
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of atropodiastereomers of heterodimeric Amaryllidaceae alkaloids: narcipavline and narcikachnine†‡
Souvik
Pal
 a,
Satyajit
Majumder
a,
Sovan
Niyogi
b,
Pranay
Shyamal
b,
Debabrata
Mondal
b,
Bishnu
Das
b and
Alakesh
Bisai
*ab
a,
Satyajit
Majumder
a,
Sovan
Niyogi
b,
Pranay
Shyamal
b,
Debabrata
Mondal
b,
Bishnu
Das
b and
Alakesh
Bisai
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhopal 462 066, Madhya Pradesh, India. E-mail: alakeshb@gmail.com
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, Nadia, Kalyani, 741 246, West Bengal, India. E-mail: alakesh@iiserkol.ac.in
First published on 5th November 2024
Abstract
We report the first asymmetric total synthesis of recently isolated heterodimeric Amaryllidaceae alkaloids, narcipavlines A (1a) and B (1b), and narcikachnines A (2a) and B (2b), thereby confirming their absolute stereochemistry. These alkaloids showcase a unique heterodimeric structure, amalgamating two distinct types of Amaryllidaceae alkaloids: the cis-hydrodibenzofuran containing tetracyclic galantamine core (6a) and the galanthindole core (7) featuring a biaryl axis. The presence of this biaryl axis, coupled with the substantial galantamine core (6a) at the ortho substituents, imposes constraints on free rotation around the C–C axis, resulting in atropisomerism, an exceedingly rare phenomenon in nature. Key steps in the synthesis encompass the utilization of a one-pot double reductive amination approach for the establishment of C–N–C bonds to merge both the galantamine (6a) and galanthindole (7) cores. Additionally, the Mitsunobu reaction and intramolecular Heck cyclization have emerged as pivotal techniques for crafting the tricyclic hydrodibenzofuran core [(−)-13], incorporating an all-carbon quaternary stereogenic center.
Introduction
The Amaryllidaceae family, particularly plants in the Narcissus genus, have a rich history in traditional and Western medicine.1 Dating back to the 4th century BC, famous Greek physician Hippocrates used extracted oil from Narcissus poeticus L. to treat uterine tumors. These plants are renowned for producing structurally and biologically significant Amaryllidaceae alkaloids2 (AAs). The identification of lycorine in 1877 marked the start of an extraordinary voyage, leading to the discovery of over 600 AAs since then, highlighting a wide array of biological activities.2,3 Of particular interest are AAs with a cis-hydrodibenzofuran structure, featuring tetracyclic skeletons with vicinal quaternary and tertiary stereogenic centers, which show potential medicinal properties such as acetylcholinesterase (AChE) inhibition,4a crucial in Alzheimer's disease (AD) treatment, as well as anti-malarial, anti-infective, and anti-cancer2 effects. Amaryllidaceae plants naturally synthesize these AAs from L-tyrosine through a series of oxidation and reduction reactions.3 Galantamine (6a), among the most well-known Amaryllidaceae alkaloids, has been utilized to treat mild to moderate Alzheimer's disease5 under the trade name Razadyne©, approved by the FDA in 2001. AD, a chronic and progressive neurodegenerative disorder, represents a significant global health concern affecting over 36 million individuals.6Given the urgent need for effective therapies, research into novel treatments for AD is imperative. Currently all treatments of AD are only symptomatic aiming to boost acetylcholine levels through the inhibition of acetylcholinesterase using specific cholinesterase inhibitors like galantamine (6a), donepezil, and rivastigmine.7 An enzyme called butyrylcholinesterase (BuChE) is also able to hydrolyze AChE.8 Studies have indicated that in later stages of AD, patients experience a significant increase in butyrylcholinesterase levels by up to 50–70% while acetylcholinesterase expression decreases. This suggests that butyrylcholinesterase could be a promising therapeutic target, not only in restoring brain acetylcholine levels but also in serving as a disease-modifying agent in the prodromal stages of the disease.
Recently, a series of intricate heterodimeric Amaryllidaceae alkaloids have been freshly isolated (Fig. 1), showcasing atropodiastereomeric relationships. narcipavline (1) and narcikachnine (2) were isolated in 2018 from Narcissus poeticus cv. Pink Parasol,9a existing as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1.2 diastereomeric mixtures respectively. They are a combination of two structural types of Amaryllidaceae alkaloids: cis-hydrodibenzofuran containing lycoramine (6b) and bi-aryl axis containing galanthindole9c (7) core (Fig. 1). Narcimatuline (3) was isolated from Narcissus pseudonarcissus bulbs,9b existing as a 1:1.1 diastereomeric mixture, featuring a keto functionality at the cis-hydrodibenzofuran skeleton and N-methylindoline at the atropodiastereomeric position. Similar structural type narcielline (4) and narciabduline (5), isolated from Zapharynthis citrina10a and Narcissus pseudonarcissus10b respectively, in 2021, also exist as inseparable diastereomeric mixtures. While the stereochemistry and atropodiastereomerism were confirmed for narcielline (4) and narciabduline (5) by isolation chemists,10a they still remained unclear for narcipavline (1) and narcikachnine (2). Therefore, we have proposed the structures of the diastereomeric mixture of narcipavline (1) as narcipavlines A (1a) and B (1b), and narcikachnine (2) as narcikachnines A (2a) and B (2b), considering them as naturally occurring atropodiastereomers. Importantly, these newly isolated alkaloids exhibit excellent BuChE inhibition, with narcipavlines (1) showing an IC50 of 24.4 ± 1.2 μM, and narcimatulines (3) exhibiting an IC50 of 5.90 ± 0.87 μM. Narcipavlines [i.e. narcipavlines A (1a) and B (1b)] also demonstrate antiplasmodial activity against malaria parasites, suggesting their potential for the development of new drugs. Biological activity for narcikachnines 2 [i.e. narcikachnines A (2a) and B (2b)] is yet to be discovered.
:1 and 1:1.2 diastereomeric mixtures respectively. They are a combination of two structural types of Amaryllidaceae alkaloids: cis-hydrodibenzofuran containing lycoramine (6b) and bi-aryl axis containing galanthindole9c (7) core (Fig. 1). Narcimatuline (3) was isolated from Narcissus pseudonarcissus bulbs,9b existing as a 1:1.1 diastereomeric mixture, featuring a keto functionality at the cis-hydrodibenzofuran skeleton and N-methylindoline at the atropodiastereomeric position. Similar structural type narcielline (4) and narciabduline (5), isolated from Zapharynthis citrina10a and Narcissus pseudonarcissus10b respectively, in 2021, also exist as inseparable diastereomeric mixtures. While the stereochemistry and atropodiastereomerism were confirmed for narcielline (4) and narciabduline (5) by isolation chemists,10a they still remained unclear for narcipavline (1) and narcikachnine (2). Therefore, we have proposed the structures of the diastereomeric mixture of narcipavline (1) as narcipavlines A (1a) and B (1b), and narcikachnine (2) as narcikachnines A (2a) and B (2b), considering them as naturally occurring atropodiastereomers. Importantly, these newly isolated alkaloids exhibit excellent BuChE inhibition, with narcipavlines (1) showing an IC50 of 24.4 ± 1.2 μM, and narcimatulines (3) exhibiting an IC50 of 5.90 ± 0.87 μM. Narcipavlines [i.e. narcipavlines A (1a) and B (1b)] also demonstrate antiplasmodial activity against malaria parasites, suggesting their potential for the development of new drugs. Biological activity for narcikachnines 2 [i.e. narcikachnines A (2a) and B (2b)] is yet to be discovered.
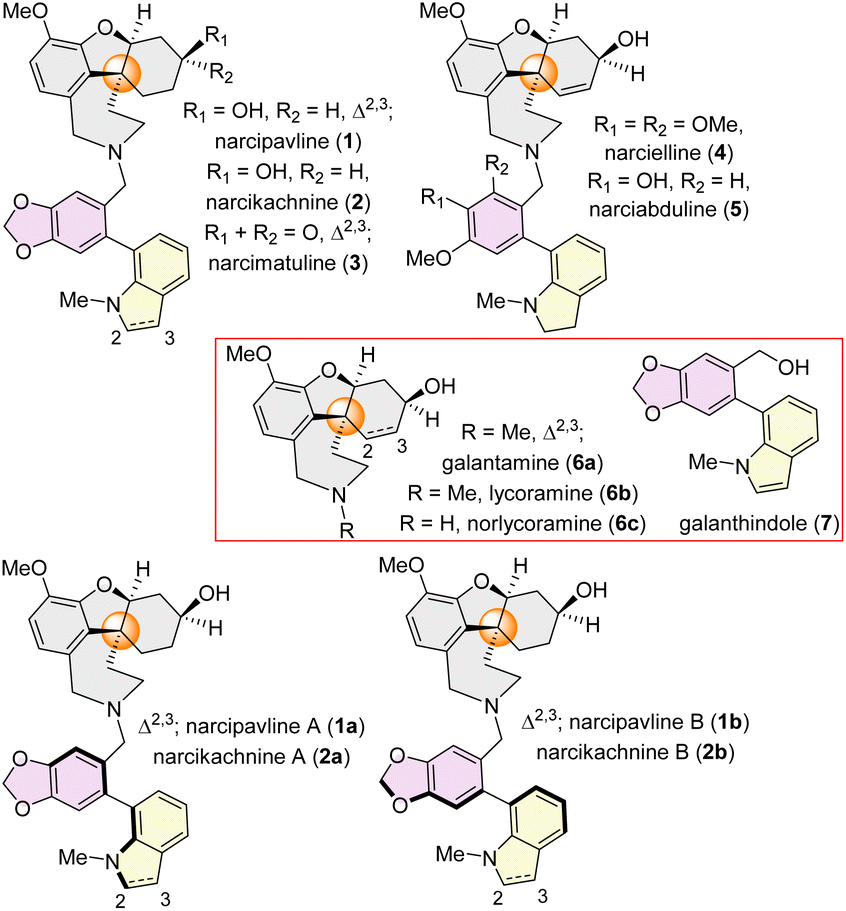 | ||
| Fig. 1 Dimeric atropodiastereomeric Amaryllidaceae alkaloids (1–5) and monomeric congeners (6–7) sharing a cis-hydrodibenzofuran core. | ||
Structurally, these alkaloids possess three chiral centers, a cis-hydrodibenzofuran core, along with an all-carbon quaternary stereogenic center11 and a chiral bi-aryl axis which also leads to complicated rare atropisomerism, makes them impressively challenging structures.12 Atropisomerism, a form of dynamic chirality, occurs due to restricted rotation between the single bond between two sp2-hybridized atoms, yielding atropisomers. These are classified into three categories, based on the rotational barrier (ΔErot) and half-life (t1/2), class I (ΔErot < 20 kcal mol−1, t1/2 ≈ ns-ms) with rapidly equilibrating conformers considered as achiral; class II (ΔErot ≈ 20–30 kcal mol−1, t1/2 ≈ min-hours per days), considered high-risk in drug development due to moderate stability and class III high rotational barrier isomers (ΔErot > 30 kcal mol−1, t1/2 ≈ years), highly stable and often developed as single atropisomers, although very challenging to synthesize.12b–e Only very few syntheses have been reported for other atropodiastereomeric natural products.12a–d Biosynthesis could be hypothesized from a combination of two different Amaryllidaceae alkaloids nor-lycoramine (6c) and galanthindole (7) via enzymatic alkylation, where both can be derived from L-tyrosine.3
Although there are many elegant approaches for the synthesis of galantamine (6a) and related tetracyclic scaffolds containing a cis-hydrodibenzofuran core,13–15 there has been no synthesis reported for the newly isolated complex heterodimeric Amaryllidaceae alkaloids to date. We hypothesized that classical reductive amination could be a useful tool to construct the C–N–C bond of the seven membered azepine ring in one pot and will be worth exploring in this regard. Due to their impressive biological activity and structurally challenging arrays, we envisioned a catalytic enantioselective unified approach via reductive amination to synthesize narcipavline (1) [i.e. the atropodiastereomers narcipavlines A (1a) and B (1b)] and narcikachnine (2) [i.e. the atropodiastereomers narcikachnines A (2a) and B (2b)] along with galantamine (6a) and lycoramine (6b).
Results and discussion
Retrosynthetically, narcikachnine (2) could be derived from narcipavline (1) via reduction of the indole ring. Narcipavline (1) consists of two parts: the northern part is the lycoramine core and southern part is a galanthindole (7) core. Initially, we envisioned accessing atropodiastereomers of narcipavline (1) [i.e. (1a) and (1b)] from an advanced intermediate 8via allylic oxidation16 and hydrogenation (Scheme 1). To construct the seven membered azepine core, a one-pot double reductive amination of galanthindolyl benzylamine (12) with tricyclic di-aldehyde 10 was postulated (Scheme 1). The all-carbon quaternary stereogenic center11 of tricycle 13 bearing an aldehyde and ester could be constructed via intramolecular Heck cyclization of alkyl aryl ether 14, which further could be accessed via Mitsunobu reaction of the phenol derivative 15 with enantioenriched α-substituted cyclohex-2-en-1-ol 16 (Scheme 1).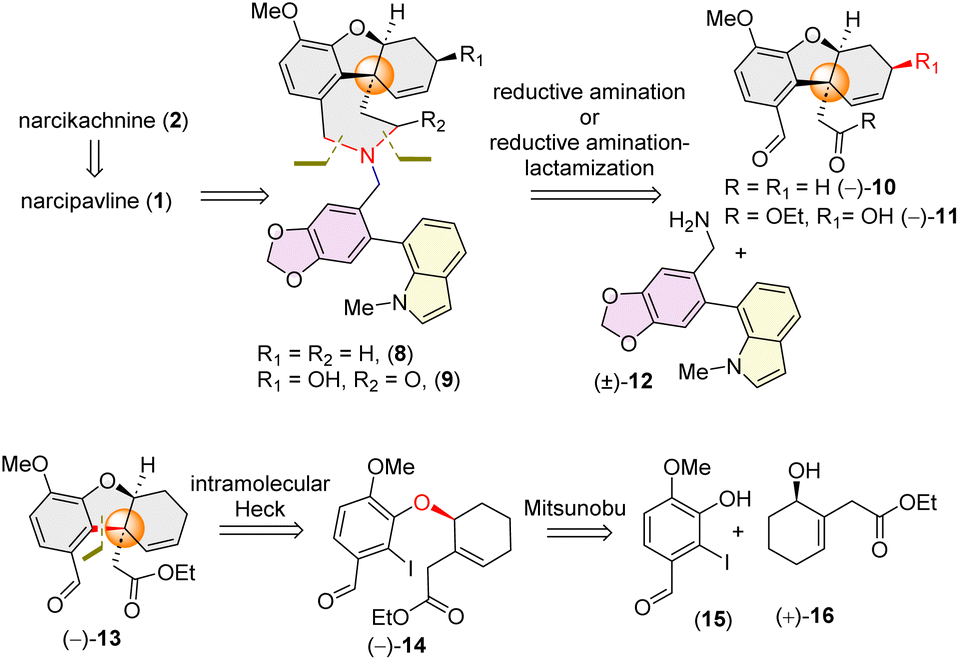 | ||
| Scheme 1 Retrosynthetic analysis of narcipavline (1) and narcikachnine (2). | ||
Our journey commenced with the preparation of the galanthindole (7) core and its corresponding benzylic amine 12. To this end, we synthesized the known biaryl aldehyde 17, and galanthindole (7) following a modified procedure of Hsieh's protocol (see ESI‡ for details).17 Subsequently, we transformed the biaryl aldehyde 17 into the corresponding benzylamine (±)-12via a reductive amination using ammonium formate (Scheme 2).
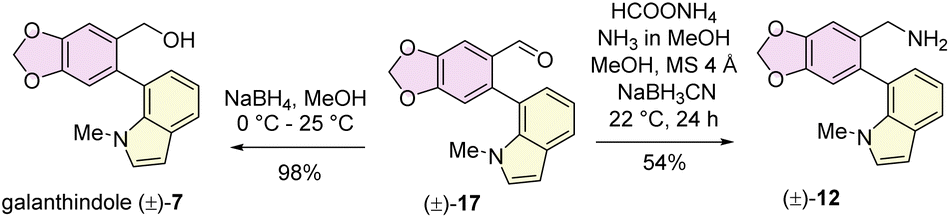 | ||
| Scheme 2 Synthesis of galanthindolyl benzyl amine. | ||
The isolation chemists did not provide any optical rotation data for galanthindole (7).9c However, due to its structural features, including a biphenyl axis and ortho-substituents, it can exhibit axial isomerism. Indeed, when subjected to HPLC analysis, galanthindole showed a 50:50 mixture of these two atropisomers, confirming its racemic nature [denoted as (±)-7]. For the synthesis of the galantamine core, we began with the synthesis of enantioenriched (96% ee) α-substituted cyclohex-2-en-1-ol1816via a catalytic enantioselective Corey–Bakshi–Shibata (CBS) reduction19 of a known Trost's enone20 [see ESI‡ for details].
Inspired by Banwell's protocol,14d we explored the Mitsunobu reaction for the synthesis of enantioenriched 14. Extensive optimization with different phosphines and azodicarboxylates revealed that allyl alcohol (+)-16 (96% ee) and 2-iodophenol 15 afforded (−)-14 in 93% ee in the presence of diethyl azodicarboxylate (DEAD) and tributylphosphine (nBu3P) [see ESI‡ for detailed optimization]. Gratifyingly, the stereochemistry of (−)-14 was confirmed by single crystal X-ray analysis [CCDC 2384521].
With enantioenriched (−)-14 (93% ee) in hand, we moved forward towards the intramolecular Heck cyclization to construct the cis-fused tricyclic hydrodibenzofuran core (Scheme 3). To our delight, charging iodoarene (−)-14 under Heck coupling conditions using catalytic Pd(OAc)2 smoothly afforded tricyclic product (−)-13 with excellent dr (dr > 20:1), with generating the all carbon quaternary stereogenic centre. By employing our strategy, we have been able to achieve the hydrodibenzofuran core in 93% ee (see ESI‡ for HPLC analysis) in 4 steps from commercially available cyclohex-2-en-1-one which is superior to the previous reports.13b–d,14d Further, lithium aluminium hydride reduction of the ester group of 13 followed by oxidation using Dess–Martin periodinane (DMP) afforded di-aldehyde (−)-10 in 85% yield over 2 steps (Scheme 3).21 A one step reduction using DIBAL-H proved to be difficult as it is associated with over-reduced diol in 35% yield in addition to the desired dialdehyde 10 (see ESI‡ for details).21 Next, reacting equimolar amounts of dialdehyde (−)-10 and benzylamine (±)-12 in the presence of sodium cyanoborohydride (NaBH3CN) and a catalytic amount of trifluoroacetic acid afforded the desired product (−)-8 in 84% yield (Scheme 3). Interestingly, 1H NMR analysis of the pure product revealed the presence of two diastereomers in a ∼1:1 ratio, attributable to atropisomerism around the biphenyl axis, resulting in two distinct configurations of atropodiastereomers 8a and 8b (Scheme 3). The separation of these atropodiastereomers was a tedious job, as the TLC shows a single spot, making them inseparable in column chromatography.
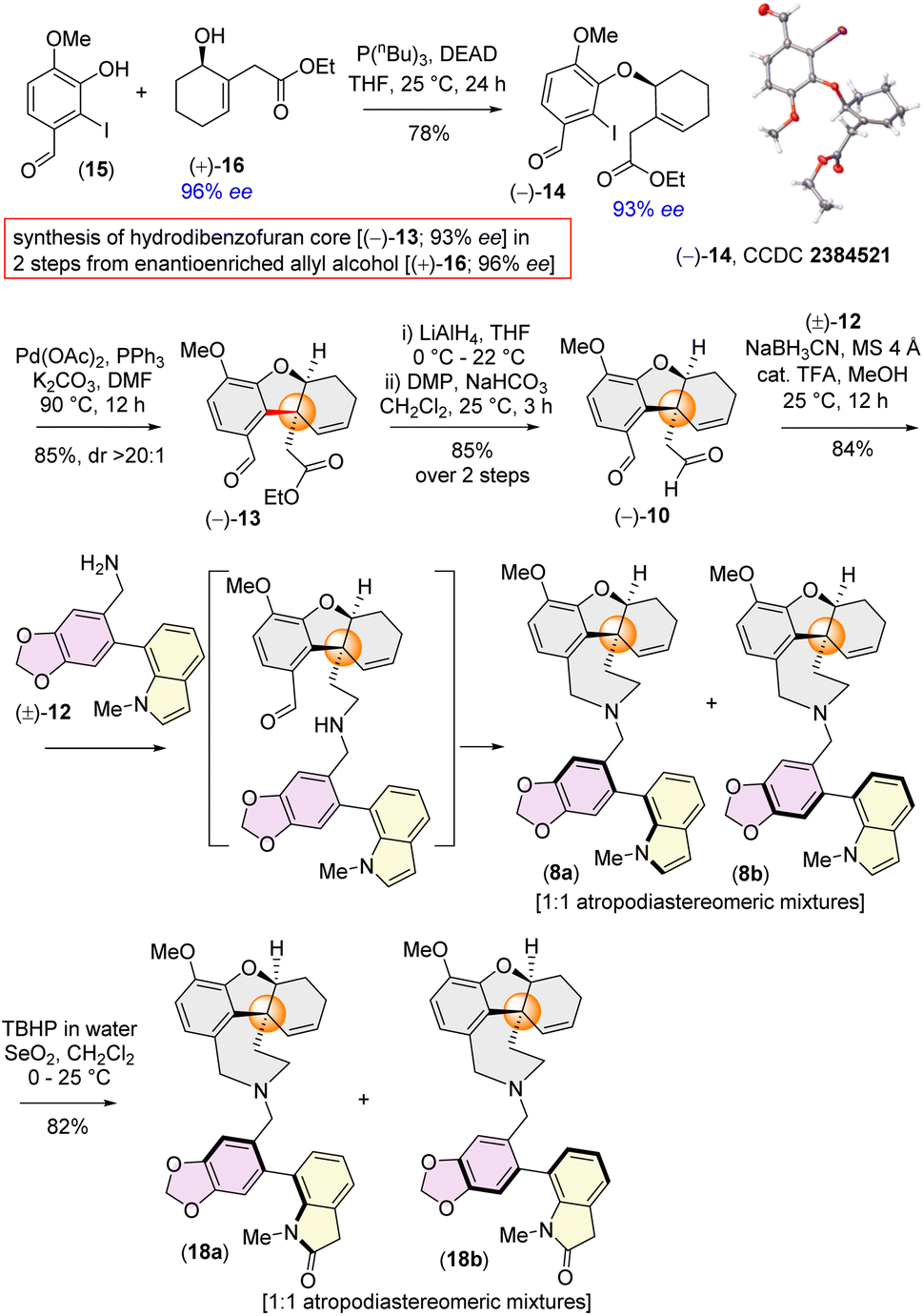 | ||
| Scheme 3 Forward synthesis towards narcipavline. | ||
At this stage, only a diastereoselective allylic oxidation was required to complete the total synthesis of narcipavlines (1). However, all efforts to perform allylic oxidation were unsuccessful and seemed to be very challenging (Scheme 3). When the mixture of 8a and 8b was charged under allylic oxidation conditions using selenium dioxide and TBHP, it was observed that the indole ring of 8 is highly susceptible to oxidation compared to the allylic position. Even under mild conditions using selenium dioxide and tert-butyl hydrogen peroxide (TBHP), we could observe the formation of ∼1:1 atropodiastereomers of 2-oxindoles, 18a and 18b (Scheme 3). Therefore, we recognized the necessity of installing the allylic alcohol early in the tricyclic core (−)-13.
Therefore, employing Trost's procedure,13c we successfully executed the allylic oxidation of (−)-13 using selenium dioxide (SeO2) with sand as a solid support in refluxing 1,4-dioxane to afford a mixture of 11 and 11a (79% yield, dr ∼6:1 in favour of 11) (Scheme 4). The diastereoselectivity observed in case of allylic oxidation was attributed to the approach of SeO2 from the non-traditional more hindered concave face of tricycle (−)-13 (Scheme 4). The mechanism involves an Ene-type reaction and the stereoelectronic requirements of the Ene reaction dictate specific spatial arrangements, where approach through the pseudo-axial H rather than the pseudo-equatorial H of cyclohexene results in the formation of 11 as the major diastereomer (Scheme 4).
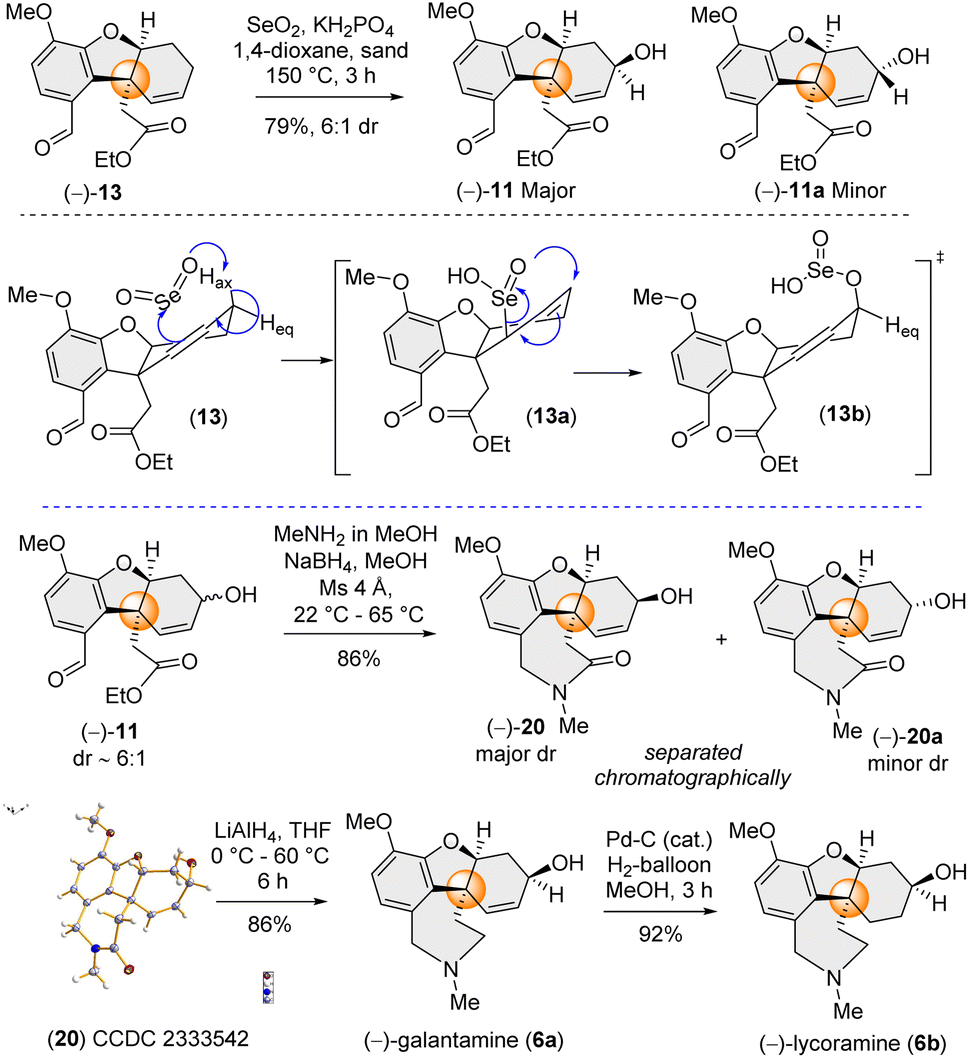 | ||
| Scheme 4 Diastereoselective allylic oxidation and synthesis of (−)-galantamine (6a) and (−)-lycoramine (6b). | ||
Next, we focused on the one-pot reductive amination-lactamization sequence of aldehyde and ester groups present in (−)-11 (Scheme 4). It was observed that (−)-11 (∼6:1 dr) in the presence of MeNH2 in MeOH followed by reduction using NaBH4 afforded lactams (−)-20 (as the major dr) and 20a (see ESI‡ for details). Gratifyingly, these two diastereomers were separable in column chromatography. The structure of major diastereomer 20, having the hydroxyl group above the plane, was confirmed by X-ray single crystal analysis (CCDC 2333542). Next, with tetracycle (−)-20 in hand, we completed the total synthesis of naturally occurring (−)-galantamine [(−)-6a] just by LiAlH4 reduction (86% yield). Further, hydrogenation of (−)-6a completed the total synthesis of (−)-lycoramine [(−)-6b].
We next turned our attention to the synthesis of the azepine core of narcipavline (1) via a similar one-pot reductive amination-lactamization cascade. However, this proved to be very challenging with the sterically demanding benzylamine (±)-12 (Scheme 5).
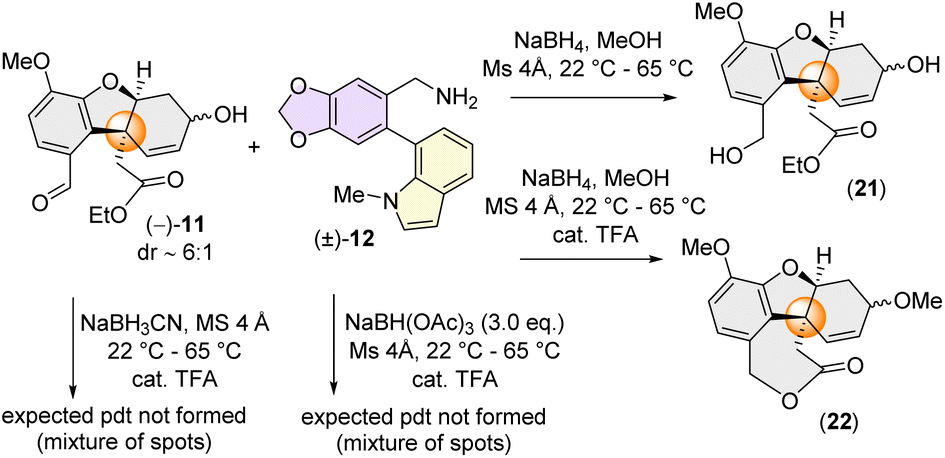 | ||
| Scheme 5 Failed attempt towards reductive amination lactamization with galanthindolyl benzylic amine. | ||
A condensation reaction of (−)-11 with benzylic amine 12, followed by reduction using NaBH4 didn't afford the required azepine core; instead, we could isolate benzyl alcohol 21 and ε-lactone 22 (through acid-catalysed cyclization) in a few specific cases (Scheme 5). This fact is probably attributed to the strongly electron-donating nature of the p-methoxy group on (−)-11 which could diminish the electrophilicity of benzaldehyde hindering the crucial imine formation (Scheme 5).
So, we first protected the secondary alcohol group of 11 as TBS-ether to access (−)-23 and (−)-23a (Scheme 6). Delightfully, we could separate the major diastereomers 23via column chromatography. Next, the ester and the aldehyde group both were reduced and again re-oxidized using Dess–Martin periodinane (DMP) to afford dialdehyde (−)-24 in 85% yield over 2 steps (Scheme 6). Subsequently, a one-pot sequential reductive amination was successfully conducted with (−)-24 and galanthindolyl benzylamine (±)-12 in the presence of NaBH3CN and catalytic trifluoroacetic acid to furnish 25 as a mixture of two atropodiastereomers, 25a and 25b with dr ∼1:1 (see ESI‡ for detailed characterization). Next, deprotection of the silyl ether group was achieved using tetra-butyl ammonium fluoride (TBAF) yielding dehydronarcipvaline (26a and 26b) as 1:1 atropodiastereomers. It is pertinent to mention that, a racemic crystal structure of (±)-26b (CCDC 2363948) (prepared from (±)-24 in a different sequence) unequivocally proved the all-bond connections present in dehydronarcipvaline 26. With a few milligrams of crystals in hand, a detailed 1H-NMR analysis was undertaken. It is interesting to note that the 1H-NMR of (±)-26b showed the signals for a single atropodiastereomer (see ESI‡ for details). However, when 13C-NMR of (±)-26b was recorded after a day (∼24 h), the signal of each carbon appeared to be doubled, thereby confirming the rotation along the axis happening at room temperature. Further, the 1H-NMR of the same sample after ∼24 h provided the signals for ∼1:1 atropodiastereomers [(±)-26a and (±)-26b]. Thus, it may be concluded that the atropodiastereomer (±)-26b has a very short half-life period and could be easily converted to (±)-26a, leading to the mixtures of atropodiastereomers at room temperature (see ESI‡ for details).
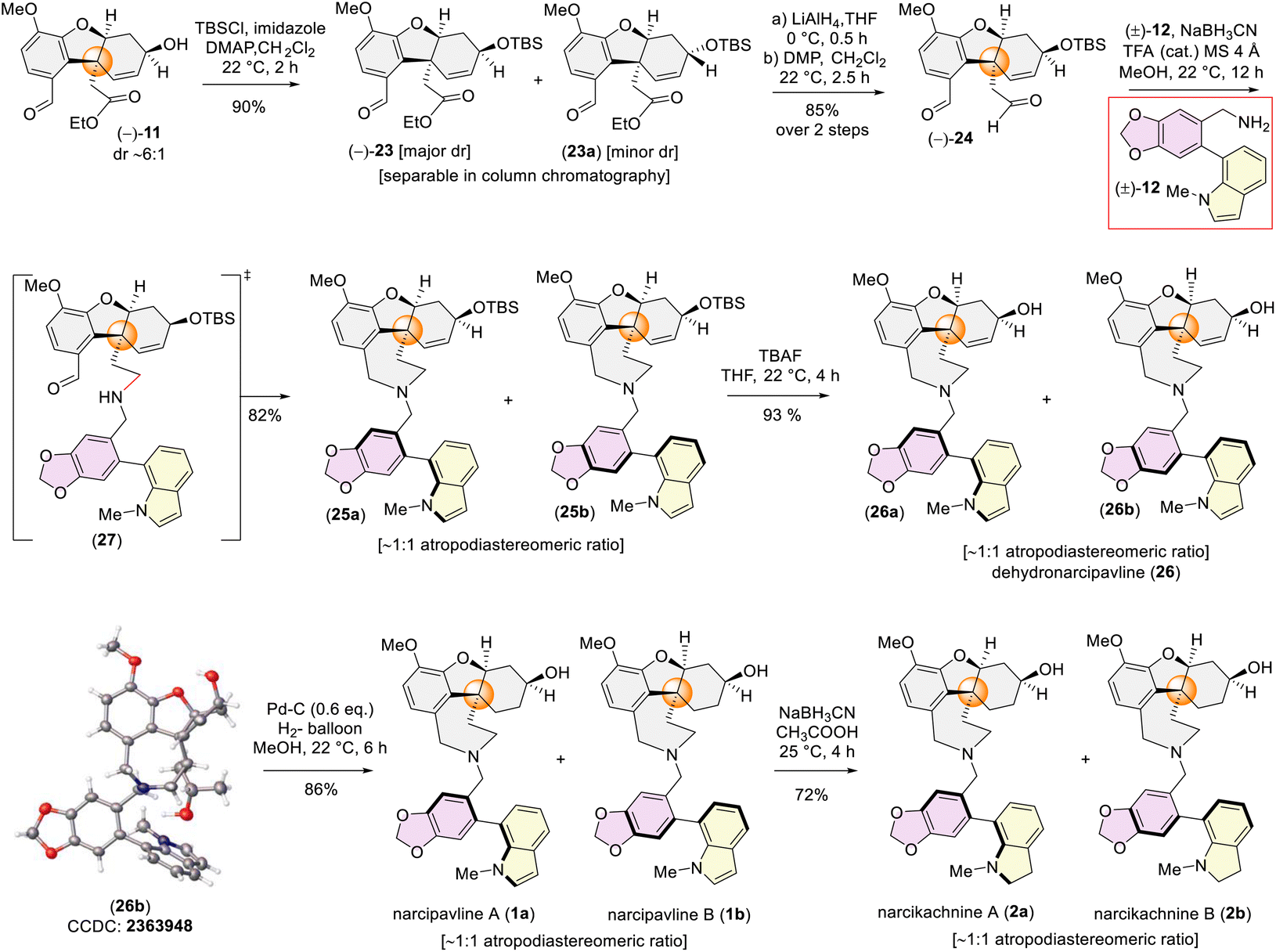 | ||
| Scheme 6 Total synthesis of atropodiastereomers narcipavline A, B and narcikachnine A, B. | ||
Next, the atropodiastereomers of dehydronarcipavline 26 (∼1:1 dr) were hydrogenated using Pd–C at 1 atm. H2-gas afforded the first total synthesis of atropodiastereomers of novel heterodimeric Amaryllidaceae alkaloids, narcipavlines A (1a) and B (1b) as ∼1:1 dr (Scheme 6). Efforts towards the separation of the atropodiastereomers proved to be unsuccessful at room temperature using column chromatography. Nevertheless, all the corresponding peaks of 1H- and 13C-NMR are in good agreement with the isolation report of narcipavlines (1a:1b ∼1:1) by Cahlíková et al.9a The structure was confirmed through 2D NMR analysis, which revealed a negative specific rotation value for the diastereomeric mixture. Notably, compound 1 and lycoramine (6b) exhibited similar NMR spectra, particularly in the region of the secondary alcohol within the cis-hydrodibenzofuran core, and displayed negative optical rotation values. Further, HPLC analysis using a Chiralpak OD-H column (40% iPrOH/n-hexane) revealed two pairs of enantiomers [93% ee] corresponding to the atropodiastereomers narcipavlines A (1a) and B (1b) [∼50:50 ratio] (see ESI‡ for details). These findings strongly support the proposed structure of atropodiastereomers 1a and 1b, resembling (−)-lycoramine (6b) as a cis-hydrodibenzofuran core, where the secondary alcohol group resides above the plane (Fig. 1).
We next turned our attention to the total synthesis of atropodiastereomeric alkaloids, narcikachnines A (2a) and B (2b) (Scheme 6). Their structures are nearly identical, with the sole difference being an indoline ring in narcikachnine (2) instead of an indole in narcipavline (1). After a quick optimization, we were pleased to see that the indole ring of narcipavline (1) could be reduced with NaBH3CN in AcOH to form indolines of narcikachnines A (2a) and B (2b) in 72% yield in ∼1:1 dr (Scheme 6). A plausible mechanism of reductive amination of indole to indoline is shown in Scheme 7.
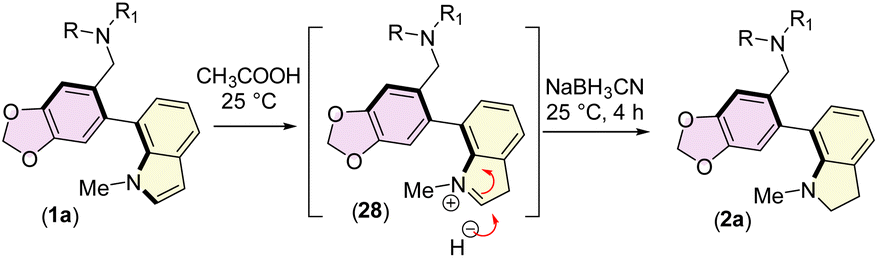 | ||
| Scheme 7 Probable mechanism for the reduction of the indole ring. | ||
Once again, all the corresponding peaks of 1H- and 13C-NMR are in good agreement with the isolation report of narcikachnines (2a:2b ∼1:1) by Cahlíková et al.9a The diastereomeric mixture of narcikachnines (2a:2b) shows a negative specific rotation value. The cause of the doubling of some signals in NMR was found to be due to axial isomerism through the sterically hindered single bond rotation. Accordingly, VT NMR analysis was performed to demonstrate the atropodiastereomerism at different temperatures. Few milligrams of (2) and (26) were dissolved in DMSO-d6 having a sufficiently high boiling point. At the highest experimental temperature, (up to 70 °C for 2, and 125 °C for 26), the proton resonances coalesced for both diastereomers (see ESI‡ for details).
Further, we performed DFT calculations to understand the rotational barriers of these naturally occurring atropodiastereomers (Fig. 1). In particular, the rotational energy barriers of narcipavline 1, narcikachnine 2, and galanthindole 7 were analyzed to understand their conformational dynamics by simulating a 360-degree rotation around their biaryl C–C bond in 36 steps.22 For narcipavline 1, the energy profile reveals that conformations 1a and 1b are different, indicating that these conformations are atropodiastereomers of each other. The high energy barriers of 35.35 kcal mol−1 at TS1 and 28.09 kcal mol−1 at TS2 indicate significant intramolecular forces and steric hindrance during rotation. This substantial energy requirement suggests that 1 is structurally rigid, making it probably less likely to undergo spontaneous conformational changes. This rigidity could facilitate the distinct separation of conformations 1a and 1b under special circumstances (Fig. 2).
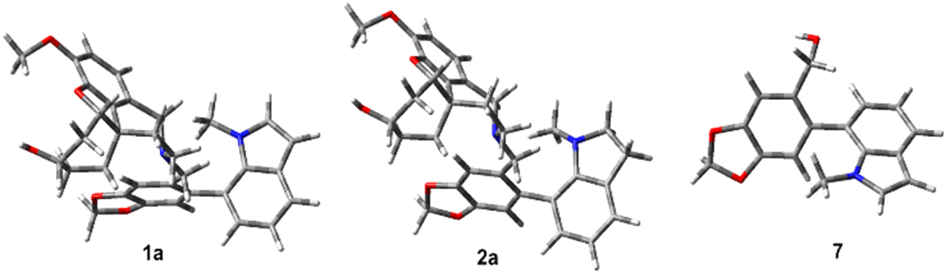 | ||
| Fig. 2 Energy minimized structure of 1a, 2a and 7 using the B3LYP/6-31G(d) method. | ||
In contrast, narcikachnine 2 exhibits a low energy barrier at TS1 (25.08 kcal mol−1) and TS2 (18.90 kcal mol−1), indicating greater rotational flexibility. The energy profile shows that conformations 2a, 2b, and 2a′ are all different. This suggests that the molecule undergoes significant structural changes throughout the rotation. The lower energy peaks at TS1 and TS2 suggest weaker intramolecular interactions, allowing 2 to transition between conformations with less resistance, which could lead to interconversion between individual conformers more easily as compared to conformations 1a and 1b (Fig. 3).
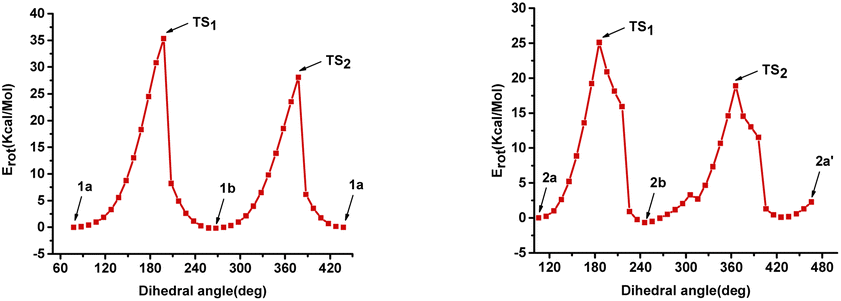 | ||
| Fig. 3 Rotational energy barrier diagram for narcipavline 1 and narcikachnine 2. | ||
Galantindole 7 presents an intermediate energy barrier, with TS1 at 25.09 kcal mol−1 and TS2 at 30.78 kcal mol−1, reflecting a balance between flexibility and rigidity. The energy peaks at TS1 and TS2 indicate that 7 experiences moderate steric hindrance during rotation. This intermediate behaviour may allow for the possibility of selectively separating the distinct conformers (S)-7, (R)-7, and (S)-7′, however under very special circumstances.
Conclusion
In summary, a concise catalytic enantioselective (93% ee) first total synthesis of heterodimeric atropodiastereomeric Amaryllidaceae alkaloids narcipavlines A (1a) and B (1b) [∼1:1 dr, 27% overall yield in 9 LLS] and narcikachnines A (2a) and B (2b) [∼1:1 dr, 19% overall yield in 10 LLS] has been achieved from enantioenriched allyl alcohol 16 (96% ee). The synthesis involves sequential reductive amination with galanthindolyl benzylamine (12) as the key steps. Through this synthesis we also confirmed the absolute structure of narcipavline A (1a) and narcipavline B (1b) as well as narcikachnine A (2a) and narcikachnine B (2b) as a mixture of atropodiastereomers. This effort also culminated in the protecting group free total synthesis of (−)-galantamine (6a) and (−)-lycoramine (6b), in six and seven steps, respectively.
Data availability
Experimental details and spectral analysis are available free of charge from the ESI‡ available with this article.Author contributions
A. B. and S. P. designed the research plan. S. P., S. M., and S. N. investigated the key synthetic processes leading to atropodiastereomers of Amaryllidaceae alkaloids, narcipavlines A (1a) and B (1b) as well as narcikachnines A (2a) and B (2b). P. S. and D. M. were actively involved in the total synthesis of (−)-galantamine (6a) and (−)-lycoramine (6b). B. D. calculated the rotational energy barrier. A. B. and S. P. wrote the manuscript with contributions from all the authors; all the authors were actively engaged in the editing of the manuscript and gave their approval of the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by grants from the SERB [SCP/2022/000486], SERB [CRG/2023/000782] and STARS-MoE [STARS/2023/0753]. SP thanks IISER Bhopal for pre-doctoral fellowships; SM and SN thank the CSIR; PS and DM thank the UGC for the research fellowships. AB is a SERB-STAR Fellow and gratefully acknowledges the SERB [STR/2020/000061] for generous support.Notes and references
- (a) Z. Jin, Nat. Prod. Rep., 2016, 33, 1318–1343 RSC; (b) Y. Ding, D. Qu, K. M. Zhang, X. X. Cang, Z. N. Kou, W. Xiao and J. B. Zhu, J. Asian Nat. Prod. Res., 2016, 19, 53–100 CrossRef PubMed.
- (a) S. Kobayashi, M. Kihara, K. Yuasa, Y. Imakura, T. Shingu, A. Kato and T. Hashomoto, Chem. Pharm. Bull., 1985, 33, 5258–5263 CrossRef CAS; (b) J. Refaat, M. S. Kamel, M. A. Ramadan and A. A. Ali, Int. J. Res. Pharm. Sci., 2013, 4, 1239–1252 CAS.
- Z. Jin and G. Yao, Nat. Prod. Rep., 2019, 36, 1462–1488 RSC.
- (a) G. M. Bores and R. W. Kosley, Drugs Future, 1996, 21, 621–635 CAS; (b) S. Lilienfeld, CNS Drug Rev., 2006, 8, 159–176 CrossRef PubMed.
- (a) R. L. Irwin and H. J. Smith III, Biochem. Pharmacol., 1960, 3, 147–148 CrossRef CAS PubMed; (b) P. Williams, A. Sorribas and M. J. R. Howes, Nat. Prod. Rep., 2011, 28, 48–77 RSC.
- E. Nichols, C. Szoeke, S. Vollset, N. Abbasi, F. Abd-Allah, J. Ebro, M. Aichour, R. Akinyemi, F. Alahdab and S. Asgedom, et al., Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016, Lancet Neurol., 2019, 18, 88–106 CrossRef PubMed.
- M. Paskaski and J. Kalman, Neurochem. Int., 2008, 53, 103–111 CrossRef PubMed.
- Y. Nicolet, O. Lockridge, P. Masson, J. C. Fontecilla-Camps and F. Nachon, J. Biol. Chem., 2003, 278, 41141–41147 CrossRef CAS PubMed.
- (a) M. Safratová, A. H. álková, D. Hulcová, K. Breiterová, V. Hrabcová, M. Machado, D. Fontnha, M. Pruděncio, J. Kuněs, J. Chlebek, D. Jan, M. Hrabinová, L. Nováková, R. Havelek, M. Seifrtová, L. Opletal and L. Cahlíková, Arch Pharm. Res., 2018, 41, 208–218 CrossRef PubMed; (b) D. Hulcová, J. Mařĺková, J. Korábečny, A. Hošťálková, D. Jun, J. Kuněs, J. Chlebek, L. Opletal, A. D. Simone, L. Nováková, V. Andrisano, A. Růžiĉka and L. Cahlíková, Phytochemisrty, 2019, 165, 112055 CrossRef PubMed; (c) Y. Yang, S. X. Huang, Y. M. Zhao, Q. S. Zhao and H. D. Sun, Helv. Chim. Acta, 2005, 88, 2550–2552 CrossRef CAS.
- (a) E. Kohelová, J. Mařĺková, J. Korábečny, D. Hulcová, T. Kučera, D. Jun, J. Chlebek, J. Jenčo, M. Safratová, M. Hrabinová, A. Ritomska, M. Malanik, R. Perinova, K. Breiterova, J. Kunes, L. Novakova, L. Opletal and L. Cahlíková, Bioorg. Chem., 2021, 107, 104567 CrossRef PubMed; (b) J. Marikova, A. Al Mamun, L. A. Shammari, J. Koranency, T. Kucera, D. Hulcova, J. Kunes, M. Malanik, M. Vaskova, E. Kohelova, L. Novakova, L. Cahlíková and M. Pour, Molecules, 2021, 26, 1279 CrossRef CAS PubMed.
- (a) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) M. K. Das, N. Kumar and A. Bisai, Org. Lett., 2018, 20, 4421–4424 CrossRef CAS PubMed.
- (a) R. Nandi, S. Niyogi, S. Kundu, V. R. Gavit, M. Munda, R. Murmu and A. Bisai, Chem. Sci., 2023, 14, 8047–8053 RSC; (b) G. Bringmann, A. Irmer, D. Feineis, T. A. M. Gulder and H. P. Fiedler, Phytochemistry, 2009, 70, 1776–1786 CrossRef CAS PubMed; (c) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC; (d) S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409–422 CrossRef CAS PubMed; (e) M. Fragkiadakis, M. Thomaidi, T. Stergiannakos, E. Chatziorfanou, M. Gaidatzi, A. M. Barakat, C. Stoumpos and C. G. Neochoritis, Chem.–Eur. J., 2024, 30, e202401461 CrossRef CAS PubMed.
- (a) B. Cheng, Q. Wang, Y. An and F. Chen, Nat. Prod. Rep., 2024, 41, 1060–1090 RSC; (b) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 11262–11263 CrossRef CAS; (c) B. M. Trost and W. Tang, Angew. Chem., Int. Ed., 2002, 41, 2795–2797 CrossRef CAS; (d) B. M. Trost, W. Tang and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 14785–14803 CrossRef CAS PubMed; (e) P. Chen, X. Bao, L. F. Zhang, M. Ding, X. J. Han, J. Li, G. B. Zhang, Y. Q. Tu and C. A. Fan, Angew. Chem., Int. Ed., 2011, 50, 8161–8166 CrossRef CAS PubMed.
- (a) V. Satcharoen, N. J. McLean, S. C. Kemp, N. P. Camp and R. C. D. Brown, Org. Lett., 2007, 9, 1867–1869 CrossRef CAS PubMed; (b) C. Guillou, J. L. Beunard, E. Gras and C. Thal, Angew. Chem., Int. Ed., 2001, 40, 4745–4746 CrossRef CAS; (c) C. A. Fan, Y. Q. Tu, Z. L. Song, E. Zhang, L. Shi, M. Wang, B. Wang and S. Y. Zhang, Org. Lett., 2004, 6, 4691–4694 CrossRef CAS PubMed; (d) J. Nugent and M. G. Banwell, Eur. J. Org Chem., 2016, 19, 5862–5867 CrossRef; (e) T. Venkatesh, P. S. Mainkar and S. T. Chandrasekhar, Org. Biomol. Chem., 2019, 17, 2192–2198 RSC.
- (a) L. Li, Q. Yang, Y. Wang and Y. Jia, Angew. Chem., Int. Ed., 2015, 54, 6255–6259 CrossRef CAS PubMed; (b) Q. Zhang, F. M. Zhang, C. S. Zhang, S. Z. Liu, J. M. Tian, S. H. Wang, X. M. Zhang and Y. Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS PubMed; (c) S. Majumder, A. Yadav, S. Pal, A. Khatua and A. Bisai, J. Org. Chem., 2022, 87, 7786–7797 CrossRef CAS PubMed; (d) J. Q. Chen, J. H. Xie, D. H. Bao, S. Liu and Q. L. Zhou, Org. Lett., 2012, 14, 2714–2717 CrossRef CAS PubMed; (e) Y. Zhang, S. Shen, H. Fang and T. Xu, Org. Lett., 2020, 22, 1244–1248 CrossRef CAS PubMed.
- B. N. Kakde, P. Kumari and A. Bisai, J. Org. Chem., 2015, 80, 9889–9899 CrossRef CAS PubMed.
- T. T. F. Chiang, H. K. Wang and J. C. Hsieh, Tetrahedron, 2016, 72, 5640–5645 CrossRef.
- A. Mondal, S. Pal, A. Khatua, A. Mondal and A. Bisai, J. Org. Chem., 2024, 89, 12485–12497 CrossRef CAS PubMed.
- (a) E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS; (b) S. P. West, A. Bisai, A. D. Lim, R. R. Narayan and R. Sarpong, J. Am. Chem. Soc., 2009, 131, 11187–11194 CrossRef CAS PubMed.
- B. M. Trost, W. Tang and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 14785–14803 CrossRef CAS PubMed.
- Reduction of (−)-13 using DIBAL-H afforded 40% yield of dialdehyde (−)-10 along with 35% yield of diol (−)-10a
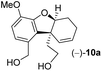 .
. - K. Kapłon, O. M. Demchuk and K. M. Pietrusiewicz, Curr. Chem. Lett., 2015, 4, 145–152 CrossRef.
Footnotes |
| † This work is dedicated respectfully to Professor Srinivasan Chandrasekaran, IISc Bangalore for his constant encouragement and inspiration. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2333542, 2363948 and 2384521. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04361h |
| This journal is © The Royal Society of Chemistry 2024 |