
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of the cryptic biosynthetic gene cluster encoding the RiPP curacozole reveals a phenylalanine-specific peptide hydroxylase†
Samantha
Hollands‡
a,
Julia
Tasch‡
b,
David J.
Simon
a,
Dimah
Wassouf
b,
Isobel
Barber
a,
Arne
Gessner
bc,
Andreas
Bechthold
 b and
David L.
Zechel
*a
b and
David L.
Zechel
*a
aDepartment of Chemistry, Queen's University, 90 Bader Lane, Kingston, Ontario K7L 3N6, Canada. E-mail: dlzechel@chem.queensu.ca; Tel: +1-613-533-3259
bPharmaceutical Biology and Biotechnology, Institute of Pharmaceutical Sciences, Albert-Ludwigs University, Freiburg, Germany
cInstitute of Experimental and Clinical Pharmacology and Toxicology, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
First published on 5th November 2024
Abstract
Curacozole is representative of a cyanobactin-like sub-family of ribosomally synthesized and post-translationally modified peptides (RiPPs). The molecule is distinguished by its small macrocyclic structure, a poly-azole sequence that includes a phenyloxazole moiety, and a D-allo-Ile residue. The enzymatic steps required for its formation are not well understood. The predicted biosynthetic gene cluster (BGC) for curacozole in Streptomyces curacoi is cryptic, but is shown to be potently activated upon constitutive expression of the bldA-specified Leu-tRNA(UUA) molecule. Heterologous expression and gene deletion studies have defined the minimum BGC as consisting of seven genes, czlA, D, E, B1, C1, F, and BC. The biosynthetic pathway is highly substrate tolerant, accepting six variants of the precursor peptide CzlA to form new curacozole derivatives. This includes replacing the phenyloxazole moiety of curacozole with indole and p-hydroxyphenyloxazole groups by conversion of the corresponding CzlA Phe18Trp and Phe18Tyr variants. In vitro experiments with purified enzymes demonstrate that CzlD and CzlBC perform cyclodehydration and dehydrogenation reactions, respectively, to form a single oxazole from Ser 22 of CzlA. The curacozole BGC is flanked by czlI, a non-essential but conserved gene of unknown function. In vitro studies demonstrate CzlI to be a non-heme iron(II) and 2-oxoglutarate-dependent dioxygenase, catalyzing the hydroxylation of Phe18 on CzlA to form the CzlA Phe18Tyr variant, which is then processed to form the p-hydroxyphenyloxazole derivative of curacozole. Overall, this work highlights the amenability of RiPP biosynthesis for engineering the production of new compounds and adds to the repertoire of known RiPP enzymes.
Introduction
Curacozole (1) is a cytotoxic cyanobactin-like molecule that was recently isolated from a mutant strain of Streptomyces curacoi.1 Cyanobactins are a large and structurally diverse class of ribosomally synthesized and post-translationally modified peptides (RiPPs)2–5 where 1 represents a biosynthetically distinct sub-family that includes aurantizolicin (2),6 YM-216391 (3),7 urukthapelstatin (4),8 and mechercharmycin9 (renamed mechercharstatin,105) (Fig. 1A). These compounds share a macrocyclic structure derived from eight amino acids, a poly-azole sequence that includes a unique phenyloxazole, and a conserved D-allo-Ile residue.114 and 5 are further distinguished by dehydroalanine and dehydrobutyrine residues, respectively. The sequentially linked oxazoles and thiazoles in these compounds are reminiscent of the cyanobactin and telomerase inhibitor, telomestatin (6).12 The BGC encoding 1 (czl)1 is highly conserved with those encoding 2 (aur)6 and 3 (ym),13 reflecting their highly similar structures (Fig. 1B). Interestingly, czl in S. curacoi does not appear to be expressed under standard cultivation conditions,6 which inspired Kaweewan and co-workers to select rifampicin-resistant mutants of this strain in order to induce production of 1.1 Compounds 1 through 5 exhibit strong anticancer activity, analogous to 6, but do not appear to target telomerase as a mode of action.8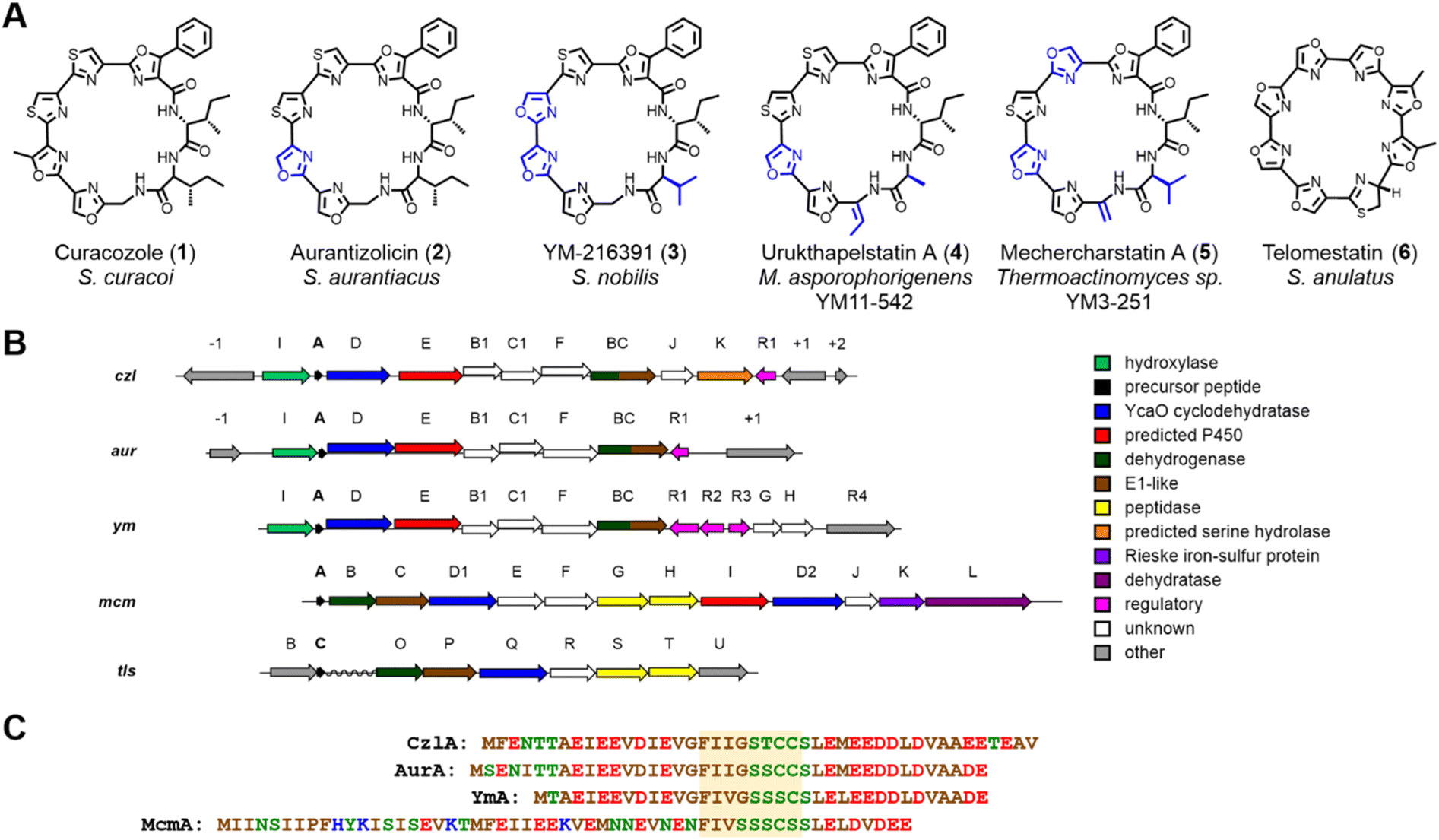 | ||
| Fig. 1 Curacozole and related cyanobactin-like molecules. (A) Structures of curacozole and sub-family relatives. (B) Comparison of the predicted biosynthetic gene clusters encoding curacozole (czl), aurantizolicin (aur), and YM-216391 (ym), mechercharstatin (mcm), and telomestatin (tls). Conserved genes share a common color. Predicted functions are shown in the legend. (C) Amino acid sequences of the precursor peptides CzlA, AurA, Yma, and McmA. The core peptide sequences are highlighted in yellow. | ||
The basic principles of cyanobactin biosynthesis are best understood for the patellamide (pat) and trunkamide (tru) pathways.3,14–16 Using nomenclature for the pat pathway, biosynthesis begins with a precursor peptide (PatE) of approximately 35 to 50 residues in length. Amino acid recognition sequences that flank a core sequence within PatE are recognized by PatD, an ATP-dependent cyclodehydratase of the YcaO superfamily,17 which catalyzes the formation of oxazoline and thiazoline rings from Ser/Thr and Cys side chains, respectively, within the core sequence. The precursor peptide is then cleaved on each side of the modified core sequence by a pair of serine-dependent proteases. One protease, PatA, cleaves on the N-terminal side of the core sequence, followed by the second protease PatG on the C-terminal side. However, unlike PatA, PatG functions as a macrocyclase by capturing the peptidyl-acyl-serine intermediate with the N-terminus of the peptide, resulting in N- to C-macrocyclization rather than hydrolysis. The macrocycle can then undergo dehydrogenation by a flavin mononucleotide (FMN)-dependent oxidase to produce oxazole and thiazole rings. In the case of patellamide biosynthesis, the FMN oxidase is present as a fusion with the macrocyclase PatG; however, there are examples of stand-alone oxidases, such as ThcOx, that will convert azolines to azoles.3,18,19
The conserved BGCs encoding 1, 2, and 3 deviate significantly from canonical cyanobactins such as trunkamides and patellamides, despite featuring similar macrocyclic structures and multiple heterocycles.17 While the czl, aur, and ym clusters all contain a gene encoding the cyclodehydratase YcaO (czlD for 1), they do not contain conserved genes encoding proteases or macrocyclases which typify the cyanobactin class of RiPPs.17 Likewise, the sequences czlE, B1, BC, C1, I, and F have no homologs in the tru/pat BGCs. Finally, while spontaneous epimerization of amino acid side chains is thought to occur in RiPPs when the corresponding α-carbon is adjacent to a thiazole or oxazole, presumably through resonance stabilization of an α-carbanion,3,20 such a mechanism is not possible to account for the formation of the D-allo-Ile residue in 1, where the α-carbon is not directly adjacent to such moieties. Intriguingly, the recently reported BGC for mechercharstatin 5 (mcm)21 appears to be a hybrid of the telomestatin 6 (tls)22 and czl/ym/aur BGCs (Fig. 1B). Most notably, the genes mcmG and mcmH encode proteases homologous to tlsS and tlsT that are proposed to mediate cleavage of the flanking peptide sequences and N- to C-terminal peptide macrocyclization. In contrast, mcmE, mcmF, and mcmI encode homologs of czlB1, czlC1, and czlE, respectively, which are conserved in the czl/ym/aur BGCs (Table S3†). Finally, the czlF gene (which is distinct from mcmF) is unique to the czl/ym/aur clusters. Overall, while these studies provide some insight into the biosynthesis of 1, a minimal BGC for 1 biosynthesis has not been defined, nor have functions been demonstrated for the conserved genes of the czl/ym/aur BGCs.
Inspired by the unique features of the BGC encoding 1, we investigated several key aspects of the biosynthesis of this molecule, including activation of the production of 1, definition of the minimal biosynthetic gene cluster, processing of precursor peptide variants, and reconstitution of reactions with purified enzymes. Overall, these studies expand the impressive repertoire of RiPP biosynthetic reactions and form a basis for further investigations of enzyme function in the biosynthesis of 1 and pathway engineering.
Results
Production of curacozole by S. curacoi is dependent on the bldA-specified Leu-tRNAUUA
We have previously demonstrated that constitutive expression of bldA in Streptomyces, even those with functional bldA sequences, can activate cryptic BGCs and alter secondary metabolite profiles.23 This includes S. curacoi DSM40107. A compound 1 with a molecular ion m/z = 741 ([M + H]+) had been observed previously in liquid cultures of S. curacoi/pTESa-bldA, a strain expressing a copy of bldA from the ermE* promoter in the integrative plasmid pTESa.23 Here we confirm that 1 corresponds to curacozole. It is also observed that bldA influences production of 1 by S. curacoi when grown on solid media. Culture extracts from S. curacoi/pTESa-bldA grown on mannitol soya agar were resolved by UPLC-DAD-MS, showing production of compound 1 (Fig. 2B). In contrast, 1 is not observed in cultures of the control strain S. curacoi/pTESa (Fig. 2A). Interestingly, production of 1 by S. curacoi/pTESa-bldA in liquid culture is highly sensitive to conditions that promote dispersed cell mass. Specifically, production of 1 is observed in shake flasks containing metal springs, which promote dispersed growth (Fig. 2D), but not in flasks lacking springs (Fig. 2C). Compound 1 was purified from a 6 L liquid culture of S. curacoi/pTESa-bldA through a sequence of solid-phase extraction, reverse phase (C18), and silica gel chromatography, yielding 251 mg of curacozole (42 mg L−1 culture purified yield). Data obtained from high-resolution MSn and multidimensional NMR spectroscopy is consistent with previously reported characterization for curacozole (Tables S1 and S2, Fig. S2 to S4†).1,11 The effect of bldA on production of 1 is notable because the native sequence of bldA on the S. curacoi chromosome (accession NZ_KQ947984.1) is predicted to encode a correctly folded Leu-tRNAUUA molecule (Fig. S1B†). Also attesting to the functionality of the native bldA gene is the ability of the wild-type strain to produce abundant spores when grown on solid media (Fig. S1A†). Additionally, the effect of bldA on expression of the czl BGC appears to be indirect, as no genes within the czl BGC contain TTA codons. The flanking czlK sequence contains a single TTA codon, specifying Leu47 of the encoded enzyme. However, as shown below, czlK is not necessary for biosynthesis of 1.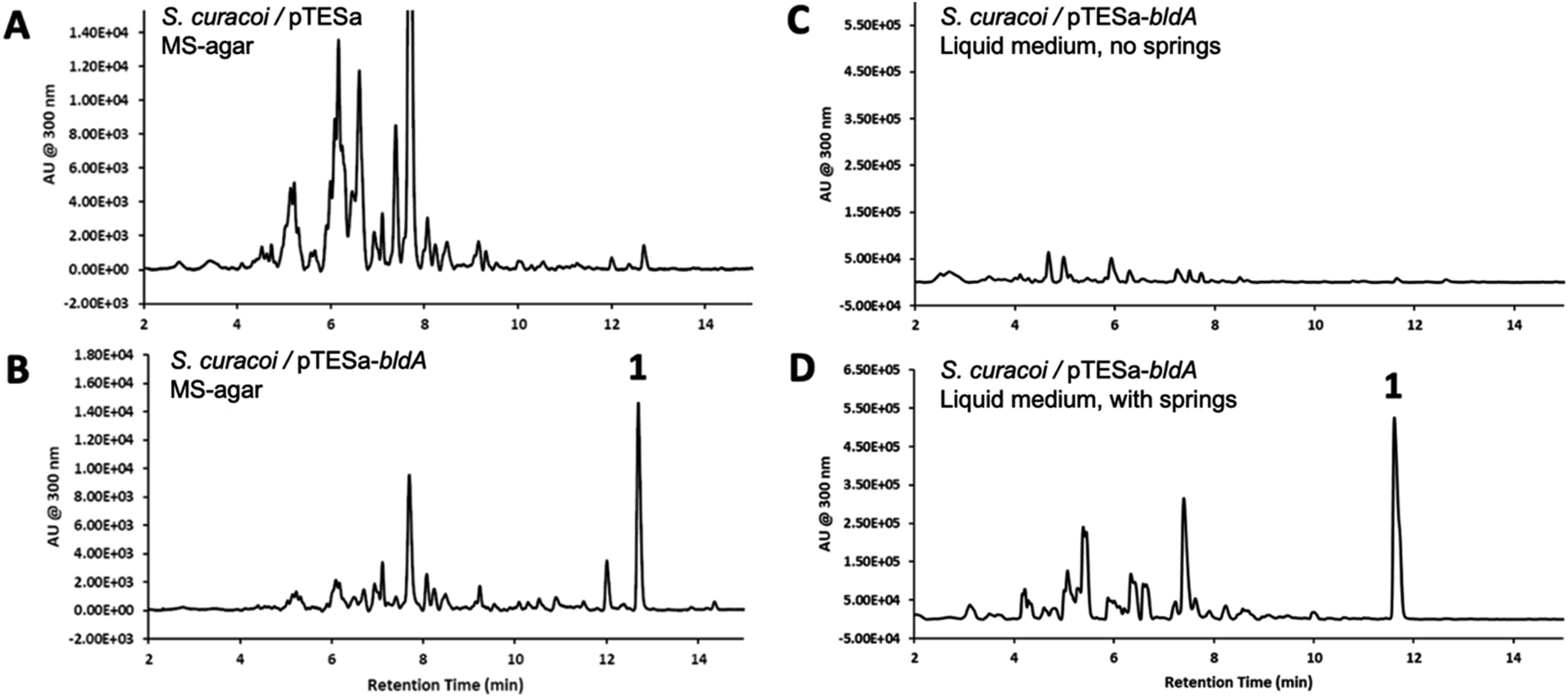 | ||
| Fig. 2 Curacozole production in S. curacoi is sensitive to bldA expression and cultivation conditions. (A) UPLC chromatogram (λ = 300 nm) of an extract from S. curacoi/pTESa grown on MS-agar. (B) Corresponding extract from and S. curacoi/pTESa-bldA. The curacozole peak is labelled as 1. (C) Extract from S. curacoi/pTESa-bldA grown in liquid culture without metal springs in the shake flask for dispersed growth (D). Extract from S. curacoi/pTESa-bldA grown in liquid culture with metal springs. | ||
