
Durable and recyclable biomimetic glycol lignin/polyolefin compounds for a circular economy†
Jonathon
Tanks
 *a,
Kenji
Tamura
b,
Kimiyoshi
Naito
ac,
Thi Thi
Nge
d and
Tatsuhiko
Yamada
d
*a,
Kenji
Tamura
b,
Kimiyoshi
Naito
ac,
Thi Thi
Nge
d and
Tatsuhiko
Yamada
d
aResearch Center for Structural Materials, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Japan 305-0047
bResearch Center for Electronic and Optical Materials, National Institute for Materials Science, Tsukuba, Japan 305-0044
cDepartment of Aerospace Engineering, Tohoku University, Sendai, Japan 980-8579
dCenter for Advanced Materials, Forestry and Forest Products Research Institute, Tsukuba, Japan 305-8687
First published on 2nd January 2024
Abstract
Polyolefins are some of the most widely used plastics in the world due to being lightweight, low cost, and resistant to water and most chemicals. However, they are highly susceptible to photo-oxidative degradation under ultraviolet radiation, leading to environmental problems such as microplastic pollution, and furthermore making it difficult to recycle. In this paper, glycol-modified lignin (GL) is blended with polypropylene to form a bio-mimetic structure similar to human skin, in which melanin absorbs UV light and sunburned tissue is removed to reveal fresh skin. We show that GL acts as a simultaneous UV absorbent, antioxidant, and reinforcement, resulting in high retention of mechanical properties even after severe UV exposure, and nearly full recovery of the original properties after mechanical recycling. This performance is attributed to the compatibility of GL with maleic anhydride-grafted PP, which is greater than other types of lignin/polyolefin blends reported in the literature. This improves the durability and in-service performance of polyolefin-based composites and makes their end-of-life recycling possible. These results demonstrate the potential for more sustainable usage of polyolefins and biomass in various applications such as automobiles, containers, household items, and building components.
Introduction
Polyolefins—a class of hydrocarbon-based plastics including polyethylene (PE) and polypropylene (PP)—have replaced metals, glass and ceramics in numerous applications such as containers, packaging, automotive and aircraft components, construction materials, household goods and appliances, and piping due to their good processability and low cost. They have become a fundamental part of modern society, representing 56% of all plastics produced annually, releasing an estimated 1.2–2.7 kg of CO2 per kg of polyolefin in the process (depending on the country).1,2 Additionally, their prevalent treatment as “single-use” materials sees them constitute half of all plastic waste at 152 million tons, of which over 60% is discarded or incinerated, while less than 20% is recycled.1 Not only does this increase pollution in oceans and forests in the form of microplastics, which refers to polymer fragments created by ultraviolet (UV) exposure,3–5 but it also represents poor resource management in a world with increasingly depleting resources. In response, various coordinated efforts among governments and industry are in development to reduce plastics waste in accordance with sustainable development goals (SDGs).Although cost-effective and high-performance “renewable” polymers (i.e., bio-based or bio-degradable) are ideal and recent progress in their development is encouraging, an immediate strategy is needed for improving the sustainability of polyolefins currently in circulation—i.e., upcycling and recycling.6,7 Owing to their continuous hydrocarbon backbone structure, polyolefins are chemically stable against a wide variety of organic solvents and reagents and thus cannot be chemically recycled—i.e., returned to monomers/oligomers—without changing the chemical structure.8,9 A common disposal route for polyolefins in the EU and USA consists of incineration (termed “thermal recovery”) for the purpose of utilizing the released thermal energy, consequently releasing additional CO2 as well.1,8 Another common strategy is to reprocess the end-of-life polyolefin into products with lesser performance due to their deteriorated material properties; this sub-optimal mechanical recycling approach is considered downcycling, or so-called open-loop recycling.1,8,9 Thus, a closed-loop mechanical recycling strategy that achieves nearly 100% retention of material performance upon reprocessing at end-of-life is vital for realizing a circular economy, but it has yet to be achieved.
Two of the toughest hurdles to the effective recycling of polyolefins, particularly PP, is (1) the suppression of UV degradation during use and (2) restoration of the material structure at end-of-life after degradation has occurred.8,9 UV-induced degradation is primarily a process called photooxidation (Fig. 1(a)), in which UV radiation initiates radicals in the material through hydrogen abstraction, often caused by trace impurities such as residual catalysts, and this leads to the diffusion of highly reactive oxygen radicals throughout the polymer.10 The body of literature devoted to the photooxidation of polyolefins is vast, and the mechanisms and kinetics are well understood.10–18 Various carbonyl groups—carboxylic acids, esters, ketones, lactones—are formed by oxidation of the hydrocarbon backbone and—given sufficient oxygen and irradiation energy—eventually extensive chain scission occurs. Termination occurs when peroxide or polymer radicals mutually couple, quenching the radical and leaving an inactive product. Drastic reductions in molecular weight (over 70% loss) and surface cracking can be observed as early as two months of sunlight exposure (equivalent to a UV irradiation dose of roughly 45 MJ m−2), leading to severely reduced mechanical behavior (over 60% and 95% loss in strength and failure strain, respectively13).
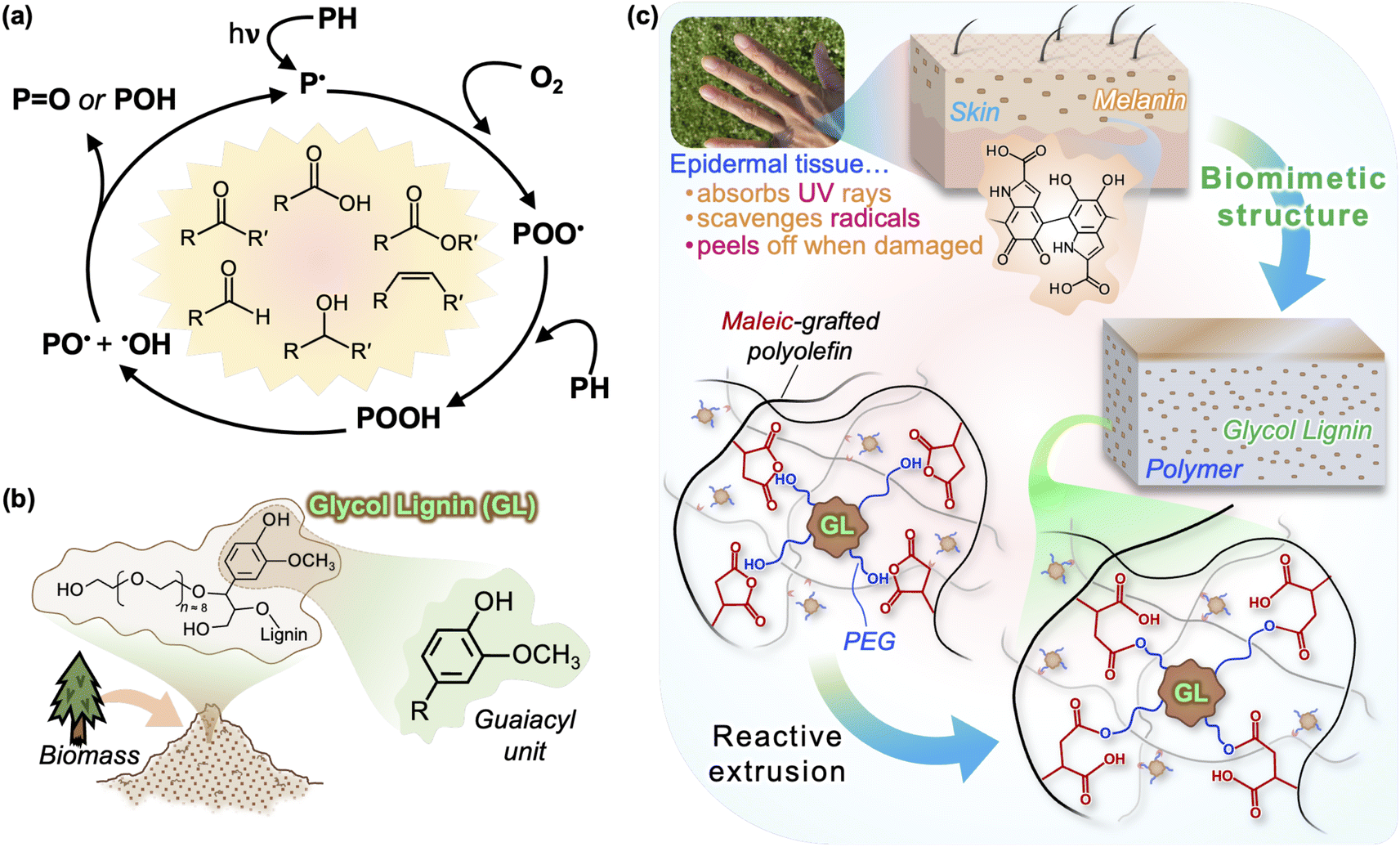 | ||
| Fig. 1 (a) Typical photooxidation pathway of polyolefins (including PP) and its reaction products. (b) Structure of glycol-modified lignin (GL) used in this study, emphasizing the predominant guaiacyl units. (c) Schematic depiction of proposed structure of biomimetic GL/PP compounds produced by reactive extrusion. The well-dispersed lignin particles imitate melanin in human skin, acting simultaneously as UV absorbent, antioxidant and reinforcement. | ||
Hindered phenol- and hindered amine-based UV absorbent and antioxidant additives are commonly used in polyolefins to prevent photooxidation, such as commercial products known as the Irganox series, Tinuvin series, UV series, and others. While these stabilizer additives provide a certain amount of protection against photooxidation, they inevitably leach out of plastics during or after the service life and many have been labeled toxic to wildlife and even humans.19,20 In an attempt to reduce microplastics pollution, a different form of pollution is created. Thus, a more environmentally-friendly alternative is needed in order to increase the sustainability of polyolefins, primarily by reducing pollution and improving mechanical recyclability.6,21
Lignin, an aromatic macromolecule that forms a sheath around cellulose in plant tissue, constitutes roughly 20% of all biomass and has long been regarded as a waste byproduct in the production of paper.22 Research focused on integrating lignin with conventional polymers has made great progress in recent years, with the aim of reducing plastic use, reducing biomass waste, and improving polymer performance. Lignin does not leach out of the polymer due to its size, but it poses no harm to humans or wildlife even in the case leaching does occur. Studies have shown that the addition of lignin can enhance the thermal- and UV-stability of polyolefins, citing its radical scavenging ability.23–28 In particular, the syringyl and guaiacyl units of lignin are similar to hindered phenols typically found in synthetic stabilizers and are capable of hydrogen atom transfer within the aromatic structure that prevents radical propagation;29 softwood lignin contains mostly guaiacyl units (Fig. 1(b)).
