
Improving upon rechargeable battery technologies: on the role of high-entropy effects
Zihao
Zhou
a,
Yuan
Ma
*b,
Torsten
Brezesinski
 c,
Ben
Breitung
c,
Yuping
Wu
*b and
Yanjiao
Ma
*a
c,
Ben
Breitung
c,
Yuping
Wu
*b and
Yanjiao
Ma
*a
aSchool of Energy and Mechanical Engineering, Nanjing Normal University, Nanjing 210023, China. E-mail: yanjiao.ma@njnu.edu.cn
bConfucius Energy Storage Lab, Key Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy and Environment & Z Energy Storage Center, Southeast University, Nanjing 211189, Jiangsu, China. E-mail: yuan.ma@seu.edu.cn; wuyp@seu.edu.cn
cInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), 76131, Karlsruhe, Germany
First published on 15th November 2024
Abstract
In recent years, high-entropy methodologies have garnered significant attention in the field of energy-storage applications, particularly in rechargeable batteries. Specifically, they can impart materials with unique structures and customized properties, thereby showcasing new attributes and application potential. In this review, we describe the various influences that the high-entropy concept exert on electrochemical performance of materials. We begin by introducing the concept and the basic effects, namely structural stabilization, lattice distortion, high defect density, and cocktail effects. Then, we provide a comprehensive overview of the fundamental advantages of high entropy or compositional/occupational disorder in battery materials design, including anodes, cathodes, and electrolytes. The summary of these effects is crucial for understanding how entropy affects the electrochemical properties of materials (redox activity, cyclability, etc.). Additionally, we outline the challenges encountered in this area of research and discuss the critical factors for rationally designing novel electrode materials, as well as the potential future directions for high-entropy strategies in the field of electrochemical energy storage.
Broader contextHigh-entropy materials (HEMs) show much promise in battery development because of their distinct features. However, the fundamental notion of the high-entropy strategy and its role in batteries remain poorly understood. This review explores the theoretical concept of configurational entropy and presents six fundamental advantages of high-entropy battery materials (HEBMs). These advantages, derived from the primary effects in HEMs, significantly enhance the electrochemical performance. Seminal discoveries discussed in this review demonstrate the transformative potential of high-entropy concepts in battery materials design, including structural stabilization and defect engineering achieved through increased configurational entropy. The implications for research span various scales, from molecular-level lattice distortions, facilitating ion diffusion, to macroscopic-level improvements in cyclability and stability. Future directions include addressing the challenges in synthesizing and designing HEBMs, tailoring element selection, and exploring theoretical models to guide materials development. |
1. Introduction
The global energy demand continues to rise, propelling the transition from the fossil fuel era to the renewable energy era. While renewable energy sources offer cleanliness and renewability benefits, challenges such as unsustainable energy supply and low energy efficiency impede their practical application. Overcoming these obstacles requires the development of high-performance energy-storage devices. Batteries, as crucial components of energy-storage devices, have become a focal point of research in energy applications.1 Significant progress has been achieved in rechargeable battery research,2–9 however, limitations in capacity, stability, and sustainability still exist. Therefore, exploring more efficient, stable, and sustainable battery materials is imperative to meet the increasing energy demand.In recent years, high-entropy materials (HEMs) have emerged as a novel concept in materials science, finding applications in various fields, including catalysis,10–13 thermoelectrics,14 and electrochemical energy storage.15–27 Unlike traditional materials, HEMs typically encompass single-phase, multicomponent (solid-solution) materials, wherein the entropy-driven effect resulting from the introduction of multiple elements plays a pivotal role in enhancing performance. In practical applications, utilizing high-entropy strategies enables the customization of performance and achieving breakthroughs in overcoming the limitations of battery materials.28–52 Overall, the advent of high-entropy methods presents a new avenue for the design of high-performance battery materials.
In 2016, Bérardan et al.53 synthesized a new solid-state electrolyte with a rock-salt type structure, (Mg,Co,Ni,Cu,Zn)1−xLi(Na)xO, introducing the high-entropy method to battery materials for the first time. Subsequently, Sarkar et al.54 synthesized a micron-sized high-entropy oxide, (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O, and used it as an anode material in lithium-ion batteries. Fig. 1 illustrates that the advancement of rock-salt type materials has inspired research into HEMs in various categories,55,56 including layered, spinel, perovskite, Prussian blue analogue (PBA), garnet, and argyrodite structures. For example, Nax(FeMnNiCuCo)[Fe(CN)6] is a high-entropy PBA cathode material that exhibits a high specific capacity (in the potential range between 2.0 and 4.2 V vs. Na+/Na) in the first cycle and maintains 94% of its initial capacity after 150 cycles.57 The high-entropy spinel-type oxide anode material, (CrNiMnFeCu)3O4, showed enhanced structural stability and cycling performance, resulting in high cell capacities and good cycling stability when lithium is inserted.58 In solid-state electrolytes with an argyrodite structure, occupational disorder induced by increasing chemical complexity promotes ionic conductivity and enables fast battery charging and discharging.59
 | ||
| Fig. 1 The historical development of HEBMs with different structural characteristics, including rock-salt, layered, spinel, perovskite, PBA, NASICON, garnet, and argyrodite phases. | ||
The emergence of high-entropy strategies has opened up new possibilities for designing battery materials and has propelled the advancement of the energy-storage sector.60–79 Nevertheless, until now, only a few studies have thoroughly summarized the impact of high-entropy effects on improving electrochemical characteristics. For this reason, this review aims at providing an overview of the various improvements achieved to battery materials through high-entropy effects. We are categorizing according to these improvements and identify seven fundamental advantages. The review further discusses the principles of formation of HEMs, including synthesis techniques used, and theoretical calculations applied in developing high-entropy battery materials (HEBMs). Moreover, it provides general recommendations for HEM design and other potential directions in the battery field, where high-entropy strategies should be considered.
2. Theoretical considerations
Entropy is a complex physical quantity that describes the tendency of a system to evolve in the direction of increasing disorder, therefore also defining reversibility and irreversibility of reactions. It determines the energy of formation of every compound, and there are plenty of different definitions, namely, Clausius, Shannon, Renyi, etc.80 There are also various entropy contributions, e.g., stemming from the configuration of the incorporated elements (configurational entropy) in a single-phase structure. Aside from the configurational entropy, the entropy of mixing is a relevant fraction of entropy necessary to describe the evolving features of HEMs. The entropy of mixing refers to the alteration in a system's entropy caused by the number and type of incorporated elements.In thermodynamics, the mixing entropy of a solid solution typically encompasses several aspects, the above-mentioned configurational entropy, vibrational entropy, magnetic dipole entropy, and electronic random entropy, to name a few.81,82 In the case of a solid-solution with M components, the system's configurational entropy can be determined by applying the following equation:82
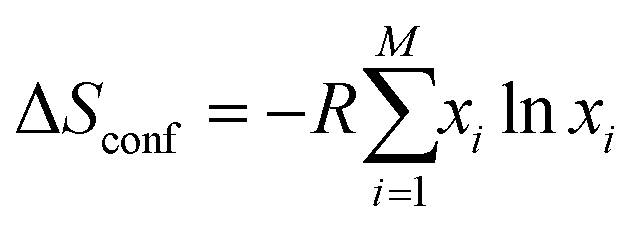 | (1) |
ΔSconf = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnM lnM | (2) |
Given that certain materials, such as rock-salt oxides, possess both anions and cations, the subsequent equation can be employed for the purpose of computation:54,83
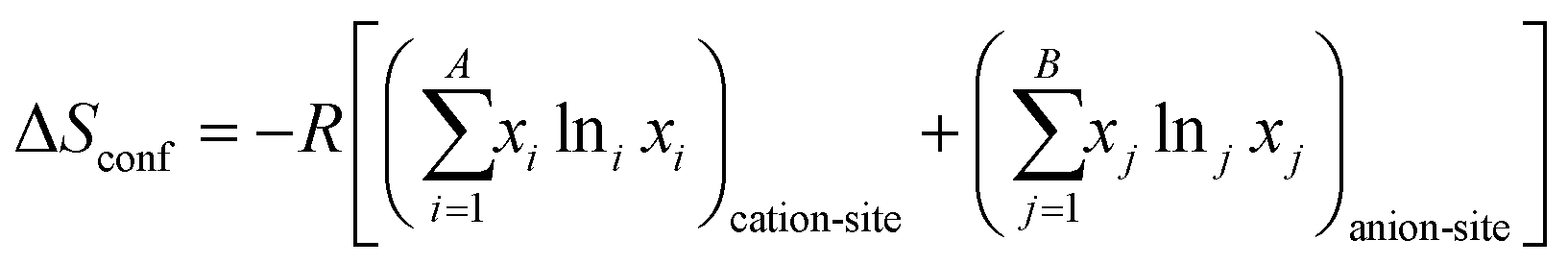 | (3) |
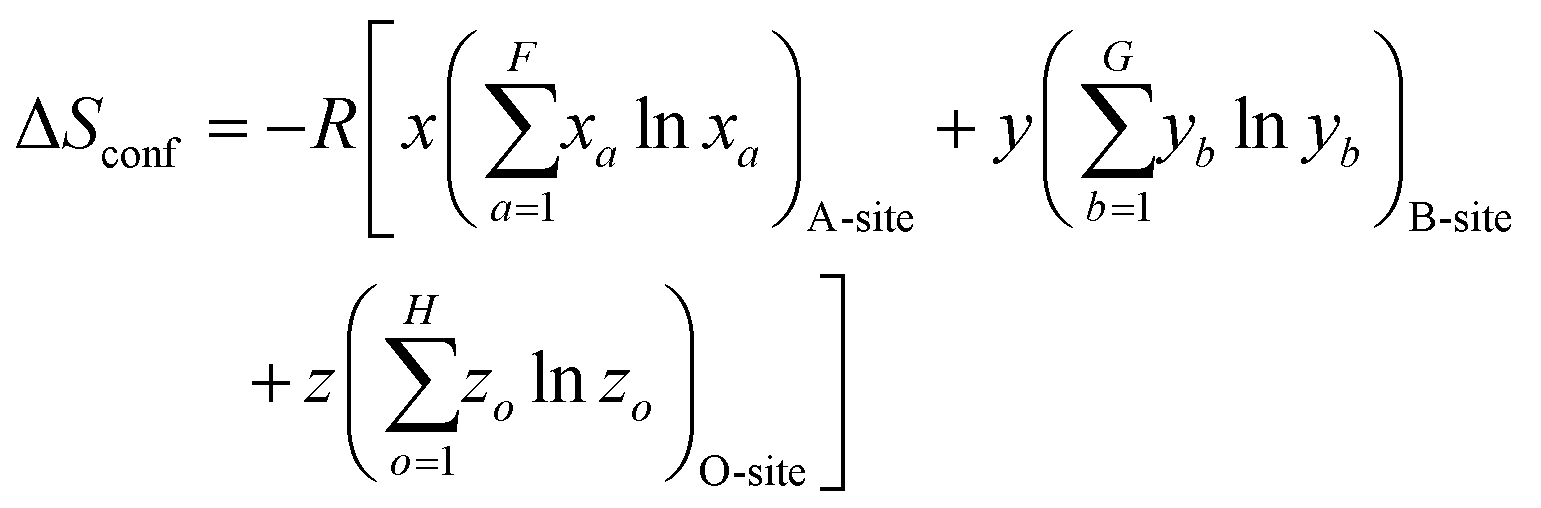 | (4) |
Given the complexity of accurately determining configurational entropy and the significance of elemental composition in defining HEMs, it is crucial to determine stoichiometric ratios with precision. Accurate stoichiometry ensures the correct application of the above equations, facilitating the calculation of entropy, which is essential for the successful synthesis of HEMs. To achieve this, determining the elemental distribution on the atomic scale becomes a critical step in HEM synthesis.86 Consequently, several characterization techniques, both qualitative and quantitative, are commonly employed in their design. Inductively coupled plasma (ICP) measurements are often combined with techniques such as OES (optical emission spectroscopy)57,78 and MS (mass spectrometry)87 to perform quantitative analyses of elements in materials based on the position and intensity of characteristic spectral lines. For instance, Ma et al.57 utilized ICP-OES for stoichiometric analysis of the synthesized cathode material, revealing that the M-site cations were present in equimolar amounts, consistent with the design. Energy-dispersive X-ray spectroscopy (EDS) mapping allows researchers to obtain distribution information of different elements within a sample, with spatial resolution reaching the micrometer or even nanometer scale.86 This is particularly useful for studying compositional differences, analyzing fine particles, and understanding the microstructure and composition of materials. When combined with scanning electron microscopy (SEM) and transmission electron microscopy (TEM), EDS can analyze the chemical composition of different phases, making it highly beneficial for studying various phases in multiphase materials or identifying trace impurities and inclusions. Hu's research group87 utilized this technique to characterize a series of high-entropy alloy nanoparticles (ranging from five to eight elements, HEA-NPs), revealing that they were solid solutions uniformly dispersed on a carbon support. Additionally, other commonly used elemental analysis techniques such as X-ray photoelectron spectroscopy (XPS), X-ray absorption near edge structure (XANES), and extended X-ray absorption fine structure (EXAFS) are often employed to investigate the redox states of electrode materials and provide local structural information.57,78
To date, high-entropy strategies have been extended to alloys,88–99 oxides,100–132 carbides,133–135 borides,136 nitrides,137 and other materials,138–151 experiencing sustained growth in popularity. Compared to conventional materials, HEMs possess more complex compositions and exhibit higher disorder within the system. As a result, they manifest numerous surprising physical and chemical properties.
In 2004, Yeh et al.152 made a groundbreaking discovery by introducing multiple elements into alloys at equimolar concentrations, designing a novel multiprincipal element alloy. The increased configurational entropy of the alloy facilitated the synthesis of single-phase solid solutions, thereby laying the foundation for high-entropy alloys (HEAs). Since then, the term HEAs has been used to describe solid-solution alloys that contain five or more elements and each with an atomic percentage mentioned above. Compared to traditional alloys, HEAs exhibit significantly higher melting points,153 excellent corrosion resistance,154 and enhanced mechanical properties,155,156 resulting from mixing multiple elements. Subsequently, the development of high-entropy functional ceramics quickly followed suit.157 High-entropy ceramics (HECs), like HEAs, are characterized by the presence of five or more cations or anions in sublattices of a solid solution, resulting in a high configurational entropy.158 By augmenting the quantity of principal elements, HECs exhibit distinctive characteristics, including improved hardness,159 elevated melting point,136 enhanced durability under extreme conditions,160 and resistance to wear and corrosion.161
Implementing high-entropy strategies in the design of electrode materials presents a novel approach to improving performance and durability in rechargeable batteries.162–210 By tailoring properties through the introduction of different components, batteries using HEMs can potentially achieve superior energy densities,211 long lifetimes,212 and accelerated charging rates.213 This has great potential to advance breakthroughs in battery technology.214
3. Core effects of “high entropy/compositional disorder” in rechargeable batteries
The intricate interactions of multiple elements give rise to the unique properties of HEMs, which distinguish them from traditional functional materials, and can be summarized as cocktail effects, lattice distortion, high defect density, and structural stabilization.49(1) Structural stabilization:21,49 it is important to distinguish the concept of structural stabilization from entropy stabilization. Entropy stabilization arises when the entropy becomes the dominant factor in the thermodynamic landscape, modulating the structure and phase behavior. Notably, the rock-salt-type oxide, (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O, developed by Rost and co-workers is one of the few materials that has been proven to be entropy-stabilized. In most HEMs, while configurational entropy contributes to their formation and stability, it is not always the primary factor compared to enthalpy. Other factors such as native defects also play a significant role in influencing stability.
(2) Lattice distortion:21,215 the presence of several elements simultaneously occupying the same (sub)lattice sites will unavoidably cause a geometric distortion. The degree of distortion is determined by the size, valence, and electronegativity of atoms or ions that coexist within the same (sub)lattice. Severe distortions can cause collapse of the structure, which in turn affects the properties of the material. On the other hand, moderate lattice distortions have been proven to change the energy landscape of ion-diffusion pathways, resulting in the creation of a percolating network that facilitates mobility.
(3) Defect density:21,49 defects include point defects (e.g., vacancies and interstitials), line defects (e.g., dislocations), and surface defects (e.g., grain and phase boundaries). Compared to conventional materials, HEMs usually exhibit higher defect densities, which are closely related to their multicomponent and highly disordered nature. High defect densities have a significant impact on the physical, chemical, and mechanical properties of HEMs, especially the catalytic activity, diffusion properties, and electrical behavior.
(4) Cocktail effects:21,215 the synergy of different components during the mixing process leads to the formation of HEMs, which may exhibit improved performance. This improvement cannot be solely attributed to the qualities of any individual component, but rather arises from the intricate interplay between the numerous constituents. Minor alterations in the kind of elements and ratios of their quantities can result in significant modifications in the functioning of the system. Hence, during the practical implementation, the performance of materials can be tailored.
Lately, high-entropy strategies have garnered significant attention among battery researchers, and an increasing number of studies have demonstrated that HEBMs exhibit superior performance over traditional materials.216 The improved performance of these electrode materials is closely linked to the benefits of the system's increased configurational entropy and compositional disorder. Herein, we describe six fundamental advantages of HEBMs, which are an expansion of the four primary effects discovered in HEMs (Fig. 2). These benefits encompass (1) zero or low volumetric strain, (2) suppression of adverse phase transitions, (3) facilitation of conversion processes, (4) constructing transport channels via lattice distortions, (5) disorder-facilitated ion diffusion, and (6) synergies induced by cocktail effects. It should be noted that these basic advantages do not exist independently, rather they often coexist or several are present within the same high-entropy system. In the following, these major advantages are introduced individually to provide readers with a clearer understanding of HEBMs.
 | ||
| Fig. 2 Fundamental advantages of HEMs in battery materials design. | ||
In addition to HEMs, certain entropy-modulation strategies have shown significant potential in material modification/functionalization. Although the entropy value of these materials with complex compositions (five elements or more) does not reach the high-entropy threshold (1.5R), the interactions between the individual elements are substantial and even profound. While we do not recommend classifying them as HEMs, the coupling of these elements can lead to unprecedented properties. To further explore the role of configurational entropy in enhancing the performance of materials, we will include these samples in our discussion.
3.1. Structural stabilization in HEMs
The expression of the change in Gibbs free energy during mixing in a multicomponent system can be represented by the Gibbs–Helmholtz equation:| ΔGmix = ΔHmix − TΔSmix | (5) |
In cases when ΔHmix is positive, the TΔSmix term indeed affects the change in Gibbs free energy. However, it is crucial to note that higher positive values of ΔSmix do not always lead to a decrease in ΔGmix and subsequent stabilization. While some literature reports attribute the improvement in battery performance, particular the cycling stability, to entropy-stabilization effects, discussing entropy stabilization without proper validation clearly lacks scientific rigour, and the concept of entropy stabilization is still controversial in the community. Rost's groundbreaking work aimed to provide such evidence by synthesizing materials exclusively at high temperatures, which resulted in multiphase compounds at lower temperatures. However, it is important to recognize the contextual limitations of this phenomenon, as it may not universally apply to high-entropy systems. While entropy may contribute to stability, it is not the sole determinant. System stability encompasses various factors beyond entropy, and merely increasing ΔSmix does not guarantee stability. Despite its prevalence in the literature, we must emphasize the complex nature of stabilization mechanisms in multicomponent systems.
While the formation conditions for entropy stabilization are rigorous and the number of truly entropy-stabilized HEMs is limited, it is undeniable that the remaining non-entropy-stabilized materials of complex composition are generally more stable than single-component materials, and that entropy still plays a positive role in the structural stability during battery operation of these systems.
 | ||
| Fig. 3 (a) Schematic illustration of zero or low volumetric strain evolution during cycling. (b)–(f) HE-LNMO cathode.216 (b) An atomic model of HE-LNMO with an O3-type lattice. (c) HAADF-STEM images of delithiated NMC-811 (upper panel) and HE-LNMO (lower panel) after in situ heating. (d) Long-term cycling performance. (e) and (f) Cross-sectional SEM images of NMC-811 and HE-LNMO secondary particles. (g)–(j) HE-PBA cathode.57 (g) First-cycle voltage profiles at 0.01 A g−1 (left) and comparison of specific energies (right) of HE-PBA and ME-PBAs. (h) Initial cyclic voltammograms at 0.05 mV s−1. (i) Multirate galvanostatic cycling. (j) Operando XRD analysis of the electrochemical extraction/insertion of Na+ ions from/into HE-PBA. (k) HE-HCF sample.212 Contour plot of in situ heating XRD patterns from room temperature to 305 °C. (l)–(n) NVMP cathode.219 (l) Working principle of the NVMP//HC cell. (m) Galvanostatic charge/discharge curves of the NVMP cathode and HC anode. (n) Cycling performance of the full cell at 0.2C rate. | ||
3.1.1.1. Layered cathode materials. The strong oxidation potential of Ni4+ and the release of oxygen from Ni-rich cathode materials result in poor thermal stability. Additionally, they experience large volume changes during cycling, leading to structural degradation that affects battery stability.220 While the introduction of Co into battery materials has been found to stabilize the structure and enhance rate performance, the limited available reserves and geopolitical issues constrain the commercialization and practical application of Ni-rich cathode materials. Zhang et al.216 synthesized a Co-free, layered cathode material, LiNi0.8Mn0.13Ti0.02Mg0.02Nb0.01Mo0.02O2 (HE-LNMO), using a high-entropy doping strategy (Fig. 3b). The initial Coulomb efficiency of HE-LNMO reached 94%, and it exhibited a capacity retention of 98% after 50 cycles. In single-layer pouch cell tests, HE-LNMO demonstrated exceptional cycling performance with a capacity retention of 95% in a voltage range of 2.8–4.2 V after 500 cycles (Fig. 3d). Furthermore, HE-LNMO showed an almost zero-strain performance, significantly reducing lattice distortion-induced crack formation (Fig. 3e and f), thus improving cycling stability. Additionally, the pinning effect of the dopants helps stabilize the lattice and ensures good thermal stability (Fig. 3c).
3.1.1.2. PBA cathode materials. The distinctive framework structure of high-entropy Prussian blue analogues (HE-PBAs) provides exceptional sodium-storage capabilities. However, as cathode materials, (irreversible) phase transitions of PBAs during cycling negatively affect the reversible specific capacity.221 Ma et al.57 introduced five different elements at the transition-metal (TM) sites in equimolar ratios to create the HE-PBA, Nax(FeMnNiCuCo)[Fe(CN)6]. The latter material demonstrated superior electrochemical performance and good cycling stability (Fig. 3g–i). Operating at a specific current of 0.01 A g−1 (2.0–4.2 V vs. Na+/Na), HE-PBA delivered an initial specific discharge capacity of 120 mA h g−1, while medium-entropy PBAs (ME-PBAs), derived by removing one of the metal elements (–Mn, –Co, –Cu, –Ni, and –Fe), displayed varying degrees of capacity decrease. Moreover, after 150 cycles, the capacity retention of HE-PBA was 94%, surpassing that of other medium-entropy PBAs. In situ X-ray diffraction (XRD) characterization revealed that HE-PBA experiences almost zero strain upon battery operation (Fig. 3j). Multiple active redox centers and the high-entropy configuration contribute to its superior cycling stability and rate performance.
Peng et al.212 synthesized a high-entropy hexacyanoferrate (HE-HCF), Na1.41Mn0.32Fe0.11Co0.28Ni0.32Cu0.32[Fe(CN)6], comprising five transition metals. The HE-HCF demonstrated good cycling and thermal stability, retaining 95% of its capacity after 10000 cycles, corresponding to a decay rate of 0.0005% per cycle. Even after 50000 cycles, the capacity retention remained at 80%. The performance testing of HE-HCF at 60 °C further revealed good stability at 1.5C rate, delivering a specific capacity of up to 93 mA h g−1 and retaining 86% after 200 cycles. In situ heating XRD indicated a transition of HE-HCF from cubic to tetragonal structure at 165 °C, which remained stable in the range of 165–265 °C. Beyond 265 °C, the crystal structure collapsed (Fig. 3k). The high performance due to quasi zero-strain (de)sodiation behavior and the structural and thermal stability of HE-HCF can be attributed to its distinctive high-entropy configuration.
3.1.1.3. NASICON-type cathode materials. Na3VM(PO4)3 undergoes irreversible phase transitions at high potentials and exhibits limited thermal stability, impeding its practical implementation.222,223 Li et al.218 synthesized a high-entropy material, Na3.4Fe0.4Mn0.4V0.4Cr0.4Ti0.4(PO4)3 (HE-NASICON), using the sol–gel method. HE-NASICON addresses the irreversible lattice evolution experienced by traditional polyanionic materials during the multi-sodium insertion/extraction processes. Specifically, it delivered a high specific capacity of 161.3 mA h g−1 (1.5–4.5 V vs. Na+/Na). Additionally, HE-NASICON demonstrated good cycling stability, with a capacity retention of 93% after 100 cycles at 0.5C rate and 85% after 1000 cycles at 5C. HE-NASICON also maintains lattice stability and undergoes minimal volume changes during cycling.
Similarly, Li et al.219 reported about a high-entropy NASICON-type cathode material of composition Na3VAl0.2Cr0.2Fe0.2In0.2Ga0.2(PO4)3 (NVMP), achieving a reversible specific capacity of 102 mA h g−1 at 0.1C rate. Even at 20C, NVMP demonstrated good cycling stability, retaining 87% of its capacity after 5000 cycles. The performance improvement is attributed to the small volume expansion of only 1.1% upon cycling. Additionally, when paired with a hard carbon anode (Fig. 3l), the NVMP//HC cells exhibited good electrochemical performance (Fig. 3m and n).
Similar to cathode materials, certain types of anodes, particularly alloy-based ones, experience significant volumetric strain during ion insertion and extraction, leading to irreversible structural evolution and rapid capacity degradation.224 Recently, Zhao and colleagues225 discovered that by mixing multiple elements to increase the configurational entropy of the system, substantial potential is shown in alleviating the volumetric expansion/contraction issues associated with alloy electrodes. Based on this, they designed a high-entropy alloy, SnSbMnBiTe (HE-SSMBT). Research indicated that HE-SSMBT exhibits strain-adaptive capabilities; after multiple charge/discharge cycles, it consistently showed lower lattice strain levels compared to control samples, maintaining a reversible alloying process. Thanks to these advantages, it is capable of delivering a specific capacity of 2838.5 mA h cm−3 at a specific current of 0.1 A g−1 after 200 cycles and 1017.4 mA h cm−3 at 5 A g−1, outperforming medium-entropy and low-entropy materials.
In general, the presence of multiple elements creates a pronounced anisotropic lattice strain field within the system. This heterogeneity may enhance the material's adaptability to local stress, preventing abrupt lattice expansion/contraction during ion insertion/extraction. The underlying mechanism of this kind of buffering effect is complex, with factors such as ionic radius, valence state, bond strength, grain boundaries, and defects playing significant roles.
 | ||
| Fig. 4 (a) Schematic illustration of the suppression of adverse phase transitions. (b) and (c) The high-entropy composition randomly assigns redox elements, with the host's structural variations accommodating these changes and delaying phase transitions.229 (b) Conventional O3-type Na-ion cathodes with three different types of TM species and (c) the proposed HEO cathodes. (d)–(i) Na-deficient O3-type cathodes.230 (d) Sodium diffusion pathways in MTS15 (top) and MTS45 (bottom). (e) Configurational entropy versus O–Na–O slab spacing. (f) Radar chart displaying the performance comparison. (g) and (h) Impedance spectra of the electrodes in the fully discharged state after 10 cycles. (i) Arrhenius plot of activation energy for the MTS15 and MTS45 cathodes. (j)–(l) NaMNO2 and CuMgTi-571 cathodes.231 (j) Schematic of the design strategy for optimizing P2-type cathodes. (k) and (l) Contour plots of desodiated CuMgTi-571 (left) and NaMNO2 (right) during heating from 30 to 450°C at 5°Cmin−1. (m) and (n) T-LRM and E-LRM cathodes.232 (m) Capacity retention of T-LRM and E-LRM after 100 cycles at 0.1C (1C = 250 mA g−1). (n) Normalized capacity–voltage curves of E-LRM cycled at 1C rate. | ||
At present, the primary focus of research on the inhibition of irreversible phase transitions in layered materials revolves around two key factors. The first involves lowering the cutoff voltage to prevent adverse structural changes at high potentials,233 albeit at the cost of sacrificing capacity. The second strategy includes material modification methods such as doping234–238 to promote reversible phase transitions and enhance electrochemical performance. Recent studies have demonstrated that employing high-entropy strategies can facilitate a tailored design of lattice structures.239 This approach may effectively mitigate irreversible phase transitions in layered electrodes,229–232,240–244 thereby increasing the cycling stability.
3.1.2.1. O3-type layered cathode materials. Zhao et al.229 incorporated nine elements at the TM sites to fabricate a sodium layered cathode material, NaNi0.12Cu0.12Mg0.12Fe0.15Co0.15Mn0.1Ti0.1Sn0.1Sb0.04O2, which successfully addressed the issue of irreversible phase transitions in traditional O3-type oxides.245 The material demonstrated a capacity retention of about 83% after 500 cycles. Microstructural characterization revealed that the phase transition between O3 and P3 structures is highly reversible during cycling, with some hysteresis in the phase evolution, enabling the O3-type phase to retain over 60% of the total capacity. The research showed that HEOs, unlike typical O3-type oxides, contain several TM species that can adapt to changes in local interactions during Na+ (de)intercalation (Fig. 4b and c).
In a study by Wang et al.,230 sodium-deficient O3-type layered cathodes, Na0.83Li0.1Ni0.25Co0.2MnxTixSn0.45−2xO2−δ (MTS45, x = 0; MTS35, x = 0.05; MTS25, x = 0.1; and MTS15, x = 0.15), have been designed by adjusting the stoichiometry of inert cations (Mn4+, Ti4+, and Sn4+) while maintaining the stoichiometry of the active cations constant. MTS45, MTS35, MTS25, and MTS15 exhibit configurational entropies of 1.26R, 1.57R, 1.71R, and 1.75R, respectively (Fig. 4e). The study indicated that MTS15 has a larger O–Na–O layer spacing, and adding Mn and Ti enhances lattice stability, resulting in a highly reversible O3–P3–O3 phase transition in MTS15. Consequently, MTS15 demonstrated good rate performance and cycling stability (Fig. 4f), with a maximum capacity retention of 77% at 10C and good retention at other rates. Furthermore, MTS15 still provided a capacity retention of 87% after 200 cycles at 2C. Experimental findings further indicated that the change in compositional complexity plays a significant role in the reversible phase change from O3 to P3. In addition, the use of high-entropy methods is promising in relieving diffusion energy barriers (Fig. 4d and i) and improving the diffusion kinetics of Na+ (Fig. 4g and h).
Similarly, Lin et al.240 fabricated a high-entropy O3-type layered cathode material, NaCu0.1Ni0.3Fe0.2Mn0.2Ti0.2O2 (NCNFMT), using the Pechini method. NCNFMT deliverd a reversible specific capacity of 130 mA h g−1, with 87% capacity retention after 100 cycles at 0.1C and about 71% after 500 cycles at 0.5C. Moreover, in quasi solid-state NCNFMT|polyethylene oxide-Na3.2Ca0.1Zr1.9Si2PO12|Na cells, the discharge capacity reached 112 mA h g−1, with a capacity retention of 78% after 100 cycles. Operando synchrotron XRD analysis revealed a delayed transition of the O3 phase, resulting in improved rate performance and cycling stability of NCNFMT.
3.1.2.2. P2-type layered cathode materials. Fu et al.231 synthesized P2-type Na0.62Mn0.67Ni0.23Cu0.05Mg0.07Ti0.01O2 (CuMgTi-571) layered cathodes through a hydrothermal method. They demonstrated that entropy modulation has a positive effect on stabilizing the crystal structure, which is crucial for improving the reversibility of the sodium extraction/insertion processes. Additionally, the presence of a greater number of {010} facets enhanced the diffusion rate of Na+ (Fig. 4j). In comparison with Na0.62Mn0.67Ni0.37O2 (NaMNO2), the optimized CuMgTi-571 cathode demonstrated good structural (thermal) stability (Fig. 4k and l) and fast kinetics, enabling high charging and discharging rates of up to 10C and providing a cycle life exceeding 2000 cycles.
3.1.2.3. Layered Li-rich cathode materials. Lithium-rich materials (LRMs) have the potential to significantly enhance the energy density of batteries through redox reactions of both anions and cations,246 positioning them as some of the most promising candidates for next-generation cathodes in secondary batteries. However, the irreversible structural evolution of LRMs can result in capacity decay. Song et al.232 successfully applied the high-entropy strategy to a Li-rich layered cathode and synthesized Li1.0[Li0.15Mn0.50Ni0.15Co0.10Fe0.025Cu0.025Al0.025Mg0.025]O2 (E-LRM), which exhibited a specific capacity of 260 mA h g−1 at a specific current of 25 mA g−1, with 93% of its capacity retained after 100 cycles (Fig. 4m). Notably, the energy retention was nearly doubled compared to that of Li1.20Mn0.54Ni0.13Co0.13O2 (T-LRM) after three months of electrochemical testing. The study suggests that the high-entropy strategy significantly enhances the capacity and voltage stability of E-LRM (Fig. 4n). Furthermore, the diverse array of elements in E-LRM introduces variability into the local structure of the system, alleviating the evolution from layered to spinel structure, ultimately leading to improved stability of the material.
Phase transitions in layered oxides are induced by dislocation slippage between the TM and alkali metal layers, where factors such as the alkali metal content in the host structure, the local environment of alkali metal ions during deintercalation, and ion/vacancy ordering significantly affect the interlayer slippage and occurrence of irreversible phase transitions.247,248 During (de)intercalation, some redox-inactive species do not participate in charge compensation, which may lead to subtle interactions with ions in the alkali metal layers, retaining more ions within the structure.249 This retention partially disrupts ion/vacancy ordering, thereby suppressing phase transitions in layered oxide materials. Moreover, in high-entropy systems, the interactions among ions/atoms are more complex, with microscale forces such as electrostatic repulsion, van der Waals forces, and Coulombic attraction/repulsion all playing crucial roles in inhibiting detrimental phase transitions.250–252
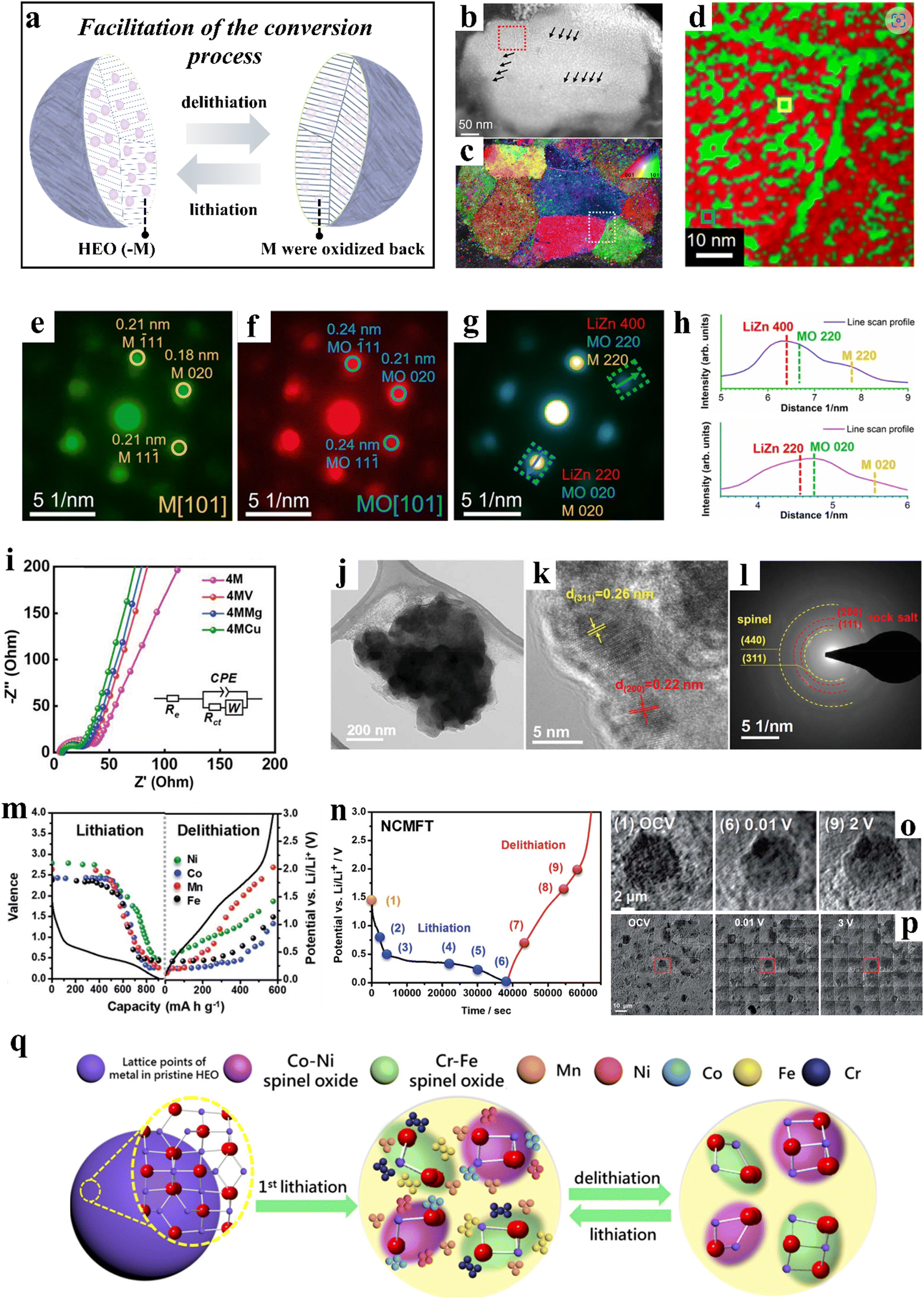 | ||
| Fig. 5 (a) Schematic illustration of the facilitation of the conversion process. (b)–(h) Orientation and phase distribution analyses of the discharged (1st cycle) sample.258 (b) HAADF-STEM image. (c) Orientation map obtained by indexing the diffraction patterns of 4D-STEM data. (d) Typical phase map. (e) and (f) Two exemplary diffraction patterns. (g) Diffraction pattern. (h) Intensity profiles of the corresponding phases. (i)–(l) Co-free, spinel-type HEOs.58 (i) Impedance spectra. (j)–(l) (High-resolution) TEM analysis of 4MCu after 400 cycles. (m)–(p) NCMFT anode.257 (m) Valence changes of Fe, Co, Mn, and Ni at different potentials. (n)–(p) Operando transmission X-ray microscopy of NCMFT. (q) Schematic illustration of the microstructure evolution of (CrMnFeCoNi)3O4 during lithiation/delithiation.256 | ||
Wang et al.258 developed a rock-salt-type anode material of composition Mg0.2Co0.2Ni0.2Cu0.2Zn0.2O. This five-cation oxide is characterized by a diversity of features that enable the metal and oxide phases to be semi-coherently entangled on the nanoscale level, exhibiting superior properties compared to conventional oxides. Although it is clear that the high-entropy structure of the material is not maintained after the first discharge, the highly disordered chemical environment of the synthesized HEO is essential for the entanglement of the nanophases (Fig. 5b–h). This entanglement can greatly facilitate the conversion process, consequently resulting in enhanced electrochemical properties.
Patra et al.58 incorporated a fifth element, Cu, into (CrNiMnFe)3O4, resulting in the formation of a single-phase spinel material, namely (CrNiMnFeCu)3O4 (4MCu). Experimental results demonstrated that 4MCu displays fast diffusion kinetics (Fig. 5i) and exceptional structural stability (Fig. 5j–l). At a specific current of 2000 mA g−1, 4MCu was capable of delivering a specific capacity of 480 mA h g−1, with minimal decay observed after 400 cycles. Huang et al.256 conducted a study on the transformation behavior of spinel-type (CrMnFeCoNi)3O4 during cycling at the atomic scale. During the initial lithiation to 0.5 V vs. Li+/Li, two new spinel phases, CrxFe3−xO4 and LiNixCo1−xO2, precipitated from the pristine phase and served as seeds to engulf the surrounding metal nanoparticles during delithiation. These phases remained present throughout lithiation/delithiation, acting as structural stabilizers and contributing to the good cyclability observed. High-resolution melting (HRM) analysis revealed the formation of Mn nanocrystals during the first lithiation, which, along with Fe, Cr, Co, and Ni metal particles formed during further lithiation, “redissolved” into the spinel material during delithiation (Fig. 5q).
Chen et al.257 synthesized spinel-type (Ni0.2Co0.2Mn0.2Fe0.2Ti0.2)3O4 (NCMFT) using a simple solid-state method. NCMFT achieved a specific capacity of 560 mA h g−1 at 100 mA g−1 and maintained a capacity retention of 100% after 100 cycles. The lithium-storage mechanism of NCMFT was proposed by employing a series of operando and ex situ techniques, including synchrotron XRD, XANES, EXAFS, etc. The lithiation process led to the transformation of the Ni, Co, Mn, and Fe ions into the metallic state (Fig. 5m), while the Ti ions remained in the spinel structure and produced LiTi2O4, which in turn helped to stabilize the structure. During the delithiation process, the majority of the metal nanoparticles underwent oxidation and reintegrated into the spinel structure. Moreover, the material exhibited negligible volume variations during cycling (Fig. 5n–p).
Unlike insertion-type anodes such as hard carbon, spinel oxides primarily deliver capacity through conversion and alloying reactions. During cycling, conversion reactions can occur in the initial reduction step, where metal elements may further form alloys with Li.259 Consequently, conversion-type electrode materials typically experience phase decomposition during battery operation, transforming into nanoparticles, with reduced metal species forming amorphous or crystalline nanoparticles that are distributed around the Li alloy(s).260 Although the initial single-phase high-entropy structure may not be fully preserved throughout the reaction, the subsequent processes of alloying and metal reduction have been found to be highly reversible in high-entropy spinel oxides.261 Moreover, the introduction of multiple elements mitigates the significant volume changes associated with single-component materials, alleviating the structural degradation often caused by the substantial volume expansion seen in conventional transition-metal oxide anodes during cycling. These advantages highlight the potential of high-entropy strategies to promote conversion reactions and enhance the cycling stability of spinel oxides.
3.2. Constructing transport channels via lattice distortions
Solid-state electrolytes provide greater electrochemical stability and safety compared to organic electrolytes.262–265 However, attaining ionic conductivities similar to those of current liquid electrolytes is challenging. Nevertheless, there are methods available to enhance the ionic conductivity of solids, such as employing ion-stuffing strategies, i.e., increasing the mobility by adding more alkali ions (Li+ or Na+) to the compound.266,267 Yet, these solutions continue to face challenges due to the intricate process of synthesis268 and ion depletion during heat treatment.269The presence of several components in HEMs causes distortions in the lattice structure as a result of variations in size and bonding energy among the elements. Through the implementation of occupational disorder, researchers discovered that a moderate degree of lattice distortion can create low-energy pathways for ion diffusion in solid-state electrolytes (Fig. 6a), thereby enhancing the ionic conductivity of the materials. For example, Zeng et al.270 designed several high-entropy electrolytes with NASICON- and garnet-type structures while keeping the alkali ion concentration constant. They found that the presence of a high configurational entropy leads to an overlapping distribution of energy sites (Fig. 6b and c). This, in turn, facilitates the construction of percolation networks with low energy barriers (Fig. 6d), which are beneficial to the ion mobility. The prepared high-entropy electrolytes, Li(Ti,Zr,Sn,Hf)2(PO4)3, Na(Ti,Zr,Sn,Hf)2(PO4)3, and Li3(La,Pr,Nd)3(Te,W)2O12, showed an increase in ionic conductivity by several orders of magnitude (Fig. 6e). In addition, taking into account that the mobile ion may also modify the site energies, the researchers calculated the energy difference with various metal species in Li-NASICON, Na-NASICON, and Li-garnet (Fig. 6f). To further optimize the ionic conductivity, they synthesized five high-entropy NASICON compounds by combining the high-entropy method with the alkali metal-stuffing strategy. All five compounds exhibited fast ion-diffusion characteristics (Fig. 6g).
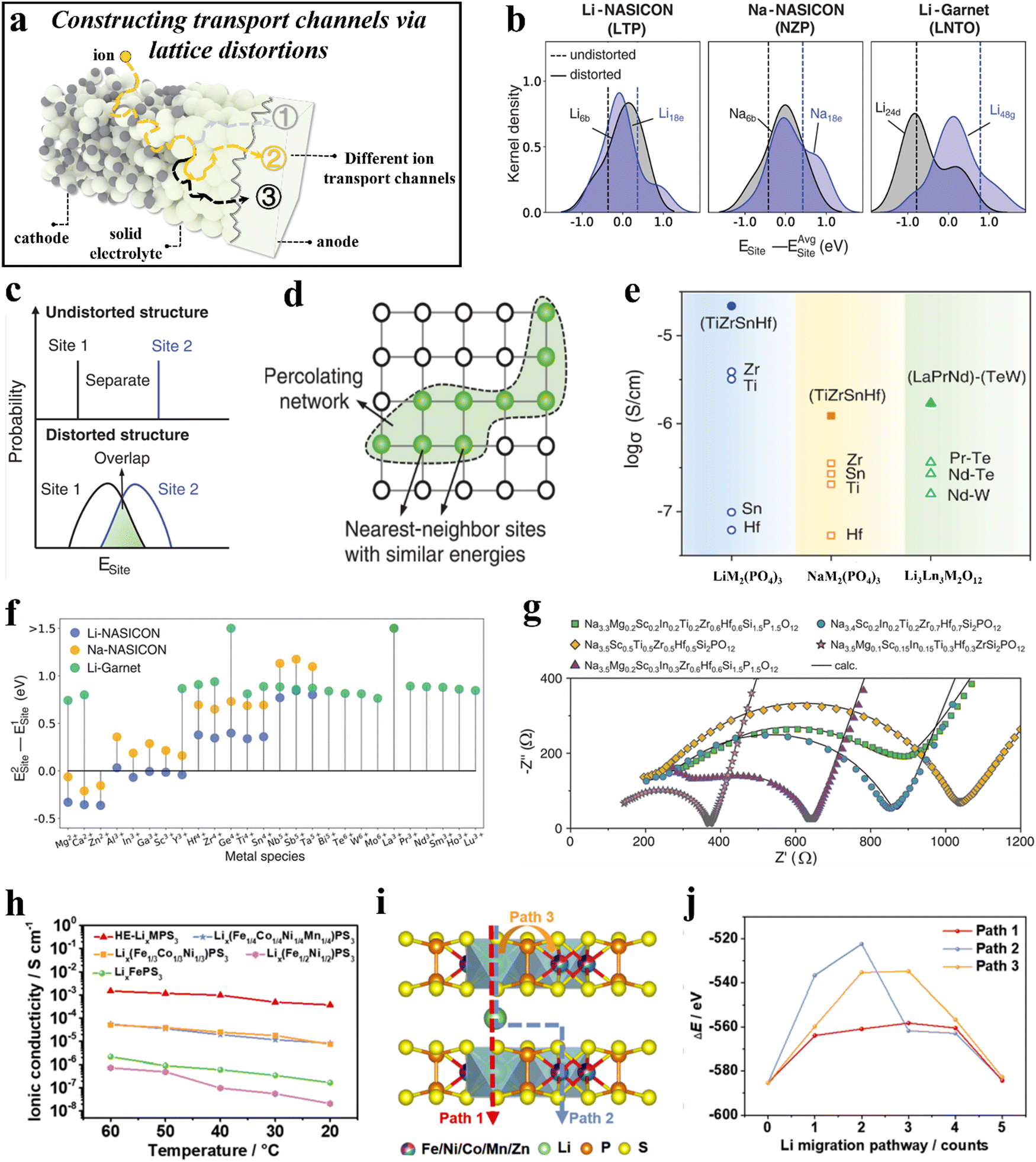 | ||
| Fig. 6 (a) Schematic illustration of constructing transport channels via lattice distortions. (b)–(g) High-entropy electrolytes.270 (b) Calculated site energies of three baseline materials. (c) Schematic showing how local distortions create overlapping site-energy distributions. (d) Schematic of the percolation network structure. (e) Ionic conductivities of LTZSHPO, NTZSHPO, and LLPNTWO as well as of various reference samples. (f) Calculated site-energy differences for various metal species in Li-NASICON, Na-NASICON, and Li-garnet. (g) Impedance spectra of Na-stuffed, high-entropy compounds. (h)–(j) LixMPS3 and HE-LixMPS3 electrolytes.271 (h) Temperature-dependent ionic conductivity of HE-LixMPS3 and other LixMPS3 electrolytes. (i) and (j) DFT calculations for the three possible paths (i) and corresponding Li+ migration energies (j). | ||
Utilizing high-entropy-induced lattice distortions to create pathways that facilitate the movement of ions has also been employed in the development of two-dimensional (2D) transition-metal phosphorus sulfides. Lix(Fe1/5Co1/5Ni1/5Mn1/5Zn1/5)PS3 (HE-LixMPS3) was designed to have a high lattice distortion and a large number of cation vacancies.271 These distinct characteristics facilitate the transport of Li+ ions between the 2D [P2S6]2− layers, resulting in an ionic conductivity of 5 × 10−4 S cm−1 at room temperature, significantly exceeding that of other LixMPS3 electrolytes (10−5–10−8 S cm−1, see Fig. 6h). After being stored under ambient conditions for 6 h, the ionic conductivity of HE-LixMPS3 remained at 4.99 × 10−4 S cm−1, and it maintained relatively stable even after three days of room-temperature storage. To gain further insight into the mechanism of lithium migration in the HE-LixMPS3 electrolytes, density-functional theory (DFT) calculations were carried out. The results disclosed the intralayer–interlayer–intralayer ion diffusion through the faces of the edge-sharing octahedra of Li+ (Fig. 6i and j). Additionally, all-solid-state Li-metal batteries using a composite electrolyte (HE-LixMPS3/NBR), a LiFePO4 (LFP) cathode, and a lithium anode were capable of achieving 2000 cycles at 5C rate, with a Coulomb efficiency of 99.8%.
The ion diffusion in materials is closely related to the characteristics of the transport pathways, and constructing additional effective transport channels enhances ion diffusion within the system. This conclusion was drawn by Fu et al.,231 who showed that the increased {010} facets in P2-type layered oxides create more favorable transport pathways, resulting in fast diffusion kinetics. In solid-state electrolytes, high-entropy strategies introduce multiple elements into the same (sub)lattice, leading to lattice distortions that disrupt the local site energy landscape. This effect generates additional transport pathways, where ions preferentially migrate along the most favorable (lowest energy barrier) diffusion routes, thereby enhancing ionic conductivity.272
Moreover, a stable and broad transport channel structure can ensure reversible ion diffusion. For intercalation-type materials, ions diffuse by continuously intercalating and deintercalating, and an expanded interlayer spacing ensures the reversibility of these intercalation/deintercalation processes, ultimately facilitating kinetics. Similarly, in 2D layered transition-metal phosphorus sulfides, the presence of multiple TM species induces significant lattice distortions within the material. These distortions effectively increase the c-parameter (layer spacing), thereby optimizing ion migration between the layers.
3.3. Disorder-facilitated ion diffusion
The term “high entropy” encompasses both the quantitative measure of a system's entropy and the inherent stochastic and disordered nature of the system, which generally intensifies as the entropy increases. HEMs, in contrast to (ordered) conventional materials, display greater disorder due to their more complex compositions. This increased occupational disorder has been found to be advantageous and actively contributes to enhancing the diffusion kinetics of electrode materials (Fig. 7a).211,273–275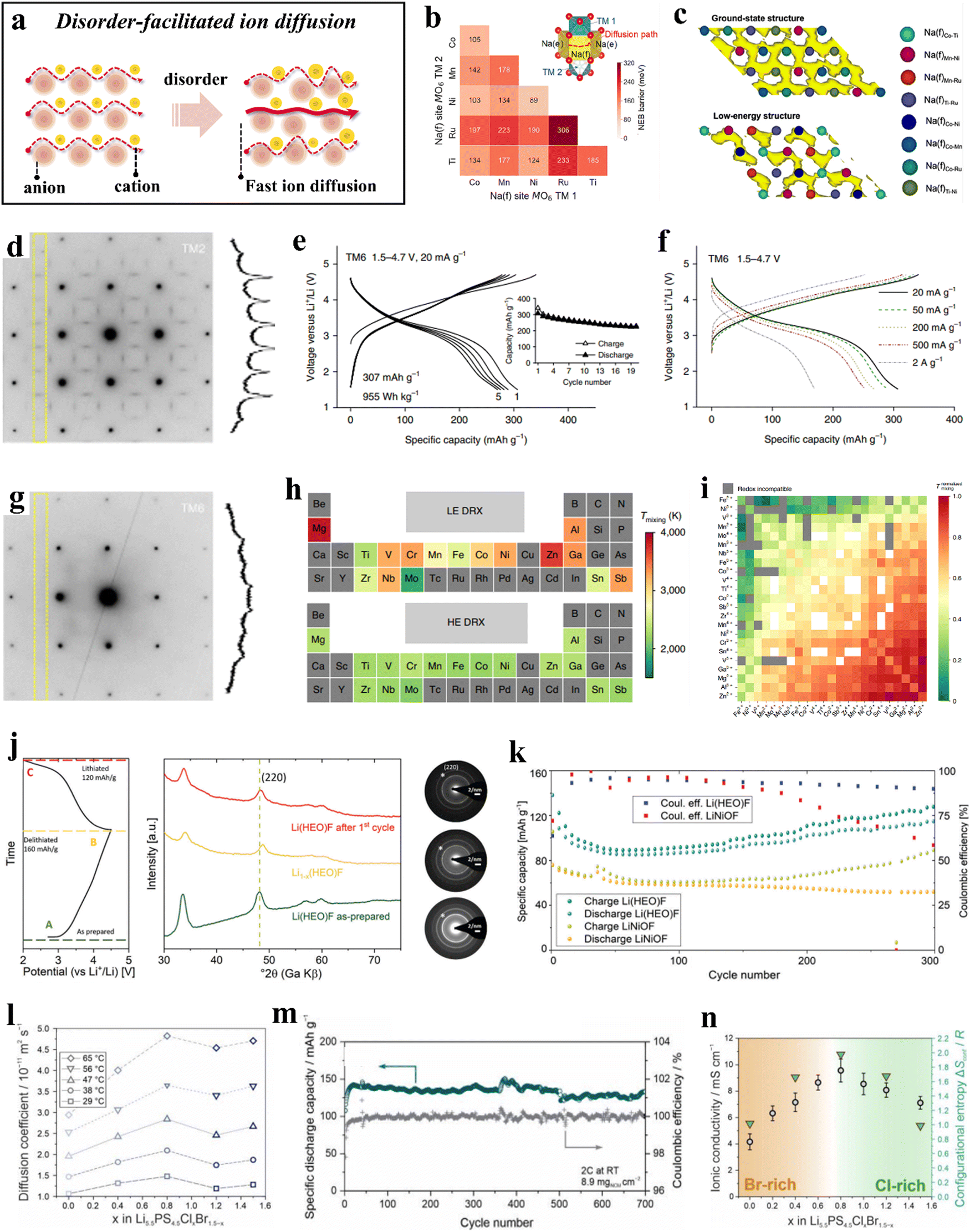 | ||
| Fig. 7 (a) Schematic illustration of disorder-facilitated ion diffusion. (b) and (c) Theoretical calculations for P2-TMCNR.273 (b) Na+ migration barriers of different Na(f)M−M′. (c) AIMD trajectories for the ground-state structure and a low-energy structure. (d)–(i) DRX compounds.211 (d) ED pattern collected from TM2 along the [100] zone axis. (e) Voltage profiles of TM6 within the potential window of 1.5–4.7 V at 20 mA g−1. (f) Rate capability of TM6. (g) ED pattern collected from TM6 along the [100] zone axis. (h) Periodic table-type heat map of mixing temperatures of different elements in LE and HE DRXs. (i) Normalized mixing temperatures. (j) and (k) Li(HEO)F and LiNiOF cathodes.276 (j) Initial charge/discharge curve at C/10 (20 mA g−1) in the potential range between 2.0 and 4.5 V (left) and the corresponding ex situ XRD patterns (middle), as well as ED data (right). (k) Comparison of capacity retentions at C/10 of Li(HEO)F and LiNiOF. (l)–(n) Li5.5PS4.5ClxBr1.5−x electrolytes.274 (l) Lithium-diffusion coefficients at different temperatures. (m) Capacity retention and Coulomb efficiencies. (n) Ionic conductivity as a function of x. | ||
The random distribution of cations can result in a corresponding improvement in transport properties by creating additional pathways for ion migration. In layered oxide cathode materials, mixing TM ions in approximately equimolar ratios not only promotes stabilization of the crystal structure, but also facilitates ion mobility by alterating the activation energy barrier. Yang et al.273 synthesized a high-entropy layered transition-metal oxide, Na0.6(Ti0.2Mn0.2Co0.2Ni0.2Ru0.2)O2 (P2-TMCNR), as a positive electrode material for sodium-ion batteries using high-energy ball milling. The specific discharge capacity at 0.1C rate was 164 mA h g−1; at 86C (15000 mA g−1), it reached 68 mA h g−1, surpassing the performance of most previously reported layered oxide cathode materials. Experimental and computational studies revealed that the cation disorder in the TM layers (creation of a percolating low-barrier diffusion network; see Fig. 7b and c) leads to improved rate performance of P2-TMCNR.
Cation-disordered rock-salt oxides (DRXs) feature three-dimensional (3D) lithium diffusion channels and undergo minor volume changes during cycling. However, the presence of short-range ordering (SRO) in DRXs can adversely affect the transport properties, leading to inferior rate performance. In a study reported by Lun et al.,211 two, four, and six transition metals were introduced into DRX cathode materials, resulting in the preparation of Li1.3Mn0.43+Ti0.3O1.7F0.3 (TM2), Li1.3Mn0.22+Mn0.23+Ti0.1Nb0.2O1.7F0.3 (TM4), and Li1.3Mn0.12+Co0.12+Mn0.13+Cr0.13+Ti0.1Nb0.2O1.7F0.3 (TM6). Comparison of these materials revealed that TM6 can provide a specific capacity of 307 mA h g−1 (955 W h kg−1) at a specific current of 20 mA g−1 (Fig. 7e) and over 170 mA h g−1 at 2 A g−1 (Fig. 7f), both significantly higher than those of TM2 and TM4. Electron diffraction indicated that as the variety of TM increases from two to six, the intensity of the SRO pattern decreases, suggesting that cation ordering in the DRX structure is suppressed (Fig. 7d and g). This suppression of SRO preserves accessible 0-TM percolation pathways, thereby enhancing ion mobility and enabling the electrode to operate at high current rates. Additionally, investigations into various chemical compositions and TM compatibility revealed that the chemical variation of the mixing temperature in high-entropy DRX (HE DRX) structures is significantly smaller than that in low-entropy counterparts (Fig. 7h), likely due to the increased entropy associated with a greater variety of elements. Moreover, all redox-active species, with the exception of Cr3+, exhibited good chemical compatibility when redox compatibility is met. However, elements such as Mg2+, Zn2+, Ga3+, Al3+, and Sn4+ showed poor compatibility with other TMs and should be maintained at low concentrations in the design (Fig. 7i). These studies will further facilitate the rational design of HE DRX compounds.
Wang et al.276 developed a cathode active material of composition Lix(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OFx [Li(HEO)F] by integrating different anions into a multication compound. The conversion mechanism of Li(HEO)F was proven by ex situ XRD and selected-area electron diffraction (SAED) measurements. The rock-salt structure was preserved during electrochemical cycling, and the process of Li insertion/extraction was highly reversible (Fig. 7j). Compared to LiNiOF, the structure of Li(HEO)F allows for a larger amount of lithium to be utilized during cycling, resulting in an increase in specific capacity (Fig. 7k). The experimental results demonstrated that the anion lattice also plays a role in the configurational entropy, leading to an increased gain in structural stabilization.
Research has demonstrated that both the anion and cation sublattices can indeed enhance configurational entropy, leading to increased disorder in the system. This increase in disorder can positively impact diffusion kinetics, thereby benefiting solid-state electrolyte design.277–281 Li et al.274 synthesized a series of high-entropy argyrodite solid electrolytes, Li5.5PS4.5ClxBr1.5−x (with 0 ≤ x ≤ 1.5). Notably, Li5.5PS4.5Cl0.8Br0.7 exhibited fast ion mobility (Fig. 7l) and achieved a high ionic conductivity of 22.7 mS cm−1 at room temperature. Furthermore, when combined with single-crystal LiNi0.9Co0.06Mn0.04O2 (s-NCM90) to form an all-solid-state battery, minimal capacity decay was observed over more than 700 cycles (Fig. 7m). The authors propose that the fast lithium diffusion in Li5.5PS4.5Cl0.8Br0.7 can be attributed to its increased configurational entropy (ΔSconf = 1.98R), due to the high degree of anion sublattice disorder (Fig. 7n).
As the number of elements introduced at the cation/anion sites increases, the configurational entropy of these sites also increases, resulting in a disordered arrangement of ions within the material. The latter has been shown to directly affect the transport characteristics. In high-entropy layered oxides, the presence of five or more TMs sharing the same sites typically leads to a disordered arrangement. This TM disorder generally induces (Li+) Na+/vacancy disorder, thereby facilitating ion diffusion.282,283 For DRXs, the reduced tetrahedral height leads to strong electrostatic repulsion acting on Li+, which in turn renders the 1-TM pathways inactive. Although the 0-TM pathways are more active, they appear with less frequency. Excess lithium can introduce additional channels within the 0-TM percolation network, thus increasing network's connectivity.284 However, SRO significantly limits the percolation through the material. By employing high-entropy strategies, the distribution of cations within the system becomes more random, thus suppressing SRO and enhancing transport properties.211 In lithium argyrodite solid-state electrolytes, halogen substitution increases the configurational entropy within the anion sublattice, with S2−/X− site disorder leading to unique transport pathways and increasing ion mobility.274
3.4. Synergies induced by cocktail effects
HEMs display a broad spectrum of (complex) constituents. The harmonious coexistence of various elements can lead to synergistic enhancements, referred to as cocktail effects (Fig. 8a). The effect of synergy being observed is a complex phenomenon that can be attributed to multiple aspects such as the composition, microstructure, electronic structure, and other relevant properties of the components involved. By adjusting the type and content of constituents during the design phase, cocktail effects enable the modification of the overall performance of HEMs. As a positive gain resulting from the combination of multiple elements, cocktail effects are widespread in HEBMs, and some particular instances will be discussed in this section.54,258,285,286 | ||
| Fig. 8 (a) Schematic illustration of the synergies induced by cocktail effects. (b)–(e) NaLFCNM cathode.285 (b) and (c) Ex situ high-resolution TEM images of NaLFCNM at 4.5 V (b) and 2.0 V (c). (d) Schematic illustration of the phase evolution of NaLFCNM upon charge and discharge. (e) The TM–O (TM = Ni, Fe, Co, and Mn) bond length changes during charging from 2.0 to 4.5 V. (f)–(h) (Co0.2Mg0.2Cu0.2Ni0.2Zn0.2)O anode.54 (f) Operando XRD data obtained in the first full lithiation/delithiation cycle (left) and corresponding ED patterns (right). (g) and (h) ED patterns of the lithiated HEO. (i)–(n) Analysis of the 3D metallic network.258 (i) Volume rendering based on tomographic reconstruction. (j)–(n) Slices through the reconstructed volume normal to the viewing direction in panel (i). | ||
Joshi et al.286 designed a Co-free layered cathode material, Na0.9Li0.1Ni0.42+Fe0.23+Mn0.42+Ti0.044+Mn0.044+Mg0.022+O1.9F0.1 (referred to as Na0.9Li0.1), using a solid-state reaction method. The introduction of F inhibits the loss of oxygen during cycling. Partial replacement of Na with Li increases the configurational entropy of the system, improving the structural stability. Additionally, the introduction of Li creates Na+ vacancies, enhancing ion diffusion. The O3-type structure with Mg reduces the layer spacing and mitigates the negative effects of water entering the TMO2 layer, thus increasing the material's stability in air. Higher concentrations of Ni2+, Fe3+, and Mn2+ maintain the redox activity of the electrode. By optimizing the material composition, Na0.9Li0.1 delivered an initial specific capacity of 144 mA h g−1 (2.0–4.3 V vs. Na+/Na) and exhibited high discharge capacities from 0.06C to 3C, showcasing good rate performance. After rate capability testing, it achieved 98% capacity retention over 70 cycles at 0.2C. Even after 300 cycles, Na0.9Li0.1 was capable of maintaining 80% of its capacity.
Cocktail effects, which entail the combination and mutual coupling of various elements, may lead to synergistic enhancements in the performance of HEMs.282 This is not merely a simple additive outcome of multiple elements' properties, but instead achieves a nonlinear improvement in material characteristics through careful selection of element types and tailoring of stoichiometric ratios.21,49 As previously discussed, cocktail effects enabled by high-entropy strategies exhibit excellent performance-tuning potential. By combining elements with diverse electrochemical properties, it is possible to simultaneously enhance capacity and provide additional structural stability to the material. Furthermore, in practical materials development, certain physical and chemical properties of specific elements should also be considered in the process of optimizing element combinations. This consideration facilitates better regulation of the synergistic effects produced by cocktail effects. Details on the selection of elements are provided in Section 5.1.1 of this review article.
4. Disscussion
In alkali-metal-ion batteries, some cathode materials such as layered oxides, PBAs, and polyanionic compounds experience significant lattice strain during the insertion/extraction processes. As this strain accumulates over multiple cycles, structural degradation occurs, ultimately leading to material failure. Additionally, the occurrence of irreversible phase transitions upon battery operation is another key factor contributing to the limited cycling stability of cathode materials. By leveraging the positive effects of entropy increase, high-entropy strategies introduce additional structural stability to the system. This enhanced stability plays a crucial role in reducing lattice strain and suppressing adverse phase transitions.Spinel-type oxides as conversion-type anode materials typically undergo severe structural degradation during cycling due to conversion or alloying reactions, leading to rapid capacity fading and poor reversibility. The introduction of multiple elements into spinel oxide systems may improve structural stability by mitigating the large volume variations, thereby ensuring reversibility of the electrochemical reaction(s). It is important to note that the unique structural stability exhibited by HEMs is still poorly understood, as the underlying principles remain largely unclear. The results result from a complex interplay of various factors, necessitating further research to elucidate the mechanisms at play. In the future, the integration of advanced characterization techniques will be essential to better understand the principles behind high-entropy strategies.
As for solid-state electrolytes, both the creation of favorable percolation networks through disorder and the construction of additional transport channels via lattice distortions show significant promise for optimizing ion mobility, opening up new possibilities for enhancing conductivity across various structure types. However, a challenge remains in tailoring the degree of disorder and optimizing the extent of distortions, which continues to be a critical issue for the rational design of high-entropy electrolytes. Furthermore, exploration of these improvements for application with multivalent charge carriers is still lacking.
5. Future research directions
Currently, investigations on HEMs are in an early stage. While the high-entropy approach has been extensively utilized in the advancement of high-performance electrochemical energy-storage devices, there are still obstacles to overcome in practical implementations. These include the need for rational design of element combinations, optimization of synthetic techniques, and innovation in theoretical calculations. Hence, additional research efforts are required to address the aforementioned issues and facilitate the actual implementation of HEMs in the energy sector (Fig. 9). Below we provide an analysis of potential development directions for HEMs, focusing on insights from current research. | ||
| Fig. 9 Overview of general approaches to the study of HEBMs. | ||
5.1. Materials design
Using high-entropy strategies for battery design is still in the exploratory phase, predominantly relying on experimental experience without a well-defined design criterion. Multiple studies have indicated that the valence state, ionic radius, and electronegativity of the elements have diverse impacts on battery performance while implementing multielement doping to increase the system's configurational entropy.21,33,49 In the following, we aim at establishing pertinent design principles for battery materials. This includes delineating criteria for element selection, optimizing stoichiometric ratios, and identifying appropriate doping/substitution sites.5.1.1.1. Electronegativity of the crystallographic site. Typically, when many elements are included at the same sites, it is necessary to maintain charge neutrality. Excessive electronegativity can induce electron transfer, resulting in the gain or loss of electrons and subsequent changes in valence state. For example, when Mn3+ and V3+ coexist on the same crystallographic sites, V3+ undergoes oxidation to V5+, while Mn3+ undergoes reduction to Mn2+. This does not imply that charge transfer is always detrimental. In some cases, one can utilize the shift in electronegativity of lattice sites to trigger charge transfer, resulting in unforeseen properties in the material. Liang et al.287 synthesized a Ni-rich, Co-free, high-entropy layered cathode material, LiNi0.8Mn0.12Al0.02Ti0.02Cr0.02Fe0.02O2 (HE-WD), by substituting Co with low electronegativity cations, based on the commercial lithium-ion battery material LiNi0.8Co0.1Mn0.1O2 (NCM811). HE-WD delivered a high specific capacity of 203.6 mA h g−1 in the potential range of 2.5–4.5 V vs. Li+/Li and retained a capacity of 80% after 300 cycles. The study suggests that HE-WD, with a higher degree of cation disorder, facilitates the anchoring of lattice oxygen, and the low electronegativity of the TM species prompts charge transfer, thereby enhancing the electron density of O in the TM–O octahedral configuration. Hence, in the design of material compositions, it is imperative to tailor the electronegativity of dopant/substitution sites in accordance with the functional characteristics of the target material.
5.1.1.2. Electrochemical properties of elements. The selection can be conducted based on the different properties of elements. Electrochemically active elements such as Co3+, Cu+, Mn2+, Ni2+, and Fe3+ can serve as redox centers to enhance the capacity of the electrode.288 For instance, Konarovet et al.289 conducted a study where they partially replaced Mn with Ni in Na2/3MnO2 and, subsequently, observed an increase in specific capacity in the high-voltage region, achieved by Ni2+/Ni4+ redox. Similarly, Nguyen et al.290 employed a sol–gel method to synthesize a new material, Na3(VO)Fe(PO4)2F2, which exhibited higher specific capacity than the material without Fe. Their findings indicated that the additional capacity results from the redox reaction of Fe2+/Fe3+ within the 1.5–4.3 V potential window. On the other hand, electrochemically inactive ions such as Ti4+, Sn4+, Mn4+, and Mg2+ can function as stabilizers for the host structure during cycling.291,292 For example, Wang et al.293 designed a structurally optimized NaNi0.5Mn0.5O2 cathode by partially substituting Ti4+ for Mn4+. This substitution inhibited phase transitions above 3 V, leading to improved structural stability. The NaNi0.5Mn0.5O2 cathode demonstrated 78% capacity retention after 200 cycles at 1C rate in the potential range of 2.0–4.2 V vs. Na+/Na. Considering the structural degradation of the P2-type layered cathode, Na0.67Ni0.33Mn0.67O2, during high-voltage operation, which leads to capacity degradation, Yuan et al.294 synthesized a new material, Na0.67Ni0.23Mg0.1Mn0.65Sn0.02O2 (NMMS2), using a one-pot solid-phase sintering method. The introduction of Mg helped to inhibit the P2-O2 phase transition at high potentials, and the co-substitution of Mg/Sn facilitated the reversible transformation of the P2-Z phase, thus significantly improving structural reversibility of the NMMS2. As a result, it demonstrated 80% capacity retention after 500 cycles at a specific current of 500 mA g−1. Moreover, Li+ and Mg2+ ions doped into the material can trigger anion redox to contribute additional capacity during cycling operation.295,296
Notably, the electrochemical properties exhibited by the same element can vary across different electrode systems. For instance, while Ni generally serves as an electrochemically active element in layered oxides,297 it often does not participate in redox reactions in PBA cathodes.57 Furthermore, even within the same system, the properties displayed by identical elements may differ across various battery materials, despite consistent elemental combinations and concentrations. For example, Mn, Fe, Ni, Cu, and Co are among the most common elements in high-entropy PBA cathodes across a range of battery systems, including sodium- and potassium-ion batteries. Among these, all elements, except Ni, typically serve as redox centers, thereby contributing to the capacity.57,298,299 However, in aqueous aluminum- and zinc-ion batteries, Ni300 and Co301 exhibit opposing electrochemical behaviors in their respective systems.
5.1.1.3. Physical and chemical properties of elements. It is generally recommended that the elements doped/substituted at the same lattice sites have similar ionic radii. This similarity allows for the random distribution of different ions at each site, thereby increasing disorder in the system and enhancing conductivity. A significant difference in ionic radii between dopants/substituents can induce significant lattice distortions, potentially leading to structural collapse and a decline in performance. In specific electrode materials, such as high-entropy P2-type layered oxides, the differences in the ionic radius within the TM layer can influence the degree of disorder.283,302 A suitable range of ionic radii facilitates the formation of a disordered structure, which may promote (Li+) Na+/vacancy disorder and increase kinetics.282 Beyond the consideration of differences in the ionic radius, parameters such as electronegativity,303 Fermi level,304 and valence electron concentration305 are closely related to properties like bonding characteristics, conductivity, and electrochemical reactivity. For instance, elements with higher electronegativity exhibit a stronger affinity for electrons, making them more likely to attract and retain electrons, while elements with lower electronegativity behave in the opposite manner. In layered oxides, for example, incorporating F—an element with high electronegativity—into the O layer has been shown to reduce the covalency of TM–O bonds. This modification alleviates electron cloud repulsion experienced by the oxygen atoms within the crystal structure, consequently decreasing O–O repulsion and resulting in a more stable layer spacing.306 Additionally, tailoring the Fermi level can help adjust the charge-compensation centers, modify the bandgap width, and enhance the electronic conductivity, thus improving charge-transfer kinetics. In layered oxides, lower-valency cations such as Li can effectively facilitate this.306 While a relationship exists between the valence electron concentration and electronegativity, they are not directly correlated concepts. Their connection primarily arises from the element's nuclear charge and electron configuration. Elements with a high valence electron concentration, typically metals or transition metals (e.g., Ni, Co, and Fe), tend to have a lower electronegativity.307 These elements often possess a higher number of valence electrons that are relatively free and can participate in metallic bonding or delocalized electron formation, thereby enhancing the material's conductivity and electrochemical activity. In addition, properties such as mixing enthalpy, mixing entropy, electronegativity, valence electron concentration, and atomic size differences significantly affect the structure/phase of materials.308,309 Based on these properties, parameters such as the Ω,310 Δχ,311 VEC,312δ,313SE,314 and θ315 have been developed to guide the phase-engineering design of HEMs.
5.1.1.4. Practical application and commercialization potential. Although the extended cycle life and capability to operate under extreme conditions endow HEMs with durability, increasing their potential for practical application, challenges such as high cost, poor recyclability, and limited environmental friendliness continue to hinder their commercialization. However, through careful selection and design of constituent elements, these challenges can be partially mitigated, promoting greater feasibility for practical application.
The cost of raw materials for HEMs might be higher than that of typical materials used in energy-storage devices. This increase is sometimes driven by objective variables such as geopolitical issues. Hence, it is crucial to discover substitute elements that uphold performance and dependability while being cost-efficient for logical component design to ensure commercial viability. Co is recognized for its ability to enhance structural stability and improve rate performance of layered cathode materials. However, practical application is hindered due to high cost and Co-related geopolitical issues. In light of these factors, Dang et al.316 prepared a high-entropy layered oxide, NaFe0.2Cu0.1Ni0.2Mn0.3Ti0.2O2 (HEO), effectively eliminating cobalt in the cathode material without compromising performance. The HEO cathode exhibited high sodium-storage performance, with a specific capacity of 121 mA h g−1, robust cycling stability, and improved water resistance.
In addition to raw material cost, the design of HEMs should prioritize sustainable elements such as Mn, Fe, and Al, which have a minimal environmental impact during extraction and processing. This approach can reduce the ecological footprint and enhance the sustainability of the materials. Compared to toxic elements like Co, Cd, etc., these eco-friendly elements also simplify later-stage separation and recycling, leading to lower maintenance cost and contributing to a more balanced commercialization cost. Nevertheless, it is important to note that the multicomponent nature of HEMs complicates the overall battery recycling process. The latter typically involves multiple steps, including collection, transportation, sorting, separation, material re-extraction, and waste disposal, adding to the complexity of recycling. To advance the practical application and commercialization of multicomponent materials, more efficient and cost-effective recycling technologies must be developed in the future (Fig. 9).
Furthermore, in the design of HEMs, varying stoichiometric ratios may lead to different effects on the increase in configurational entropy of the system, even if the total concentration and type of elements remain constant. According to eqn (1), the entropy of the system is maximized when the components are mixed in equal molar ratios. This suggests that the favorable factors resulting from the increase in entropy may operate at their maximum level.
5.1.3.1. Transition-metal sites. Researchers have primarily focused on doping/substituting TM sites in layered oxides. Their objective is to improve material performance by leveraging the synergistic effects resulting from the presence of different elements at the same sites. Du et al.317 introduced eight different species into the TM sites of O3-NaNi0.5Mn0.5O2, resulting in the development of a novel layered oxide, namely NaNi0.1Mn0.15Co0.2Cu0.1Fe0.1Li0.1Ti0.15Sn0.1O2 (HEO). Their findings indicate that the synergistic effect of each element effectively mitigates detrimental phase transitions during Na+ intercalation, thereby bolstering the structural resilience. Remarkably, after 1000 cycles, the HEO demonstrated a high capacity retention of 83%, underscoring its superior stability. Furthermore, the high-entropy strategy also conferred enhanced humidity stability and mechanical performance. Ding et al.318 fabricated O3-NaNi0.25Mg0.05Cu0.1Fe0.2Mn0.2Ti0.1Sn0.1O2 (HEO424) through high-temperature calcination. During electrochemical performance testing, the energy density of HEO424 exceeded that of O3-NaNi0.4Fe0.2Mn0.4O2 (NFM424) at all current densities, and its initial Coulomb efficiency (91%) was higher than that observed for NFM424 (87%). After 500 cycles at 1C rate, NFM424 showed significant capacity degradation, whereas HEO424 maintained 75% of its capacity. The study suggests that the high-entropy configuration of the TMO2 layer in HEO424 widens the Na+ transport channels, promotes reversible sodium storage, delays phase transitions, and improves the overall structural stability of the layered cathode material.
5.1.3.2. Alkali-metal sites. Compared to the TM sites, the doping/substitution of the alkali-metal sites is mainly focused on Mg2+, Ca2+, and K+, which have a unique “pillar” effect, inhibiting TM migration and mitigating detrimental phase transitions. Wang et al.319 introduced larger K+ into the Na sites and designed a new P2-type layered cathode material, Na0.612K0.056MnO2. The K+ was pinned into the Na+ vacancy, and the enhanced Mn–O bonding suppressed the sliding of the TM layer, stabilized the lattice structure, and mitigated irreversible phase transitions upon cycling. Na0.612K0.056MnO2 delivered specific capacities of up to 241 mA h g−1, with an energy density of 654 W h kg−1 and a capacity retention of 98% after 100 cycles. Shen et al.320 introduced a high-performance cathode material, P2-Na0.76Ca0.05[Ni0.23□0.08Mn0.69]O2, exhibiting dual cation and anion redox activities. Their study revealed that incorporating Ca2+ into the Na sites disrupts the ordering of Na+ vacancies, thereby augmenting the migration rate of Na+ ions. Moreover, Ca2+ acts as a stabilizing “pillar,” impeding the sliding of the TM layer and consequently inhibiting the P2–O2 phase transition. This innovative design manifests in superior electrochemical performance and long-term cycling stability. Notably, Na0.76Ca0.05[Ni0.23□0.08Mn0.69]O2 delivered specific capacities of 154 and 75 mA h g−1 at rates of 0.1C and 20C, respectively. Even after 50 cycles, the material retained 87% of its initial capacity.
5.1.3.3. Oxygen sites. Doping at the oxygen sites in layered oxides can efficiently modify the bonding energy between the elements, thereby having a substantial impact on material properties. Current research indicates that F doping at the oxygen sites can relieve structural changes.321 Ding et al.306 designed a high-entropy cathode material, Na0.95Li0.07Cu0.11Ni0.11Fe0.3Mn0.41O1.97F0.03 (LCNFMF), which demonstrated a notable improvement in capacity at high voltage (capacity enhancement by about 29%) and good stability (80% capacity retention after 300 cycles at 1C rate). They showed that Li+ enhances the high-voltage redox activity of the TM species. Additionally, further substitution of F decreased the covalency of the TM–O bonds and improved the stability of the lattice structure.
5.1.3.4. Dual-site doping. Dual-site doping/substitution not only allows for the tailoring of ion diffusivity, but also alters the phase-transition properties.322,323 This greater tunability offers a broader range for battery materials design and optimization. Wang et al.322 developed a new cathode material for sodium-ion batteries, Na0.7Mg0.05[Mn0.6Ni0.2Mg0.15]O2 (MNM-2), by introducing Mg2+ ions into both the Na and TM sites. Studies have shown that the Mg2+ ions in the Na layer play a role in stabilizing the structure via pillaring, preventing the P2–O2 phase transition. During high-voltage charging, the Mg2+ ions in the TM layer disrupt the charge ordering. In addition, they can form “Na–O–Mg” and “Mg–O–Mg” type structures in both the Na and TM layers, which places these O 2p states on top of those that interact with the transition metals in the oxygen valence band. As a result, reversible oxygen redox is promoted. MNM-2 exhibited good performance and high structural stability, providing a specific capacity of 57 mA h g−1 at 25C rate and a capacity retention of 79% after 1000 cycles at 1C. Peng et al.323 designed a new layered cathode material, [Na0.67Zn0.05]Ni0.18Cu0.1Mn0.67O2, by doping Cu ions at the TM sites and Zn ions at the Na sites and showed that the Cu doping helps stabilize the TM layer. In contrast, the Zn2+ ions as a “pillar” increased the force between the TM layers, thus inhibiting the formation of O2 phases during deep desodiation. This unique cathode showed good cycling performance, with a capacity retention of 81% after 2000 cycles at a high rate of 10C. In addition, full cells with [Na0.67Zn0.05]Ni0.18Cu0.1Mn0.67O2 as a cathode and commercial hard carbon as an anode achieved an energy density of 218 W h kg−1 and showed good longevity (over more than 1000 cycles), suggesting that it holds great potential for practical applications.
5.2. Synthesis methods
HEMs can be synthesized using various methods, primarily categorized into solid-, liquid-, and gas-phase approaches. Among these, the ball-milling technique, a solid-state method, has become widely adopted for the synthesis of high-entropy oxides due to its relatively low energy consumption, cost-effectiveness, and simplicity. The synthesis steps are as follows: metal precursors, typically five or more in specific stoichiometric ratios, are ground and blended through ball milling, compacted, and then annealed at high temperatures to obtain the final sample.324 For example, Xiao et al.325 successfully utilized the high-energy ball-milling method to produce Ni-free spinel-type HEOs, such as (FeCoCrMnZn)3O4 and (FeCoCrMnMg)3O4, by wet-mixing Fe2O3, Co3O4, ZnO (or MgO), Cr2O3, and MnO2. The resulting nanoscale HEOs exhibited uniform elemental distribution and good crystallinity. On the other hand, the co-precipitation method, a widely used wet-chemical approach, involves adding a precipitant to a solution, making it suitable for large-scale production. As a primary synthesis method for PBAs, the co-precipitation technique is commonly conducted by mixing different solutions. This method allows for uniform reaction and stable precipitate formation by controlling the stirring and mixing rates. The precipitate is then separated from the mother liquor through centrifugation and undergoes multiple washing steps to remove excess salts, effectively eliminating impurities. After washing, the precipitate is dried and ready for use. This synthesis process does not require high-temperature calcination, making it straightforward and convenient.57 Ma et al.57 employed this method and successfully synthesized various high-entropy PBAs, including Nax(FeMnNiCuCo)[Fe(CN)6] (HE-PBA) and Nax(FeMnNiCo)[Fe(CN)6] (ME-PBA(-Cu)), as well as conventional NaxFe[Fe(CN)6]. The materials produced at room temperature showed high purity and uniform elemental distribution.In addition, sol–gel processing, chemical vapor deposition (CVD), and self-propagating high-temperature synthesis (SHS) are widely used as effective methods for synthesizing HEMs. The sol–gel method involves the gelation of a multicomponent colloidal suspension in air, followed by drying and calcination under a specific atmosphere to yield the product. This technique offers high flexibility in the synthesis process, with advantages in material uniformity and controllability of the morphology, making it a mainstream approach for synthesizing HEOs.324 CVD, primarily conducted at elevated temperatures, uses precursor gases to form a high-density, solid film on a substrate surface. This method is favored for synthesizing high-entropy 2D layered materials due to its precise control over film thickness and uniformity.326 SHS, a solid-state method, relies on the exothermic reactions between reactants to drive the synthesis process. Specifically, metal precursors are blended and initially heated to initiate the reaction. Once started, the exothermic reaction self-sustains, creating high-temperature zones that propagate through the remaining reactants, ultimately forming the desired sample. Compared to conventional solid-state synthesis methods, SHS requires minimal external energy input and is frequently used in the preparation of HEAs and HECs.327,328
Although traditional synthesis processes have achieved numerous success, there is still significant room for optimization. Ball-milling methods relying on high-temperature post-sintering are energy-intensive, often susceptible to impurity contamination, and produce coarse powders, thus making it challenging to approach nanoscale materials. Additionally, synthesis parameters such as temperature, mixing conditions, and heating/cooling rates are critical factors that affect the phase formation in the final sample. Poorly optimized parameters may lead to phase separation and uneven elemental distribution.
The co-precipitation method requires precise control over the synthesis rate and reaction time to avoid heterogeneous phase formation. More importantly, aqueous synthesis processes inevitably introduce water into the material structure, potentially affecting performance.329,330 In PBA systems, for example, the incorporated lattice water tends to decompose upon cycling, leading to structural degradation and limited stability.331 The sol–gel method faces challenges in completely removing solvents and moisture, which can compromise purity and performance of the final product.332 In CVD, the high temperatures required demand thermally stable substrates and can lead to additional post-treatment processing steps.333 SHS routes, while effective, suffer from poorly controllable reactions; once initiated, they proceed rapidly, making it difficult to accurately control temperature and the reaction rate, which can negatively affect phase purity and uniformity of the product.334
Aside from that, traditional synthesis methods are labor-intensive and time-consuming, limiting their suitability for rapid performance screening.335 Consequently, developing new synthesis methods is crucial for improving the preparation efficiency and advancing battery technology development.336–345 Recently, some new methods such as the non-equilibrium thermal carbon impact method,346 the Joule-heating method,347 and the ultrafast high-temperature sintering method348–350 have been successfully employed.
Li et al.346 synthesized uniformly distributed single-phase medium-entropy oxide nanoparticles using the non-equilibrium carbothermal shock method, known for its fast heating rate, enabling high temperatures to be reached within milliseconds to tens of seconds. This method mitigates particle aggregation and oxide reduction, effectively promoting the synthesis of nanoscale particles. Meanwhile, Wu et al.347 prepared HEOs utilizing the Joule-heating method, which rapidly pyrolyzes precursors in nickel foils within tens of seconds. This method demonstrated its universality in preparing HEOs such as (MgFeCoNiZn)O, (MgMnCoNiZn)O, (MgMnCoNiZn)Fe2O4, (CrMnFeCoNi)3O4−x, and La(CrMnFeCoNi)O3−x, showcasing its potential in HEM synthesis. Additionally, Guo et al.349 successfully densified various bulk oxide ceramics using ultrafast high-temperature sintering (UHS), reducing the densification time from several hours to less than 30 seconds and controlling the sintering cycle in just a few minutes. The emergence of UHS significantly enhances the preparation efficiency.
5.3. Theoretical calculations
Currently, DFT and ab initio molecular dynamics (AIMD) are frequently utilized in traditional theoretical calculations to forecast the structure and phase stability of materials.351,352 However, both face several inherent limitations when applied to HEMs. Designing the optimal composition of HEMs as electrode materials through trial-and-error experimental approaches is time-consuming. Additionally, the structure of HEMs has a high degree of disorder and complexity.353 These factors make traditional theoretical calculations unsuitable for large-scale data screening.354 Thus, there is an urgent need to develop new (theoretical) computational methods for materials design.In recent years, there has been a growing focus on applying cluster-expansion Monte Carlo (CEMC) simulation,211 machine learning (ML),355–360 high-throughput calculation (HTC),361–363 and the special quasi-random structure (SQS) method364,365 to high-entropy systems. This is due to the inherent advantages over traditional modeling approaches, facilitating discovering and better understanding these complex systems.366 It should be noted that although certain techniques such as HTC, ML and SQS have thus far been primarily applied to HEAs, they present promising new avenues for designing future HEBMs.
Lun et al.211 introduced the CEMC simulation technique into the design of HEMs, successfully synthesizing a series of DRX compounds. They found that the SRO is reduced when more TM cations are combined, leading to improved energy density and rate capability. Rao et al.367 proposed an active learning loop strategy, integrating machine learning with DFT, thermodynamic calculations, and experiments. Employing the active learning loop, the team characterized 17 new alloys out of millions of compositions in just a few months, ultimately identifying two high-entropy Invar alloys. Kaufmann et al.368 introduced a novel high-throughput adaptive machine learning model (ML-HEA) for predicting the ability of solid-solution formation in HEAs. This model combines thermodynamic/chemical characterization with a random forest ML approach. The model successfully predicted 436 tetragonal components and 91 pentagonal components in 1110 tetragonal and 130 pentagonal component systems. Experimental results confirmed that these tetragonal/pentagonal components could form face-centered cubic or body-centered cubic solid solutions. The author highlighted that the model can continuously enhance prediction accuracy through regular database updates. Feng et al.369 combined a high-throughput computational approach based on Calculation of Phase Diagrams (CALPHAD), multiscale modeling, and experimental demonstration to efficiently screen lightweight HEAs from thousands of compositions. This innovative, high-throughput computational approach significantly reduces the time and labor cost required for data screening and improves design efficiency. The SQS method is employed to create a small crystal structure that accurately depicts the randomness inherent to multi-component solid solutions. This involves optimizing the arrangement of atoms and minimizing the correlation function to imitate the disordered state of a small supercell.370 Zuo et al.365 and Niu et al.364 have demonstrated that this approach is effective in the design of HEAs.
It is important to note that while the computing approaches mentioned above have significant potential in predicting and screening HEMs, they require fundamental theoretical computational methods for large-scale computation. Hence, while optimizing traditional computing methods, it is critical to continuously improve fundamental theories to ensure the reliability and accuracy of novel computational approaches.
5.4. Further applications of high-entropy strategies
Most current efforts have mainly concentrated on enhancing electrode materials and improving electrolytes. However, limited attention has been given to interface engineering for improving charge transport and ion diffusion. Furthermore, current doping techniques typically employ only a limited number of primary components, and the exploration of high-entropy doping as a novel modification approach remains incomplete. The following section will focus on high-entropy methodologies applied to battery surface coating and doping, broadening the practical application range of high-entropy strategies (Fig. 10). | ||
| Fig. 10 Extended applications of high-entropy strategies. | ||
Yuan et al.378 combined NCM811 microspheres with (La0.2Nd0.2Sm0.2Eu0.2Gd0.2)2Zr2O7 nanoparticles obtained via ball milling. The resultant HEO-coated NCM811 showed effective suppression of side reactions at the electrode|electrolyte interface, reduced the formation of Li2CO3 and LiOH, and helped prevent electrode corrosion. Notably, at 5 wt.% coating content, the polarization was significantly reduced, with the capacity retention after 300 cycles at 1C reaching 74%, marking a 17% increase compared to that of uncoated NCM811, while the rate performance remained stable. Various characterization confirmed that the HEO coating on the NCM811 surface suppresses side reactions between the electrode and the electrolyte, ultimately enhancing cycling performance. Consequently, the HEO coating presents a novel approach for surface modification, expanding the practical application of high-entropy strategies and providing new ideas for the development of advanced lithium-ion batteries and even other types of batteries.
It is worth mentioning that the definition of high entropy is empirical and used to distinguish between HEMs and materials with “simpler” compositions.379 While certain materials may not meet the requirements for high entropy, the synergistic effect resulting from the mixing of elements will still act on these multicomponent materials. Motivated by the entropy effect, Zhao et al.380 present an (epitaxial) entropy-assisted coating method for LiNi0.9Co0.05Mn0.05O2 (NCM90) cathodes by means of an orientated attachment-driven reaction between the layered oxide cathode and Wadsley–Roth-based oxides. Improved long-term cycling performance of NCM90 was provided by the fast transport characteristics of the entropy-assisted coating, together with its high cracking and corrosion resistance. Surface-modified NCM90 was thus able to deliver a specific capacity of 187 mA h g−1 and showed a capacity retention of 96% after 200 cycles at 1C rate, both of which were far greater compared to those of the uncoated NCM90 (138 mA h g−1 and 79%).
Ma et al.395 leveraged the high-entropy doping strategy, entailing five cation substitutions to fabricate Na0.667Mn0.667Ni0.167Co0.117Ti0.01Mg0.01Cu0.01Mo0.01Nb0.01O2 (HE-NMNC). This cathode material was capable of overcoming the detrimental phase transition that the traditional P2-type layered oxide Na0.667Mn0.667Ni0.167Co0.167O2 (NMNC) is experiencing during cycling. HE-NMNC delivered a reversible specific capacity of 111 mA h g−1 at 5C and maintained about 130 mA h g−1 after 100 cycles at 1C, with a cutoff potential of 4.5 V vs. Na+/Na. The high-entropy doping enlarged the voltage region of the reversible phase transition of HE-NMNC, concurrently enhancing its rate performance and cycling stability. The transition metals and lattice oxygen were found to contribute to the charge compensation. Ge et al.396 employed high-entropy doping to design nanoscale Na4Fe2.85(Ni,Co,Mn,Cu,Mg)0.03(PO4)2P2O7 (NFPP-HE) as a cathode material by in situ substituting the Fe atoms. NFPP-HE exhibited a specific capacity of 122 mA h g−1 at 0.1C, and even at 50C, it maintained 85 mA h g−1. Also, after 1500 cycles at 10C, the capacity retention amounted to 82%.
Wang et al.402 discovered that the addition of numerous salts to a single solvent can create a disordered solvation structure dominated by entropy. A high-entropy electrolyte (HE-DME) was designed to have numerous anionic groups. This resulted in a significant variation in the solvation structure, leading to a decrease in the solvation intensity between lithium ions and the solvent/anions. Consequently, the HE-DME demonstrated improved ion diffusion compared to each individual salt electrolyte at the same concentration. Similarly, the researchers developed another liquid electrolyte of high entropy by utilizing a multisalt approach, which likewise demonstrated enhanced kinetics for ion diffusion.403
Unlike the process of combining multiple salts, solvent mixing is also a commonly used approach to address the limitations of individual solvents. This involves adding additional functional solvents as additives to achieve specific properties.406,407 Furthermore, the increase in the number of solvents greatly influences the mixing of the electrolyte and enhances the uniformity of the system.408 Kim et al.404 introduced a lithium salt into a multisolvent system and found that the increased entropy of mixing can affect the thermodynamic equilibrium, promoting the mixing process and preventing the formation of clusters. The high-entropy liquid electrolyte, which has been specifically engineered, had the smallest ionic clusters and the highest diffusion coefficient. This electrolyte enabled cycling up to 2C in anode-less NCM pouch cells.
6. Conclusions
As an innovative approach, the high-entropy method opens up new avenues for developing high-performance battery materials. By increasing the configurational entropy (compositional complexity), it has realized various positive effects in battery materials design. This work summarizes these positive effects and presents six fundamental advantages of HEBMs, which may be viewed as extensions to the four major effects in HEMs: (1) zero or low volumetric strain, (2) suppression of adverse phase transitions, (3) facilitation of conversion processes, (4) constructing transport channels via lattice distortions, (5) disorder-facilitated ion diffusion, and (6) synergies induced by cocktail effects. It must be mentioned that these fundamental advantages do not exist independently. Rather, they often coexist or several of these advantages coexist within the same high-entropy system. Additionally, this study offers overarching perspectives and solutions to the challenges encountered during the synthesis and design of HEBMs, encompassing the selection of elements, preparation methods, and guidance from theoretical considerations. At last, we outline several future directions for the advancement of high-entropy methods in battery materials, aiming at stimulating new ideas and possibilities.Author contributions
Y.-J. M. and Y. M. proposed the topic of the review. Z. Z. and Y. M. conducted the literature search. Z. Z. and Y.-J. M. designed the figures and co-wrote the manuscript. T. B., B. B., and Y. W. discussed and revised the manuscript.Data availability
No primary research results, software, or code have been included in this review article, and no new data were generated or analyzed as part of this study. All data discussed within this review are sourced from previously published studies, which are appropriately cited throughout the article. Therefore, no new datasets were generated, and no additional data are available.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by the National Key Research and Development Program of China (no. 2021YFB2400400), the National Natural Science Foundation of China (no. 52073143 and 22279016), Key Project (no. 52131306), the Project on Carbon Emission Peak and Neutrality of Jiangsu Province (no. BE2022031-4), Research Start-up Fund of Nanjing Normal University (184080H201B41) and a Research Start-up Fund of Southeast University (RF1028624081).References
- P. G. Bruce, B. Scrosati and J. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- Y. Li, H. Jia, U. Ali, H. Wang, B. Liu, L. Li, L. Zhang and C. Wang, Adv. Energy Mater., 2023, 13, 2301643 CrossRef CAS.
- W. Huang, S. Wang, X. Zhang, Y. Kang, H. Zhang, N. Deng, Y. Liang and H. Pang, Adv. Mater., 2023, 35, 2310147 CrossRef CAS.
- X. Pang, H. Geng, S. Dong, B. An, S. Zheng and B. Wang, Small, 2023, 19, 2205525 CrossRef CAS.
- F. Karcher, M. Uhl, T. Geng, T. Jacob and R. Schuster, Angew. Chem., Int. Ed., 2023, 62, e202301253 CrossRef CAS.
- W. Ren, M. Qin, Y. Zhou, H. Zhou, J. Zhu, J. Pan, J. Zhou, X. Cao and S. Liang, Energy Storage Mater., 2023, 54, 776–783 CrossRef.
- R. Hou, S. Guo and H. Zhou, Adv. Energy Mater., 2023, 13, 2300053 CrossRef CAS.
- L. Zhu, M. Wang, S. Xiang, D. Sun, Y. Tang and H. Wang, Adv. Energy Mater., 2023, 13, 2302046 CrossRef CAS.
- Z.-Y. Yu, Q. Sun, H. Li, Z.-J. Qiao, W.-J. Li, S.-L. Chou, Z.-J. Zhang and Y. Jiang, Rare Met., 2023, 42, 2982–2992 CrossRef CAS.
- Y. Wang, X. Zhang, H. He, J. Chen and B. Liu, Adv. Energy Mater., 2023, 14, 2303923 CrossRef.
- S. L. Fereja, Z. Zhang, Z. Fang, J. Guo, X. Zhang, K. Liu, Z. Li and W. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 38727–38738 CrossRef CAS.
- M. Xie, X. Xiao, D. Wu, C. Zhen, C. Wu, W. Wang, H. Nian, F. Li, M. D. Gu and Q. Xu, Nano Res., 2024, 17, 5288–5297 CrossRef CAS.
- M. Tamtaji, M. G. Kim, J. Wang, P. R. Galligan, H. Zhu, F. Hung, Z. Xu, Y. Zhu, Z. Luo, W. A. Goddard and G. Chen, Adv. Sci., 2024, 2309883 CrossRef CAS.
- B. Jiang, Y. Yu, J. Cui, X. Liu, L. Xie, J. Liao, Q. Zhang, Y. Huang, S. Ning, B. Jia, B. Zhu, S. Bai, L. Chen, S. J. Pennycook and J. He, Science, 2021, 371, 830–834 CrossRef CAS PubMed.
- Y. Li, Z. Chen, J. Liu, R. Liu, C. Zhang and H. Li, Ceram. Int., 2023, 49, 38439–38447 CrossRef CAS.
- A. Chatterjee, D. Ganguly, R. Sundara and S. S. Bhattacharya, Energy Technol., 2024, 12, 2300576 CrossRef CAS.
- J. Mu, T. Cai, W. Dong, C. Zhou, Z. Han and F. Huang, Chem. Eng. J., 2023, 471, 144403 CrossRef CAS.
- B. Ouyang and Y. Zeng, Nat. Commun., 2024, 15, 973 CrossRef CAS PubMed.
- Z.-Y. Liu, Y. Liu, Y. Xu, H. Zhang, Z. Shao, Z. Wang and H. Chen, Green Energy Environ., 2023, 8, 1341–1357 CrossRef CAS.
- Y. Wang, M. J. Robson, A. Manzotti and F. Ciucci, Joule, 2023, 7, 848–854 CrossRef CAS.
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC.
- D. Du, H. He, R. Zheng, L. Zeng, X. Wang, C. Shu and C. Zhang, Adv. Energy Mater., 2024, 14, 2304238 CrossRef CAS.
- Y. Ma, Y. Ren, D. Sun, B. Wang, H. Wu, H. Bian, J. Cao, X. Cao, F. Ding, J. Lu and X. Meng, J. Mater. Sci. Technol., 2024, 188, 98–104 CrossRef CAS.
- J. M. Gonçalves, É. A. Santos, P. R. Martins, C. G. Silva and H. Zanin, Energy Storage Mater., 2023, 63, 102999 CrossRef.
- C. Triolo, S. Schweidler, L. Lin, G. Pagot, V. Di Noto, B. Breitung and S. Santangelo, Energy Adv., 2023, 2, 667–678 RSC.
- S. Lee, L. Bai, J. Jeong, D. Stenzel, S. Schweidler and B. Breitung, Electrochim. Acta, 2023, 463, 142775 CrossRef CAS.
- B. Ran, H. Li, R. Cheng, Z. Yang, Y. Zhong, Y. Qin, C. Yang and C. Fu, Adv. Sci., 2024, 11, 2401034 CrossRef CAS PubMed.
- Y. Li, S. Song, H. Kim, K. Nomoto, H. Kim, X. Sun, S. Hori, K. Suzuki, N. Matsui, M. Hirayama, T. Mizoguchi, T. Saito, T. Kamiyama and R. Kanno, Science, 2023, 381, 50–53 CrossRef CAS PubMed.
- P. Basumatary, J. Hyeok Choi, M. Emin Kilic, D. Konwar and Y. Soo Yoon, ChemSusChem, 2024, 17, e202300801 CrossRef CAS PubMed.
- C. Yang, J. Xia, C. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. Xu, W. Song, E. Hu, X. Ji, S. Hou, X. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X.-Q. Yang, H. Wang, O. Borodin and C. Wang, Nat. Sustainable, 2023, 6, 325–335 CrossRef.
- X. He, Z. Zhu, G. Wen, S. Lv, S. Yang, T. Hu, Z. Cao, Y. Ji, X. Fu, W. Yang and Y. Wang, Adv. Mater., 2024, 36, 2307599 CrossRef CAS.
- J. Xu, Mater. Futures, 2023, 2, 047501 CrossRef CAS.
- W. Zheng, G. Liang, Q. Liu, J. Li, J. A. Yuwono, S. Zhang, V. K. Peterson and Z. Guo, Joule, 2023, 7, 2732–2748 CrossRef CAS.
- Q. Zheng, Z. Ren, Y. Zhang, X. Liu, J. Ma, L. Li, X. Liu and L. Chen, J. Phys. Chem. Lett., 2023, 14, 5553–5559 CrossRef CAS.
- X. Gao, X. Zhang, X. Liu, Y. Tian, Q. Cai, M. Jia and X. Yan, Small Methods, 2023, 7, 2300152 CrossRef CAS.
- X. Tan, Y. Zhang, S. Xu, P. Yang, T. Liu, D. Mao, J. Qiu, Z. Chen, Z. Lu, F. Pan and W. Chu, Adv. Energy Mater., 2023, 13, 2300147 CrossRef CAS.
- J. Dai, S. Tan, L. Wang, F. Ling, F. Duan, M. Ma, Y. Shao, X. Rui, Y. Yao, E. Hu, X. Wu, C. Li and Y. Yu, ACS Nano, 2023, 17, 20949–20961 CrossRef PubMed.
- Y.-J. Liao, W.-W. Shen, C.-B. Chang and H.-Y. Tuan, Chem. Eng. J., 2023, 469, 143942 CrossRef CAS.
- P. Zhou, Z. Che, J. Liu, J. Zhou, X. Wu, J. Weng, J. Zhao, H. Cao, J. Zhou and F. Cheng, Energy Storage Mater., 2023, 57, 618–627 CrossRef.
- Z. Zhou, Z. Chen, H. Lv, Y. Zhao, H. Wei, G. Huai, R. Xu and Y. Wang, J. Mater. Chem. A, 2023, 11, 5883–5894 RSC.
- C.-B. Chang, Y.-R. Lu and H.-Y. Tuan, Energy Storage Mater., 2023, 59, 102770 CrossRef.
- Z. Wang, S. Zhang, X. Fu, R. Huang, L. Huang, J. Zhang, W. Yang, F. Fu and S. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 2378–2388 CrossRef CAS PubMed.
- Q. Liang, S. Wang, X. Lu, X. Jia, J. Yang, F. Liang, Q. Xie, C. Yang, J. Qian, H. Song and R. Chen, ACS Nano, 2024, 18, 2395–2408 CrossRef CAS PubMed.
- B. Liu, Q. Zhang, L. Zhang, X. Yong, L. Li and C. Wang, Energy Storage Mater., 2024, 66, 103221 CrossRef.
- X. Liu, Y. Yu, K. Li, Y. Li, X. Li, Z. Yuan, H. Li, H. Zhang, M. Gong, W. Xia, Y. Deng and W. Lei, Adv. Mater., 2024, 36, 2312583 CrossRef CAS.
- N. Ci, Y. Hu, Q. Li, J. Cheng, H. Zhang, D. Li, K. Li, K. M. Reddy, L. Ci, G. Xie, X. Liu and H. Qiu, Small Methods, 2023, 2301322 Search PubMed.
- S. Wang, K. Wang, Y. Zhang, Y. Jie, X. Li, Y. Pan, X. Gao, Q. Nian, R. Cao, Q. Li, S. Jiao and D. Xu, Angew. Chem., Int. Ed., 2023, 62, e202304411 CrossRef PubMed.
- S. Han, Z. Wang, Y. Ma, Y. Miao, X. Wang, Y. Wang and Y. Wang, J. Adv. Ceram., 2023, 12, 1201–1213 CrossRef CAS.
- S. Schweidler, M. Botros, F. Strauss, Q. Wang, Y. Ma, L. Velasco, G. Cadilha Marques, A. Sarkar, C. Kübel, H. Hahn, J. Aghassi-Hagmann, T. Brezesinski and B. Breitung, Nat. Rev. Mater., 2024, 9, 266–281 CrossRef.
- L. Oroszová, D. Csík, G. Baranová, G. Bortel, R. Džunda, L. Temleitner, M. Hagarová, B. Breitung and K. Saksl, Crystals, 2024, 14, 218 CrossRef.
- M. Qiu, P. Sun, K. Han, Z. Pang, J. Du, J. Li, J. Chen, Z. L. Wang and W. Mai, Nat. Commun., 2023, 14, 601 CrossRef CAS PubMed.
- Y. Yao, J. Chen, R. Niu, Z. Zhao and X. Wang, Metals, 2023, 13, 833 CrossRef CAS.
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. De Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- P. Wang, Y. You, Y. Yin and Y. Guo, Adv. Energy Mater., 2018, 8, 1701912 CrossRef.
- Y. He, S. L. Dreyer, Y. Ting, Y. Ma, Y. Hu, D. Goonetilleke, Y. Tang, T. Diemant, B. Zhou, P. M. Kowalski, M. Fichtner, H. Hahn, J. Aghassi-Hagmann, T. Brezesinski, B. Breitung and Y. Ma, Angew. Chem., Int. Ed., 2024, 63, e202315371 CrossRef CAS PubMed.
- Y. Ma, Y. Ma, S. L. Dreyer, Q. Wang, K. Wang, D. Goonetilleke, A. Omar, D. Mikhailova, H. Hahn, B. Breitung and T. Brezesinski, Adv. Mater., 2021, 33, 2101342 CrossRef CAS PubMed.
- J. Patra, T. X. Nguyen, C. Tsai, O. Clemens, J. Li, P. Pal, W. K. Chan, C. Lee, H. T. Chen, J. Ting and J. Chang, Adv. Funct. Mater., 2022, 32, 2110992 CrossRef CAS.
- W. Li, Z. Chen, Y. Chen, L. Zhang, G. Liu and L. Yao, Adv. Funct. Mater., 2024, 34, 2312832 CrossRef CAS.
- X. Lei, Y. Wang, J. Wang, Y. Su, P. Ji, X. Liu, S. Guo, X. Wang, Q. Hu, L. Gu, Y. Zhang, R. Yang, G. Zhou and D. Su, Small Methods, 2024, 8, 2300754 CrossRef CAS.
- J. Feng, Y. Liu, D. Fang and J. Li, Nano Energy, 2023, 118, 109030 CrossRef CAS.
- X. L. Wang, E. M. Kim, T. G. Senthamaraikannan, D.-H. Lim and S. M. Jeong, Chem. Eng. J., 2024, 484, 149509 CrossRef CAS.
- Y.-H. Tseng, Y.-C. Lin, Y.-H. Wu, J.-M. Ting and S.-H. Chung, J. Energy Storage, 2023, 68, 107767 CrossRef.
- A. N. Singh, M. Islam, A. Meena, M. Faizan, D. Han, C. Bathula, A. Hajibabaei, R. Anand and K. Nam, Adv. Funct. Mater., 2023, 33, 2304617 CrossRef CAS.
- S. Zhou, Y. Sun, T. Gao, J. Liao, S. Zhao and G. Cao, Angew. Chem., Int. Ed., 2023, 62, e202311930 CrossRef CAS.
- G. Ma, Y. Zheng, F. Meng and R. Hu, Energy Adv., 2023, 2, 1685–1692 RSC.
- R. Kang, D. Zhang, Y. Du, C. Sun, W. Zhou, H. Wang, J. Wan, G. Chen and J. Zhang, Small, 2024, 20, 2305998 CrossRef CAS.
- N. Jiang, X. Wang, H. Zhou, Y. Wang, S. Sun, C. Yang and Y. Liu, Small, 2024, 20, 2308681 CrossRef CAS.
- Z. Wang, L. Fang, X. Fu, S. Zhang, H. Kong, H. Chen and F. Fu, Chem. Eng. J., 2024, 480, 148130 CrossRef CAS.
- Y. Ma, T. Brezesinski, B. Breitung and Y. Ma, Matter, 2023, 6, 313–315 CrossRef CAS.
- S. Chu, C. Shao, J. Tian, J. Wang, Y. Rao, C. Xu, H. Zhou and S. Guo, ACS Nano, 2024, 18, 337–346 CrossRef CAS.
- P.-W. Chien, C.-B. Chang and H.-Y. Tuan, Energy Storage Mater., 2023, 61, 102853 CrossRef.
- S. Liu, Y. Wang, T. Jiang, S. Jin, M. Sajid, Z. Zhang, J. Xu, Y. Fan, X. Wang, J. Chen, Z. Liu, X. Zheng, K. Zhang, Q. Nian, Z. Zhu, Q. Peng, T. Ahmad, K. Li and W. Chen, ACS Nano, 2024, 18, 4229–4240 CrossRef CAS.
- H. Zhang, Y. Wang, J. Huang, W. Li, X. Zeng, A. Jia, H. Peng, X. Zhang and W. Yang, Energy Environ. Mater., 2023, 7, e12514 CrossRef.
- Q. Wang, M. Zhao, H. Yu, Y. Xie and T.-F. Yi, Inorg. Chem. Front., 2024, 11, 4249–4262 RSC.
- F. Ding, Y. Lu, L. Chen and Y.-S. Hu, Electrochem. Energy Rev., 2024, 7, 16 CrossRef.
- Y.-Y. Ting, R. Ye, E. Dashjav, Q. Ma, S. Taminato, D. Mori, N. Imanishi, M. Finsterbusch, M. H. Eikerling, O. Guillon, P. Kaghazchi and P. M. Kowalski, Front. Energy Res., 2024, 12, 1393914 CrossRef.
- Y. Ma, Y. Hu, Y. Pramudya, T. Diemant, Q. Wang, D. Goonetilleke, Y. Tang, B. Zhou, H. Hahn, W. Wenzel, M. Fichtner, Y. Ma, B. Breitung and T. Brezesinski, Adv. Funct. Mater., 2022, 32, 2202372 CrossRef CAS.
- Y. Zhou, G. Xu, J. Lin, J. Zhu, J. Pan, G. Fang, S. Liang and X. Cao, Nano Energy, 2024, 128, 109812 CrossRef CAS.
- A. Wehrl, Rep. Math. Phys., 1991, 30, 119–129 CrossRef.
- B. S. Murty, J.-W. Yeh and S. Ranganathan, High-entropy alloys, Elsevier Inc., Londres, 2014 Search PubMed.
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef.
- A. Sarkar, B. Breitung and H. Hahn, Scr. Mater., 2020, 187, 43–48 CrossRef CAS.
- R. Carroll, C. Lee, C.-W. Tsai, J.-W. Yeh, J. Antonaglia, B. A. W. Brinkman, M. LeBlanc, X. Xie, S. Chen, P. K. Liaw and K. A. Dahmen, Sci. Rep., 2015, 5, 16997 CrossRef CAS.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Mater. Today, 2016, 19, 349–362 CrossRef CAS.
- M. C. Gao, D. B. Miracle, D. Maurice, X. Yan, Y. Zhang and J. A. Hawk, J. Mater. Res., 2018, 33, 3138–3155 CrossRef CAS.
- J. Tian, Y. Rao, W. Shi, J. Yang, W. Ning, H. Li, Y. Yao, H. Zhou and S. Guo, Angew. Chem., Int. Ed., 2023, 62, e202310894 CrossRef CAS.
- Z. Wang, H. Ge, S. Liu, G. Li and X. Gao, Energy Environ. Mater., 2023, 6, e12358 CrossRef CAS.
- Q. Zhang, K. Lian, Q. Liu, G. Qi, S. Zhang, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 646, 844–854 CrossRef CAS PubMed.
- R. He, L. Yang, Y. Zhang, X. Wang, S. Lee, T. Zhang, L. Li, Z. Liang, J. Chen, J. Li, A. Ostovari Moghaddam, J. Llorca, M. Ibáñez, J. Arbiol, Y. Xu and A. Cabot, Energy Storage Mater., 2023, 58, 287–298 CrossRef.
- R. He, L. Yang, Y. Zhang, D. Jiang, S. Lee, S. Horta, Z. Liang, X. Lu, A. Ostovari Moghaddam, J. Li, M. Ibáñez, Y. Xu, Y. Zhou and A. Cabot, Adv. Mater., 2023, 35, 2303719 CrossRef CAS.
- J. Wang, W. Liu, Y. Wang, Y. Guo, M. Liu, C. Ye, S. Wang and Q. Zou, Chem. Eng. J., 2023, 477, 146884 CrossRef CAS.
- J. Wang, Y. Wang, X. Lu, J. Qian, C. Yang, I. Manke, H. Song, J. Liao, S. Wang and R. Chen, Adv. Mater., 2024, 36, 2308257 CrossRef CAS.
- D.-H. Wu, M. Ul Haq, L. Zhang, J.-J. Feng, F. Yang and A.-J. Wang, J. Colloid Interface Sci., 2024, 662, 149–159 CrossRef CAS PubMed.
- S. Alvi, A. P. Black, I. Jozami, C. Escudero, F. Akhtar and P. Johansson, Batteries Supercaps, 2024, 7, e202300585 CrossRef CAS.
- X. Li, J. Wang, L. Yang, T. Liu, S. Huang, B. Ho, H. Hsueh, J. Chen, L. He, Z. Guo, M. Liu and W. Li, Adv. Mater., 2024, 2409278 CrossRef CAS.
- L. Tang, Z. Li, K. Chen, C. Li, X. Zhang and L. An, J. Am. Ceram. Soc., 2021, 104, 1953–1958 CrossRef CAS.
- J. Dąbrowa, M. Stygar, A. Mikuła, A. Knapik, K. Mroczka, W. Tejchman, M. Danielewski and M. Martin, Mater. Lett., 2018, 216, 32–36 CrossRef.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS.
- M. Song, X. Zhang, S. Wan, G. Wang, J. Liu, W. Li, H. Dong, C. Lou, Z. Chen, B. Chen and H. Zhang, JACS Au, 2024, 4, 592–606 CrossRef CAS.
- S. Li, Z. Peng and X. Fu, J. Adv. Ceram., 2023, 12, 59–71 CrossRef CAS.
- Q. Zheng, Z. Ren, Y. Zhang, T. Qin, J. Qi, H. Jia, L. Jiang, L. Li, X. Liu and L. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 4643–4651 CrossRef CAS.
- Y.-T. Chen, J.-T. Lee, T. Y. Liang, Y.-H. Song and J. M. Wu, Electrochim. Acta, 2023, 456, 142459 CrossRef CAS.
- T. X. Nguyen, J. Patra, C. Tsai, W. Xuan, H. T. Chen, M. S. Dyer, O. Clemens, J. Li, S. B. Majumder, J. Chang and J. Ting, Adv. Funct. Mater., 2023, 33, 2300509 CrossRef CAS.
- C. Zhao, Z. Yao, Q. Wang, H. Li, J. Wang, M. Liu, S. Ganapathy, Y. Lu, J. Cabana, B. Li, X. Bai, A. Aspuru-Guzik, M. Wagemaker, L. Chen and Y.-S. Hu, J. Am. Chem. Soc., 2020, 142, 5742–5750 CrossRef CAS.
- C. Liu, J. Bi, L. Xie, X. Gao and J. Rong, J. Energy Storage, 2023, 71, 108211 CrossRef.
- S. Chung, Y. Wu, Y. Tseng, T. X. Nguyen and J. Ting, ChemSusChem, 2023, 16, e202300135 CrossRef CAS PubMed.
- Y. Cai, W. Liu, F. Chang, S. Jin, X. Yang, C. Zhang, L. Bai, T. Masese, Z. Li and Z.-D. Huang, ACS Appl. Mater. Interfaces, 2023, 15, 48277–48286 CrossRef CAS PubMed.
- W. Bian, H. Li, Z. Zhao, H. Dou, X. Cheng and X. Wang, Electrochim. Acta, 2023, 447, 142157 CrossRef CAS.
- H. Deng, L. Liu and Z. Shi, Mater. Lett., 2023, 340, 134113 CrossRef CAS.
- Y. Wang, J. Liu, Y. Song, J. Yu, Y. Tian, M. J. Robson, J. Wang, Z. Zhang, X. Lin, G. Zhou, Z. Wang, L. Shen, H. Zhao, S. Grasso and F. Ciucci, Small Methods, 2023, 7, 2201138 CrossRef CAS.
- H. Nan, S. Lv, Z. Xu, Y. Feng, Y. Zhou, M. Liu, T. Wang, X. Liu, X. Hu and H. Tian, Chem. Eng. J., 2023, 452, 139501 CrossRef CAS.
- X. Ji, F. Yang, Y. Du, J. Li, J. Li and Q. Hu, J. Mater. Sci. Technol., 2024, 168, 71–78 CrossRef CAS.
- X.-H. Xiao, D. M. Kabtamu, A. M. Demeku, G.-C. Chen, Y.-T. Ou, Z.-J. Huang, N.-Y. Hsu, H.-H. Ku, Y.-M. Wang and C.-H. Wang, J. Power Sources, 2024, 597, 234178 CrossRef CAS.
- L. Tian, Z. Zhang, S. Liu, G. Li and X. Gao, Nano Energy, 2023, 106, 108037 CrossRef CAS.
- K. Du, Y. Liu, Y. Yang, F. Cui, J. Wang, M. Han, J. Su, J. Wang, X. Han and Y. Hu, Adv. Mater., 2023, 35, 2301538 CrossRef CAS.
- H. Ge, L. Zheng, G. Yuan, W. Shi, J. Liu, Y. Zhang and X. Wang, J. Am. Chem. Soc., 2024, 146, 10735–10744 CrossRef CAS.
- J. Wang, Y. Cui, Q. Wang, K. Wang, X. Huang, D. Stenzel, A. Sarkar, R. Azmi, T. Bergfeldt, S. S. Bhattacharya, R. Kruk, H. Hahn, S. Schweidler, T. Brezesinski and B. Breitung, Sci. Rep., 2020, 10, 18430 CrossRef CAS.
- L. Dong, Y. Tian, C. Luo, W. Zhao, C. Qin and Z. Wang, Materials, 2024, 17, 1542 CrossRef CAS.
- W. Wang, W. Song, Y. Li, Y. Guo, K. Yang, L. Yu, F. Xie, Q. Ren, K. He, S. Wang and Y. Yuan, Nano Energy, 2024, 124, 109482 CrossRef CAS.
- J. Wang, D. Stenzel, R. Azmi, S. Najib, K. Wang, J. Jeong, A. Sarkar, Q. Wang, P. A. Sukkurji, T. Bergfeldt, M. Botros, J. Maibach, H. Hahn, T. Brezesinski and B. Breitung, Electrochem, 2020, 1, 60–74 CrossRef.
- S. Chen, M. Bao, Y. Jia, X. Shao, Y. Guo, S. Li, A. Mao, J. Tan and X. Liu, J. Alloys Compd., 2024, 990, 174480 CrossRef CAS.
- Q. Wang, A. Sarkar, Z. Li, Y. Lu, L. Velasco, S. S. Bhattacharya, T. Brezesinski, H. Hahn and B. Breitung, Electrochem. Commun., 2019, 100, 121–125 CrossRef CAS.
- L. Lin, K. Wang, A. Sarkar, C. Njel, G. Karkera, Q. Wang, R. Azmi, M. Fichtner, H. Hahn, S. Schweidler and B. Breitung, Adv. Energy Mater., 2022, 12, 2103090 CrossRef CAS.
- L. Lin, K. Wang, R. Azmi, J. Wang, A. Sarkar, M. Botros, S. Najib, Y. Cui, D. Stenzel, P. Anitha Sukkurji, Q. Wang, H. Hahn, S. Schweidler and B. Breitung, J. Mater. Sci., 2020, 55, 16879–16889 CrossRef CAS.
- S. Schweidler, Y. Tang, L. Lin, G. Karkera, A. Alsawaf, L. Bernadet, B. Breitung, H. Hahn, M. Fichtner, A. Tarancón and M. Botros, Front. Energy Res., 2022, 10, 983979 CrossRef.
- D. Stenzel, B. Zhou, C. Okafor, M. V. Kante, L. Lin, G. Melinte, T. Bergfeldt, M. Botros, H. Hahn, B. Breitung and S. Schweidler, Front. Energy Res., 2022, 10, 942314 CrossRef.
- A. Sarkar, P. K. Mannava, L. Velasco, C. Das, B. Breitung, S. S. Bhattacharya, R. Kruk and H. Hahn, Scr. Mater., 2022, 207, 114273 CrossRef CAS.
- S. Schweidler, S. L. Dreyer, B. Breitung and T. Brezesinski, Sci. Rep., 2021, 11, 23381 CrossRef CAS.
- Y. Leng, Z. Zhang, H. Chen, S. Du, J. Liu, S. Nie, Y. Dong, P. Zhang and S. Dai, Chem. Commun., 2021, 57, 3676–3679 RSC.
- E. Castle, T. Csanádi, S. Grasso, J. Dusza and M. Reece, Sci. Rep., 2018, 8, 8609 CrossRef.
- K. Lu, J.-X. Liu, X.-F. Wei, W. Bao, Y. Wu, F. Li, F. Xu and G.-J. Zhang, J. Eur. Ceram. Soc., 2020, 40, 1839–1847 CrossRef CAS.
- J. Gild, Y. Zhang, T. Harrington, S. Jiang, T. Hu, M. C. Quinn, W. M. Mellor, N. Zhou, K. Vecchio and J. Luo, Sci. Rep., 2016, 6, 37946 CrossRef CAS.
- T. Jin, X. Sang, R. R. Unocic, R. T. Kinch, X. Liu, J. Hu, H. Liu and S. Dai, Adv. Mater., 2018, 30, 1707512 CrossRef PubMed.
- M. J. Theibault, C. R. McCormick, S. Lang, R. E. Schaak and H. D. Abruña, ACS Nano, 2023, 17, 18402–18410 CrossRef CAS.
- X. Wang, G. Liu, C. Tang, H. Tang, W. Zhang, Z. Ju, J. Jiang, Q. Zhuang and Y. Cui, J. Alloys Compd., 2023, 934, 167889 CrossRef CAS.
- H. Wang, Y. Yuan, Q. Jia, A. Shao, M. Zhang, Z. Wang, L. Cheng, X. Tang, S. Li and Y. Ma, Adv. Funct. Mater., 2024, 34, 2314186 CrossRef CAS.
- Y. Wei, Y. Zhao, Y. Chen, M. Zhang, Z. Zhang, J. Kang, X. Ma, Y. Jiang and Y. Zhang, Chem. Eng. J., 2023, 476, 146881 CrossRef CAS.
- D. Stenzel, I. Issac, K. Wang, R. Azmi, R. Singh, J. Jeong, S. Najib, S. S. Bhattacharya, H. Hahn, T. Brezesinski, S. Schweidler and B. Breitung, Inorg. Chem., 2021, 60, 115–123 CrossRef CAS PubMed.
- S. Schweidler, S. L. Dreyer, B. Breitung and T. Brezesinski, Coatings, 2022, 12, 402 CrossRef CAS.
- B. Zhou, S. An, D. Kitsche, S. L. Dreyer, K. Wang, X. Huang, J. Thanner, M. Bianchini, T. Brezesinski, B. Breitung, H. Hahn and Q. Wang, Small Struct., 2024, 2400005 CrossRef CAS.
- M. R. Chellali, A. Sarkar, S. H. Nandam, S. S. Bhattacharya, B. Breitung, H. Hahn and L. Velasco, Scr. Mater., 2019, 166, 58–63 CrossRef CAS.
- Y. Cui, P. A. Sukkurji, K. Wang, R. Azmi, A. M. Nunn, H. Hahn, B. Breitung, Y.-Y. Ting, P. M. Kowalski, P. Kaghazchi, Q. Wang, S. Schweidler and M. Botros, J. Energy Chem., 2022, 72, 342–351 CrossRef CAS.
- L. Lin, Z. Ding, G. Karkera, T. Diemant, M. V. Kante, D. Agrawal, H. Hahn, J. Aghassi-Hagmann, M. Fichtner, B. Breitung and S. Schweidler, Small Struct., 2023, 4, 2300012 CrossRef CAS.
- P. A. Sukkurji, Y. Cui, S. Lee, K. Wang, R. Azmi, A. Sarkar, S. Indris, S. S. Bhattacharya, R. Kruk, H. Hahn, Q. Wang, M. Botros and B. Breitung, J. Mater. Chem. A, 2021, 9, 8998–9009 RSC.
- W. Li, Y. Li, J.-H. Wang, S. Huang, A. Chen, L. Yang, J. Chen, L. He, W. K. Pang, L. Thomsen, B. Cowie, P. Xiong, Y. Zhou, G. Jang, D. H. Min, J. S. Byun, L. Xu, J.-Q. Huang, K. C. Roh, S. H. Kang, M. Liu, X. Duan and H. S. Park, Energy Environ. Sci., 2024, 17, 5387–5398 RSC.
- Y. Zhang, J. Y. Xing, M. Chen and Q. Jin, Chem. Eng. J., 2024, 479, 147709 CrossRef CAS.
- W. Li, J.-H. Wang, Y. Li, H. Hsueh, X. Liu, Y. Zhao, S. Huang, X. Li, H.-M. Cheng, X. Duan and H. S. Park, J. Am. Chem. Soc., 2024, 146, 21320–21334 CrossRef.
- J.-W. Yeh, S.-K. Chen, S.-J. Lin, J.-Y. Gan, T.-S. Chin, T.-T. Shun, C.-H. Tsau and S.-Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- S. Wei, S. J. Kim, J. Kang, Y. Zhang, Y. Zhang, T. Furuhara, E. S. Park and C. C. Tasan, Nat. Mater., 2020, 19, 1175–1181 CrossRef CAS PubMed.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534 CrossRef CAS.
- Y. Lu, Y. Dong, H. Jiang, Z. Wang, Z. Cao, S. Guo, T. Wang, T. Li and P. K. Liaw, Scr. Mater., 2020, 187, 202–209 CrossRef CAS.
- K. M. Youssef, A. J. Zaddach, C. Niu, D. L. Irving and C. C. Koch, Mater. Res. Lett., 2015, 3, 95–99 CrossRef CAS.
- S. Akrami, P. Edalati, M. Fuji and K. Edalati, Mater. Sci. Eng., R, 2021, 146, 100644 CrossRef.
- C. Toher, C. Oses, D. Hicks and S. Curtarolo, npj Comput. Mater., 2019, 5, 69 CrossRef.
- L. Chen, B. Li, J. Guo, Y. Zhu and J. Feng, J. Adv. Ceram., 2022, 11, 556–569 CrossRef CAS.
- D. Ni, Y. Cheng, J. Zhang, J.-X. Liu, J. Zou, B. Chen, H. Wu, H. Li, S. Dong, J. Han, X. Zhang, Q. Fu and G.-J. Zhang, J. Adv. Ceram., 2022, 11, 1–56 CrossRef CAS.
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 5, 295–309 CrossRef CAS.
- Y. Pang, Y. Wang, X. Ding, Y. Xin, Q. Zhou, C. Jiang, B. Chen, H. Liu, F. Wu and H. Gao, ACS Sustainable Chem. Eng., 2024, 12, 8203–8213 CrossRef CAS.
- X. Liu, X.-Y. Liu, N. Zhang, P.-F. Wang, Z.-L. Liu, J.-H. Zhang, J. Shu, Y. Sun, C.-S. Li and T.-F. Yi, Chem. Eng. J., 2024, 496, 153743 CrossRef CAS.
- X. Ding, X. Yang, Y. Yang, L. Liu, Y. Xiao and L. Han, J. Energy Chem., 2024, 97, 429–437 CrossRef CAS.
- Y. Pang, Y. Wang, C. Jiang, X. Ding, Y. Xin, Q. Zhou, B. Chen, H. Liu, P. Singh, Q. Wang and H. Gao, ChemSusChem, 2024, e202400768 CrossRef.
- K. Tian, Y. Dang, Z. Xu, R. Zheng, Z. Wang, D. Wang, Y. Liu and Q. Wang, Energy Storage Mater., 2024, 73, 103841 CrossRef.
- C. Liu, S. Li, Y. Zheng, M. Xu, H. Su, X. Miao, Y. Liu, Z. Zhou, J. Qi, B. Yang, D. Chen, C.-W. Nan and Y.-H. Lin, Prog. Mater. Sci., 2025, 148, 101385 CrossRef CAS.
- J. Zhan, J. Huang, Z. Li, J. Yuan, S.-X. Dou, H.-K. Liu and C. Wu, Nano Lett., 2024, 24, 9793–9800 CrossRef CAS PubMed.
- Y. Chen, X. Liao, M. Xie, P. Wang, J. Chen, X. Zhang, H. Xie, Q. Zheng and D. Lin, ACS Sustainable Chem. Eng., 2024, 12, 13568–13577 CrossRef CAS.
- J. Patra, T. X. Nguyen, A. Panda, S. B. Majumder, C.-C. Yang, T.-Y. Wu, Y.-S. Su, C.-T. Hsieh, J.-M. Ting and J.-K. Chang, Electrochim. Acta, 2024, 498, 144653 CrossRef CAS.
- Z. Liu, X. Wu, Z. Song, J. Wang, P. Hu, J. Wen, C. Chen and C. Shang, J. Energy Storage, 2024, 94, 112537 CrossRef.
- Z. Hao, X. Shi, W. Zhu, Z. Yang, X. Zhou, C. Wang, L. Li, W. Hua, C.-Q. Ma and S. Chou, ACS Nano, 2024, 18, 9354–9364 CrossRef CAS.
- C. Jin, Y. Wang, P. Lu, H. Dong, Y. Wei, R. Nan, Z. Jian, Z. Yang and Q. Ding, Electrochim. Acta, 2024, 506, 145004 CrossRef CAS.
- J. Shen, Z. Zeng and W. Tang, SusMat, 2024, 4, e215 CrossRef CAS.
- Y. Zhang, R. Wang, W. Song, M. Lei, Y. Zhang, Z. Lei, Q. Wei, X. Zhang and X. Wang, Chem. Eng. J., 2024, 500, 157005 CrossRef CAS.
- Y. Li, J.-H. Wang, T.-Y. Liu, X. Li, Z. Guo, M. Liu and W. Li, Energy Environ. Sci., 2024, 17, 6533–6547 RSC.
- W. Song, D. Liu, B. Zhu, Y. He, S. Dou, X. Huang, M. Li and B. Zhong, J. Colloid Interface Sci., 2025, 677, 795–803 CrossRef CAS PubMed.
- J. Park, Y. Yang, H. Park, A. Sundar, S. Lee, T. L. Kinnibrugh, S. Son, E. Lee, P. Zapol, R. F. Klie and J. J. Kim, ACS Appl. Mater. Interfaces, 2024, 16, 57151–57161 CrossRef CAS.
- B. Li, J. Zhong, H. Wang, J. Gu, F. Lyu, S. Chen, H. Wu, L. Li, C. Zhi, J. Lu and Y. Y. Li, Angew. Chem., Int. Ed., 2024, e202410978 CAS.
- H. Yan, D. Chai, X. Li and Y. Fu, Small, 2024, 2404039 CrossRef CAS PubMed.
- X. Hu, H. Li, Z. Wang, M. Liu, Y. Lu, Y. Zhang, J. Li, K. Ding, H. Liu, Z. Ma and Y. Wang, Adv. Funct. Mater., 2024, 2412730 CrossRef.
- C. Chang, Y. Tseng, Y. Lu, Y. Yang and H. Tuan, Adv. Funct. Mater., 2024, 2411193 CrossRef.
- Q. He, J. Li, W. Liu, J. Zhang, K. Wang, J. Liu, J. Hui and H. Zhang, J. Power Sources, 2024, 619, 235207 CrossRef CAS.
- T. G. Ritter, S. Pappu and R. Shahbazian-Yassar, Batteries, 2024, 10, 96 CrossRef CAS.
- S. Hou, J. Luo, W. Gong, Y. Xie, X. Zhou, F. Yue, J. Shen, C. Li, L. Wei, F. Xu and Q. Zhang, ACS Nano, 2024, 18, 31524–31536 CrossRef CAS.
- Y. Qiu, Q. Shi, X. Yu, Y. Liu, Y. Liu, W. Feng, J. Wang and Y. Zhao, Chem. Eng. Sci., 2024, 300, 120671 CrossRef CAS.
- Z. Zeng, A. Abulikemu, J. Zhang, Z. Peng, Y. Zhang, Y. Uchimoto, J. Han and Q.-C. Wang, Nano Energy, 2024, 128, 109813 CrossRef CAS.
- Z. Yang, X. Xiang, J. Yang and Z.-Y. Zhao, Mater. Futures, 2024, 3, 042103 CrossRef CAS.
- W. Tanmathusorachai, S. Aulia, M. Rinawati, L.-Y. Chang, C.-Y. Chang, W.-H. Huang, M.-H. Lin, W.-N. Su, B. Yuliarto and M.-H. Yeh, ACS Appl. Mater. Interfaces, 2024, 16, 62022–62032 CrossRef CAS.
- J. Xu, M. Galib, Z. Wu, L. Tao, Y. Shao, Y. Zhang, X. Guo, E. J. Hansen, Y. Chen, Z. Wang, C. Liu, M. Ponga and J. Liu, Nano Energy, 2024, 132, 110373 CrossRef.
- Z. Qiu, X. Guo, S. Cao, M. Du, Q. Wang, Y. Pi and H. Pang, Angew. Chem., Int. Ed., 2024, e202415216 Search PubMed.
- Z. Huang, S. Wang, X. Guo, F. Marlton, Y. Fan, W. Pang, T. Huang, J. Xiao, D. Li, H. Liu, Q. Gu, C. Yang, C. Dong, B. Sun and G. Wang, Adv. Mater., 2024, 2410857 CrossRef PubMed.
- C. Hong, R. Tao, S. Tan, L. A. Pressley, C. A. Bridges, H. Li, X. Liu, H. Li, J. Li, H. Yuan, X. Sun and J. Liang, Adv. Funct. Mater., 2024, 2412177 CrossRef.
- S. Zhao, F. Ning, X. Yu, B. Guo, R. F. Teófilo, J. Huang, Q. Shi, S. Wu, W. Feng and Y. Zhao, Angew. Chem., Int. Ed., 2024, e202416290 Search PubMed.
- Y. He, S. L. Dreyer, T. Akçay, T. Diemant, R. Mönig, Y. Ma, Y. Tang, H. Wang, J. Lin, S. Schweidler, M. Fichtner, H. Hahn, T. Brezesinski, B. Breitung and Y. Ma, ACS Nano, 2024, 18, 24441–24457 CrossRef CAS.
- D. Wang, X. Li, A. Zhang, X. Wen, Q. Wang, Y. Liu, X. Qi and Z. Wang, J. Environ. Chem. Eng., 2024, 12, 114085 CrossRef CAS.
- V. Lokhande, D. Malavekar, C. Kim, A. Vinu and T. Ji, Energy Storage Mater., 2024, 72, 103718 CrossRef.
- L. Dong, C. Luo, Y. Wang, S. Meng, H. Yu, W. Zhao, C. Qin and Z. Wang, J. Energy Storage, 2024, 98, 113185 CrossRef.
- L. Duan, Y. Zhang, H. Tang, J. Liao, G. Zhou and X. Zhou, Adv. Mater., 2024, 2411426 CrossRef.
- X. Li, W. Zhang, K. Lv, J. Liu and A. Bayaguud, J. Power Sources, 2024, 620, 235259 CrossRef CAS.
- Y. Su, X. Lei, W. Chen, Y. Su, H. Liu, S. Ren, R. Tong, Y. Lin, W. Jiang, X. Liu, D. Su and Y. Zhang, Chem. Eng. J., 2024, 500, 157197 CrossRef CAS.
- H. Cheng, H. Xu, J. Shang, Y. Xu, H. Zong, W. Yao, Z. Fang, W. Dou, L. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2024, e202414302 Search PubMed.
- Z. Qian, R.-J. Luo, C. Ma, C.-Y. Du, J. Zeng, X. Xu, Z. Mei, Z.-T. Zhou, Z.-W. Fu and Y.-N. Zhou, Chem. Eng. J., 2024, 500, 156767 CrossRef CAS.
- Y. Jiang, W. Li and K. Luo, ACS Sustainable Chem. Eng., 2024, 12, 8051–8060 CrossRef CAS.
- F. Ding, P. Ji, Z. Han, X. Hou, Y. Yang, Z. Hu, Y. Niu, Y. Liu, J. Zhang, X. Rong, Y. Lu, H. Mao, D. Su, L. Chen and Y.-S. Hu, Nat. Energy, 2024, 1–11 Search PubMed.
- H. Li, X. Sun and H. Huang, Prog. Mater. Sci., 2025, 148, 101382 CrossRef CAS.
- P. Hou, M. Gong, M. Dong, Z. Lin, J. Huang, H. Zhang and F. Li, Energy Storage Mater., 2024, 72, 103750 CrossRef.
- Y. Lin, S. Luo, W. Zhao, Q. Sun, J. Cong, P. Li, P. Li and S. Yan, J. Energy Chem., 2024, 98, 441–471 CrossRef CAS.
- S. Hou, Y. Qi, J. Bao, J. Shen, Y. Han, D. Yuan, R. Fu, J. Luo, Q. Zhang and F. Xu, Nano Energy, 2024, 130, 110121 CrossRef CAS.
- K. Karami, S. Amjad-Iranagh, K. Dehghani, M. H. Saznaghi and R. Riahifar, J. Energy Storage, 2024, 101, 113823 CrossRef.
- Z. Lun, B. Ouyang, D.-H. Kwon, Y. Ha, E. E. Foley, T.-Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clément, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS.
- J. Peng, B. Zhang, W. Hua, Y. Liang, W. Zhang, Y. Du, G. Peleckis, S. Indris, Q. Gu, Z. Cheng, J. Wang, H. Liu, S. Dou and S. Chou, Angew. Chem., Int. Ed., 2023, 62, e202215865 CrossRef CAS.
- M. T. Ahsan, D. Qiu, Z. Ali, Z. Fang, W. Zhao, T. Shen and Y. Hou, Adv. Energy Mater., 2024, 14, 2302733 CrossRef CAS.
- C. Li, X. Chen, J. Li, Y. Xiang, Y. Yao, W. Liao, W. Xue, X. Zhang and Y. Xiang, Energy Storage Mater., 2024, 103468 CrossRef.
- J.-W. Yeh, Ann. Chim.: Sci. Mater., 2006, 31, 633–648 CrossRef CAS.
- R. Zhang, C. Wang, P. Zou, R. Lin, L. Ma, L. Yin, T. Li, W. Xu, H. Jia, Q. Li, S. Sainio, K. Kisslinger, S. E. Trask, S. N. Ehrlich, Y. Yang, A. M. Kiss, M. Ge, B. J. Polzin, S. J. Lee, W. Xu, Y. Ren and H. L. Xin, Nature, 2022, 610, 67–73 CrossRef CAS.
- X. Zhao and G. Ceder, Joule, 2022, 6, 2683–2685 CrossRef.
- H. Li, M. Xu, H. Long, J. Zheng, L. Zhang, S. Li, C. Guan, Y. Lai and Z. Zhang, Adv. Sci., 2022, 9, 2202082 CrossRef CAS PubMed.
- M. Li, C. Sun, Q. Ni, Z. Sun, Y. Liu, Y. Li, L. Li, H. Jin and Y. Zhao, Adv. Energy Mater., 2023, 13, 2203971 CrossRef CAS.
- X. Feng, D. Ren, X. He and M. Ouyang, Joule, 2020, 4, 743–770 CrossRef CAS.
- Y. Wang, N. Jiang, C. Yang, J. Liu, S. Sun, X. Wang, J. Yang and Y. Liu, J. Mater. Chem. A, 2024, 12, 5170–5180 RSC.
- Y. Zhou, G. Xu, J. Lin, Y. Zhang, G. Fang, J. Zhou, X. Cao and S. Liang, Adv. Mater., 2023, 35, 2304428 CrossRef CAS PubMed.
- X. Ma, X. Cao, Y. Zhou, S. Guo, X. Shi, G. Fang, A. Pan, B. Lu, J. Zhou and S. Liang, Nano Res., 2020, 13, 3330–3337 CrossRef CAS.
- Y. Wei, R. Yao, X. Liu, W. Chen, J. Qian, Y. Yin, D. Li and Y. Chen, Adv. Sci., 2023, 10, 2300271 CrossRef CAS.
- J. Zhao, Z. Wei, N. Chen, F. Meng, R. Tian, Y. Zeng and F. Du, Energy Storage Mater., 2024, 65, 103127 CrossRef.
- H. Liu, Y. Wang, X. Ding, Y. Wang, F. Wu and H. Gao, Sustainable Energy Fuels, 2024, 8, 1304–1313 RSC.
- H. Gao, J. Zeng, Z. Sun, X. Jiang and X. Wang, Mater. Today Energy, 2024, 42, 101551 CrossRef CAS.
- J. Liu, W. Huang, R. Liu, J. Lang, Y. Li, T. Liu, K. Amine and H. Li, Adv. Funct. Mater., 2024, 36, 2315437 CrossRef.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. Hu, Angew. Chem., Int. Ed., 2020, 59, 264–269 CrossRef CAS.
- H. Wang, X. Gao, S. Zhang, Y. Mei, L. Ni, J. Gao, H. Liu, N. Hong, B. Zhang, F. Zhu, W. Deng, G. Zou, H. Hou, X.-Y. Cao, H. Chen and X. Ji, ACS Nano, 2023, 17, 12530–12543 CrossRef CAS PubMed.
- F. Fu, X. Liu, X. Fu, H. Chen, L. Huang, J. Fan, J. Le, Q. Wang, W. Yang, Y. Ren, K. Amine, S.-G. Sun and G.-L. Xu, Nat. Commun., 2022, 13, 2826 CrossRef CAS PubMed.
- J. Song, F. Ning, Y. Zuo, A. Li, H. Wang, K. Zhang, T. Yang, Y. Yang, C. Gao, W. Xiao, Z. Jiang, T. Chen, G. Feng and D. Xia, Adv. Mater., 2023, 35, 2208726 CrossRef CAS.
- X. Wang, Q. Zhang, C. Zhao, H. Li, B. Zhang, G. Zeng, Y. Tang, Z. Huang, I. Hwang, H. Zhang, S. Zhou, Y. Qiu, Y. Xiao, J. Cabana, C.-J. Sun, K. Amine, Y. Sun, Q. Wang, G.-L. Xu, L. Gu, Y. Qiao and S.-G. Sun, Nat. Energy, 2024, 9, 184–196 CrossRef CAS.
- P. Zou, L. Yao, C. Wang, S. J. Lee, T. Li and H. L. Xin, Angew. Chem., 2023, 135, e202304628 CrossRef.
- B. Zhang, Y. Zhang, X. Wang, H. Liu, Y. Yan, S. Zhou, Y. Tang, G. Zeng, X. Wu, H.-G. Liao, Y. Qiu, H. Huang, L. Zheng, J. Xu, W. Yin, Z. Huang, Y. Xiao, Q. Xie, D.-L. Peng, C. Li, Y. Qiao and S.-G. Sun, J. Am. Chem. Soc., 2023, 145, 8700–8713 CrossRef CAS.
- W.-J. Shi, Y.-W. Yan, C. Chi, X.-T. Ma, D. Zhang, S.-D. Xu, L. Chen, X.-M. Wang and S.-B. Liu, J. Power Sources, 2019, 427, 129–137 CrossRef CAS.
- Y. J. Park, J. U. Choi, J. H. Jo, C. Jo, J. Kim and S. Myung, Adv. Funct. Mater., 2019, 29, 1901912 CrossRef.
- J. Wang, X. Lei, S. Guo, L. Gu, X. Wang, A. Yu and D. Su, Renewables, 2023, 1, 316–340 CrossRef.
- Z. Liu, R. Liu, S. Xu, J. Tian, J. Li, H. Li, T. Yu, S. Chu, A. M. D’Angelo, W. K. Pang, L. Zhang, S. Guo and H. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202405620 CrossRef CAS.
- C.-C. Lin, H.-Y. Liu, J.-W. Kang, C.-C. Yang, C.-H. Li, H.-Y. T. Chen, S.-C. Huang, C.-S. Ni, Y.-C. Chuang, B.-H. Chen, C.-K. Chang and H.-Y. Chen, Energy Storage Mater., 2022, 51, 159–171 CrossRef.
- X. Wang, Y. Zuo, Y. Qin, X. Zhu, S. Xu, Y. Guo, T. Yan, L. Zhang, Z. Gao, L. Yu, M. Liu, Y. Yin, Y. Cheng, P. Wang and Y. Guo, Adv. Mater., 2024, 2312300 CrossRef CAS PubMed.
- B. Wang, J. Ma, K. Wang, D. Wang, G. Xu, X. Wang, Z. Hu, C. Pao, J. Chen, L. Du, X. Du and G. Cui, Adv. Energy Mater., 2024, 2401090 CrossRef CAS.
- J. Wang, S. L. Dreyer, K. Wang, Z. Ding, T. Diemant, G. Karkera, Y. Ma, A. Sarkar, B. Zhou, M. V. Gorbunov, A. Omar, D. Mikhailova, V. Presser, M. Fichtner, H. Hahn, T. Brezesinski, B. Breitung and Q. Wang, Mater. Futures, 2022, 1, 035104 CrossRef CAS.
- J. Wang, Y. Cui, Q. Wang, K. Wang, X. Huang, D. Stenzel, A. Sarkar, R. Kruk, H. Hahn, T. Brezesinski and B. Breitung, Sci. Rep., 2020, 10, 18430 CrossRef CAS PubMed.
- K. Kubota and S. Komaba, J. Electrochem. Soc., 2015, 162, A2538–A2550 CrossRef CAS.
- S. Hu, A. S. Pillai, G. Liang, W. K. Pang, H. Wang, Q. Li and Z. Guo, Electrochem. Energy Rev., 2019, 2, 277–311 CrossRef CAS.
- C. Cheng, H. Hu, C. Yuan, X. Xia, J. Mao, K. Dai and L. Zhang, Energy Storage Mater., 2022, 52, 10–18 CrossRef.
- O. Arcelus and J. Carrasco, ACS Appl. Mater. Interfaces, 2019, 11, 12562–12569 CrossRef CAS.
- Y. Wang, Y. Wang, Y. Xing, C. Jiang, Y. Pang, H. Liu, F. Wu and H. Gao, J. Mater. Chem. A, 2023, 11, 19955–19964 RSC.
- A. Gao, S. Shen, T. Shang, Y. Shi, H. Zhang, W. Lin, S. Wang, T. Lin, P. Ji, Y. Wang, Y. Chen, B. Yu, X. Lu, W. Zhong, Q. Zhang and L. Gu, Sci. Adv., 2024, 10, eadp4906 CrossRef CAS PubMed.
- H. Ren, Y. Li, Q. Ni, Y. Bai, H. Zhao and C. Wu, Adv. Mater., 2022, 34, 2106171 CrossRef CAS PubMed.
- A. J. Perez, D. Batuk, M. Saubanère, G. Rousse, D. Foix, E. McCalla, E. J. Berg, R. Dugas, K. H. W. Van Den Bos, M.-L. Doublet, D. Gonbeau, A. M. Abakumov, G. Van Tendeloo and J.-M. Tarascon, Chem. Mater., 2016, 28, 8278–8288 CrossRef CAS.
- X. Luo, J. Patra, W. Chuang, T. X. Nguyen, J. Ting, J. Li, C. Pao and J. Chang, Adv. Sci., 2022, 9, 2201219 CrossRef CAS.
- T. X. Nguyen, C.-C. Tsai, J. Patra, O. Clemens, J.-K. Chang and J.-M. Ting, Chem. Eng. J., 2022, 430, 132658 CrossRef CAS.
- B. Xiao, G. Wu, T. Wang, Z. Wei, Y. Sui, B. Shen, J. Qi, F. Wei and J. Zheng, Nano Energy, 2022, 95, 106962 CrossRef CAS.
- C.-Y. Huang, C.-W. Huang, M.-C. Wu, J. Patra, T. Xuyen Nguyen, M.-T. Chang, O. Clemens, J.-M. Ting, J. Li, J.-K. Chang and W.-W. Wu, Chem. Eng. J., 2021, 420, 129838 CrossRef CAS.
- T.-Y. Chen, S.-Y. Wang, C.-H. Kuo, S.-C. Huang, M.-H. Lin, C.-H. Li, H.-Y. T. Chen, C.-C. Wang, Y.-F. Liao, C.-C. Lin, Y.-M. Chang, J.-W. Yeh, S.-J. Lin, T.-Y. Chen and H.-Y. Chen, J. Mater. Chem. A, 2020, 8, 21756–21770 RSC.
- K. Wang, W. Hua, X. Huang, D. Stenzel, J. Wang, Z. Ding, Y. Cui, Q. Wang, H. Ehrenberg, B. Breitung, C. Kübel and X. Mu, Nat. Commun., 2023, 14, 1487 CrossRef CAS PubMed.
- C. Wu, S. Dou and Y. Yu, Small, 2018, 14, 1703671 CrossRef.
- S. Yu, S. H. Lee, D. J. Lee, Y. Sung and T. Hyeon, Small, 2016, 12, 2146–2172 CrossRef CAS.
- S. Hou, L. Su, S. Wang, Y. Cui, J. Cao, H. Min, J. Bao, Y. Shen, Q. Zhang, Z. Sun, C. Zhu, J. Chen, Q. Zhang and F. Xu, Adv. Funct. Mater., 2024, 34, 2307923 CrossRef CAS.
- A. K. Das, P. Gami, H. N. Vasavan, S. Saxena, N. Dagar, S. Deswal, P. Kumar and S. Kumar, ACS Appl. Energy Mater., 2024, 7, 8301–8307 CrossRef CAS.
- D. Li, D. Yu, G. Zhang, A. Du, Z. Ye, Y. Jia, W. Hou, T. Xu, F. Li, S. Chi, Y. Zhu and C. Yang, Angew. Chem., 2024, e202419735 Search PubMed.
- X. Kong, R. Gu, Z. Jin, L. Zhang, C. Zhang, W. Xiang, C. Li, K. Zhu, Y. Xu, H. Huang, X. Liu, R. Peng and C. Wang, Nat. Commun., 2024, 15, 7247 CrossRef CAS PubMed.
- S. Yu, Y. Li, J. Luo, D. Chen, L. Yang, Y. Wei, D. Li, Y. Li and Y. Chen, J. Energy Chem., 2024, 96, 414–423 CrossRef CAS.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Adachi, J. Electrochem. Soc., 1990, 137, 1023–1027 CrossRef CAS.
- M. P. O’Callaghan, A. S. Powell, J. J. Titman, G. Z. Chen and E. J. Cussen, Chem. Mater., 2008, 20, 2360–2369 CrossRef.
- Y. Xiao, K. Jun, Y. Wang, L. J. Miara, Q. Tu and G. Ceder, Adv. Energy Mater., 2021, 11, 2101437 CrossRef CAS.
- A. Paolella, W. Zhu, G. Bertoni, S. Savoie, Z. Feng, H. Demers, V. Gariepy, G. Girard, E. Rivard, N. Delaporte, A. Guerfi, H. Lorrmann, C. George and K. Zaghib, ACS Appl. Energy Mater., 2020, 3, 3415–3424 CrossRef CAS.
- Y. Zeng, B. Ouyang, J. Liu, Y.-W. Byeon, Z. Cai, L. J. Miara, Y. Wang and G. Ceder, Science, 2022, 378, 1320–1324 CrossRef CAS PubMed.
- Q. Zhao, Z. Cao, X. Wang, H. Chen, Y. Shi, Z. Cheng, Y. Guo, B. Li, Y. Gong, Z. Du and S. Yang, J. Am. Chem. Soc., 2023, 145, 21242–21252 CrossRef CAS PubMed.
- M. Botros and J. Janek, Science, 2022, 378, 1273–1274 CrossRef CAS PubMed.
- L. Yang, C. Chen, S. Xiong, C. Zheng, P. Liu, Y. Ma, W. Xu, Y. Tang, S. P. Ong and H. Chen, JACS Au, 2021, 1, 98–107 CrossRef CAS.
- S. Li, J. Lin, M. Schaller, S. Indris, X. Zhang, T. Brezesinski, C. Nan, S. Wang and F. Strauss, Angew. Chem., Int. Ed., 2023, 62, e202314155 CrossRef CAS.
- J. Lin, M. Schaller, S. Indris, V. Baran, A. Gautam, J. Janek, A. Kondrakov, T. Brezesinski and F. Strauss, Angew. Chem., Int. Ed., 2024, e202404874 CAS.
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC.
- J. Lin, G. Cherkashinin, M. Schäfer, G. Melinte, S. Indris, A. Kondrakov, J. Janek, T. Brezesinski and F. Strauss, ACS Mater. Lett., 2022, 4, 2187–2194 CrossRef CAS.
- J. Lin, M. Schaller, G. Cherkashinin, S. Indris, J. Du, C. Ritter, A. Kondrakov, J. Janek, T. Brezesinski and F. Strauss, Small, 2024, 20, 2306832 CrossRef CAS PubMed.
- J. Du, J. Lin, R. Zhang, S. Wang, S. Indris, H. Ehrenberg, A. Kondrakov, T. Brezesinski and F. Strauss, Batteries Supercaps, 2024, e202400112 CrossRef CAS.
- F. Strauss, J. Lin, M. Duffiet, K. Wang, T. Zinkevich, A.-L. Hansen, S. Indris and T. Brezesinski, ACS Mater. Lett., 2022, 4, 418–423 CrossRef CAS.
- F. Strauss, M. Botros, B. Breitung and T. Brezesinski, J. Appl. Phys., 2024, 135, 120901 CrossRef CAS.
- Y. Dong, Z. Zhou, Y. Ma, H. Zhang, F. Meng, Y. Wu and Y. Ma, ACS Energy Lett., 2024, 9, 5096–5119 CrossRef CAS.
- Y. Wang, R. Xiao, Y.-S. Hu, M. Avdeev and L. Chen, Nat. Commun., 2015, 6, 6954 CrossRef CAS PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS.
- L. Yao, P. Zou, C. Wang, J. Jiang, L. Ma, S. Tan, K. A. Beyer, F. Xu, E. Hu and H. L. Xin, Adv. Energy Mater., 2022, 12, 2201989 CrossRef CAS.
- A. Joshi, S. Chakrabarty, S. H. Akella, A. Saha, A. Mukherjee, B. Schmerling, M. Ejgenberg, R. Sharma and M. Noked, Adv. Mater., 2023, 35, 2304440 CrossRef CAS PubMed.
- P. Liang, K. Qi, S. Chen, X. Ding, X. Wu, C. Wu and Y. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202318186 CrossRef CAS PubMed.
- R. Guo, Y. Yang, C. Zhao, F. Huo, J. Xue, J. He, B. Sun, Z. Sun, H. K. Liu and S. X. Dou, Adv. Funct. Mater., 2024, 34, 2313168 CrossRef CAS.
- A. Konarov, J. U. Choi, Z. Bakenov and S.-T. Myung, J. Mater. Chem. A, 2018, 6, 8558–8567 RSC.
- L. H. B. Nguyen, J. Olchowka, S. Belin, P. Sanz Camacho, M. Duttine, A. Iadecola, F. Fauth, D. Carlier, C. Masquelier and L. Croguennec, ACS Appl. Mater. Interfaces, 2019, 11, 38808–38818 CrossRef CAS.
- C. Triolo, W. Xu, B. Petrovičovà, N. Pinna and S. Santangelo, Adv. Funct. Mater., 2022, 32, 2202892 CrossRef CAS.
- Y. Hou, J. Jin, C. Huo, Y. Liu, S. Deng and J. Chen, Energy Storage Mater., 2023, 56, 87–95 CrossRef.
- P. Wang, H. Yao, X. Liu, J. Zhang, L. Gu, X. Yu, Y. Yin and Y. Guo, Adv. Mater., 2017, 29, 1700210 CrossRef.
- S. Yuan, L. Yu, G. Qian, Y. Xie, P. Guo, G. Cui, J. Ma, X. Ren, Z. Xu, S.-J. Lee, J.-S. Lee, Y. Liu, Y. Ren, L. Li, G. Tan and X. Liao, Nano Lett., 2023, 23, 1743–1751 CrossRef CAS PubMed.
- Q. Liu, W. Zheng, G. Liu, J. Hu, X. Zhang, N. Han, Z. Wang, J. Luo, J. Fransaer and Z. Lu, ACS Appl. Mater. Interfaces, 2023, 15, 9324–9330 CrossRef CAS.
- Q. Liu, W. Zheng, X. Su, X. Zhang, N. Han, Z. Wang, J. Luo, Z. Lu and J. Fransaer, Chem. Eng. J., 2023, 452, 139337 CrossRef CAS.
- A. Gomez-Martin, F. Reissig, L. Frankenstein, M. Heidbüchel, M. Winter, T. Placke and R. Schmuch, Adv. Energy Mater., 2022, 12, 2103045 CrossRef CAS.
- C. Ma, C. Lin, N. Li, Y. Chen, Y. Yang, L. Tan, Z. Wang, Q. Zhang and Y. Zhu, Small, 2024, 20, 2310184 CrossRef CAS.
- B. Ran, R. Cheng, Y. Zhong, X. Zhang, T. Zhao, Z. Yang, C. Yang, J. Zhang and C. Fu, Energy Storage Mater., 2024, 71, 103583 CrossRef.
- K. Du, Y. Liu, Y. Zhao, H. Li, H. Liu, C. Sun, M. Han, T. Ma and Y. Hu, Adv. Mater., 2024, 36, 2404172 CrossRef CAS.
- J. Xing, Y. Zhang, Y. Jin and Q. Jin, Nano Res., 2023, 16, 2486–2494 CrossRef CAS.
- L. Wu, Y. Zhang, Z. Wu, J. Tian, H. Wang, H. Zhao, S. Xu, L. Chen, X. Duan, D. Zhang, H. Guo, Y. You and Z. Zhu, Adv. Sci., 2023, 10, 2304067 CrossRef CAS.
- Y.-S. Kim, T.-H. Kim, H. Lee and H.-K. Song, Energy Environ. Sci., 2011, 4, 4038 RSC.
- Q. He, B. Yu, Z. Li and Y. Zhao, Energy Environ. Mater., 2019, 2, 264–279 CrossRef CAS.
- H. Hafiz, K. Suzuki, B. Barbiellini, N. Tsuji, N. Yabuuchi, K. Yamamoto, Y. Orikasa, Y. Uchimoto, Y. Sakurai, H. Sakurai, A. Bansil and V. Viswanathan, Nature, 2021, 594, 213–216 CrossRef CAS.
- F. Ding, H. Wang, Q. Zhang, L. Zheng, H. Guo, P. Yu, N. Zhang, Q. Guo, F. Xie, R. Dang, X. Rong, Y. Lu, R. Xiao, L. Chen and Y.-S. Hu, J. Am. Chem. Soc., 2023, 145, 13592–13602 CrossRef CAS PubMed.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- A. Amiri and R. Shahbazian-Yassar, J. Mater. Chem. A, 2021, 9, 782–823 RSC.
- X. Chang, M. Zeng, K. Liu and L. Fu, Adv. Mater., 2020, 32, 1907226 CrossRef CAS.
- X. Yang and Y. Zhang, Mater. Chem. Phys., 2012, 132, 233–238 CrossRef CAS.
- S. Guo and C. T. Liu, Prog. Nat. Sci.: Mater. Int., 2011, 21, 433–446 CrossRef.
- E. J. Pickering and N. G. Jones, Int. Mater. Rev., 2016, 61, 183–202 CrossRef CAS.
- Y. Zhang, Y. J. Zhou, J. P. Lin, G. L. Chen and P. K. Liaw, Adv. Eng. Mater., 2008, 10, 534–538 CrossRef CAS.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Intermetallics, 2015, 59, 75–80 CrossRef CAS.
- Y. F. Ye, Q. Wang, J. Lu, C. T. Liu and Y. Yang, Scr. Mater., 2015, 104, 53–55 CrossRef CAS.
- Y. Dang, Z. Xu, H. Yang, K. Tian, Z. Wang, R. Zheng, H. Sun, Y. Liu and D. Wang, Appl. Surf. Sci., 2023, 636, 157856 CrossRef CAS.
- X.-Y. Du, Y. Meng, H. Yuan and D. Xiao, Energy Storage Mater., 2023, 56, 132–140 CrossRef.
- F. Ding, C. Zhao, D. Xiao, X. Rong, H. Wang, Y. Li, Y. Yang, Y. Lu and Y.-S. Hu, J. Am. Chem. Soc., 2022, 144, 8286–8295 CrossRef CAS.
- C. Wang, L. Liu, S. Zhao, Y. Liu, Y. Yang, H. Yu, S. Lee, G.-H. Lee, Y.-M. Kang, R. Liu, F. Li and J. Chen, Nat. Commun., 2021, 12, 2256 CrossRef CAS PubMed.
- Q. Shen, Y. Liu, X. Zhao, J. Jin, Y. Wang, S. Li, P. Li, X. Qu and L. Jiao, Adv. Funct. Mater., 2021, 31, 2106923 CrossRef CAS.
- X. Cui, S. Wang, X. Ye, X. Fan, C. Gao, Y. Quan, S. Wen, X. Cai, J. Huang and S. Li, Energy Storage Mater., 2022, 45, 1153–1164 CrossRef.
- Q.-C. Wang, J.-K. Meng, X.-Y. Yue, Q.-Q. Qiu, Y. Song, X.-J. Wu, Z.-W. Fu, Y.-Y. Xia, Z. Shadike, J. Wu, X.-Q. Yang and Y.-N. Zhou, J. Am. Chem. Soc., 2019, 141, 840–848 CrossRef CAS.
- B. Peng, Y. Chen, F. Wang, Z. Sun, L. Zhao, X. Zhang, W. Wang and G. Zhang, Adv. Mater., 2022, 34, 2103210 CrossRef CAS.
- W. Bao, H. Shen, Y. Zhang, C. Qian, G. Zeng, K. Jing, D. Cui, J. Xia, H. Liu, C. Guo, F. Yu, K. Sun and J. Li, J. Mater. Chem. A, 2024, 12, 23179–23201 RSC.
- B. Xiao, G. Wu, T. Wang, Z. Wei, Z. Xie, Y. Sui, J. Qi, F. Wei, X. Zhang, L. Tang and J. Zheng, ACS Appl. Mater. Interfaces, 2023, 15, 2792–2803 CrossRef CAS.
- T. Ying, T. Yu, Y.-S. Shiah, C. Li, J. Li, Y. Qi and H. Hosono, J. Am. Chem. Soc., 2021, 143, 7042–7049 CrossRef CAS PubMed.
- G. Tallarita, R. Licheri, S. Garroni, R. Orrù and G. Cao, Scr. Mater., 2019, 158, 100–104 CrossRef CAS.
- N. Kashaev, V. Ventzke, N. Stepanov, D. Shaysultanov, V. Sanin and S. Zherebtsov, Intermetallics, 2018, 96, 63–71 CrossRef CAS.
- I. H. Gul, A. Z. Abbasi, F. Amin, M. Anis-ur-Rehman and A. Maqsood, J. Magn. Magn. Mater., 2007, 311, 494–499 CrossRef CAS.
- K. Petcharoen and A. Sirivat, Mater. Sci. Eng., B, 2012, 177, 421–427 CrossRef CAS.
- A. Zhou, W. Cheng, W. Wang, Q. Zhao, J. Xie, W. Zhang, H. Gao, L. Xue and J. Li, Adv. Energy Mater., 2021, 11, 2000943 CrossRef CAS.
- L. Fu, H. Liu, C. Li, Y. Wu, E. Rahm, R. Holze and H. Wu, Prog. Mater. Sci., 2005, 50, 881–928 CrossRef CAS.
- X. Wang and G. Yushin, Energy Environ. Sci., 2015, 8, 1889–1904 RSC.
- X. Su, F. Fu, Y. Yan, G. Zheng, T. Liang, Q. Zhang, X. Cheng, D. Yang, H. Chi, X. Tang, Q. Zhang and C. Uher, Nat. Commun., 2014, 5, 4908 CrossRef CAS.
- R.-Z. Zhang and M. J. Reece, J. Mater. Chem. A, 2019, 7, 22148–22162 RSC.
- Y. Bai, J. Li, H. Lu, J. Liu, C. Ma, B. Wang, X. Zhao and J. Deng, J. Adv. Ceram., 2023, 12, 1857–1871 CrossRef CAS.
- B. Wang, J. Tang, S. Jia, Z. Xing, S. Chen, Y. Deng, X. Meng and S. Tang, Adv. Funct. Mater., 2024, 2315836 CrossRef CAS.
- D. Lai, L. Ling, M. Su, Q. Kang, F. Gao and Q. Lu, Chem. Sci., 2023, 14, 1787–1796 RSC.
- P. K. Tyagi and S. K. Jha, Materialia, 2024, 33, 101967 CrossRef CAS.
- R. Ren, Y. Xiong, Z. Xu, J. Zhang, Y. Zhang, G. Zhu, K. Yin and S. Dong, Chem. Eng. J., 2024, 479, 147896 CrossRef CAS.
- Y. Huang, X. Zhang, L. Ji, L. Wang, B. B. Xu, M. W. Shahzad, Y. Tang, Y. Zhu, M. Yan, G. Sun and Y. Jiang, Energy Storage Mater., 2023, 58, 1–8 CrossRef.
- D. Shin, S. Chae, S. Park, B. Seo and W. Choi, NPG Asia Mater., 2023, 15, 54 CrossRef CAS.
- X. Liu, R. Tao, C. Li, J. Wang, S. Yao, C. Hong, H. Li, J. Geng and J. Liang, Chem. Eng. J., 2024, 484, 149791 CrossRef CAS.
- H. K. Beere, N. S. Reddy, P. Kulkarni, K. Samanta, H. Y. Jung and D. Ghosh, J. Energy Storage, 2024, 80, 110325 CrossRef.
- Q. Wang, L. Velasco, B. Breitung and V. Presser, Adv. Energy Mater., 2021, 11, 2102355 CrossRef CAS.
- T. Li, Y. Yao, Z. Huang, P. Xie, Z. Liu, M. Yang, J. Gao, K. Zeng, A. H. Brozena, G. Pastel, M. Jiao, Q. Dong, J. Dai, S. Li, H. Zong, M. Chi, J. Luo, Y. Mo, G. Wang, C. Wang, R. Shahbazian-Yassar and L. Hu, Nat. Catal., 2021, 4, 62–70 CrossRef CAS.
- H. Wu, Q. Lu, Y. Li, J. Wang, Y. Li, R. Jiang, J. Zhang, X. Zheng, X. Han, N. Zhao, J. Li, Y. Deng and W. Hu, Nano Lett., 2022, 22, 6492–6500 CrossRef CAS.
- Y. Feng, L. Yang, Z. Yan, D. Zuo, Z. Zhu, L. Zeng, Y. Zhu and J. Wan, Energy Storage Mater., 2023, 63, 103053 CrossRef.
- R.-F. Guo, Scr. Mater., 2021, 193, 103–107 CrossRef CAS.
- Y. Li, J. Chen, P. Cai and Z. Wen, J. Mater. Chem. A, 2018, 6, 4948–4954 RSC.
- M. Gao and J. Qiao, Metals, 2018, 8, 108 CrossRef.
- D. A. H. Hanaor, Computational Design of Battery Materials, Springer International Publishing, Cham, 2024, vol. 150 Search PubMed.
- J. M. Rickman, H. M. Chan, M. P. Harmer, J. A. Smeltzer, C. J. Marvel, A. Roy and G. Balasubramanian, Nat. Commun., 2019, 10, 2618 CrossRef CAS.
- M. Widom, J. Mater. Res., 2018, 33, 2881–2898 CrossRef CAS.
- R. Machaka, Comput. Mater. Sci., 2021, 188, 110244 CrossRef CAS.
- K. Kaufmann, D. Maryanovsky, W. M. Mellor, C. Zhu, A. S. Rosengarten, T. J. Harrington, C. Oses, C. Toher, S. Curtarolo and K. S. Vecchio, npj Comput. Mater., 2020, 6, 42 CrossRef.
- Z. Pei, J. Yin, J. A. Hawk, D. E. Alman and M. C. Gao, npj Comput. Mater., 2020, 6, 50 CrossRef.
- W. Huang, P. Martin and H. L. Zhuang, Acta Mater., 2019, 169, 225–236 CrossRef CAS.
- D. Dai, T. Xu, X. Wei, G. Ding, Y. Xu, J. Zhang and H. Zhang, Comput. Mater. Sci., 2020, 175, 109618 CrossRef CAS.
- Y. Zhang, C. Wen, C. Wang, S. Antonov, D. Xue, Y. Bai and Y. Su, Acta Mater., 2020, 185, 528–539 CrossRef CAS.
- M. Moorehead, K. Bertsch, M. Niezgoda, C. Parkin, M. Elbakhshwan, K. Sridharan, C. Zhang, D. Thoma and A. Couet, Mater. Des., 2020, 187, 108358 CrossRef CAS.
- A. Van De Walle and M. Asta, MRS Bull., 2019, 44, 252–256 CrossRef CAS.
- M. Kumbhakar, A. Khandelwal, S. K. Jha, M. V. Kante, P. Keßler, U. Lemmer, H. Hahn, J. Aghassi-Hagmann, A. Colsmann, B. Breitung, L. Velasco and S. Schweidler, Adv. Energy Mater., 2023, 13, 2204337 CrossRef CAS.
- C. Niu, A. J. Zaddach, A. A. Oni, X. Sang, J. W. Hurt, J. M. LeBeau, C. C. Koch and D. L. Irving, Appl. Phys. Lett., 2015, 106, 161906 CrossRef.
- T. Zuo, M. C. Gao, L. Ouyang, X. Yang, Y. Cheng, R. Feng, S. Chen, P. K. Liaw, J. A. Hawk and Y. Zhang, Acta Mater., 2017, 130, 10–18 CrossRef CAS.
- J. Schuetzke, S. Schweidler, F. R. Muenke, A. Orth, A. D. Khandelwal, B. Breitung, J. Aghassi-Hagmann and M. Reischl, Adv. Intell. Syst., 2024, 6, 2300501 CrossRef.
- Z. Rao, P.-Y. Tung, R. Xie, Y. Wei, H. Zhang, A. Ferrari, T. P. C. Klaver, F. Körmann, P. T. Sukumar, A. Kwiatkowski Da Silva, Y. Chen, Z. Li, D. Ponge, J. Neugebauer, O. Gutfleisch, S. Bauer and D. Raabe, Science, 2022, 378, 78–85 CrossRef CAS PubMed.
- K. Kaufmann and K. S. Vecchio, Acta Mater., 2020, 198, 178–222 CrossRef CAS.
- R. Feng, C. Zhang, M. C. Gao, Z. Pei, F. Zhang, Y. Chen, D. Ma, K. An, J. D. Poplawsky, L. Ouyang, Y. Ren, J. A. Hawk, M. Widom and P. K. Liaw, Nat. Commun., 2021, 12, 4329 CrossRef CAS.
- P. Wei, Y. Fan, W. Zhang, X. Li, S. Zhu, J. Wang, C. Wang, T. Zhang, C. Chen and S. Guan, J. Magn. Magn. Mater., 2021, 519, 167432 CrossRef CAS.
- J. Li, H. Yang, Q. Deng, W. Li, Q. Zhang, Z. Zhang, Y. Chu and C. Yang, Angew. Chem., 2024, 136, e202318042 CrossRef.
- X. Liu, Q. Han, Q. Ma, Y. Wang and C. Liu, Small, 2022, 18, 2203327 CrossRef CAS.
- L. Hong, X. Wu, L.-Y. Wang, M. Zhong, P. Zhang, L. Jiang, W. Huang, Y. Wang, K.-X. Wang and J.-S. Chen, ACS Nano, 2022, 16, 6906–6915 CrossRef CAS.
- P. Liang, J. Yi, X. Liu, K. Wu, Z. Wang, J. Cui, Y. Liu, Y. Wang, Y. Xia and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908528 CrossRef CAS.
- Y. Ma, J. H. Teo, F. Walther, Y. Ma, R. Zhang, A. Mazilkin, Y. Tang, D. Goonetilleke, J. Janek, M. Bianchini and T. Brezesinski, Adv. Funct. Mater., 2022, 32, 2111829 CrossRef CAS.
- D. Csík, G. Baranová, R. Džunda, D. Zalka, B. Breitung, M. Hagarová and K. Saksl, Coatings, 2023, 13, 1219 CrossRef.
- S. Sun and Y. Bai, Appl. Phys. Lett., 2024, 124, 171602 CrossRef CAS.
- K. Yuan, T. Tu, C. Shen, L. Zhou, J. Liu, J. Li, K. Xie and G. Zhang, J. Adv. Ceram., 2022, 11, 882–892 CrossRef CAS.
- S. Schweidler, T. Brezesinski and B. Breitung, Nat. Energy, 2024, 9, 240–241 CrossRef CAS.
- C. Zhao, C. Wang, X. Liu, I. Hwang, T. Li, X. Zhou, J. Diao, J. Deng, Y. Qin, Z. Yang, G. Wang, W. Xu, C. Sun, L. Wu, W. Cha, I. Robinson, R. Harder, Y. Jiang, T. Bicer, J.-T. Li, W. Lu, L. Li, Y. Liu, S.-G. Sun, G.-L. Xu and K. Amine, Nat. Energy, 2024, 9, 345–356 CrossRef CAS.
- X. Zuo, K. Chang, J. Zhao, Z. Xie, H. Tang, B. Li and Z. Chang, J. Mater. Chem. A, 2016, 4, 51–58 RSC.
- L. Fu, K. Tang, K. Song, P. A. Van Aken, Y. Yu and J. Maier, Nanoscale, 2014, 6, 1384–1389 RSC.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Adv. Energy Mater., 2020, 10, 2000927 CrossRef CAS.
- D. Xu, C. Chen, J. Xie, B. Zhang, L. Miao, J. Cai, Y. Huang and L. Zhang, Adv. Energy Mater., 2016, 6, 1501929 CrossRef.
- J. Yang, X. Zhou, D. Wu, X. Zhao and Z. Zhou, Adv. Mater., 2017, 29, 1604108 CrossRef.
- W. Shi, H. Dong, X. Feng, J. Yin, W. Sun, Y. Cheng and X. Xu, Electrochim. Acta, 2023, 464, 142945 CrossRef CAS.
- Y. Ma, Z. Zhou, T. Brezesinski, Y. Ma and Y. Wu, Research, 2024, 7, 0503 CrossRef PubMed.
- T. Gao, S.-Y. Zhou, P. Dai, J. Song, J. Liao, Y. Sun, C. Yu, S.-X. Zhao and G. Cao, ACS Appl. Energy Mater., 2024, 7, 6961–6971 CrossRef CAS.
- S. Zeng, Y. Zhu, J. Si, H. Liu, Y. Wang, Y. Hu and C. Chen, J. Power Sources, 2025, 626, 235726 CrossRef CAS.
- N. Zhang, X. Dong, Q. Yan, J. Wang, F. Jin, J. Liu, D. Wang, H. Liu, B. Wang and S. Dou, Energy Storage Mater., 2024, 72, 103734 CrossRef.
- G. Su, Y. Wang, J. Mu, Y. Ren, P. Yue, W. Ji, L. Liang, L. Hou, M. Chen and C. Yuan, Adv. Energy Mater., 2024, 2403282 CrossRef.
- Y. Kang, X. Lin, S. Tong, W. Zhu, Y. Wang, M. Jia and X. Zhang, Chem. Commun., 2024, 60, 2512–2515 RSC.
- H. Jia, Y. Li, U. Ali, B. Liu, Z. Jin, L. Li, Y. Chen, L. Zhang, T. Wang and C. Wang, Nano Energy, 2024, 122, 109348 CrossRef CAS.
- T. Cai, M. Cai, J. Mu, S. Zhao, H. Bi, W. Zhao, W. Dong and F. Huang, Nano-Micro Lett., 2024, 16, 10 CrossRef CAS PubMed.
- S. Ma, P. Zou and H. L. Xin, Mater. Today Energy, 2023, 38, 101446 CrossRef CAS.
- X. Ge, H. Li, J. Li, C. Guan, X. Wang, L. He, S. Li, Y. Lai and Z. Zhang, Small, 2023, 19, 2302609 CrossRef CAS PubMed.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- M. Wang, M. Zheng, J. Lu and Y. You, Joule, 2024, 8, 2467–2482 CrossRef CAS.
- X. Zhao, Z. Fu, X. Zhang, X. Wang, B. Li, D. Zhou and F. Kang, Energy Environ. Sci., 2024, 17, 2406–2430 RSC.
- Q. Wang, J. Wang, J. R. Heringa, X. Bai and M. Wagemaker, ACS Energy Lett., 2024, 9, 3796–3806 CrossRef PubMed.
- B. Xie, C. Zheng, H. Lang, M. Li, Q. Hu, X. Tan, Q. Zheng, Y. Huo, J. Zhao, J.-L. Yang, Z.-Y. Gu, D. Lin and X.-L. Wu, Energy Environ. Sci., 2024, 17, 7281–7293 RSC.
- Q. Wang, C. Zhao, J. Wang, Z. Yao, S. Wang, S. G. H. Kumar, S. Ganapathy, S. Eustace, X. Bai, B. Li and M. Wagemaker, Nat. Commun., 2023, 14, 440 CrossRef CAS PubMed.
- Q. Wang, C. Zhao, Z. Yao, J. Wang, F. Wu, S. G. H. Kumar, S. Ganapathy, S. Eustace, X. Bai, B. Li, J. Lu and M. Wagemaker, Adv. Mater., 2023, 35, 2210677 CrossRef CAS.
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Z. Yu, S. T. Oyakhire, W. Zhang, M. S. Kim, D. T. Boyle, J. Qin and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef CAS.
- K.-F. Ren, H. Liu, J.-X. Guo, X. Sun, F. Jiang, C. Guo, W. Bao, F. Yu, G. Kalimuldina, L. Kong, X.-B. Cheng and J. Li, ACS Energy Lett., 2024, 2960–2980 CrossRef CAS.
- Q. Wang, Z. Yao, C. Zhao, T. Verhallen, D. P. Tabor, M. Liu, F. Ooms, F. Kang, A. Aspuru-Guzik, Y.-S. Hu, M. Wagemaker and B. Li, Nat. Commun., 2020, 11, 4188 CrossRef PubMed.
- X. Dong, Y. Lin, P. Li, Y. Ma, J. Huang, D. Bin, Y. Wang, Y. Qi and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 5623–5627 CrossRef CAS PubMed.
- C. Yang, X. Liu, Y. Lin, L. Yin, J. Lu and Y. You, Adv. Mater., 2023, 35, 2301817 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |