
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplification-free miRNA detection with CRISPR/Cas12a system based on fragment complementary activation strategy†
Shuang
Zhao‡
a,
Qiuting
Zhang‡
a,
Ran
Luo‡
a,
Jiudi
Sun
a,
Cheng
Zhu
 b,
Dianming
Zhou
*c and
Xiaoqun
Gong
*a
b,
Dianming
Zhou
*c and
Xiaoqun
Gong
*a
aSchool of Life Sciences, Faculty of Medicine, Tianjin University and Tianjin Engineering Center of Micro-Nano Biomaterials and Detection-Treatment Technology (Tianjin), Tianjin, 300072, China. E-mail: gongxiaoqun@tju.edu.cn; Fax: +86-022-27403906
bSchool of Life Sciences, Faculty of Medicine, Tianjin University and Tianjin Key Laboratory of Function and Application of Biological Macromolecular Structures, Tianjin 300072, China
cDepartment of Toxicology, Tianjin Centers for Disease Control and Prevention, NHC Specialty Laboratory of Food Safety Risk Assessment and Standard Development (Tianjin), Tianjin Key Laboratory of Pathogenic Microbiology of Infectious Disease, Tianjin, 300011, China. E-mail: dianmingzhou@foxmail.com; Fax: +86-022-24755561
First published on 17th October 2024
Abstract
CRISPR/Cas12a systems have been repurposed as powerful tools for developing next-generation molecular diagnostics due to their trans-cleavage ability. However, it was long considered that the CRISPR/Cas12a system could only recognize DNA targets. Herein, we systematically investigated the intrinsic trans-cleavage activity of the CRISPR/Cas12a system (LbCas12a) and found that it could be activated through fragmented ssDNA activators. Remarkably, we discovered that the single-stranded DNA (ssDNA) activators in the complementary crRNA-distal domain could be replaced by target miRNA sequences without the need for pre-amplification or specialized recognition mechanisms. Based on these findings, we proposed the “Fragment Complementary Activation Strategy” (FCAS) and designed reverse fluorescence-enhanced lateral flow test strips (rFLTS) for the direct detection of miRNA-10b, achieving a limit of detection (LOD) of 5.53 fM and quantifying the miRNA-10b biomarker in clinical serum samples from glioma patients. Moreover, for the first time, we have developed the FCAS-based CRISPR/Cas12a system for miRNA in situ imaging, effectively recognizing tumor cells. The FCAS not only broadens the scope of CRISPR/Cas12a system target identification but also unlocks the potential for in-depth studies of CRISPR technology in many diagnostic settings.
Introduction
MicroRNAs (miRNAs, RNA = ribonucleic acid) are a class of small noncoding RNA molecules (18–24 nucleotides) that play pivotal roles in regulating gene expression.1,2 Dysregulation of miRNA expression is intricately associated with the onset and progression of many types of tumors, making them promising diagnostic biomarkers and therapeutic targets.3,4 Therefore, convenient, accurate, and rapid nucleic acid tests are essential for early diagnosis, monitoring, routine management, and prognosis of diseases.5,6 However, due to the inherent limitations of miRNAs, such as short sequences, low abundances, high sequence homology among family members, and susceptibility to degradation, traditional analytical methods face significant challenges in detecting miRNAs.7,8 During the past several decades, the quantitative real-time polymerase chain reaction (qPCR) has been the gold-standard tool for measuring relative gene expression.9 Unfortunately, the qPCR-based diagnostic approach has relied heavily on well-established laboratories, expensive instrumentation, and time-consuming processes, hindering their widespread applications to clinical diagnostics, especially for point-of-care testing (POCT).10,11Recently, researchers have sought to utilize simple and cost-effective diagnosis tools to address the above issues. One of them, the Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins, an adaptive immune system, have attracted extensive interest and gradually evolved into a pioneering biotechnology tool in various areas, including gene editing,12–14 transcription regulation,15,16 gene therapy,17 and most recently molecular diagnostic uses.18–20 Among numerous accessible Cas family members, the Class 2 system, Cas12a (previously referred to as Cpf1, subtype V-A), is often considered an ideal option for nucleic acid detection due to its distinctive collateral cleavage activity, which ensures the precise identification of target sequences and facilitates the detection of low concentrations targets.21–23 The Cas12a has a single RuvC structural domain that can recognize single-stranded DNA (ssDNA) targets or dsDNA targets with T nucleotide-rich protospacer adjacent motif (PAM) sequences (5′-TTTV-3′) under the guidance of a single crRNA.23,24 The CRISPR/Cas12a system can realize the signal amplification by activating the trans-cleavage activity upon target recognition and cleaving nontarget fluorophore quencher (FQ)-labeled ssDNA reporters.25–27 With owning the profits of simplicity, cost-effectiveness, reproducibility and efficiency, CRISPR/Cas12a systems have offered powerful molecular recognition tools for tracking gene expression in point-of-care diagnostic28 and live cells.29 Nevertheless, the existing CRISPR/Cas12a system activation modes are confined to ssDNA or dsDNA targets, and direct recognition of RNA in disease-associated nucleic acid molecules presents significant challenges.
For the RNA targets, the CRISPR/Cas12a system often recognizes them through additional reverse transcription steps and/or DNA pre-amplification steps, including recombinase polymerase amplification (RPA),30 loop-mediated isothermal amplification (LAMP),31 catalytic hairpin assembly (CHA),32 “invading stacking primer” amplification reaction (ISAR),33 ligase chain reaction (LCR)34 and rolling circle amplification (RCA).35 However, these methods often require two independent pre-amplification and CRISPR-mediated readout steps, leading to a long turnaround time and increased risk of aerosol contamination. To address all the above limitations, researchers have increasingly focused on investigating the performance of the CRISPR/Cas12a system, particularly in the direct detection of RNA in recent years. Jain et al.36 found that the CRISPR/Cas12a system could be activated by using a pair of split DNA/RNA targets and proposed a SAHARA-based method to directly detect RNA with Cas12a. Nevertheless, this method was insufficient for complex clinical sample analysis due to only binding to 12 nt of the target RNA and low sensitivity. In addition, Liu et al.9 have reported that the detection sensitivity could be improved through a competitive reaction between full-size crRNA and split crRNA in the CRISPR/Cas12a system. However, this method still requires an artificial DNA activator and two different crRNAs to enable direct detection of RNA by the Cas12a system, leading to increased design complexity and cost for the assays. Therefore, it is necessary to develop a novel strategy for the highly sensitive and specific detection of miRNA using the CRISPR/Cas12a system.
In this work, we systematically investigated the trans-cleavage activity of the CRISPR/Cas12a system (LbCas12a) based on a pair of split ssDNA activators. We found that the trans-cleavage activity of CRISPR/Cas12a system could only be effectively activated by the co-existence of two splits ssDNA activators, and either of them is too short to be activated. Most importantly, the direct detection of miRNA could be achieved by simply providing a short DNA sequence complementary to the crRNA-proximal hairpin domain, eliminating the need for reverse transcription or additional recognition mechanisms. Based on these findings, we developed an assay named “Fragment Complementary Activation Strategy” (FCAS) for detecting miRNA directly. As proof-of-concept potential applications, we designed reverse fluorescence-enhanced lateral flow test strips (rFLTS) for the detection of miRNA biomarkers in glioma patients, with the limit of detection that could achieve femtomole levels and enable the detection of clinical serum samples. Importantly, we have applied the FCAS-based CRISPR/Cas12a system for the first time to intracellular imaging, which could effectively distinguish between tumors and normal cells. These pivotal discoveries are poised to transcend the existing limitations of the CRISPR/Cas12a system, thereby broadening the toolbox for Cas12a-based molecular diagnostics.
Results and discussion
CRISPR/Cas12a system tolerates fragmented ssDNA activator inputs for trans-cleavage activity
Doudna et al.37 found that the cleavage activity of the CRISPR/Cas12a system was mainly dependent on the complementary recognition between crRNA and target DNA. Based on this, we systematically investigate the effect of the fragmented ssDNA activator inputs on the CRISPR/Cas12a system and explored the detailed split positions, including between the 5th and 6th bases, 6th and 7th bases, until the 16th and 17th bases, respectively (Fig. S1†). The research indicated that the CRISPR/Cas12a system could be efficiently activated by hybridizing two fragmented ssDNA activators to the crRNA, showing no significant differences compared to full-length ssDNA (Fig. 1a), except for the split position between the 8th and 9th bases (ssDNA1–8,9–30) (Fig. 1b). The reason may be that Lys897 and Lys900 in LbCas12a play a key role in stabilizing the phosphate group between 8th and 9th bases on template DNA through electrostatic interactions (Fig. 1c). Breaking the phosphodiester bond at this position could affect protein-substrate interactions. The split positions of the input ssDNA were in the hairpin “central” structural domain (between the 10th and 11th bases), and the CRISPR/Cas12a system exhibited the optimal trans-cleavage activity. Consistent with previously reported results,36,38 when the length of preposition ssDNA (pre-ssDNA) exceeded 11 nucleotides (nt), the activation efficiency increased gradually, and the CRISPR/Cas12a system could be completely activated when the pre-ssDNA was longer than 14 nt (Fig. 1d). For the rear position ssDNA (rear-ssDNA), activation of the CRISPR/Cas12a system only happened when the rear-ssDNA lacked 5–7 bases of pairing with the 5′-end of crRNA (ssDNA6–30, ssDNA7–30, ssDNA8–30) (Fig. 1e). To sum up, the CRISPR/Cas12a system exhibited a remarkable capability to accept two distinct ssDNA target sequences (each corresponding to a different position on the same crRNA), thereby initiating the trans-cleavage activity.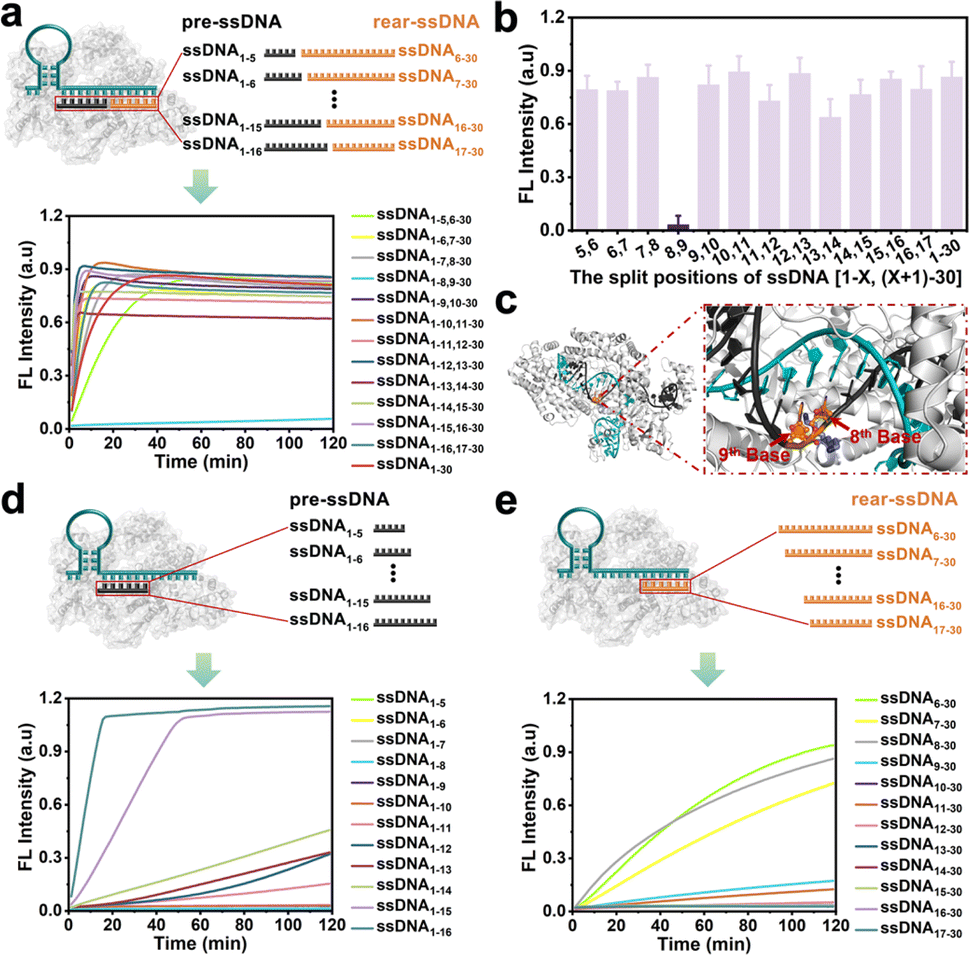 | ||
| Fig. 1 CRISPR/Cas12a system tolerates a combination of fragmented ssDNA activator inputs. (a) Schematic and real-time fluorescence analysis of Cas12a/crRNA complex binding two fragmented ssDNA activators (total 30 nt); (b) fluorescence intensity of input two fragmented ssDNA activators reactions at 40 min (n = 3) (c) diagram of the Cas12a protein structure at special sites. Schematic and real-time fluorescence analysis of Cas12a/crRNA complex binding to different ssDNA activators: (d) pre-ssDNA (5–16 nt); (e) rear-ssDNA (14–25 nt). | ||
Activating the CRISPR/Cas12a system with fragmented RNA/DNA activators
The CRISPR/Cas12a system has been proven the precise recognition of ssDNA and dsDNA targets. Nevertheless, direct RNA detection increased the difficulty and complexity of this system. Based on the finding that the two fragmented ssDNA activator inputs could effectively activate the CRISPR/Cas12a system, we further explored the effect of rear-RNA instead of rear-DNA on the trans-cleavage activity of the CRISPR/Cas12a system (Fig. 2a). The system was efficiently activated with cleavage efficiency comparable to that of fragmented ssDNA (Fig. 2b). Meanwhile, the activation efficiency improved with the increasing length of ssDNA. Besides, only the pre-ssDNA or rear-RNA had difficulty activating the CRISPR/Cas12a system (Fig. 2c and d). We also investigated the pre-ssDNA replaced with pre-RNA and found that it was unable to activate the trans-cleavage activity of the CRISPR/Cas12a system (Fig. S2†). To minimize background signal interference, the ssDNA1–10 and RNA11–30 were selected for synergistic activation of the CRISPR/Cas12a system in subsequent applications. The above phenomena suggested that the CRISPR/Cas12a system could be directly applied to RNA detection without reverse transcription.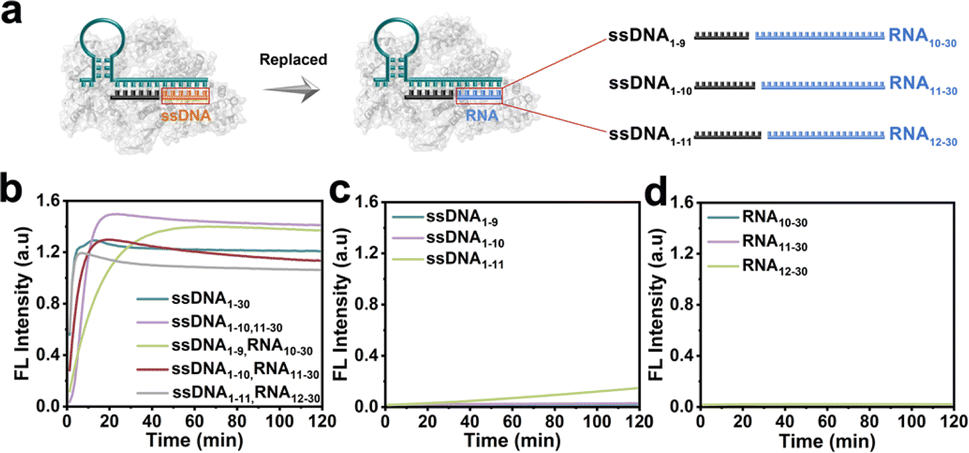 | ||
| Fig. 2 Detection of RNA substrates using split activators in the CRISPR/Cas12a system. (a) Schematic representation of the CRISPR/Cas12a system activated by inputting replaced RNA. Real-time fluorescence of the Cas12a/crRNA complex binding to different activators (b) fragmented ssDNA and RNA (total 30 nt); (c) ssDNA (9–11 nt); (d) RNA (19–21 nt). | ||
In most cases, the target DNA to be detected often involves a specific segment of the entire nucleic acid chain rather than precisely at the terminal positions. Consequently, the 3′ end of the rear-ssDNA often had protruding sequences. Subsequently, we simulated this situation by adding the 5T and 10T sequences to the 3′-end of rear-ssDNA respectively, to investigate the trans-cleavage activity of the CRISPR/Cas12a system. The results in Fig. S3a† revealed that the activation efficiency of the CRISPR/Cas12a system decreased compared to that with two fragmented ssDNA activators (ssDNA1–10,11–30). The protrusion at the 3′-end of rear-ssDNA produces steric hindrance, affecting the activation efficiency of the CRISPR/Cas12a system, we speculate that long-chain targets may not be effectively detected in this case. In addition, deletions of 1 to 7 bases (Gap) between the pre-ssDNA and rear-ssDNA also hardly activate the trans-cleavage activity of the CRISPR/Cas12a system (Fig. S3b†).
FCAS-based CRISPR/Cas12a system for amplification-free miRNA-10b detection using rFLTS
Based on the above findings, the CRISPR/Cas12a system could be activated with DNA and RNA heteroduplexes, we proposed an assay named “Fragment Complementary Activation Strategy” (FCAS) for direct detection of miRNA. MiRNA-10b was overexpressed in gliomas compared to normal tissues and gradually increased with disease progression.39,40 Therefore, rapid, sensitive, and accurate detection of miRNA-10b was important for early diagnosis and prognosis of diseases. Hence, the full-sized crRNA (crRNAmiRNA-10b) was first designed that specifically bind to miRNA-10b and ssDNA1–10 activators. The rear position ssDNA was substituted with miRNA-10b could efficiently activate the CRISPR/Cas12a system (Fig. S4†). Next, we constructed rFLTS to detect miRNA-10b using the FCAS-based CRISPR/Cas12a system (Fig. 3a). In the absence of miRNA-10b, the trans-cleavage activity of the CRISPR/Cas12a system was not activated, preventing the cleavage of T3 reporters. The 30 nm of AuNPs labeled probes (AuNPs@T2) could conjugate with the T3 reporters and were hitched by the Cy5-SA-biotin-T1 modified on the T-line, resulting in a red colorimetric signal and quenched fluorescence signal (quenching of Cy5 dyes) (Fig. S5†). In the presence of miRNA-10b, the CRISPR/Cas12a system was activated, leading to the efficient and continuous cleavage of T3 reporters, which prevents their hitch by Cy5-SA-biotin-T1 on the T-line. To obtain optimum analytical performance, several key experimental parameters were first optimized (Fig. S6 and S7†). Under the optimal conditions, different concentrations of miRNA-10b standard solutions (0–10−15 mol L−1) were prepared to investigate the sensitivity of the proposed FCAS-based rFLTS (Fig. 3b). The red signal on the T-line enhanced with the decreasing concentrations of miRNA-10b, as well as the fluorescence signal decreased (Fig. 3c and d). A standard curve designated for miRNA-10b in the dynamic range from 2.5 × 10−14 mol L−1 to 5 × 10−13 mol L−1 was fitted as Y = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 577.42 + 26.06X (Y was the T-line fluorescence intensity, and X was the concentrations of miRNA-10b), with a correlation coefficient (R2) of 0.98027 (Fig. 3e). In addition, the limit of detection (LOD) was calculated to be 1.17 pM and 5.53 fM in the naked-eye and fluorescence modes, respectively (Fig. S8†). Compared with other miRNA detection methods (Table S2†), the constructed FCAS-based rFLTS biosensor platform has good sensitivity in detecting miRNAs with low expression levels and different application requirements.
577.42 + 26.06X (Y was the T-line fluorescence intensity, and X was the concentrations of miRNA-10b), with a correlation coefficient (R2) of 0.98027 (Fig. 3e). In addition, the limit of detection (LOD) was calculated to be 1.17 pM and 5.53 fM in the naked-eye and fluorescence modes, respectively (Fig. S8†). Compared with other miRNA detection methods (Table S2†), the constructed FCAS-based rFLTS biosensor platform has good sensitivity in detecting miRNAs with low expression levels and different application requirements.
 | ||
| Fig. 3 Analytical sensitivity of FCAS-based rFLTS for miRNA-10b detection. (a) Schematic diagram of the FCAS-based rFLTS for detecting miRNA-10b. (b) Naked eyes visualization and fluorescent images in response to different concentrations of miRNA-10b. Response of the (c) naked eye visualization and the (d) fluorescent intensity to miRNA-10b at different concentrations under FCAS-based rFLTS. (e) Calibration curve showing the relationship between T-line fluorescent intensity and miRNA-10b concentrations in the range of 2.5 × 10−14 mol L−1 to 5 × 10−13 mol L−1. Error bars represent the standard deviation of the values. | ||
The specificity of the proposed FCAS-based rFLTS was further assessed by monitoring five different types of miRNAs (miRNA-21, miRNA-5010, miRNA-331, miRNA-141, and miRNA-122 at 10−7 mol L−1), and the results demonstrated excellent selectivity (Fig. 4a and b). Subsequently, the accuracy and practicability of the FCAS-based rFLTS were assessed through the detection of miRNA-10b in spiked healthy human serums using the standard addition method. The calibration curve of fluorescence intensity versus the different concentrations of miRNA-10b was shown in Fig. 4c, and the detected levels of miRNA-10b in each sample were consistent with the added (Fig. 4d). Furthermore, the average recoveries were 107.77–112.02% with relative standard deviation (RSD) values lower than 6.83%, indicating an acceptable precision for the detection of miRNA-10b in complex sample matrices (Table S3†).
 | ||
| Fig. 4 Rapid and specific detection of miRNA-10b through FCAS-based rFLTS. (a) Photographs and (b) corresponding histogram of specificity results for detecting miRNA-10b. Numbers 1–6 represent miRNA-10b, miRNA-21, miRNA-141, miRNA-5010, miRNA-31 and miRNA-122. (c) Calibration curve of fluorescence intensity versus concentration of miRNA-10b in human serum samples. (d) Serum analysis of five healthy samples using standard addition method for the detection of miRNA-10b. (e) Detection results of miRNA-10b using FCAS-based rFLTS in clinical serum samples. (f) Identification of miRNA-10b in 20 clinical patient samples through FCAS-based rFLTS and the RT-qPCR assay. (g) Correlation assay between the developed FCAS-based rFLTS and the RT-qPCR assay. | ||
To further evaluate the practical applicability of this biosensing approach, the FCAS-based rFLTS and the reverse-transcription quantitative real-time polymerase chain reaction (RT-qPCR) were applied to the detection of miRNA-10b in serum samples (collected from the Tianjin Medical University Affiliated General Hospital) for the clinical monitoring of glioma. The detection results of miRNA-10b in different clinical samples were consistent with the assessment results of RT-qPCR, with an R2 of 0.977 (Fig. 4e–g and S9†). Based on these results, the developed biosensor served as a promising and accurate quantitative platform for early and rapid detection of miRNA-10b.
Implementation of the FCAS-based CRISPR/Cas12a system in glioma cells
Inspired by the prominent performance of the developed FCAS-based CRISPR/Cas12a system for in vitro detection of miRNA-10b, we subsequently endeavored to employ this system to monitor miRNA-10b expression in living cells. Previous studied39 have proved that miRNA-10b was highly expressed in U87 cells (human glioma cell lines), so we selected U87 cells as positive cells and HT22 cells (mouse hippocampal neuron cell lines) as negative cells. The commercial transfection reagent jetPRIME was chosen to transport all components of the biosensor (crRNAmiRNA-10b, ssDNA1–10, and AF488-sssDNA-BHQ1) into cells (Fig. 5a). Meanwhile, the AuNPs-PEG was selected to transfer Cas12a proteins into cells through electrostatic adsorption (Fig. S10†). First, the potential cytotoxicity of the AuNPs-PEG to U87 and HT22 cells was evaluated using cell viability analysis. After 24 h of incubation with different concentrations of the AuNPs-PEG (1–40 μM), the viabilities of the cells were all maintained above 90%, indicating low toxicity of AuNPs-PEG to cells (Fig. S11†). Subsequently, we optimized the incubation time of the FCAS-based CRISPR/Cas12a system in U87 cells through high-resolution laser confocal microscope (Fig. S12†). A noticeable AF488 fluorescence signal enhanced continuously with longer incubation times, until the fluorescence intensity reached a maximum at 3 h. So, the optimal incubation time was selected as 3 h. Subsequently, we further explored the miRNA-10b sensing ability of the FCAS-based CRISPR/Cas12a system in living cells. U87 cells showed strong fluorescence signals due to overexpressed miRNA-10b, while HT22 cells exhibited weak fluorescence, suggesting the selective miRNA-10b sensing performance of this system (Fig. 5b–d). Collectively, these results indicated our FCAS-based CRISPR/Cas12a system could successfully monitor miRNA-10b in tumor cells. | ||
| Fig. 5 Exploration of mRNA-10b imaging capabilities using the FCAS-based CRISPR/Cas12a system. (a) Illustration of design of the FCAS-based CRISPR/Cas12a system for in situ imaging to monitor miRNA-10b. (b) Fluorescence images of U87 and HT22 cells treated with the FCAS-based CRISPR/Cas12a system. U87 cells and HT22 cells were treated with or without Cas12a proteins, respectively. Scale bar: 10 μm. (c) Corresponding flow cytometry analysis. (d) Statistical histogram analysis of U87 and HT22 cells. | ||
Experimental
Ethical statement
All experiments were approved by the ethics committee of the Tianjin Medical University Affiliated General Hospital. The ethical approval number was IRB2020-KY-097. All methods were performed in accordance with the Declaration of Helsinki and the relevant guidelines and regulations. All serum samples were collected from anonymized patients with written informed consent under the agreement of the responsible ethical committees. All assays were conducted and adhered to legal requirements and ethical guidelines.Assay procedure of FCAS-based CRISPR/Cas12a system
2 pM Cas12a, 1×NEBuffer 2.1, 1 μM fluorescence-quencher reporters, 0.1 μM crRNA and different combinations of ssDNA activators (Final concentration was 0.1 μM) were prepared and complemented with DNase/RNase-free water to make a final volume of 10 μL. Reactions were allowed to proceed for 2 h at 37 °C on a real-time PCR system with fluorescence signals monitored.Synthesis of the fluorescence quenched AuNPs@T2 probes
The traditional AuNPs synthesized were shown in ESI.† The AuNPs were conjugated with sulfhydryl-modified T2 nucleic acid chains (SH-T2) via the Au–S covalent bonds. Briefly, the 100 μL of SH-T2 (10 μM) was mixed with 100 μL of TCEP (1 mM) and incubated for 30 min at ambient temperature. Then, the activated SH-T2 was added to 1 mL of AuNPs with a uniform particle size of 30 nm and incubated for 60 min. Subsequently, 1 M NaCl solution was slowly added to the above mixture until the final concentration reached 0.3 M. The labeled probes were stored at 4 °C for 24 h. The final products, the fluorescence quenched AuNPs@T2 probes, were recovered through centrifugation (6950g, 10 min) and then redispersed in a 0.01 M PBS buffer (pH 7.4).Preparation of the rFLTS
SA and N-hydroxysuccinimide ester-Cyanine 5 (Cy5-NHS) were mixed in 500 μL of boric acid solution (0.1 M, pH 8) (molar mass ratio of 1:10) and incubated for 6 h at room temperature. Then, the biotin-T1 was added to the above mixture (molar mass ratio of 1:5) and incubated for 30 min. The supernatant was removed by centrifugation at 10010g for 40 min at 4 °C using a 10 kD millipore ultrafiltration tube. The final product (Cy5-SA-biotin-T1) was recovered through centrifugation and fixed to 1.5 mg mL−1 with 0.01 M PBS buffer (pH 7.4). Similarly, 2 mg mL−1 of SA-biotin-C1 was also prepared.
The reverse fluorescence-enhancement test strips (rFLTS) contain five major parts: NC membranes with a test line (T-line) and a control line (C-line), sample pads, conjugate pads, polyvinyl chloride (PVC) backing cards, and absorbent pads. Among them, the conjugate pads were pre-blocked with 4 mL immune buffer (5 mM PVP-10000, 0.15 M sucrose, 0.45 mM BSA, 2% Tween-20 in 0.01 M PBS) and dried at 37 °C for 8 h. The SA-biotin-C1 (2 mg mL−1) and Cy5-SA-biotin-T1 (1.5 mg mL−1) were applied onto the NC membranes at different densities of 0.6 and 0.5 μL cm−1 as the C-line and T-line, respectively. The interval of two lines was separated at a 4.0 mm interval, then the NC membranes were dried completely at 37 °C. Finally, different pads were constructed on a PVC backing card with an overlap of ∼2 mm and then were cut into 3.5 mm individual strips by a guillotine cutter. The prepared rFLTS were placed in aluminum foil bags and stored at 4 °C for the subsequent assays.
Detection of miRNA-10b with the rFLTS
2 pM Cas12a, 1×NEBuffer 2.1, 0.1 μM crRNAmiRNA-10b, 0.1 μM ssDNA1–10, 6 μM T3 reporters and a series of different concentrations miRNA-10b standard solutions (10−7, 10−8, 10−9, 10−10, 10−11, 10−12, 10−13, 10−14, 10−15 and 0 mol L−1) were prepared and complemented with DNase/RNase-free water to make a final volume of 10 μL. After reaction at 37 °C for 40 min, the above products and 6 μL of AuNPs@T2 probes were added to 60 μL of PBS solution (0.01 M, pH 7.4). After mixing, the strips were immersed into the solution and incubated for 10 min. Then strips were taken out, and photos were obtained using a smartphone and Azure C600 with default parameters, respectively. The intensity of the T-line was measured using ImageJ software with the following procedure: open the ImageJ software and open an image in “File”; transform images into 32 bit type; subtract background; set measurements to take analysis of integrated density; invert color; use the rectangle tool to obtain the integrated density of the T-line. The T-line intensity was obtained by integrating the peaks. The limit of detection (LOD) was calculated as follows:where y was the LOD, xb1 was the blank mean, k was the constant data related to confidence (usually k = 3 was taken), and Sb1 was the standard deviation of the blank value.
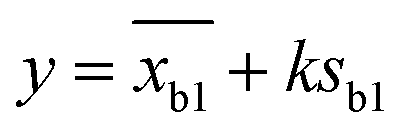
Furthermore, miRNA-10b (10−8 mol L−1) and five different types of miRNAs (10−7 mol L−1), including miRNA-21, miRNA-5010, miRNA-331, miRNA-141 and miRNA-122 were used to assess the specificity.
Serum miRNA extraction and detection in clinical samples by rFLTS
Informed written consent from all participants or next of kin was obtained before the research. All serum samples were obtained from Tianjin Medical University Affiliated General Hospital (Tianjin, China), and all methods were conducted in accordance with the approved guidelines. Total miRNAs in 200 μL serum per sample were extracted using the serum/plasma miRNA extraction kit. All samples were purified according to the manufacturer's instructions. The eluted miRNAs were stored in 20 μL DNase/RNase-free water at −20 °C until needed. These samples were analyzed following the procedures mentioned above.Intracellular imaging
:1) for the labeling reaction for 4 h. Then, the precipitate (AuNPs-PEG probes) was recovered by centrifugation (10010g, 10 min) and redispersed in PBS solution (0.01 M, pH 7.4). Afterward, the Cas12a proteins were placed in the prepared AuNPs-PEG probes for 2 h. Finally, the unloaded Cas12a proteins were discarded by centrifugation and the AuNPs-PEG@Cas12a was redispersed in 0.01 M PBS solution for cell imaging.
The U87 and HT22 cells were inoculated seeded and cultured in a confocal dish with a cell density of about 1 × 105 cells, respectively. After cultured for 24 h in the sterile incubator, the fully dispersed AuNPs-PEG@Cas12a in DMEM medium containing 10% FBS was cultured with U87 and HT22 cells for 3 h at 37 °C. Then the cells were transfected with crRNAmiRNA-10b, ssDNA1–10, and reporter molecules (AF488-ssDNA-BHQ1) using the jetPRIME reagent for a period of 4 h, washed with 0.01 M PBS solution three times and stained with Hoechst 33342 (10 μg mL−1) for 20 min. Then the cells were washed again with 0.01 M PBS solution three times. Cell imaging was performed on a high-resolution laser confocal microscope with a 100× oil dipping objective less with a 488 nm laser channel.
000 useable events, and the data were analyzed by the FlowJo software.
Conclusions
The CRISPR/Cas12a system is known to recognize DNA targets and exhibits distinctive collateral ssDNA cleavage activity. In this research, we found that the trans-cleavage activity of the CRISPR/Cas12a system could be directly activated by hybridizing two fragmented ssDNA activators to the crRNA, except the split position between the 8th and 9th bases. Notably, the direct detection of miRNA sequences could be achieved after providing a short DNA sequence complementary to the crRNA-proximal hairpin domain without the need for reverse transcription and special recognition mechanisms. Meanwhile, we also found the trans-cleavage activity of CRISPR/Cas12a was affected by the 3′ protruding ends when detecting longer nucleic acid targets. By replacing one fragment of ssDNA activators (rear-ssDNA) with miRNA-10b, we proposed a split-activator-based method called FCAS and designed rFLTS, achieving highly sensitive (LOD of 5.53 fM) and selectivity for the direct detection of miRNAs targets. With the FCAS-based rFLTS, we successfully detected the miRNA-10b biomarker in clinical serum samples from glioma patients, achieving performance consistent with RT-qPCR methods. Importantly, for the first time, a miRNA in situ imaging platform based on the FCAS was developed to identify tumor cells. In conclusion, the FCAS-based CRISPR/Cas12a system could be expanded to other CRISPR-Cas enzymes for developing advanced molecular diagnostic tools and broadening its application fields.Data availability
All relevant data is presented in the article and the ESI.†Author contributions
Shuang Zhao: conceptualization, methodology, formal analysis, writing – original draft. Qiuting Zhang: conceptualization, methodology, formal analysis. Ran Luo: conceptualization, methodology, formal analysis. Jiudi Sun: validation, visualization. Cheng Zhu: validation, visualization. Dianming Zhou: conceptualization, writing – review & editing, supervision, project administration. Xiaoqun Gong: conceptualization, writing – review & editing, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (U2233206), National Natural Science Foundation of China (62105242), and the National Science Foundation of Tianjin (21ZXGWSY00060).Notes and references
- J. J. Kim, J. S. Hong, H. Kim, M. Choi, U. Winter, H. Lee and H. Im, CRISPR/Cas13a-assisted amplification-free miRNA biosensor dark-field imaging and magnetic gold nanoparticles, Sens. Diagn., 2024, 3, 1310–1318 RSC
.
- Y. C. Zhao, J. Z. Xiang, H. Cheng, X. J. Liu and F. Li, Flexible photoelectrochemical biosensor for ultrasensitive microRNA detection based on concatenated multiplex signal amplification, Biosens. Bioelectron., 2021, 194, 113581 CrossRef CAS PubMed
- Y. C. Han, J. Zhou, F. Liu, Y. Ouyang, R. Yuan and Y. Q. Chai, pH-Stimulated Self-Locked DNA Nanostructure for the Effective Discrimination of Cancer Cells and Simultaneous Detection and Imaging of Endogenous Dual-MicroRNAs, Anal. Chem., 2023, 95, 12754–12760 CrossRef CAS PubMed
- H. R. Mao, Y. Cao, Z. H. Zou, J. A. Xia and J. Zhao, An enzyme-powered microRNA discriminator for the subtype-specific diagnosis of breast cancer, Chem. Sci., 2023, 14, 2097–2106 RSC
- M. Li, F. F. Yin, L. Song, X. H. Mao, F. Li, C. H. Fan, X. L. Zuo and Q. Xia, Nucleic Acid Tests for Clinical Translation, Chem. Rev., 2021, 121, 10469–10558 CrossRef CAS PubMed
- S. C. Song, N. Li, L. P. Bai, P. P. Gai and F. Li, Photo-Assisted Robust Anti-Interference Self-Powered Biosensing of MicroRNA Based on Pt-S Bonds and the Inorganic-Organic Hybridization Strategy, Anal. Chem., 2022, 94, 1654–1660 CrossRef CAS PubMed
- Y. Yao, W. Liu, J. P. Guan, Y. J. Cheng, Z. L. Wu, Q. Liu and X. Q. Chen, Synergy of Target-Induced Magnetic Network and Single-Drop Chromogenic System for Ultrasensitive “All-in-Tube” Detection of miRNA in Whole Blood, Anal. Chem., 2024, 96, 12012–12021 CrossRef CAS PubMed
- H. Y. Li, Q. T. Yang, Z. X. Wang and F. Li, Iridium Complex with Specific Intercalation in the G-Quadruplex: A Phosphorescence and Electrochemiluminescence Dual-Mode Homogeneous Biosensor for Enzyme-Free and Label-Free Detection of MicroRNA, ACS Sens., 2023, 8, 1529–1535 CrossRef CAS PubMed
- J. Moon and C. C. Liu, Asymmetric CRISPR enabling cascade signal amplification for nucleic acid detection by competitive crRNA, Nat. Commun., 2023, 14, 7504 CrossRef CAS PubMed
- J. R. Choi, K. W. Yong, R. H. Tang, Y. Gong, T. Wen, F. Li, B. Pingguan-Murphy, D. Bai and F. Xu, Advances and challenges of fully integrated paper-based point-of-care nucleic acid testing, TrAC, Trends Anal. Chem., 2017, 93, 37–50 CrossRef CAS
- L. M. Yang, H. Guo, Q. Gao, T. Hou, J. A. Zhang, X. J. Liu and F. Li, Integrating Reliable Pt-S Bond-Mediated 3D DNA Nanomachine with Magnetic Separation in a Homogeneous Electrochemical Strategy for Exosomal MicroRNA Detection with Low Background and High Sensitivity, Anal. Chem., 2023, 95, 17834–17842 CrossRef CAS PubMed
- J. B. Miller, S. Zhang, P. Kos, H. Xiong, K. Zhou, S. S. Perelman, H. Zhu and D. J. Siegwart, Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA, Angew. Chem., Int. Ed., 2016, 56, 1059–1063 CrossRef PubMed
- Y. J. Sun, W. D. Chen, J. Liu, J. J. Li, Y. Zhang, W. Q. Cai, L. Liu, X. J. Tang, J. Hou, M. Wang and L. Cheng, A Conformational Restriction Strategy for the Control of CRISPR/Cas Gene Editing with Photoactivatable Guide RNAs, Angew. Chem., Int. Ed., 2022, 62, e202212413 CrossRef PubMed
- Y. Zhang, Q. Ren, X. Tang, S. Liu, A. A. Malzahn, J. Zhou, J. Wang, D. Yin, C. Pan, M. Yuan, L. Huang, H. Yang, Y. Zhao, Q. Fang, X. Zheng, L. Tian, Y. Cheng, Y. Le, B. McCoy, L. Franklin, J. D. Selengut, S. M. Mount, Q. Que, Y. Zhang and Y. Qi, Expanding the scope of plant genome engineering with Cas12a orthologs and highly multiplexable editing systems, Nat. Commun., 2021, 12, 1944 CrossRef CAS PubMed
- N. S. McCarty, A. E. Graham, L. Studená and R. Ledesma-Amaro, Multiplexed CRISPR technologies for gene editing and transcriptional regulation, Nat. Commun., 2020, 11, 1281 CrossRef CAS PubMed
- L. M. Hochrein, H. Y. Li and N. A. Pierce, High-Performance Allosteric Conditional Guide RNAs for Mammalian Cell-Selective Regulation of CRISPR/Cas, ACS Synth. Biol., 2021, 10, 964–971 CrossRef CAS PubMed
- X. R. Song, C. Liu, N. Wang, H. Huang, S. Y. He, C. Y. Gong and Y. Q. Wei, Delivery of CRISPR/Cas systems for cancer gene therapy and immunotherapy, Adv. Drug Delivery Rev., 2021, 168, 158–180 CrossRef CAS PubMed
- C. M. Green, J. Spangler, K. Susumu, D. A. Stenger, I. L. Medintz and S. A. Díaz, Quantum Dot-Based Molecular Beacons for Quantitative Detection of Nucleic Acids with CRISPR/Cas(N) Nucleases, ACS Nano, 2022, 16, 20693–20704 CrossRef CAS PubMed
- D. Wang, X. D. Wang, F. D. Ye, J. Zou, J. X. Qu and X. Y. Jiang, An Integrated Amplification-Free Digital CRISPR/Cas-Assisted Assay for Single Molecule Detection of RNA, ACS Nano, 2023, 17, 7250–7256 CrossRef CAS PubMed
- L. T. Nguyen, B. M. Smith and P. K. Jain, Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified nucleic acid detection, Nat. Commun., 2020, 11, 4906 CrossRef CAS PubMed
- Y. F. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Exploring the Trans-Cleavage Activity of CRISPR-Cas12a (cpf1) for the Development of a Universal Electrochemical Biosensor, Angew. Chem., Int. Ed., 2019, 58, 17399–17405 CrossRef CAS PubMed
- Y. Huang, Q. Wen, Y. Xiong, Y. N. Chen, W. Li, J. L. Ren and H. Y. Zhong, Nanomaterials driven CRISPR/Cas-based biosensing strategies, Chem. Eng. J., 2023, 474, 145615 CrossRef CAS
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System, Cell, 2015, 163, 759–771 CrossRef CAS PubMed
- D. C. Swarts and M. Jinek, Mechanistic Insights into the cis- and trans-Acting DNase Activities of Cas12a, Mol. Cell, 2019, 73, 589–600 CrossRef CAS PubMed
- I. Fonfara, H. Richter, M. Bratovic, A. Le Rhun and E. Charpentier, The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA, Nature, 2016, 532, 517–521 CrossRef CAS PubMed
- W. R. Su, J. R. Li, C. Ji, C. S. Chen, Y. Z. Wang, H. L. Dai, F. Q. Li and P. F. Liu, CRISPR/Cas systems for the detection of nucleic acid and non-nucleic acid targets, Nano Res., 2023, 16, 9940–9953 CrossRef CAS PubMed
- S. Y. Li, Q. X. Cheng, J. K. Liu, X. Q. Nie, G. P. Zhao and J. Wang, CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA, Cell Res., 2018, 28, 491–493 CrossRef CAS PubMed
- Y. Li, Y. Wu, R. Xu, J. Guo, F. Quan, Y. Zhang, D. Huang, Y. Pei, H. Gao, W. Liu, J. Liu, Z. Zhang, R. Deng, J. Shi and K. Zhang, In vivo imaging of mitochondrial DNA mutations using an integrated nano Cas12a sensor, Nat. Commun., 2023, 14, 7722 CrossRef CAS PubMed
- X. X. Cheng, X. S. Li, Y. X. Kang, D. C. Zhang, Q. B. Yu, J. M. Chen, X. Y. Li, L. Du, T. T. Yang, Y. Gong, M. Yi, S. Z. Zhang, S. S. Zhu, S. J. Ding and W. Cheng, Rapid in situ RNA imaging based on Cas12a thrusting strand displacement reaction, Nucleic Acids Res., 2023, 51, e111 CrossRef CAS PubMed
- W. Feng, H. Y. Peng, J. Y. Xu, Y. M. Liu, K. Pabbaraju, G. Tipples, M. A. Joyce, H. A. Saffran, D. L. Tyrrell, S. Babiuk, H. Q. Zhang and X. C. Le, Integrating Reverse Transcription Recombinase Polymerase Amplification with CRISPR Technology for the One-Tube Assay of RNA, Anal. Chem., 2021, 93, 12808–12816 CrossRef CAS PubMed
- O. Mukama, J. H. Wu, Z. Y. Li, Q. X. Liang, Z. J. Yi, X. W. Lu, Y. J. Liu, Y. M. Liu, M. Hussain, G. G. Makafe, J. X. Liu, N. Xu and L. W. Zeng, An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids, Biosens. Bioelectron., 2020, 159, 112143 CrossRef CAS PubMed
- S. Peng, Z. Tan, S. Y. Chen, C. Y. Lei and Z. Nie, Integrating CRISPR-Cas12a with a DNA circuit as a generic sensing platform for amplified detection of microRNA, Chem. Sci., 2020, 11, 7362–7368 RSC
- M. H. Chen, R. Luo, S. H. Li, H. X. Li, Y. Qin, D. M. Zhou, H. Liu, X. Q. Gong and J. Chang, Paper-Based Strip for Ultrasensitive Detection of OSCC-Associated Salivary MicroRNA via CRISPR/Cas12a Coupling with IS-Primer Amplification Reaction, Anal. Chem., 2020, 92, 13336–13342 CrossRef CAS PubMed
- X. R. Yan, J. Y. Zhang, Q. Jiang, D. Jiao and Y. Q. Cheng, Integration of the Ligase Chain Reaction with the CRISPR-Cas12a System for Homogeneous, Ultrasensitive, and Visual Detection of microRNA, Anal. Chem., 2022, 94, 4119–4125 CrossRef CAS PubMed
- H. Yan, Y. J. Wen, Z. M. Tian, N. Hart, S. Han, S. J. Hughes and Y. Zeng, A one-pot isothermal Cas12-based assay for the sensitive detection of microRNAs, Nat. Biomed. Eng., 2023, 7, 1583–1601 CrossRef CAS PubMed
- S. R. Rananaware, E. K. Vesco, G. M. Shoemaker, S. S. Anekar, L. S. W. Sandoval, K. S. Meister, N. C. Macaluso, L. T. Nguyen and P. K. Jain, Programmable RNA detection with CRISPR-Cas12a, Nat. Commun., 2023, 14, 5409 CrossRef CAS PubMed
- J. S. Chen, E. B. Ma, L. B. Harrington, M. Da Costa, X. R. Tian, J. M. Palefsky and J. A. Doudna, CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity, Science, 2018, 360, 436–439 CrossRef CAS PubMed
- Q. Li, Z. L. Song, Y. X. Zhang, L. A. Zhu, Q. Yang, X. F. Liu, X. F. Sun, X. X. Chen, R. M. Kong, G. C. Fan and X. L. Luo, Synergistic Incorporation of Two ssDNA Activators Enhances the Trans-Cleavage of CRISPR/Cas12a, Anal. Chem., 2023, 95, 8879–8888 CrossRef CAS PubMed
- G. Gabriely, M. Yi, R. S. Narayan, J. M. Niers, T. Wurdinger, J. Imitola, K. L. Ligon, S. Kesari, C. Esau, R. M. Stephens, B. A. Tannous and A. M. Krichevsky, Human Glioma Growth Is Controlled by MicroRNA-10b, Cancer Res., 2011, 71, 3563–3572 CrossRef CAS PubMed
- J. Shen, S. A. Stass and F. Jiang, MicroRNAs as potential biomarkers in human solid tumors, Cancer Lett., 2013, 329, 125–136 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05647g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |