmacol. Rev., 2004, 56(3), 387–437 CrossRef CAS PubMed.
- C. Pasero and M. McCaffery, Selective COX-2 inhibitors, Am. J. Nurs., 2001, 101, 55–56 CrossRef CAS PubMed.
- A. Chakraborti, S. Garg, R. Kumar, H. Motiwala and P. Jadhavar, Progress in COX-2 inhibitors: a journey so far, Curr. Med. Chem., 2010, 17(15), 1563–1593 CrossRef CAS PubMed.
- B. Demircan, Anti-inflammatory effects of selective COX-2 inhibitors, Pol. J. Pharmacol., 2004, 56, 775–780 Search PubMed.
- J. Uddin, P. N. P. Rao and E. E. Knaus, Design and synthesis of novel celecoxib analogues as selective cyclooxygenase-2 (COX-2) inhibitors: replacement of the sulfonamide pharmacophore by a sulfonylazide bioisostere, Bioorg. Med. Chem., 2003, 11, 5273–5280 CrossRef PubMed.
- M. W. Farrar and J. Lerman, Novel concepts for analgesia in pediatric surgical patients: cyclo-oxygenase-2 inhibitors, α2-agonists, and opioids, Anesthesiol. Clin. North. Am., 2002, 20(1), 59–82 CrossRef CAS PubMed.
- P. K. Deb, R. P. Mailabaram, M. J. Saadh and B. Al-Jaidi, Molecular basis ofbinding interactions of NSAIDS and computer-aided drug design approaches in the pursuit ofthe development of cyclooxygenase-2 (COX-2) selective inhibitors, IntechOpen, 2017, 1, 1–144 Search PubMed.
- D. A. Finn, G. Orlans and R. Davenport, COX-2 inhibitors, Lancet, 1999, 353, 978–979 CrossRef PubMed.
- T. Grosser, The pharmacology of selective inhibition of COX-2, Thromb. Haemostasis, 2006, 96(4), 393–400 CAS.
- V. Sharma, P. Bhatia, O. Alam, M. J. Naim, F. Nawaz and A. A. Sheikh, et al., Bioorganic chemistry recent advancement in the discovery and development of COX-2 inhibitors: insight into biological activities and SAR studies, Bioorg. Chem., 2019, 89, 1–45, DOI:10.1016/j.bioorg.2019.103007.
- M. M. Ibrahim, T. Elsaman and M. Y. Al-Nour, Synthesis, anti-inflammatory activity, and in silico study of novel diclofenac and isatin conjugates, Int. J. Med. Chem., 2018, 18, 1–11 Search PubMed.
- F. A. Fitzpatrick, Cyclooxygenase enzymes: regulation and function, Curr. Pharm. Des., 2004, 10, 577–588 CrossRef CAS PubMed.
- A. S. Kalgutkar, B. C. Crews, S. W. Rowlinson, A. B. Marnett, K. R. Kozak and R. P. Remmel, et al., Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors, Proc. Natl. Acad. Sci. U. S. A., 1999, 97, 925–930 CrossRef PubMed.
- S. Bombardier, L. Laine, A. Reicin, D. Shapiro, R. Burgos-Vargas, B. Davis, R. Day, M. B. Ferraz and C. J. Hawkey, Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis, N. Engl. J. Med., 2000, 2, 1520–1528 CrossRef PubMed.
- L. Glen, M. John, L. G. Cleland and M. J. James, Cardiovascular safety of rofecoxib (Vioxx): lessons learned and unanswered questions, Med. J. Aust., 2007, 182, 197–198 Search PubMed.
- P. Hjemdahl, Lessons from 20 years with COX-2 inhibitors: importance of dose – response considerations and fair play in comparative trials, Assoc. Publ. J. Int. Med., 2022, 4, 557–574 Search PubMed.
- D. J. Watson, S. E. Harper, P. Zhao, H. Quan, J. A. Bolognese and T. J. Simon, Gastrointestinal tolerability of the selective cyclooxygenase-2 (COX-2) inhibitor rofecoxib compared with nonselective COX-1 and COX-2 inhibitors in osteoarthritis, Arch. Int. Med., 2000, 160, 2998–3003 CrossRef CAS PubMed.
- G. A. FitzGerald and C. Patrono, The coxibs, selective inhibitors of cyclooxygenase-2, N. Engl. J. Med., 2001, 345, 433–442 CrossRef CAS PubMed.
- J. P. Killen, The coxibs, selective inhibitors of cyclooxygenase-2, N. Engl. J. Med. Corresp., 2001, 345, 1706–1712 CrossRef PubMed.
- D. O. Stichtenoth and J. C. Frölich, The second generation of COX-2 inhibitors: what advantages do the newest offer?, Drugs, 2003, 63, 33–45 CrossRef CAS PubMed.
- L. G. Howes, Selective COX-2 inhibitors, NSAIDs and cardiovascular events – is celecoxib the safest choice, Ther. Clin. Risk Manage., 2007, 3, 831–845 CAS.
- N. Yamakawa, K. Suzuki, Y. Yamashita, T. Katsu and K. Hanaya, Structure – activity relationship of celecoxib and rofecoxib for the membrane permeabilizing activity, Bioorg. Med. Chem., 2014, 22, 2529–2534, DOI:10.1016/j.bmc.2014.02.032.
- M. Wadman, Merck settles vioxx lawsuits for $4.85 billion, Nature, 2007, 450, 68–71 CrossRef PubMed.
- H. Krumholz, What have we learnt from vioxx, BMJ, 2007, 334, 120–123 CrossRef PubMed.
- J. Eric and M. D. Topol, Failing the public health — rofecoxib, merck, and the FDA, N. Engl. J. Med., 2004, 351, 1707–1709 CrossRef PubMed.
- R. Matthew, M. D. Weir, S. Rhoda, M. D. Sperling, M. D. Alise Reicin, J. Barry and M. D. Gertz, Selective COX-2 inhibition and cardiovascular effects: a review of the rofecoxib development program, Am. Heart J., 2003, 146, 591–604 CrossRef PubMed.
- B. J. Orlando and M. G. Malkowski, Crystal structure of rofecoxib bound to human research communications, Acta Cryst., 2016, 72, 772–776 CrossRef CAS PubMed.
- X. Liu, P. Yue, Z. Zhou, F. R. Khuri, S. Sun and B. Celecoxib, Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells, J. Natl. Cancer Inst., 2004, 96, 1769–1780 CrossRef CAS PubMed.
- D. Wei, L. Wang and Y. He, Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of sp1 transcription factor activity, Am Assoc Cancer Res, 2004, 64, 2030–2038 CrossRef CAS PubMed.
- M. J. Schwarz, S. Dehning, A. Douhe and A. Cerovecki, The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine, Nature, 2006, 11, 680–684 Search PubMed.
- V. S. Kamanna and M. L. Kashyap, Nicotinic acid (niacin) receptor agonists: will they be useful therapeutic agents?, Am. J. Cardiol., 2007, 100, 53–61 CrossRef PubMed.
- S. M. Sondhi, S. Rajvanshi, N. Singh, S. Jain and A. M. Lahoti, Heterocyclic inflammation inhibitors, Cent. Eur. J. Chem., 2004, 2, 141–187 CAS.
- I. Melnikova, Future of COX2 inhibitors, Nat. Rev. Drug Discov., 2005, 4(6), 453–454 CrossRef CAS PubMed.
- R. G. Kurumbail, A. M. Stevens, J. K. Gierse, J. J. Mcdonald, R. A. Stegeman and J. Y. Pak, et al., By anti-inflammatory agents, Nature, 1996, 384, 538–541 CrossRef PubMed.
- S. Shorvon, New drug classes pyrrolidone derivatives, Lancet, 2001, 358, 1885–1892 CrossRef CAS PubMed.
- M. Fereidoni, A. Ahmadiani, S. Semnanian and M. Javan, An accurate and simple method for measurement of paw edema, J. Pharmacol. Toxicol. Methods, 2000, 43, 9–12 CrossRef PubMed.
- E. Mohammed, J. Mohsen, N. A. Salih, A. Wheed and R. Atiya, Nicotinic acid derivatives: application and uses, review, J. Res. Chem., 2021, 2, 51–56 Search PubMed.
- M. Gund, F. R. N. Khan, A. Khanna and V. Krishnakumar, Nicotinic acid conjugates of nonsteroidal anti-inflammatory drugs (NSAID's) and their anti-inflammatory properties, Eur. J. Pharm. Sci., 2013, 49(2), 227–232 CrossRef CAS PubMed.
- H. Bundgaard, Novel chemical approaches in prodrug design, Drugs Future, 1991, 16, 443–458 Search PubMed.
- N. A. Baeian, M. J. Anafrooz, M. A. Bbaspour, V. Z. Iaee, A. D. Alvandi and A. S. Hafiee, Synthesis and analgesic activity of 2-phenoxybenzoic acid and N-phenylanthranilic acid hydrazides, Biol. Pharm. Bull., 2006, 29, 1180–1185 CrossRef PubMed.
- A. Shafiee, A. Kheradmand, L. Navidpour, H. Shafaroodi and G. Saeedi-Motahar, Design and synthesis of niflumic acid-based N-acylhydrazone derivatives as novel anti-inflammatory and analgesic agents, Med. Chem. Res., 2013, 22, 2411–2420 CrossRef.
- M. B. Labib, A. M. Fayez, E. S. EL-Nahass, M. Awadallah and P. A. Halim, Novel tetrazole-based selective COX-2 inhibitors: design, synthesis, anti-inflammatory activity, evaluation of PGE2, TNF-α, IL-6 and histopathological study, Bioorg. Chem., 2020, 104, 104–308 CrossRef PubMed.
- L. Navidpour, H. Shafaroodi, G. Saeedi-Motahar and A. Shafiee, Synthesis, anti-inflammatory and analgesic activities of arylidene-2-(3-chloroanilino)nicotinic acid hydrazides, Med. Chem. Res., 2014, 23(6), 2793–2802 CrossRef CAS.
- F. Kazunaga, K. Osamu, H. Morihide, M. Noriyuki and H. yoshikazU, A method for evaluating analgesic agents in rats, J. Pharmacol. Methods, 1980, 4, 251–259 CrossRef PubMed.
- Y. El-Dash, N. A. Khalil, E. M. Ahmed and M. S. A. Hassan, Synthesis and biological evaluation of new nicotinate derivatives as potential anti-inflammatory agents targeting COX-2 enzyme, Bioorg. Chem., 2021, 107, 104–610 CrossRef PubMed.
- J. Grover, N. Bhatt, V. Kumar, N. K. Patel, B. J. Gondaliya, M. E. Sobhia, K. K. Bhutani and S. M. Jachak, 2,5-Diaryl-1,3,4-oxadiazoles as selective COX-2 inhibitors and anti-inflammatory agents, R. Soc. Chem., 2015, 18, 1–29 Search PubMed.
- C. Viegas-junior, A. Danuello, S. Bolzani, E. J. Barreiro, C. Alberto and M. Fraga, Molecular hybridization: a useful tool in the design of new drug prototypes, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef CAS PubMed.
- A. G. Banerjee, N. Das, S. A. Shengule, P. A. Sharma, R. Shyam and S. Kumar, Design, synthesis, evaluation and molecular modelling studies of some heterocyclic moieties as potential COX-2 inhibitors: a hybrid pharmacophore approach, Bioorg. Chem., 2016, 69, 102–120, DOI:10.1016/j.bioorg.2016.10.003.
- H. S. Abd-ellah, M. Abdel-aziz, M. E. Shoman, E. A. M. Beshr, T. Kaoud and A. F. F. Ahmed, New 1,3,4-oxadiazole/oxime hybrids: Design, synthesis, anti- inflammatory, COX inhibitory activities and ulcerogenic liability, Bioorg. Chem., 2017, 74, 15–29, DOI:10.1016/j.bioorg.2017.06.003.
- Y. A. Ammar, E. E. Fayed, A. Bayoumi and M. Saleh, Design and synthesis of pyridine-amide based compounds appended naproxen moiety as anti-microbial and anti- inflammatory agents, Am. J. PharmTech. Res., 2015, 6, 245–273 Search PubMed.
- D. S. Gonçalvesa, S. M. Melo, A. P. Jacomini, M. J. V. Silva, K. E. Pianoskia, F. Q. Ames, R. P. Aguiar, A. F. Oliveira and H. Volpato, et al., Synthesis of novel 3,5,6-trisubstituted 2-pyridone derivatives and evaluation for their anti-inflammatory activity, Bioorg. Med. Chem., 2020, 28, 1–14, DOI:10.1016/j.bmc.2020.115549.
- R. H. Bensimon, Croton oil peels, Aesthetic Surg. J., 2008, 28, 33–45 CrossRef CAS PubMed.
- A. Negrusz, C. Moore, D. Deitermann, D. Lewis, K. Kaleciak and R. Kronstrand, et al., Highly sensitive micro-plate enzyme immunoassay screening and NCI-GC-MS confirmation of flunitrazepam and its major metabolite 7-aminoflunitrazepam in hair, J. Anal. Toxicol., 1999, 23, 429–435 CrossRef CAS PubMed.
- M. Helal, S. El-Awdan, M. Salem, T. Abd-Elaziz, Y. Moahamed, A. El-Sherif and G. Mohamed, Synthesis, biological evaluation and molecular modeling of novel series of pyridine derivatives as anticancer, anti-inflammatory and analgesic agents, Acta Part A Mol. Biomol. Spectrosc., 2015, 135, 764–773 CrossRef CAS PubMed.
- M. Abdel-tawab, H. Zettl and M. Schubert-zsilavecz, Nonsteroidal anti-inflammatory drugs: a critical review on current concepts applied to reduce gastrointestinal toxicity, Curr. Med. Chem., 2009, 16, 2042–2063 CrossRef CAS PubMed.
- E. M. Ahmed, A. E. Kassab, A. A. El-Malah and M. S. A. Hassan, Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents, Eur. J. Med. Chem., 2019, 171, 25–37 CrossRef CAS PubMed.
- E. M. Ahmed, M. S. A. Hassan, A. A. El-Malah and A. E. Kassab, New pyridazine derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents; design, synthesis and biological evaluation, Bioorg. Chem., 2020, 95, 1–24 CrossRef PubMed.
- R. Pratap and V. J. Ram, Natural and synthetic chromenes, fused chromenes, and versatility of dihydrobenzo [h] chromenes in organic synthesis, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed.
- J. L. Wang, D. Limburg, M. J. Graneto, J. Springer, J. R. B. Hamper and S. Liao, et al., The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life, Bioorg. Med. Chem. Lett., 2010, 20(23), 7159–7163 CrossRef CAS PubMed.
- A. M. Gouda, H. I. Ali, W. H. Almalki, M. A. Azim, M. A. S. Abourehab and A. H. Abdelazeem, Design, synthesis, and biological evaluation of some novel pyrrolizine derivatives as COX inhibitors with anti-inflammatory/analgesic activities and low ulcerogenic liability, Molecules, 2016, 21(2), 1–21 CrossRef PubMed.
- W. Shahid, S. Abida, M. Saleem, M. Ahmed, J. Rahman and N. Riaz, et al., Identification of NSAIDs as lipoxygenase inhibitors through highly sensitive chemiluminescence method, expression analysis in mononuclear cells and computational studies, Bioorg. Chem., 2021, 110, 10–48 CrossRef PubMed.
- S. Yaqoob, N. Nasim, R. Khanam, Y. Wang, A. Jabeen and U. Qureshi, et al., Synthesis of highly potent antiinflammatory compounds ( ROS Inhibitors) from isonicotinic acid, Molecules, 2021, 2, 1–8 Search PubMed.
- B. Irena and E. M. Corinne, 7 - Detection Methods Using Chemiluminescence, Molecular Methods for Virus Detection, ed. D. L. Wiedbrauk, D. H. Farkas, Academic Press, 1995, pp. 147–174, DOI:10.1016/B978-012748920-9/50008-X.
- L. W. Mohamed, M. A. Shaaban, A. F. Zaher, S. M. Alhamaky and A. M. Elsahar, Synthesis of new pyrazoles and pyrozolo [3,4-b] pyridines as anti- inflammatory agents by inhibition of COX-2 enzyme, Bioorg. Chem., 2019, 83, 47–54 CrossRef CAS PubMed.
- B. Seaver and J. R. Smith, Inhibition of COX isoforms by nutraceuticals, J. Herb Pharmacother., 2004, 4, 11–18 CrossRef CAS PubMed.
- H. F. Rizk and S. A. Ibrahim, Synthesis of some novel heterocyclic dyes derived from pyrazole derivatives, Arab. J. Chem., 2011, 4, 37–44, DOI:10.1016/j.arabjc.2010.06.012.
- N. R. Thakare, A. K. Dhawas, P. S. Ganoskar and P. D. Kale, Synthesis, characterization of some new 3,5-dimethyl azopyrazoles and its derivatives, J. Chem. Pharm. Res., 2012, 4, 3329–3332 CAS.
- H. Wu, X. Lu, J. Xu, X. Zhang, Z. Li and X. Yang, Design, synthesis and fungicidal activity of N-(thiophen-2-yl) nicotinamide derivatives, Molecules, 2022, 27, 4–15 Search PubMed.
- K. Paruch, A. Biernasiuk, D. Khylyuk, R. Paduch, M. Wujec and Ł. Popiołek, Synthesis, biological activity and molecular docking studies of novel nicotinic acid derivatives, Int. J. Mol. Sci., 2022, 23, 1–23 Search PubMed.
- E. M. Ahmed, N. A. Khalil, E. Ramadan, T. Tharwat, M. A. Ali and Z. Mahmoud, Synthesis and biological evaluation of new nicotinic acid derivatives as potential anti-inflammatory agents with enhanced gastric safety profile, Bioorg. Chem., 2024, 144, 107–136, DOI:10.1016/j.bioorg.2024.107136.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Zeinab Mahmoud
and
Zeinab Mahmoud