DOI:
10.1039/D4RA04730C
(Review Article)
RSC Adv., 2024,
14, 27657-27696
FDA-approved drugs containing dimethylamine pharmacophore: a review of the last 50 years
Received
29th June 2024
, Accepted 23rd August 2024
First published on 2nd September 2024
Abstract
Dimethylamine (DMA) derivatives represent a promising class of compounds with significant potential in the field of medicinal chemistry. DMA derivatives exhibit a diverse range of pharmacological activities, including antimicrobial, antihistaminic, anticancer, and analgesic properties. Their unique chemical structure allows for the modulation of various biological targets, making them valuable candidates for the treatment of numerous diseases. Synthetic strategies for the preparation of DMA derivatives vary depending on the desired biological activity and target molecule. Common synthetic routes involve the modification of the DMA scaffold through functional group manipulation, scaffold hopping, or combinatorial chemistry approaches. Therapeutically, DMA derivatives have shown promise in the treatment of infectious diseases, especially bacterial infections. Additionally, by focusing on particular biochemical pathways involved in tumor growth and metastasis, DMA-based drugs have shown anticancer activity. In addition to their direct pharmacological effects, DMA derivatives can serve as valuable tools in drug delivery systems, prodrug design, and molecular imaging techniques, enhancing their utility in medicinal chemistry research. Overall, DMA derivatives represent a versatile class of compounds with immense potential in medicinal chemistry. Further research and development efforts are warranted to explore their full therapeutic capabilities and optimize their clinical utility in the treatment of various diseases. This article outlines the pharmacological properties, synthetic strategies, and therapeutic applications of DMA derivatives of FDA approved drugs, highlighting their importance in drug discovery and development.
1. Introduction
Dimethylamine (DMA) is an organic compound having molecular formula NH(CH3)2. Chemically DMA is a secondary amine, in which two methyl (–CH3) groups are directly attached to the nitrogen atom. It is a colourless gas that, at lower concentrations, smells like fish and, in higher concentrations, smells like ammonia.1 DMA gas is corrosive and easily dissolves in water to produce combustible solutions that are corrosive. Because the DMA gas is heavier than air, it can cause asphyxia through air displacement. DMA gas ignites easily and releases harmful oxides of nitrogen. DMA is a weak base and it reacts with acid to form salts such as dimethylamine hydrochloride.2 DMA is present in a broad variety of animals and plants and can be found in many foods at low concentrations.3 DMA is a precursor of many industrially important compounds. When it combines with carbon disulfide, it produces dimethyl dithiocarbamate, which is a precursor to zinc bis(diethyldithiocarbamate) and other compounds that are needed to vulcanize rubber with sulfur. DMA is the source of the solvents such as dimethylformamide and dimethylacetamide. DMA is also used to create rocket fuel such as unsymmetrical dimethylhydrazine.4 DMA is a precursor to several significant pharmaceutically active substances that are used to treat a wide range of diseases, which comprise agents used in the treatment of CNS disease,5,6 anti-cancer agents,7 antihistaminic agent,8,9 analgesic,10 anti-obesity,11 anti-malarial,12 and antimuscarinic agent.13 DMA is an electron-donating group (EDG). The loan pair of nitrogen atoms of DMA participates in many nucleophilic reactions, for example bis(dimethylamino)methane was formed when DMA reacted with formaldehyde. Similar to DMA, the majority of the drugs containing DMA (such as tramadol, tripelennamine, imipramine, trimeprazine, amitriptyline, and ranitidine) are water-soluble and remain in the bloodstream for an extended period. These drugs offer key advantages such as high solubility, limited side effects, and increased bioavailability and effectiveness. Dimethylamine is a weak base, and when it interacts with acidic compounds, it forms salts. These salts are often more water-soluble than the non-ionic forms of the compounds. DMA has a hydrophilic (water-attracting) nature, due to the presence of the nitrogen atom, and the ability to form hydrogen bonds with water molecules. This property also enhances its solubility in water. In 2019, concerns arose about the risk of N-nitrosodimethylamine (NDMA) formation associated with ranitidine and its potential link to cancer. NDMA is a chemical compound considered a probable human carcinogen. It is not intentionally added to products but can emerge as a by-product of specific chemical reactions. NDMA can form in drugs containing dimethylamine when exposed to nitrites, particularly during manufacturing, storage, and breakdown processes. Conditions that promote nitrosation, such as elevated temperatures or acidic environments, can worsen this issue (Fig. 1). To avoid the formation of NDMA from DMA, it's essential to control factors that promote its synthesis, such as nitrites, reaction conditions, and storage conditions. Additionally, adding antioxidants like ascorbic acid (vitamin C) and alpha-tocopherol (vitamin E) can inhibit the nitrosation reaction. DMA derivatives are a prevalent building block of natural products and bioactive molecules used in materials, medicines, and agrochemical industries. Thus, there has been a lot of interest in the synthesis of useful DMA molecules, which are discussed below.
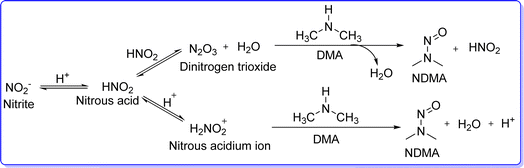 |
| | Fig. 1 Formation of NDMA from DMA and nitrite. | |
1.1 General synthesis of DMA
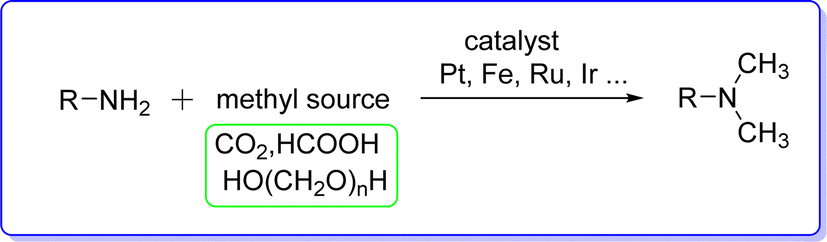
The Eschweller–Clarke reaction is the conventional synthetic method of preparing DMA in industry, which is accomplished by methylation of alkyl amine by HCOOH/HCHO (ref. 14) (Fig. 2).
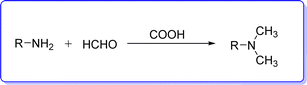 |
| | Fig. 2 Conventional synthesis of DMA. | |
DMA is also synthesized by a substitution reaction of alkyl amine and methyl halide15 or dimethyl sulfate16 (Fig. 3).
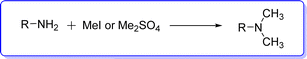 |
| | Fig. 3 Synthesis of DMA. | |
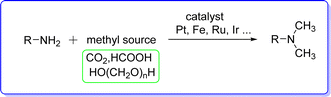
Nevertheless, these reactions have a number of drawbacks, including the use of hazardous chemicals, a high waste production rate, and frequently poor selectivity. In order to overcome these problems, transition metal catalyzed N-methylation of amines with methyl reagents17 like formic acid,18 carbon dioxide,19 or paraformaldehyde20 has been developed effectively (Fig. 4).
 |
| | Fig. 4 Transition metal catalyzed synthesis of DMA. | |
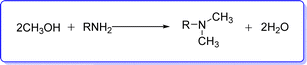
These reactions have low selectivity, high temperature and pressure requirements, and necessitate the use of a reductant. Therefore, employing amines and alcohols in a catalytic “borrowing hydrogen strategy” to create a C–N bond has been considered a clean and atom efficient technique.21 The entire reaction is comprised of multiple sequential stages, such as transfer hydrogenation, imine production, and dehydrogenation. H2O is the only harmless byproduct is produced during the reaction. Numerous reports of catalytic N-methylation utilizing methanol to produce N,N-dimethylamine derivatives have been made based on this procedure22,23 (Fig. 5).
 |
| | Fig. 5 Atom-efficient synthetic route to DMA. | |
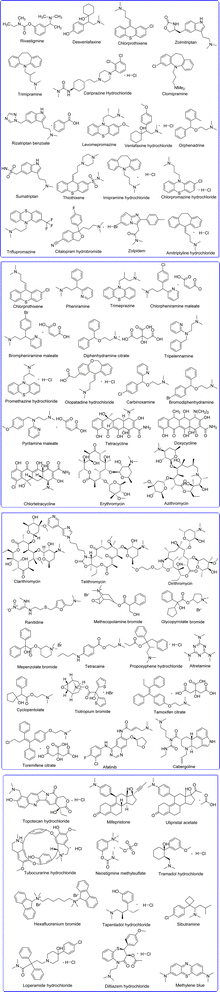
In this review article, we exclusively focus on the DMA moieties containing drugs approved by FDA over past 50 years, and we document the synthesis of drug molecule, and their clinical importance as antihistaminic, antibiotic, antimuscarinic, antidiarrheal, anti-obesity, anticancer, analgesics, muscle relaxant, local anesthetics, and contraceptive agents (Fig. 6). We also highlighted the DMA-containing drugs that are specifically employed in treatment of peptic ulcer, CNS disease, and cardiovascular disease. The current article aids to researchers in development of new drugs that incorporate the DMA pharmacophore.
 |
| | Fig. 6 List of FDA approved drugs containing dimethylamine. | |
2. Drug used in treatment of CNS disease
2.1 Rivastigmine
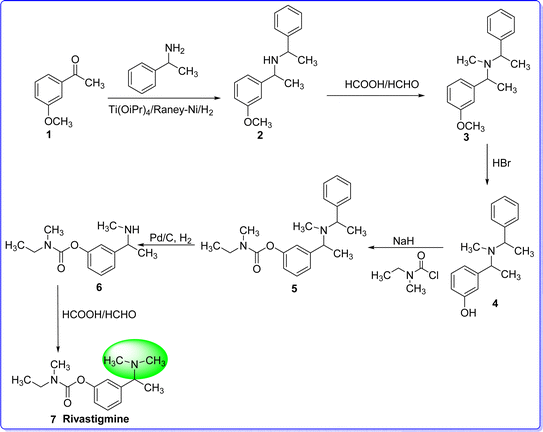
Rivastigmine is a brain-region selective acetylcholinesterase inhibitor with a prolonged duration of action.24 The medication can be applied topically or sublingually and the application of the medication topically decreases the potential of adverse effects, like nausea and vomiting.25,26 The United States Food and Drug Administration (USFDA) approved rivastigmine for the treatment of Alzheimer's disease (AD) on April 21, 2000. The chemical name of rivastigmine is (S)-3-(1-(dimethylamino)ethyl)phenyl ethyl(methyl)carbamate. Rivastigmine is synthesized by reacting 3-methoxyacetophenone 1 with methylbenzylamine using the combination of Ti(OiPr)4/Raney-Ni/H2 as a catalyst to give compound 2. Compound 2 underwent N-methylation using formic acid and formaldehyde, resulting in compound 3. Demethylation of the methoxy function of compound 3 in the presence of HBr to give compound 4. On reacting compound 4 with N-ethyl-N-methyl carbamoyl chloride to give compound 5. The hydrogenolysis of compound 5 in the presence of Pd/C, H2 results in compound 6. The N-methylation of compound 6 by HCOOH/HCHO to give rivastigmine 7 (ref. 27) (Fig. 7).
 |
| | Fig. 7 Synthesis of rivastigmine. | |
2.2 Desvenlafaxine
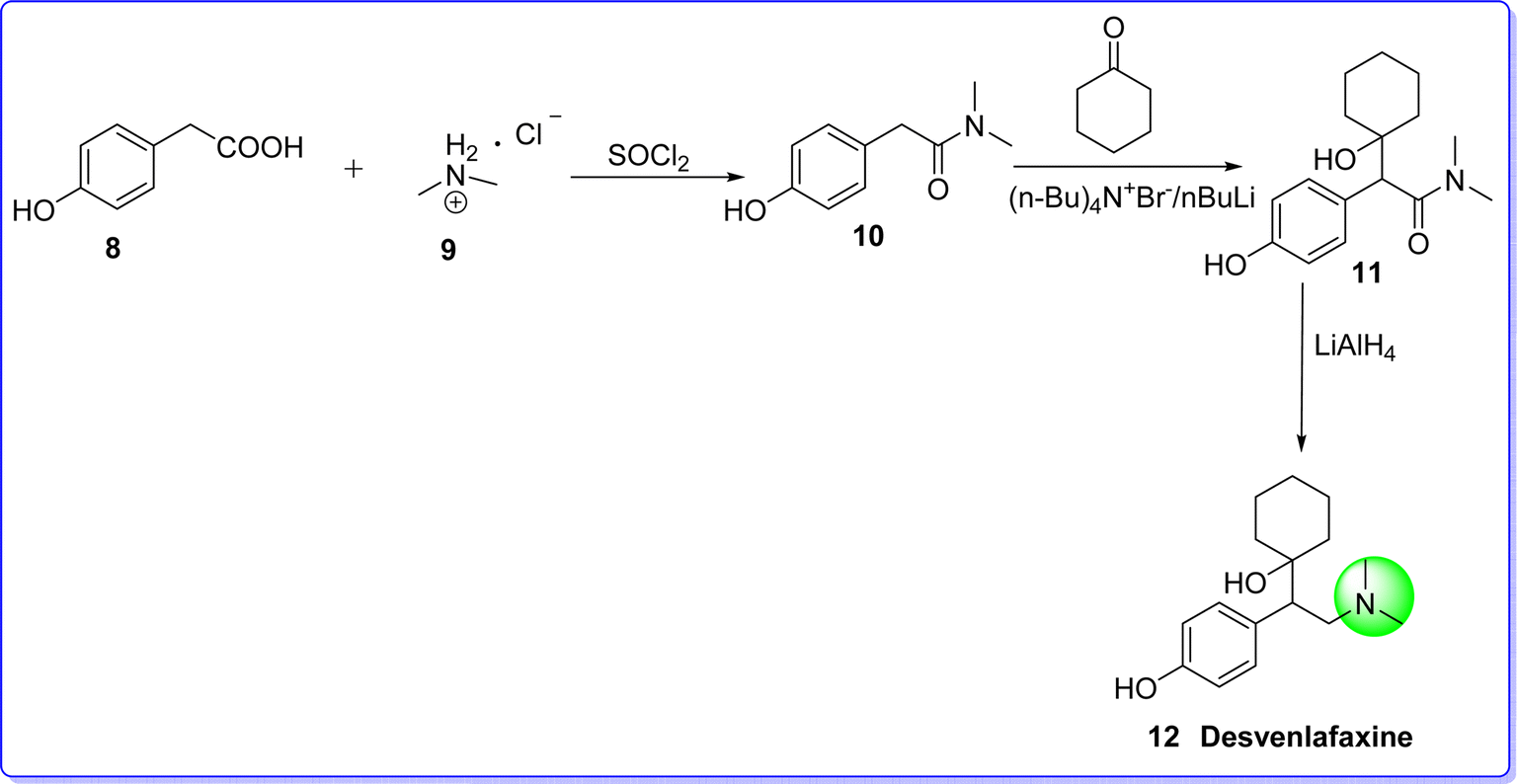
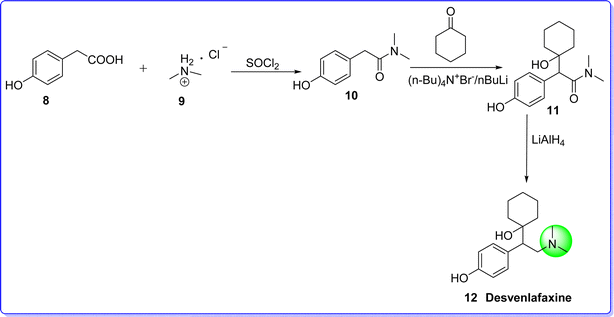
Desvenlafaxine is an antidepressant drug classified as a serotonin–norepinephrine reuptake inhibitor (SNRI). It is used to treat mild to severe depression, anxiety, and other mental disorders, offering superior clinical advantages.28 It was approved by FDA on 29 February 2008. Chemically it is known as 4-(2-(dimethylamino)-1-(1-hydroxycyclohexyl)ethyl)phenol. It is synthesized by reacting p-hydroxyphenyl acetic acid 8 and dimethylammonium 9 in the presence of sulphonyl chloride to give compound 10, which react with cyclohexanone in the presence of n-butyl lithium to give compound 11. On reduction of compound 11 with LiAlH4 produces desvenlafaxine 12 (ref. 29) (Fig. 8).
 |
| | Fig. 8 Synthesis of desvenlafaxine. | |
2.3 Chlorprothixene
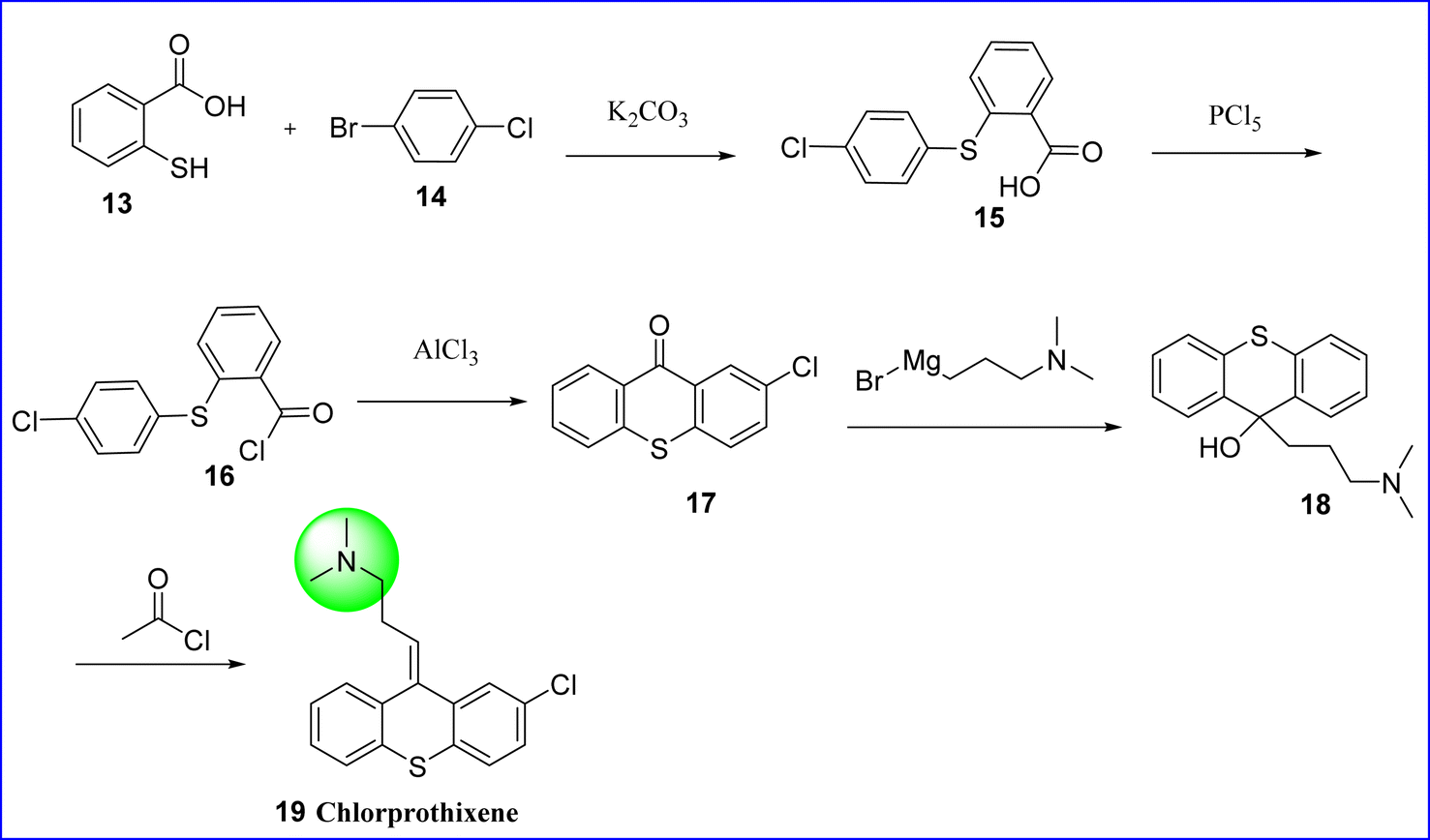
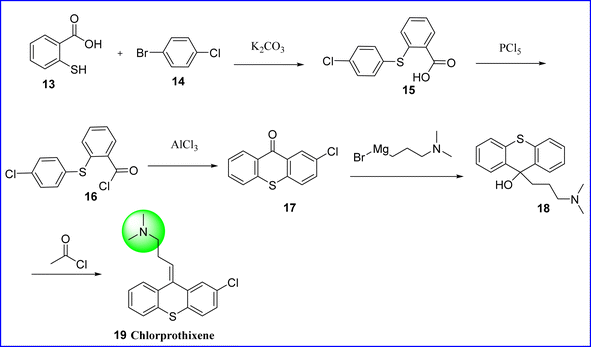
Chlorprothixene, a thioxanthene antipsychotic, acted through immunomodulation instead of direct radical scavenging to prevent activated macrophages from producing reactive oxygen species.30,31 The chemical name of chlorprothixene is (3Z)-3-(2-chlorothioxanthen-9-ylidene)-N,N-dimethylpropan-1-amine. When chlorprothixene was initially licensed in the late 1950s, clinical trials concentrated on side effects such as extrapyramidal symptoms rather than metabolic adverse events.32 Its antipsychotic efficacy is modest, ranging from half to two-thirds that of chlorpromazine. Although chlorprothixene has shown antiemetic properties, its antidepressant and analgesic benefits have not been fully proven. It is employed in the treatment of emotional, mental, and neurological disorders. Chlorprothixene is synthesized by reacting 2-mercaptobenzoic acid 13 with 1-bromo-4-chlorobenzene 14 to form 2-(4-chlorophenylthio) benzoic acid (compound 15). Phosphorous pentachloride is further reacted with compound 15 to convert the former into corresponding acid chloride (compound 16). Through intramolecular cyclization, compound 16 is converted to 2-chlorthioxantone (compound 17) by using aluminum chloride. Tertiary alcohol (compound 18) is produced when compound 17 reacts with 3-dimethylaminopropyl magnesium bromide as a carbonyl component. Finally, the tertiary hydroxyl group on dehydration followed by acylation using acetyl chloride and on pyrolysis of formed acetate gives chlorprothixene 19 (ref. 33) (Fig. 9).
 |
| | Fig. 9 Synthesis of chlorprothixene. | |
2.4 Zolmitriptan
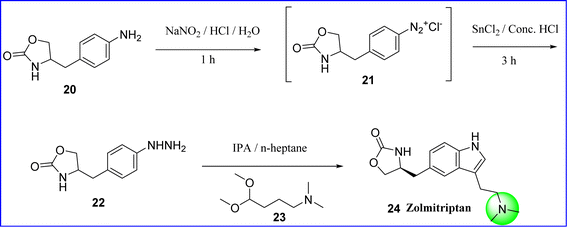
Zolmitriptan is a class of the triptan family of selective serotonin receptor agonist (SSRA), specifically targeting 5-HT1B/1D receptors. Chemical it is known (S)-4-((3-(2-(dimethylamino)ethyl)-1H-indol-5-yl)methyl)oxazolidin-2-one. On November 25, 1997, the US FDA approved zolmitriptan to cure acute migraine in adults. Since zolmitriptan is highly significant in the treatment of migraine headaches, it binds to the serotonin (5-HT1B) receptors. This binding results in the inhibition of nociceptive transmission, constriction of cranial vessels, and a decrease in vessel pulsation. The synthesis was started with diazotisation of 4-(4-amino benzyl) oxazolidine-2-one (compound 20) to provide the diazonium chloride salt (compound 21) by using concentrated HCl and aqueous sodium nitrite at −5 to 0 °C. The hydrazine hydrochloride salt (compound 22) was obtained by reducing (compound 21) with stannous chloride dihydride and concentrated HCl and then adjusting the pH of the reaction mass to 1.7–1.85 using NaOH solution. By reflux condensation of compound (compound 22) with 4-4-dimethoxy-N,N-dimethylbutane-1-amine (compound 23), zolmitriptan is produced. The obtained product was recrystallized from IPA/n-heptane yields pure zolmitriptan 24 (ref. 34) (Fig. 10).
 |
| | Fig. 10 Synthesis of zolmitriptan. | |
2.5 Trimipramine
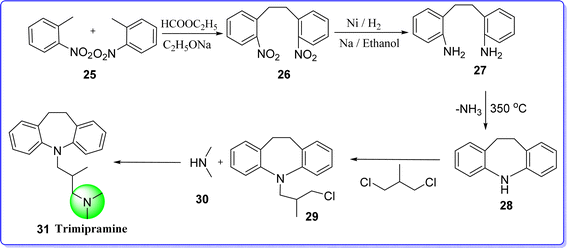
Trimipramine, also known as surmontil, is an antidepressant that also acts as a sedative to lessen anxiety. Its exact mechanism of action on the CNS is unknown. However, it primarily does not function by stimulating the CNS, unlike substances of the amphetamine class. Chemically it is known as 3-(10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-N,N,2-trimethylpropan-1-amine. In 1982, trimipramine (surmontil) capsules containing 25 mg, 50 mg, and 100 mg were approved by the US FDA. It was authorized in 1982 for the 100 mg formulation. In 2006, all three formulations were approved generic versions by the FDA.35 Trimipramine was synthesized by reacting 2 molecules of o-nitrotoluene (compound 25) in the presence of ethylformate and sodium ethoxide resulting in the formation of compound 26 which on reduction gives compound 27 followed by cyclization produces compound 28. The compound 28 further reacted with 1,3-dichloro-2-methyl propane (compound 29) to give compound 30, which finally reacted with dimethyl amine (compound 31) resulting in the formation of trimipramine 31 (ref. 36) (Fig. 11).
 |
| | Fig. 11 Synthesis of trimipramine. | |
2.6 Cariprazine hydrochloride
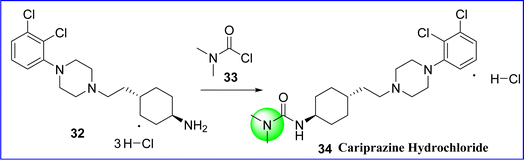
Cariprazine is a derivative of piperazine and an atypical antipsychotic medication. It functions as an antagonist at serotonin 5-HT2A receptors and as a partial agonist at central dopamine D2, D3, and 5-HT1A receptors. Numerous mental conditions, such as major depressive disorder, schizophrenia, and bipolar disorder have been studied in relation to cariprazine. In September 2015, the US FDA authorized cariprazine for use worldwide for the first time. Health Canada also approved the drug in April 2022. Currently, it is used to treat manic or mixed episodes, depressive episodes linked to bipolar I illness, and schizophrenia.37 The chemical name for cariprazine hydrochloride is 3-((1R,4R)-4-(2-(4-(2,3-dichlorophenyl)piperazin-1-yl)ethyl)cyclohexyl)-1,1-dimethylurea hydrochloride. It was synthesized by reacting trans – 4-(2-(4-(2,3-chlorophenyl) piperazine – 1-yl) ethyl) cyclohexylamine trihydrochloride (compound 32) with N,N-dimethylcarbarnoylchloride (compound 33) in the presence of dichloromethane and triethylamine to produce cariprazine 34 (ref. 38) (Fig. 12).
 |
| | Fig. 12 Synthesis of cariprazine hydrochloride. | |
2.7 Clomipramine hydrochloride
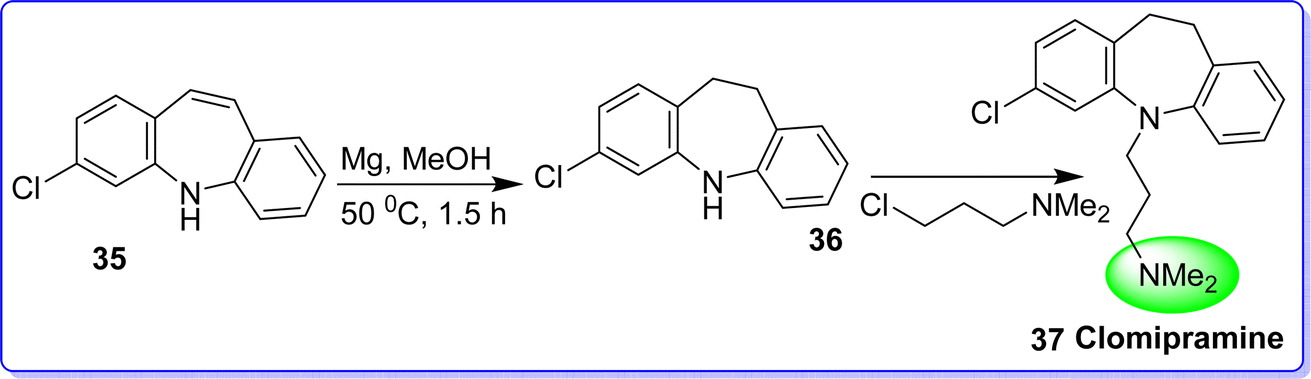
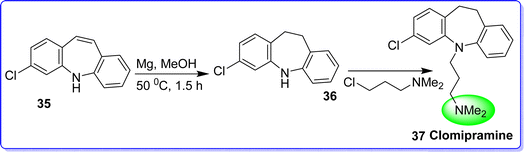
Clomipramine hydrochloride is a tertiary amine that is categorized as a class of tricyclic antidepressant (TCA) drugs. Clomipramine is a SSRI that has a higher affinity for the serotonin transporter (SERT) in comparison with other SSRI. As a result, clomipramine enhances noradrenergic and serotonergic transmission.39 The chemical name of clomipramine is (3-chloro-5-[3-dimethylaminopropyl]-10,11-dihydro-5H-dibenz[b,f]azepine). On September 19, 1995, US FDA approved clomipramine hydrochloride to cure obsessive compulsive disorder (OCD).40 Clomipramine is only licensed by the FDA to treat OCD in patients 10 years of age and older. The synthesis of clomipramine starts with 3-chloro-5H-dibenzo[b,f]azepine(35). This compound is then easily converted to the corresponding dihydro compound with a 95% yield through a straightforward reduction of the conjugated double bond using magnesium in methanol at 50 °C for 1.5 hours to compound (36). Finally, 3-chloro-N,N-dimethylpropan-1-amine is used in an alkylation reaction to produce the target compound, clomipramine 37 (ref. 41) (Fig. 13).
 |
| | Fig. 13 Synthesis of clomipramine. | |
2.8 Rizatriptan benzoate
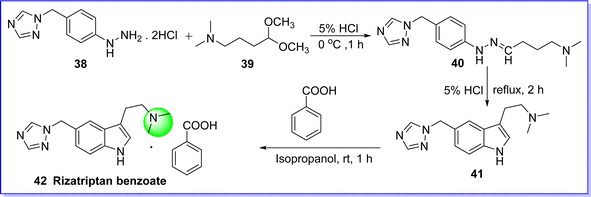
Rizatriptan benzoate is a selective 5-hydroxy tryptamine receptor antagonist. It is used in management of migraine and severe headache. Vascular constriction occurs in the brain when rizatriptan benzoate binds to serotonin receptors. Rizatriptan reduces pain by narrowing blood arteries in the brain. Rizatriptan is approved by FDA in 1998 and it is sold under the brand name Maxalt in tablet form. The chemical name of rizatriptan benzoate is 2-(5-((1H-1,2,4-triazol-1-yl)methyl)-1H-indol-3-yl)-N,N-dimethylethan-1-amine benzoate. It is synthesized by reacting 1-(4-hydrazino phenyl) methyl-1,2,4-triazole dihydrochloride 38 with 4-N,N-dimethyl amino butanal dimethyl acetal 39 in the presence of 5% HCl to produce compound 40, which undergoes cyclization under reflux to rizatriptan 41. The target molecule rizatriptan benzoate is obtained by reacting compound 41 with benzoic acid 42 (ref. 42 and 43) (Fig. 14).
 |
| | Fig. 14 Synthesis of rizatriptan benzoate. | |
2.9 Levomepromazine
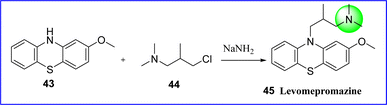
Levomepromazine is a phenothiazine neuroleptic drug, commonly referred to as methotrimeprazine. It is a powerful analgesic, hypnotic, antiemetic and antipsychotic with modest strength (about half that of chlorpromazine), mostly used in palliative care. It showed antihistaminic effects on the CNS that are similar to those of chlorpromazine. The chemical name for levomepromazine is (2R)-3-(2-methoxyphenothiazine-10-yl-)-N,N,2-trimethyl propanamine. Levomepromazine 45 was synthesized by reacting 1-methoxy phenothiazine (compound 43) with 3-dimethylamino-2-methyl propyl chloride (compound 44) in the presence of sodium amide44 (Fig. 15).
 |
| | Fig. 15 Synthesis of levomepromazine. | |
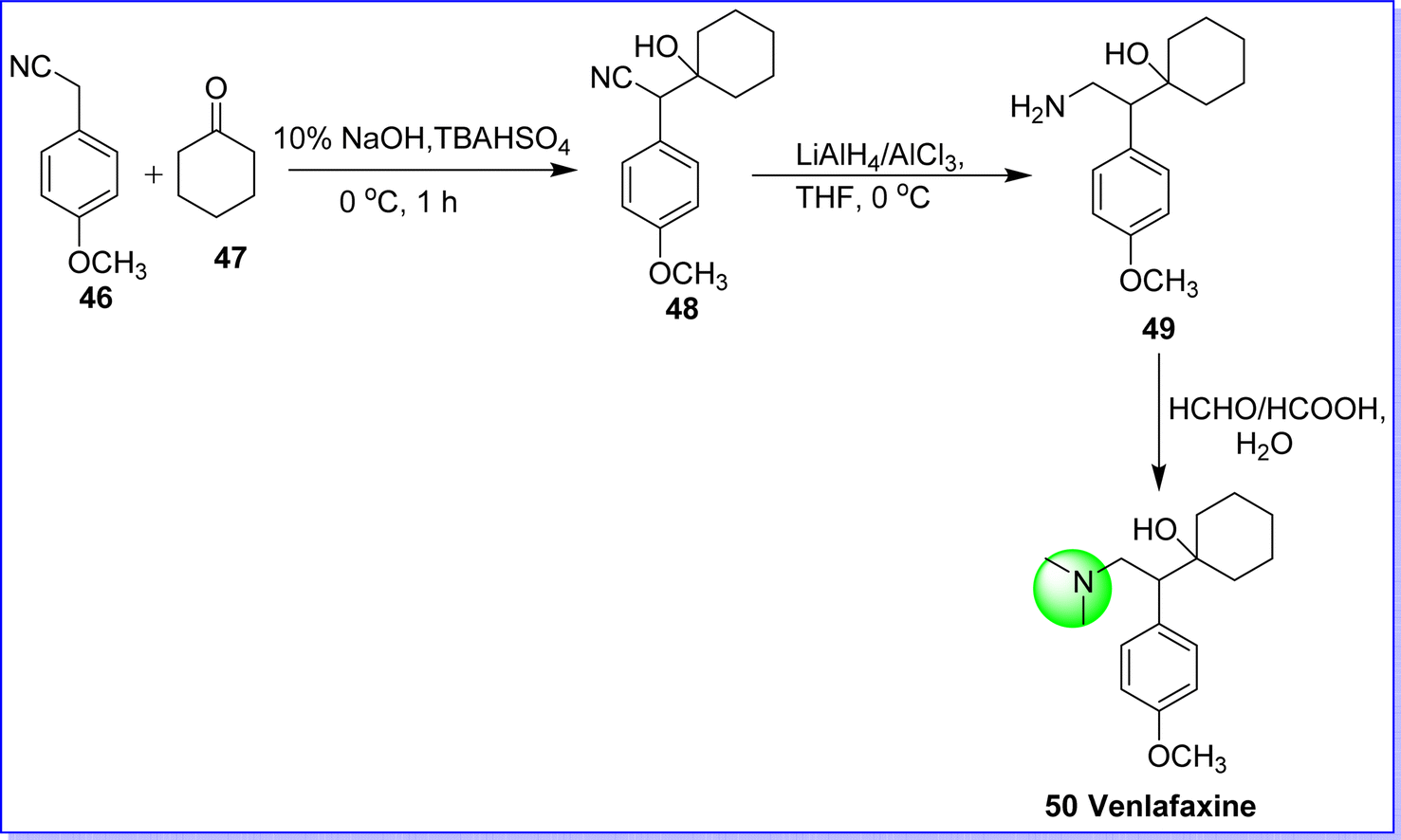
2.10 Venlafaxine hydrochloride
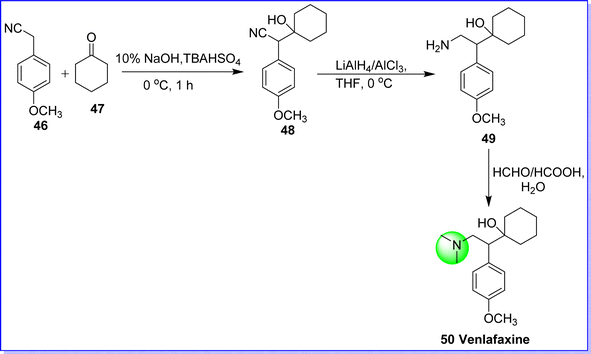
Venlafaxine hydrochloride, classified as an antidepressant drug, is a SNRI. It is used to treat major depressive disorder, panic disorder, generalized anxiety disorder, and social anxiety disorder.45 It was approved by the FDA in 1993. It is sold under the brand name Effexor. Chemically it is known as 1-(2-(dimethylamino)-1-(4-methoxyphenyl)ethyl)cyclohexan-1-ol. It was synthesized by reacting p-methoxyphenyl acetonitrile 46 with cyclohexanone 47 to form cyanoalcohol 48 in the present of base NaOH/KOH, which undergoes a reduction in the presence of LiAlH4/AlCl3 in THF to produce amine derivative 49. The target molecule venlafaxine hydrochloride 50 was obtained by alkylation of compound 49 by HCHO/HCOOH45 (Fig. 16).
 |
| | Fig. 16 Synthesis of venlafaxine. | |
2.11 Orphenadrine
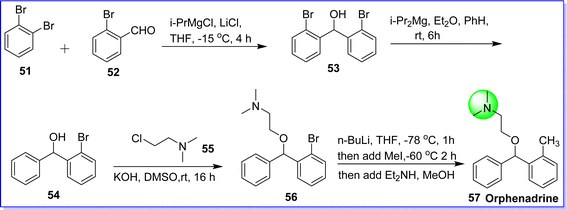
Orphenadrine is an anticholinergic and antihistaminic drug. It acts as a muscle relaxant, aiding in the treatment of muscle pain, and contributes to motor control in Parkinson's disease. It was approved by the FDA on 29 Jan 1999. It is sold under the brand name Disipal. The chemical name of orphenadrine is N,N-dimethyl-2-(phenyl(o-tolyl)methoxy)ethan-1-amine.46 It was synthesized by reacting 2-bromobenzaldehyde 51 with 1,2-bromobenzene 52 in the presence of Grignard reagent in THF to form compound 53, which reacts with alkyl magnesium in diethyl ether (Et2O) to form compound 54. On reacting compound 54 with 2-chloro-N,N-dimethylethan-1-amine 55 in the presence of KOH in DMSO, compound 56 was obtained. The target molecule orphenadrine 57 was obtained by alkylation of compound 56 in the presence of n-butyl lithium in THF47 (Fig. 17).
 |
| | Fig. 17 Synthesis of orphenadrine. | |
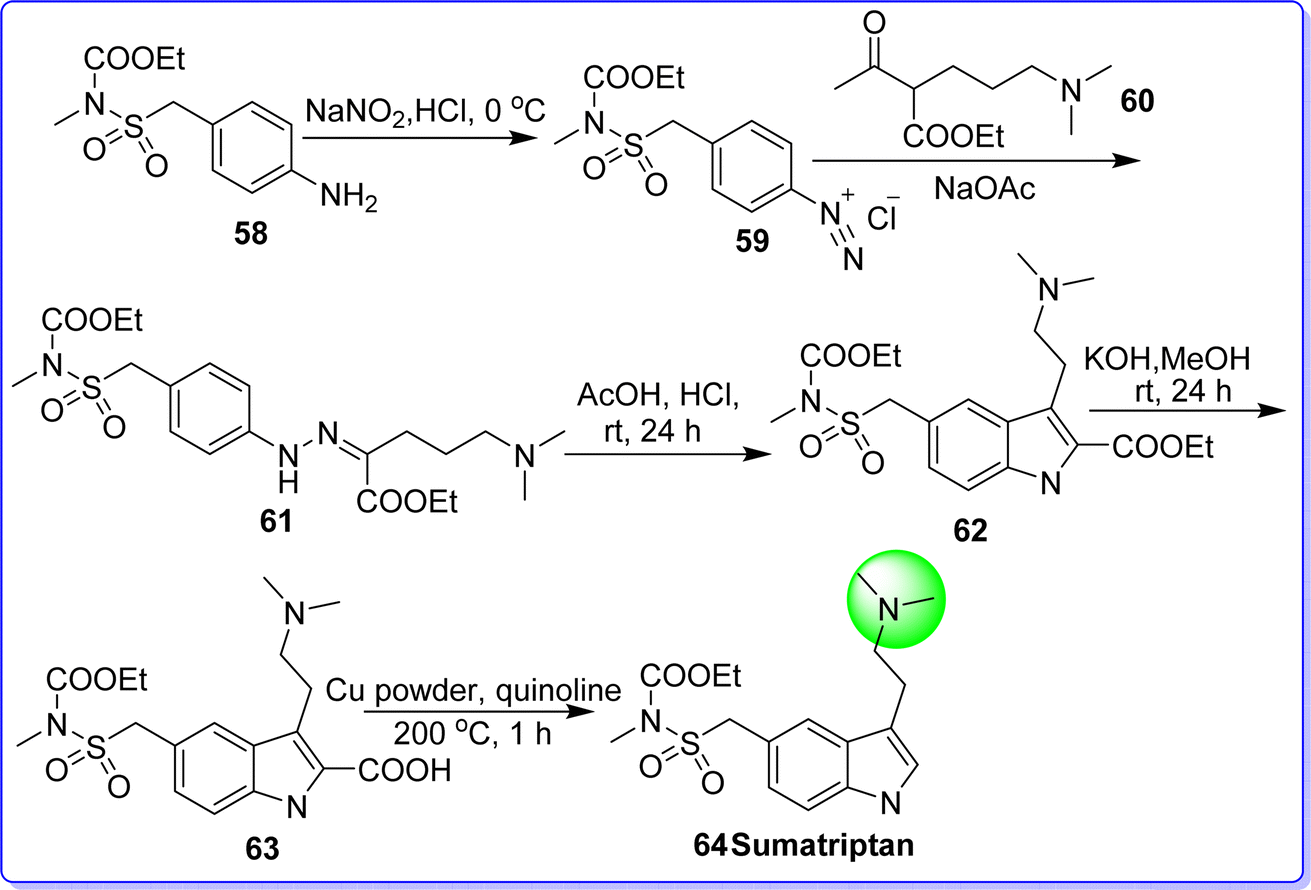
2.12 Sumatriptan
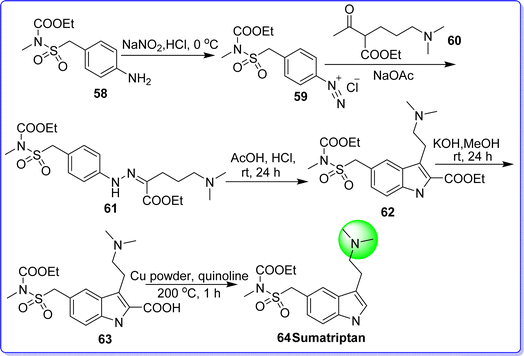
Sumatriptan is a selective serotonin agonist, used in the management of migraine. It was approved by the FDA in July 2009. It is sold under the brand name Imitrex. Chemically it is known as 1-(3-(2-(dimethylamino)ethyl)-1H-indol-5-yl)-N-methylmethanesulfonamide. It is synthesized by reacting ethyl (((3-amino-1H-indol-5-yl)methyl)sulfonyl)(methyl)carbamate 58 with NaNO2 in the presence of HCl to form diazonium salt 59, which reacts with ethyl 2-acetyl-5-(dimethylamino)pentanoate 60 to give compound 61. On reacting compound 61 with AcOH/HCl give compound 62. The reaction of compound 62 with MeOH in the presence of KOH resulted in compound 63. The compound 63 on decarboxylation in the presence of copper in quinolone at 200 °C to form sumatriptan 64 (ref. 48 and 49) (Fig. 18).
 |
| | Fig. 18 Synthesis of sumatriptan. | |
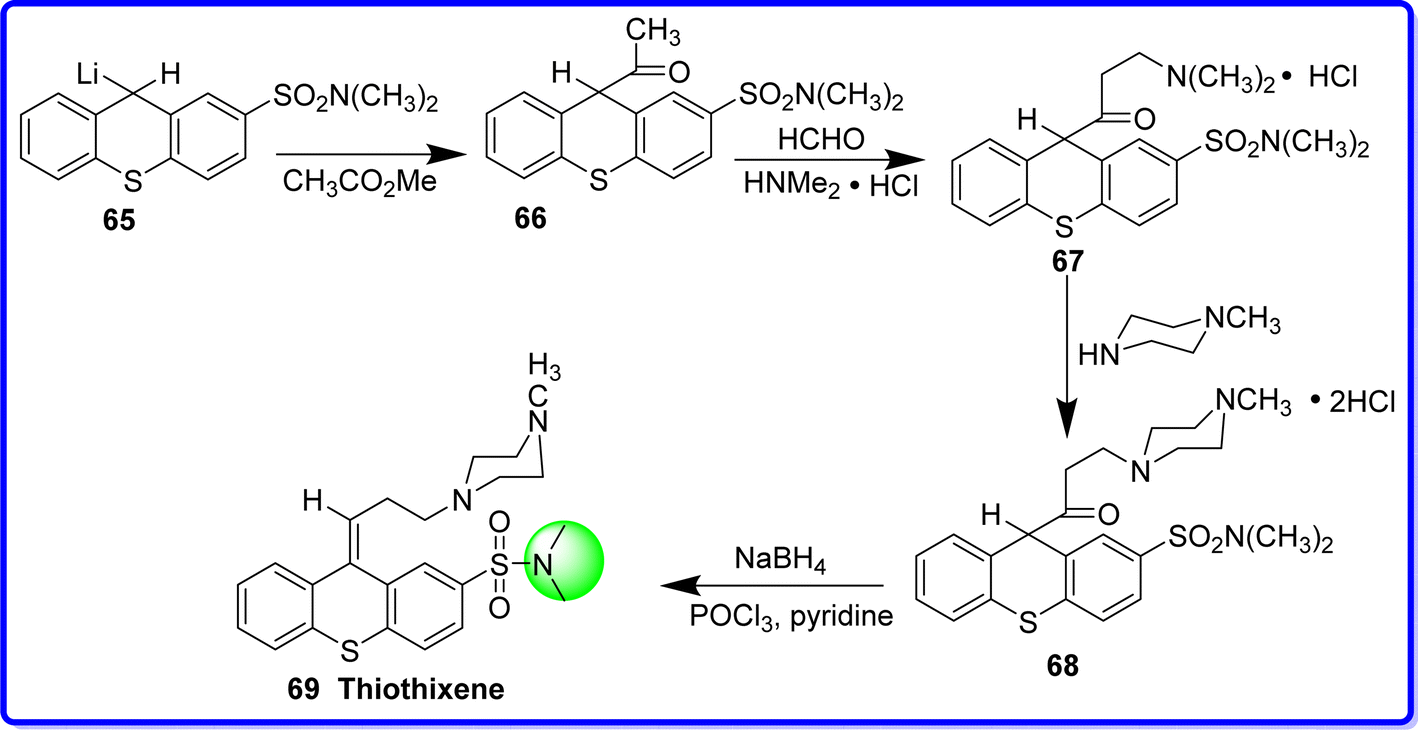
2.13 Thiothixene
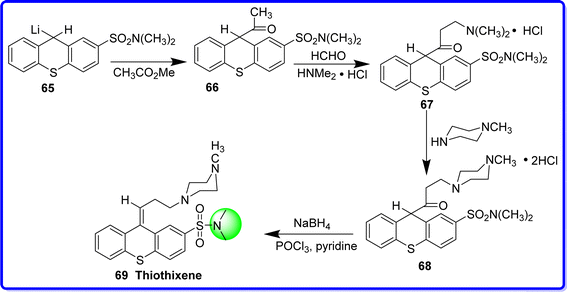
Thiothixene is tricyclic psychotherapy substance called thioxanthene-2-sulfonamide has antipsychotic action similar to some popular phenothiazine medications.50 Chemically thiothixene is known as (Z)-N,N-dimethyl-9-(3-(4-methylpiperazin-1-yl)propylidene)-9H-thioxanthene-2-sulfonamide. In 1967, thiothixene—the third of several derivatives of thioxanthene—was produced. It is thought that while the sulfonamide pharmacophore amplifies the tranquilizer activity, and the piperazine moiety increases lipid solubility. It was synthesized by acetylation of 9-with-N,N-dimethyl thioxanthene-2-sulfonamide 65 could reliably give the critical intermediate 66 in a yield of 30–39%. The compound 66 on mannich condensation using Me2NH·HCl and HCHO produced compound 67. The compound 68 in basically quantifiable yield was obtained by amine exchange with 1-methyl piperazine. The target molecule 69 was obtained by reduction of the keto group of compound 68 with NaBH4 followed by POCl3 in the presence of pyridine51 (Fig. 19).
 |
| | Fig. 19 Synthesis of thiothixene. | |
2.14 Imipramine hydrochloride
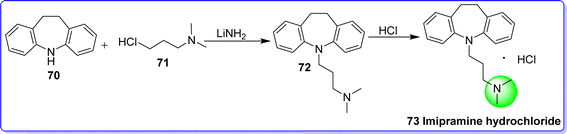
Imipramine hydrochloride is a dibenzoazepine tricyclic antidepressant. Imipramine was discovered to have a particular impact on the treatment of depression.52 The first study on imipramine treatment for depressed conditions was published by Kuhn in 1957.53 It was approved by the FDA in 1959. It is marketed under various brand names such as Tofranil, SK-Pramine, and Janimine. Chemically it is known as 3-(10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-N,N-dimethylpropan-1-amine. It is synthesized by reacting 10,11-dihydro-5H-dibenzo[b,f]azepine 70 with 3-chloro-N,N-dimethylpropan-1-amine 71 in the presence of LiNH2 to give imipramine base 72, which on reaction with hydrochloric acid, impramine hydrochloride 73 was obtained54 (Fig. 20).
 |
| | Fig. 20 Synthesis of imipramine hydrochloride. | |
2.15 Chlorpromazine hydrochloride
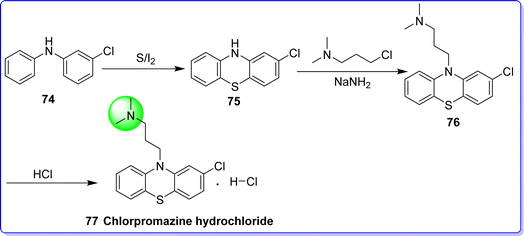
Chlorpromazine hydrochloride exhibits similarities with promethazine hydrochloride and other substances that are employed in management of Parkinson's disease. In addition to being demonstrated to stop apomorphine-induced vomiting, it has been asserted to have an analgesic effect. It is also acting as an antipsychotic agent and it employed in management of psychotic disorders such as schizophrenia and bipolar disorder. It was approved by the FDA on 12/30/1999. It is sold under the brand names Thorazine and Largactil. The chemical name of chlorpromazine hydrochloride is 3-(2-chloro-10H-phenothiazin-10-yl)-N,N-dimethylpropan-1-amine hydrochloride. It is synthesized by reacting 3-chloro-N-phenylbenzenamine 74 with sulfur to give 2-chloro-10H-phenothiazine 75, which reacts with 3-dimethylaminopropylchloride in the presence of NaNH2 to give chlorpromazine base 76. On reacting compound 76 with hydrochloric acid, chlorpromazine hydrochloride 77 was obtained55 (Fig. 21).
 |
| | Fig. 21 Synthesis of chlorpromazine hydrochloride. | |
2.16 Triflupromazine
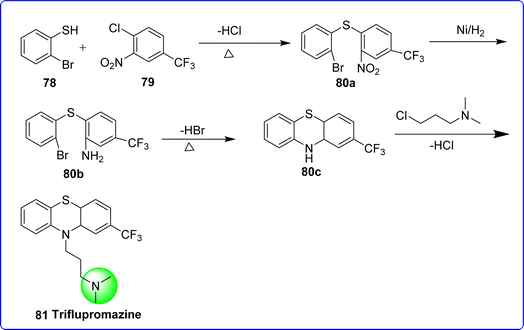
Triflupromazine comes under the category of phenothiazine derivatives, which are antagonists of the dopamine D1/D2 receptor. It is especially utilized to manage aggressive conduct when psychotic illnesses are experiencing acute episodes. It can also be used to manage moderate to severe pain in some hospitalized patients, severe hiccups, and extreme nausea and vomiting. The chemical name for triflupromazine is N,N-dimethyl-3-[2-(trifluoromethyl) phenothiazin-10-yl] propan-1-amine. On 26th March 2001, the US FDA approved triflupromazine for the management of schizophrenia. Triflupromazine was synthesized by reacting 2-bromobenzenethiol 78 with compound 79 to give compound 80a, which on catalytic reduction nickel give compound 80b. Cyclization of compound 80b yield compound 80c, which on reaction with 3-chloro-N,N-dimethylpropan-1-amine yield 81 triflupromazine56 (Fig. 22).
 |
| | Fig. 22 Synthesis of triflupromazine. | |
2.17 Citalopram hydrobromide
Citalopram is a SSRIs antidepressant that is commonly used to cure the symptoms of depression. Its antidepressant effects due to the increase in serotonergic activity in the CNS. The chemical name for citalopram is 1-[3-(dimethylamino) propyl]-1-(4-fluorophenyl)-3H-2-benzofuran-5-carbonitrile. Citalopram received FDA approval in 1998. The reaction of 5-bromophthalide (compound 82) with 4-fluorophenyl magnesium bromide (compound 83) produced 4-bromo-4′-fluoro-2-(hydroxymethyl)-benzophenone (compound 84), which is then reduced in to 4-bromo4′-fluoro-2-(hydroxymethyl) benzhydrol (compound 85) using LiAlH4 or NaBH4. The product of cyclizing compound 85 at 100 °C with 60% H3PO4, TsOH, or H2SO4 is 5-bromo-1-(4-fluoropheny) phthalan (compound 86). Following that, 1-(4-fluorophenyl)-5-phthalancarbonitrile (compound 87), was produced by reacting the resulting compound 86 with cuprous cyanide. Compound 87 was finally condensed using 3-(dimethylamino) propyl chloride (compound 88) utilizing DMSO and NaH, to yield citalopram 87 (ref. 57) (Fig. 23).
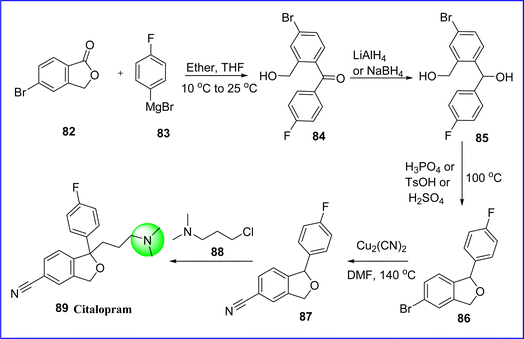 |
| | Fig. 23 Synthesis of citalopram. | |
2.18 Zolpidem tartrate
Zolpidem is a member of the class of drugs known as non-benzodiazepine hypnotics. Its effects include sedation, hypnosis, and anticonvulsants (control seizures). It functions by binding to the GABA receptor chloride channel macromolecular complex, which is a chemical messenger that reduces brain neuron activity and promotes sleep. The chemical name for zolpidem tartrate is (2R,3R)-2,3-dihydroxy butanedioic acid; N,N-dimethyl-2-[6-methyl-2-(4-methyl phenyl)imidazo[1,2-a]pyridin-3-yl]acetamide. On 16th December 1992, the US FDA approved zolpidem tartrate for treating insomnia. In the synthesis of zolpidem, 2-amino-5-methylpyridine (compound 90) and 2-bromo-4-methylacetophenone (compound 91) combine to produce imidazopyridine (compound 92), which is then aminomethylated to form the amine (compound 93). After methylating the resultant, dimethylamino derivative (compound 93) with methyl iodide forms as its quaternary ammonium salt (compound 94). Quaternary ammonium salt and sodium cyanide reacted resulting in nitrile (compound 95), which underwent acidic hydrolysis to get the equivalent acid chloride. Heating the phosphoryl chloride with dimethylamine yields the desired compound zolpidem 94 (ref. 58) (Fig. 24).
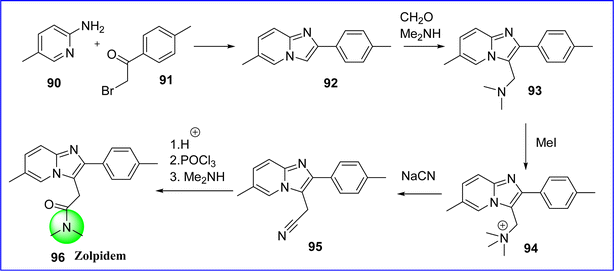 |
| | Fig. 24 Synthesis of zolpidem. | |
2.19 Amitriptyline hydrochloride
Amitriptyline is a tricyclic antidepressant commonly used in treatment disorder such as migraine, headache, fibromyalgia, and neuropathic pain. It acts by inhibiting reuptake of norepinephrine and serotonin. Scientists at Merck made the discovery of amitriptyline in the late 1950s, and the US FDA approved it in 1961.59 Currently, it is marketed as a tablet for combination therapy, which helps in its absorption by being a hydrochloride salt. However, when it is given via intramuscular injection in a hospital environment, it is commonly presented in the form of an embonate (pamoate) salt. In 2016, amitriptyline ranked as the 88th most prescribed drug in the United States. It was written for more than 8.6 million prescriptions, with patients paying more than $60 million out of pocket for the drug.60 Chemically amitriptyline is known as 3-(10,11-dihydro-5H-dibenzo[a,d][7]annulen-5-ylidene)-N,N-dimethylpropan-1-amine. Amitriptyline was synthesized by reacting dibenzosuberone 97 with Grignard reagent in THF to form compound 98, which react with hydrochloric acid in present of acetic acid to produce compound 99. The target molecule amitriptyline 100 was obtained by reacting compound 99 with dimethylamine in benzene at 95 °C for 18 h (ref. 61) (Fig. 25).
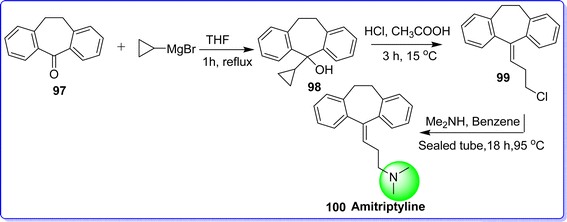 |
| | Fig. 25 Synthesis of amitriptyline. | |
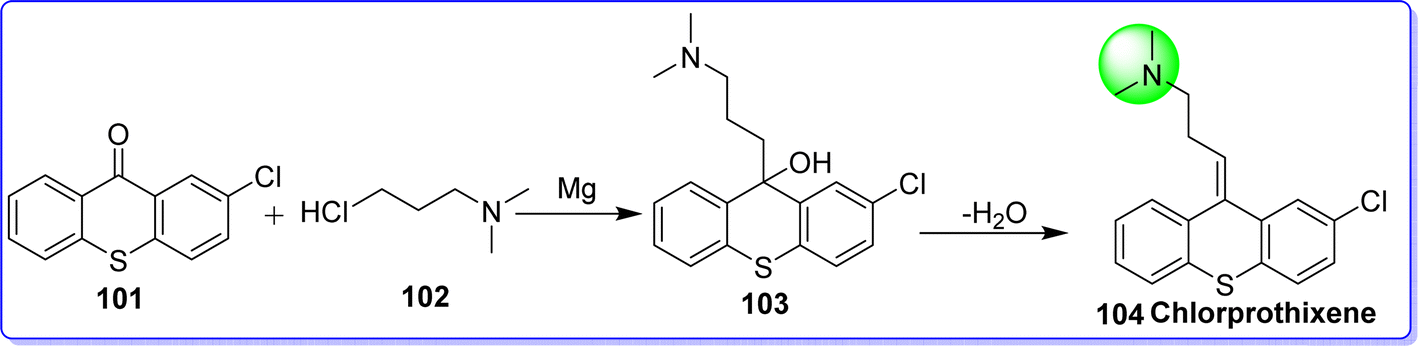
2.20 Chlorprothixene
Chlorprothixene, a first-generation antipsychotic medication with low potency, was introduced to the market in 1959 and remains available in 16 countries, despite the increasing availability of second-generation antipsychotics for clinical use.62 Different countries have different licensed indications for chlorprothixene. In Denmark, the approved indications include schizophrenia and other psychoses,63 in Norway, they also include feelings of anxiety and withdrawal, in Germany, they include manic symptoms.64 Chlorprothixene is still a widely used antipsychotic in Scandinavia and several other countries with significant off-label use, despite its limited indications.65 Chlorprothixene has a strong affinity for 5-HT2C, H1, and M3 receptors, in contrast to the majority of other first-generation antipsychotics. The antagonistic action at these receptors has been associated with the development of metabolic abnormalities (dyslipidemia or poor glucose metabolism), as well as some of the anxiolytic and hypnotic effects of chlorprothixene. It was approved by the FDA in 1950 and sold under the brand name Truxal. Chemically chlorprothixene is known as (E)-3-(2-chloro-9H-thioxanthen-9-ylidene)-N,N-dimethylpropan-1-amine. It was synthesized by reacting 2-chlorothioxanthone 101 with 3-chloro-N,N-dimethylpropan-1-amine 102 in the presence of magnesium to give compound 103, which on dehydration gives chlorprothixene 104 (ref. 63) (Fig. 26).
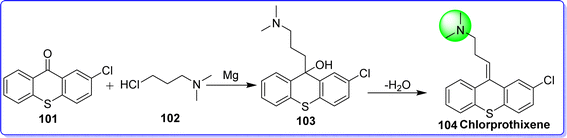 |
| | Fig. 26 Synthesis of chlorprothixene. | |
3. Antihistaminic agents
3.1 Pheniramine
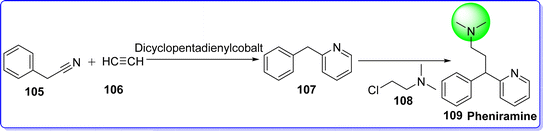
Pheniramine, categorized as an alkylamine derivative, is a potent H1 antagonist and an antihistaminic drug. It exhibits a lower tendency compared to other H1 antagonists to induce drowsiness, making it more suitable for daytime administration. When pheniramine is taken in lethal doses, it can cause cardiac arrhythmias, dilated pupils, urine retention, tachycardia, dry flushed skin, decreased bowel noises, confusion, and a little rise in body temperature. The majority of the side effects occurs due to its antimuscarinic action.66 It is used in the treatment of allergies, hay fever, and common cold. It was approved by the FDA in 1948. The chemical name of pheniramine is N,N-dimethyl-3-phenyl-3-(pyridin-2-yl)propan-1-amine. It is synthesized by reacting benzyl cyanide 105 with acetylene gas 106 in the presence of diclopentadienylcobalt as a catalyst, under high pressure and temperature to give 2-benzylpyridine 107, which react with 2-chloro-N,N-dimethylethan-1-amine 108 to give pheniramine 109 (ref. 67) (Fig. 27).
 |
| | Fig. 27 Synthesis of pheniramine. | |
3.2 Trimeprazine tartrate
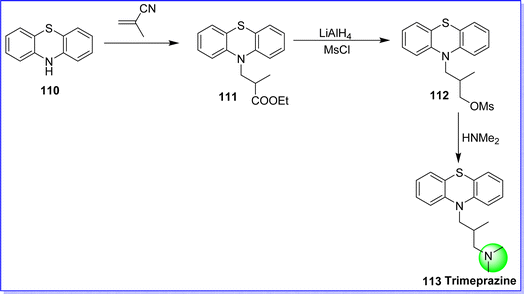
Trimeprazine is also known as alimemazine. Trimeprazine is an antihistamine agent, is a derivative of phenothiazine, similar to chlorpromazine.68 Trimeprazine and free histamine compete with each other for binding to H1-receptor sites. This reduces the negative symptoms caused by histamine H1-receptor binding and inhibits the effects of histamine on H1-receptors. It is mostly used because of its significant ability to relieve pruritus. It exhibits strong sedative, antihistamine, and slightly antimuscarinic properties. Additionally, trimeprazine can be used as a cough suppressant. It is taken orally, the usual daily dose of trimeprazine is 5–40 mg.69 It is sold under the trade name, Nedeltran, Therafene, Repeltin, Theraligene, Panectyl, Theralen, Vallergan, and Vanectyl and Temaril and it is commonly provided as a tartrate salt. Chemically trimeprazine was known as N,N,2-trimethyl-3-(10H-phenothiazin-10-yl)propan-1-amine. The synthesis of trimeprazine is started with the reaction between phenothiazine 110 and methacrylonitrile to produce compound 111, which on reduction with LiAlH4 followed by alkylation with methanesulfonyl chloride (MsCl) to from compound 112. On reacting compound 112 with dimethylamine, the target molecule trimeprazine 113 was formed70 (Fig. 28).
 |
| | Fig. 28 Synthesis of trimeprazine. | |
3.3 Chlorpheniramine maleate
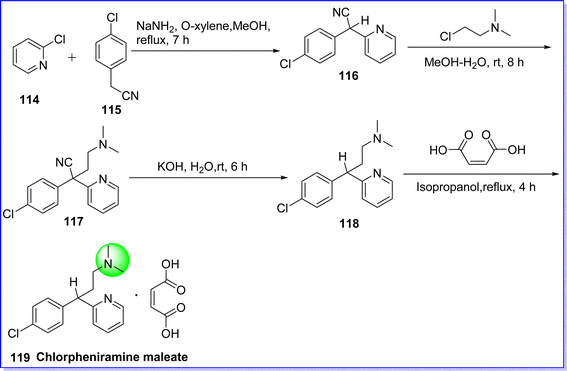
Chlorpheniramine maleate (CPM) is a first-generation antihistaminic agent, classified as an alkylamine derivative, and acts by antagonizing the H1 receptor.71 It is employed in the treatment of allergies, hay fever, and common cold. While subcutaneous, intramuscular, and intravenous administration have been reported, oral administration of this antihistamine is the usual method. Recent research has investigated this medication's intranasal routes, particularly in the context of its antiviral properties against the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2).72 The chemical name of CPM is 3-(4-chlorophenyl)-N,N-dimethyl-3-(pyridine-2-yl)propan-1-amine maleate. It is synthesized by reacting 2-chloropyridine 114 with 2-(4-chlorophenyl) acetonitrile 115 in the presence of NaNH2 in O-xylene to give compound 116, which undergoes condensation with 2-chloro-N,N dimethylethanamine at room temperature for 8 h to give compound 117. After that, the cyano base of compound 117 is subsequently decyanated with flakes of caustic potash (KOH) to produce chlorphenamine 118 with a good yield. Compound 118 is further treated with maleic acid in presence of isopropanol under reflux condition for 4 h to give CPM 119 (ref. 73) (Fig. 29).
 |
| | Fig. 29 Synthesis of chlorpheniramine maleate. | |
3.4 Brompheniramine maleate
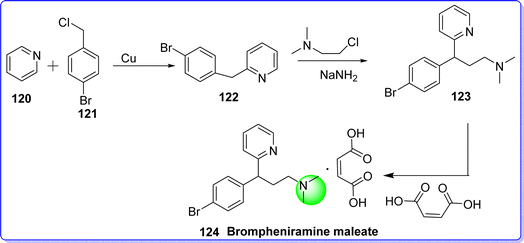
Brompheniramine maleate is a first-generation antihistaminic agent, that acts by blocking the H1 receptor. It is an alkylamine derivative antihistaminic drug. It is offered for sale using the brand Dimetapp among others.74 It is used in the treatment of common colds and allergies. The chemical name of brompheniramine maleate is 3-(4-bromophenyl)-N,N-dimethyl-3-(pyridin-2-yl)propan-1-amine maleate. It is synthesized by reacting pyridine 120 with 4-bromobenzylcloride 121 to give 2-(4-bromobenzyl)pyridine 122, which reacts with 2-dimethylaminoethylchloride in the presence of NaNH2 to give brompheniramine 123. Brompheniramine reacts with maleic acid to produce brompheniramine maleate 124 (ref. 75) (Fig. 30).
 |
| | Fig. 30 Synthesis of brompheniramine maleate. | |
3.5 Diphenhydramine citrate
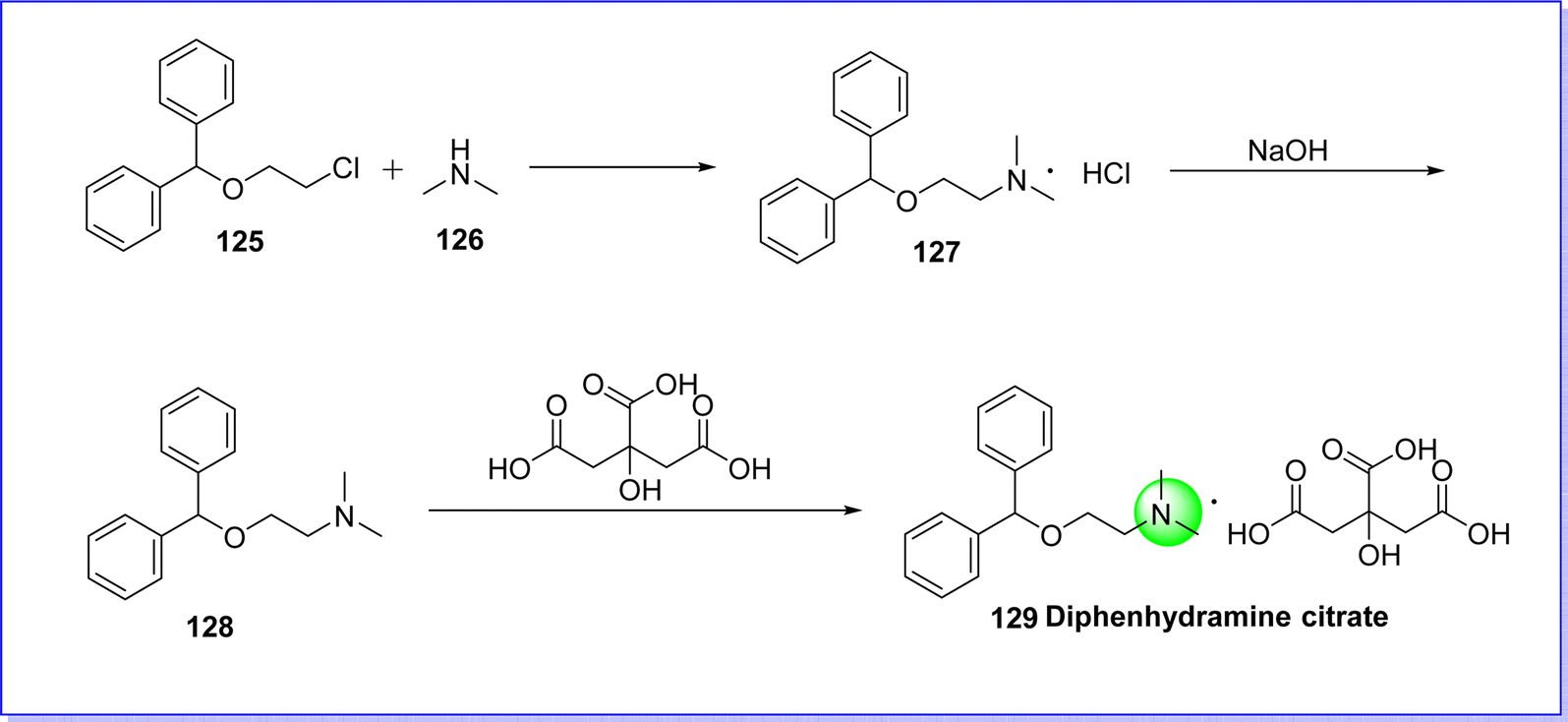
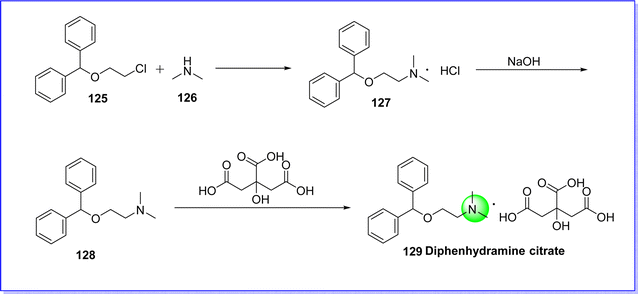
Diphenhydramine citrate is an antihistaminic drug, used to treat allergies, hay fever, insomnia, and common cold-like symptoms.76 It was discovered in 1943 by George Rieveschl and it became the first prescription antihistaminic drug approved by FDA in 1946. The chemical name is 2-(benzhydryloxy)-N,N-dimethylethan-1-amine 2-hydroxypropane-1,2,3-tricarboxylate. It is synthesized by reacting ((2-chloroethoxy)methylene)dibenzene 125 with dimethylamine 126 to give diphenhydramine hydrochloride 127, which reacts with NaOH to give diphenhydramine 128. On reacting compound 128 with 2-hydroxypropane-1,2,3-tricarboxylic acid to produce diphenhydramine citrate 129 (ref. 77) (Fig. 31).
 |
| | Fig. 31 Synthesis of diphenhydramine citrate. | |
3.6 Tripelennamine
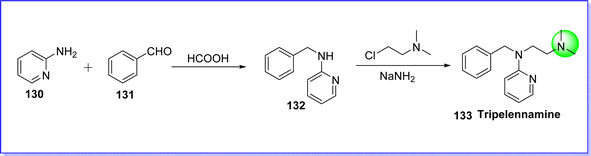
Tripelennamine is an antihistaminic drug that comes under the alkylamine derivative H1 antagonist. It is used to treat antipruritic, hay fever, asthma, and rhinitis.78 On 19 Feb 1948, it is approved by the FDA. It is sold under the brand name Pyribenzamine. The chemical name of tripelennamine is N1-benzyl-N2, N2-dimethyl-N1-(pyridine-2-yl)ethane-1,2-diamine. It is synthesized by reacting 2-aminopyridine 130 with benzaldehyde 131 in the presence of formic acid to give compound 132, which reacts with 2-dimethylaminoethylchloride in the presence of NaNH2 to form tripelennamine 133 (ref. 79) (Fig. 32).
 |
| | Fig. 32 Synthesis of tripelennamine. | |
3.7 Promethazine hydrochloride
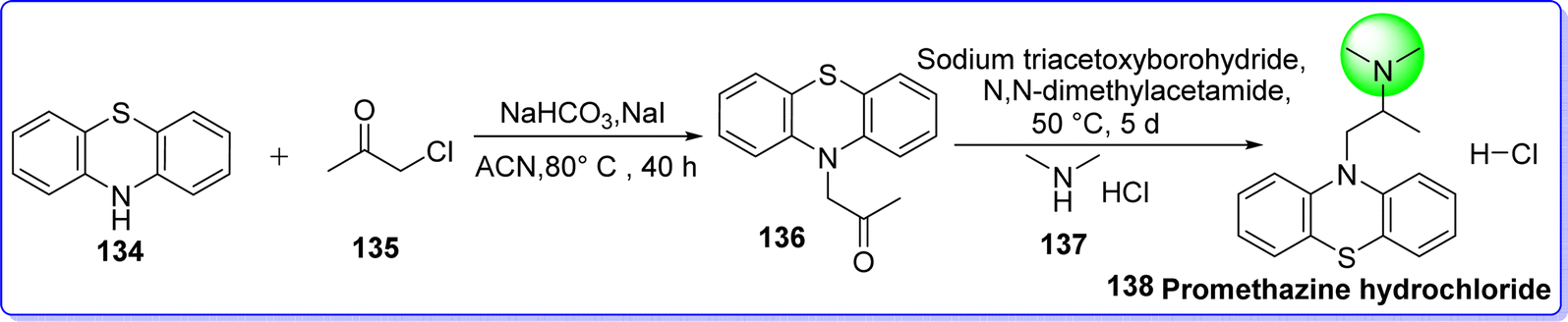
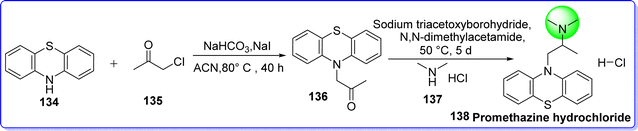
Promethazine hydrochloride is a strong antihistaminic H1 receptor blocker with a bitter taste that is frequently used for treating motion sickness, nausea, dizziness, and vomiting.80 On 29 March 1951, it was approved by the FDA. Chemically it is known as N,N-dimethyl-1-(10H-phenothiazin-10-yl)propan-2-amine hydrochloride. Promethazine belongs to the phenothiazine category hence; its synthesis starts from phenothiazine. It is synthesized by alkylation of secondary amine of phenothiazine 134 with chloroacetone 135 in presence of NaHCO3 in refluxing acetonitrile to form compound 136, which react with dimethylamine hydrochloride 137 in presence of sodium triacetoxyborohydride as reducing agent in N,N dimethylacetamide to form promethazine hydrochloride 138 (Fig. 33).81
 |
| | Fig. 33 Synthesis of promethazine hydrochloride. | |
3.8 Olopatadine hydrochloride
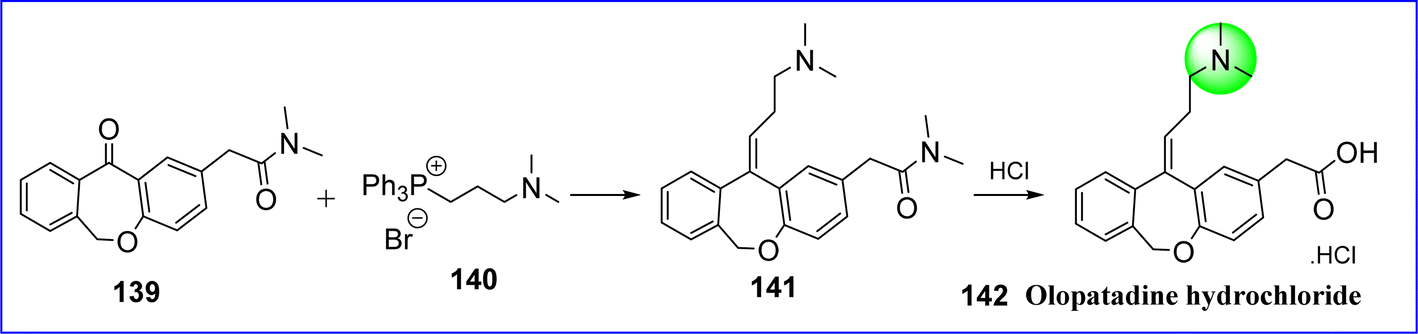
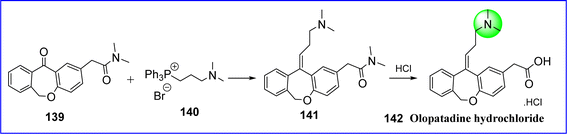
Olopatadine is a mast cell stabilizer and selective H1 antagonist that reduces allergy and inflammatory responses. The FDA and the European Union authorized olopatadine as an ophthalmic solution in 1996 and 2002, respectively, for the management of seasonal and perennial allergic conjunctivitis. The chemical name for olopatadine is (Z)-2-(11-(3-(dimethylamino)propylidene)-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetic acid hydrochloride. Olopatadine is employed in the management of symptoms of seasonal allergic rhinitis in intranasal formulations and ocular irritation related to allergic conjunctivitis in ophthalmic formulations. It is synthesized by reacting methyl-2-[llZ-(3-mesyloxypropylidene)-6H-benzo[c][2]benzoxepin-2-yl] acetate (compound 139) with dimethylamine (compound 140) in the presence of a phase transfer catalyst to give compound 141. Moreover, it undergoes hydrolysis in an inorganic acid and agitation in a water-immiscible solvent to produce olopatadine hydrochloride. Following this, it crystallizes from a water mixture into a solvent that is miscible with water, resulting in the Z isomer of olopatadine hydrochloride 142 (ref. 82) (Fig. 34).
 |
| | Fig. 34 Synthesis of olopatadine hydrochloride. | |
3.9 Carbinoxamine
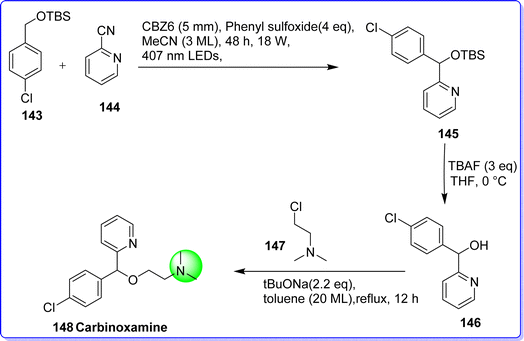
Carbinoxamine is a first-generation antihistaminic H1 receptor antagonist. It is also acting as an anticholinergic agent. It is used in the treatment of rhinitis, hay fever, urticaria, dermatographism, and allergic conjunctivitis.83 The chemical name of carbinoxamine is 2-((4-chlorophenyl)(pyridine-2-yl)methoxy)-N,N-dimethylethan-1-amine. It was synthesized by reacting tert-butyl((4-chlorobenzyl)oxy) dimethyl silane 143 and 2-cyanopyridine 144 in the presence of phenyl sulfoxide and non donor–acceptor type organic photoreductant CBZ6 in methyl cyanide to form compound 145, which on reacting with TBAF in THF form compound 146. The target molecule carbinoxamine 148 was obtained by reacting compound 146 with 2-chloro-N,N-dimethylethan-1-amine 147 (ref. 84) (Fig. 35).
 |
| | Fig. 35 Synthesis of carbinoxamine. | |
3.10 Bromodiphenhydramine
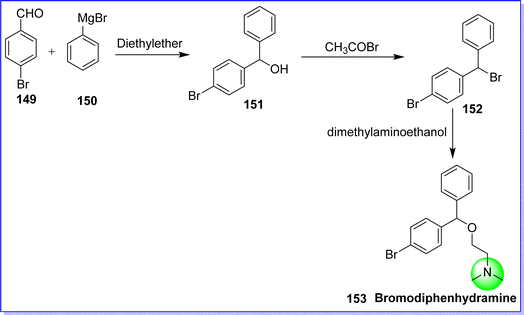
Bromodiphenhydramine is also known as bromazine. It is an antihistaminic (H1 antagonist) anticholinergic agent used in the treatment of hay fever and allergy.85 It is marketed under the brand names Ambodryl, Ambrosial, and Deserol. The chemical name of bromodiphenhydramine is 2-((4-bromophenyl)(phenyl)methoxy)-N,N-dimethylethan-1-amine. It is synthesized by reacting bromobenzaldehyde 149 with phenyl magnesium bromide 150 to form (4-bromophenyl)(phenyl)methanol 151, which on bromination form 1-bromo-4-(bromo (phenyl)methyl)benzene 152. On reacting dimethylaminoethanol with compound 152, the target molecule bromodiphenhydramine 153 was obtained86 (Fig. 36).
 |
| | Fig. 36 Synthesis of bromodiphenhydramine. | |
3.11 Pyrilamine maleate
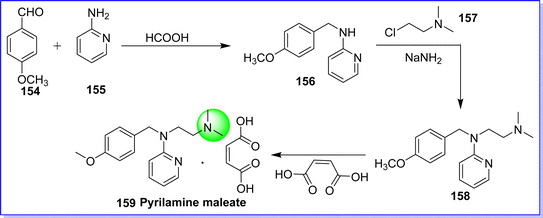
Pyrilamine is also known as mepyramine. It is an antihistaminic, H1 receptor antagonist. Pyrilamine rapidly permeates the brain and causes drowsiness.87 It was patented in 1943 and used in medicine in 1949. It is sold under the brand names Prefrin-A, Neo-Pyramine, Histadyl, Neo-Antergan, and Nisaval.88 The chemical name of pyrilamine is N1-(4-methoxybenzyl)-N2,N2-dimethyl-N1-(pyridine-2-yl)ethane-1,2-diamine. It is synthesized by reacting 4-methoxy benzaldehyde 154 with pyridine-2-amine 155 in presence of formic acid to form compound 156, which on reacting with 2-chloro-N,N-dimethylethan-1-amine 157 to give pyrilamine 158. On reacting compound 158 with maleic acid target molecule pyrilamine 159 maleate was obtained89 (Fig. 37).
 |
| | Fig. 37 Synthesis of pyrilamine. | |
4. Antibiotics
4.1 Tetracycline hydrochloride
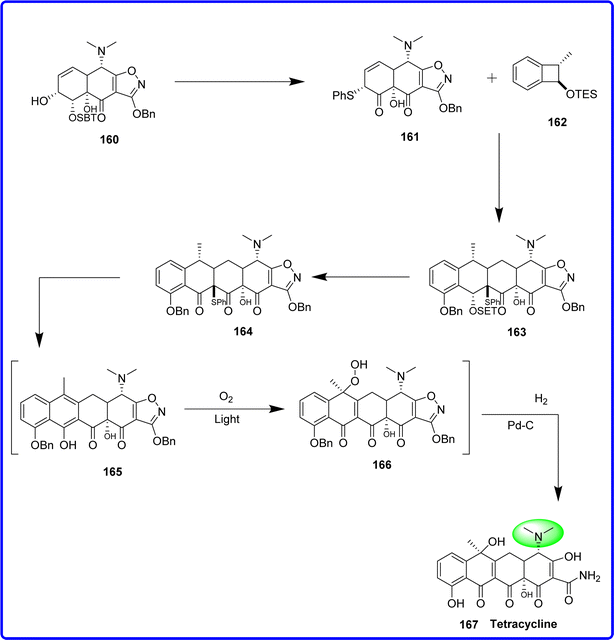
Tetracyclines are broad-spectrum antibiotics that work against intracellular chlamydiae, mycoplasmas, and rickettsiae as well as Gram negative and Gram-positive bacteria. In contemporary times, tetracyclines have been employed to combat eukaryotic protozoan parasites and to address an array of noninfectious ailments, frequently at subtherapeutic dosages (like acne), and for prolonged durations (like ∼1 year). Tetracyclines were first identified in the 1940s, and their therapeutic application started in the 1950s due to their minimal serious side effects with the exception of small children and expectant mothers.90 The Pfizer–Woodward group revealed the key structural characteristics of the molecules in a seminal work titled “The Structure of Aureomycin” in 1954. It was discovered that Aureomycin's C7 chlorine was not the source of its activity and that it could be altered or eliminated to create an analogue which was more active and had better antibacterial properties. The C7 dichloro derivative, which was later given the name tetracycline, was created by catalytic hydrogenation of aureomycin using palladium metal and hydrogen. This process produced a molecule with greater potency, a superior solubility profile, and good pharmacological activity. As the first new tetracycline, this substance was licensed by the FDA for clinical use in 1954.91 The synthesis of tetracycline was started with transforming the alcohol 160 into the enone 161. The cyclobutane ring of compound 162 was then opened to form an o-quinone methide intermediate, which underwent a Diels–Alder cycloaddition with compound 161, resulting in the formation of the endo adduct 163. Subsequent deprotection and oxidation of 163 produced compound 164. This compound was further oxidized to form the sulfoxide. Eliminating the sulfoxide yielded the naphthalene derivative 165, which spontaneously oxidized to form 166. Finally, reductive deprotection of 166 led to the synthesis of tetracycline 167 (ref. 92) (Fig. 38).
 |
| | Fig. 38 Synthesis of tetracycline. | |
4.2 Doxycycline hyclate
Doxycycline, a member of the tetracycline antibiotic family, is a versatile medication effective against mycoplasma, protozoa, and a wide range of both Gram-negative and Gram-positive bacteria.93 Synthetically, doxycycline (α-6-deoxy-5-oxytetracyclin) was produced from oxytetracycline. It is a bacteriostatic agent that prevents the synthesis of bacterial proteins by interfering with the transfer and messenger RNA at the ribosomal sites.94 Doxycycline was initially made from methacycline by a team of Pfizer chemists under the direction of Robert Blackwood. It was approved by FDA in 1967, and sold under the brand name Vibramycin. Among the safest and most efficient antibiotics for the human body system, it is one of the medications on the WHO essential pharmaceuticals list.95 Doxycycline is an isomer of tetracycline with one hydroxyl group different from the other. Chemically it is also known as (4S,4aR,5S,5aR,6R,12aS)-4-(dimethylamino)-3,5,10,12,12a-pentahydroxy-6-methyl-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide. Oxytetracycline is the precursor of doxycycline 168. The oxidation step of the homoallyl system, with the exception that N-chlorosuccinimide is employed as the oxidant. This produces a derivative of naphtha centetrahydrofuran 169, which disintegrates when it reacts with HF to produce an 11a-chloro-6-exomethylene derivative 170. Employing sodium thiosulfate for reductive dichlorination yields the intermediate methacycline 171. Subsequently, thiophenol is coupled to the methyl group responsible for radical reactions, resulting in the derivative 172. Doxycycline 173 is produced by reducing compound 172 with hydrogen in presence of Raney nickel catalyst, which causes reductive desulfurization96 (Fig. 39).
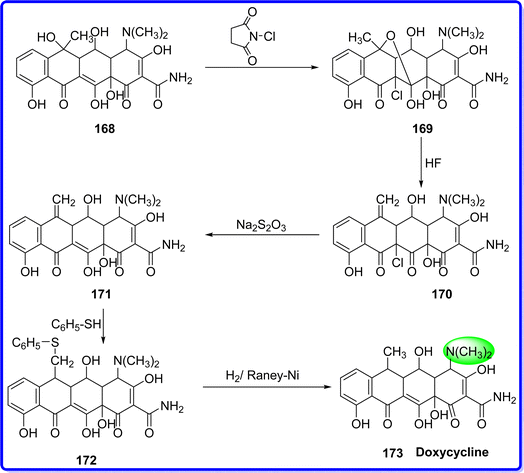 |
| | Fig. 39 Synthesis of doxycycline. | |
4.3 Chlortetracycline hydrochloride
Chlortetracycline hydrochloride (CTC-HCl) is a compound derived from the antibiotic substance generated through the cultivation of streptomyces aureofaciens, a member of the streptomycetaceae family. Its discovery is credited to Duggar in 1948. A broad-spectrum antibiotic and antiprotozoal, CTC-HCl is effective against a variety of big viruses, spirochetes, amebae, Gram-negative and Gram-positive bacteria. Antibiotic resistant strains of Staphylococci can be discovered amid other bacterial genera and species that are normally vulnerable to the antibiotic, despite the fact that the bacteria has become generally resistant to it97 (Fig. 40).
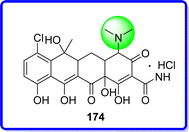 |
| | Fig. 40 Structure of chlortetracycline hydrochloride. | |
4.4 Erythromycin
Erythromycin stands as the primary macrolide antibiotic today. Originating from the actinomycete Streptomyces erythreus, it was first utilized in clinical practice in 1952 under the guidance of McGuire and colleagues. Structurally, erythromycin consists of a macrocyclic lactone ring, known as a macrolide, linked to two sugar components.98 The antibacterial action involves attaching to the 50S subunit of bacterial ribosomes, which disrupts RNA dependent protein production by impeding transpeptidation and/or translocation processes99 (Fig. 41).
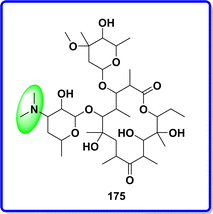 |
| | Fig. 41 Structure of erythromycin. | |
4.5 Azithromycin
Azithromycin is a broad-spectrum antibiotic of the second generation. Its impact on chronic human diseases and host-defense responses has made it a subject of increasing study in recent times. Similar to other macrolide antibiotics, it works by inhibiting the growth of microorganisms.100 Azithromycin is a macrolide antibiotic that is semisynthetic and acid-stable. Azithromycin's structural makeup is a 15-membered lactone ring with a tertiary amino group linked to two sugar moieties; it is distinct from erythromycin in that it has an enlarged macrolide nucleus that is amino-substituted.101 The US FDA authorized azithromycin for use in medicine in 1992.102 It was synthesized by converting erythromycin A into azithromycin by treating the erythromycin 176 in CH3OH with hydroxylamine hydrochloride and a base at reflux temperature for 10 hours to form oxime 177. The oxime was isolated, purified, and then underwent Beckmann's rearrangement to produce the intermediate compound (6,9-imino ether) 178 in acetone in the presence of p-toluenesulfonyl chloride and base for two hours at 5 °C. The iminoether was then reduced to the secondary amine 179 with NaBH4 in CH3OH solvents. The target molecule azithromycin 180 was obtained by methylation of compound 179 (ref. 103) (Fig. 42).
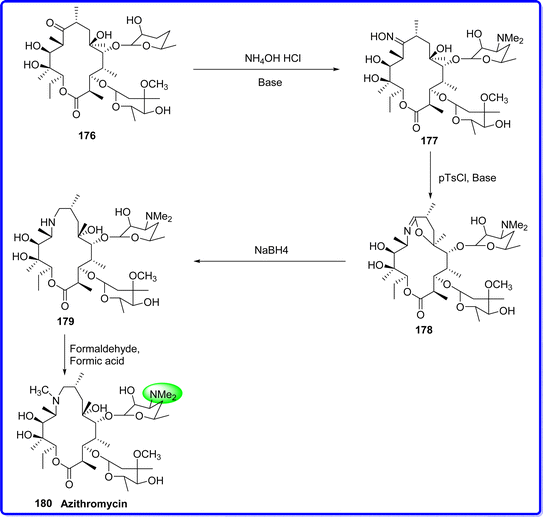 |
| | Fig. 42 Synthesis of azithromycin. | |
4.6 Clarithromycin
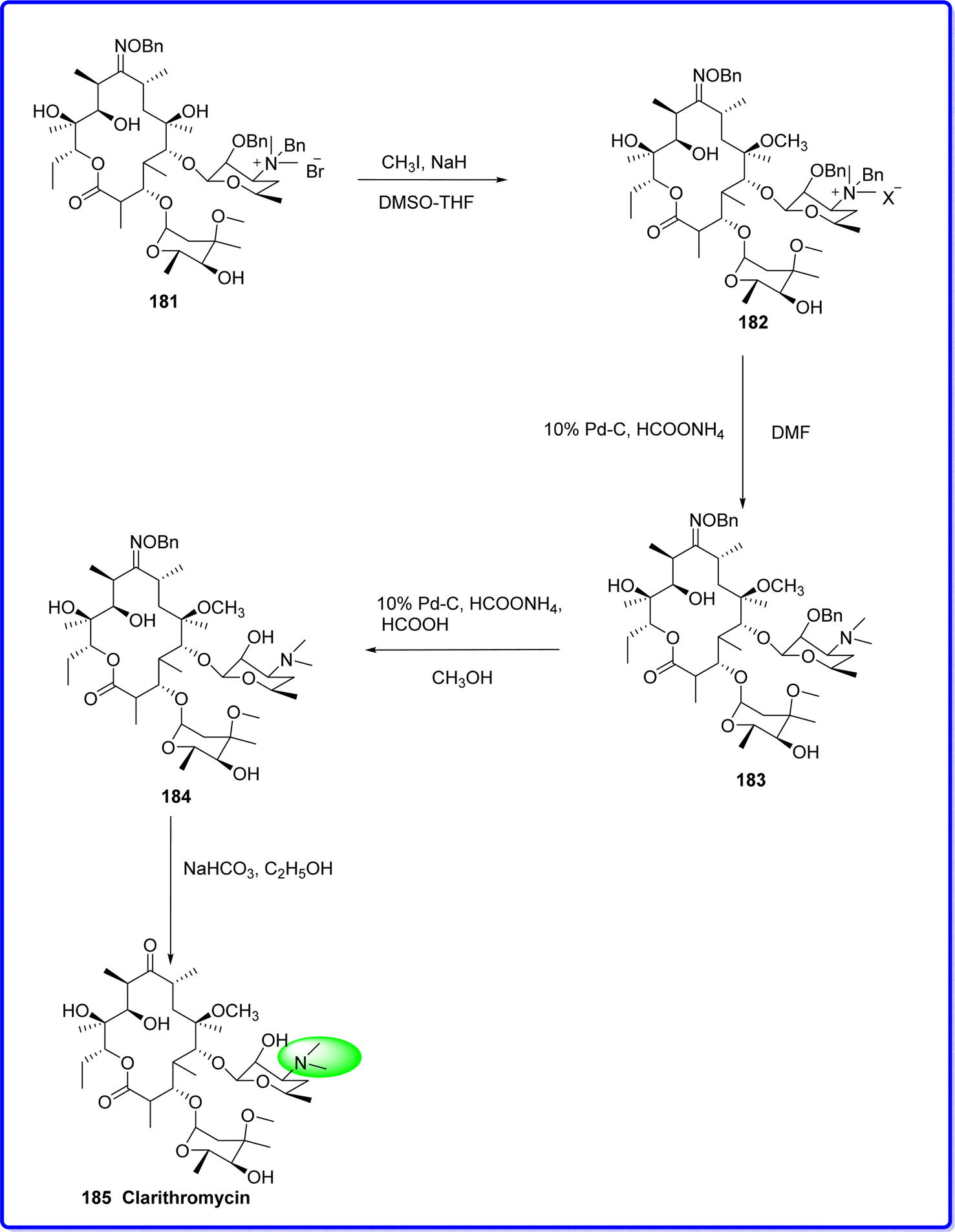
Clarithromycin is a semisynthetic acid-stable macrolide antibiotic. Clarithromycin is structurally different from erythromycin in that it has an o-methyl substitution at 6th position of the lactone ring, which results in enhanced antibacterial and pharmacokinetic qualities as well as acid stability. Clarithromycin is composed of a 14-membered lactone ring connected to two sugar moieties.104 It was synthesized by the methylation of 2′-0,3′-N-bis(benzyloxycarbonyl)-N-dimethyl-erythromycin A 181 with methyl iodide (CH3I) and sodium hydride (NaH) in a mixture of dimethyl sulfoxide and tetrahydrofuran, yielding compound 182. Compound 182 was then reacted with ammonium formate and palladium on carbon (Pd–C) to produce compound 183. Next, compound 183 was treated with formic acid, ammonium formate, and methanol to transform it into compound 184. Finally, compound 184 was reacted with sodium bicarbonate in ethanol to synthesize the final product, clarithromycin 185 (ref. 105) (Fig. 43).
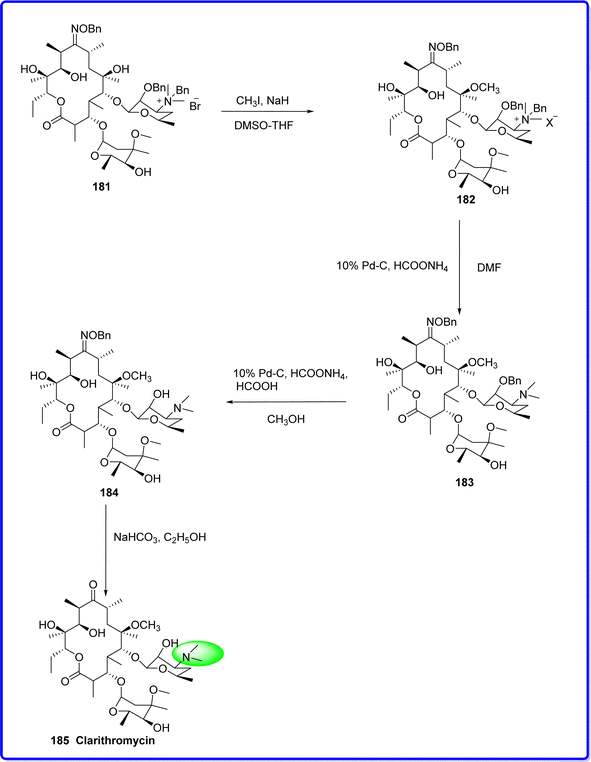 |
| | Fig. 43 Synthesis of clarithromycin. | |
4.7 Telithromycin
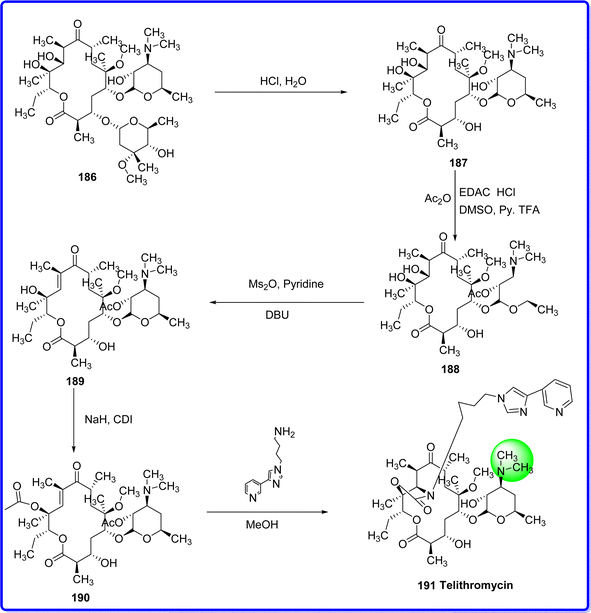
Telithromycin is the first medication in a novel class of semisynthetic ketolide-lincosamide streptogramin-B (MLSB) antibiotics. Ketolides are 14-membered ring macrolides with a methoxy substituent at position C6 and a 3-keto structure in place of the macrolides' L-cladinose moiety. Furthermore, at locations C11 and C12, telithromycin has a carbamate extension.106 Telithromycin, akin to macrolides, operates by engaging with the 50S subunit within the bacterial ribosome, thereby impeding ribosome assembly and protein synthesis. Its antibacterial effect stems from its binding to domains II and V of the 23S rRNA found within the 50S ribosomal subunit.107 Telithromycin holds the distinction of being the inaugural ketolide to progress through clinical development and gain approval for use in Germany, with its initial launch dating back to 2001. It is subsequently obtained the approval for use in the European Union and Canada, with the latter occurring by January 2003.108 Clarithromycin 186 undergoes a series of chemical transformations to produce telithromycin. Initially, hydrochloric acid is employed to remove the cladinose ring from clarithromycin at the C-3 position, resulting in a compound 187. Subsequently, selective acetylation of the 2′-hydroxy group in 187 and oxidation of the 3-hydroxy group yields a ketolide compound 188. Following this, the 11-hydroxy group of 188 is specifically mesylated, followed by a base-induced β-elimination reaction to yield an α,β-unsaturated ketone compound 189. The compound is then treated with sodium hydride and carbonyldiimidazole to form a 12-O-acyl imidazole derivative 190. Finally, 190 undergoes a stereoselective cyclization reaction with (4-(3-pyridinyl)-imidazole-1-yl)-butylamine, followed by deprotection of the 2′-hydroxy group, resulting in the synthesis of telithromycin 191 (ref. 109) (Fig. 44).
 |
| | Fig. 44 Synthesis of telithromycin. | |
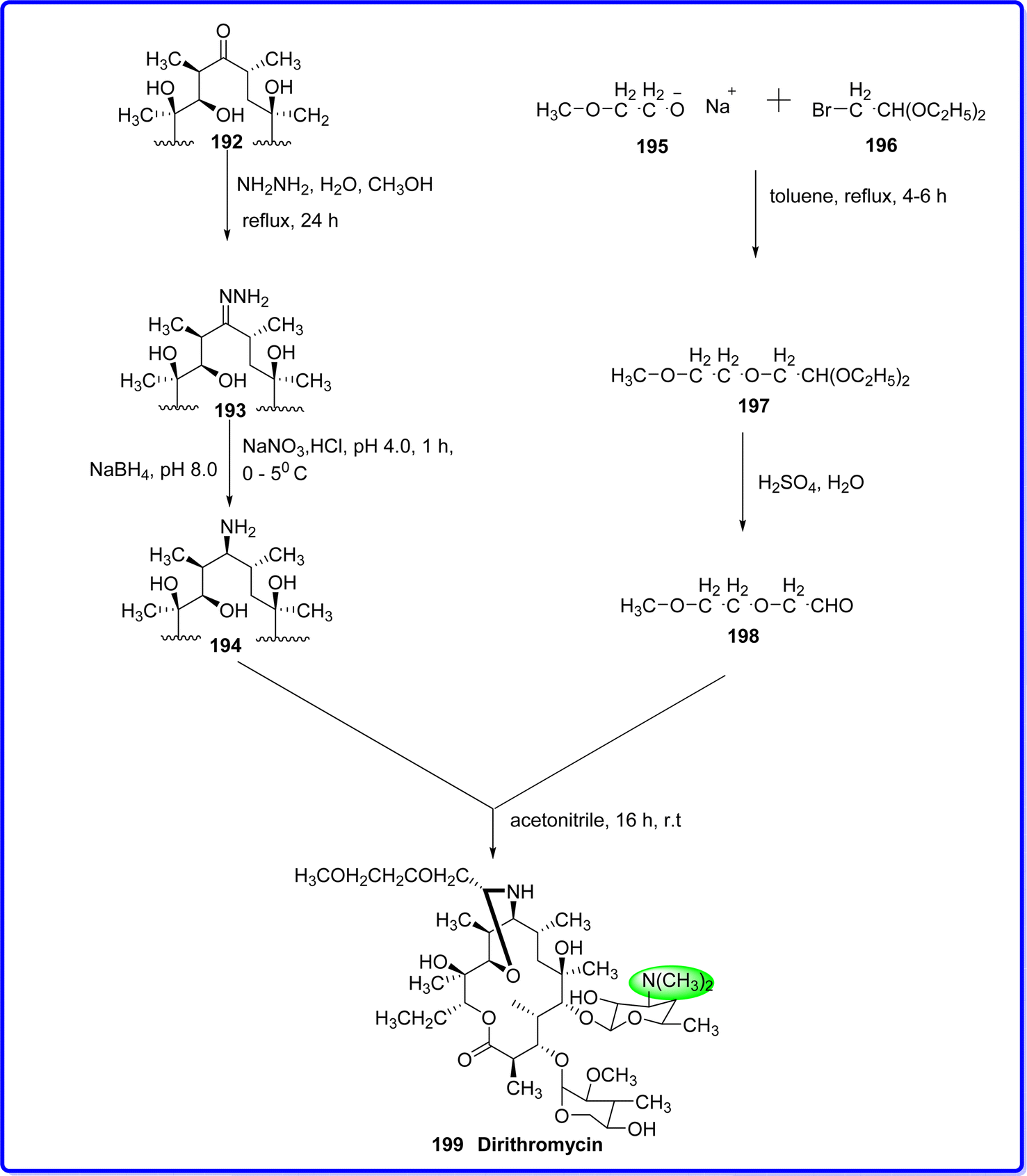
4.8 Dirithromycin
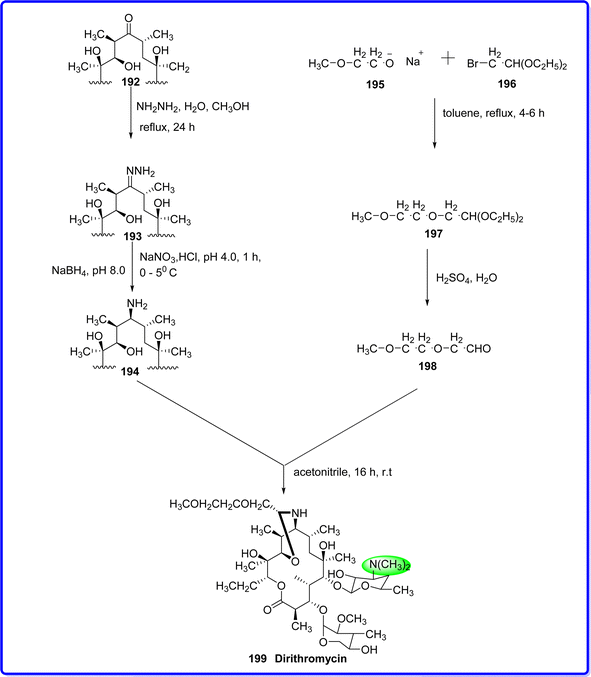
Dirithromycin, an orally administered macrolide antibiotic, features a 14-membered lactone ring and is synthesized through the condensation of erythromycylamine and an aliphatic aldehyde. While its in vitro antibacterial range and potency closely resemble those of erythromycin, dirithromycin exhibits comparable spectrum of activity.109 Dirithromycin is a derivative of erythromycin known as a 9-N-11-O-oxazine. Like other macrolide antibiotics, its mechanism of action involves reversible binding to the 23S component of the 50S ribosomal subunit. This binding disrupts the association of peptidyl-tRNA with the ribosome during the elongation phase of protein synthesis, leading to the inhibition of RNA-dependent protein synthesis and ultimately hindering bacterial growth.110 Dirithromycin was introduced by Lilly in September 1993, initially in Spain, and gained FDA approval in August 1995. Its official launch in the United States followed shortly after, in October 1995.111 Erythromycin 192 is initially reacted with hydrazine hydrate to form erythromycin hydrazone 193. This hydrazone is then used to synthesize erythromycylamine 194, which is subsequently reacted with 2-methoxyethoxy acetaldehyde 198 to produce dirithromycin 199 (ref. 112) (Fig. 45).
 |
| | Fig. 45 Synthesis of dirithromycin. | |
5. Drug used in treatment of peptic ulcer
5.1 Ranitidine hydrochloride
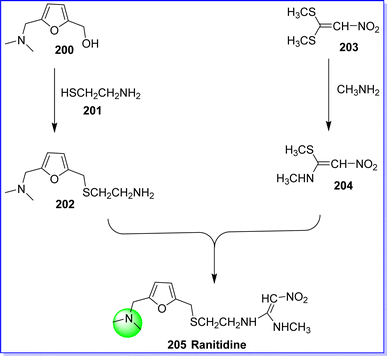
Ranitidine belongs to the class of H2 receptor antagonists and reduces the hydrochloric acid and pepsin excretion by stomach membrane cells. It is employed in the management of Zollinger–Ellison syndrome, peptic ulcer, and gastroesophageal reflux.113 It was approved by the FDA on 08/31/2004. It is marketed in various brand names such as Aciloc, Zantac and Rantac. The chemical name of ranitidine is (E)-N-(2-(((5-((dimethylamino)methyl)furan-2-yl)methyl)thio)ethyl)-N′-methyl-2 nitroethane-1,1-diamine hydrochloride. It is synthesized by reacting 5-dimethylaminomethyl-2-furanyl-methanol 200 with 2-mercaptoethylamine 201 in presence of hydrochloric acid to form 2-[(5-dimethyl-aminomethyl-2-furanyl)methyl] thiolethaneamine 202, which is reacted with N-methyl-1-methylthio-2-nitro etheneamine 204 at 50 °C for 4 h to produce ranitidine 205. The compound 204 was obtained by reacting 1,l-bis(methylthio)-2-nitroethane 203 with methylamine in ethanol114 (Fig. 46).
 |
| | Fig. 46 Synthesis of ranitidine. | |
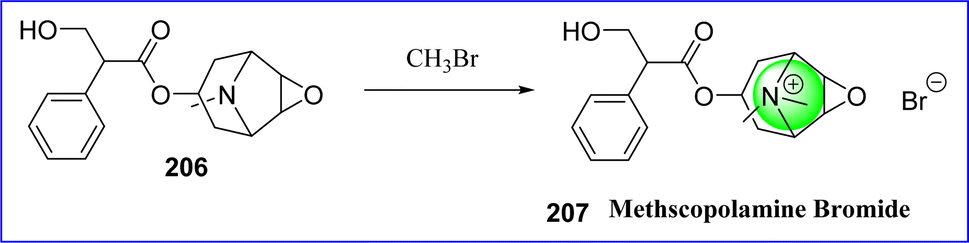
5.2 Methscopolamine bromide
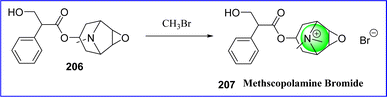
Methylscopolamine, also known as methscopolamine, is often supplied as a nitrate or bromide salt. It acts by decreasing the production of stomach acid and is used in conjunction with other drugs to treat peptic ulcers. Methscopolamine blocks acetylcholine's muscarinic function in postganglionic parasympathetic effector areas. The majority of the pharmacologic effects of that drug class are exhibited by methscopolamine bromide, an anticholinergic agent. Chemically it is known as 3-oxa-9-azoniatricyclo [3.3.1.0 2,4] nonane, 7-(3-hydroxy-1-oxo-2-phenylpropoxy)-9, 9-dimethyl-, bromide, [7(S)-(1α, 2β, 4β, 5α, 7β)]. In 1952, the FDA authorized methscopolamine. In June 2001, a lactose-free version of Pamine(R) received approval. The synthesis of methscopolamine involves reacting scopolamine (compound 206) with methyl bromide, and occasionally adding silver nitrate to replace the bromide ion with a nitrate ion115 (Fig. 47).
 |
| | Fig. 47 Synthesis of methscopolamine bromide. | |
5.3 Glycopyrrolate
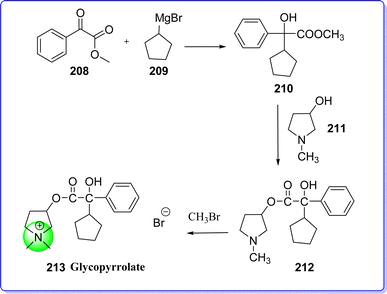
Glycopyrrolate is an anticholinergic drug, which is also referred to as glycopyrronium.116 Children between the age of 3 and 16 who have specific medical disorders that cause them to drool are treated with glycopyrrolate to lessen their saliva and drooling. It works by inhibiting the function of a certain natural substance in the body, which lowers the production of saliva and stomach acid. The chemical name for glycopyrrolate is (1,1-dimethylpyrrolidin-1-ium-3-yl) 2-cyclopentyl-2-hydroxy-2-phenylacetate bromide. On 28th July 2010, the US FDA approved glycopyrrolate for treating peptic ulcers in adults. Glycopyrrolate was synthesized by reacting methyl 2-oxo-2-phenylacetate 208 with Grignard reagent 209 to produce compound 210, which was react with 1-methylpyrrolidin-3-ol 211 to give α-cyclopentylmandelic acid 212. The compound 212 was react with methyl bromide to form glycopyrrolate 213 (ref. 117) (Fig. 48).
 |
| | Fig. 48 Synthesis of glycopyrrolate. | |
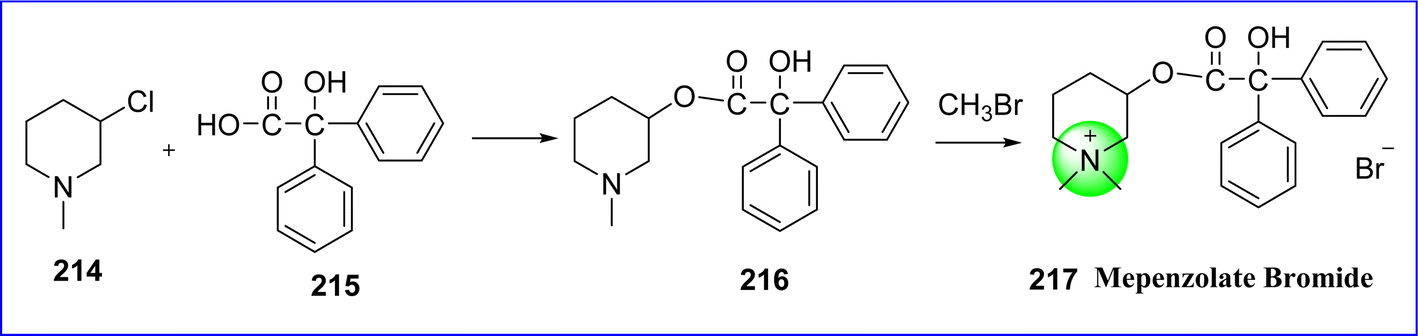
5.4 Mepenzolate bromide
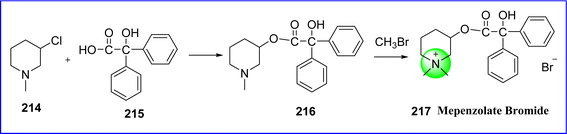
Mepenzolate is a parasympathetic post-ganglionic inhibitor. It reduces the colon's spontaneous contractions and lowers the release of pepsin and stomach acid. The chemical name for mepenzolate bromide is (1,1-dimethylpiperidin-1-ium-3-yl) 2-hydroxy-2,2-diphenylacetate; bromide.118 Mepenzolate, is prepared by reacting 3-chloro-1-methylpiperidine (compound 214) with 2-hydroxy-2,2-diphenylacetic acid (compound 215) to produce ester (compound 216), the resulting ester is further reacted with methyl bromide to form mepenzolate bromide 217 (ref. 119) (Fig. 49).
 |
| | Fig. 49 Synthesis of mepenzolate bromide. | |
6. Local anesthetics
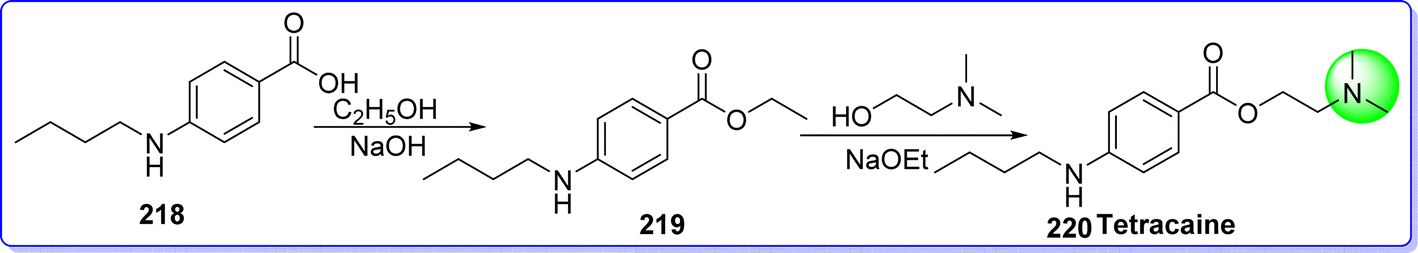
6.1 Tetracaine
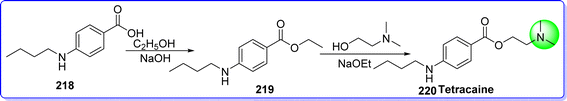
Tetracaine is a long-acting ester derivative used as a local anesthetic agent.120 In 2016 tetracaine was approved by the FDA. It is sold under the brand name Pontocaine, Ametop, and Dicaine. The chemical name of tetracaine is 2-(dimethylamino)ethyl 4-(butylamino)benzoate. It is synthesized by reacting 4-(butylamino)benzoic acid 218 with ethanol in the presence of HCl to give compound 219, which reacts with 2-(dimethylamino)ethan-1-ol in the presence of NaOEt to form tetracaine 220 (ref. 121) (Fig. 50).
 |
| | Fig. 50 Synthesis of tetracaine. | |
6.2 Propoxyphene hydrochloride
An opioid analgesic called propoxyphene is used to relieve minor pain symptoms. It has local anesthetic and antitussive properties. Propoxyphene has been taken off the market in both Europe and the US due to the potential for cardiac arrhythmias and overdose, which might result in death. The chemical name for propoxyphene hydrochloride is 4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl propionate. Propoxyphene received FDA approval in 1957. When phenyl propenyl ketone and secondary amine are combined, an amino ketone known as beta-dimethyl amino butrophenone (compound 221) was produced. Compound 222 (benzyl magnesium chloride) and amino ketone 221 undergo the Grignard reaction, which produces compound 223. Propoxyphene 224 was produced when propionic anhydride is added to compound 223 and heated to reflux122 (Fig. 51).
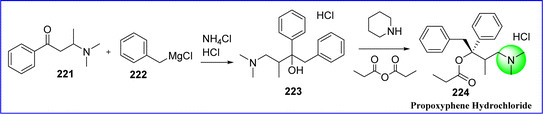 |
| | Fig. 51 Synthesis of propoxyphene hydrochloride. | |
7. Anti muscarinic agents
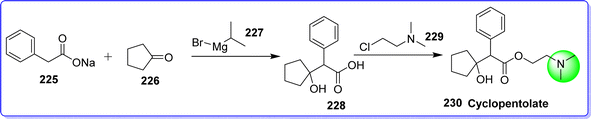
7.1 Cyclopentolate
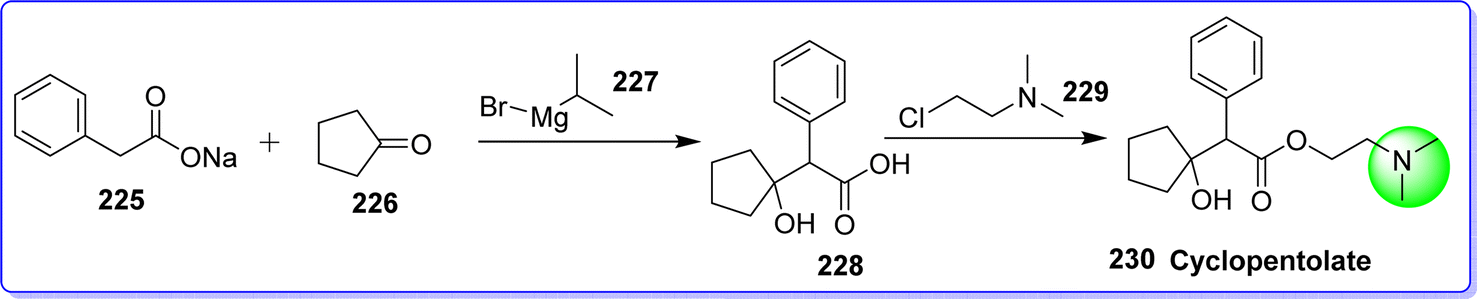
Cyclopentolate is an antimuscarinic agent used in eye examination in pediatric to dilate the eye (mydriatic) and prevent the eye from cycloplegic.123 It is sold under the brand name AK-Pentolate. The chemical name of cyclopentolate is 2-(dimethylamino)ethyl 2-(1-hydroxycyclopentyl)-2-phenylacetate. It is synthesized by reacting sodium 2-phenylacetate 225 with cyclopentanone 226 in presence of isopropylmagnesium bromide 227 to form compound 228. On reacting compound 228 with 2-chloro-N,N-dimethylethan-1-amine 229, target molecule cyclopentolate 230 was obtained (Fig. 52).124
 |
| | Fig. 52 Synthesis of cyclopentolate. | |
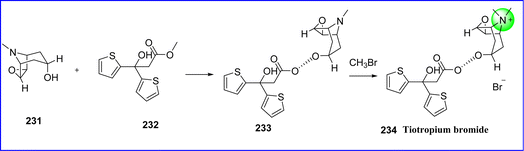
7.2 Tiotropium bromide
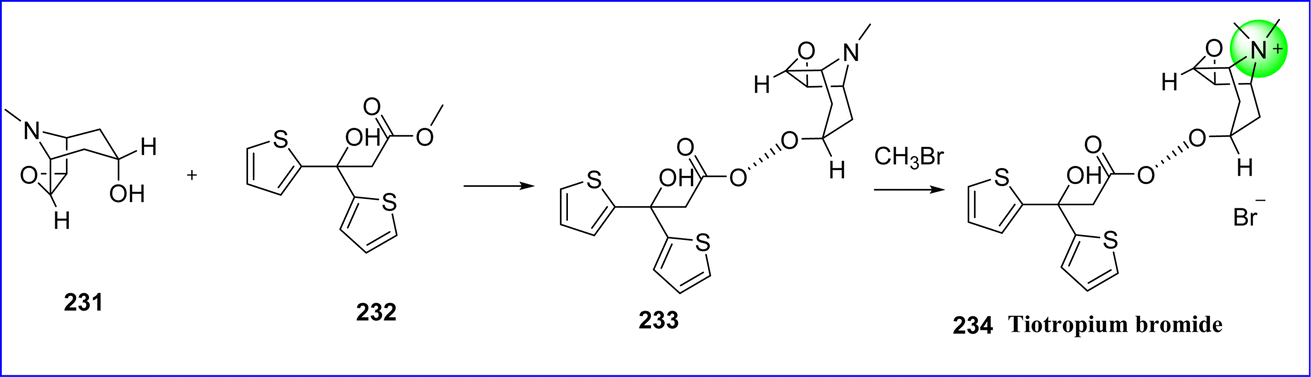
Tiotropium is an antimuscarinic bronchodilator that acts slowly and is applied to the management of chronic obstructive pulmonary disease (COPD) and asthma. Tiotropium produces bronchodilation and smooth muscle relaxation primarily by activating M3 muscarinic receptors in the airways. On January 30, 2004, the FDA approved tiotropium. The chemical name for tiotropium bromide is (1R,2R,4S,5S)-7-{[2-hydroxy-2,2-bis(thiophen-2-yl)acetyl]oxy}-9,9-dimethyl-3-oxa-9-azatricyclo[3.3.1.0{2,4}]nonan-9-ium bromide. Tiotropium bromide is prepared by first transesterifying scopine (compound 231) and methyl di-(2-dithienyl)glycolate (compound 232) to produce N-demethyltiotropium (compound 233), and then reacting N-demethyltiotropium with bromomethane to produce tiotropium bromide 234 (ref. 125) (Fig. 53).
 |
| | Fig. 53 Synthesis of tiotropium bromide. | |
8. Anticancer drugs
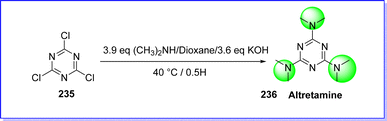
8.1 Altretamine
Altretamine is an anti-cancer (antineoplastic) chemotherapy drug. This drug is classified as an alkylating agent. It is initially invented by the National Cancer Institute (NCI), it has been classified as a group C medication for almost 16 years (restricted availability for particular indications to physicians registered with the NCI as investigators).126 But later, in 1990 Altretamine was approved by FDA under the brand name Hexalen for the treatment of ovarian cancer. This drug is less toxic than other anticancer drugs used in treatment of ovarian cancer.127 However, the exact mechanism by which altretamine inhibits cancer is uncertain.128 The chemical name of altretamine is N,N,N′,N′,N′′,N′′-hexamethyl-1,3,5-triazine-2,4,6-triamine. It is also known as hexamethyl melamine (HMM). The altretamine was synthesized by reacting cyanuric chloride 235 with a solution of 40% dimethylamine and KOH in dioxane at 40 °C for 0.5 h. after this, the reaction mixture was cooled by adding ice-cooled water. The resulting altretamine 236 was filtered and recrystallized by ethanol129 (Fig. 54).
 |
| | Fig. 54 Synthesis of altretamine. | |
8.2 Tamoxifen citrate
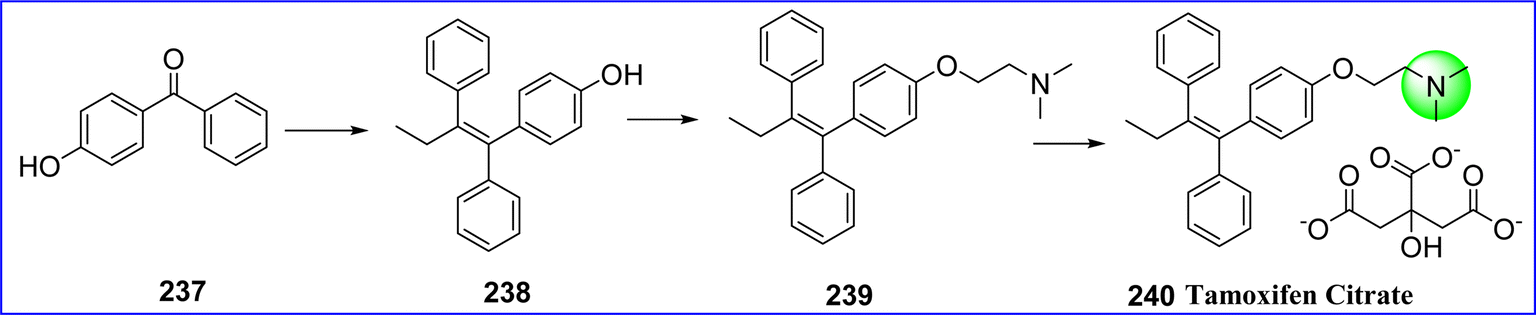
Tamoxifen is a non-steroidal anti-estrogen that is used to treat breast cancers and to lower the likelihood of developing the disease in those who are at high risk. In these therapies, tamoxifen is administered either as a stand-alone medication or as an adjuvant. With patients usually having higher survival rates, side effect profiles, and anastrozole compliance, tamoxifen may no longer be the recommended therapy for certain kinds of malignancies. The FDA approved tamoxifen on December 30, 1977. The chemical name for tamoxifen citrate is (Z)-2-(4-(1,2-diphenylbut-1-en-1-yl)phenoxy)-N,N-dimethylethan-1-amine 2-hydroxypropane-1,2,3-tricarboxylate.130 Using the easily accessible and basic raw material 4-hydroxybenzophenone (compound 237), tamoxifen citrate is made by performing a coupling reaction to give compound 238, which undergoes alkylation reactions to yield a crude product 2-[4-(1,2-diphenyl1-butenyl)-phenoxy]-N,N-dimethylethylamine (compound 239), a direct isomerization reaction is performed, followed by purification and salinization with citric acid to yield the high-purity tamoxifen citrate 240 effortlessly131 (Fig. 55).
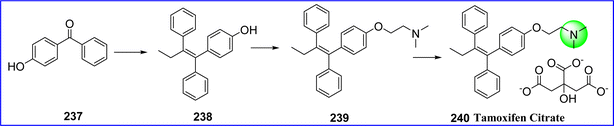 |
| | Fig. 55 Synthesis of tamoxifen citrate. | |
8.3 Toremifene citrate
Toremifene (TOR) functions as a selective estrogen receptor modulator (SERM) in the management of breast cancer. Its molecular structure differs from tamoxifen by only one chlorine atom.132 The main way that toremifene inhibits the growth of tumors is by selectively and aggressively blocking the binding of estrogen to its receptor. It seems to have a similar binding affinity to tamoxifen for the estrogen receptor. After estrogen binds to its receptor, an active hormone-receptor complex is created that can interact with nuclear estrogen response elements to regulate specific mRNA transcription, protein synthesis, expression of genes regulated by estrogen, and ultimately, cell growth. Through methods unrelated to the estrogen receptor, toremifene may also prevent the proliferation of tumor cells. The mode of action of toremifene is similar to mechanisms of action of tamoxifen.133 Toremifene was created in the chemical research laboratory of Farmos in Oulu, Finland.134 Since 1988, toremifene has been sold in Finland, and in 1997, the drug received approval for use in the US.135 It was synthesized by McMurry reaction, where compound 241 reacts with compound 242 in the presence of zinc and titanium tetrachloride, leading to the formation of compound 243. Following this, compound 243 undergoes a specific alkylation of the phenolic hydroxyl group using the hydrochloride form of compound 244, to produce compound 245. Finally, the synthesis is completed by treating compound 245 with dichlorosulfoxide, resulting in the production of toremifene 246 (ref. 136) (Fig. 56).
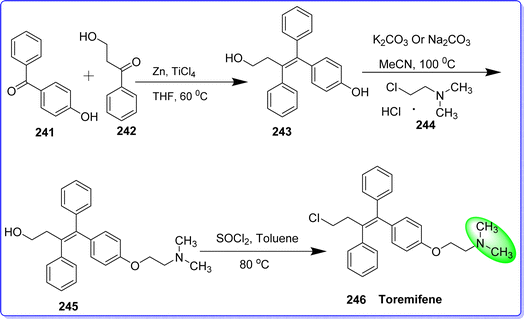 |
| | Fig. 56 Synthesis of toremifene. | |
8.4 Afatinib dimaleate
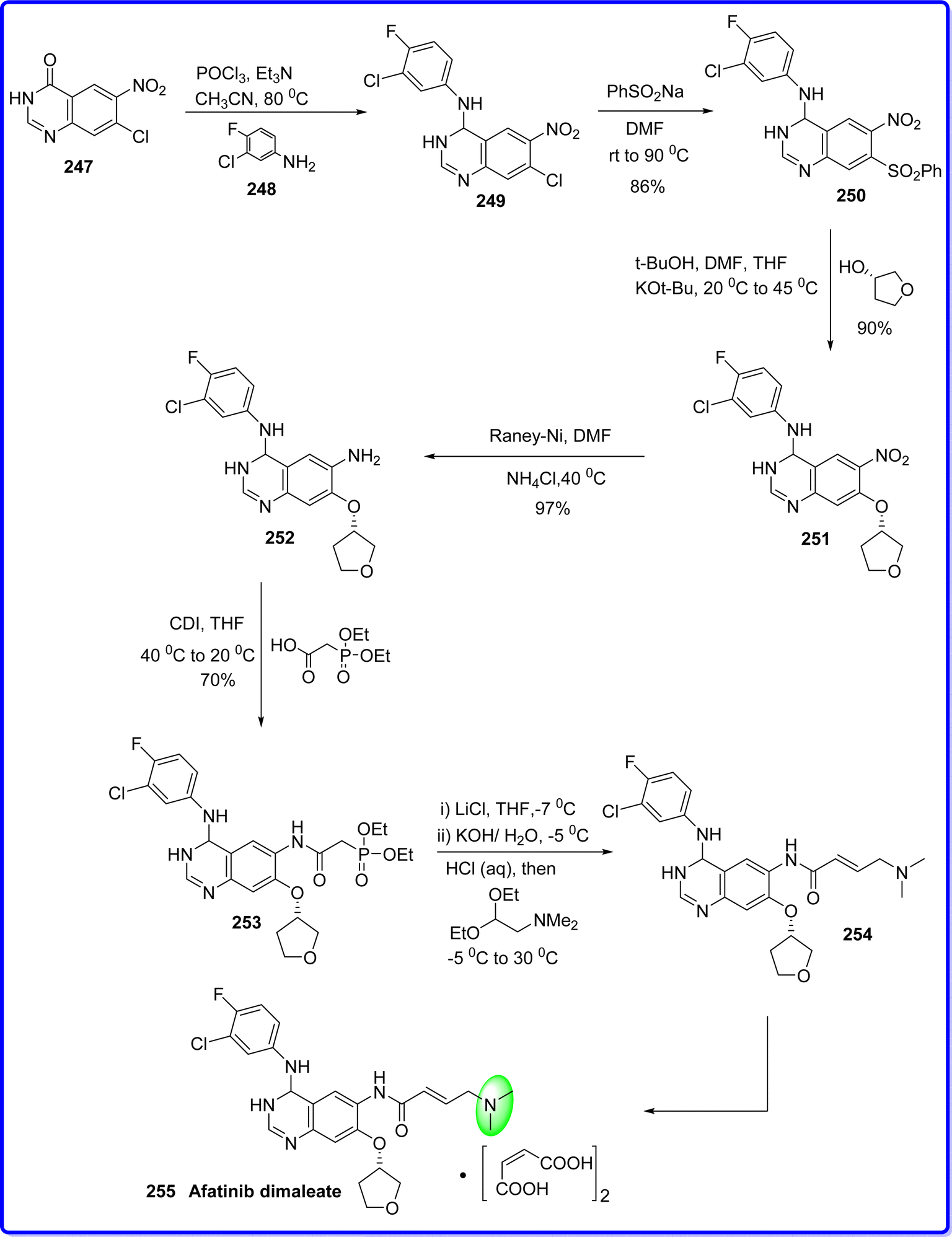
Afatinib, has an irreversible activity and is a second-generation inhibitor of the ErbB family of tyrosine kinases. This compound is an aniline–quinazoline, effectively diminishes ErbB signalling by forming covalent bonds with the kinase domains of EGFR, HER2, and HER4. Consequently, it leads to the permanent inhibition of tyrosine kinase autophosphorylation. Moreover, afatinib also blocks the transphosphorylation of HER3.137 On July 12, 2013, the US FDA approved afatinib for the first time globally as a first-line therapy for metastatic non-small cell lung cancer (NSCLC) with EGFR mutation.138 Commercially available nitroquinazolinone 247 was first chlorinated using POCl3, and then it was treated with 3-chloro-4-fluoroaniline 248 to produce compound 249 with a 90% yield, which react with PhSO2Na to give compound of 250 with 86% yield. After substituting 250 react with (S)-tetrahydrofuran-3-ol, to produce 251 with 90% yield. Final side-chain functionalization was made possible by the Raney-Nickel reduction of the nitro group into amino group 252 in 90% yield. Compound 252 was reacted with 2-(diethoxyphosphoryl) acetic acid and N,N′-carbonyldiimidazole (CDI) to give 253 in a 70% isolated yield. Subsequently, the (E)-olefin 254 was obtained quantitatively using a Horner–Wadsworth–Emmons homologation, and 92% yield of the afatinib 255 was then delivered by maleate salt production139 (Fig. 57).
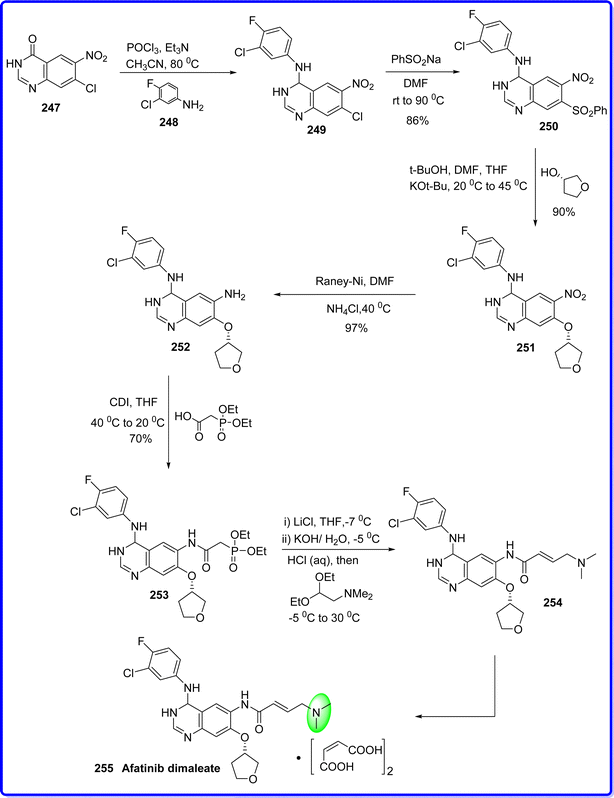 |
| | Fig. 57 Synthesis of afatinib dimaleate. | |
8.5 Cabergoline
Cabergoline (CAB) is a synthetic ergoline compound known for its high affinity and specificity towards the dopamine D2 receptor. Many medical conditions that arise from the overproduction of the hormone prolactin are treated with cabergoline. It is effective in treating some menstruation issues, issues with male and female infertility, and pituitary prolactinomas (pituitary gland tumours). It is an effective and very long-acting prolactin secretion inhibitor. Cabergoline exhibits a strong affinity and selectivity for the dopamine D2 receptor. Prolactin production is inhibited by antagonist activity at this lactotroph receptor.140 It is possible that CAB directly influences insulin sensitivity. Consequently, a reduction in prolactin levels accompanied by an increase in insulin sensitivity and the direct effects of CAB treatment may be reflected in the other metabolic markers,141 following the 2007 New England Journal of Medicine (NEJM) findings that pergolide and cabergoline were linked to valvular disease in men with Parkinson's disease, a number of organizations released guidelines and warnings requiring echocardiography at baseline and then during follow-up in patients receiving cabergoline for pituitary disorders. In 2011, the FDA changed the safety label to recommend routine echocardiography in patients receiving cabergoline at a “recommended frequency every 6 to 12 months or as clinically indicated”.142 N-acyl urea functionality is an uncommon feature of cabergoline, which is attached to the C-8 carboxylic acid of N-allyl dihydro lysergic acid (cabergoline acid). This group is introduced into the synthetic process through the interaction between amide group of 256 and ethyl isocyanate. This procedure offers a straightforward way to prepare cabergoline 257 (ref. 143) (Fig. 58).
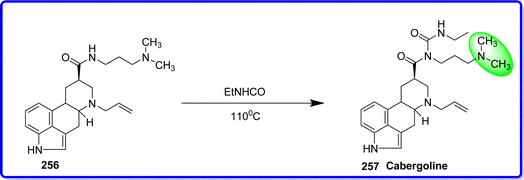 |
| | Fig. 58 Synthesis of cabergoline. | |
8.6 Topotecan hydrochloride
Topotecan is an alkaloid obtained from camptothecin, which is semisynthetic and originally sourced from plants. Topotecan is a 100 kDa monomeric protein that selectively and potently inhibits the nuclear enzyme topoisomerase I. It functions by combining with the DNA/topoisomerase I aggregate to produce a stable covalent complex, or “cleavable complex”, which gives topotecan its cytotoxic characteristics. The skeleton of topotecan contain a lactone ring. Topotecan is soluble in water because it has a stable basic side chain at 9th position of the A ring. The biologically active form of topotecan is lactone, which is hydrolyzed reversibly and, in a pH-dependent manner into the carboxylate form (dihydroxy carboxylic acid) at the E ring.144 In humans, topotecan is effective against a variety of solid tumor malignancies, including small-cell and non-small-cell lung cancer, ovarian cancer, and esophageal cancer.145 In 1996, hycamtin/topotecan, or topotecan hydrochloride, was licensed for the management of patients with ovarian cancer who had not responded to initial or subsequent chemotherapy. The topotecan dose-limiting effect is bone marrow suppression, mainly neutropenia.146 It was synthesized by adding isopropanol, dichloromethane, and 10-hydroxy-camptothecin 258 in an appropriate reactor. In the reactor, N,N-dimethylmethyleneiminium chloride 259 was introduced. After that, the mixture was added to triethylamine and agitated for at least 12 hours at 20 to 35 °C to give compound 260. Following the completion of the reaction, isopropanol and hydrochloric acid were added to the resultant mixture. Following the completion of the addition, the mixture was agitated, filtered, and washed. After drying the moist particles, crude topotecan hydrochloride 261 was obtained147 (Fig. 59).
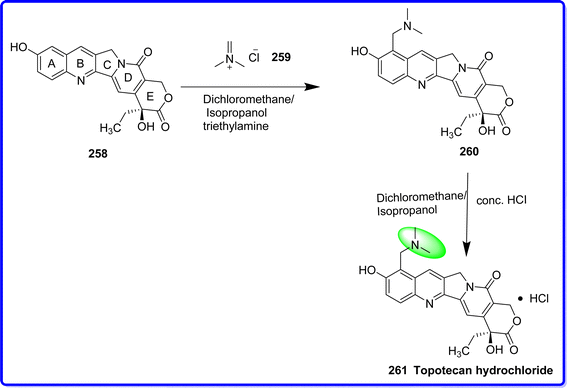 |
| | Fig. 59 Synthesis of topotecan hydrochloride. | |
9. Contraceptive agents
9.1 Mifepristone
Mifepristone, usually referred to as RU486; is a synthetic 19-nor steroid derived from norethindrone. Mifepristone operates as an antagonist to progestational and glucocorticoid activities because it binds strongly to both progesterone and glucocorticoid receptors. Mifepristone functions as a powerful abortifacient, and when combined with prostaglandin, it becomes much more effective.148 The FDA approved the use of mifepristone and misoprostol together on September 28, 2000, and on November 16, 2000, the medication was made available for purchase. This was the first FDA-licensed technique of medical abortion in the United States. Since the late 1980s, mifepristone has been approved for usage in France, Sweden, the UK, and China. Mifepristone was also authorized by the European Drug Agency in 1999 for use in the Netherlands, Austria, Belgium, Denmark, Finland, Greece, and Spain.149 The primary structural feature of mifepristone is the perpendicular grafting of a phenyl-aminomethyl group onto the steroidal structure at position 11β150 (Fig. 60).
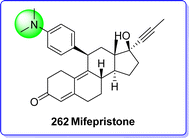 |
| | Fig. 60 Mifeprestone. | |
9.2 Ulipristal acetate
Ulipristal acetate (UPA), also referred to as CDB/VA-2914, is a tissue-specific combined progesterone agonist and antagonist that acts on the myometrial and endometrial tissues. It is a selective progesterone receptor modulator (PRM). Because of the unopposed estrogen effect that this progesterone antagonistic action may provide, the endometrium may develop malignant lesions and hyperplasia.151 The European Medicines Agency (EMA) approved the use of ulipristal for emergency contraception in 2009; the US FDA followed suit in 2010 and Asia. Although there is strong evidence that the primary mechanism of action of UPA is the suppression of ovulation, the precise pharmacological mechanism of action remains unclear.152 Compound 1,3-ethylenedioxy-estra-5(10),9(11)-diene-17-one 263, underwent epoxidation with H2O2, yielding two diastereoisomers: compound 264a and 264b in a ratio of 80/20. Without separation, this mixture was reacted with sodium acetylide in anhydrous THF to produce compound 265a and 265b in a similar ratio. Both compounds react with Grignard reagent in presence of copper catalyst to yield compound 266 at 0 °C. Compound 266 was then treated with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethanol and sulfuric acid solution at 70 °C to produce compound 267. Compound 267 underwent a reaction with phenylsulfenyl chloride in the presence of triethylamine in anhydrous THF at 78 °C, resulting in the formation of compound 268. Compound 268 was then combined with sodium methoxide in methanol and treated with trimethyl phosphate at 70 °C to yield compound 269. Subsequent hydrolysis in the presence of diluted HCl in CH3OH at room temperature led to the formation of compound 270. Finally, a solution of compound 270 in methylene chloride was slowly added to a mixture of acetic acid and perchloric acid at 30 °C to obtain the ulipristal acetate 271 (ref. 153) (Fig. 61).
:1 mixture of ethanol and sulfuric acid solution at 70 °C to produce compound 267. Compound 267 underwent a reaction with phenylsulfenyl chloride in the presence of triethylamine in anhydrous THF at 78 °C, resulting in the formation of compound 268. Compound 268 was then combined with sodium methoxide in methanol and treated with trimethyl phosphate at 70 °C to yield compound 269. Subsequent hydrolysis in the presence of diluted HCl in CH3OH at room temperature led to the formation of compound 270. Finally, a solution of compound 270 in methylene chloride was slowly added to a mixture of acetic acid and perchloric acid at 30 °C to obtain the ulipristal acetate 271 (ref. 153) (Fig. 61).
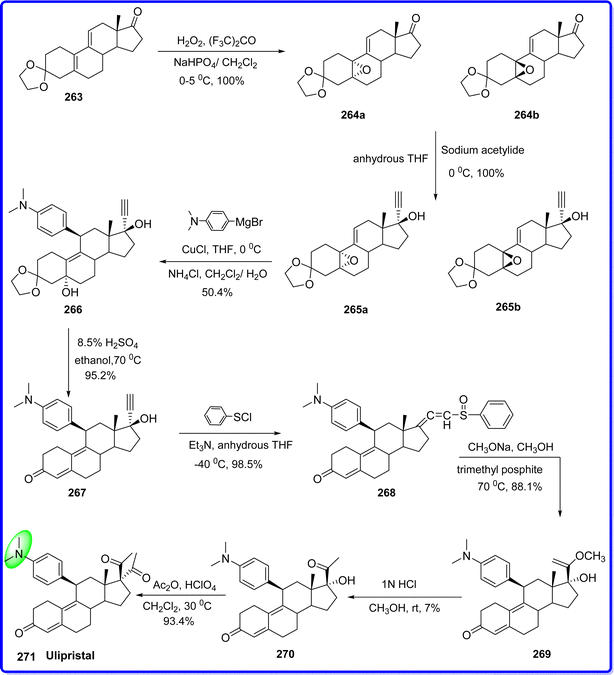 |
| | Fig. 61 Synthesis of ulipristal. | |
10. Muscle relaxant
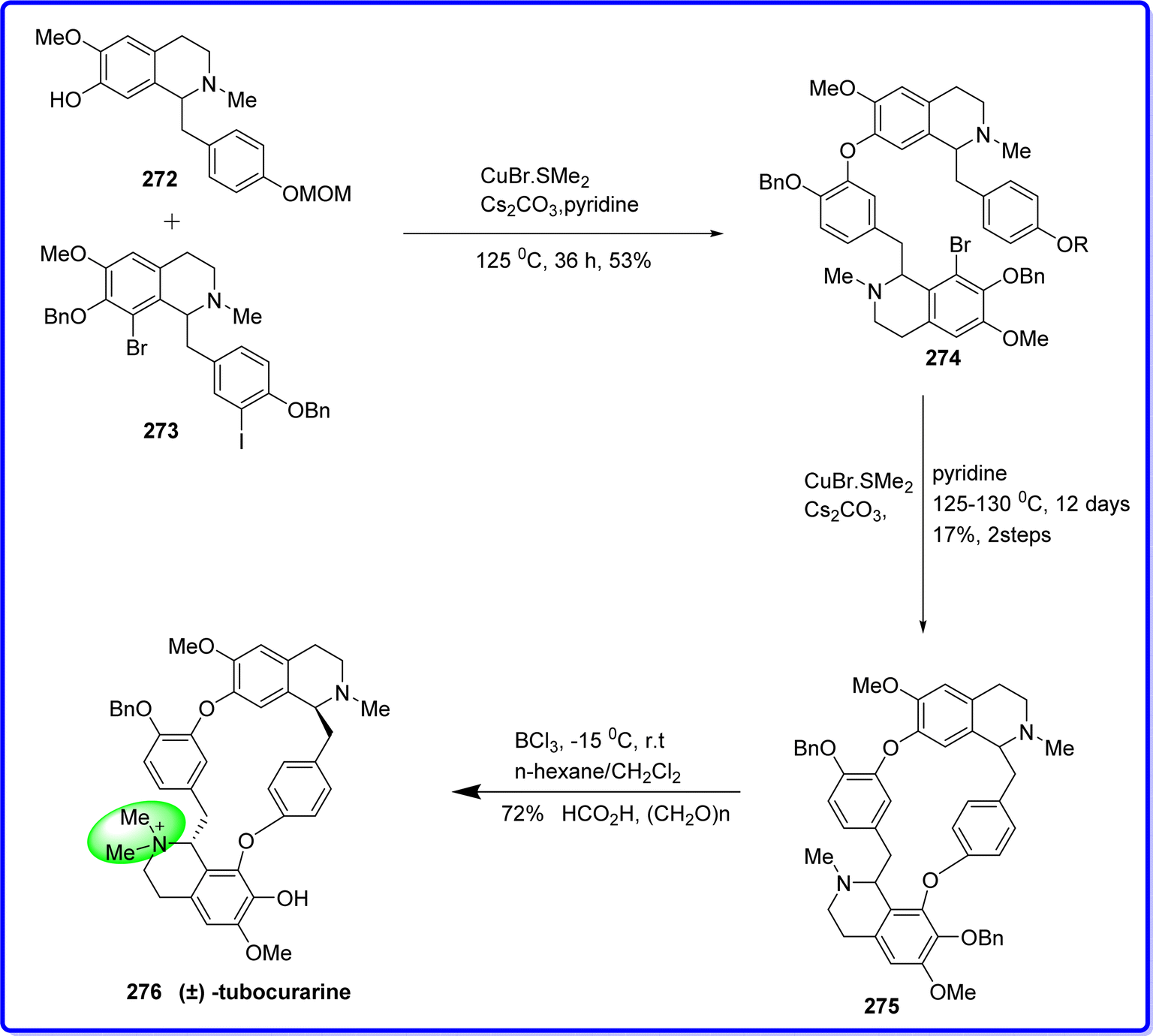
10.1 Tubocurarine chloride
Tubocurarine, a toxic alkaloid historically utilized as an arrow poison, is derived from the chondrodendron tomentosum plant. Initially, it was believed that the dextrorotatory form was effective, but subsequent research revealed that its levorotatory isomer, extracted by Chinese scientists from Cyclea hainansis and Cyclea barbata Miers (Menispermaceae), demonstrated efficacy. Tubocurarine functions by binding to the N2 cholinergic receptor located on the motor nerve endplate, competitively obstructing acetylcholine-mediated depolarization, thereby inducing skeletal muscle relaxation. The medication is primarily employed in abdominal surgery and was previously utilized to treat conditions such as tremor paralysis, tetanus, rabies, and poisoning.154 It is synthesized by reacting methoxymethyl ether (MOM) protecting benzoisoquinoline derivative 272 with benzyltetrahydroisoquinoline 273 in the presence of cesium carbonate and pyridine to form compound 274, which then converts in to compound 275. Compound 275 is then reacted with boron trichloride in the presence of hexane or dichloromethane to yield the final product, tubocurarine 276 (ref. 155) (Fig. 62).
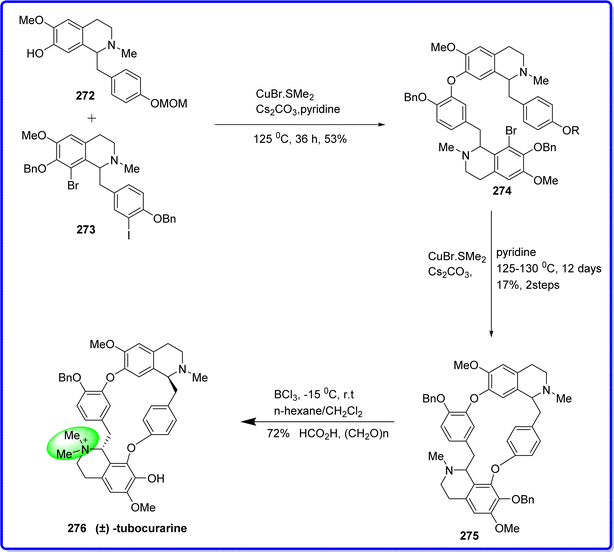 |
| | Fig. 62 Synthesis of tubocurarine. | |
10.2 Neostigmine methylsulfate
Neostigmine is a quaternary nitrogen-containing cholinesterase inhibitor that has less toxicity on cholinesterase inhibition in the brain since it has difficulty crossing the blood–brain barrier. The quaternary nitrogen atom in the neostigmine makes it different than other cholinesterase inhibitors because it direct stimulates cholinergic receptors. Neostigmine works by competing acetylcholine for the binding site on acetylcholinesterase, and this is the mechanism by which it produces its effects. Neostigmine increases the effects of acetylcholine at both nicotinic and muscarinic receptors by obstructing its enzymatic breaking down. On 31 may 2013 Neostigmine methylsulfate was approved by FDA under the brand name Bloxiverz for the treatment reversal of nondepolarizing muscle relaxants. It is primarily used in the management of myasthenia gravis and acute exacerbations of the disease.156 The chemical name of neostigmine methylsulfate is 3-((dimethylcarbamoyl)oxy)-N,N,N-trimethylbenzenaminium methanesulfonate. It is synthesized by reacting 3-dimethylaminophenol 277 with dimethyl carbamic chloride 278 to give dimethyl carbamate 279, which on alkylation by dimethylsulfate gives neostigmine methylsulfate 280 (ref. 157) (Fig. 63).
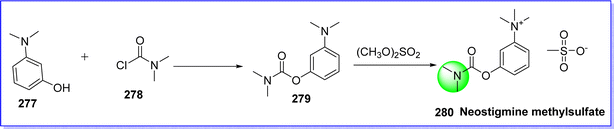 |
| | Fig. 63 Synthesis of neostigmine methylsulfate. | |
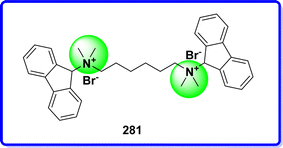
10.3 Hexafluorenium bromide
Hexafluorenium (HFL) is a bis-quaternary ammonium compound that has anticholinesterase activity. It is used as a muscle relaxant. Although HFL was less successful in inhibiting real cholinesterase, it was a strong inhibitor of plasma pseudocholinesterase.158 A fortuitous discovery occurred when it was observed that administering a small amount of succinylcholine after hexafluorenium significantly extended the duration of succinylcholine's effects159 (Fig. 64).
 |
| | Fig. 64 Structure of hexafluorenium bromide. | |
11. Analgesics
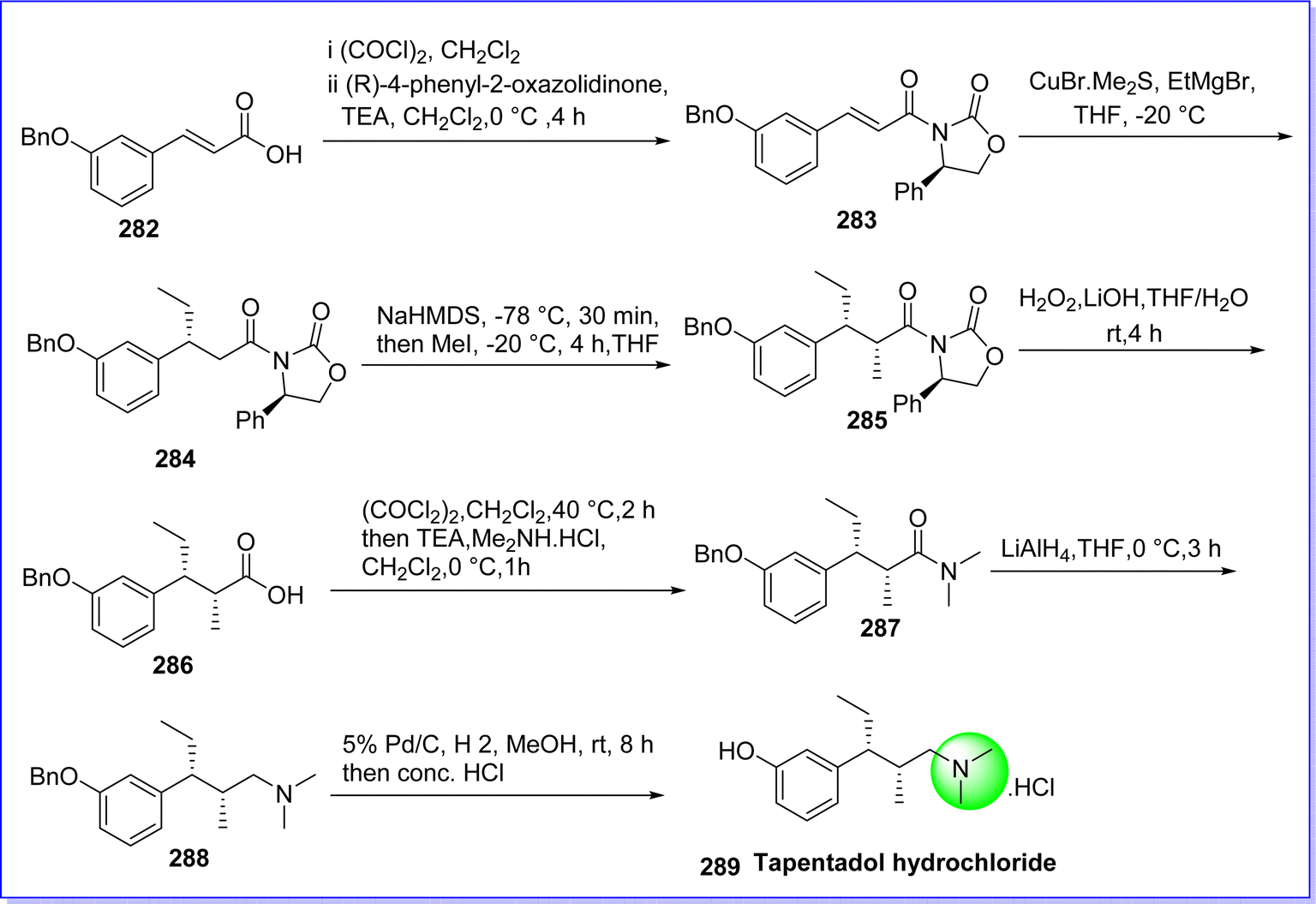
11.1 Tapentadol hydrochloride
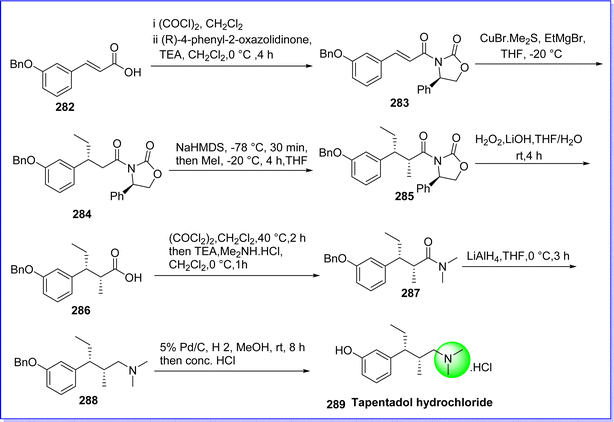
Tapentadol, a centrally acting analgesic, operates through dual mechanisms: firstly, by acting as an agonist on opioid receptors, and secondly, by inhibiting the reuptake of noradrenaline. Recent reports suggest that tapentadol exhibits analgesic effects in patients experiencing moderate to severe acute inflammatory pain and chronic neuropathy.160 On 20 November 2008 tapentadol was approved by FDA under the brand name Nucynta. The chemical name of tapentadol is 3-((2S,3R)-1-(dimethylamino)-2-methylpentan-3-yl)phenol. It was synthesized by reacting (E)-3-(3-(benzyloxy)phenyl)acrylic acid 282 and (R)-4-phenyloxazolidin-2-one as a chiral auxiliary and acylated by 3-benzyloxy cinnamoyl chloride in CH2Cl2 in the presence of triethylamine (TEA) to give compound 283. The compound 283 was reacted to organocuprate, prepared from CuBr·Me2S complex and ethyl magnesium bromide in tetrahydrofuran (THF) at −20 °C to give compound 284. The compound 284 reacts with NaHMDS to form enolate at −78 °C which on alkylation by CH3I gives compound 285. The compound 286 α,β-disubstituted acid was obtained by removing the chiral auxiliary under base hydrolysis conditions (LiOH/H2O2 in H2O/THF). The compound 287 was produced by treating compound 286 with oxalyl chloride, followed by dimethylamine hydrochloride in the presence of TEA in CH2Cl2. The reduction of compound 287 with LiAlH4 in THF gives (benzyl-tapentadol) compound 288. The compound 288 on debenzylation under a hydrogen atmosphere in presence of Pd/C as catalyst gives tapentadol hydrochloride 289 (ref. 161) (Fig. 65).
 |
| | Fig. 65 Synthesis of tapentadol hydrochloride. | |
11.2 Tramadol hydrochloride
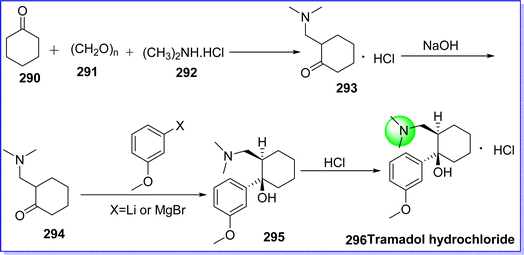
Tramadol is an atypical synthetic analgesic that acts centrally. It works by two mechanisms, first is inhibition of the reuptake of serotonin and norepinephrine and second, it acts as an agonist of the μ-opioid receptor. It is used to treat mild to severe pain.162 In March 1995 It was approved by FDA. it is sold under the brand name Ultram. The chemical name of tramadol is (1R,2R)-2-((dimethylamino)methyl)-1-(3-methoxyphenyl)cyclohexan-1-ol. It is synthesized by reacting cyclohexanone 290, paraformaldehyde 291 and dimethylamine hydrochloride 292 (Mannich reaction) to give 2-dimethylaminomethyl-cyclohexanone hydrochloride 293, which reacts with NaOH to give intermediate 294. On reacting intermediate 294 with m-methoxyphenyl magnesium bromide or m-methoxyphenyl lithium, tramadol base 295 was obtained, which reacted with HCl to form tramadol hydrochloride 296 (ref. 163) (Fig. 66).
 |
| | Fig. 66 Synthesis of tramadol hydrochloride. | |
12. Antidiarrheal agents
12.1 Loperamide hydrochloride
Loperamide hydrochloride is strongest active ingredient in pharmaceuticals known to cure diarrhea. Chemically it is known as 4-(4-(4-chlorophenyl)-4-hydroxypiperidin-1-yl)-N,N-dimethyl-2,2-diphenylbutanamide hydrochloride. It is marketed as an over-the-counter medication under the Imodium brand. Although the substance is an opioid, it does not exhibit the analgesic, miotic, or respiratory depression side effects associated with opioids.164 Since its 1977 introduction, loperamide has been categorized as an opioid and subject to abuse; as a result, it is listed in schedule V of the US Controlled Substances Act. The medicine was available without a prescription in 1982, and later volunteer tests showed a low risk of physical dependence and abuse.165 It is synthesized by reacting α-(2-bromoethyl)-α-phenylbenzeneacetonitrile 297 with acetic acid to form compound 298, which react with hydrogen bromide to produce 299. In presence of SO2Cl2 299 converted to compound 300. The condensation of compound 300 with 4-(4-chlorophenyl)-4-piperidinol 303 form loperamide hydrochloride 304. The compound 303 was obtained by reacting ethyl 4-oxo-1-piperidinecarboxylate 301with para dichlorobenzene compound 302 (ref. 166) (Fig. 67).
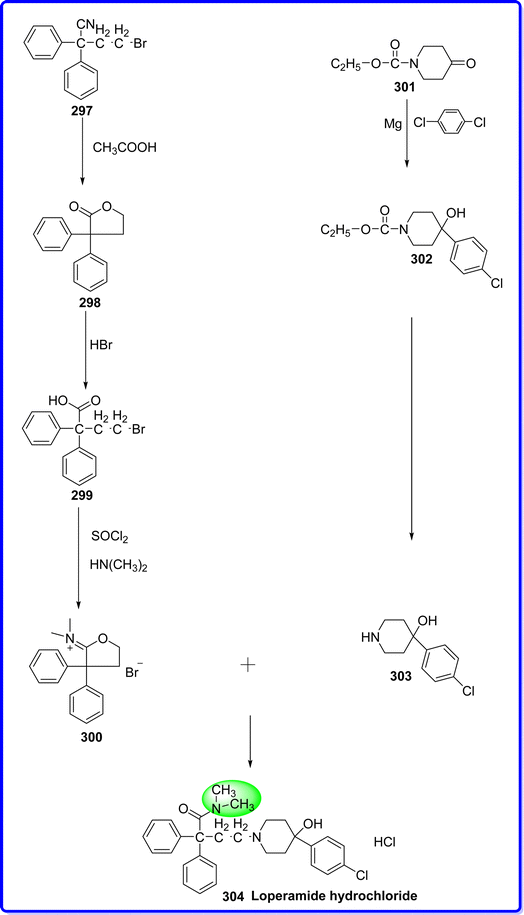 |
| | Fig. 67 Synthesis of loperamide hydrochloride. | |
13. Drug used in the treatment of cardiovascular disease
13.1 Diltiazem hydrochloride
Diltiazem hydrochloride is a member of the benzothiazepine chemical class. It was initially created in the Japanese laboratories of Tanabe Seiyaku Co., Ltd, and its first patent was issued in 1969. Diltiazem was first mentioned in non-patent literature in 1970.167 The cis form of a salt derivative of benzothiazepine hydrochloride is called diltiazem. Over the last two decades, these medications have been created and are currently used in treatment of various cardiovascular disease. These include coronary spasm, angina, hypertension, hypertrophic cardiomyopathy, and both supraventricular and ventricular arrhythmias. The compound is known as (2S,3S)-5-(2-(dimethylamino)ethyl)-2-(4-methoxyphenyl)-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]thiazepin-3-yl acetate hydrochloride.168 Slow calcium channels are blocked by diltiazem in a concentration-dependent way. Even though the medication may result in atrioventricular nodal conduction delay and sinus node depression. When administered intravenously, diltiazem is a mildly negative inotrope with strong vasodilatory effects.169 The methyl ester of 3-(4-methoxyphenyl)-glycidylic acid 307 is produced by condensation of 4-methoxybenzaldehyde 305 with methylchoroacetate 306 in the presence of sodium methoxide under Darzens reaction conditions. It opens the epoxide ring when it reacts with 2-aminothiophenol, yielding compound 308. When the resultant product 308 is hydrolyzed with an alkali, a racemic mixture containing the corresponding acid 309 is formed. When 309 is boiled in a solution of acetic anhydride, dimethylformamide, and pyridine, the thiazepine ring cycles and the hydroxyl group is concurrently acylated, resulting in the formation of compound 310. Diltiazem 311 is produced by alkylating the compound 310 using 2,2-dimethylaminoethyl chloride169 (Fig. 68).
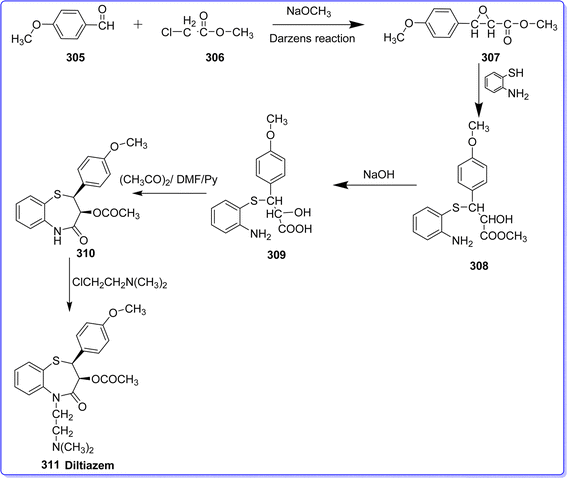 |
| | Fig. 68 Synthesis of diltiazem. | |
14. Anti-obesity drugs
14.1 Sibutramine
Sibutramine, also known as Meridia, has sympathomimetic effects by centrally inhibiting the neuronal uptake of norepinephrine and serotonin and activating peripheral β3-adrenergic receptors to promote satiety. Nevertheless, there are noticeable elevations in heart rate and systolic and diastolic blood pressure. Sibutramine is a SNRI and used in the treatment of obesity.170 In November 1997, it was approved by FDA for management of obesity. It is sold under the brand name Meridia and Reductil.171 The chemical name of sibutramine is 1-(1-(4-chlorophenyl)cyclobutyl)-N,N,3-trimethylbutan-1-amine. It is synthesized by reacting 4-chlorophenyl acetonitrile 312 with 1, 3-dibromopropane 313 to produce cyclobutane derivative 314. The reaction of compound 314 with Grignard's reagent 315, prepared from isobutyl bromide form give Imine derivatives 316, which on reduction with LiAlH4 give amine derivatives 317. Bis-N-methylation of compound 317 in the presence of formic acid/formaldehyde (Clark–Eshweiler reaction) produces sibutramine 318 (ref. 172) (Fig. 69).
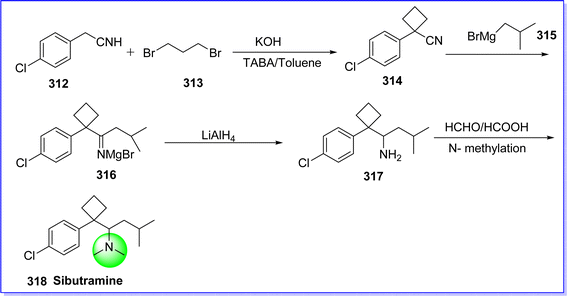 |
| | Fig. 69 Synthesis of sibutramine. | |
15. Miscellaneous
15.1 Methylene blue
Methylene blue is a synthetic phenothiazine derivative dye. It is used in treatment of malarial,173 methemoglobinemia174 and potassium cyanide poisoning.175 The chemical name of methylene blue is 3,7-bis(dimethylamino)phenothiazin-5-ium. It is synthesized by electro-organic method. In this method, in a divided cell with carbon rods serving as the anode and Pt-cathode, 70 ml of hydrochloric acid (0.1 M) was pre-electrolyzed at 0.6 V. After that, 1 mmol of sodium sulphide and 1 mmol of N,N-dimethyl-p-phenylenediamine were added to the cell. When the rate of current degradation exceeded 95%, the electrolysis was stopped. After electrolysis, the cell was refrigerated for the entire night. Filtration was used to collect the solid precipitate. Following filtration, column chromatography was used to obtained pure methylene blue 326 (ref. 176) (Fig. 70).
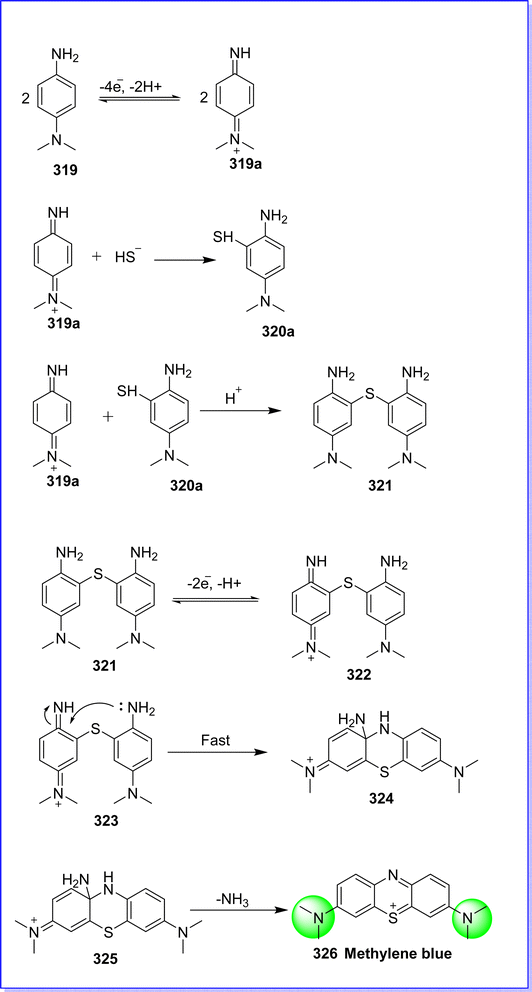 |
| | Fig. 70 Synthesis of methylene blue. | |
16. Conclusion
DMA is an organic compound with the molecular formula NH(CH3)2. Chemically, DMA is a secondary amine, characterized by two methyl (–CH3) groups directly attached to a nitrogen atom. It is a colorless gas that emits a fish-like odor at lower concentrations and an ammonia-like smell at higher concentrations. DMA plays a vital role in the pharmaceutical industry, particularly as a component of several FDA-approved drugs. Its unique chemical properties enable the synthesis of active pharmaceutical ingredients (APIs) that are crucial for treating a wide range of diseases. In this article, we summarized the DMA-containing drugs approved by FDA over last 50 years. We documented the synthesis of these drug molecules and their clinical application in various therapeutic areas, including antibacterial, antimuscarinic, antidiarrheal, anti-obesity, anticancer, analgesic, muscle relaxant, local anaesthetic, and contraceptive drugs. We also highlighted DMA-containing medications that are specifically used to treat cardiovascular, neurological, and peptic ulcer conditions. The synthesis of drugs containing DMA faces a major challenge due to the potential formation of NDMA, a known carcinogen. NDMA can form during both the synthesis and storage of these drugs, especially when exposed to nitrites, acidic conditions, and high temperatures. This issue came to prominence in 2019 when ranitidine, a popular antacid, was withdrawn from the market due to detected NDMA and its potential cancer risk. To prevent NDMA formation from DMA, it is crucial to control factors that promote its synthesis, such as nitrites, reaction conditions, and storage conditions. Additionally, incorporating antioxidants like ascorbic acid and alpha-tocopherol can inhibit the nitrosation reaction. This article provides a comprehensive overview that can aid researchers in development of innovative DMA-based drugs, ensuring continued progress in the field of pharmaceuticals. There are various methods to synthesize DMA-containing drugs, such as the Eschweiler–Clarke reaction and the substitution reaction of alkylamine with methyl halide. However, these reactions have several drawbacks, including the use of hazardous chemicals, high waste production, and often poor selectivity. To address these issues, transition metal-catalyzed N-methylation of amines with methyl sources such as formic acid, carbon dioxide, and paraformaldehyde has been employed. However, this approach also has limitations, including low selectivity, the need of high temperatures and pressures, and solvent wastage. Therefore, researchers should focus on atom-efficient methods for synthesizing DMA-containing drugs by using alcohol and amine to create the DMA moiety, as discussed in the introduction.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts of interest to declare.
References
- A. B. van Gysel and W. Musin, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- D. R. Corbin, S. Schwarz and G. C. Sonnichsen, Catal. Today, 1997, 37, 71–102 CrossRef CAS.
- G. B. Neurath, M. Dünger, F. G. Pein, D. Ambrosius and O. Schreiber, Food Cosmet. Toxicol., 1977, 15, 275–282 CrossRef CAS.
- J. Schirmann and P. Bourdauducq, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2001 Search PubMed.
- R. J. Polinsky, Clin. Ther., 1998, 20, 634–647 CrossRef CAS PubMed.
- K. J. Klamerus, K. Moloney, R. L. Rudolph, S. F. Sisenwine, W. J. Jusko and S. T. Chiang, J. Clin. Pharmacol., 1992, 32, 716–724 CrossRef CAS PubMed.
- R. G. Van Der Hoop, M. E. L. Van Der Burg, W. W. B. Ten Huinink, J. C. Van Houwelingen and J. P. Neijt, Cancer, 1990, 66, 1697–1702 CrossRef CAS.
- K. Venugopal, M. Reddy, M. Bharathraj, K. Jaligidad and D. Kushal, Toxicol. Int., 2014, 21, 319 CrossRef CAS PubMed.
- D. M. Paton and D. R. Webster, Clin. Pharmacokinet., 1985, 10, 477–497 CrossRef CAS.
- J. Schneider, U. Jahnel and K. Linz, Drug Dev. Res., 2010, 71, 197–208 CrossRef CAS.
- N. Bunya, K. Sawamoto, S. Uemura, R. Kyan, H. Inoue, J. Nishida, H. Kouzu, N. Kokubu, T. Miura and E. Narimatsu, Acute Med. Surg., 2017, 4, 334–337 CrossRef PubMed.
- R. H. Schirmer, B. Coulibaly, A. Stich, M. Scheiwein, H. Merkle, J. Eubel, K. Becker, H. Becher, O. Müller, T. Zich, W. Schiek and B. Kouyaté, Redox Rep., 2003, 8, 272–275 CrossRef CAS PubMed.
- J. V. Lovasik, Optom. Vis. Sci., 1986, 63, 787–803 CrossRef CAS PubMed.
- W. Eschweiler, Ber. Dtsch. Chem. Ges., 1905, 38, 880–882 CrossRef.
- C. Chiappe, P. Piccioli and D. Pieraccini, Green Chem., 2006, 8, 277–281 RSC.
- M. Prashad, D. Har, B. Hu, H.-Y. Kim, O. Repic and T. J. Blacklock, Org. Lett., 2003, 5, 125–128 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 7956–7966 RSC.
- L. Zhu, L.-S. Wang, B. Li, W. Li and B. Fu, Catal. Sci. Technol., 2016, 6, 6172–6176 RSC.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- H. Wang, Y. Huang, X. Dai and F. Shi, Chem. Commun., 2017, 53, 5542–5545 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- G. Choi and S. H. Hong, ACS Sustain. Chem. Eng., 2019, 7, 716–723 CrossRef CAS.
- M. A. R. Jamil, A. S. Touchy, Md. N. Rashed, K. W. Ting, S. M. A. H. Siddiki, T. Toyao, Z. Maeno and K. Shimizu, J. Catal., 2019, 371, 47–56 CrossRef CAS.
- R. J. Polinsky, Clin. Ther., 1998, 20, 634–647 CrossRef CAS PubMed.
- B. Winblad, G. Grossberg, L. Frölich, M. Farlow, S. Zechner, J. Nagel and R. Lane, Neurology, 2007, 69(4), 14–22, DOI:10.1212/01.wnl.0000281847.17519.e0.
- F. Inglis, Int. J. Clin. Pract., Suppl., 2002, 45–63 CAS.
- M. Hu, F.-L. Zhang and M.-H. Xie, Synth. Commun., 2009, 39, 1527–1533 CrossRef CAS.
- K. J. Klamerus, K. Moloney, R. L. Rudolph, S. F. Sisenwine, W. J. Jusko and S. T. Chiang, J. Clin. Pharmacol., 1992, 32, 716–724 CrossRef CAS PubMed.
- S. Yang, S. Chen, C. Wang, S. Zhang, S. Li, X. Yuan, F. Peng and Y. He, Front. Chem., 2022, 10, 860292, DOI:10.3389/fchem.2022.860292.
- V. Hadjimitova, T. Traykov, R. Bakalova, V. Petrova, I. Lambev, H. Ohba, M. Ishikawa and Y. Baba, Luminescence, 2004, 19, 319–321 CrossRef CAS PubMed.
- J. Kim and J.-H. Song, Eur. J. Pharmacol., 2016, 779, 31–37 CrossRef CAS PubMed.
- M. Højlund, C. B. Wagner, R. Wesselhoeft, K. Andersen, A. Fink-Jensen and J. Hallas, Basic Clin. Pharmacol. Toxicol., 2022, 130, 501–512 CrossRef.
- R. S. Vardanyan and V. J. Hruby, in Synthesis of Essential Drugs, Elsevier, 2006, pp. 83–101 Search PubMed.
- Y. Kumar, M. Prasad and A. Nath, Process for the synthesis of zolpidem field of the invention, WO2005010002A1, 2005.
- M. Berger and M. Gastpar, Eur. Arch. Psychiatry Clin. Neurosci., 1996, 246, 235–239 CrossRef CAS.
- Pharmacology in Anesthesia Practice, ed. A. Gupta and N. Singh-Radcliff, Oxford University Press, 2013, pp. 349–352 Search PubMed.
- R. El-Mallakh, L. J. Findlay, P. L. El-Mallakh and R. J. Roberts, Neuropsychiatr. Dis. Treat., 2016, 12, 1837–1842 CrossRef PubMed.
- P. K. Neela, G. Batthini, S. K. Sankineni, H. R. Bingi, R. R. Maddi and S. Meenakshisunderam, A Process for the Preparation of Cariprazine Hydrochloride Foreign Application Priority Data, US20200375983A1, 2017.
- F. Cavaliere, A. Fornarelli, F. Bertan, R. Russo, A. Marsal-Cots, L. A. Morrone, A. Adornetto, M. T. Corasaniti, D. Bano, G. Bagetta and P. Nicotera, Sci. Rep., 2019, 9, 4881 CrossRef PubMed.
- Obsessive-compulsive disorder, in Neurobiology of psychiatric disorders, ed. T. E. Schlaepfer and C. B. Nemeroff, Elsivier Science Publishers B. V., 2012, pp. 375–390 Search PubMed.
- A. Kanwal, U. Afzal, M. Zubair, M. Imran and N. Rasool, RSC Adv., 2024, 14, 6948–6971 RSC.
- P. Seetharama Sarma, C. Nageswar Rao, M. V. Surayanarayana, P. Pratap Reddy, M. Khalilluah and C. Praveen, Synth. Commun., 2008, 38, 603–612 CrossRef.
- C. Pramanik, R. Bhumkar, G. Karhade, P. Khairnar, N. K. Tripathy and M. K. Gurjar, Org. Process Res. Dev., 2012, 16, 507–511 CrossRef CAS.
- N. L. Agarwal, P. P. Mistri and N. M. Patel, An Improved Process for the Preparation of Levomepromazine Maleate, EP2743263A1, 2014.
- A. T. Harvey, R. L. Rudolph and S. H. Preskorn, Arch. Gen. Psychiatry, 2000, 57, 503 CrossRef CAS PubMed.
- J. Kornhuber, C. G. Parsons, S. Hartmann, W. Retz, S. Kamolz, J. Thome and P. Riederer, J. Neural Transm., 1995, 102, 237–246 CrossRef CAS PubMed.
- D. Sälinger and R. Brückner, Synfacts, 2009, 2009, 1309 CrossRef.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- D. C. Hobbs, J. Pharm. Sci., 1968, 57, 105–111 CrossRef CAS PubMed.
- J. F. Muren and B. M. Bloom, J. Med. Chem., 1970, 13, 17–23 CrossRef CAS PubMed.
- H. F. Garrison, J. Am. Med. Assoc., 1962, 179, 456 CrossRef PubMed.
- R. Kuhn, Schweiz. Med. Wochenschr., 1957, 87, 1135–1140 CAS.
- D. N. Kender and R. E. Schiesswohl, Anal. Profiles Drug Subst. 14, 1985, pp. 37–75 Search PubMed.
- E. M. Glaser and P. S. B. Newling, Br. J. Pharmacol. Chemother., 1955, 10, 429–433 CrossRef CAS PubMed.
- A. P. Feinberg and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1899–1903 CrossRef CAS PubMed.
- R. Vedantham, V. P. R. Vetukuri, A. Boini, M. Khagga and R. Bandichhor, Org. Process Res. Dev., 2013, 17, 798–805 CrossRef CAS.
- N. Fajkis, M. Marcinkowska, B. Gryzło, A. Krupa and M. Kolaczkowski, Molecules, 2020, 25, 3161 CrossRef CAS PubMed.
- P. Fangmann, H.-J. Assion, G. Juckel, C. Á. González and F. López-Muñoz, J. Clin. Psychopharmacol., 2008, 28, 1–4 CrossRef PubMed.
- M.-S. Brueckle, E. T. Thomas, S. E. Seide, M. Pilz, A. I. Gonzalez-Gonzalez, T. S. Dinh, F. M. Gerlach, S. Harder, P. P. Glasziou and C. Muth, PLoS One, 2023, 18, e0284168 CrossRef CAS PubMed.
- E. W. McClure and R. N. Daniels, ACS Chem. Neurosci., 2021, 12, 354–362 CrossRef CAS PubMed.
- J. Ravn, Nord. Psykiatr. Tidsskr., 1963, 17, 69–82 CrossRef.
- M. Højlund, J. H. Andersen, K. Andersen, C. U. Correll and J. Hallas, Epidemiol. Psychiatr. Sci., 2021, 30, e28 CrossRef PubMed.
- M. Højlund, C. B. Wagner, R. Wesselhoeft, K. Andersen, A. Fink-Jensen and J. Hallas, Basic Clin. Pharmacol. Toxicol., 2022, 130, 501–512 CrossRef PubMed.
- M. Højlund, A. Pottegård, E. Johnsen, R. A. Kroken, J. Reutfors, P. Munk-Jørgensen and C. U. Correll, Br. J. Clin. Pharmacol., 2019, 85, 1598–1606 CrossRef PubMed.
- K. Venugopal, M. Reddy, M. Bharathraj, K. Jaligidad and D. Kushal, Toxicol. Int., 2014, 21, 319 CrossRef CAS PubMed.
- T. L. Lemke, D. A. Williams, V. F. Roche and S. W. Zito, Foye's Principles of Medicinal Chemistry, 7th edn, 2013 Search PubMed.
- P. Kintz, F. Berthault, A. Tracqui and P. Mangin, J. Anal. Toxicol., 1995, 19, 591–594 CrossRef CAS PubMed.
- D. Wingate, Gut, 1989, 30, 1804 CrossRef.
- E. Rosen and J. V Swintosky, J. Pharm. Pharmacol., 2011, 12, 237T–244T CrossRef PubMed.
- D. M. Paton and D. R. Webster, Clin. Pharmacokinet., 1985, 10, 477–497 CrossRef CAS PubMed.
- S. A. A. Rizvi, G. Ferrer, U. A. Khawaja and M. A. Sanchez-Gonzalez, Curr. Rev. Clin. Exp. Pharmacol., 2024, 19, 137–145 CrossRef CAS PubMed.
- M. Raad, K. F. Alsamarrai and Y. K. Al-Bayati, J. Soc. Pure Appl. Nat. Sci., 2017, 30, 44 Search PubMed.
- D. Wisher, J. Med. Libr. Assoc., 2012, 100, 75–76 CrossRef.
- R. S. Vardanyan and V. J. Hruby, in Synthesis of Essential Drugs, Elsevier, 2006, pp. 219–235 Search PubMed.
- E. J. Scharman, A. R. Erdman, P. M. Wax, P. A. Chyka, E. Martin Caravati, L. S. Nelson, A. S. Manoguerra, G. Christianson, K. R. Olson, A. D. Woolf, D. C. Keyes, L. L. Booze and W. G. Troutman, Clin. Toxicol., 2006, 44, 205–223 CrossRef CAS PubMed.
- S. M. Miller and K. L. Cumpston, in Encyclopedia of Toxicology, Elsevier, 2014, pp. 195–197 Search PubMed.
- S. Y. Yeh, G. D. Todd, R. E. Johnson, C. W. Gorodetzky and W. R. Lange, Clin. Pharmacol. Ther., 1986, 39, 669–676 CrossRef CAS PubMed.
- H. G. Piskorik, Anal. Profiles Drug Subst. 14, 1985, pp. 107–133 Search PubMed.
- H. S. Alyami, D. K. Ali, Q. Jarrar, A. Jaradat, H. Aburass, A. A. Mohammed, M. H. Alyami, A. H. Aodah and E. Z. Dahmash, Molecules, 2023, 28, 748 CrossRef CAS PubMed.
- C. A. Whitmore, M. I. Boules, W. J. Behof, J. R. Haynes, D. Koktysh, A. J. Rosenberg, M. N. Tantawy and W. Pham, Molecules, 2021, 26, 2182 CrossRef CAS PubMed.
- K. Nishimura and M. Kinugawa, Org. Process Res. Dev., 2012, 16, 225–231 CrossRef CAS.
- W. Xu, S. Xia, J. Pu, Q. Wang, P. Li, L. Lu and S. Jiang, Front. Microbiol., 2018, 9, 2643, DOI:10.3389/fmicb.2018.02643.
- X.-C. Cui, H. Zhang, Y.-P. Wang, J.-P. Qu and Y.-B. Kang, Chem. Sci., 2022, 13, 11246–11251 RSC.
- F. Kalpaklioglu and A. Baccioglu, Anti-Allergy Agents Med. Chem., 2012, 11, 230–237 CrossRef CAS PubMed.
- A. Ahmadi, M. Khalili, R. Hajikhani, N. Safari and B. Nahri-Niknafs, Med. Chem. Res., 2012, 21, 3532–3540 CrossRef CAS.
- H. A. Al-Haboubi and I. J. Zeitlin, Eur. J. Pharmacol., 1982, 78, 175–185 CrossRef CAS PubMed.
- W. E. Thornton, J. Am. Coll. Emerg. Physicians, 1977, 6, 408–412 CrossRef CAS.
- R. L. Mayer, J. Allergy, 1946, 17, 153–165 CrossRef CAS PubMed.
- M. C. Roberts, Clin. Infect. Dis., 2003, 36, 462–467 CrossRef CAS PubMed.
- M. L. Nelson and S. B. Levy, Ann. N. Y. Acad. Sci., 2011, 1241, 17–32 CrossRef CAS PubMed.
- R. Ramachanderan and B. Schaefer, ChemTexts, 2021, 7, 18 CrossRef CAS.
- L. A. Meijer, K. G. F. Ceyssens, B. I. J. A. C. de Grève and W. de Bruijn, Vet. Q., 1993, 15, 1–5 CrossRef CAS PubMed.
- T. Phaechamud and J. Charoenteeraboon, AAPS PharmSciTech, 2008, 9, 829–835 CrossRef CAS PubMed.
- M. R. Khan, M. A. Khan, K. Ahmad, A. Hamad, M. Sajid-ur-Rehman, H. M. Asif, M. Younus and Y. Kamal, RADS J. Pharm. Pharm. Sci., 2020, 7, 215–226 CrossRef.
- R. S. Vardanyan and V. J. Hruby, Synthesis of Essential Drugs, Elsevier, 2006 Search PubMed.
- G. Schwartzman, L. Wayland, T. Alexander, K. Furnkranz and G. Selzer, Anal. Profiles Drug Subst. 8, 1979, pp. 101–137 Search PubMed.
- M. J. Gribble and A. W. Chow, Med. Clin. North Am., 1982, 66, 79–89 CrossRef CAS PubMed.
- C. W. Derrick and K. M. Reilly, Pediatr. Clin. North Am., 1983, 30, 63–69 CrossRef PubMed.
- M. J. Parnham, V. E. Haber, E. J. Giamarellos-Bourboulis, G. Perletti, G. M. Verleden and R. Vos, Pharmacol. Ther., 2014, 143, 225–245 CrossRef CAS PubMed.
- D. H. Peters, H. A. Friedel and D. McTavish, Drugs, 1992, 44, 750–799 CrossRef CAS PubMed.
- J. M. Zuckerman, Infect. Dis. Clin. N. Am., 2004, 18, 621–649 CrossRef PubMed.
- A. H. H. Bakheit, B. M. H. Al-Hadiya and A. A. Abd-Elgalil, Profiles Drug Subst. Excip. Relat. Methodol. 39, 2014, pp. 1–40 Search PubMed.
- D. H. Peters and S. P. Clissold, Drugs, 1992, 44, 117–164 CrossRef CAS PubMed.
- H. F. Anwar, M. Andrei and K. Undheim, Bioorg. Med. Chem., 2017, 25, 2313–2326 CrossRef CAS PubMed.
- J. A. Barman Balfour and D. P. Figgitt, Drugs, 2001, 61, 815–829 CrossRef PubMed.
- K. Wellington and S. Noble, Drugs, 2004, 64, 1683–1694 CrossRef CAS PubMed.
- K. M. Speirs and M. J. Zervos, Expert Rev. Anti-Infect. Ther., 2004, 2, 685–693 CrossRef CAS PubMed.
- E. Scholar, in xPharm: the Comprehensive Pharmacology Reference, Elsevier, 2009, pp. 1–8 Search PubMed.
- S. M. Wintermeyer, S. M. Abdel-Rahman and M. C. Nahata, Ann. Pharmacother., 1996, 30, 1141–1149 CrossRef CAS PubMed.
- I. Shinkai and Y. Ohta, Bioorg. Med. Chem., 1996, 4, 521–522 CrossRef CAS PubMed.
- F. T. Counter, P. W. Ensminger, D. A. Preston, C. Y. Wu, J. M. Greene, A. M. Felty-Duckworth, J. W. Paschal and H. A. Kirst, Antimicrob. Agents Chemother., 1991, 35, 1116–1126 CrossRef CAS PubMed.
- R. N. Brogden, A. A. Carmine, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1982, 24, 267–303 CrossRef CAS PubMed.
- M. Hohnjec, J. Kuftinec, M. Malnar, M. Škreblin, F. Kajfež, A. Nagl and N. Blažević, Anal. Profiles Drug Subst. 15, 1986, pp. 533–561 Search PubMed.
- R. S. Vardanyan and V. J. Hruby, Synthesis of Essential Drugs, Elsevier, 2006 Search PubMed.
- D. Zanon, C. Tumminelli, A. M. C. Galimberti, L. Torelli, A. Maestro, E. Barbi and N. Maximova, Ital. J. Pediatr., 2021, 47, 222 CrossRef CAS PubMed.
- F. Ji, F. Huang, A. Juhasz, W. Wu and N. Bodor, ChemInform, 2010, 55(3), 187–191, DOI:10.1002/chin.200031123.
- K.-I. Tanaka, T. Ishihara, T. Sugizaki, D. Kobayashi, Y. Yamashita, K. Tahara, N. Yamakawa, K. Iijima, K. Mogushi, H. Tanaka, K. Sato, H. Suzuki and T. Mizushima, Nat. Commun., 2013, 4, 2686 CrossRef PubMed.
- Z. Clarke, in xPharm: the Comprehensive Pharmacology Reference, Elsevier, 2007, pp. 1–3 Search PubMed.
- M. İ. Han and N. İmamoğlu, ACS Omega, 2023, 8, 9198–9211 CrossRef CAS PubMed.
- W. Li, J. Zhao and Y. Cui, IOP Conf. Ser. Earth Environ. Sci., 2017, 64, 012108 CrossRef.
- D.-P. Hydrochloride, J. Am. Chem. Soc., 1953, 75, 3400–3401 CrossRef.
- J. V. Lovasik, Optom. Vis. Sci., 1986, 63, 787–803 CrossRef CAS PubMed.
- J. K. Aronson, Meyler's Side Effects of Drugs, Elsevier, 2016, pp. 783–784 Search PubMed.
- L. Sobral, M. Temtem, R. Antunes and B. Nunes, US20150018556A1, 2015.
- L. A. Hansen and T. E. Hughes, Drug Intell. Clin. Pharm., 1991, 25, 146–152 CAS.
- C. R. Lee and D. Faulds, Drugs, 1995, 49, 932–953 CrossRef CAS PubMed.
- G. Damia and M. D'Incalci, Clin. Pharmacokinet., 1995, 28, 439–448 CrossRef CAS PubMed.
- V. B. Duong, P. Van Hien, T. T. Ngoc, P. D. Chau and T. K. Vu, Lett. Org. Chem., 2020, 17, 628–630 CrossRef CAS.
- V. Hug, G. N. Hortobagyi, B. Drewinko and M. Finders, J. Clin. Oncol., 1985, 3, 1672–1677 CrossRef CAS PubMed.
- Y. Zhang, B. Li, Z. Li, N. xia, H. Yu and Y. Zhang, J. Photochem. Photobiol., B, 2018, 180, 68–71 CrossRef CAS PubMed.
- X. Li, Z. Li, L. Li, T. Liu, C. Qian, Y. Ren, Z. Li, K. Chen, D. Ji, M. Zhang and J. Wang, Cancer Res. Treat., 2024, 56, 134–142 CrossRef CAS PubMed.
- L. R. Wiseman and K. L. Goa, Drugs, 1997, 54, 141–160 CrossRef CAS PubMed.
- H. Sipilä, L. Kangas, L. Vuorilehto, A. Kalapudas, M. Eloranta, M. Södervall, R. Toivola and M. Anttila, J. Steroid Biochem., 1990, 36, 211–215 Search PubMed.
- C. L. Vogel, M. A. Johnston, C. Capers and D. Braccia, Clin. Breast Cancer, 2014, 14, 1–9 CrossRef CAS PubMed.
- K. Nagren, T. Takahashi, P. Lehikoinen and J. Bergman, J Labelled Comp Radiopharm, 1991, 29, 1085–1089 CrossRef CAS.
- G. M. Keating, Drugs, 2014, 74, 207–221 CrossRef CAS PubMed.
- R. T. Dungo and G. M. Keating, Drugs, 2013, 73, 1503–1515 CrossRef CAS PubMed.
- H. X. Ding, C. A. Leverett, R. E. Kyne, K. K.-C. Liu, S. J. Fink, A. C. Flick and C. J. O'Donnell, Bioorg. Med. Chem., 2015, 23, 1895–1922 CrossRef CAS PubMed.
- C. P. Rains, H. M. Bryson and A. Fitton, Drugs, 1995, 49, 255–279 CrossRef CAS PubMed.
- R. S. Auriemma, L. Granieri, M. Galdiero, C. Simeoli, Y. Perone, P. Vitale, C. Pivonello, M. Negri, T. Mannarino, C. Giordano, M. Gasperi, A. Colao and R. Pivonello, Neuroendocrinology, 2013, 98, 299–310 CrossRef CAS PubMed.
- L. B. Nachtigall, Endocrine, 2017, 57, 3–5 CrossRef CAS PubMed.
- S. W. Ashford, K. E. Henegar, A. M. Anderson and P. G. M. Wuts, J. Org. Chem., 2002, 67, 7147–7150 CrossRef CAS PubMed.
- C. Kollmannsberger, K. Mross, A. Jakob, L. Kanz and C. Bokemeyer, Oncology, 1999, 56, 1–12 CrossRef CAS PubMed.
- F. G. Vogt, P. C. Dell'Orco, A. M. Diederich, Q. Su, J. L. Wood, G. E. Zuber, L. M. Katrincic, R. L. Mueller, D. J. Busby and C. W. DeBrosse, J. Pharm. Biomed. Anal., 2006, 40, 1080–1088 CrossRef CAS PubMed.
- M. V. Blagosklonny, Cell Cycle, 2004, 3, 1033–1040 CrossRef.
- T.-C. Hu and P.-J. Harn, US7977483B2, 2011.
- N. N. Sarkar, Eur. J. Obstet. Gynecol. Reprod. Biol., 2002, 101, 113–120 CrossRef CAS PubMed.
- R. M. DeHart and M. S. Morehead, Ann. Pharmacother., 2001, 35, 707–719 CrossRef CAS PubMed.
- P. F. Cadepond, M. P. A. Ulmann and M. P. E.-E. Baulieu, Annu. Rev. Med., 1997, 48, 129–156 CrossRef PubMed.
- I. De Milliano, D. Van Hattum, J. C. F. Ket, J. A. F. Huirne and W. J. K. Hehenkamp, Eur. J. Obstet. Gynecol. Reprod. Biol., 2017, 214, 56–64 CrossRef CAS PubMed.
- E. Rosato, M. Farris and C. Bastianelli, Front. Pharmacol, 2016, 6, 315, DOI:10.3389/fphar.2015.00315.
- Y. Yu, Y. He, Y. Zhao, L. Hai and Y. Wu, Steroids, 2013, 78, 1293–1297 CrossRef CAS PubMed.
- Z. Clarke, in xPharm: the Comprehensive Pharmacology Reference, Elsevier, 2007, pp. 1–4 Search PubMed.
- N. Otto, D. Ferenc and T. Opatz, J. Org. Chem., 2017, 82, 1205–1217 CrossRef CAS PubMed.
- A. S. Habib and T. J. Gan, CNS Drugs, 2006, 20, 821–839 CrossRef CAS PubMed.
- C. Suresh, D. R. Devi, K. Basavaiah, K. V. V. Prasada rao, B. Aegurla, G. A. Kumar, D. Bhalerao and A. S. Kumar Reddy, Chem. Pap., 2023, 77, 5713–5719 CrossRef CAS.
- R. Rizzi and E. Galeotto, Anesth. Analg., 1956, 13, 245–257 CAS.
- C. J. Cavallito and F. F. Foldes, Drug Metab. Rev., 1984, 15, 591–618 CrossRef CAS PubMed.
- J. V. Pergolizzi, R. Taylor, J. A. LeQuang, R. B. Raffa and J. Bisney, Pain Ther., 2018, 7, 37–57 CrossRef PubMed.
- Q. Zhang, J.-F. Li, G.-H. Tian, R.-X. Zhang, J. Sun, J. Suo, X. Feng, D. Fang, X.-R. Jiang and J.-S. Shen, Tetrahedron: Asymmetry, 2012, 23, 577–582 CrossRef CAS.
- S. Sarkar, N. Nebhinani, S. M. Singh, S. K. Mattoo and D. Basu, Indian J. Psychol. Med., 2012, 34, 283–285 CrossRef PubMed.
- R. Smyj, X.-P. Wang and F. Han, Profiles Drug Subst. Excip. Relat. Methodol., 2013, pp. 463–494 Search PubMed.
- J. Brüning, D. Podgorski, E. Alig, J. W. Bats and M. U. Schmidt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, o111–o113 CrossRef PubMed.
- P. E. Wu and D. N. Juurlink, Ann. Emerg. Med., 2017, 70, 245–252 CrossRef PubMed.
- Carles Estevez Company, N. B. Ferrer, J. C. Boliart and B. E. Beistegui, WO2008080601A2, 2008.
- M. Prasad, S. Vidyadhara, R. L. Sasidhar, T. Balakrishna and P. Trilochani, J. Adv. Pharm. Technol. Res., 2013, 4, 101 CrossRef PubMed.
- B. J. McAuley and J. S. Schroeder, Pharmacotherapy, 1982, 2, 121–131 CrossRef CAS PubMed.
- D. J. Mazzo, C. L. Obetz and J. Shuster, Anal. Profiles Drug Subst. Excip. 23, 1994, pp. 53–98 Search PubMed.
- N. Bunya, K. Sawamoto, S. Uemura, R. Kyan, H. Inoue, J. Nishida, H. Kouzu, N. Kokubu, T. Miura and E. Narimatsu, Acute Med. Surg., 2017, 4, 334–337 CrossRef PubMed.
- J. R. Araujo and F. Martel, Curr. Neuropharmacol., 2012, 10, 49–52 CrossRef CAS PubMed.
- J. E. Jeffery, F. Kerrigan, T. K. Miller, G. J. Smith and G. B. Tometzki, J. Chem. Soc., Perkin Trans. 1, 1996, 2583 RSC.
- R. H. Schirmer, B. Coulibaly, A. Stich, M. Scheiwein, H. Merkle, J. Eubel, K. Becker, H. Becher, O. Müller, T. Zich, W. Schiek and B. Kouyaté, Redox Rep., 2003, 8, 272–275 CrossRef CAS PubMed.
- B. Modarai, Emerg. Med. J., 2002, 19, 270 CrossRef CAS PubMed.
- P. Haouzi, M. McCann, J. Wang, X. Zhang, J. Song, I. Sariyer, D. Langford, M. Santerre, N. Tubbs, A. Haouzi-Judenherc and J. Y. Cheung, Ann. N. Y. Acad. Sci., 2020, 1479, 108–121 CrossRef CAS PubMed.
- A. Maleki and D. Nematollahi, Electrochem. Commun., 2009, 11, 2261–2264 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *f and
Bijo Mathew
*f and
Bijo Mathew