Definition and mutagenesis of a minimal curacozole BGC
A sequence of approximately 11 kbp spanning the putative BGC for 1 (genes czlI through czlK) was PCR amplified from S. curacoi genomic DNA and cloned into the integrative plasmid pTESa, yielding pTESa-czl (Tables S4 and S5†). Plasmid pTESa-czl was introduced into S. coelicolor CH999 via intergeneric conjugation. Cultivation of S. coelicolor/pTESa-czl in liquid media followed by LC-MS analysis of a culture extract revealed a peak with the same retention time and m/z value (741, [M + H]+) as 1 produced by S. curacoi/pTESa-bldA (Fig. 3, I and II). No such peak is observed in the control strain containing the empty plasmid, S. coelicolor/pTESa (Fig. 3, III). To further refine the boundaries of the czl BGC, the flanking genes czlI, czlJ, and czlK were deleted from pTESa-czl. The gene czlI, which is conserved in the ym and aur BGCs, is predicted to encode an enzyme of the cupin superfamily and resides immediately upstream of the pre-peptide gene czlA. LC-MS analysis of extracts from S. coelicolor/pTESa-czlΔI revealed continued production of 1 (Fig. 3, IV), indicating that czlI is not essential for biosynthesis. CzlK and czlJ, encoding a serine hydrolase and a protein of unknown function, respectively, appear at the opposite end of the czl cluster relative to czlI and are not conserved in the ym and aur BGCs. Production of 1 is also observed in the culture extract of S. coelicolor/pTESa-czlΔJK (Fig. 3, V), indicating that czlJ and czlK are not necessary for biosynthesis. In contrast, deletion of czlD, czlC1, czlF, and czlBC in S. coelicolor/pTESa-czl led to loss of production of 1 (Fig. 3, VI to IX), as did disruption of czlE and czlB1 in S. curacoi-bldA (Fig. 3, X and XI). These experiments demonstrate that the minimum BGC for biosynthesis of 1 consists of the seven genes: czlA, D, E, B1, C1, F, and BC.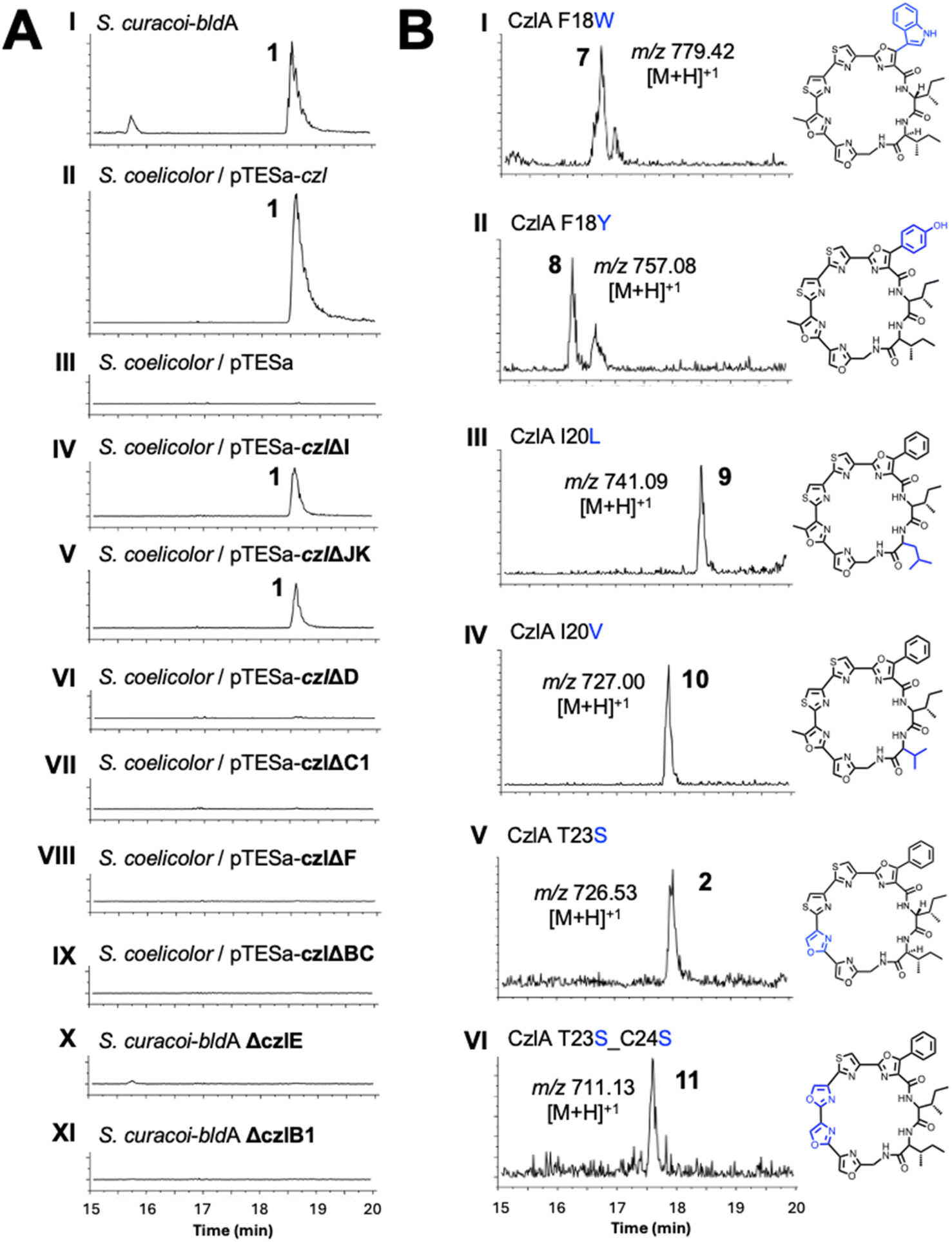 | ||
| Fig. 3 (A) Mutational analysis of the curacozole BGC in S. curacoi-bldA and the heterologous host S. coelicolor/pTESa-czl. Normalized extracted-ion chromatograms for m/z = 740–742 corresponding to curacozole are shown. (B) Extracted ion chromatograms for heterologously produced CzlA variants yielding curacozole derivatives. Observed m/z values are indicated for the numbered peak in each chromatogram. S. lividans ΔYA9 was used to produce 7 and 9, while S. coelicolor CH999 was used to produce 8, 10, 2, and 11. | ||
The substrate flexibility of the biosynthetic pathway leading to 1 was examined by expressing variants of the precursor peptide CzlA. Mutant czlA sequences encoding substitutions within the core peptide sequence FIIGSTCC18–26 were introduced into pTESa-czl, which in turn was transformed into S. coelicolor CH999 and S. lividans ΔYA9. Six CzlA variants with core peptide sequences WIIGSTCC, YIIGSTCC, FILGSTCC, FIVGSTCC, FIIGSSCC and FIIGSSSC (substituted residues underlined) were successfully processed by the pathway to form the expected curacozole derivatives 7, 8, 9, 10, 2, and 11, as detected by LC-MS analysis of culture extracts (Fig. 3B). It is notable that while CzlA F18W and F18Y variants are successfully processed by the pathway, yielding indole and 4-hydroxyphenyloxazole derivatives 7 and 8, the CzlA F18V variant did not yield a product (data not shown). Therefore, the presence of an aromatic amino acid at position 18 in the CzlA sequence appears to be essential for full pathway processing.
Co-dependent cyclodehydratase and dehydrogenase activities of CzlD and CzlBC
The czlD sequence encodes a hypothetical YcaO family cyclodehydratase,17 while czlBC encodes a hypothetical E1-like/dehydrogenase fusion enzyme (Table S3†). It has been hypothesized that CzlD will form the oxazoline/thiazoline rings of curacozole, followed by oxidation by CzlBC to form the final oxazole/thiazole moieties.7,13 To assign specific functions to these enzymes, we first modelled the structures of CzlD and CzlBC using AlphaFold2.24 The top ranked models were submitted to the DALI Server25 to search the Protein Data Bank (PDB) for structural homologs. From the search with CzlD, the top hit was LynD (PDB ID: 4VIV, RMSD = 3.4 Å, Z = 31.8), a YcaO family cyclodehydratase from the aestuaramide pathway (Fig. 4A, Fig. S9A and B†).26,27 Interestingly, the peptide-binding domain observed in the structure of LynD is not present in CzlD, which consists solely of the catalytic YcaO domain. A comparison of the ATP-binding site of LynD with the CzlD model reveals several conserved residues involved in ATP and Mg2+ binding (Fig. 4A, Fig. S9C and S10†). The binding of the precursor peptide CzlA to CzlD was also modelled using AlphaFold2 (Fig. S9D and E†).24 This model revealed that the STCC22–56 sequence of CzlA, which are converted to oxazoles and thiazoles in 1, is positioned adjacent to the predicted ATP-binding site of CzlD. This predicted binding mode of CzlA is consistent with the predicted ATP-dependent cyclodehydratase activity of CzlD. To test this prediction, CzlD and CzlA were expressed and purified from E. coli (Table S6, Fig. S5 and S6†), then incubated together in the presence of ATP. However, no reaction with CzlA was observed over 24 hours at 25 °C by UPLC-MS analysis (Fig. S13†). This indicates that the CzlD/CzlA model is not representative of a reactive complex.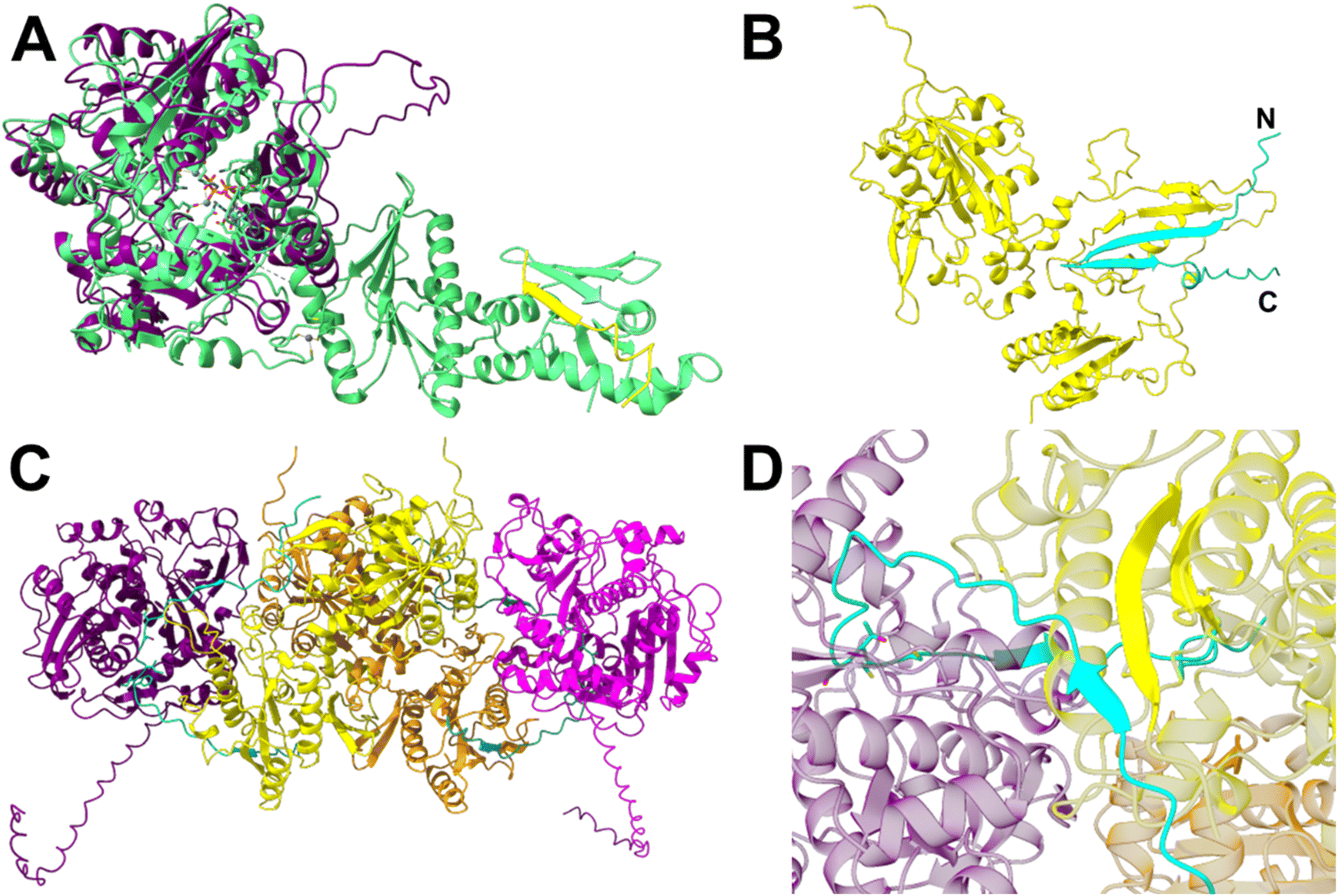 | ||
| Fig. 4 CzlD and CzlBC structure predictions from AlphaFold2 (A and B) and AlphaFold3 (C and D). (A) CzlD (purple) aligned with LynD (PDB ID: 4V1V; RMSD = 3.4 Å, Z = 31.8). The precursor peptide bound to LynD is shown in yellow. Active site residues of LynD and the bound ATP analog ANP are shown as sticks. Metal ions Mg2+ and Zn2+ are shown as spheres. (B) CzlA (cyan) complexed with CzlBC (yellow). (C) Model of the (CzlD/BC)2 complex bound to two CzlA peptides. CzlD monomers shown in purple and magenta; CzlBC in yellow and orange; CzlA in cyan and teal. CzlA residues S22, T23, C24, and C25 are shown as sticks. (D) The beta-sheet interaction between CzlA and CzlBC in the (CzlD/BC/A)2 complex. | ||
We next examined the model of the putative dehydrogenase, CzlBC. A DALI25 search of the PDB identified the top structural homolog as ThcOx (PDB ID: 5LQ4; RMSD = 3.8 Å, Z = 24.8).19 ThcOx is a stand-alone cyanobactin oxidase comprised of an N-terminal, E1-like domain that is predicted to bind the leader sequence of the peptide substrate, and a C-terminal FMN-dependent catalytic domain.18,19 CzlBC shares both the peptide-binding and FMN-binding domains of ThcOx, along with several conserved FMN-binding residues (Fig. S9F–H, K and S12†). A complex of the precursor peptide CzlA bound to CzlBC was modelled using AlphaFold2,24 revealing a beta-strand formed by CzlA residues 9–29 bound to the N-terminal E1-like domain of CzlBC (Fig. 4B, S9I and J†). In this model the N-terminal sequence of CzlA (IEEVDIEVGFII9–20) is predicted to form a β-strand that continues the 2-stranded antiparallel β-sheet of CzlBC's E1-like domain (residues 176–192). This is followed by a variable loop containing the middle of the core peptide (GS21, 22) and then a second, short β-strand (TCCSLEM23–29). Overall, the predicted binding mode of CzlA to the N-terminal E1-domain of CzlBC collectively creates a 4-stranded antiparallel β-sheet (Fig. 4B) which resembles the peptide binding domain architecture of cyclodehydratases such as PatD or LynD.17 The predicted binding mode places the core peptide sequence of CzlA approximately 30 Å from the FMN co-factor binding site of CzlBC. Therefore, a conformational change in CzlBC or an alternative binding mode for CzlA would be necessary for oxidation to occur, which may be induced by complex formation with CzlD.
Because CzlD alone failed to convert the pre-peptide CzlA (Fig. S13†), the combined activities of CzlD and CzlBC towards CzlA were examined in vitro. CzlBC was expressed and purified from E. coli (Fig. S7†). A reaction mixture consisting of 5 μM CzlD, 5 μM CzlBC, 50 μM CzlA, 50 μM FMN, 1 mM ATP, 5 mM MgCl2 (pH 7.5) was incubated at 25 °C for 24 hours. The peptide product was then proteolyzed using endoproteinase GluC to release the CzlA16–28 sequence encompassing the core peptide then analyzed by UPLC-MS. The calculated mass of CzlA16–28 is 1327.6152 Da, and the corresponding M + 1 ion at 1328.6 m/z was observed at 6.3 min (Fig. 5A, upper panel). After the reaction, an additional peak at 1308.6 m/z was observed at 6.6 min (Fig. 5A, lower panel). This mass change corresponds to a single cyclodehydration (−18 Da) and desaturation (−2 Da) of a Ser, Thr, or Cys side chain to form an oxazole or thiazole moiety. Reaction of CzlD and CzlBC with the CzlA variants C24S, C25S, and C24S/C25S also produced a 20 Da mass loss, indicating that heterocyclization was not occurring at either Cys residue (Tables S7 and S8, Fig. S14†). Next, MS/MS based peptide sequencing was used to identify the heterocyclized residue. Reaction products derived from CzlA and CzlA C24S/C25S were treated with GluC to release the CzlA16–28 sequence. The MS/MS fragmentation spectrum of the CzlA16–28 sequence indicates that Ser22 in both variants was converted to an oxazole (Fig. 5B and S15†).
