
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThymol and carvacrol derivatives as anticancer agents; synthesis, in vitro activity, and computational analysis of biological targets†
Mohammed A. Alamri *a,
Maged S. Abdel-Kaderb,
Mohamad Ayman Salkinib and
Mubarak A. Alamric
*a,
Maged S. Abdel-Kaderb,
Mohamad Ayman Salkinib and
Mubarak A. Alamric
aDepartment of Pharmacology, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 16273, Saudi Arabia. E-mail: ma.alamri@psau.edu.sa
bDepartment of Pharmacognosy, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 16273, Saudi Arabia
cDepartment of Pharmaceutical Chemistry, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 16273, Saudi Arabia
First published on 25th September 2024
Abstract
Various thymol and carvacrol derivatives have been synthesized to test the anticancer activity potential. Computational methods including network pharmacology and molecular docking approaches were utilized to identify and assess the potential biological targets relating to cancer. Amongst the synthesized derivatives the ethoxy-cyclohexyl analogues were consistently the most active against a panel of 10 different cancer cell lines covering a variety of origins. Biological target predictions revealed the AKT1 protein to be a core and central target of the most active compounds. Molecular docking identified a binding pocket within this protein in which the most active compounds bind. The incorporation of computational analysis methods and conventional structure–activity approaches identified analogues of thymol and carvacrol with the highest anticancer potential, and analyzed their possible biological targets in a comprehensive manner.
1. Introduction
Thymol is a monoterpene phenol appendant in Thymus species including T. vulgaris as well as Trachyspermum ammi fruits.1,2 Carvacrol is structural isomer to thymol naturally occurring in T. vulgaris and Origanum species.3 Both compounds are simple in structure with one reactive site that make derivatization generally straightforward, and hence, many derivatives were reported.4 Many essential oils contain thymol, which exhibits many biological activities including antimicrobial,5 anti-inflammatory,6 wound healing,7 antioxidant,8 and due to its minimal toxicity, it shows promise as a botanical pesticide.9 Thymol and thymol derivatives were reported to exert anticancer activity via various mechanisms including producing intracellular reactive oxygen species, suppressing cell growth, depolarizing mitochondrial membrane potential, inducing apoptosis, and activating the proapoptotic mitochondrial proteins Bax, interaction with cysteine aspartases (caspases) or poly ADP ribose polymerase.10–12 Carvacrol possess antimicrobial, antioxidant, analgesic, antispasmodic, and anti-inflammatory activities amongst others.13–15 Carvacrol has also been reported to possess anticancer activity via decreasing the expressions of matrix metalloprotease 2 and 9.16,17 Furthermore, carvacrol-derived Schiff base derivatives were reported to have anti-lung cancer activity as well.18 These promising reports incentivize the pursuit of more active thymol and carvacrol derivatives. In this study, we synthesized various analogues and assessed the in vitro anticancer activity of these analogues in addition to the parent compounds. Furthermore, computational network pharmacology approaches were utilized to predict and analyze the possible cancer-related gene targets of these molecules.19 In addition, the most integral biological gene target of the analogues with the highest anticancer activity was studied using molecular docking techniques to further elucidate the observed anticancer activity.2. Materials and methods
2.1 General procedure for organic synthesis and analysis
Solvents and reagents were used as is without further treatment. Reactions were conducted under nitrogen gas. Thymol and carvacrol were acquired from Aktin Chemicals, Inc. (Chengdu, China). All solvents and remaining reagents were acquired from Sigma Aldrich (St. Louis, Missouri, United States of America). 1500-Micron preparative thin layer chromatography plates were acquired from ANALTECH (Newark, Delaware, United States of America). Nuclear magnetic resonance analysis was performed using CDCl3 (chloroform-d3) on a Bruker Ultrashield Plus 500 MHz spectrometer at 24 °C. Chemical shifts (δ) are relative to TMS as ppm. Chemical shifts multiplicity are reported as “s” for singlet, “d” for doublet, “t” for triplet, and “m” for multiplet, and broad shifts are preceded by “b”. Chloroform was used as a solvent to dilute samples in order to obtain 1 ppm concentration. Injection of 1 μL of the solutions to Agilent GC/MS instrument Model 7890 MSD were performed with the help of autosampler adjusted to splitless mode. Helium (99.999% purity) was the carrier gas adjusted to flow rate = 1.2 mL min−1. HP5MS 30 m length column with 0.25 mm i.d. and 0.25 μm thickness was used in the analysis. The injector temperature was set at a constant temperature of 280 °C. The instrument was programmed with a start temperature of 70 °C for 5 min, then gradual raising of the temperature at a rate of 2 °C min−1 till 120 °C where it was held for 2 min, followed by increasing rate of 15 °C min−1 to 290 °C that was kept for 2 min. The ionization in the mass spectrometer was performed at 70 eV, mass range was set between 30–600 daltons. The ion-source temperature was adjusted to 280 °C. High performance liquid chromatography (HPLC) analysis was used for compounds TN and CN due fragmentation when using GC. HPLC was carried out using a 250 × 4.6 mm C18 column, a UV detector (254 nm), and a mobile phase consisting of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 methanol and water (0.1% formic acid). HPLC samples were injected at a volume of 5 μL at a concentration of 1 mg mL−1. The synthesis scheme and brief reaction conditions are depicted in Fig. 1. All analysis spectra are listed in the ESI.†
:25 methanol and water (0.1% formic acid). HPLC samples were injected at a volume of 5 μL at a concentration of 1 mg mL−1. The synthesis scheme and brief reaction conditions are depicted in Fig. 1. All analysis spectra are listed in the ESI.†
 | ||
| Fig. 1 Synthesis scheme and carbon numbering assignment of target compounds. Conditions: (i) potassium hydroxide, acetonitrile, 90 °C, 1 hour. (ii) triethylamine, tetrahydrofuran, 25 °C. | ||
2.2 Preparation of 2-isopropyl-1-(2-methoxy-ethoxy)-5-methyl-benzene (TE)
This compound was formed by reacting 0.33 mmole (50 mg) of thymol with 0.66 mmole (37 mg) of potassium hydroxide, and 1 mmole (94 μL) of 2-bromoethyl methyl ether in acetonitrile for 1 hour at 90 °C. Following this, water and a mixture of n-hexane and ethyl acetate (1:1 v/v) were added and the organic phase was collected, dried over anhydrous sodium sulphate, and condensed before purification using preparative TLC and a mobile phase consisting of n-hexane and ethyl acetate (2:1 v/v, Rf = 0.72). The product was collected as a colorless oil (17 mg, 0.082 mmole, 24.85%).
MS calculated: 208.146, found: 208.2.
GC AUC: >99%.
1H NMR (500 MHz, CDCl3) δ 7.13 (1H, d, J = 7.2 Hz, H-3), 6.79 (1H, bd, J = 6.3 Hz, H-4), 6.71 (1H, bs, H-6), 4.15 (2H, s, H-1′), 3.81 (2H, s, H-2′), 3.50 (3H, s, H-3′), 3.35 (1H, m, H-7), 2.35 (3H, s, H-10), 1.39 (6H, d, J = 6.7 Hz, H-8, H-9).
13C NMR (125 MHz, CDCl3) δ 21.36 (C-7), 22.82 (C-8, C-9), 26.54 (C-10), 59.25 (C-3′), 67.65 (C-2′), 71.32 (C-1′), 112.68 (C-6), 121.50 (C-4), 125.93 (C-3), 134.38 (C-2), 136.28 (C-5), 149.54 (C-1).
2.3 Preparation of 3-isopropyl-1-(2-methoxy-ethoxy)-6-methyl-benzene (CE)
The reaction was carried out by reacting 0.33 mmole (52 μL) of carvacrol with 1 mmole (94 μL) of 2-bromoethyl methyl ether, and 0.66 mmole (37 mg) of potassium hydroxide in acetonitrile at 90 °C for 1 h. Following this, water was added, and the product was extracted using a mixture of n-hexane and ethyl acetate (1:1 v/v). The organic phase was dried over anhydrous sodium sulphate and dried under heat. Purification was performed using preparative thin-layer chromatography using a mobile phase consisting of n-hexane and ethyl acetate (2:1 v/v, Rf = 0.72) yielding a colorless oil (30.3 mg, 0.146 mmole, 44.24%).
MS calculated: 208.146, found: 208.2.
GC AUC: >99%.
1H NMR (500 MHz, CDCl3) δ 7.11 (1H, d, J = 7.4 Hz, H-3), 6.80 (1H, bd, J = 7.6 Hz, H-4), 6.77 (1H, bs, H-6), 4.19 (2H, m, H-1′), 3.83 (2H, m, H-2′), 3.53 (3H, s, H-3′), 2.94 (1H, m, H-7), 2.27 (3H, s, H-10), 1.29 (6H, d, J = 7.1 Hz, H-8, H-9).
13C NMR (125 MHz, CDCl3) δ 15.91 (C-10), 24.19 (C-8, C-9), 34.16 (C-7), 59.35 (C-3′), 67.73 (C-2′), 71.34 (C-1′), 109.95 (C-2), 118.43 (C-4), 124.39 (C-6), 130.51 (C-5), 147.88 (C-3), 156.94 (C-1).
2.4 Preparation of 1-(2-cyclohexyl-ethoxy)-2-isopropyl-5-methyl-benzene (TH)
This compound was synthesized by reacting 0.33 mmole (50 mg) of thymol with 0.66 mmole (37 mg) of potassium hydroxide and 0.66 mmole of (103.3 μL) 1-bromo-2-cyclohexylethane in acetonitrile for at 90 °C for 1 hour. After this, water was added and the product was extracted by adding a mixture of ethyl acetate and n-hexane (1:1 v/v). The organic phase was dried using anhydrous sodium sulphate and dried under heat. Preparative thin-layer chromatography was used to purify the compound with a mobile phase of n-hexane and ethyl acetate (2:1 v/v, Rf = 0.73). The product was collected as a colorless oil (39.1 mg, 0.15 mmole, 45.45%).
MS calculated: 260.214, found: 260.3.
GC AUC: >99%.
1H NMR (500 MHz, CDCl3) δ 7.16 (1H, d, J = 7.5 Hz, H-3), 6.80 (1H, bd, J = 6.5 Hz, H-4), 6.73 (1H, bs, H-6), 4.05 (2H, t, J = 6.9 Hz, H-1′), 3.38 (1H, m, H-7), 2.39 (3H, s, H-10), 1.27 (6H, d, J = 6.7 Hz, H-8, H-9 overlapped), 1.05, 1.34, 1.62, 1.78, 1.85 (H-2′–H-8′).
13C NMR (125 MHz, CDCl3) δ 21.43 (C-7), 22.82 (C-8, C-9), 26.38 (C-10), 65.76 (C-1′), 26.65, 33.37, 34.74, 36.87 (C-2′–C-8′), 112.14 (C-6), 120.84 (C-4), 125.82 (C-3), 134.08 (C-2), 136.26 (C-5), 156.24 (C-1).
2.5 Preparation of 1-(2-cyclohexyl-ethoxy)-3-isopropyl-6-methyl-benzene (CH)
This compound was synthesized by reacting 0.33 mmole (52 μL) of carvacrol with 0.66 mmole (37 mg) of potassium hydroxide and 0.66 mmole of (103.3 μL) 1-bromo-2-cyclohexylethane in acetonitrile for 1 hour at 90 °C. Following this, water and a mixture of n-hexane and ethyl acetate (1:1 v/v) were added to the reaction, and the organic phase was collected, dried using anhydrous sodium sulphate, then condensed under heat. Preparative TLC was used to purify the product using a mobile phase of n-hexane and ethyl acetate (2:1 v/v, Rf = 0.73). The product was collected as a colorless oil (38.7 mg, 0.149 mmole, 45.03%).
MS calculated: 260.214, found: 260.2.
GC AUC: >99%.
1H NMR (500 MHz, CDCl3) δ 7.13 (1H, d, J = 7.5 Hz, H-5), 6.80 (1H, bd, J = 7.6 Hz, H-4), 6.78 (1H, bs, H-2), 4.08 (2H, t, J = 6.5 Hz, H-1′), 2.95 (1H, m, H-7), (3H, s, H-10), (6H, d, J = 6.7 Hz, H-8, H-9 overlapped), 1.06, 1.62, 1.79, 1.78, 1.87 (H-2′–H-8′).
13C NMR (125 MHz, CDCl3) δ 15.98 (C-7), 24.25 (C-8, C-9), 34.26 (C-7), 26.41, 26.66, 33.44, 34.82, 36.91 (C-2′–C-8′), 65.84 (C-1′), 109.38 (C-2), 117.77 (C-4), 124.16 (C-6), 130.38 (C-5), 147.86 (C-3), 157.26 (C-1).
2.6 Preparation of acetic acid 2-isopropyl-5-methyl-phenyl ester (TA)
This reaction was conducted by reacting 0.666 mmole (100 mg) thymol with 2.663 mmole (189 μL) acetyl chloride and 2.663 mmole (370 μL) triethylamine in anhydrous THF at room temperature. Following this immediate reaction, water was added, and the product was extracted using a mobile phase of n-hexane and ethyl acetate (2:1 v/v). The organic phase was dried using anhydrous sodium sulphate, then dried under heat. Purification was performed using preparative TLC and a mobile phase of n-hexane and ethyl acetate (2:1 v/v, Rf = 0.49) to yield a colorless oil (37.6 mg, 0.196 mmole, 29.37%).
MS calculated: 192.115, found: 192.1.
GC AUC: >99%.
1H NMR (500 MHz, CDCl3) δ 7.26 (1H, d, J = 7.4 Hz, H-3), 7.09 (1H, bd, J = 6.5 Hz, H-4), 6.88 (1H, bs, H-6), 3.04 (1H, m, H-7), 2.37 (3H, s, H-10), 2.38 (3H, s, OCOCH3), 1.27 (6H, d, J = 6.6 Hz, H-8, H-9).
13C NMR (125 MHz, CDCl3) δ 20.88 (C-7), 20.99 (OCOCH3), 23.11 (C-8, C-9), 27.19 (C-10), 122.80 (C-6), 126.52 (C-4), 127.25 (C-3), 136.63 (C-2), 137.05 (C-5), 147.92 (C-1), 169.88 (OCOCH3).
2.7 Preparation of acetic acid 3-isopropyl-6-methyl-phenyl ester (CA)
Synthesis of this compound was carried out by reacting 0.647 mmole (102 μL) of carvacrol with 2.588 mmole (184 μL) acetyl chloride in tetrahydrofuran with 2.588 mmole (360 μL) triethylamine. The reaction is completed at room temperature and within seconds. Following this, water is added and the product is extracted with a mixture of n-hexane and ethyl acetate (2:1 v/v), dried using anhydrous sodium sulphate, then condensed under heat. Purification was carried out using preparative TLC and a mobile phase consisting of n-hexane and ethyl acetate (2:1 v/v, Rf = 0.49) to yield the product as a colorless oil (24.8 mg, 0.129 mmole, 19.94%).
MS calculated: 192.115, found: 192.2.
GC AUC: >99%.
1H NMR (500 MHz, CDCl3) δ 7.20 (1H, d, J = 7.8 Hz, H-3), 7.08 (1H, bd, J = 7.8 Hz, H-4), 6.93 (1H, bs, H-6), 2.96 (1H, m, H-7), 2.36 (3H, s, H-10), 2.20 (3H, s, OCOCH3), 1.29 (6H, d, J = 7.0 Hz, H-8, H-9).
13C NMR (125 MHz, CDCl3) δ 15.82 (C-10), 20.87 (OCOCH3), 23.95 (C-8, C-9), 33.60 (C-7), 119.81 (C-2), 124.24 (C-4), 127.22 (C-6), 130.93 (C-5), 148.11 (C-3), 149.32 (C-1), 169.37 (OCOCH3).
2.8 Preparation of 4-nitro-benzoic acid 2-isopropyl-5-methyl-phenyl ester (TN)
This compound was prepared by reacting 0.666 mmole (100 mg) thymol, 1.332 mmole (247 mg) 4-nitrobenzoyl chloride, and 1.294 mmole (180 μL) triethylamine in anhydrous THF at room temperature. Following this, water and a mixture of n-hexane and ethyl acetate (4:1 v/v) were added, and the organic phase was collected, then dried using anhydrous sodium sulphate. The mixture was condensed under heat, then purified using preparative TLC using a mobile phase consisting of n-hexane and ethyl acetate (4:1 v/v, Rf = 0.62) to yield a colorless oil (47.8 mg, 0.16 mmole, 23.98%).
MS calculated: 299.1158, found: 299.1.
HPLC AUC: 96.96%.
1H NMR (500 MHz, CDCl3) δ 8.42 (4H, m, 4-nitro-benzoyl), 7.30 (1H, d, J = 7.0 Hz, H-3), 7.14 (1H, bd, J = 6.4 Hz, H-4), 6.99 (1H, bs, H-6), 3.04 (1H, m, H-7), 2.39 (3H, s, H-10), 1.25 (6H, d, J = 6.7 Hz, H-8, H-9).
13C NMR (125 MHz, CDCl3) δ 20.90 (C-7), 23.09 (C-8, C-9), 27.35 (C-10), 122.54 (C-6), 123.84 (C-3′, C-5′), 126.73 (C-4), 127.73 (C-3), 131.29 (C-2′, C-6′), 135.00 (C-2), 136.96 (C-5), 147.76 (C-4′), 150.91 (C-1), 163.60 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
2.9 Preparation of 4-nitro-benzoic acid 3-isopropyl-6-methyl-phenyl ester (CN)
This analogue was prepared by reacting 0.647 mmole (102 μL) of carvacrol, 0.776 mmole (144 mg) 4-nitrobenzoyl chloride, and 1.294 mmole (180 μL) triethylamine in anhydrous THF at room temperature. Following this, water was added, and the product was collected using an organic phase composed of n-hexane and ethyl acetate (4:1 v/v, Rf = 0.62). After drying using anhydrous sodium sulphate, the mixture was condensed under heat, then purified using preparative TLC and a mobile phase of n-hexane and ethyl acetate (4:1 v/v) to yield a colorless oil (33.2 mg, 0.111 mmole, 17.16%).
MS calculated: 299.1158, found: 299.1.
HPLC AUC: 96.61%.
1H NMR (500 MHz, CDCl3) δ 8.43 (4H, m, 4-nitro-benzoyl), 7.25 (1H, d, J = 7.7 Hz, H-3), 7.13 (1H, bd, J = 7.6 Hz, H-4), 7.04 (1H, bs, H-6), 2.95 (1H, m, H-7), 2.23 (3H, s, H-10), 1.29 (6H, d, J = 7.0 Hz, H-8, H-9).
13C NMR (125 MHz, CDCl3) δ 15.88 (C-10), 23.95 (C-8, C-9), 33.64 (C-7), 119.58 (C-2), 123.79 (C-3′, C-4′), 124.76 (C-4), 127.09 (C-6), 131.30 (C-2′, C-6′), 134.98 (C-5), 148.44 (C-3), 149.09 (C-1), 150.89 (C-4′), 163.11 (CO).
2.10 In vitro anticancer activity screening
Anticancer activity was assessed based on a sulforhodamine-B assay and a variety of cancer cell lines of different origins including: A375 (melanoma), A549 (alveolar basal epithelial cell adenocarcinoma), DU145 (prostate cancer), HeLa (epithelial-like cervical cancer cells), Hep2 (head and neck tumor epithelial cell), HepG2 (Hepatocellular carcinoma), HT29 (colorectal adenocarcinoma), MCF7 (breast adenocarcinoma), PANC1 (pancreatic ductal carcinoma), and SCOV3 (ovarian adenocarcinoma). Cancer cell lines were acquired from Nawah Scientific Inc., (Mokatam, Cairo, Egypt). Cells were maintained in DMEM (A549, Hep2, HepG2, A375DU145, MCF7) or RPMI (HT29, PANC1, SCOV3, HeLa) media including streptomycin (100 mg mL−1), penicillin (100 units per mL), and 10% fetal bovine serum in humidified, 5% (v/v) CO2 atmosphere at 37 °C. 100 μL cell suspension (5 × 103 cells) were transferred to 96-well plates and incubated in corresponding media for 24 hours. Following this, cells were treated with additional media (100 μL) containing drugs at 10 and 100 μM concentrations. After 48 hours, media were replaced by fresh media containing 150 μL 10% TCA, then incubated for 1 hour at 4 °C to fix the cells. Following this, TCA-containing media were removed, and the cells were washed 5 times with distilled water. After this, aliquots of 70 μL SRB solution (0.4% w/v) were added and incubated at room temperature for 10 minutes away from direct light. Following this, cells were washed with 1% acetic acid 3 times, and were left to dry overnight. Next, 150 μL of TRIS solution (10 mM) was added to dissolve bound SRB stain. Absorbance was measured using an Infinite F50 microplate reader at 540 nm (TECAN, Switzerland). Viability was calculated relative to control (average of 3 wells), and error was calculated as standard deviation. Doxorubicin was tested for comparison.2.11 Determination of physicochemical and pharmacokinetic properties
Physicochemical properties and pharmacokinetic interaction predictions were obtained using the SwissADME database, which computes these parameters based on structural similarities.202.12 Virtual screening of cancer-related genes
Identification of cancer-related genes was predicted by using the GeneCards® The Human Gene Database, using the key term “cancer” to identify cancer-related genes.21 Microsoft Excel spreadsheet was utilized to export and manage data for further study.2.13 Virtual screening of compounds' potential gene targets
Possible gene targets of the parent and synthesized analogues were identified by utilizing the “SwissTargetPrediction” database.22 Predictions are based on chemical structure similarity and the number of similar compounds to other known molecules indexed within the database, and a probability score is given on that basis. All gene targets with a probability higher than 0 were considered for this study.2.14 Identification of overlapping genes between cancer and the test compounds
Detection of common gene targets between the test compounds and cancer was performed using Venn diagrams (Venny 2.1.0 online database).23 Overlapping genes for all test compounds as a whole, and for individual compounds were highlighted.2.15 Enrichment analysis of molecular function pathways
Analysis of the cancer-related genes overlapping with the most active compounds was performed based on gene ontology (GO) to determine the molecular functions of these genes. This analysis was conducted by the ShinyGO database.24 The data acquired takes into consideration the molecular functions, the number of genes, and the statistical significance, defined as false discovery rate (FDR), where a lower FDR indicates a stronger prediction of the involvement of the genes in the corresponding molecular function.2.16 Analysis of protein–protein interactions
Gene targets of the most active compounds (TH and CH) were assessed for potential protein–protein interactions (PPIs) by utilizing the “STRING: functional protein association networks” database.25 This analysis helps identify core cancer-related gene targets of the test compounds. All genes with a confidence score ≥0.4 were considered for this study.2.17 Network construction
To further analyze molecular interactions between the genes, and identify the genes with the highest relation to cancer and the most active test compounds (TH and CH), a network of the previously identified PPI genes was constructed by the Cytoscape bioinformatics software 3.10.1.26 This offers a comprehensive overview of the highest potential cancer-related, and compound-related genes.2.18 Molecular docking
Compounds TH and CH were considered for molecular docking within the highest potential, cancer-related gene target from the PPI data. Test compounds were drawn in ChemDraw Ultra 7.0 and saved in .Mol format. Before docking, the molecular structures of compounds energy minimized and converted into .pdb using BIOVIA Discovery Studio Visualizer 2019 tool. The crystal structure of the top anticancer core target obtained from the PPI results; AKT1 (PDB ID: 3o96) was downloaded from the Protein Data Bank (PDB) in PDB format.27 The protein structure was prepared by removing any water molecules and co-crystalized ligands – compounds that may bind to the protein. The grid-box with dimension of 20 Å was generated to cover all essential restudies within the active site of AKT1 using Autodock tools (center_x = 8.018 Å, center_y = −7.694 Å, center_z = 11.973 Å). The docking process was carried out using Autodock Vina software.28 Following this, the compounds were converted into .pdbqt format, which is compatible with the Autodock Vina software. Finally, results were visualized and analyzed using BIOVIA Discovery Studio Visualizer 2019 and Pymol programs.3. Results and discussion
3.1 Synthesis
The synthesis of the ether bond-containing analogues (TE, CE, TH, and CH) was performed via “Williamson ether synthesis”. These analogues form successfully in anhydrous tetrahydrofuran containing potassium hydroxide and the corresponding alkyl bromides. Interestingly, synthesis of other analogues using alkyl chlorides proved difficult, even with modulation of reaction conditions (temperature and time) and molar ratios. The synthesis of the ester-containing analogues (TA, CA, TN, and CN) was rapidly performed in anhydrous acetonitrile containing triethylamine and the corresponding acid chlorides. In general, the proximity of the reacting phenolic hydroxy to the isopropyl moiety in carvacrol did not show an apparent effect on the product yields when compared to the thymol analogues.3.2 Anticancer and pharmacokinetic properties
Cell viability after a 48 hour incubation at a concentration of 100 μM revealed compounds TH and CH to be the most active following doxorubicin which proved to be potent at that concentration (Fig. 2). In some cell lines, the nitrobenzene derivatives (TN and CN) had some anticancer activity. Furthermore, some cell lines displayed high resistance such as A375 and HT29, while others showed more susceptibility such as HepG2, PANC1, MCF7, and DU145. Testing at a concentration of 10 μM did not show any note-worthy anticancer activity, with the exception of doxorubicin (ESI†).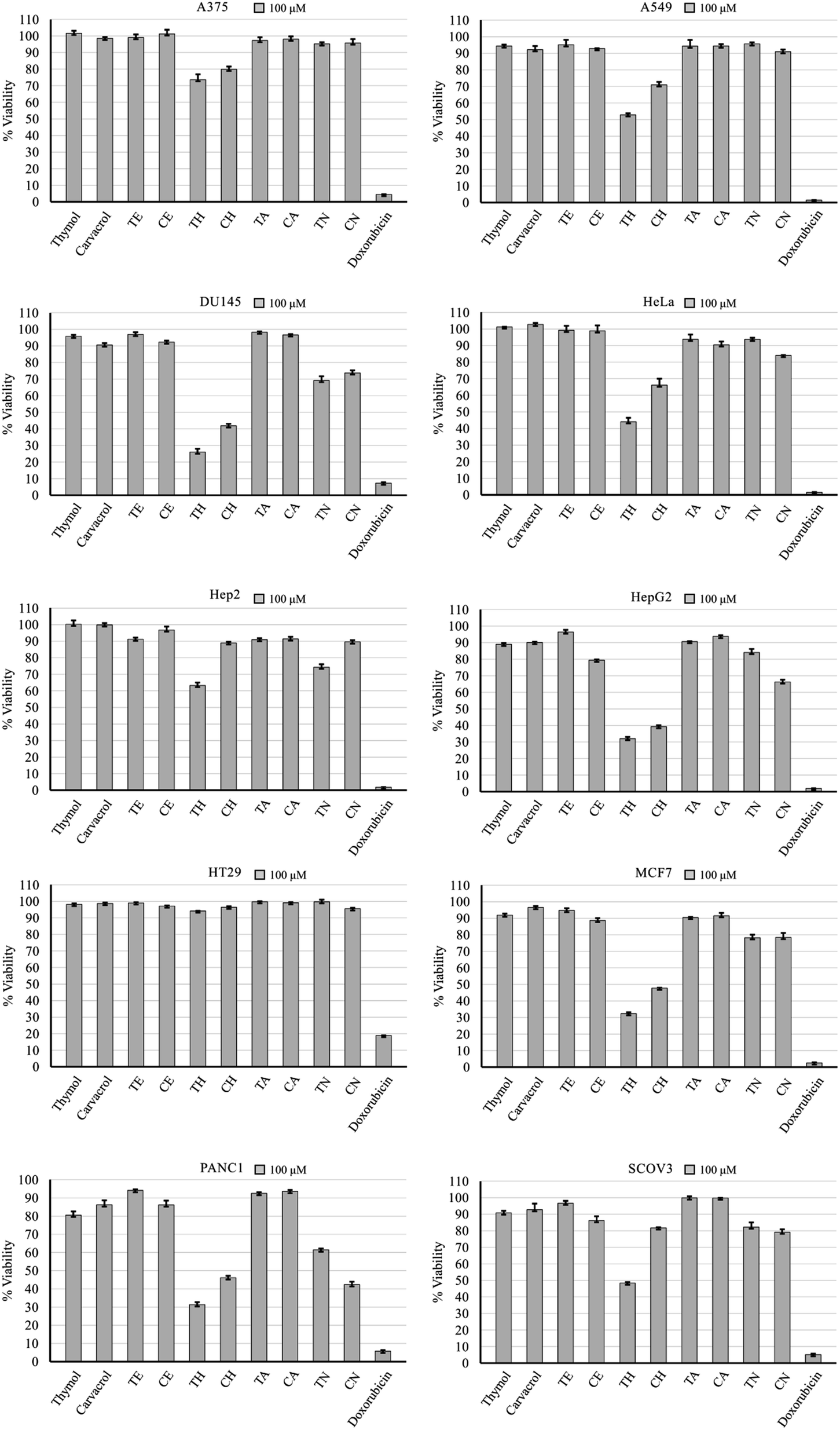 | ||
| Fig. 2 Anticancer activity of test compounds and doxorubicin against 10 cell lines after 48 hour treatment at 100 μM. Error bars: standard deviation. | ||
The cyclohexyl analogues (TH and CH) demonstrated the highest anticancer activity on average, followed by the para-nitrobenzene analogues (Table 1). The cyclohexyl moiety has been reported previously to induce this effect.29 Data retrieved from the SwissADME database predicts no interaction with P-glycoprotein, which is a cancer-related, non-specific efflux pump.30 Pharmacokinetic and physicochemical properties had no apparent link with the observed anticancer activity of compounds TH and CH, except for lipid solubility (logP), which was the highest in these two analogues. This may play a role in the observed anticancer activity, but further investigation is required.
| Cell viability average | log P | H-bond acceptors | H-bond donors | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | |
|---|---|---|---|---|---|---|---|---|---|---|
| Thymol | 94.38% | 2.82 | 1 | 1 | No | Yes | No | No | No | No |
| Carvacrol | 94.83% | 2.8 | 1 | 1 | No | Yes | No | No | No | No |
| TE | 96.27% | 3.17 | 2 | 0 | No | Yes | No | No | Yes | No |
| CE | 91.97% | 3.19 | 2 | 0 | No | Yes | No | No | Yes | No |
| TH | 49.85% | 5.18 | 1 | 0 | No | Yes | No | No | Yes | No |
| CH | 65.95% | 5.25 | 1 | 0 | No | Yes | No | No | Yes | No |
| TA | 94.79% | 3.02 | 2 | 0 | No | No | No | No | No | No |
| CA | 94.90% | 3.1 | 2 | 0 | No | No | No | No | No | No |
| TN | 83.41% | 3.5 | 4 | 0 | No | Yes | Yes | Yes | No | No |
| CN | 79.59% | 3.46 | 4 | 0 | No | Yes | Yes | Yes | No | No |
3.3 Cancer-related and potential compounds targets
The top 25000 cancer-related genes were retrieved from the GeneCards database, which utilizes data for over 350000 genes to detect genes related to the search query, which was the term “cancer” for this study. The potential gene targets of the test compounds were identified using the SwissTargetPrediction database, which identifies possible targets based on a probability score that is proportional to degree of structural similarity with other known molecules within the database. This probability score is also proportional to the number of similar molecules as well. A total of 262 possible gene targets of the test compounds were used for further study. By utilizing Venn diagrams, 241 common genes between cancer and all the test compounds as whole were identified (Fig. 3).
 | ||
| Fig. 3 The number of common genes between the test compounds and cancer. | ||
Interestingly, when considering individual compound targets, the data revealed the most active compounds (TH and CH) to have the least number of possible cancer-related targets compared to the other semi-synthetic analogues (Table 2). Since compounds TH and CH had the most anticancer activity, only their gene targets will be considered for the following steps. Full lists of genes are in the ESI section.†
| Gene targets | Overlapping with 25000 cancer-related genes |
|
|---|---|---|
| Thymol | 27 | 27 |
| Carvacrol | 23 | 20 |
| TE | 48 | 46 |
| CE | 45 | 43 |
| TH | 28 | 26 |
| CH | 28 | 25 |
| TA | 46 | 40 |
| CA | 33 | 31 |
| TN | 93 | 84 |
| CN | 100 | 93 |
| All compounds | 262 | 241 |
3.4 Enrichment analysis of molecular function pathways
Enrichment analysis of the overlapping genes between cancer and compounds TH and CH revealed a high number of genes (14) relating to various signaling receptors which is a wide category including membrane receptors for neurotransmitters, cytokines, and nuclear receptors as well (Fig. 4).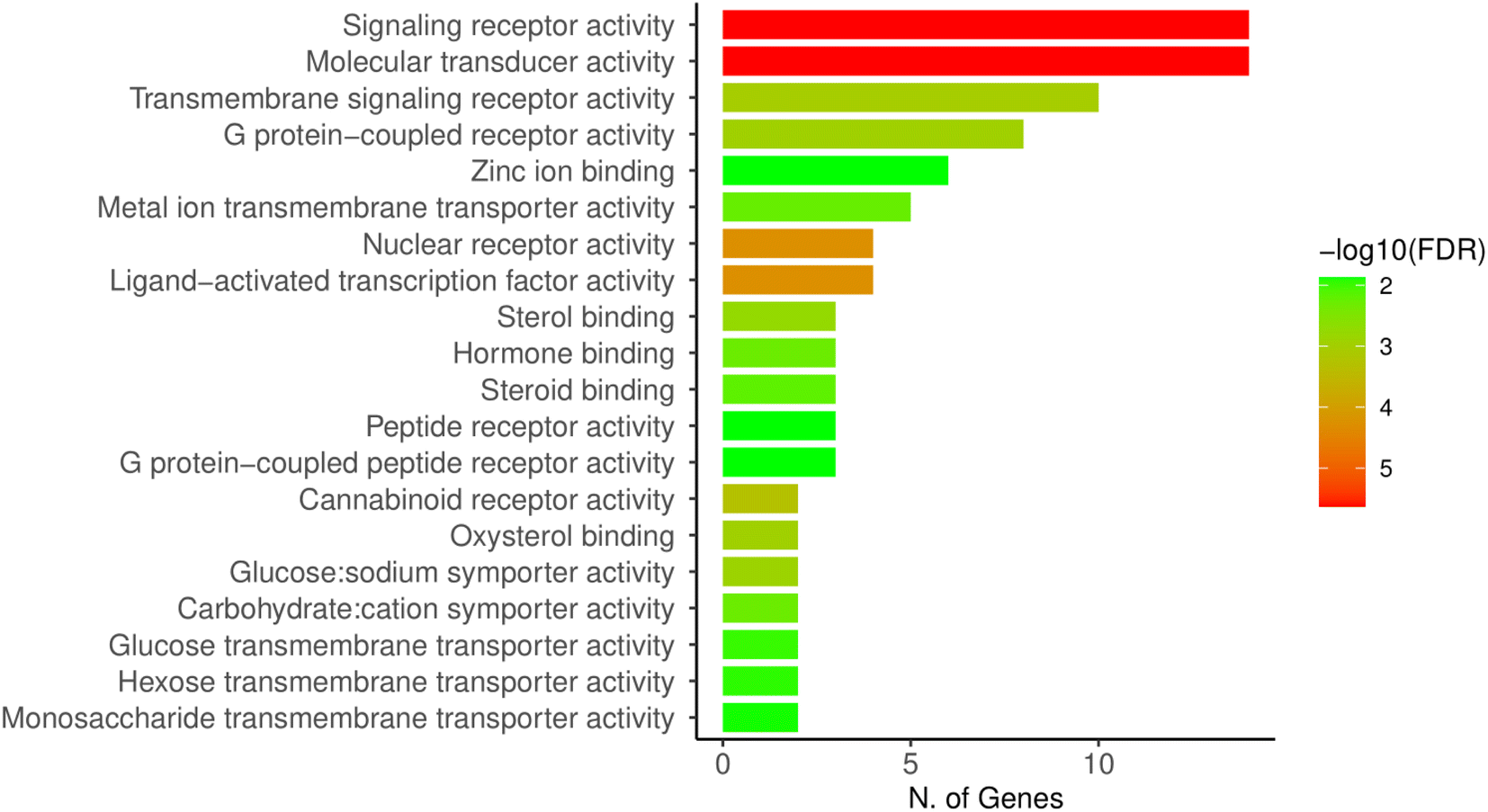 | ||
| Fig. 4 ShinyGO analysis of molecular functions of the cancer-related gene targets of TH and CH. | ||
In addition, molecular transduction was also found to be a molecular function linked to a high number of genes (14). This class of activity also includes a wide variety or receptors and biological effector functions, and is often overlapping with the “signaling receptor activity” category.
3.5 Protein–protein interactions and network construction
A network of the cancer-related, and potential compound TH and CH target genes (total of 31) was constructed using the STRING database (Fig. 5). This is performed to highlight known interactions within these genes. This analysis had a small PPI enrichment p-value of 5.31 × 10−7, indicating a significant relationship between some of these genes. | ||
| Fig. 5 PPI network of compounds TH and CH cancer-related gene targets constructed by the STRING database. | ||
A network of the previous PPI genes was constructed using Cytoscape software to highlight the genes with the most known interactions to other genes, totaling 26 genes and excluding 5 genes with no known interactions (Fig. 6). Based on the degree of centrality, which represents high interaction with other genes, the gene encoding AKT1 was found to be the most important, which is a protein kinase highly involved in the regulation of cell division in normal and cancer states.31 Other prominent genes within this network are highlighted in Fig. 5 as well.
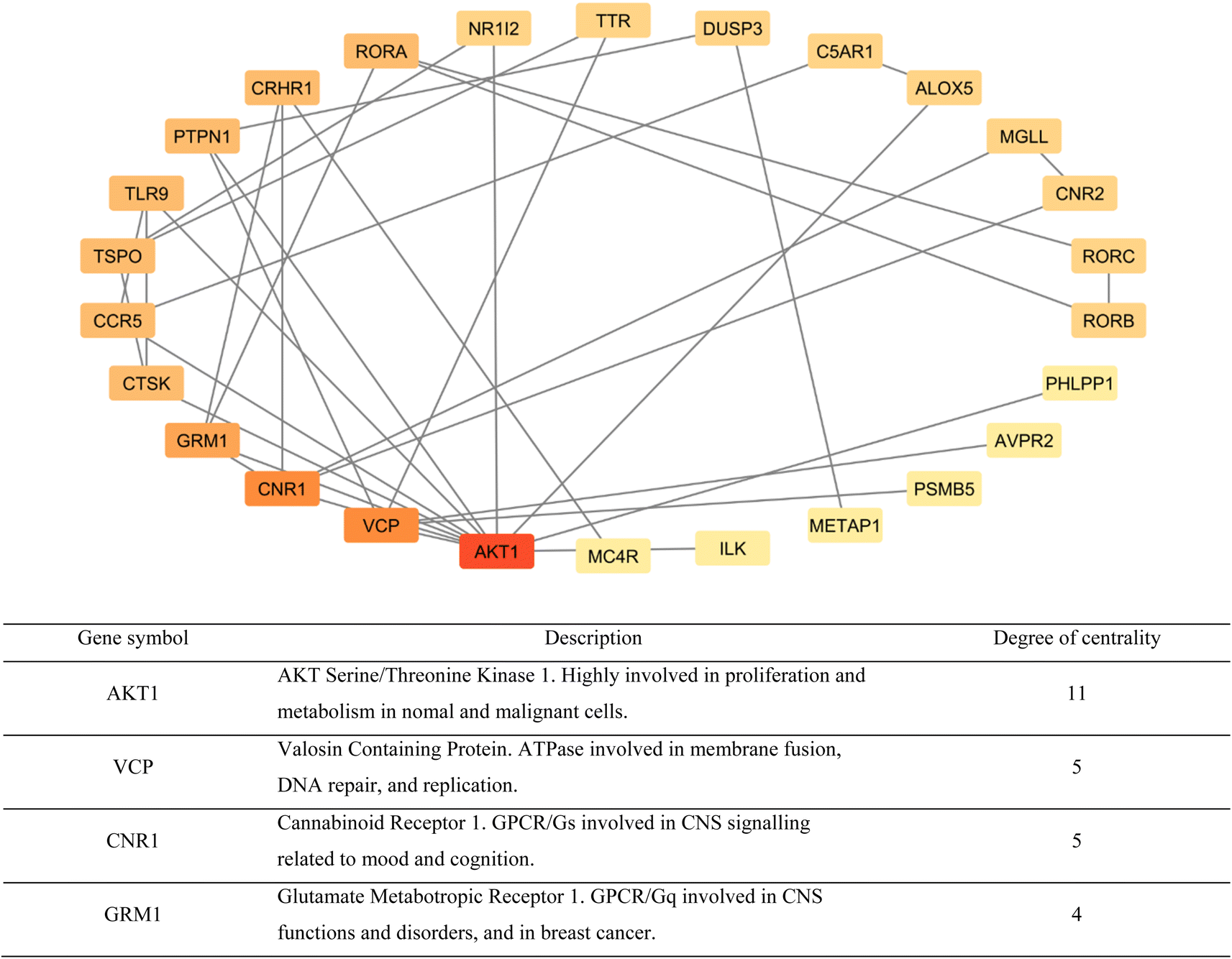 | ||
| Fig. 6 Cytoscape network of protein–protein interactions highlighting known interactions between the cancer-related targets of compounds TH and CH (from red to light yellow: highest to lowest). In addition, a brief description of the top 4 genes based on degree of centrality (bottom). | ||
The Cytoscape software was used to display a network of the gene targets of compounds TH and CH to highlight common, and exclusive, gene targets of each (Fig. 7). Interestingly, the top 4 genes from the previous PPI network were common to both compounds. Furthermore, the slight structural difference between compounds TH and CH was able to elicit some differences in potential biological targets. This highlights the importance of testing structurally similar analogues to better understanding the corresponding alteration of potential biological targets.
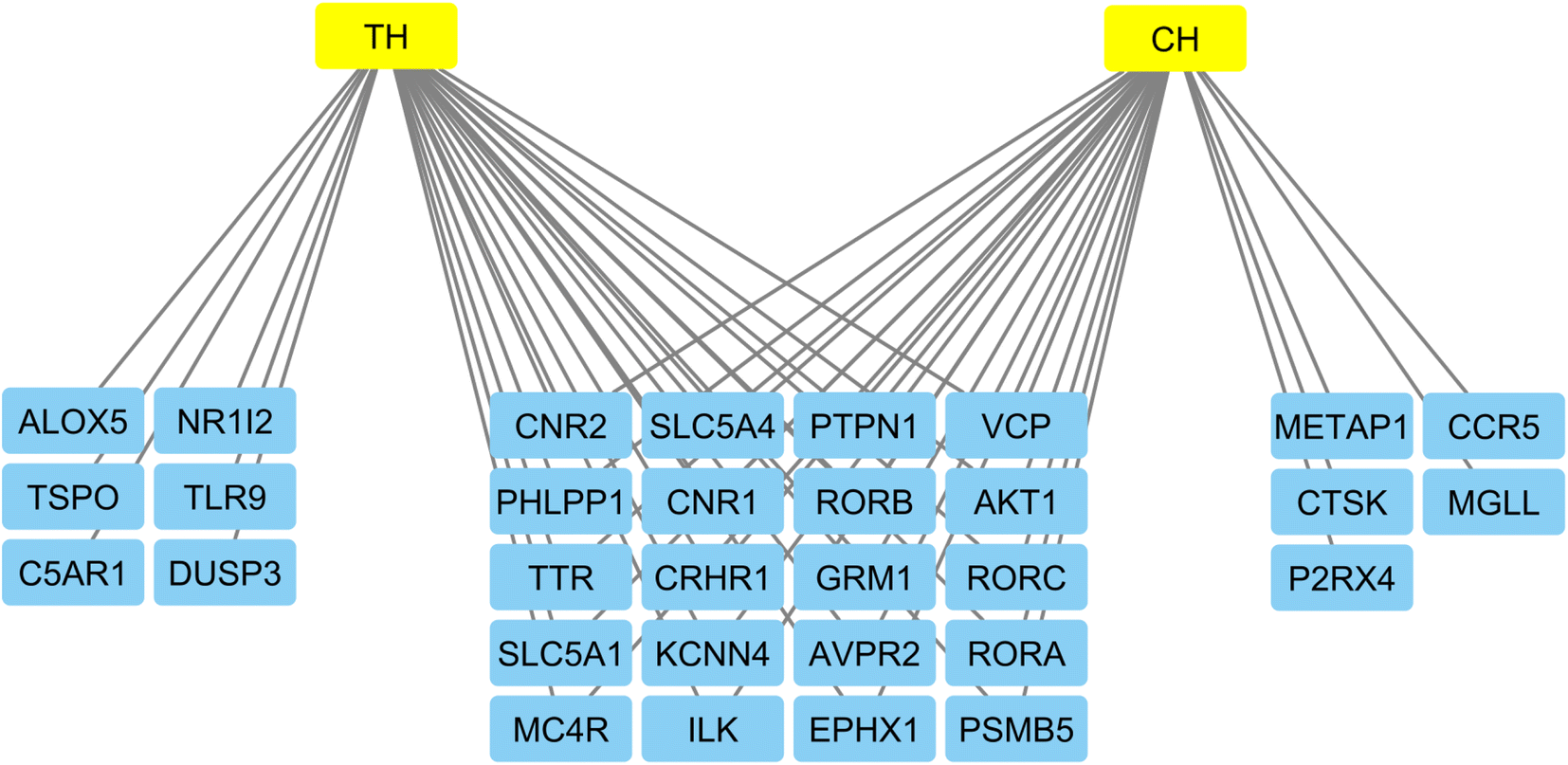 | ||
| Fig. 7 Network of gene targets of the most active compounds constructed by the Cytoscape software to highlight common, and exclusive targets of both TH and CH compounds. | ||
3.6 Molecular docking
The top anti-cancer core target (AKT1) was molecularly docked with the two compounds TH and CH. Both compounds adapted a stable conformation deeply inside the active site of AKT1 (Fig. 8A). In terms of interaction, CH formed multiple hydrophobic interactions with Val27, Trp80, Leu210, Leu264, Lys268 within the active site of AKT1 (Fig. 8B). Similarly, TH interacts with Val27, Trp80, Leu210, Leu264, Lys268 and Trp272 via hydrophobic bonds at the active site chamber (Fig. 8C). Additionally, both molecules exhibit strong binding ability towards AKT1. The binding energy scores of CH and TH were −8.8 and −9.0 kcal mol−1, respectively. | ||
| Fig. 8 Docking and interaction mechanism of CH and TH with AKT1. (A) Binding mode of CH (green) and TH (orange) inside the active site of AKT1 (purple). (B) Amino acid interactions with CH. (C) Amino acid interactions with TH. | ||
4. Conclusion
The synthesis of various thymol and carvacrol analogues was performed to test in vitro anticancer activity and to identify commonality and differences in potential biological targets related to cancer. Within the test compounds, the cyclohexylethane variants (TH and CH) displayed the highest anticancer activity when compared to the other analogues. Computational methods identified gene targets of the test compounds which were also cancer-related. The analysis of these genes revealed that the most central of them was AKT1, which was reported to be highly involved in proliferation and cancer. Furthermore, structural differences between the most active compounds revealed differences in some biological targets. Molecular docking study of compounds TH and CH revealed that they localize within a deep binding pocket within the AKT1 protein with strong binding characteristics facilitated via interaction with several residues within the protein. Taken collectively, thymol and carvacrol may have potential as anticancer agents if modified accordingly, and the observed anticancer activity could be mediated via interaction with AKT1 protein. Lastly, the use of network pharmacology and molecular docking approaches can help assess, and guide conventional SAR studies to achieve more potent analogues, and to better understand the biological targets and molecular functions involved.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no known financial or personal competing interest.Acknowledgements
This study was supported via funding from Prince Sattam Bin Abdulaziz University project number (PSAU/2024/R/1446).References
- S. J. Lee, K. Umano, T. Shibamoto and K. G. Lee, Identification of volatile components in basil (Ocimum basilicum L.) and thyme leaves (Thymus vulgaris L.) and their antioxidant properties, Food Chem., 2005, 91(1), 131–137 CrossRef CAS
.
- I. M. Chung, T. Dangkhanh, O. K. Lee and A. Ahmad, Chemical Constituents from Ajwain Seeds (Trachyspermum ammi) and Inhibitory Activity of Thymol, Lupeol and Fatty Acids on Barnyardgrass and Radish Seeds, Asian J. Chem., 2010, 19(2), 1524–1534 Search PubMed
- B. Tohidi, M. Rahimmalek, A. Arzani and M. R. Sabzalian, Thymol, carvacrol, and antioxidant accumulation in Thymus species in response to different light spectra emitted by light-emitting diodes, Food Chem., 2020, 307, 125521 CrossRef CAS PubMed
- K. H. Baser, Biological and pharmacological activities of carvacrol and carvacrol bearing essential oils, Curr. Pharm. Des., 2008, 14(29), 3106–3119 CrossRef CAS PubMed
- H. Miladi, T. Zmantar, B. Kouidhi, Y. Chaabouni, K. Mahdouani, A. Bakhrouf and K. Chaieb, Use of carvacrol, thymol, and eugenol for biofilm eradication and resistance modifying susceptibility of Salmonella enterica of serovar Typhimurium strains to nalidixic acid, Microb. Pathog., 2017, 104, 56–63 CrossRef CAS PubMed
- M. A. Botelho, G. Barros, D. B. Queiroz, C. F. Carvalho, J. Gouvea, L. Patrus, M. Bannet, D. Patrus, A. Rego and I. Silva, Nanotechnology in Phytotherapy: Antiinflammatory Effect of a Nanostructured Thymol Gel from Lippia sidoides in Acute Periodontitis in Rats, Phytother. Res., 2016, 30(1), 152–159 CrossRef CAS PubMed
- K. Riella, R. Marinho, J. Santos, R. Pereira-Filho, J. Cardoso, R. Albuquerque-Junior and S. Homazzi, Anti-inflammatory and cicatrizing activities of thymol, a monoterpene of the essential oil from Lippia gracilis, in rodents, J. Ethnopharmacol., 2012, 143(2), 656–663 CrossRef CAS PubMed
- L. L. Deng, M. Taxipalati, F. Que and H. Zhang, Physical characterization and antioxidant activity of thymol solubilized Tween 80 micelles, Sci. Rep., 2016, 6, 38160–38168 CrossRef CAS PubMed
- T. Gao, H. Zhou, W. Zhou, L. Hu, J. Chen and Z. Shi, The fungicidal activity of thymol against Fusarium graminearum via inducing lipid peroxidation and disrupting ergosterol biosynthesis, Molecules, 2016, 21(6), 770 CrossRef PubMed
- S. H. Kang, Y. S. Kim, E. K. Kim, J. W. Hwang and J. H. Jeong, Anticancer effect of thymol on AGS human gastric carcinoma cells, J. Microbiol. Biotechnol., 2016, 26(1), 28–37 CrossRef CAS PubMed
- B. Salehi, A. P. Mishra, I. Shukla, M. Sharifi-Rad and M. D. M. Contreras, Thymol, thyme, and other plant sources: health and potential uses, Phytother. Res., 2018, 32(9), 1688–1706 CrossRef PubMed
- B. Salehi, P. Zucca, M. Sharifi-Rad, R. Pezzani and S. Rajabi, Phytotherapeutics in cancer invasion and metastasis, Phytother. Res., 2018, 32(8), 1425–1449 CrossRef PubMed
- D. Zhang, R. Gan, Y. Ge, Q. Yang, J. Ge, H. Li and H. Corke, Research Progress on the antibacterial mechanisms of carvacrol: A Minireview, Bioactive Compounds in Health and Disease, 2018, 1(6), 71–81 Search PubMed
- B. Tohidi, M. Rahimmalek, A. Arzani and M. R. Sabzalian, Thymol, carvacrol, and antioxidant accumulation in Thymus species in response to different light spectra emitted by light-emitting diodes, Food Chem., 2020, 307, 125521 CrossRef CAS PubMed
- M. Zielińska-Błajet and J. Feder-Kubis, Monoterpenes and Their Derivatives-Recent Development in Biological and Medical Applications, Int. J. Mol. Sci., 2020, 21(19), 7078 CrossRef PubMed
- F. B. Rezvi and A. Roy, Carvacrol: A well-known phytochemical for modern dentistry, Drug Invent. Today, 2019, 11(6), 1460–1463 Search PubMed
- A. G. Bayir, H. S. Kiziltan and A. Kocyigit, Plant Family, Carvacrol, and Putative Protection in Gastric Cancer, in Dietary Interventions in Gastrointestinal Diseases, ed. R. R. Watson and V. R. Preedy, Academic Press, 2019, pp. 3–18 Search PubMed
- A. Bansal, M. M. Saleh-E-In, P. Kar, A. Roy and N. R. Sharma, Synthesis of Carvacrol Derivatives as Potential New Anticancer Agent against Lung Cancer, Molecules, 2022, 27(14), 4597 CrossRef CAS PubMed
- P. Zhang, D. Zhang, W. Zhou, L. Wang, B. Wang, T. Zhang and S. Li, Network pharmacology: towards the artificial intelligence-based precision traditional Chinese medicine, Briefings Bioinf., 2024, 25(1), bbad518 CrossRef PubMed
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed
- M. Safran, I. Dalah, J. Alexander, N. Rosen, T. I. Stein, M. Shmoish, N. Nativ, I. Bahir, T. Doniger, H. Krug, A. Sirota-Madi, T. Olender, Y. Golan, G. Stelzer, A. Harel and D. Lancet, GeneCards Version 3: the human gene integrator, Database, 2010, 2010, baq020 CrossRef PubMed
- A. Daina, O. Michielin and V. Zoete, SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules, Nucleic Acids Res., 2019, 47(W1), W357–W364 CrossRef CAS PubMed
- J. C. Oliveros, Venny. An Interactive Tool for Comparing Lists with Venn's Diagrams, 2015, https://bioinfogp.cnb.csic.es/tools/venny/index.html Search PubMed
- S. X. Ge, D. Jung and R. Yao, ShinyGO: a graphical gene-set enrichment tool for animals and plants, Bioinform, 2020, 36(8), 2628–2629 CrossRef CAS PubMed
- D. Szklarczyk, R. Kirsch, M. Koutrouli, K. Nastou, F. Mehryary, R. Hachilif, A. L. Gable, T. Fang, N. T. Doncheva, S. Pyysalo, P. Bork, L. J. Jensen and C. V. Mering, The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest, Nucleic Acids Res., 2023, 51(D1), D638–D646 CrossRef CAS PubMed
- M. Kohl, S. Wiese and B. Warscheid, Cytoscape: Software for Visualization and Analysis of Biological Networks, Methods Mol. Biol., 2011, 696, 291–303 CrossRef CAS PubMed
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28(1), 235–242 CrossRef CAS PubMed
- S. Dallakyan and A. J. Olson, Small-molecule library screening by docking with PyRx, Methods Mol. Biol., 2015, 1263, 243–250 CrossRef CAS PubMed
- S. Benetti, M. Dalla Pozza, L. Biancalana, S. Zacchini, G. Gasser and F. Marchetti, The beneficial effect of cyclohexyl substituent on the in vitro anticancer activity of diiron vinyliminium complexes, Dalton Trans., 2023, 52(17), 5724–5741 RSC
- C. P. Heming, W. Muriithi, L. W. Macharia, P. N. Filho, V. Moura-Neto and V. Aran, P-glycoprotein and cancer: what do we currently know?, Heliyon, 2022, 8(10), e11171 CrossRef PubMed
- A. Alwhaibi, A. Verma, M. S. Adil and P. R. Somanath, The unconventional role of Akt1 in the advanced cancers and in diabetes-promoted carcinogenesis, Pharmacol. Res., 2019, 145, 104270 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: All analysis spectra and complete lists of genes are listed in the ESI material. See DOI: https://doi.org/10.1039/d4ra03941f |
| This journal is © The Royal Society of Chemistry 2024 |