Despite the clear potential for lignin to be utilized as a UV absorbent/antioxidant, there are still several challenges left to be addressed and considerable room for improvement. Successful incorporation of higher lignin content (10–50 wt%) with both excellent UV resistance and mechanical properties has yet to be reported; nearly all past attempts to reach these higher loadings involve compounding the lignin into polyolefins at conventional temperatures (160–190 °C), and either employ alkyl modification of the lignin or no modification, resulting in poor dispersion and no improvement in mechanical properties.30–35 While the UV stabilization performance of lignin has been studied in films, its behavior in bulk (thick) structures like those found in automotive and aircraft components or building materials have not been investigated. Furthermore, to the authors' knowledge, there are no studies that demonstrate recyclability of lignin-based polyolefin blends; in some cases “recycling” is used to refer to simple re-processing without any kind of degradation protocol,36–38 which is not realistic in the context of establishing a framework for end-of-life processing and reuse.
Recently, a glycol-modified lignin (GL) was developed from softwood biomass via acid-catalyzed PEG solvolysis.39,40 In our previous work, we showed that a combination of using GL and higher extrusion temperatures was the key to effectively integrating lignin with PP, producing biomass-containing compounds with unprecedented mechanical properties and processability.41 In the current study, we propose a GL/PP compound that exhibits excellent UV resistance by mimicking the melanin/tissue structure of human skin, which simultaneously acts as a UV absorbent and antioxidant to protect tissue from harmful irradiation through its phenolic structure.42 If the skin is exposed to excessive irradiation, a thin surface layer of dead tissue is created which can be peeled off to reveal fresh regenerated tissue beneath; as shown in Fig. 1(c), we applied this concept to design a material that is durable and also fully recyclable using a simple mechanical process to remove the sun-damaged layer (similar to “peeling”). Chemical and physical changes in neat PP and GL blends after accelerated UV irradiation are compared and discussed, particularly focusing on the behavior of thick specimens rather than films, and we show that GL makes closed-loop recycling possible—even after UV irradiation equivalent to 1 year of sunlight.
Experimental methods
Materials
The GL used in this study was obtained from glycol solvolysis of softwood biomass, where about 26 wt% PEG400 was grafted onto lignin macromolecules;40 this translates to roughly 0.65 mmol g−1 and 2.5 mmol g−1 of glycol-OH and phenolic-OH per unit mass of lignin, respectively. A commercial maleic-grafted PP known as ADMER™ QE800E (MFI = 9.7 g/10 min, density = 0.91 g cm−3, maleic anhydride content = 0.04 wt% (ref. 43)) was received from Mitsui Chemicals. A hydroxyphenyl benzotriazole-type commercial stabilizer (Tinuvin 1130, BASF) and acetylated solvent lignin (Guangzhou Yinnovator Biotech) were also included for comparison.Compounding of GL/PP blends
Biomass/polyolefin blends were prepared using the method described in our previous report.41 Briefly, raw materials were melt-mixed at 240–250 °C using a twin-screw extruder, after which the collected pellets were dried at 80 °C overnight and injection molded into ISO 527-2 dumbbell specimens (nominal cross-section 5 × 2 mm2). The compositions were varied from 0 to 30 wt% of GL, all of which exhibited good processability. The commercial UV absorber (UVA, Tinvin 1130) was compounded with PP at 0.1, 0.5 and 1 wt%, while a 20 wt% acetyl lignin (AcL) compound was also prepared.UV irradiation conditions
Dumbbell specimens were attached to vertical holder plates around a metal halide lamp (MV3000, Suga) with a longpass filter that cuts wavelengths below 295 nm, corresponding to a maximum irradiance of 530 W m−2 (monitored over 300–400 nm, Fig. S1†). To avoid complications that may arise when moisture and heat combine with UV light, we set the relative humidity and chamber temperature at 10% and 37 °C, respectively. Since photooxidation is accelerated by the irradiation energy and the irradiance and spectrum varies for different lamps, we selected the exposure durations by cumulative irradiation energy rather than time: 100, 300, and 1000 MJ m−2 (equivalent to roughly 4, 12, and 40 months of sunlight exposure, respectively). Only one side of the specimens were exposed for the entire duration in order to simulate an actual in-service structure.Recycling protocol
Neat PP and GL-10 wt% were selected to demonstrate the recyclability of GL-containing compounds (Fig. S2†). The extruded pellets were exposed to 300 MJ m−2 of UV irradiation before being mechanically recycled via one of the following procedures: (i) remolding via injection molding without any other treatment (Remolding); (ii) mechanically removing the degraded portion of the pellets using a household rice grinder (RCI-B5) for 4 min, washing with water/ethanol, then remolding (Grinding + Remolding); (iii) treating the pellets with alkali solution (NaOH, 1 M) at 90 °C for 24 h, washing, then remolding (Alkali + Remolding); (iv) treating the pellets with alkali solution as above, followed by grinding for 4 min, washing, then remolding (Alkali + Grinding + Remolding). Morphology, chemical structure, and tensile properties were assessed.Characterization
Fourier transform infrared (FTIR) spectra of films were recorded over the range 400–4000 cm−1 in ATR mode (IR Affinity, Shimadzu), and UV-vis spectra were recorded in transmission mode (V-770, Jasco) between 200 and 800 nm. Dumbbell specimens exposed to 1000 MJ m−2 of UV radiation were microtomed to create a smooth cross-section, from which through-thickness FTIR spectra were recorded over the range 650–4000 cm−1 using an IR microscope in ATR mode (AIMsight, Shimadzu). A measurement area of 50 × 50 μm2 was averaged over 100 μm increments from the surface to the center of the cross-section. The change in the carbonyl index (ΔCI) was used as a qualitative measure of photo-oxidation according to the following:where Ax is the integrated area of the peaks for carbonyls (x: C
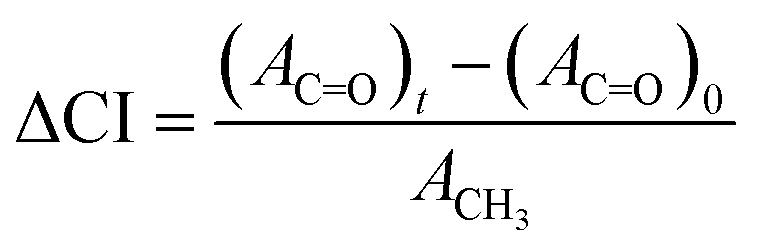
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) between 1710 and 1770 cm−1 or methylene (x: CH3) at 1458 cm−1. The contributions of the carbonyls native to maleic-grafted PP and lignin in the initial state were subtracted to isolate new carbonyl formation during irradiation.
O) between 1710 and 1770 cm−1 or methylene (x: CH3) at 1458 cm−1. The contributions of the carbonyls native to maleic-grafted PP and lignin in the initial state were subtracted to isolate new carbonyl formation during irradiation.
Differential scanning calorimetry (DSC7020, Hitachi Hi Tech) was performed on roughly 1.5–2 mg of sample under N2 flow over the range 0–200 °C at a heating and cooling rate of 10 °C min−1, and the degree of crystallinity χc was approximated with the enthalpy of fusion ΔHm taken from the melting peak on the first heating scan:
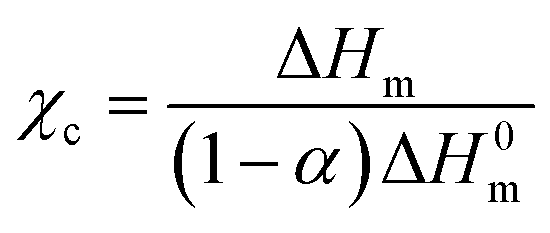
Results and discussion
Processing and structure of GL/PP blends
The extrusion conditions used in this work resulted in uniformly blended compounds that show good moldability even at 30 wt% loading. The expected structure is depicted in Fig. 1(c), where sub-micron lignin particles41 are grafted to the matrix by ester bonds formed between the PEG and maleic anhydride groups in GL and PP, respectively. This was confirmed by FTIR (Fig. S3†), where the carbonyl peaks associated with maleic anhydride change to those of ester bonds,41,44 and the intensities of the characteristic GL peaks change depending on the loading. Thermal analyses confirmed that the blends exhibit similar melting/crystallization behavior as neat PP, meaning that they remain thermoplastic on the macroscale. Dynamic mechanical analysis revealed that lignin affects the segmental dynamics of polypropylene by introducing structural heterogeneity, which is only possible if the lignin is chemically integrated into the polymer nanostructure through covalent bonding. The resulting material imitates the melanin-containing epidermis layer of human tissue, with well-dispersed particles acting as UV absorbent and antioxidant. This brief description of the fabricated materials is included to aid in understanding the following sections, but readers interested in further details should refer to our previous report.41UV-induced changes in structure and mechanical behavior
Thermal transitions (e.g., melting/crystallization) are affected by UV irradiation through the decrease in polymer molecular weight (Mw) caused by chain scission, while the amorphous regions undergo chemi-crystallization due to increased mobility after chain scission.16,17Fig. 2(a) shows the change in melting temperature Tm through the thickness at different irradiation energies for each blend. While neat PP shows extensive reduction in Mw near the surface at lower energies, prolonged radiation leads to drastic reduction through the entire thickness.18 In contrast, GL suppresses the degradation process such that the Mw reduction is contained within the surface layer. The effect is more pronounced at higher loadings, with a degradation layer of only ∼100 μm for 30 wt% GL even after 1000 MJ m−2 of irradiation (equivalent to ∼3 years of natural sunlight). Tc of the neat PP steadily decreases in the same manner as Tm, which is attributed to the significant degree of chain scission and carbonyl formation (Fig. S4†). However, while changes in Tm are directly related to Mw, changes in crystallization temperature Tc may be influenced by various factors such as cooling rate, nucleation agents and interfaces.7 The presence of GL retards the crystallization process in unexposed specimens, while it appears to facilitate crystallization upon irradiation. Heterogeneous microdomains can act as nucleation sites in PP,45 but the influence of UV irradiation on this process in lignin compounds remains unclear based on our current data and thus requires further study. Meanwhile, the crystallinity χc (i.e., crystalline fraction) of the PP matrix exhibits changes attributed with chemi-crystallization starting at the outer surface and progressing inward as irradiation energy increases (Fig. 2(a)). Severe degradation of the chemical structure in the surface layer at longer exposure times leads to an apparent decrease in χc due to the drastically reduced Mw and abundant carbonyl groups. This evolution is suppressed in GL blends because the chemi-crystallization phenomenon becomes increasingly more limited to the surface layer as GL content increases, with no significant changes in χc at depths greater than ∼250 μm.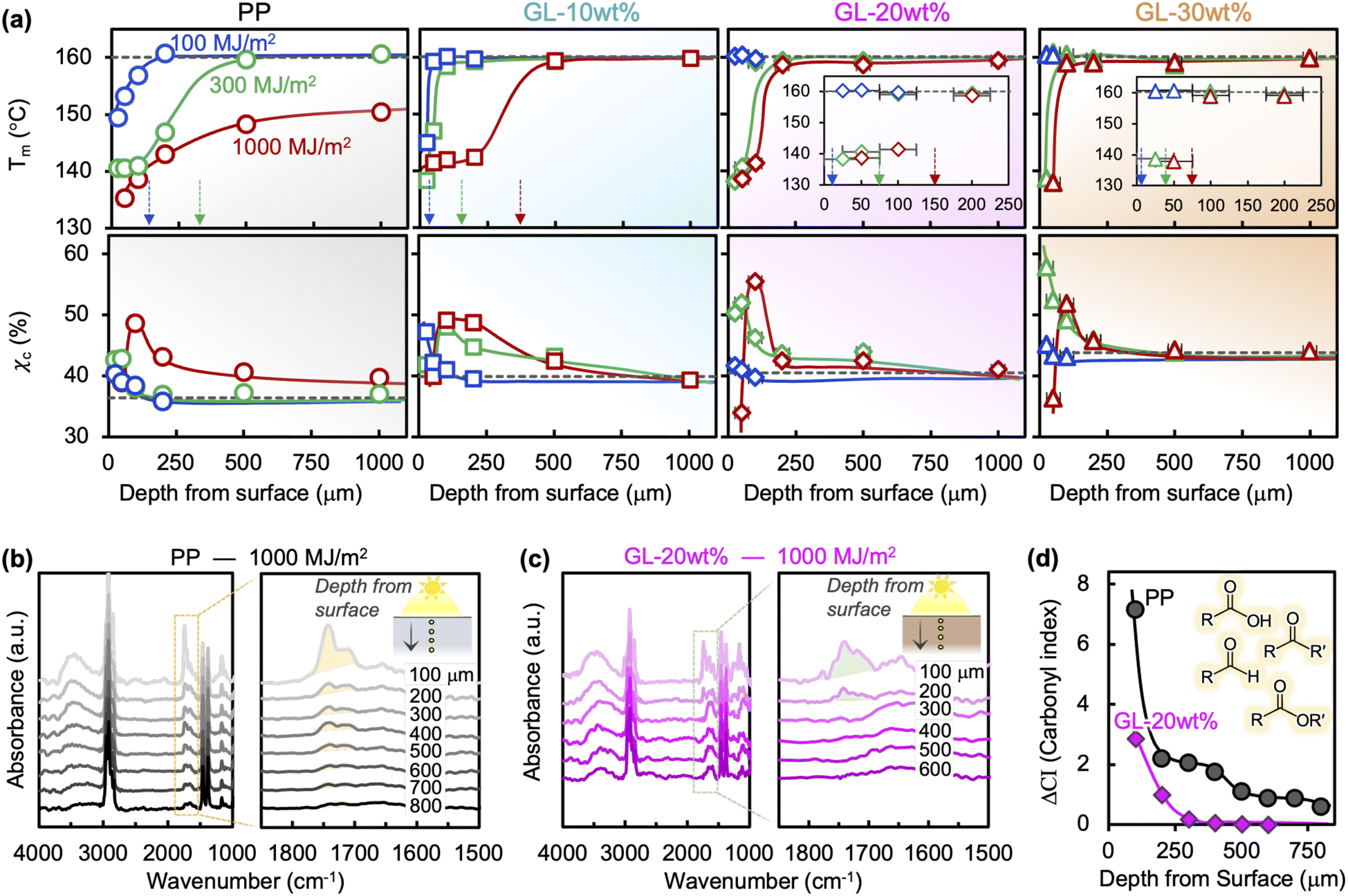 | ||
| Fig. 2 (a) Melting temperature (Tm) and crystallinity (χc) of GL/PP blends; curved lines are only included to guide the eye. Dashed black lines represent the pre-exposure values, and dashed colored arrows point to the approximate depths where Mw appears unchanged. Through-thickness FTIR spectra of (b) neat PP and (c) GL-20 wt% after 1000 MJ m−2 of irradiation, and (d) comparison of carbonyl index (CI) values taken from the integrated areas of the region 1690–1780 cm−1 (shaded in (b) and (c)). | ||
Fig. 2(b) and (c) show the through-thickness FTIR spectra for neat PP and GL-20 wt% exposed to 1000 MJ m−2 of irradiation, with carbonyl peaks (between 1710 and 1750 cm−1) growing larger near the surface. The formation of these carbonyl groups represents the early stage of chain scission and can be quantified by the carbonyl index (CI), shown in Fig. 2(d). Although the validity of the CI as a quantitative measure of chemical degradation is under debate,4,15,16 it does serve to identity the difference in photooxidation behavior between materials under the same conditions. Not only is the CI significantly lower throughout the bulk of the GL compound compared to neat PP, but the depth at which carbonyl groups are no longer detected is also smaller, meaning that the GL restricts photooxidation to the surface region. This is consistent with the Tm profiles in Fig. 2(a), which show a transition depth of around ∼150 μm (Tm) vs. ∼250 μm (CI) for GL-20 wt%, and >1000 μm (Tm) vs. >1000 μm (CI) for neat PP. Note that lignin may also have barrier properties that would limit oxygen diffusion into the polymer, but these effects are ignored in the current analysis while keeping irradiation conditions equal for all materials. Since the carbonyl formation stage of photooxidation precedes chain scission, it is reasonable that the transition depth for CI is greater than Tm (i.e., Mw). Chemi-crystallization also depends on chain scission and thus on carbonyl formation; no studies to-date have reported on the effects of carbonyl groups on chemi-crystallization nor its associated crystal phase.17
The formation of a gradient in χc over the depth corresponds to a density gradient as well, which causes differential stresses near the surface. In combination with decreased strength and toughness caused by chain scission itself, this stress can lead to microcracking.18 LSM images of the specimen surfaces are shown in Fig. 3(a) and S8,† where surface microcracks grow in size and density with irradiation energy for all blends. However, it is clear from the images that GL modifies this behavior so that the cracks become smaller, more closely spaced, and more unidirectional as the GL content increases. The relationship between surface crack depth dc and crack spacing hc appears linear on a log–log plot (Fig. 3(b)), indicating a decreasing trend where dc reaches zero when hc is very large (i.e., undamaged material). Thus, it is clear that GL prevents crack growth, diverting the UV-induced damage toward the formation of new surface cracks. Inferring the changes in Mw from Tm, this is because significant chain scission and chemi-crystallization occurs only in the surface layer of GL blends, which should suppress through-thickness crack growth. This phenomenon is illustrated in Fig. 3(b), where lignin acts as a shield against structural and morphological changes during UV irradiation via the formation of a distinct “sunburned” surface layer.
 | ||
| Fig. 3 (a) LSM images of neat PP and GL blends after different UV irradiation energies (scale bars are 100 μm). (b) Inverse relationship between dc and hc for different GL loadings, showing that GL suppresses UV damage through the formation of distributed small cracks. (c) Illustration of the degradation process for neat PP vs. GL blends: more surface microcracking occurs in GL blends to release shrinkage stress caused by chemi-crystallization and chain scission, whereas neat PP exhibits more severe degradation through the entire bulk over time. | ||
This brings us to examine the tensile behavior of GL blends, summarized in Fig. 4 and S5.† In accord with the literature, neat PP exhibits typical ductile behavior before irradiation, having a strength of 38 MPa and failure strain exceeding 600%, but chemical degradation and microcracking cause significant decreases in strength (Fig. 4(c)), modulus (Fig. 4(d)), and failure strain (Fig. 4(e)) after only 100 MJ m−2 of irradiation.13,18,25 A uniform state of degradation throughout the polymer is only possible for thin films (<100 μm) due to oxygen diffusion limited reactions, so differentiating the individual effects of chemical degradation (i.e., Mw reduction) and surface crack formation on the bulk material properties of molded specimens remains difficult. As discussed in the previous section, photooxidation of PP results in volatile products such as acetone which diffuse out of the material, as well as the formation of carbonyl and alkene groups in the main chain; some of these groups may recombine to form crosslink points which—combined with increased crystallinity—temporarily increase the apparent elastic modulus at short irradiation times, as seen for neat PP in Fig. 4(d). Ultimately, with sufficient irradiation energy the chain scission leads to reduced Mw and surface cracking, which contributes to a crack growth-dominated failure mode denoted by a brittle fracture surface (Fig. 4(b) and S6†), and nearly constant failure strain accompanied by reduced strength and modulus.
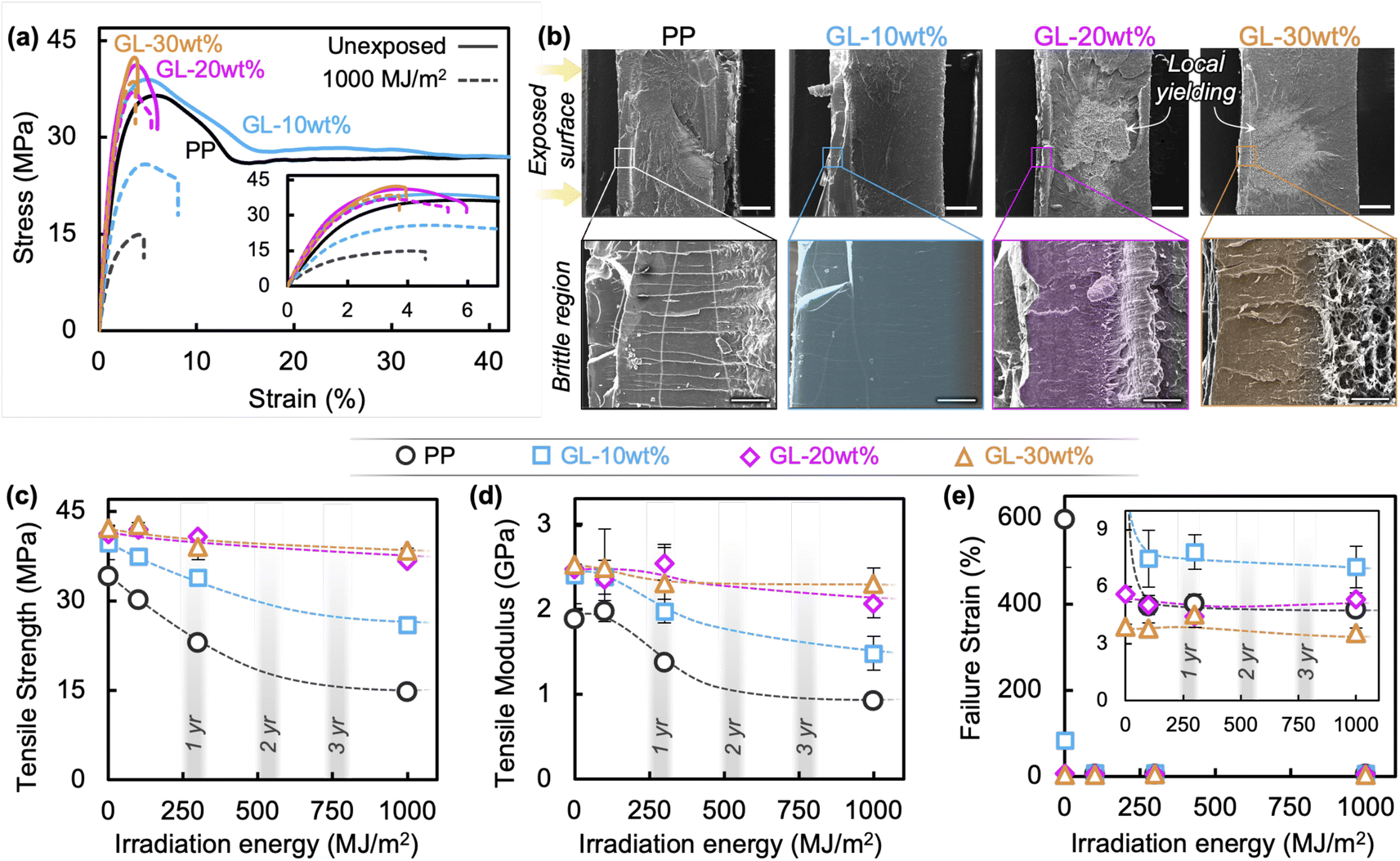 | ||
| Fig. 4 (a) Stress–strain curves for specimens before and after irradiation (1000 MJ m−2). (b) SEM images of fracture surfaces of specimens after 1000 MJ m−2 of irradiation, showing a transition from brittle surface crack growth to post-yield fracture behavior (scale bars are 500 μm for top, 50 μm for bottom). Summaries of (c) tensile strength, (d) elastic modulus, and (e) failure strain at different irradiation energies and their equivalent sunlight exposure times; lines are intended to guide the eyes. | ||
Conversely, tensile properties of the blends change as GL content increases, with 10 wt% showing improvement over neat PP while 20 and 30 wt% appear to reach a saturated level of property retention. It is important to note that the pre-exposure failure strains were substantially lower at higher GL loadings due to the large amount of rigid aromatic molecules and possible agglomeration. However, the tensile properties were negligibly affected at higher GL loadings even after 1000 MJ m−2 of irradiation, and local yielding was still visible away from the crack tip (Fig. 4(b)). Recalling that surface cracking occurs under these conditions, the high retention of mechanical properties in these blends indicates that GL mechanically reinforces the polymer so that surface crack growth does not determine bulk behavior; this is discussed further below.
Role of GL in improving UV resistance
We assert that GL, like melanin in skin, plays three major roles in improving the durability of polyolefin blends: (i) UV absorption, (ii) oxygen radical scavenging, and (iii) mechanical reinforcing. As pure PP homopolymer exhibits negligible UV absorption, impurities from manufacturing are often responsible for initiating the photo-oxidation process. Molecules with relatively stronger UV absorption than the host polymer can serve to divert photonic energy from polymer chains.46,47Fig. 5(a) shows the UV-vis spectra of GL and its blends, where a strong absorbance is observed between 200 and 400 nm for GL powder, compared to the low absorbance in neat PP above 275 nm. This absorption behavior is due to the highly aromatic structure of lignin, particularly the methoxyphenol-based guaiacyl unit absorbing in the 240–280 nm region, as well as the π → π* and n → π* transitions associated with conjugated molecules in the 250–350 nm region.46 Corresponding to the pure GL spectrum, the GL blends show stronger absorbance with increasing lignin content. While 20 and 30 wt% show maximum UV absorbance over the experimental irradiation range, 10 wt% is sub-maximum and suggests minor susceptibility to photo-induced radical formation.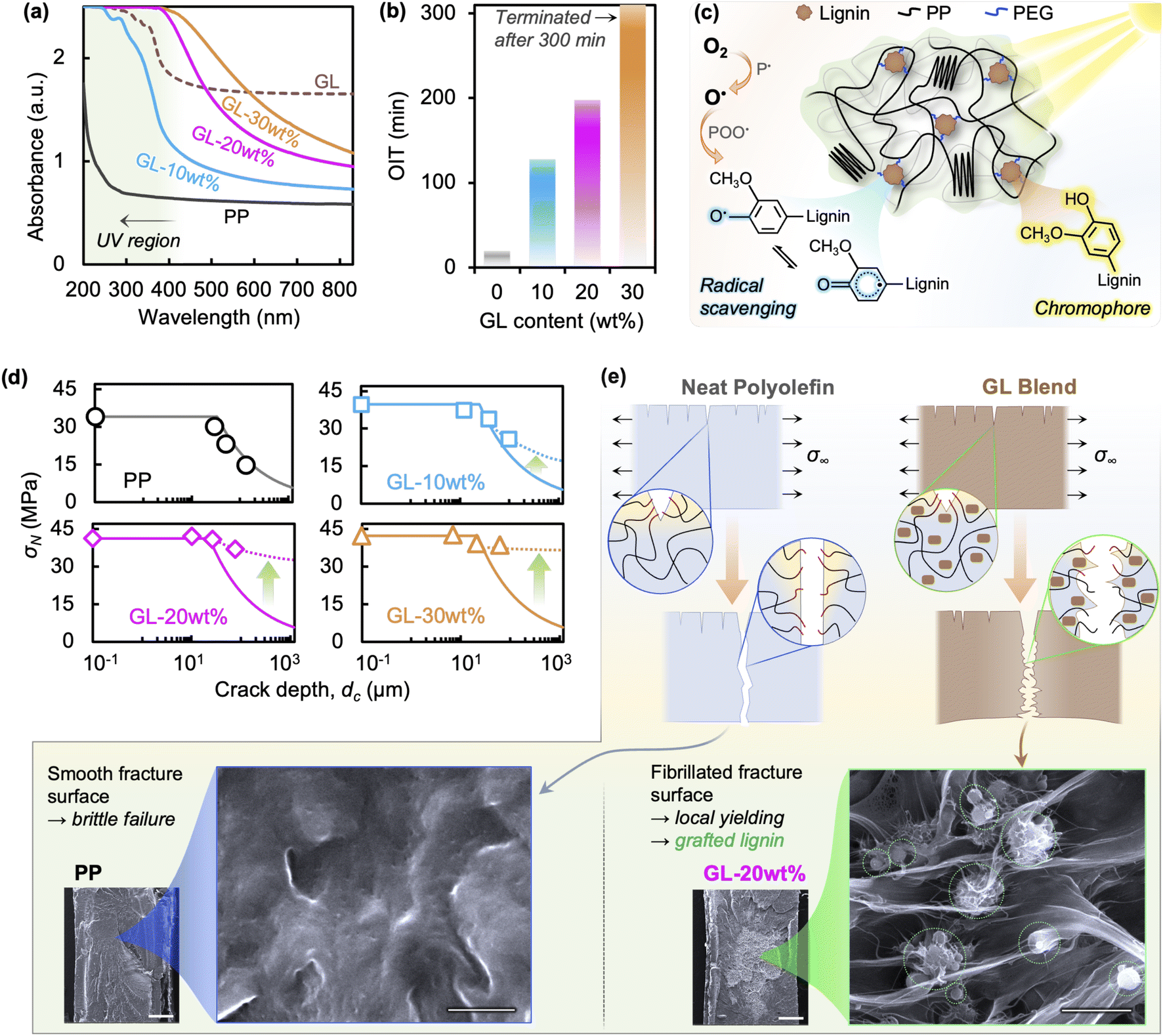 | ||
| Fig. 5 (a) UV-vis spectra and (b) OIT results for GL/PP blends, and (c) illustration of the simultaneous UV absorbent/antioxidant behavior of GL in polyolefin blends. (d) Comparison of experimental (symbols, dotted lines) and analytical (solid lines) values of nominal tensile strength (σN) of neat PP and GL blends as a function of surface crack depth (dc). The deviation from LEFM predictions (KIC ∼ 3.5 MPa√m) indicates that brittle crack growth does not determine the nominal tensile strength of irradiated GL blends. (e) Illustration of the mechanical reinforcement/toughening effect of GL in polyolefin blends. With sufficient GL loading, well-dispersed lignin particles (circled in green) that are covalently bonded to PP can simultaneously increase stiffness through composite action while blunting crack tips, leading to bulk yielding even when surface cracks are present (see SEM images; scale bars are 2 μm). | ||
We selected the oxidation induction time (OIT) test as a simple way to demonstrate the radical scavenging ability of GL in polyolefin blends, with results summarized in Fig. 5(b). The results clearly indicate that GL protects the polymer matrix from oxidation—through radical scavenging and possibly impedance of oxygen diffusion—with growing efficacy as GL content increases, reaching an unprecedented 1000% improvement for 20 wt% loading. Despite having a lower phenolic –OH content than some unmodified lignins (∼2.5 mmol g−1vs. ∼4.5 mmol g−1 (ref. 23 and 29)), GL shows superior compatibility with maleic-grafted PP and the uniform dispersion leads to lower surface energy and more effective radical scavenging ability.23,24 This reinforces the notion that effective compatibilization and dispersion of biomass in the host polymer is crucial for improving overall performance. These dual melanin-like UV absorbent/antioxidant functions are illustrated in Fig. 5(c).
Finally, GL acts as mechanical reinforcement at the micron and sub-micron scale via composite action between the polymer matrix and well-dispersed particles of rigid aromatic biomolecules that increase the bulk tensile strength and modulus.25,35,41 This is a key feature of GL that differentiates it from other reported lignin blends and is vital for creating durable and recyclable composites. Here we use a simple stress analysis based on linear elastic fracture mechanics (LEFM) to show that GL is responsible for limiting the effect of surface cracks on the apparent strength after UV irradiation; see ESI† for details. The nominal strength σN—i.e., the applied load averaged over the nominal specimen cross sectional area—is equal to the bulk tensile strength when surface crack depths dc are very small but decreases when the cracks become larger. This is because the applied load required to propagate a surface crack of a certain length exceeds the critical value corresponding to the fracture toughness (KIC) of the material before yielding can occur.
The predictions of nominal strength using LEFM are shown in Fig. 5(d), assuming a typical value of KIC ∼ 3.5 MPa√m;48 calculations using a wider range of values are compared in Fig. S9.† A detailed analysis using elastic–plastic fracture mechanics (EPFM) is required for accurate predictions in ductile polymers; however, we only intend this analysis to demonstrate whether the fracture behavior is brittle (i.e., controlled by UV-induced surface cracks) or ductile (i.e., unchanged from initial behavior). Overestimation of experimental values by LEFM may be due to reduction of the bulk material properties by chemical degradation, which is expected for neat PP but not for GL blends. More notably, the drastic underestimation seen for GL blends strongly indicates that crack size alone does not dominate bulk tensile properties. In addition to the suppression of crack formation during irradiation via UV absorption and radical scavenging (Fig. S8†), GL also reinforces the polymer such that surface cracks formed by chemical degradation are hindered from growing before bulk yielding occurs (Fig. 5(e)). This novel toughening behavior has not been reported in lignin-based blends to date.
Other commercially available UV stabilizers, a hydroxyphenyl benzotriazole-type Tinuvin 1130 (UVA) and acetyl lignin (AcL), provide varying degrees of UV resistance, but neither can compare to GL's performance. A relatively high loading of 1 wt% of UVA only has a marginal effect on Mw reduction (Fig. 6(a)) after 300 MJ m−2 of UV exposure, with no discernible effect at practical loadings of 0.1–0.5 wt% (Fig. S10†). Despite this, while not providing any reinforcement, it does show higher retention of tensile strength than neat PP (65% vs. 45%), as seen in Fig. 6(b) and S11.† In contrast, AcL-20 wt% seems to be just as effective at suppressing Mw reduction as GL-20 wt% through absorbent/antioxidant action (Fig. 6(a)). However, the lack of covalent bonding with maleic-grafted PP and inadequate dispersibility result in negligible reinforcing capability and only moderate retention of mechanical properties (Fig. 6(b)). The chemical structures of all three additives in this study are shown in Fig. 6(c), along with qualitative comparison of their overall performance. These results support our assertion that GL blends possess superior mechanical and durability performance due to GL's simultaneous roles as absorbent, antioxidant, and reinforcement, which is essential for creating a class of durable and recyclable polyolefins.
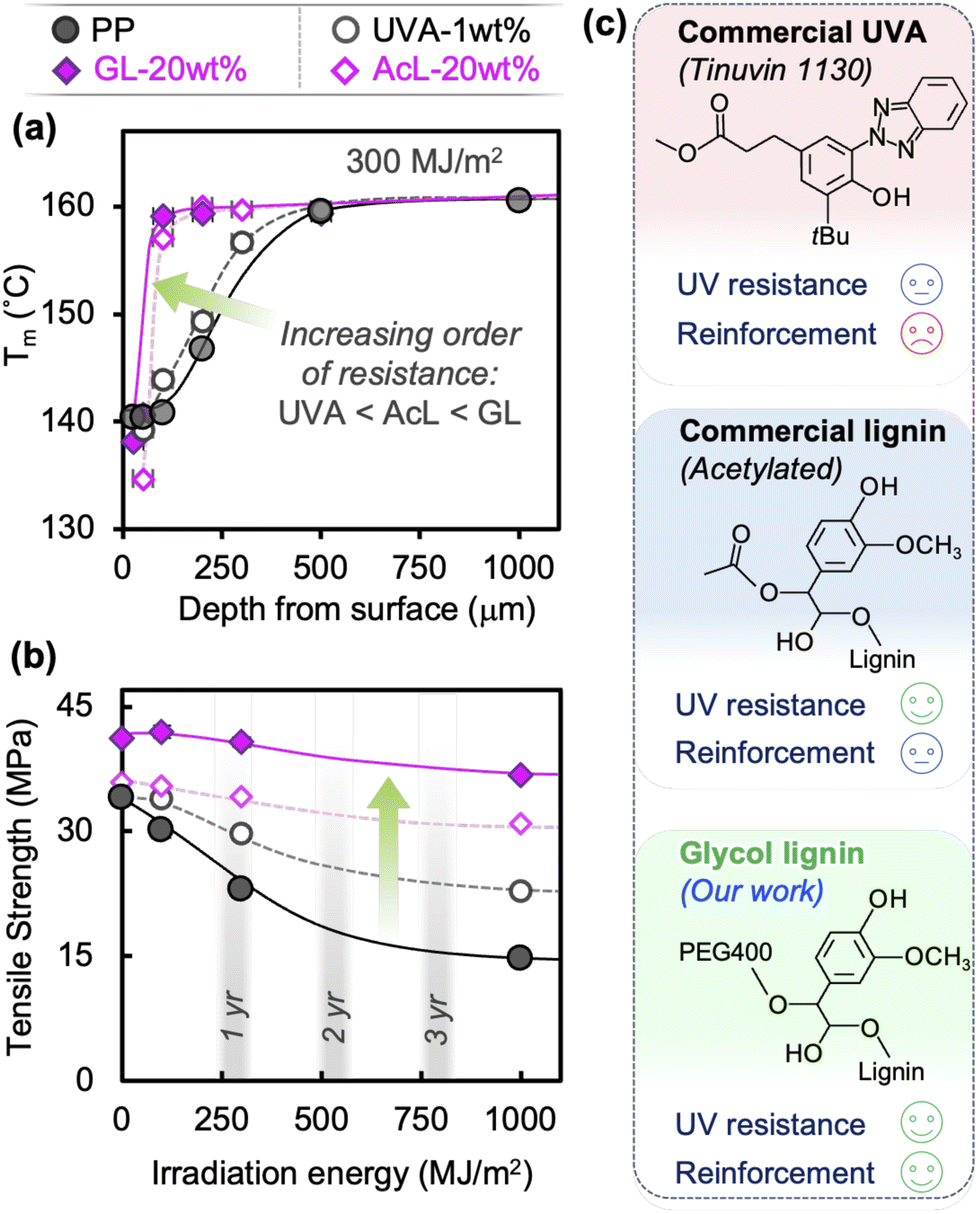 | ||
| Fig. 6 (a) Through-depth profiles of Tm after 300 MJ m−2 of irradiation and (b) tensile strength at different UV irradiation energies for commercial UVA and acetyl lignin. (c) Chemical structures and qualitative performance comparison of three additives. | ||
Closed-loop recyclability of GL blends
Although thermoplastic polymers such as polyolefins are “remoldable” in terms of processability, this does not mean that they can be successfully recycled by simple mechanical/physical approaches (i.e., re-granulation and remolding) due to chemical degradation that occurs over time in the service environment.36–38 This problem can be addressed by the GL-based compounds in this study owing to their drastically improved UV resistance and mechanical properties over the neat polyolefin. The formation of a distinct “sunburned” surface layer after prolonged UV irradiation not only improves the in-service performance, but also facilitates recycling by suppressing changes to the chemical structure throughout the bulk and creating an easily-removed surface layer. This is a commonly observed phenomenon in nature, such as epidermis (human skin) and periderm (tree bark), which regenerate by peeling the outer damaged layer to expose the fresh tissue beneath;42 similarly, metals such as aluminum rapidly form a thin oxidize layer on the surface while the bulk is protected from further corrosion. Thus, as depicted in Fig. 7(a), our compounds aim to mimic biological structures by utilizing the UV absorption and antioxidant properties of lignin.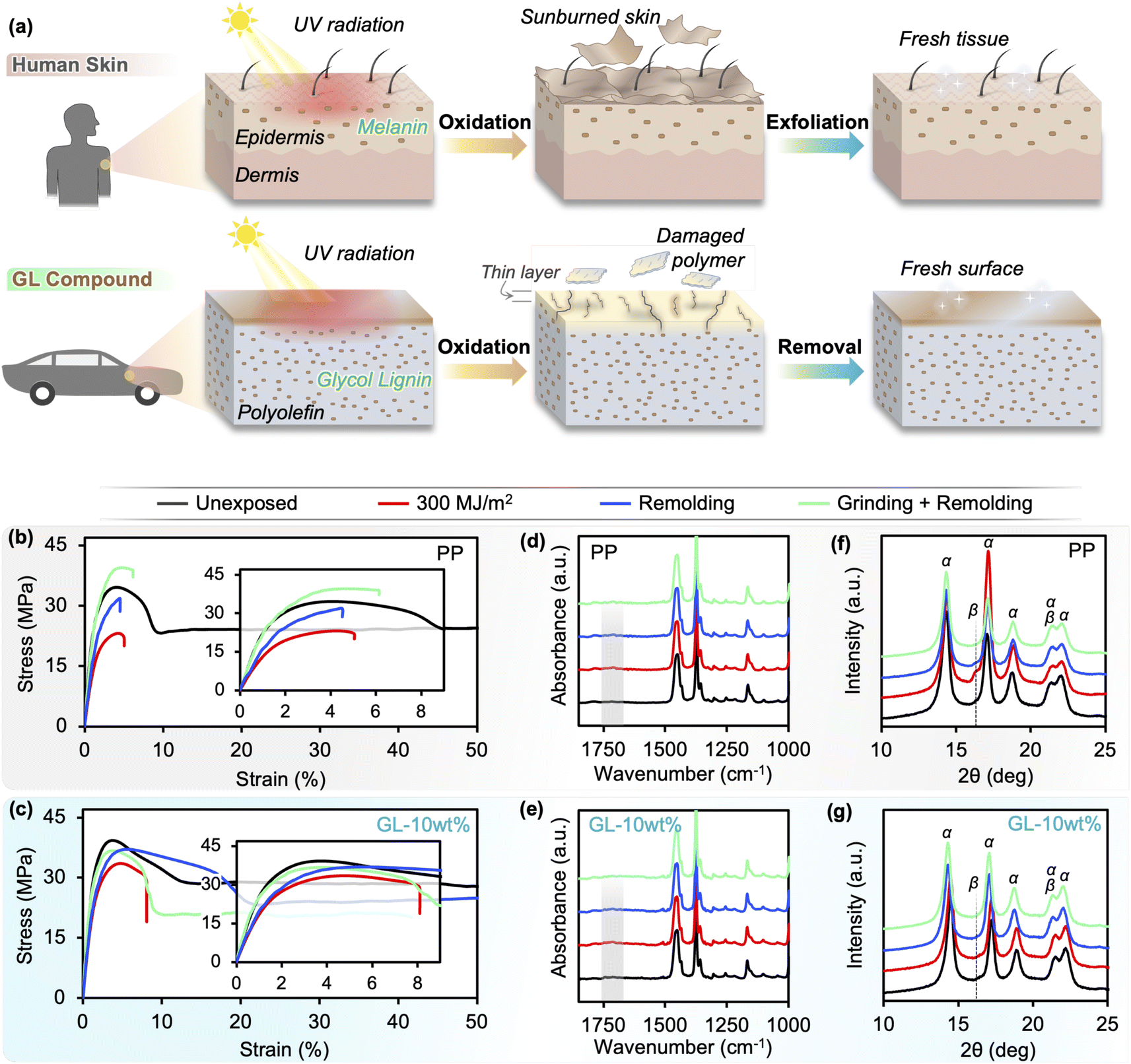 | ||
| Fig. 7 (a) Illustration depicting the biomimetic nature of GL blends: human skin contains melanin which functions as both a UV absorbent and antioxidant, restricting UV-induced sunburn to the outer surface layer which can then be exfoliated to reveal pristine tissue underneath. Likewise, lignin particles that are effectively dispersed in maleic-grafted polyolefins with the aid of PEG modification act as UV absorbent, antioxidant, and reinforcement to restrict photooxidation damage to the outer layer while retaining high mechanical performance, enabling closed-loop recycling on a large scale. (b and c) Tensile behavior, (d and e) FTIR spectra, and (f and g) XRD patterns of the neat PP and 10 wt% GL blend subjected to 300 MJ m−2 of UV irradiation and then recycled. | ||
To demonstrate recyclability, we chose the GL-10 wt% compound because it shows considerably improved durability over neat PP with relatively low lignin content, and selected an irradiation energy of 300 MJ m−2 in order to sufficiently degrade the material (equivalent to 12–14 months of sunlight exposure) for a valid assessment. Two mechanical recycling approaches were compared: (i) remolding the irradiated pellets without any further processing (Remolding), and (ii) mechanical processing of the irradiated pellets to remove the surface degradation layer before remolding (Grinding + Remolding). The surface removal was accomplished by grinding the pellets in a commercial rice polishing machine for roughly 4 min, then washing with water and ethanol and drying. The amount of surface material removed was 7.2% and 2.5% (by mass) for neat PP and GL-10 wt%, respectively (Fig. S12(a)†). Further optimization of the recycling conditions should be considered, including a comparison of different techniques depending on the part size and geometry (e.g., grinding, sand-blasting, milling). Unexposed pellets subjected to the same treatment did not exhibit any surface removal. Additionally, we included an alkali pre-treatment before grinding to assess the efficacy of chemically extracting the carbonyl-containing irradiated polymer phase, but this appeared to promote lignin agglomeration during remolding and thus ductility was not achieved: furthermore, the grinding treatment was repeated for five cycles to check the removal efficiency, but no significant difference in material properties was observed (Fig. S12(a)†).
As discussed in previous sections, the tensile behavior of neat PP drastically changes after UV exposure (residual strength and modulus of 68% and 73%, respectively; Fig. 4). When the irradiated PP is remolded without any other processing, the low Mw surface layer is incorporated into the bulk and results in reduced average Mw and altered chain structure, which explains why the tensile modulus does not recover to its pre-exposure value (Fig. 7(b)). This is supported by the FTIR spectrum (Fig. 7(d)) in which carbonyl peaks around 1710–1750 cm−1 seen in the as-irradiated specimen are still visible at lower intensity for the remolded PP specimen. In addition, XRD revealed that the irradiated specimens contain newly formed β-phase crystals (Fig. 7(f)) which are nucleated by the carboxyl groups populating the surface layer.49 A weak β-phase peak remains in the remolded specimen as well, corresponding to the carbonyl groups from the surface layer that are mixed into the bulk during remolding. However, remolding does remove the surface cracks and other morphological heterogeneities that develop during irradiation, which partially increases the tensile strength of neat PP until the degraded chemical structure causes premature failure (Fig. 7(b)). This occurs at a critical strain value equal to the failure strain of the as-irradiated material before remolding (around 5%). When the degraded surface layer of neat PP is removed by grinding before remolding, the strength fully recovers but the failure strain remains unchanged, indicating that the chemical structure cannot be fully regenerated to the pristine state using the processing conditions of this study. The carbonyl peaks in the FTIR spectrum decreased but are still visible, while the β-phase disappears from the XRD pattern. It may be possible to completely remove all degraded PP with extensive mechanical processing if the irradiation energy is low enough, but Fig. 2 shows that irradiation energies greater than 300 MJ m−2 result in irreversible degradation throughout the entire bulk of the neat PP specimens and thus makes recycling unfeasible.
Conversely, GL-10 wt% shows higher retention than neat PP after UV irradiation (residual strength and modulus of 87% and 83%, respectively), which makes the starting point more advantageous for recycling. As mentioned above, remolding removes the surface cracks and homogenizes the morphology which brings the tensile strength nearly to the pre-exposure value, while the incorporation of low Mw polymer from the surface layer into the bulk prevents significant change in the modulus from the as-irradiated state (Fig. 7(c)). The most remarkable difference compared to neat PP is that the failure strain of remolded GL-10 wt% nearly reaches the pre-exposure value, an unprecedented recovery of ductility. Coincidentally, the relatively small carbonyl peaks found in the irradiated GL-10 wt% specimens were no longer visible after remolding (Fig. 7(e)), and no β-phase was detected before nor after irradiation and remolding (Fig. 7(g)). This suggests that GL blends may be successfully recycled without surface treatment for low levels of UV degradation, which could reduce waste and simplify the process for large scale implementation. Similar to the restoration of epidermis by exfoliating sunburned tissue, removing the degraded surface layer before remolding restores the pristine material structure of the lignin/polymer blend, and the tensile modulus, strength, and failure strain all return to their pre-exposure values. These results demonstrate that a fully closed-loop recyclable biomass/polymer compound can be easily attained even at relatively low GL content, while higher contents can guarantee high performance even after extensive UV irradiation. We used commercially available pristine PP pellets in this study, but we assert that the same material design and compounding approach can be applied to other polyolefins including waste plastics, which would open the door to a dual upcycling/recycling framework for a circular economy (Fig. 8). Especially in the case of maleic-grafted polyolefins, the ester linkages formed with GL provide a means for transesterification or ester thesis during melt processing, allowing the material to behave macroscopically as a thermoplastic despite forming covalent bonds between the particles and matrix.6,50
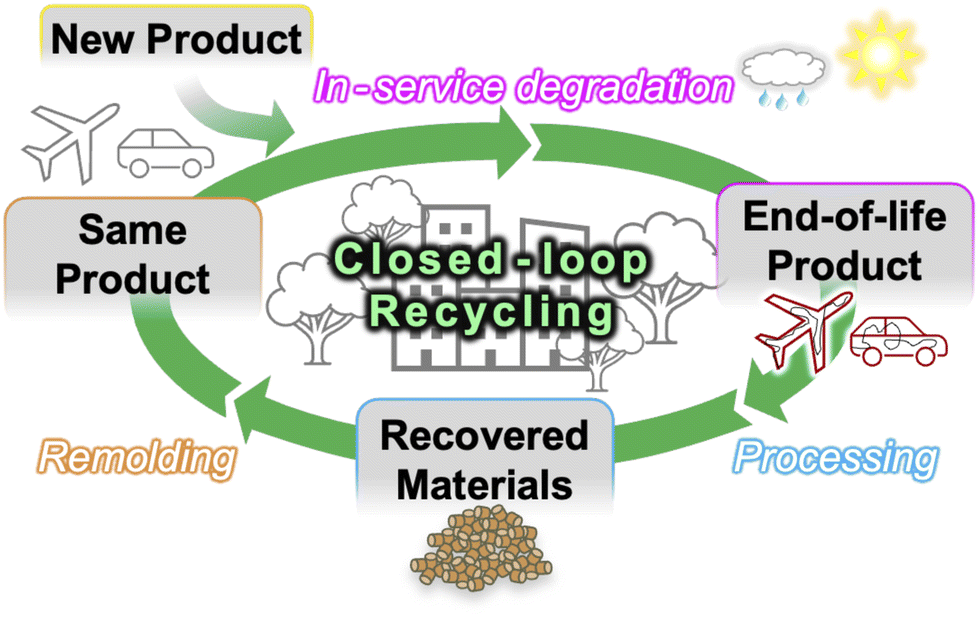 | ||
| Fig. 8 Scheme of closed-loop recycling enabled by biomass-based biomimetic materials. New products are manufactured with GL/polyolefin compounds and retain high performance during their service life, after which they are mechanically processed (e.g., sandblasting, pulverization, etc.) and pelletized, then re-manufactured into the same—or similar—product with the original material properties. | ||
Conclusions
We have presented a scalable, sustainable strategy for integrating polyolefins with renewable biomass to produce cost-effective and fully recyclable compounds. By mimicking the exfoliation of thin degraded surface layers found in biological structures such as human skin and tree bark, in which melanin and polyphenol compounds are primarily responsible for UV absorption and radical scavenging, GL successfully acts as a simultaneous UV absorbent, antioxidant, and mechanical reinforcement. This is achieved by utilizing a glycol modification that allows the lignin particles to be effectively dispersed and covalently bonded to the maleic-grafted polyolefin—a key feature of the material's design. Not only does this enhance the mechanical properties of the host polymer in the pristine state, but it drastically improves the performance during UV irradiation with less than 10% reduction in tensile strength and modulus after 1000 MJ m−2 (equivalent to ∼3 years of sunlight exposure). We show that changes in chemical structure and morphology are limited to the surface layer, ranging from depths of ∼80 μm (GL-30 wt%) to ∼400 μm (GL-10 wt%) after extensive irradiation. This coincides with smaller surface crack depths with higher GL content, supporting our assertion that GL also suppresses chain scission via its chemical structure and not just mechanically. The mechanical reinforcing ability of GL in polyolefins was elucidated by showing that irradiated blends do not follow surface crack-dominated brittle failure.Finally, closed-loop recyclability was demonstrated by remolding the irradiated materials, either with or without a mechanical processing step that involves removing the degraded surface layer. Unlike neat PP, which could not be successfully returned to its pristine state, compounds containing only 10 wt% GL exhibited nearly identical behavior to the pristine material even after 300 MJ m−2 of irradiation. Furthermore, this biomimetic polyolefin/biomass compounding strategy is not limited to polypropylene nor pristine starting materials, but can also be extended to the upcycling of waste plastics to reduce waste and pollution while producing value-added materials. We assert that these closed-loop recyclable materials will have a significant impact on in-service performance and end-of-life reprocessing in various applications from automobiles to household goods and more.
Author contributions
J. T.: investigation, methodology, writing – original draft. K. T.: conceptualization, funding acquisition, supervision, methodology, writing – original draft. K. N.: resources, writing – review & editing. T. T. N.: resources, writing – review & editing. T. Y.: project administration, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MAFF (Ministry of Agriculture, Forestry and Fisheries), Japan commissioned project “Development of Glycol Lignin–Based High-Value-Added Materials”, Grant Number J008722. The authors thank T. Akagawa and M. Kamon (NIMS) for assistance with sample preparation and tensile testing.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 1700782 CrossRef PubMed.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- W. Li, W. Zhao, H. Zhu, Z.-J. Li and W. Wang, J. Mater. Chem. A, 2023, 11, 2503–2527 RSC.
- E. Syranidou, K. Karkanorachaki, D. Barouta, E. Papadaki, D. Moschovas, A. Avgeropoulos and N. Kalogerakis, Environ. Sci. Technol., 2023, 57, 8130–8138 CrossRef CAS PubMed.
- F. Julienne, F. Lagarde and N. Delorme, Polym. Degrad. Stab., 2019, 170, 109012 CrossRef.
- G. P. Kar, M. O. Saed and E. M. Terentjev, J. Mater. Chem. A, 2020, 8, 24137–24147 RSC.
- Q. Wu, X. Wang, M. Nie and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 860–867 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- C. W. S. Yeung, J. Y. Q. Teo, X. J. Loh and J. Y. C. Lim, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef CAS.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustain. Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- A. Francois-Heude, E. Richaud, E. Desnoux and X. Colin, J. Photochem. Photobiol., A, 2015, 296, 48–65 CrossRef CAS.
- B. Gewert, M. M. Plassmann and M. MacLeod, Environ. Sci.: Processes Impacts, 2015, 17, 1513–1521 RSC.
- A. K. Rodriguez, B. Mansoor, G. Ayoub, X. Colin and A. A. Benzerga, Polym. Degrad. Stab., 2020, 180, 109185 CrossRef CAS.
- S. Morlat, B. Mailhot, D. Gonzalez and J.-L. Gardette, Chem. Mater., 2004, 16, 377–383 CrossRef CAS.
- S. Therias, G. Rapp, C. Masson and J.-L. Gardette, Polym. Degrad. Stab., 2021, 183, 109443 CrossRef CAS.
- C. Rouillon, P.-O. Bussiere, E. Desnoux, S. Collin, C. Vial, S. Therias and J.-L. Gardette, Polym. Degrad. Stab., 2016, 128, 200–208 CrossRef CAS.
- M. S. Rabello and J. R. White, Polymer, 1997, 38, 6379–6387 CrossRef CAS.
- I. Yakimets, D. Lai and M. Guigon, Polym. Degrad. Stab., 2004, 86, 59–67 CrossRef CAS.
- C. M. Rochman, M. A. Browne, B. S. Halpern, B. T. Hentschel, E. Hoh, H. K. Karapanagioti, L. M. Rios-Mendoza, H. Takada, S. The and R. C. Thompson, Nature, 2012, 494, 169–171 CrossRef PubMed.
- J. H. Bridson, R. Abbel, D. A. Smith, G. L. Northcott and S. Gaw, J. Hazard. Mater., 2023, 459, 132303 CrossRef CAS PubMed.
- M. K. Singh, A. K. Mohanty and M. Misra, Composites, Part B, 2023, 263, 110852 CrossRef CAS.
- A. K. Mohanty, S. Vivekanandhan, J.-M. Pin and M. Misra, Science, 2018, 362, 536–542 CrossRef CAS PubMed.
- D. Ye, S. Li, X. Lu, X. Zhang and O. J. Rojas, ACS Sustain. Chem. Eng., 2016, 4, 5248–5257 CrossRef CAS.
- R. Gadioli, W. R. Waldman and M. A. De Paoli, J. Appl. Polym. Sci., 2016, 133, 43558 CrossRef.
- F. Chen, W. Liu, S. I. S. Shahabadi, J. Xu and X. Lu, ACS Sustain. Chem. Eng., 2016, 4, 4997–5004 CrossRef CAS.
- J. A. Sirviö, M. Y. Ismail, K. Zhang, M. V. Tejesvi and A. Ämmälä, J. Mater. Chem. A, 2020, 8, 7935–7946 RSC.
- A. Guilhen, R. Gadioli, F. C. Fernandes, W. R. Waldman and M. A. De Paoli, J. Appl. Polym. Sci., 2017, 134, 45219 CrossRef.
- D. Ye, J. Kong, S. Gu, Y. Zhou, C. Huang, W. Xu and X. Zhang, Int. J. Biol. Macromol., 2018, 108, 775–781 CrossRef CAS PubMed.
- P. C. Eklund, O. K. Långvik, J. P. Wärnå, T. O. Salmi, S. M. Willför and R. E. Sjöholm, Org. Biomol. Chem., 2005, 3, 3336–3347 RSC.
- P. Buono, A. Duval, P. Verge, L. Averous and Y. Habibi, ACS Sustain. Chem. Eng., 2016, 4, 5212–5222 CrossRef CAS.
- F. Chen, H. Dai, X. Dong, J. Yang and M. Zhong, Polym. Compos., 2011, 32, 1019–1025 CrossRef CAS.
- J.-S. Yeo, D.-W. Seong and S.-H. Hwang, J. Ind. Eng. Chem., 2015, 31, 80–85 CrossRef CAS.
- O. A. T. Dias, M. Sain, I. Cesarino and A. L. Leão, Polym. Adv. Technol., 2019, 30, 70–78 CrossRef CAS.
- X. Xu, Z. He, S. Lu, D. Guo and J. Yu, Macromol. Res., 2014, 22, 1084–1089 CrossRef CAS.
- C. Miao and W. Y. Hamad, J. Appl. Polym. Sci., 2017, 134, 44669 CrossRef.
- Y. Tang, M. Jean, S. Pourebrahimi, D. Rodrigue and Z. Ye, Can. J. Chem. Eng., 2021, 99, 27–38 Search PubMed.
- A. S. Babetto, M. C. Antunes, S. H. P. Bettini and B. C. Bonse, J. Polym. Environ., 2020, 28, 699–712 CrossRef CAS.
- M. Aldas, A. Paladines, V. Valle, M. Pazmiño and F. Quiroz, Int. J. Polym. Sci., 2018, 10, 2474176 Search PubMed.
- T. T. Nge, T. Yamada, Y. Tobimatsu, M. Yamamura, R. Ishii, O. Tanaike and T. Ebina, ACS Sustain. Chem. Eng., 2021, 9, 756–764 CrossRef CAS.
- T. T. Nge, Y. Tobimatsu, S. Takahashi, E. Takata, M. Yamamura, Y. Miyagawa, T. Ikeda, T. Umezawa and T. Yamada, ACS Sustain. Chem. Eng., 2018, 6, 7841–7848 CrossRef CAS.
- J. Tanks, K. Tamura, K. Naito, T. T. Nge and T. Yamada, Compos. Sci. Technol., 2023, 238, 110030 CrossRef CAS.
- W. Song, H. Yang, S. Liu, H. Yu, D. Li, P. Li and R. Xing, J. Mater. Chem. B, 2023, 11, 7528–7543 RSC.
- T. Kawamura,M. Miyamoto, Composition of a Polyolefin Resin and its Application as Gas Barrier Material, Jp. Pat., JP2016141793A, 2016 Search PubMed.
- X. Shi, P. Chen, K. Han, C. Li, R. Zhang, J. Luo and Z. L. Wang, J. Mater. Chem. A, 2023, 11, 11730–11739 RSC.
- S. Wang and J. Zhang, Sol. Energy Mater. Sol. Cells, 2013, 117, 577–584 CrossRef CAS.
- Y. Zhang and M. Naebe, ACS Sustain. Chem. Eng., 2021, 9, 1427–1442 CrossRef CAS.
- Y. Qian, X. Qiu, X. Zhong, D. Zhang, Y. Deng, D. Yang and S. Zhu, Ind. Eng. Chem. Res., 2015, 54, 12025–12030 CrossRef CAS.
- A. Salazar, P. M. Fontini and J. Rodriguez, Eng. Fract. Mech., 2014, 126, 87–107 CrossRef.
- S. Zhao, K. Liu, S. Zhou, Y. Shi and Z. Xin, Polymer, 2017, 132, 69–78 CrossRef CAS.
- S. Yang, W. Liu, J. Guo, Z. Yang, Z. Qiao, C. Zhang, J. Li, J. Xu and N. Zhao, J. Am. Chem. Soc., 2023, 145, 20927–20935 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06230a |
| This journal is © The Royal Society of Chemistry 2024 |