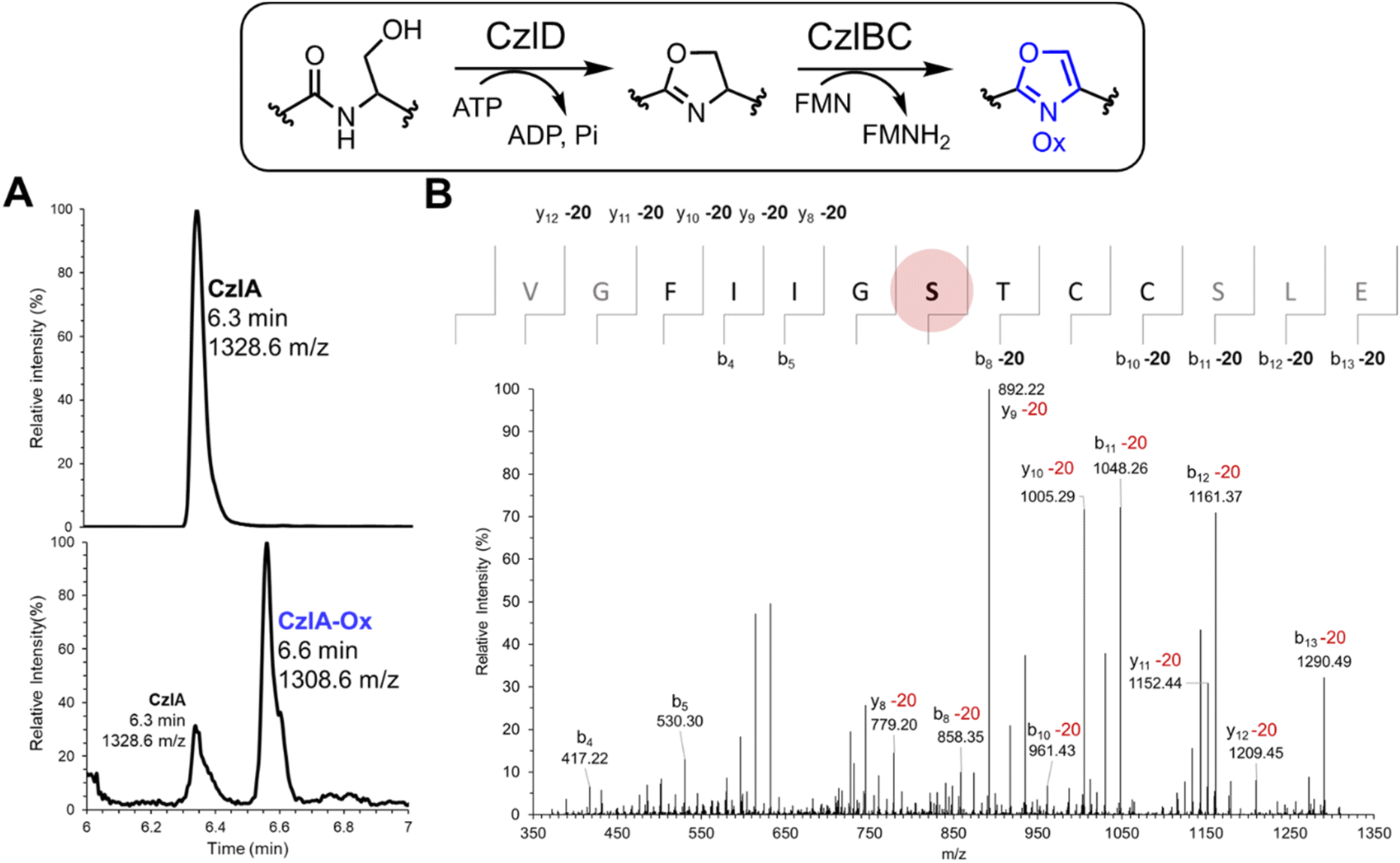 | ||
| Fig. 5 Cyclodehydratase CzlD and dehydrogenase CzlBC combine to form an oxazole on Ser22 of CzlA. (A) UPLC-MS analysis of the cyclodehydration and dehydrogenation of CzlA by CzlD and CzlBC, respectively, followed by proteolysis by GluC to produce the CzlA16–28 sequence. Upper panel shows SIR of CzlA16–28 at 1328.6 m/z. Lower panel shows 5-channel SIR at 1328.6 m/z and the expected m/z for 4 sequential heterocyclization/oxidation reactions (1308.6, 1288.6, 1268.6, and 1248.6 m/z). (B) MS/MS sequencing. The oxazole-containing CzlA was trimmed with GluC to release the CzlA16–28 sequence, which was then sequenced by MS/MS fragmentation of the molecular ion (m/z = 1308.6, [M + H]+). Ions that deviate by m/z = −20 from the calculated values for the native CzlA sequence are indicated. The modified Ser22 residue is highlighted as a red sphere. | ||
The co-dependence of CzlD and CzlBC for heterocyclization of CzlA implies formation of a reactive protein complex. A protein complex consisting of CzlD/BC/A could not be observed by size exclusion chromatography as the individual proteins alone or together eluted with the void volume (data not shown). An AlphaFold3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 model of the CzlD/BC/A complex shows a similar binding mode between CzlD and CzlA, but the β-sheet interaction between CzlA and BC is absent (Fig. S11A and B†). However, as ThcOx19 is biologically relevant as a homodimer, we surmised that including two copies of CzlBC and CzlD may represent a reactive complex. The AlphaFold328 prediction of the corresponding (CzlD/BC/A)2 complex reveals a heterotetramer of CzlD and CzlBC, where two CzlD/BC heterodimers interface through CzlBC (Fig. 4C, S11C and D†). In this model, the N-terminal E1-like domain of CzlBC maintains the β-sheet interaction with the N-terminal leader sequence of CzlA, while the core peptide sequence of CzlA projects into the active site of CzlD (Fig. 4D). Overall, CzlBC is predicted to assist catalysis of CzlD by forming a protein complex as well as by binding the precursor peptide via the E1 domain.
28 model of the CzlD/BC/A complex shows a similar binding mode between CzlD and CzlA, but the β-sheet interaction between CzlA and BC is absent (Fig. S11A and B†). However, as ThcOx19 is biologically relevant as a homodimer, we surmised that including two copies of CzlBC and CzlD may represent a reactive complex. The AlphaFold328 prediction of the corresponding (CzlD/BC/A)2 complex reveals a heterotetramer of CzlD and CzlBC, where two CzlD/BC heterodimers interface through CzlBC (Fig. 4C, S11C and D†). In this model, the N-terminal E1-like domain of CzlBC maintains the β-sheet interaction with the N-terminal leader sequence of CzlA, while the core peptide sequence of CzlA projects into the active site of CzlD (Fig. 4D). Overall, CzlBC is predicted to assist catalysis of CzlD by forming a protein complex as well as by binding the precursor peptide via the E1 domain.
CzlI is a Fe/2OG dependent dioxygenase that creates a CzlA Phe18Tyr variant
CzlI is predicted to specify a metalloenzyme belonging to the JmjC subfamily of cupin enzymes (Table S3†). To investigate the function of CzlI, the structure was first predicted using AlphaFold2 (Fig. S16A and B†).24 The top structural homolog identified by DALI25 is Bacillus subtilis YxbC, a putative Fe(II) and 2-oxoglutarate (2OG)-dependent dioxygenase (PDB ID: 1VRB, RMSD = 3.3 Å, Z = 23.7). In the active site of CzlI, His 145, Asp 147, and His 224 align with the 2 His/1 Asp motif of the YxbC which are used to bind the catalytic Fe(II) ion (Fig. 6A and S17†). Binding of peptide CzlA to CzlI was modelled using AlphaFold2.24 In this model, CzlA is observed to bind across the active site with Phe18 positioned next to the predicted site of the catalytic Fe(II) ion (Fig. 6B, C and S16C, D†).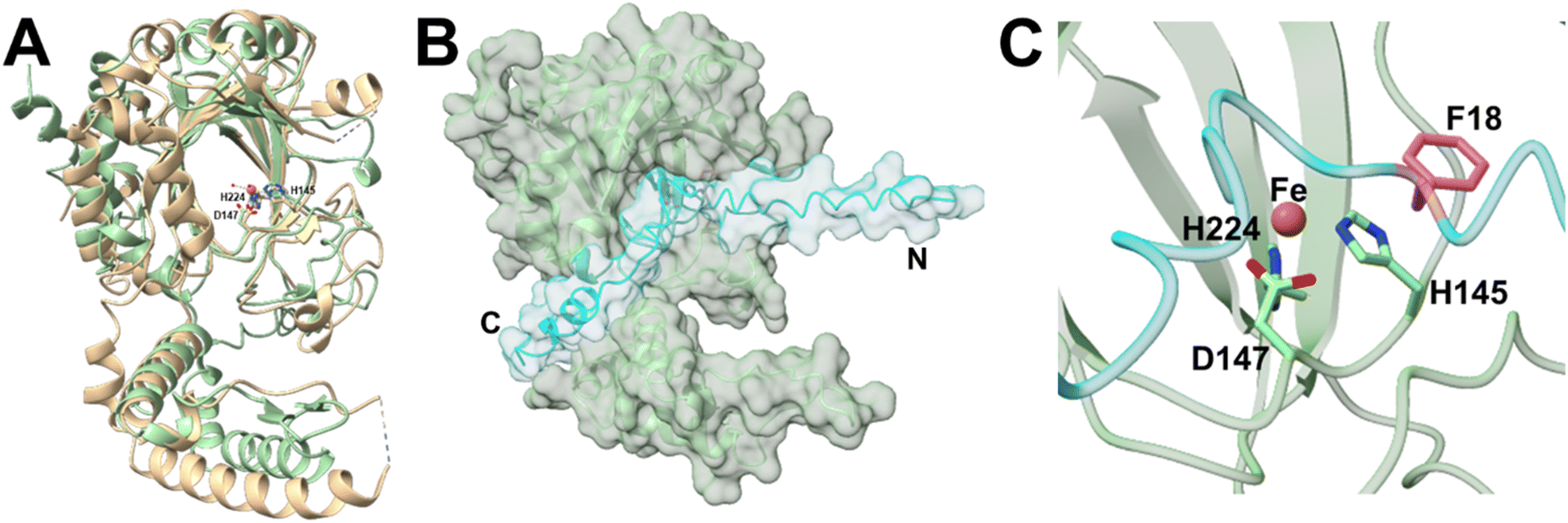 | ||
| Fig. 6 Fe/2OG dioxygenase CzlI structure predictions from AlphaFold2. (A) CzlI (green) aligned with a predicted asparaginyl hydroxylase (PDB ID: 1VRB; RMSD = 3.3 Å, Z = 23.7) shown in tan. Metal ion Fe2+ is shown as an orange sphere and water molecules are shown as smaller red spheres. Labelled iron-coordinating residues are shown as sticks. (B) CzlA (cyan) complexed with CzlI (green). (C) The putative active site residues of CzlI (green) complexed with CzlA (cyan). Metal ion Fe2+ is modelled as an orange sphere. Iron-coordinating residues are shown as sticks. Phe18 of CzlA is shown as pink sticks. | ||
Based on this structural analysis, we predicted that CzlI would perform a Fe(II) and 2OG-dependent oxidation reaction on Phe18 of CzlA. To test this hypothesis, CzlI was expressed with an N-terminal His6 tag in E. coli and purified to homogeneity by immobilized metal affinity chromatography (Table S6 and Fig. S8†). A reaction mixture consisting of 5 μM CzlI, 50 μM CzlA, 1 mM 2OG, 100 μM ammonium iron(II) sulfate, and 200 μM ascorbic acid (pH 7.5) was incubated at 25 °C for 18 hours. The peptide product was digested with GluC to release CzlA16–28, followed by UPLC-MS analysis. Compared to unmodified CzlA (Fig. 7A, upper panel), reaction with CzlI yields a new peak with 1344.6 m/z (Fig. 7A, lower panel). This corresponding increase in mass by 16 Da is consistent with incorporation of a single oxygen atom. An equivalent mass change is observed directly on the intact peptide (Fig. S18A†). No change in the mass of CzlA was observed when 2OG was omitted from the reaction (Fig. S18B, Table S9†). Likewise, no mass change was observed when CzlI was reacted with the variant CzlA F18G (Fig. S19†), implying Phe18 was the modified residue. Pre-treating CzlA with GluC followed by reaction with CzlI showed minimal conversion of the resulting CzlA16–28 peptide to the hydroxylated product (Fig. S18C†), indicating that the sequences flanking the core peptide of CzlA are also necessary for activity. MS/MS based peptide sequencing of the modified CzlA16–28 peptide confirmed that Phe18 was the site of oxygen incorporation (Fig. 7B).
 | ||
| Fig. 7 CzlI oxidatively converts CzlA to CzlA F18Y. (A) UPLC-MS analysis of the hydroxylation of CzlA by CzlI. Upper panel shows SIR of CzlA16–28 at 1328.6 m/z, with core peptide sequence shown in parentheses. Lower panel shows SIR of CzlA following reaction with CzlI with a smaller peak at 1328.6 m/z and a larger peak at 1344.6 m/z. The m/z value increases from substrate to product by a value that corresponds to addition of oxygen (+16 Da). See Table S9† for calculated and observed masses. (B) MS/MS sequencing of hydroxylated CzlA. The peptide was trimmed with GluC to release the CzlA16–28 sequence (shown above), which was then sequenced by MS/MS fragmentation of the molecular ion at 1344.6 m/z ([M + H]+). Ions that deviate by m/z = +16 from the calculated values for the native CzlA sequence are indicated. The hydroxylated Phe18 residue is highlighted with a green sphere. (C) UPLC-MS analysis of dansylated amino acids arising from acid hydrolysis of hydroxylated CzlA. The chromatograms show single ion recordings (SIR) for dansylated phenylalanine (DNS-Phe, m/z 399.13) and dansylated tyrosine (DNS-Tyr, m/z 415.12). Chromatograms (I) and (II) correspond to amino acids derived from hydroxylated CzlA, while (III) and (IV) correspond to standards of dansylated Phe and Tyr. Additional SIR chromatograms were recorded for dansylated D/L- and L-phenylserine, L-meta-Tyr, and D/L-ortho-Tyr (see Fig. S18†). (D) Extracted ion chromatogram (m/z 756–758) of an S. curacoi-bldA extract. The observed m/z value of the peak corresponding to the hydroxylated derivative 8 formed by the activity of CzlI is indicated. | ||
We next determined the position of oxygenation on Phe18 of CzlA. Hydroxylated CzlA was subjected to total acid hydrolysis followed by reaction of the liberated amino acids with dansyl chloride. The resulting dansylated amino acids were resolved by UPLC-MS along with dansylated amino acid standards. CzlI-modified CzlA produces dansylated Phe (Fig. 7C, I and III) in addition to a new peak that co-elutes with dansylated Tyr (Fig. 7C, II and IV), which was confirmed by spiking the sample with dansylated Tyr and observing an increase in the size of the peak (Fig. S20A and B†). This result is consistent with two Phe residues, Phe2 and Phe18, in the CzlA sequence, with Phe 18 being converted to Tyr. Reaction of CzlA with CzlI in the absence of the co-substrate 2OG does not produce the dansylated Tyr peak (Fig. S20C†), which is consistent with the 2OG-dependence of CzlI, and the absence of a Tyr residue in the CzlA sequence. Peaks corresponding to dansylated L-meta-Tyr, D/L-ortho-Tyr, and D/L-phenylserine are not observed as products of CzlI-modified CzlA (Fig. S21†). Because the pathway for 1 can process the CzlA F18Y variant to form the hydroxylated derivative 8 (Fig. 3B, II), we hypothesized that S. curacoi-bldA would also produce this derivative through the activity of CzlI. This proved to be the case, whereby the extracted ion chromatogram of the S. curacoi-bldA culture extract revealed a peak with the expected m/z value for hydroxylated 8 (Fig. 7D). Overall, these results indicate that CzlI oxidatively converts CzlA to form a CzlA F18Y variant, which is ultimately processed by the curacozole pathway to form a hydroxylated curacozole derivative 8.
Discussion
The czl cluster encoding 1 is an interesting example of a cryptic BGC. The bldA specified Leu-tRNAUUA molecule is necessary for translating UUA codons, which are rare in Streptomyces genomes and tend to be found in regulatory and biosynthetic genes.29 Mutations in bldA have long been shown to disrupt sporulation and secondary metabolite biosynthesis in Streptomyces.30 This has led to the hypothesis that bldA and TTA codons form a mechanism for regulating the biosynthesis of bioactive molecules in Streptomyces.31 Constitutive expression of bldA in S. curacoi and other Streptomyces has been shown to activate or enhance production of secondary metabolites, including annimycin and nucleocidin in S. calvus,23,32 peucemycin in S. peucetius,33 jadomycin in S. venezuelae,34 milbemycin A4 in Streptomyces sp. BB47,35 nosokomycin A in S. ghanaensis B38.3,36 daunorubicin in S. peucetius ATCC 27952,37 and ascamycin in Streptomyces sp. 80H647.38 In the case of S. curacoi, bldA expression has a potent effect on the production of 1, with levels of 1 rising from below detection limits in the wild-type strain to 42 mg L−1 culture in the S. curacoi/pTESa-bldA strain. In contrast, 2 mg L−1 culture of 1 was obtained from the rifampicin resistant mutant S. curacoi R25 reported by Kaweewan and co-workers.1 Because yields of cyanobactins and cyanobactin-like compounds are typically poor in native producers, methods such as heterologous expression, optimizing metabolic pathways, and genome engineering have been used to improve titres or produce derivatives, including telomestatin 6 (5 mg L−1),22 patellins (27 mg L−1),39 aurantizolicin 2 (5–10 mg L−1),40 and YM-216391 3 (4–36 mg L−1).13,41 The high titre of 1 induced by bldA expression in S. curacoi is notable in this context and serves as a good starting point for pathway engineering.The minimum BGC encoding 1 has been established through gene deletion and heterologous expression studies as the seven genes czlA, D, E, B1, C1, F, and BC. The flanking genes czlI, czlJ, and czlK, while not essential, may play a regulatory or auxiliary role in the biosynthesis of 1 in S. curacoi, which is bypassed through heterologous expression of the seven essential genes from the strong constitutive promoter, ermE*. Interestingly, studies by Guo and co-workers have shown that the ym BGC encoding 3 is transcribed as an eight-gene cistron (ymI to ymBC) under the control of a single promoter, likely that of ymI (equivalent to czlI).42In vitro studies have now assigned functions to CzlD (YcaO cyclodehydratase), CzlBC (FMN-dependent oxidase), and CzlI (peptide-specific Fe/2OG oxygenase). This leaves CzlE, B1, C1, and F with experimentally undefined roles. In the pathway for 5, deletion of mcmE, the homolog of czlB1, abolishes production of 5 and instead yields an interesting macrocycle that is larger by two amino acids and retains the phenylserine.21CzlB1 is likewise shown to be essential for biosynthesis of 1. This suggests a role for CzlB1 and McmE as cyclodehydratases to form the phenyloxazole. CzlE encodes a predicted P450 monoxygenase, and is thought to add a hydroxyl group to the β-carbon of Phe to form a phenylserine residue.1,21 As shown here, czlE is essential in the biosynthesis of 1, as is the homolog mcmI in the pathway for 5.21 However, the activity of CzlE or its homologs has not yet been reconstituted in vitro, therefore the details of this reaction remain unknown. CzlC1 and czlF encode proteins of unknown function yet are shown in this work to be essential for production of 1. While both czlC1 and czlF sequences are conserved in the ym and aur BGCs, only czlC1 has a homolog, mcmF, in the BGC for 5.21 Finally, the minimal BGC for 1 is notable for lacking the class-defining cyanobactin biosynthetic genes encoding enzymes that would catalyze proteolysis and macrocyclization to form 1. Such functions might be encoded by czlE, B1, or F. It is also possible that proteolysis and/or macrocyclization may be carried out by proteases that are endogenous to Streptomyces. A similar phenomenon is observed in the biosynthesis of 6: when tlsS and tlsT, encoding predicted peptidases (Fig. 1), are deleted, heterologous production of 6 is reduced but not abolished.22
Heterocyclization of serine, threonine, and cysteine side chains to form oxazoles and thiazoles is a distinctive feature of cyanobactins that is shared by the cyanobactin-like family represented by 1. The YcaO family cyclodehydratase CzlD, in combination with the FMN-dependent oxidase CzlBC, is shown to convert Ser22 of the precursor peptide CzlA into an oxazole. This is the first residue of the CzlA STCC22–25 sequence that yields a sequence of oxazole–methyloxazole–thiazole–thiazole in 1. In contrast, TruD has been shown to form heterocycles starting with the most C-terminal residue (in this case Cys) of the core sequence of the peptide substrate.43 CzlD and CzlBC also convert Ser22 to an oxazole when the Cys residues of the STCC22–25 sequence are substituted for Ser. However, in all cases, additional heterocycles are not formed by CzlD and CzlBC. This may stem from the requirement of other modifications to occur on CzlA before other heterocycles can be formed. Notably, heterocyclization operates by a specific order in the biosynthesis of 5, where the first three heterocycles are not formed by McmB/C/D1 on the precursor peptide until the dehydroalanine residue is created.17
CzlD was unable to form an oxazoline on CzlA in the absence of CzlBC, suggesting that CzlBC is providing a required function. The predicted structure for CzlD lacks an E1-like peptide-binding domain that is observed in other YcaO enzymes such as LynD or PatD.17 In these enzymes, the E1 domain engages the peptide substrate while the YcaO domain catalyzes ATP-dependent backbone amide activation and heterocyclization.3,44 The AlphaFold2 model for CzlD predicts binding of the core peptide sequence of CzlA within the ATP-binding site. However, the AlphaFold2 model for CzlA bound to CzlBC shows the leader peptide of the former continuing the β-sheet topology of the E1-like “C” domain of the latter. This resembles the binding mode of a precursor peptide substrate to the aestuaramide cyclodehydratase LynD, where the leader peptide binds to and extends the 3-stranded β-sheet of the E1-like domain to create a 4-stranded anti-parallel β-sheet.26 However, an AlphaFold3 model of (CzlD/BC/A)2 complex combines these interactions, with CzlBC binding the leader sequence of CzlA, while the core peptide sequence of the latter is bound to the active site of CzlD. Therefore, it is possible that CzlD requires CzlBC for recruitment of the CzlA peptide for processing. Interestingly, in the pathway for 5, the oxidase “B” and E1-like “C” components occur as separate proteins.21
CzlI is shown to be a Fe/2OG dependent dioxygenase, and the first example of a phenylalanine-specific peptide hydroxylase.45 Out of two possible Phe residues in the CzlA substrate, only Phe18 is converted to Tyr. The corresponding CzlA F18Y product is processed by the pathway to form a new curacozole derivative 8 with a p-hydroxyphenyloxazole group. Furthermore, the production of 8 through the activity of CzlI was confirmed in S. curacoi-bldA culture extracts. The activity CzlI as a peptide hydroxylase is also dependent on the sequences that flank the core peptide of CzlA, as shown by minimal conversion of a truncated CzlA16–28 sequence. Fe/2OG-dependent dioxygenases are commonly found as tailoring enzymes in biosynthetic pathways and in the post-translational modification of proteins.46–48 CzlI belongs to the Jumonji-C (JmjC) subfamily of Fe/2OG oxygenases, which includes enzymes that hydroxylate amino acid residues of proteins (e.g.: Lys, His, Asn, Asp), as well as demethylate N-methylated Lys and Arg residues of various protein targets, such as histones, as a mechanism of epigenetic regulation.49,50 CzlI also joins a small but emerging group of Fe/2OG enzymes acting on peptides during RiPP biosynthesis, including CinX (β-hydroxylation of Asp in cinnamycin),51 ThoJ and TsaJ (β-hydroxylation of dimethyl-His in thioholgamide and thiostreptamide S4),52 and CanE (β-hydroxylation of Asp in canucin A).53 Interestingly, a predicted P450 monoxygenase, TaaCYP, is proposed to generate an ortho-tyrosine residue in thioalbamide.54 Analogous to these examples, CzlI serves as a tailoring enzyme to enhance the structural diversity of products that are generated by the pathway.
Conclusions
Curacozole is representative of an outlying group of cyanobactin-like molecules that deviate biosynthetically from canonical cyanobactins such as patellamides and trunkamides. A minimal biosynthetic gene cluster has been determined for the biosynthesis of curacozole consisting of seven genes, czlA, D, E, B1, C1, F, and BC. The expression of this BGC is cryptic in the host strain Streptomyces curacoi, and shows a dramatic dependence on the bldA gene, encoding a rare Leu-tRNAUUA molecule. The high titre of curacozole produced in S. curacoi upon constitutive bldA expression, in addition to the tolerance of the pathway to substitutions in the core peptide sequence of precursor peptide CzlA, make this a promising system for producing curacozole derivatives with new bioactivities. Knowledge of the minimal curacozole BGC also sets the stage for functional analysis of the encoded enzymes and defining a biosynthetic pathway. To this end, two early steps in the biosynthesis of 1 and derivatives have been established. The cyclodehydratase CzlD, in combination with the FMN-dependent oxidase CzlBC, create a single oxazole on the precursor peptide CzlA from Ser22, while the Fe/2OG-dependent dioxygenase CzlI initiates the biosynthesis of a derivative of 1 by hydroxylating Phe18 of CzlA to form a CzlA F18Y variant. The timing and enzymes required to perform the subsequent transformations on CzlA, such as epimerization of Ile19, β-hydroxylation of Phe18, formation of two additional oxazoles and two thiazoles, and macrocyclization, remain to be determined. For this reason, the curacozole biosynthetic pathway has more to contribute to the rich repertoire of RiPP biosynthetic enzymes.Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study. The figures and tables supporting this article have been uploaded as part of the ESI.†Author contributions
DLZ and AB conceived of the study and designed experiments. SH, DSS, and IB performed biochemical assays. AG and DSS characterized the cryptic behaviour of curacozole. DSS isolated and characterized curacozole. JT and DW performed gene knock out and heterologous expression studies. DLZ, AB, SH, and JT analyzed the results and wrote the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are greatly indebted to the late Dr Françoise Sauriol for her assistance with the NMR spectroscopic characterization of curacozole. This work is supported by the Natural Sciences and Engineering Research Council of Canada and by the University of Freiburg.References
- I. Kaweewan, H. Komaki, H. Hemmi, K. Hoshino, T. Hosaka, G. Isokawa, T. Oyoshi and S. Kodani, J. Antibiot., 2019, 72, 1–7 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- W. Gu, S.-H. Dong, S. Sarkar, S. K. Nair and E. W. Schmidt, Methods Enzymol., 2018, 604, 113–163 CAS.
- J. Martins and V. Vasconcelos, Mar. Drugs, 2015, 13, 6910–6946 CrossRef CAS PubMed.
- G. Zhong, Z.-J. Wang, F. Yan, Y. Zhang and L. Huo, ACS Bio Med Chem Au, 2023, 3, 1–31 CrossRef CAS.
- M. A. Skinnider, C. W. Johnston, R. E. Edgar, C. A. Dejong, N. J. Merwin, P. N. Rees and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6343–E6351 CrossRef CAS PubMed.
- K. Sohda, K. Nagai, T. Yamori, K. Suzuki and A. Tanaka, J. Antibiot., 2005, 58, 27–31 CrossRef CAS PubMed.
- Y. Matsuo, K. Kanoh, T. Yamori, H. Kasai, A. Katsuta, K. Adachi, K. Shin-ya and Y. Shizuri, J. Antibiot., 2007, 60, 251–255 CrossRef CAS.
- K. Kanoh, Y. Matsuo, K. Adachi, H. Imagawa, M. Nishizawa and Y. Shizuri, J. Antibiot., 2005, 58, 289–292 CrossRef CAS.
- K. Kanoh, Y. Matsuo, K. Adachi, H. Imagawa, M. Nishizawa and Y. Shizuri, J. Antibiot., 2007, 60, C2 CrossRef CAS.
- A. Oberheide, S. Pflanze, P. Stallforth and H.-D. Arndt, Org. Lett., 2019, 21, 729–732 CrossRef CAS.
- K. Shin-ya, K. Wierzba, K. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Seto, J. Am. Chem. Soc., 2001, 123, 1262–1263 CrossRef CAS.
- X.-H. Jian, H.-X. Pan, T.-T. Ning, Y.-Y. Shi, Y.-S. Chen, Y. Li, X.-W. Zeng, J. Xu and G.-L. Tang, ACS Chem. Biol., 2012, 7, 646–651 CrossRef CAS PubMed.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS.
- J. Koehnke, A. F. Bent, W. E. Houssen, G. Mann, M. Jaspars and J. H. Naismith, Curr. Opin. Struct. Biol., 2014, 29, 112–121 CrossRef CAS PubMed.
- C. M. Czekster, Y. Ge and J. H. Naismith, Curr. Opin. Chem. Biol., 2016, 35, 80–88 CrossRef CAS.
- B. J. Burkhart, C. J. Schwalen, G. Mann, J. H. Naismith and D. A. Mitchell, Chem. Rev., 2017, 117, 5389–5456 CrossRef CAS.
- W. E. Houssen, A. F. Bent, A. R. McEwan, N. Pieiller, J. Tabudravu, J. Koehnke, G. Mann, R. I. Adaba, L. Thomas, U. W. Hawas, H. Liu, U. Schwarz-Linek, M. C. M. Smith, J. H. Naismith and M. Jaspars, Angew. Chem., Int. Ed., 2014, 53, 14171–14174 CrossRef CAS PubMed.
- A. F. Bent, G. Mann, W. E. Houssen, V. Mykhaylyk, R. Duman, L. Thomas, M. Jaspars, A. Wagner and J. H. Naismith, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 1174–1180 CrossRef CAS.
- B. F. Milne, P. F. Long, A. Starcevic, D. Hranueli and M. Jaspars, Org. Biomol. Chem., 2006, 4, 631–638 RSC.
- Z.-F. Pei, M.-J. Yang, K. Zhang, X.-H. Jian and G.-L. Tang, Cell Chem. Biol., 2022, 29, 650–659 CrossRef CAS PubMed .e5..
- K. Amagai, H. Ikeda, J. Hashimoto, I. Kozone, M. Izumikawa, F. Kudo, T. Eguchi, T. Nakamura, H. Osada, S. Takahashi and K. Shin-ya, Sci. Rep., 2017, 7, 3382 CrossRef.
- A. Gessner, T. Heitzler, S. Zhang, C. Klaus, R. Murillo, H. Zhao, S. Vanner, D. L. Zechel and A. Bechthold, ChemBioChem, 2015, 16, 2244–2252 CrossRef CAS.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- L. Holm, Nucleic Acids Res., 2022, 50, W210–W215 CrossRef CAS.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. E. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS PubMed.
- J. A. McIntosh, Z. Lin, M. D. Tianero and E. W. Schmidt, ACS Chem. Biol., 2013, 8, 877–883 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- N. Zaburannyy, B. Ostash and V. Fedorenko, Bioinformatics, 2009, 25, 2432–2433 CrossRef CAS.
- O. Tsypik, R. Makitrynskyy, X. Yan, H.-G. Koch, T. Paululat and A. Bechthold, Microorganisms, 2021, 9, 374 CrossRef CAS.
- K. F. Chater and G. Chandra, J. Microbiol., 2008, 46, 1–11 CrossRef CAS.
- L. Kalan, A. Gessner, M. N. Thaker, N. Waglechner, X. Zhu, A. Szawiola, A. Bechthold, G. D. Wright and D. L. Zechel, Chem. Biol., 2013, 20, 1214–1224 CrossRef CAS PubMed.
- R. T. Magar, V. T. T. Pham, P. B. Poudel, A. F. Bridget and J. K. Sohng, Appl. Microbiol. Biotechnol., 2024, 108, 107 CrossRef CAS PubMed.
- S. Qiu, B. Yang, Z. Li, S. Li, H. Yan, Z. Xin, J. Liu, X. Zhao, L. Zhang, W. Xiang and W. Wang, Metab. Eng., 2024, 81, 210–226 CrossRef CAS PubMed.
- N. Matsui, S. Kawakami, D. Hamamoto, S. Nohara, R. Sunada, W. Panbangred, Y. Igarashi, T. Nihira and S. Kitani, J. Gen. Appl. Microbiol., 2021, 67, 240–247 CrossRef CAS PubMed.
- Y. Kuzhyk, M. Lopatniuk, A. Luzhetskyy, V. Fedorenko and B. Ostash, Indian J. Microbiol., 2019, 59, 109–111 CrossRef CAS PubMed.
- A. R. Pokhrel, A. K. Chaudhary, H. T. Nguyen, D. Dhakal, T. T. Le, A. Shrestha, K. Liou and J. K. Sohng, Microbiol. Res., 2016, 192, 96–102 CrossRef CAS.
- Y. Zheng, N. Morita, H. Takagi, Y. Shiozaki-Sato, J. Ishikawa, K. Shin-ya and S. Takahashi, ACS Catal., 2024, 14, 3533–3542 CrossRef CAS.
- Ma. D. Tianero, E. Pierce, S. Raghuraman, D. Sardar, J. A. McIntosh, J. R. Heemstra, Z. Schonrock, B. C. Covington, J. A. Maschek, J. E. Cox, B. O. Bachmann, B. M. Olivera, D. E. Ruffner and E. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1772–1777 CrossRef CAS.
- Z.-F. Pei, M.-J. Yang, L. Li, X.-H. Jian, Y. Yin, D. Li, H.-X. Pan, Y. Lu, W. Jiang and G.-L. Tang, Org. Biomol. Chem., 2018, 16, 9373–9376 RSC.
- L. Li, G. Zheng, J. Chen, M. Ge, W. Jiang and Y. Lu, Metab. Eng., 2017, 40, 80–92 CrossRef CAS.
- W. Guo, Z. Xiao, T. Huang, K. Zhang, H.-X. Pan, G.-L. Tang, Z. Deng, R. Liang and S. Lin, Microb. Cell Fact., 2023, 22, 127 CrossRef CAS PubMed.
- J. Koehnke, A. F. Bent, D. Zollman, K. Smith, W. E. Houssen, X. Zhu, G. Mann, T. Lebl, R. Scharff, S. Shirran, C. H. Botting, M. Jaspars, U. Schwarz-Linek and J. H. Naismith, Angew. Chem., Int. Ed., 2013, 52, 13991–13996 CrossRef CAS PubMed.
- K. L. Dunbar, J. O. Melby and D. A. Mitchell, Nat. Chem. Biol., 2012, 8, 569–575 CrossRef CAS.
- A. K. Alexander and S. I. Elshahawi, ChemBioChem, 2023, 24, e202300372 CrossRef CAS.
- M. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS.
- S.-S. Gao, N. Naowarojna, R. Cheng, X. Liu and P. Liu, Nat. Prod. Rep., 2018, 35, 792–837 RSC.
- C. Q. Herr and R. P. Hausinger, Trends Biochem. Sci., 2018, 43, 517–532 CrossRef CAS PubMed.
- S. Markolovic, S. E. Wilkins and C. J. Schofield, J. Biol. Chem., 2015, 290, 20712–20722 CrossRef CAS PubMed.
- S. Markolovic, T. M. Leissing, R. Chowdhury, S. E. Wilkins, X. Lu and C. J. Schofield, Curr. Opin. Struct. Biol., 2016, 41, 62–72 CrossRef CAS.
- A. Ökesli, L. E. Cooper, E. J. Fogle and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 13753–13760 CrossRef PubMed.
- T. H. Eyles, N. M. Vior, R. Lacret and A. W. Truman, Chem. Sci., 2021, 12, 7138–7150 RSC.
- C. Zhang and M. R. Seyedsayamdost, ACS Chem. Biol., 2020, 15, 890–894 CrossRef CAS PubMed.
- L. Frattaruolo, R. Lacret, A. R. Cappello and A. W. Truman, ACS Chem. Biol., 2017, 12, 2815–2822 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectroscopic data for curacozole; bacterial strains and plasmids; supplementary MS data for enzyme reactions; structural sequence alignments; PDB files for AlphaFold predicted structures; experimental methods for mutagenesis of curacozole BGC, cultivation of bacteria, expression and purification of proteins, enzyme activity assays, LC-MS methods. See DOI: https://doi.org/10.1039/d4sc02262a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |