
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomogenization offers access to quinoxalines in minutes: a solvent-free, catalyst-free protocol with a near-zero E-factor†
Abboy
Chatterjee
a,
Didreekshya
Mahanta
a,
Shamima
Hussain
b,
Amrita
Chatterjee
 *a and
Mainak
Banerjee
*a
*a and
Mainak
Banerjee
*a
aDepartment of Chemistry, Birla Institute of Technology and Science Pilani, KK Birla Goa Campus, NH 17B Bypass Road, Zuarinagar, Goa 403726, India. E-mail: mainak@goa.bits-pilani.ac.in; amrita@goa.bits-pilani.ac.in; Fax: +91-832-255-7031; Tel: +91-832-2580-347 Tel: +91-832-2580-320
bUGC-DAE CSR, Kalpakkam Node, Kokilamedu 603104, TN, India
First published on 17th October 2024
Abstract
A reagent-free, catalyst-free, sustainable methodology was developed for fast and effortless synthesis of quinoxalines by mixing and homogenizing the substrates in a mini cell homogenizer. The mechanochemical agitation between several aromatic (and heteromatic) 1,2-diamines and various 1,2-dicarbonyl compounds with stainless steel balls in simple polypropylene vials at 4000 rpm afforded the corresponding quinoxalines and pyrido[2,3-b]pyrazines via cyclocondensation within a few minutes mostly in quantitative yields. The use of an equimolar ratio of substrates and complete conversion ensured quick access to quinoxalines in a sufficiently pure form offering the additional advantages of a work-up free and purification-free approach. As eliminated water molecules in the process do not contribute to waste, the E-factor of the method is practically zero.
Introduction
Over the last few decades, “mechanochemistry” has gained substantial attention as an efficient technology for organic transformations with a motto to reduce environmental hazards in pharmaco- and agro-chemical processes and assist in attaining sustainable development goals.1 It is identified by the IUPAC as one of 10 world-changing technologies.2 While a large variety of reactions have been successfully carried out under mechanochemical conditions, often demonstrating unique features that are unprecedented in conventional processes, various milling techniques and conditions have also been exploited.3 This includes mechanochemistry under cryogenic conditions,4 photomechanochemistry using LED-wrapped transparent jars,5 electromechanochemistry by using piezoelectric materials,6 thermo-mechanochemistry using heat-guns7 or even continuous-flow mechanochemistry using twin-screw extruders.8 In most cases, a mixer mill or a planetary ball mill is used for laboratory-scale demonstrations, which are limited to a few reactions at a time. There is hardly any attempt at combinatorial chemistry under mechanochemical agitation.9Quinoxalines are an important class of nitrogen-containing benzo-fused heterocycles due to their ubiquitous use in pharmaceuticals and advanced functional materials.10 Riboflavin (vitamin B2) and several antibiotics such as echinomycin, leromycin and actinomycin possess a quinoxaline scaffold (Fig. 1).10–12 Structurally diversified quinoxalines exhibit remarkable biological activities, including anti-cancer, anti-HIV, anti-dengue, antituberculosis, anti-malarial, antioxidant, anti-tumour, antidiabetic, antiulcer, and antidepressant activities, and possess enzyme inhibitory and receptor antagonist properties.10,13 In addition, quinoxaline-derived functional molecules and materials have been employed in diverse areas with commercial implications, such as electroluminescent materials, dyes, organic semiconductors, and OLEDs.14 With an additional N-atom, pyrido[2,3-b]pyrazines possess vast bioactivities as well as light-emitting properties.15,16
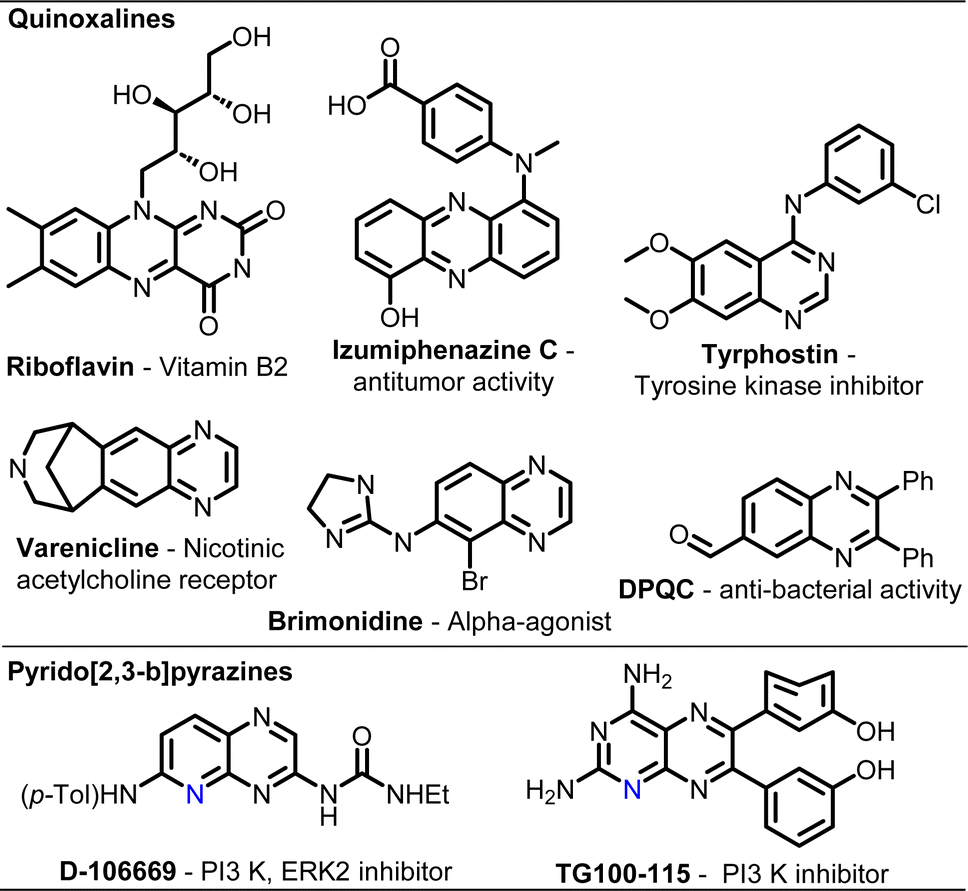 | ||
| Fig. 1 Selected bioactive quinoxalines and pyrido[2,3-a]pyrazines. | ||
The vast use of quinoxaline derivatives instigates tremendous research interest among chemists to develop efficient and sustainable synthetic methods for this scaffold.10a,17 The conventional synthetic strategies, developed by Körner18 and Hinsberg,19 involve the condensation of o-phenylenediamine with 1,2-dicarbonyl compounds or 1,2-ketoesters or oxalic acid derivatives. Several contemporary methods involving the oxidation of α-hydroxy carbonyl and condensation with o-phenylenediamine,20in situ reduction of nitroarenes followed by condensation with carbonyl,21 tandem redox reactions,22 condensation with a-halo carbonyl,23 or coupling of arynes with o-phenylenediamine are also reported.24 However, a vast literature is available for quinoxalines based on heterogeneous catalysis exploiting nanoparticles,25 silica nanomaterials,26 carbon nanomaterials,27 and solid sulfuric acid as the catalysts28 for the cyclocondensation of o-phenylenediamine with 1,2-dicarbonyl compounds, usually carried out in a benign medium (e.g. water or EtOH). The other notable greener methods include quinoxaline synthesis in micellar media,29 ionic liquids,30 deep eutectic solvents,31etc. The synthesis of quinoxalines adapting green technologies such as microwaves and sonochemistry is also reported.32,33 In all cases, even if the reaction media or the technology is benign, the requirement of catalysts or chromatographic purification steps adds to costs and environmental hazards.
Mechanochemistry has been frequently employed in synthesizing quinoxaline derivatives by grinding or milling the reaction mass, often in the presence of a catalyst and/or milling auxiliaries (Fig. 2).34,35 For example, Li et al., Singh and co-workers, and our group developed hand-grinding processes for the cyclocondensation of o-phenylenediamine and 1,2-dicarbonyl compounds in basic Al2O3, SiO2, and EtOH (for liquid-assisted grinding) respectively, as grinding auxiliaries under solvent-less conditions, but the products were extracted and purified using organic solvents.34b,d,e By a similar strategy, Carlier's group used a vibrating ball-mill for the synthesis of quinoxalines, which took several hours for complete conversion even after operating at high-frequency (50 Hz).35d In a recent study, Wu et al. used a costly metal catalyst, lanthanum dodecyl sulfate for the same transformation in a ball-mill in the presence of NaCl as the milling auxiliary.35a We envisaged that this prospective high atom-efficient strategy for quinoxaline could be more benign and cost-effective if it is achieved just by frictional force without a catalyst, solvent, or any other auxiliary to eventually achieve a near zero E-factor. We also envisioned that a multi-station cell homogenizer could offer the scope of combinatorial chemistry and parallel synthesis for organic transformations. With the intention of exploring new equipment for mechanochemistry, we, for the first time, explored the potential of homogenization, as an alternative to ball-milling, for highly efficient synthesis of quinoxalines and pyrido[2,3-b]pyrazines (Fig. 2).
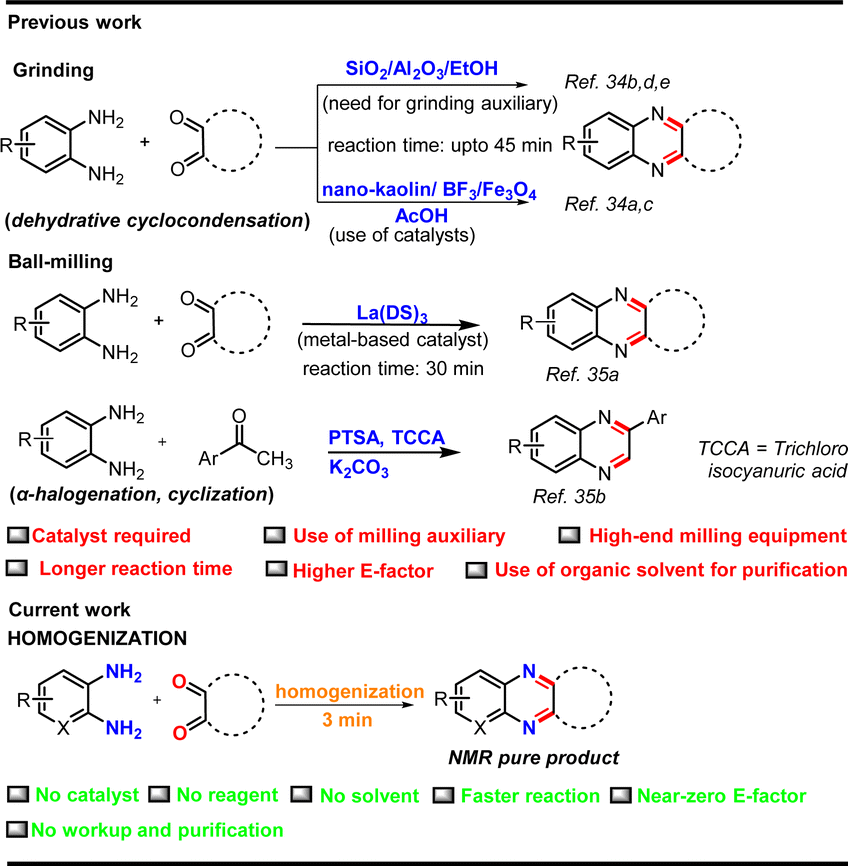 | ||
| Fig. 2 Available synthetic routes to quinoxalines. | ||
Results and discussion
Optimization of the reaction conditions
Our initial investigations were centered on optimizing the cyclocondensation under homogenization conditions using a model reaction between equimolar quantities of o-phenylenediamine (1a) and benzil (2a). The reactions were conducted using an iRupt Jr. mini-cell homogenizer in a 2 mL polypropylene vial containing 0.25 g of 2 mm (8 no.) stainless steel (SS) balls, and the mixture was homogenized at 2800 rpm. Thin-layer chromatography (TLC) analysis after 3 min indicated incomplete conversion, and further homogenization up to 15 min ensured complete conversion, which afforded quinoxaline 3a in 96% isolated yield after chromatographic purification (entry 1, Table 1). Thus, it may be noted that homogenization by SS balls, even in polypropylene containers, can offer enough frictional force to carry out chemical transformations. In order to expedite the reaction, small amounts of EtOH, PEG-400, and water were added to the reaction mixture as the liquid-assisted grinding (LAG) agent, and homogenization was conducted for several minutes (entries 2–4, Table 1). For EtOH and PEG-400 complete conversion was achieved after 9 and 6 min, respectively (entries 2 and 3, Table 1). In contrast, water as the LAG agent produced a reduced yield of 3a, presumably because of partial hydrolytic decomposition of the intermediate imine bonds (entry 4, Table 1). Altering the stoichiometric ratio of benzil (2a) did not enhance the reaction rate (entries 5 and 6, Table 1). However, in all these cases, column chromatography was required to obtain the pure product (3a). Considering that the reaction under neat conditions produced better yield, further optimization studies were conducted under neat conditions by varying the number of balls and the homogenization frequency to check its impact on yield and reaction kinetics (entries 7–9, Table 1). Quantitative conversion to 3a was observed when 1a and 2a were homogenized in a 2 mL vial with 1 g of SS balls at a frequency of 4000 rpm for 3 min (entry 9, Table 1). For a finer investigation of reaction kinetics, TLCs were checked after each minute, and it was observed that the reaction was actually completed in 1 min only (entry 10, Table 1). After air-drying the crude product, 1H and 13C NMR spectra were collected without purification, and clean spectra of 3a revealed that the crude solid is sufficiently pure, indicating no need for additional workup and purification steps. In subsequent studies, the ball size was changed to 5 mm (SS balls), the ball material was changed to zirconia and glass, and the mixture was homogenized at 4000 rpm (entries 11–13, Table 1). Although yields were comparable, the reactions took more time to complete. To understand how fast homogenization facilitates the conversion, time-scale infrared (IR) spectroscopy was conducted on the model reaction between o-phenylenediamine (1a) and benzil (2a).36 The samples from the reaction mixture were collected after (a) physical mixing and homogenization after (b) 15 s, (c) 30 s, (d) 1 min, and (e) 3 min, and solid-state IR data were collected from the reaction mixture (Fig. S2, ESI†). Furthermore, IR data were compared for pure starting materials and the product (3a). The study showed the steady disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak of benzil at 1662 cm−1 and the formation of new peaks at 770 cm−1 and 1346 cm−1, which are two prominent peaks of 3a, just after 30 s of homogenization (Fig. S2, ESI†). The time-scale IR study revealed that the homogenization quickly activates a cyclocondensation process, and quinoxaline synthesis follows fast reaction kinetics. For a comparative analysis of the effect of homogenization on reaction kinetics, several reactions were attempted under neat mixing and in the solution phase with a model reaction for the formation of 3a (Table S1, ESI†). Simple physical mixing by using a stirring bar under catalyst-free and solvent-free conditions hardly progressed even after 2 h (entry 1, Table S1, ESI†). For the catalyst-free solution phase, the reactions mostly undergo partial conversion to product 3a, and in all these cases, work-up and chromatographic purification were required for isolation purposes. However, the generality of the solution phase catalyst-free quinoxaline syntheses was not tested. Nonetheless, homogenization was found to be a much faster and more efficient method, and 1 g SS balls and 4000 rpm were set as the optimum conditions for this cyclocondensation for the validation of the method with the synthesis of a series of quinoxalines.
O stretching peak of benzil at 1662 cm−1 and the formation of new peaks at 770 cm−1 and 1346 cm−1, which are two prominent peaks of 3a, just after 30 s of homogenization (Fig. S2, ESI†). The time-scale IR study revealed that the homogenization quickly activates a cyclocondensation process, and quinoxaline synthesis follows fast reaction kinetics. For a comparative analysis of the effect of homogenization on reaction kinetics, several reactions were attempted under neat mixing and in the solution phase with a model reaction for the formation of 3a (Table S1, ESI†). Simple physical mixing by using a stirring bar under catalyst-free and solvent-free conditions hardly progressed even after 2 h (entry 1, Table S1, ESI†). For the catalyst-free solution phase, the reactions mostly undergo partial conversion to product 3a, and in all these cases, work-up and chromatographic purification were required for isolation purposes. However, the generality of the solution phase catalyst-free quinoxaline syntheses was not tested. Nonetheless, homogenization was found to be a much faster and more efficient method, and 1 g SS balls and 4000 rpm were set as the optimum conditions for this cyclocondensation for the validation of the method with the synthesis of a series of quinoxalines.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | o-Phenylene diamine (equiv.) | Benzil (equiv.) | LAG/milling auxiliary | Ball amount (no. of balls) | Ball size (ball type) | RPM | Time (min) | Yieldb (%) |
| a The reactions were carried out in an iRupt Jr. indigenous mini-cell homogenizer instrument with 32 2 mm SS balls in a 2 mL polypropylene tube to optimize the reaction conditions. o-Phenylenediamine (1) and benzil (2) were considered as the model substrates. b All yields reported here are isolated yields. c No purification was required. | ||||||||
| 1 | 1 | 1 | Neat | 0.25 g (8) | 2 mm (SS) | 2800 | 15 | 96 |
| 2 | 1 | 1 | EtOH | 0.25 g (8) | 2 mm (SS) | 2800 | 9 | 98 |
| 3 | 1 | 1 | PEG 400 | 0.25 g (8) | 2 mm (SS) | 2800 | 6 | 97 |
| 4 | 1 | 1 | H2O | 0.25 g (8) | 2 mm (SS) | 2800 | 9 | 71 |
| 5 | 1 | 1.05 | Neat | 0.25 g (32) | 2 mm (SS) | 2800 | 3 | 96 |
| 6 | 1 | 1.1 | Neat | 0.25 g (32) | 2 mm (SS) | 2800 | 3 | 94 |
| 7 | 1 | 1 | Neat | 0.5 g (16) | 2 mm (SS) | 2800 | 6 | 98 |
| 8 | 1 | 1 | Neat | 1 g (32) | 2 mm (SS) | 2800 | 3 | 98 |
| 9 | 1 | 1 | Neat | 1 g (32) | 2 mm (SS) | 4000 | 3 | 99 |
| 10 | 1 | 1 | Neat | 1 g (32) | 2 mm (SS) | 4000 | 1 | 99 |
| 11 | 1 | 1 | Neat | 1 g (2) | 5 mm (SS) | 4000 | 6 | 99 |
| 12 | 1 | 1 | Neat | 1 g (86) | 1 mm (zirconia) | 4000 | 6 | 97 |
| 13 | 1 | 1 | Neat | 1 g (116) | 2 mm (glass) | 4000 | 3 | 98 |
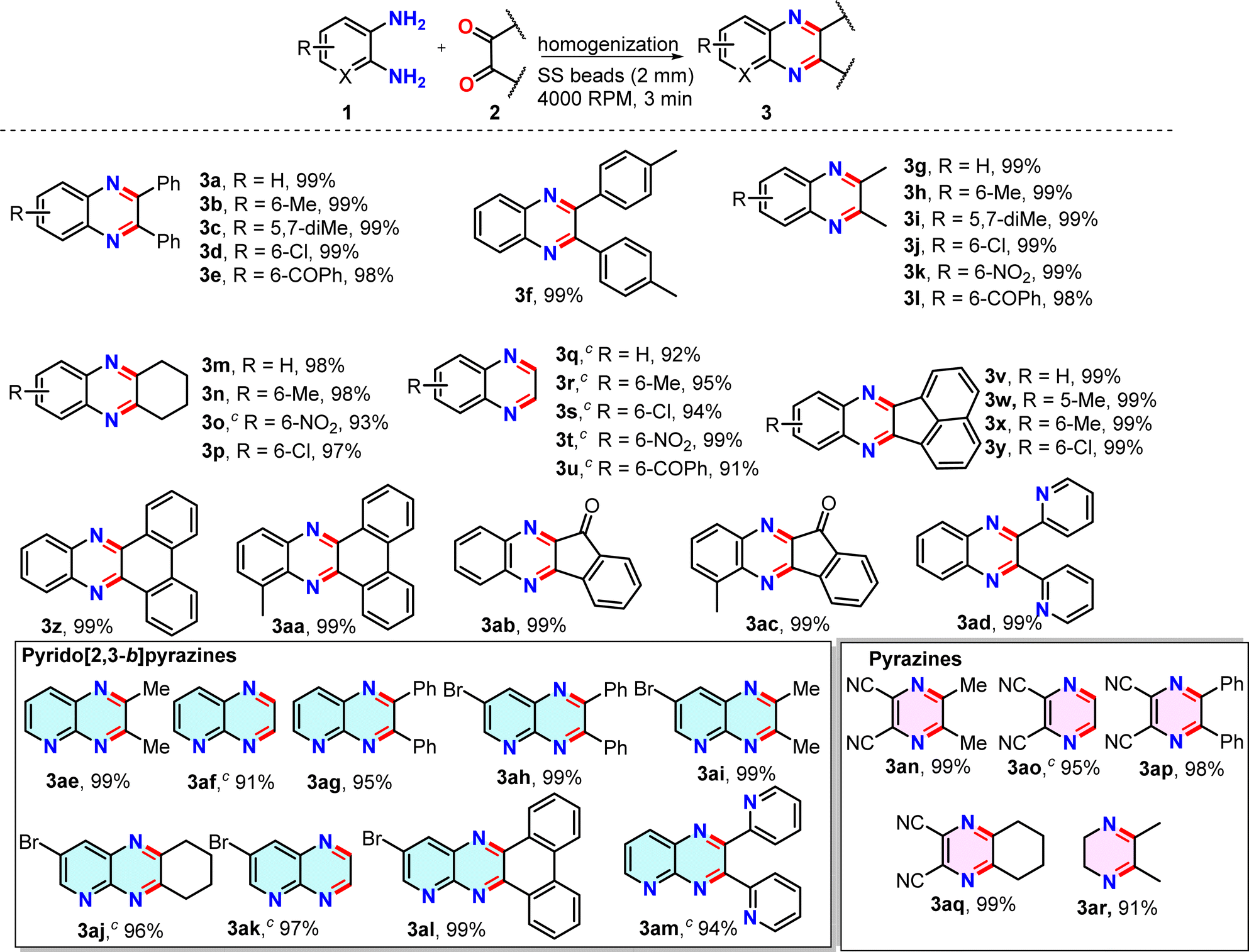
Substrate scope for the synthesis of quinoxalines (3)
To demonstrate the broad applicability of this method, a series of o-diaminoarenes (1) and 1,2-dicarbonyl compounds (2) were homogenized in a 2 mL vial containing 1 g of SS balls at 4000 rpm, setting the timer to 60 seconds for 3 min (Table 2). The progress of the reactions was monitored via TLC after each minute and the complete conversion was generally observed within a minute. In a few cases, if the reactions did not progress much after 1 min and a pasty mass was obtained upon homogenization, a few drops of EtOH were added, as indicated in the Table 2 footnote, and the reactions were further homogenized for 2 min to achieve complete conversion. After transferring the crude product from the vial, the SS balls were easily separated by using a magnet retriever, thus indicating a simple reaction set-up, fast reaction, and easy isolation of the product (Fig. S2, ESI†). The crude mixture, after air-drying, was directly subjected to NMR and ESI-MS analysis unless otherwise noted. The 1H and 13C NMR data of all the synthesized products were consistent with those of previously reported quinoxaline derivatives.34,35 Screening various o-diaminoarenes (1) having both electron-donating and electron-withdrawing groups in the ring with benzil (2a) revealed that the electronic effects of substituents in the ring did not significantly impact the reaction rate and the yield, as reactions afforded products in 99% yields within 3 min (entries 3a–3e, Table 2). Although the conversion is quantitative, a 1% loss in yield is actually because of a very small amount of powdery product sticking to the small SS balls, and we decided not to use any organic solvent for the recovery. Similarly, 4,4′-dimethyl benzil (2b) took part in the reaction (entry 3f, Table 2). When benzil was replaced by an aliphatic dicarbonyl, butanedione (2c), the series of reactions again afforded quinoxalines in quantitative yields (entries 3g–3l, Table 2). The reactions with 1,2-cyclohexanedione (2d) and glyoxal (40% in water, 2e) proceeded smoothly to completion producing the corresponding quinoxalines in excellent yields and showing no apparent electronic effect (entries 3m–3u, Table 2); however, a short-bed column purification was preferred to get glyoxal derived quinoxalines in higher purity as a small amount of polar impurities were observed in TLC. Subsequently, reactions of polyaromatic dicarbonyls such as acenaphthaquinone (2f) and phenanthraquinone (2g) afforded near quantitative yields of products (entries 3v–3aa, Table 2). Next, a couple of indeno[1,2-b]quinoxalinones were synthesized by condensing ninhydrin (2h) with o-diaminoarenes in 99% yields (entries 3ab,3ac, Table 2). We also explored the scope of the method with heteroaryl diketone using 2,2′-pyridil and obtained a nearly quantitative yield of the product (entry 3ad, Table 2). To further demonstrate the broad utility of our method, we explored the synthesis of a series of pyrido[2,3-b]pyrazines by taking 2,3-diaminopyridines and condensing them with several aromatic and aliphatic dicarbonyl compounds under the same homogenization conditions (entries 3ae–3am, Table 2). Again, the reactions afforded sufficiently pure pyrido[2,3-b]pyrazines in excellent yields. Next, a short series of pyrazine derivatives were also synthesized by the condensation of 2,3-diaminomaleonitrile and dicarbonyls (entries 3an–3aq, Table 2). In all previous cases, the homogenization was conducted between two solids or for a solid–liquid combination. We sought to verify whether two liquids, under reduced frictional force, can undergo cyclocondensation during homogenization at 4000 rpm. We were pleased to observe that the homogenization of ethylenediamine and 2,3-butanedione afforded the dihydropyrazine derivative in a good yield in 3 min (entry 3ar, Table 2).| a In each case, 0.5 mmol of 1 and 0.5 mmol of 2 were homogenized in a 2 mL polypropylene tube with 32 2 mm SS balls in an iRupt Jr. indigenous mini-cell homogenizer instrument at 4000 rpm for 3 min. b All products with 99% yield were obtained by simple removal from the tube with no further purification. c The products (3) purified by flash chromatography and isolated yields are reported. |
|---|
|
Scale-up synthesis
The scalability of the homogenization technique was investigated by using 2.5 mmols each of 1,2-dicarbonyl compounds and o-diaminoarenes for three reactions at a time (viz. synthesis of 3g, 3t, and 3ac) using 1 g of SS balls in 2 mL polypropylene tubes and homogenized at 4000 rpm (Table 3). This time, the compounds occupied about half of the total volume of the reaction tubes. Interestingly, the complete conversion was again observed in just 3 min indicating that enough space is still available for proper homogenization. At the same time, this also demonstrates a semi high-throughput synthesis to access three quinoxaline derivatives at a time. The yield in all cases was identical as compared to small-scale reactions (on the 0.5 mmol scale) needing no purification. Furthermore, scale-up in the mini-homogenizer was not feasible due to the limitation of jar size. However, this indicates that a higher version of the cell homogenizer with multiple stations and bigger space may certainly be used for gram-scale synthesis as well as high throughput screening purposes.
Comparative analysis and sustainability metrics
Quinoxaline synthesis is one of the oldest reactions, reported by Körner and Hinsberg in 1884,18,19 and being an important bioactive scaffold, it has witnessed a recent upsurge in the development of various synthetic methods, including the use of various homogeneous and heterogeneous catalysts, green technologies such as microwaves, water as media, etc. A complete comparative analysis of a few thousand available literature studies seems out of scope. Therefore, the present method was compared with selected conventional solution phase methods and green protocols (Table S2, ESI†). For the cases of proclaimed “green” protocols, the use of catalysts during the reaction or the organic solvents for chromatographic purification or recrystallization adds to costs and environmental hazards, and increase E-factors (Table S2, ESI†). The current protocol is mostly devoid of work-up and purification steps and can achieve a near-zero E-factor. For example, the estimated E-factor and ecoscale score for the formation of 3a are 0.01 and 97.5, respectively (entry 9, Table S2†). A quick comparison of the current methods with other mechanochemical methods is also conducted (Table S3, ESI†). Once again, the current method is way superior to other available mechanochemical methods for quinoxaline synthesis via cyclocondensation in terms of simplicity of the reaction set-up, reaction completion time, and various other greener attributes (Table S3, ESI†). The cost calculation revealed that the manufacturing cost for 1 mol of 3a is USD 48.7, which is similar to the cost of starting substrates only (USD 49.1) without taking the equipment and manpower costs into account.Experimental
The general information, instrumentation, synthetic procedures, the spectral characterization of all quinoxaline derivatives and copies of 1H and 13C NMR are available in the ESI.†Conclusions
To summarize, for the first time, we have demonstrated that a mini cell-homogenizer can be used for chemical transformations, taking a cyclocondensation reaction as an example, and developed a reagent-free, catalyst-free eco-friendly protocol for fast access to several quinoxaline derivatives. The homogenization of equimolar mixtures of a variety of 1,2-dicarbonyl compounds and o-diaminoarenes in polypropylene vials in the presence of 2 mm stainless steel balls at 4000 rpm resulted in the formation of the corresponding quinoxalines in excellent to quantitative yields within just 3 min. This method was further extended to the synthesis of a short series of pyrido[2,3-b]pyrazines and pyrazines using 1,2-diaminopyridines and diaminomaleonitrile. The steric and electronic effects were not significant, as in most of the cases, the full conversion took place in just 3 min under the same mechanochemical conditions. Notably, the simple, high-yielding ambient-temperature protocol is devoid of workup and purification steps, leading to a near-zero E-factor and ecoscale score in the high 90 s, which are excellent attributes for sustainability matrices. We also investigated the potential for scale-up and demonstrated the semi high-throughput enabling three reactions at a time with excellent outcomes. This work demonstrates the scope of exploring the vast potential of a bead homogenizer as a new tool for mechanochemical transformations. As 24-station homogenizers are available, combinatorial chemistry and high-throughput screening can potentially be performed.Data availability
This is to declare that the data of this work will be available on request from readers.Author contributions
MB conceived the project. Abboy and DM performed the experiments and processed the data. Writing and editing were conducted by Abboy and MB, and reviewed by AC. MB supervised the overall project. AC and SH offered time-to-time scientific input. All authors have read and approved the final version of the manuscript.Conflicts of interest
The authors declare no personal, financial, or organizational conflict of interest.Acknowledgements
M. B. is grateful to the UGC-DAE, CRS/2021-22/04/631 project for the financial support. Abboy is indebted to BITS Pilani, KK Birla Goa campus for JRFship. We acknowledge central sophisticated instrumentation facilities (CSIFs) of BITS Pilani, KK Birla Goa campus for NMR and ESI-MS analysis.Notes and references
- (a) B. Ranu and A. Stolle, Ball Milling towards Green Synthesis: Applications, Projects, Challenges, Royal Society of Chemistry, Cambridge, UK, 2015, 10.1039/9781782621980; (b) E. Colacino, G. Ennas, I. Halász, A. Porcheddu and A. Scano, Mechanochemistry: A Practical Introduction from Soft to Hard Materials, De Gruyter, Berlin, 2021 Search PubMed; (c) J.-L. Do and T. Friščić, ACS Cent. Sci., 2016, 3, 13–19, DOI:10.1021/acscentsci.6b00277; (d) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2019, 59, 1018–1029, DOI:10.1002/anie.201906755; (e) D. Tan and T. Friščić, Eur. J. Org Chem., 2017, 2018, 18–33, DOI:10.1002/ejoc.201700961; (f) E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339, DOI:10.1016/j.trechm.2021.02.008.
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17, DOI:10.1515/ci-2019-0203.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2022, 7, 51–65, DOI:10.1038/s41570-022-00442-1.
- (a) J. Andersen and J. Mack, Angew. Chem., Int. Ed., 2018, 130, 13246–13249, DOI:10.1002/anie.201805263; (b) S. B. Liang, D. P. Hu, C. Zhu and A. B. Yu, Chem. Eng. Technol., 2002, 25, 401–405, DOI:10.1002/1521-4125(200204)25:4%3C401::AID-CEAT401%3E3.0.CO;2-S.
- D. M. Baier, C. Spula, S. Fanenstich, S. Grätz and L. Borchardt, Angew. Chem., Int. Ed., 2023, 62, e202218719, DOI:10.1002/anie.202218719.
- (a) C. Schumacher, J. G. Hernández and C. Bolm, Angew. Chem., Int. Ed., 2020, 59, 16357–16360, DOI:10.1002/anie.202003565; (b) Y. Pang, J. W. Lee, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 22570–22576, DOI:10.1002/anie.202009844.
- K. Kubota, T. Endo and H. Ito, Chem. Sci., 2024, 15, 3365–3371, 10.1039/D3SC05796H.
- Q. Cao, D. E. Crawford, C. Shi and S. L. James, Angew. Chem., Int. Ed., 2020, 59, 4478–4483, DOI:10.1002/anie.201913625.
- K. Martina, L. Rotolo, A. Porcheddu, F. Delogu, S. R. Bysouth, G. Cravotto and E. Colacino, Chem. Commun., 2018, 54, 551–554, 10.1039/C7CC07758K.
- (a) G. Chawla, O. Gupta and T. Pradhan, ChemistrySelect, 2023, 8, e202301401, DOI:10.1002/slct.202301401; (b) H. Khatoon and E. Abdulmalek, Molecules, 2021, 26, 1055, DOI:10.3390/molecules26041055; (c) J. A. Pereira, A. M. Pessoa, M. N. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672, DOI:10.1016/j.ejmech.2014.06.058.
- (a) J. I. Ahamed, G. R. Ramkumaar, P. Kamalarajan, K. Narendran, M. F. Valan, T. Sundareswaran, T. A. Sundaravadivel, B. Venkatadri and S. Bharathi, J. Mol. Struct., 2022, 1248, 131418, DOI:10.1016/j.molstruc.2021.131418; (b) H. F. VanBrocklin, J. K. Lim, S. L. Coffing, D. L. Hom, K. Negash, M. Y. Ono, J. L. Gilmore, I. Bryant and D. J. Riese, J. Med. Chem., 2005, 48, 7445–7456, DOI:10.1021/jm050607w.
- (a) T. Fortier, A. Belouin, F. Vuillermet, J. Bourret and G. Pelletier, Eur. J. Org Chem., 2024, 27, e202400297, DOI:10.1002/ejoc.202400297; (b) M. S. Palanki, E. Dneprovskaia, J. Doukas, R. M. Fine, J. Hood, X. Kang, D. Lohse, M. Martin, G. Noronha, R. M. Soll, W. Wrasidlo, S. Yee and H. Zhu, J. Med. Chem., 2007, 50, 4279–4294, DOI:10.1021/jm051056c.
- (a) I. A. Bashir, A. O. Oyewale, S. Lee, A. B. Aliyu, H. Ibrahim and H. Y. Etukudoh, J. Mol. Struct., 2024, 1315, 138947, DOI:10.1016/j.molstruc.2024.138947; (b) Y. M. Syam, M. M. Anwar, S. S. Abd El-Karim, S. A. Elseginy, B. M. Essa and T. M. Sakr, RSC Adv., 2021, 11, 36989–37010, 10.1039/d1ra06799k; (c) M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147, DOI:10.1016/j.ejmech.2018.11.059; (d) T. Kaushal, G. Srivastava, A. Sharma and A. Singh Negi, Bioorg. Med. Chem., 2019, 27, 16–35, DOI:10.1016/j.bmc.2018.11.021; (e) M. Imanishi, M. Sonoda, H. Miyazato, K. Sugimoto, M. Akagawa and S. Tanimori, ACS Omega, 2017, 2, 1875–1885, DOI:10.1021/acsomega.7b00124.
- (a) D. Gedefaw, M. Prosa, M. Bolognesi, M. Seri and M. R. Andersson, Adv. Energy Mater., 2017, 7, 1700575, DOI:10.1002/aenm.201700575; (b) J. Yuan, J. Ouyang, V. Cimrová, M. Leclerc, A. Najari and Y. Zou, J. Mater. Chem. C, 2017, 5, 1858–1879, 10.1039/C6TC05381E; (c) S. Achelle, C. Baudequin and N. Plé, Dyes Pigm., 2013, 98, 575–600, DOI:10.1016/j.dyepig.2013.03.030; (d) Y. Zhang, S. Li, J. Xu, Y. Dai and J. Qiao, Adv. Opt. Mater., 2024, 12, 2400902, DOI:10.1002/adom.202400902.
- (a) D. Choudhary, S. Garg, M. Kaur, H. S. Sohal, D. S. Malhi, L. Kaur, M. Verma, A. Sharma and V. Mutreja, Polycyclic Aromat. Compd., 2022, 43, 4512–4578, DOI:10.1080/10406638.2022.2092873; (b) L. Niaz, F. A. Saddique, S. Aslam, M. Ahmad and N. ul Mohsin, Synth. Commun., 2020, 50, 2755–2786, DOI:10.1080/00397911.2020.1786123; (c) L. Kékesi, A. Sipos, G. Németh, J. Pató, N. Breza, F. Baska, L. Őrfi and G. Kéri, Bioorg. Med. Chem. Lett., 2013, 23, 6152–6155, DOI:10.1016/j.bmcl.2013.09.005.
- (a) M. Lamoria and M. D. Milton, Dyes Pigm., 2023, 213, 111182, DOI:10.1016/j.dyepig.2023.111182; (b) L. Yu, Z. Wu, G. Xie, J. Luo, Y. Zou, D. Ma and C. Yang, J. Mater. Chem. C, 2020, 8, 12445–12449, 10.1039/d0tc02412k.
- (a) B. Avula, C. K. R. Reddivari, R. M. R. Muchumarri, S. Eraganaboyina, G. V. Zyryanov and B. R. Nemallapudi, Polycyclic Aromat. Compd., 2023, 44, 634–670, DOI:10.1080/10406638.2023.2167215; (b) G. Yashwantrao and S. Saha, Org. Chem. Front., 2021, 8, 2820–2862, 10.1039/d0qo01575j; (c) R. S. Keri, D. Reddy, S. Budagumpi and V. Adimule, RSC Adv., 2023, 13, 20373–20406, 10.1039/D3RA03646D; (d) B. Borah and L. R. Chowhan, RSC Adv., 2021, 11, 37325–37353, 10.1039/D1RA06942J.
- G. Körner, Ber. Dtsch. Chem. Ges., 1884, 17, 572–573 CrossRef.
- O. Hinsberg, Ber. Dtsch. Chem. Ges., 1884, 17, 318–323 CrossRef.
- R. Taylor and R. Robinson, Synlett, 2005, 2005, 1003–1005, DOI:10.1055/s-2005-864830.
- N. Sharma, S. Swami, V. Shrivastava, R. Nair and R. Shrivastava, Mater. Today: Proc., 2021, 43, 3309–3317, DOI:10.1016/j.matpr.2021.01.968.
- A. Mondal, M. K. Sahoo, M. Subaramanian and E. Balaraman, J. Org. Chem., 2020, 85, 7181–7191, DOI:10.1021/acs.joc.0c02453.
- K. Kumar, S. R. Mudshinge, S. Goyal, M. Gangar and V. A. Nair, Tetrahedron Lett., 2015, 56, 1266–1271, DOI:10.1016/j.tetlet.2015.01.138.
- H.-R. Yang, Z.-Y. Hu, X.-C. Li, L. Wu and X.-X. Guo, Org. Lett., 2022, 24, 8392–8396 CrossRef CAS.
- (a) A. Kumar, S. kumar, A. Saxena, A. De and S. Mozumdar, Catal. Commun., 2008, 9, 778–784, DOI:10.1016/j.catcom.2007.08.021; (b) M. Kalhor, M. Shayestefar, M. Khalaj and F. Janghorban, Res. Chem. Intermed., 2022, 49, 885–900, DOI:10.1007/s11164-022-04914-3.
- A. Hasaninejad, M. Shekouhy and A. Zare, Catal. Sci. Technol., 2012, 2, 201–214, DOI:10.24200/sci.2017.4516.
- N. Shah, E. Gravel, D. V. Jawale, E. Doris and I. N. Namboothiri, ChemCatChem, 2014, 7, 57–61, DOI:10.1002/cctc.201402782.
- B. S. Kuarm, P. A. Crooks and B. Rajitha, Green Chem. Lett. Rev., 2013, 6, 228–232, DOI:10.1080/17518253.2012.752041.
- D. Kumar, K. Seth, D. N. Kommi, S. Bhagat and A. K. Chakraborti, RSC Adv., 2013, 3, 15157–15168, 10.1039/C3RA41038B.
- G. Badhani, A. Joshi and S. Adimurthy, Eur. J. Org Chem., 2021, 2021, 6705–6716, DOI:10.1002/ejoc.202101135.
- G. Lupidi, A. Palmieri and M. Petrini, Green Chem., 2022, 24, 3629–3633, 10.1039/D2GC00664B.
- L. K. Beagle, E. Horsting, J. Buechele and J. K. Beagle, Results Chem., 2023, 6, 101211, DOI:10.1016/j.rechem.2023.101211.
- A. Mishra, S. Singh, M. A. Quraishi and V. Srivastava, Org. Prep. Proced., 2019, 51, 345–356, DOI:10.1080/00304948.2019.1596469.
- (a) S. Das, P. Das, S. Maity, P. Ghosh and A. Dutta, Results Chem., 2023, 5, 100713, DOI:10.1016/j.rechem.2022.100713; (b) Z. T. Bhutia, G. Prasannakumar, A. Das, M. Biswas, A. Chatterjee and M. Banerjee, ChemistrySelect, 2017, 2, 1183–1187, DOI:10.1002/slct.201601672; (c) B. F. Mirjalili and M. D. Tafti, Sci. Iran., 2017, 24, 3014–3021, DOI:10.24200/sci.2017.4516; (d) G. C. Nandi, S. Samai, R. Kumar and M. S. Singh, Synth. Commun., 2011, 41, 417–425, DOI:10.1080/00397910903576685; (e) J. Li, D. Jiang, J. Chen, M. Liu, J. Ding and H. Wu, J. Heterocycl. Chem., 2011, 48, 403–406, DOI:10.1002/jhet.597.
- (a) Z. Wu, X. Li, P. Jiang, G. Feng, L.-A. Cao, X.-M. Wang, R. Tan and E. Feng, Appl. Catal., A, 2024, 684, 119893, DOI:10.1016/j.apcata.2024.119893; (b) H. Nagarajaiah, A. K. Mishra and J. N. Moorthy, Org. Biomol. Chem., 2016, 14, 4129–4135, 10.1039/c6ob00351f; (c) H. A. Etman, H. M. Metwally, M. M. Elkasaby, A. M. Khalil and M. A. Metwally, Am. J. Org. Chem., 2012, 1, 10–13, DOI:10.5923/j.ajoc.20110101.03; (d) L. Carlier, M. Baron, A. Chamayou and G. Couarraze, Tetrahedron Lett., 2011, 52, 4686–4689, DOI:10.1016/j.tetlet.2011.07.003; (e) G. Kaupp, M. R. Naimi-Jamal and J. Schmeyers, Chem.–Eur. J., 2002, 8, 594–600, DOI:10.1002/1521-3765(20020201)8:3%3C594::AID-CHEM594%3E3.0.CO;2-5.
- S. Saha, A. B. Pinheiro, A. Chatterjee, Z. T. Bhutia and M. Banerjee, Green Chem., 2024, 26, 5879–5889, 10.1039/D4GC00486H.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental procedures, IR study, solution phase study, comparative table, spectral data and copies of spectra. See DOI: https://doi.org/10.1039/d4mr00100a |
| This journal is © The Royal Society of Chemistry 2025 |