
Nanocluster reaction-driven in situ transformation of colloidal nanoparticles to mesostructures†‡
Paulami
Bose§
 a,
Pillalamarri
Srikrishnarka§
a,
Matias
Paatelainen
b,
Nonappa
b,
Amoghavarsha Ramachandra
Kini
a,
Anirban
Som
a and
Thalappil
Pradeep
*a
a,
Pillalamarri
Srikrishnarka§
a,
Matias
Paatelainen
b,
Nonappa
b,
Amoghavarsha Ramachandra
Kini
a,
Anirban
Som
a and
Thalappil
Pradeep
*a
aDST Unit of Nanoscience (DST UNS) and Thematic Unit of Excellence (TUE), Department of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, India. E-mail: pradeep@iitm.ac.in
bFaculty of Engineering and Natural Sciences, Tampere University, Korkeakoulunkatu 3, FI-33720, Tampere, Finland
First published on 25th September 2024
Abstract
Atomically precise noble metal nanoclusters (NCs) are molecular materials known for their precise composition, electronic structure, and unique optical properties, exhibiting chemical reactivity. Herein, we demonstrated a simple one-pot method for fabricating self-assembled Ag–Au bimetallic mesostructures using a reaction between 2-phenylethanethiol (PET)-protected atomically precise gold NCs and colloidal silver nanoparticles (Ag NPs) in a tunable reaction microenvironment. The reaction carried out in toluene at 45 °C with constant stirring at 250 revolutions per minute (RPM) yielded a thermally stable, micron-sized cuboidal mesocrystals of self-assembled AgAu@PET nanocrystals. However, the reaction in dichloromethane at room temperature with constant stirring at 250 RPM resulted in a self-assembled mesostructure of randomly close-packed AgAu@PET NPs. Using a host of experimental techniques, including optical and electron microscopy, optical absorption spectroscopy, and light scattering, we studied the nucleation and growth processes. Our findings highlight a strategy to utilize precision and plasmonic NP chemistry in tailored microenvironments, leading to customizable bimetallic hybrid three-dimensional nanomaterials with potential applications.
1. Introduction
Atomically precise noble metal nanoclusters (NCs) are an emerging class of nanomaterials known for their precise composition and tuneable chemical, physical, and optical properties.1–8 The chemistry of noble metal NCs and its versatility to facilitate the formation of precise assemblies,9–12 nanocomposites,13,14 membranes,15 colloidal frameworks,16 and hybrid nanomaterials17–19 are currently gaining prominence.20–25 NC-assembled solids are well-known for their fascinating properties, such as photoluminescence, conductivity, magnetism, mechanical strength, and catalysis.12,26–29 NCs can be solidified using different approaches, such as solvent-induced crystallization,30–32 electro-crystallization,33 and gelation.34,35 Ligand functionalization of the particle surface plays a prominent role in controlling interparticle interactions and, eventually, the frameworks of assemblies.36–38 Such assemblies are mostly driven by supramolecular forces, including H-bonding, electrostatic, dipolar, π-stacking, and van der Waals interactions.20,21,39,40 Tellurium nanowires (Te NWs) and Au32SG19 NCs (where SG refers to glutathione) react to form nanodumbell-shaped Ag–Te hybrid NWs.18 Te NWs modified with Ag44(p-MBA)30 (where p-MBA refers to para-mercaptobenzoic acid) form a crossed bilayer assembly via ligand-mediated H-bonding.19 Similarly, Ag44(p-MBA)30 NCs encapsulate gold nanorods (Au GNRs) to form cage-like nanostructures.41 However, in most of the nanocluster–nanoparticle (NC–NP) assemblies, supramolecular interactions constitute the sole driving factor, resulting in superstructures where reacting particles preserve their intrinsic properties.9,15,20 Recent studies indicate that thiol (SR)-protected noble metal NCs can facilitate spontaneous interparticle (NC–NC and NP–NC) atom exchange in solution, leading to chemically modified particles.21,42–44 For instance, a galvanic exchange interparticle reaction between Ag@SR NPs and [Au25(SR)18]− NCs results in self-assembled two-dimensional (2D) crystals of Ag–Au-bimetallic NPs (to be discussed in detail in the following section).45 In our recent publication, we reported that an [Ag25(SR)18]− NC-mediated unconventional anti-galvanic exchange reaction (AGR) in Au@SR NPs creates alloy NPs.46 A reaction of [Au25(SR)18]− NCs with CuO NPs induces the aggregation of Cu-doped Au NCs into a nanodisc-shaped superstructure.47 [Ag25(SR)18]− NCs can also enable site-selective etching of anisotropic Au nanotriangles (Au NTs) to produce tuneable core–shell Ag@Au NT nanocomposites.48 From the above examples, it is evident that the final hybrid nanostructures are the consequence of a careful selection of reacting/interacting interparticle systems in terms of particle shape, geometry, composition, and protecting ligands. However, the possibility of creating different nanoarchitectures from the same interparticle system, simply by tuning the reaction microenvironment (temperature and solvent, for instance), is relatively less explored. Additionally, there is a need to explore the feasibility of interparticle reactions further to create next generation three-dimensional (3D) materials.The hierarchical assembly of colloidal NPs represents a transformative frontier in materials science for its ability to create next generation 3D materials with tailored functionalities.49–52 Typically, metal NPs are susceptible to polydispersity, lack of directional interactions, and non-specific aggregation.53 However, size-selected metal NPs are known to self-assemble, leading to 3D crystals,54 2D arrays,55–60 supraparticles,61–65 and colloidal capsids.10,66,67 Mesocrystals are types of colloidal crystals formed by individual nanocrystals self-assembled into higher-order superstructures.68–74 This is an example of a non-classical crystallization technique.74–77 Classical crystallization involves layer-by-layer growth via nucleation and sequential addition of atomic, ionic, or molecular building blocks.78,79 In contrast, non-classical crystallization involves more complex pathways and intermediate structures, often involving larger building blocks such as NPs, complexes, oligomers, etc., resulting in hybrid crystalline materials.77,79 Nanocrystal synthesis is a complex process of controlled precipitation in which the chemical environment,58,80 solvents,81 and ligands play a crucial role.81,82 Self-assembled mesocrystals find potential applications in electronics,83,84 plasmonics,85 catalysis,86–88 energy storage,89,90 and many more.91,92 To engineer precise and configurable mesostructures, it is essential to have robust assembling protocols for their formation with a comprehensive understanding to fully harness their potential applications.
In our previous publication, we demonstrated that polydisperse 2-phenylethanethiol (PET)-protected plasmonic Ag NPs (also referred to as Ag@PET NPs) spontaneously react with atomically precise [Au25(PET)18]− NCs to create a 2D superlattice of highly monodisperse bimetallic AgAu@PET NPs.45 In the present study, we examined a strategy to utilize interparticle (NP–NC) reactions to create mesostructures of bimetallic NPs under adjustable reaction conditions (schematically illustrated in Fig. 1A). At a constant temperature and with mild agitation, the reaction resulted in the directed assembly of bimetallic nanocrystals with a platelet-like morphology into a cuboidal mesocrystal. The same reaction under ambient conditions produced a random close-packed (RCP) bimetallic NP mesostructure. In situ nucleation and growth of mesostructures are driven by interparticle reactions under given conditions. Interestingly, manipulating the reacting interparticle system along with the reaction microenvironment makes it possible to create a variety of stable nanoarchitectures of self-assembled NPs. However, obtaining microscopic-level insight into such assembling events from experiments is incredibly challenging.
 | ||
| Fig. 1 (A) Schematic illustration of the interparticle, polydisperse Ag NPs (grey spheres) and Au NCs (magenta spheres), reaction driving the formation of the mesostructures of bimetallic NPs (purple spheres). (B) Schematic representation and characterization of (a–c) [Au25(PET)18]− NCs, (d–f) AgAu@PET NPs, (g–i) Ag@PET NPs, and mesostructures obtained (j–l) with and (m–o) without heating conditions. PET refers to 2-phenylethanethiol. Color code: grey, Ag; yellow, Au; blue, S; magenta, C; H is omitted for clarity. Please note that the atomic dimensions and ligand attachments are not a true representation. Illustrations are created with a licensed version of https://www.biorender.com/. | ||
2. Results and discussion
Our previous publication was limited to the interparticle reaction between 2-phenylethanethiol (PET)-protected Ag NP–Au NCs, resulting in bimetallic NPs.45 We observed that polydisperse Ag@PET NPs and [Au25(PET)18]− NCs (1a) undergo a spontaneous interparticle atom-exchange reaction to form a self-assembled 2D superlattice of monodisperse alloy AgAu@PET NPs (1b), schematically illustrated in Fig. 1A (1a → 1b). The present study explores the self-assembling properties of the reacted AgAu@PET NPs in different microenvironments in solution, schematically presented in Fig. 1A (1b → 1d). To begin with, we utilized our previously reported protocol using atomically precise [Au25(PET)18]− NCs and polydisperse plasmonic Ag@PET NPs to prepare bimetallic AgAu@PET NPs. The schematic representation and characterization details of the particles involved in the reaction, such as [Au25(PET)18]− NCs (Fig. 1B a–c), AgAu@PET NPs (Fig. 1B d–f), and Ag@PET NPs (Fig. 1B g–i), are presented, as per their size as measured in transmission electron microscopy (TEM).30,93 The synthesized [Au25(PET)18]− NCs exhibited a molecular ion peak centered at m/z 7391 in electrospray ionization mass spectrometry (ESI MS), an average diameter of ∼1.8 nm in the TEM images, and characteristic optical absorption peaks at 675 and 450 nm in the UV-Vis spectrum (further characterization details are presented in ESI Fig. S1‡). The parent Ag@PET NPs have an average diameter of 4.4 ± 2.3 nm (d-spacing 0.21 nm) and a characteristic localized surface plasmon resonance (LSPR) feature at ∼450 nm (characterization details are presented in Fig. S2‡). It should be noted that the particle size refers to the most probable metal core diameter measured using TEM.We utilized a combination of scanning/transmission electron microscopy (S/TEM), field emission scanning electron microscopy (FESEM) imaging, optical absorption spectroscopy, and dynamic light scattering (DLS) techniques to capture the nucleation and growth of the NPs into mesostructures. Using our previously reported method, we prepared bimetallic AgAu@PET NPs by mixing known volumes of Ag@PET NP and [Au25(PET)18]− NC solutions (refer to the Experimental section in ESI for details‡).45 NP–NC reactions are stoichiometric in nature.45,94 The reacted NPs are highly monodisperse in nature, with an average diameter of 3.5 ± 0.5 nm (TEM images in Fig. 1B e and f, and further characterization data are included in Fig. S3‡). However, the parent monometallic Au@DMBT NPs were polydisperse in nature and exhibited no assembling tendency in solution (TEM image in Fig. 1B h). In a typical energy-dispersive X-ray spectroscopy (EDS) spectrum, the presence of Ag is detected using L-shell emissions (Ag L) at 2.98 keV, Au is detected using M-shell emissions (Au M) at 2.12 keV, and both S and C are detected using K-shell emissions (S K and C K) at 2.30 and 0.28 keV, respectively. Dark-field STEM coupled with EDS estimated the atomic percentages of Ag, Au, and S in the reacted NPs to be 57, 27, and 16, respectively (Fig. S4‡). Next, we stirred the reacted NP solution at a constant speed (250 revolutions per minute, RPM) and allowed the reaction to proceed for several days under different experimental conditions (see the Experimental section in ESI for details‡). Experimental conditions include reactions in (i) high boiling and less polar solvents, such as toluene with/without constant heating, and (ii) low boiling and polar solvents, such as DCM under ambient conditions. In order to prevent solvent loss over time, extra precautions were taken to ensure that the reaction vial was leakproof (Fig. S5‡). We observed that the reacted NPs undergo in situ nucleation and growth to form cuboidal mesostructures with different orders of packing under various reaction conditions, as schematically illustrated in Fig. 1B j (in toluene under heating conditions) and Fig. 1B m (in DCM under ambient conditions), respectively. Later sections will provide a detailed discussion on the morphology and composition of the as-obtained mesostructures.
The reacting particles dispersed well in toluene and remained stable. Under constant and gradual stirring at 45 °C, we observed that the reacted NP suspension required ∼100 days to fully precipitate (schematically illustrated in Fig. 2A a, with further details provided in the Experimental section of the ESI‡). We drop-cast about 10 μL from the reaction mixture onto a silicon wafer for FESEM, TEM, and optical microscopy imaging. Imaging revealed that the as-obtained precipitate was composed of multiple cuboidal mesostructures (schematically illustrated in Fig. 2A b). Each cuboid can be visualized as a collection of multiple platelet-shaped nanocrystals held together, presumably by a ligand–ligand interaction (schematically represented in Fig. 2A c–e).67,95 FESEM, HRTEM, and the corresponding fast Fourier transform (FFT) images of the isolated nanocrystals with a platelet-like shape and a dimension of 0.3 nm × 0.3 nm (length (l) × breadth (b)) are presented in Fig. S6.‡ The FESEM image of the drop-cast sample captured at a magnification of 8000× shows multiple mesocrystals with well-defined edges and flat facets (Fig. 2B a). The image was acquired using a beam energy of 10 keV and a secondary electron detector in field-free mode. We further centrifuged the reaction mixture to eliminate smaller structures, and the precipitate was imaged at 100× magnification using an optical microscope (Fig. 2B b). We observed that the precipitate was composed of several micron-sized mesostructures using a dark-field optical microscope (Fig. 2B b). One of these mesostructures was further imaged using FESEM at a magnification of 2500× (Fig. 2B c). The estimated dimensions of the cuboidal mesostructure are ∼48 μm (l) × ∼50 μm (b); information on height is unavailable. The fissured edge of the cuboid indicates layer-by-layer growth of the platelet-shaped nanocrystals (Fig. 2B d). The FESEM image of one such edge viewed at a magnification of 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× further revealed that the cuboidal mesostructure is composed of multiple layers of nanocrystals (dimensions of approximately 50 nm × 50 nm in l × b, respectively). TEM images further support the mesostructure formation involving a layer-by-layer arrangement of nanocrystal units (Fig. 2B e). We utilized atomic force microscopy (AFM) to analyze the height and surface topology of the mesocrystals (Fig. 2B f, and further details are provided in Fig. S7‡). AFM analysis of the selected mesocrystal revealed a maximum height of 0.98 μm and a root mean square (RMS) roughness of 6.24 nm, indicating a highly ordered structure with minimal surface irregularities (Fig. S7‡). The steep edges and lower roughness suggest a precise self-assembly of the nanocrystals into well-packed layer-by-layer mesocrystals.
000× further revealed that the cuboidal mesostructure is composed of multiple layers of nanocrystals (dimensions of approximately 50 nm × 50 nm in l × b, respectively). TEM images further support the mesostructure formation involving a layer-by-layer arrangement of nanocrystal units (Fig. 2B e). We utilized atomic force microscopy (AFM) to analyze the height and surface topology of the mesocrystals (Fig. 2B f, and further details are provided in Fig. S7‡). AFM analysis of the selected mesocrystal revealed a maximum height of 0.98 μm and a root mean square (RMS) roughness of 6.24 nm, indicating a highly ordered structure with minimal surface irregularities (Fig. S7‡). The steep edges and lower roughness suggest a precise self-assembly of the nanocrystals into well-packed layer-by-layer mesocrystals.
 | ||
| Fig. 2 (A) Schematic illustration of the experimental setup (a), sample preparation (b), and typical morphologies (c and d) and morphogenesis (e) of the mesocrystal obtained. (B) FESEM (a, c, and d), dark-field optical microscopy (b), TEM (e), and AFM (f) images of the mesocrystals obtained after ∼100 days of reaction. Slanted cuboidal-shaped mesocrystals as imaged under FESEM (g) and TEM (in the inset), and an optical microscope in the dark field (h) and the corresponding depth-of-field (in the inset). Color code: grey, Ag; yellow, Au; green, S; pink-blue, ligand. Illustrations are created with a licensed version of https://www.biorender.com/. | ||
We selected a mesocrystal with a slanted cuboidal shape for detailed investigation to obtain further insights into the morphology and composition of the constituent particles (Fig. 2B g and h). A slanted cuboid is one that has two of its opposing faces parallel to the line of sight, while the other four faces converge at a single point, giving the object a triangular appearance. The FESEM image of a selected mesostructure (face and edge length of ∼10 μm and 8 μm, respectively) acquired at 20000× magnification revealed layered growth at the edges, and the TEM image revealed nanocrystals as building blocks (Fig. 2B g and inset, respectively). The optical image of the selected structure appears shiny, which can be attributed to the dense packing of the individual nanocrystals (Fig. 2B h). The depth-of-field optical image of the mesocrystals further confirms a layered crystal growth (inset, Fig. 2B h). We analyzed the mesocrystals using FESEM coupled with EDS to gain insight into the elemental composition and distribution. The composition of the slanted cuboid can be schematically represented as a collection of tightly packed AgAu@PET nanocrystals, as shown in Fig. 3A. The atomic percentages of the elements present in the mesostructure are as follows: 21% for Ag, 17% for Au, 20% for S, and 42% for C (Fig. S8‡). EDS mappings of elemental gold (Au), silver (Ag), sulfur (S), and carbon (C) of the isolated mesostructure obtained using FESEM EDS are presented in Fig. 3B. Mapping data revealed a uniform distribution of Au, Ag, S, and C across the mesostructure, indicating the AgAu@PET NP crystallites as the building blocks of such mesostructures.
 | ||
| Fig. 3 (A) Schematic illustration of a slanted cuboidal-shaped mesocrystal (a) and its compositional representation (b). (B) FESEM image of such a crystalline entity (a) and the corresponding EDS maps of S (b), C (c), Ag (d), Au (e), and Ag–Au overlay (f). Illustrations are created with a licensed version of https://www.biorender.com/. | ||
To comprehend the reaction-induced aggregation behavior of the particles in solution, we combined dynamic light scattering (DLS), the Tyndall effect, and optical absorption spectroscopy as a function of time (Fig. 4). DLS measures the scattered light intensity change caused by the motion of particles suspended in a solution. We conducted the study at a ten-fold dilution of the parent particle concentrations to maintain a lower polydispersity index (PDI). DLS measurements showed that the hydrodynamic radius (RH) of the AgAu@PET NPs in solution gradually increased with reaction time (Fig. 4A). The continuous red-shift and the broadening in the DLS peak indicate that the bimetallic NPs nucleated and grew in the solution, resulting in the formation of larger structures over time. Next, we used the Tyndall effect to probe the aggregation of colloidal dispersion by focusing a 630 nm laser beam (green laser) on the solution (Fig. 4B). As shown in Fig. 4B, on the 0th day, the suspension of the well-dispersed parent Ag@PET NPs appears homogeneous as the light passes through. The Tyndall effect was seen in the colloidal AgAu@PET NPs after 1 day of reaction, with further aggregation and eventual precipitation occurring over the next 10 days. The scattering of light becomes evident when the size of the particle aggregate exceeds the wavelength of incident light. Time-dependent optical absorption spectra of the particle mixture also showed a similar aggregation trend as observed in the light scattering experiment (Fig. 4C). Here, the optical absorption spectrum on the 0th day corresponds to the optical absorption spectrum of the parent Ag@PET NPs. The red-shift in the characteristic SPR peak of the Ag@PET NPs confirms the presence of bimetallic Au–Ag NPs in the reaction mixture. We also observed a progressive broadening in the SPR peak of the reaction mixture, indicating the onset of particle aggregation in the solution over time. Furthermore, the rise in the baseline as the reaction progresses can be attributed to particle aggregation. Under similar reaction conditions, FESEM and TEM images revealed the formation of comparably smaller-sized cuboidal-shaped mesocrystals in the solution (Fig. 4D). Only the dimensions of the as-obtained mesocrystals were impacted by diluting the reaction mixture; however, the overall morphology of the mesocrystals remained unchanged, as seen in the previous case.
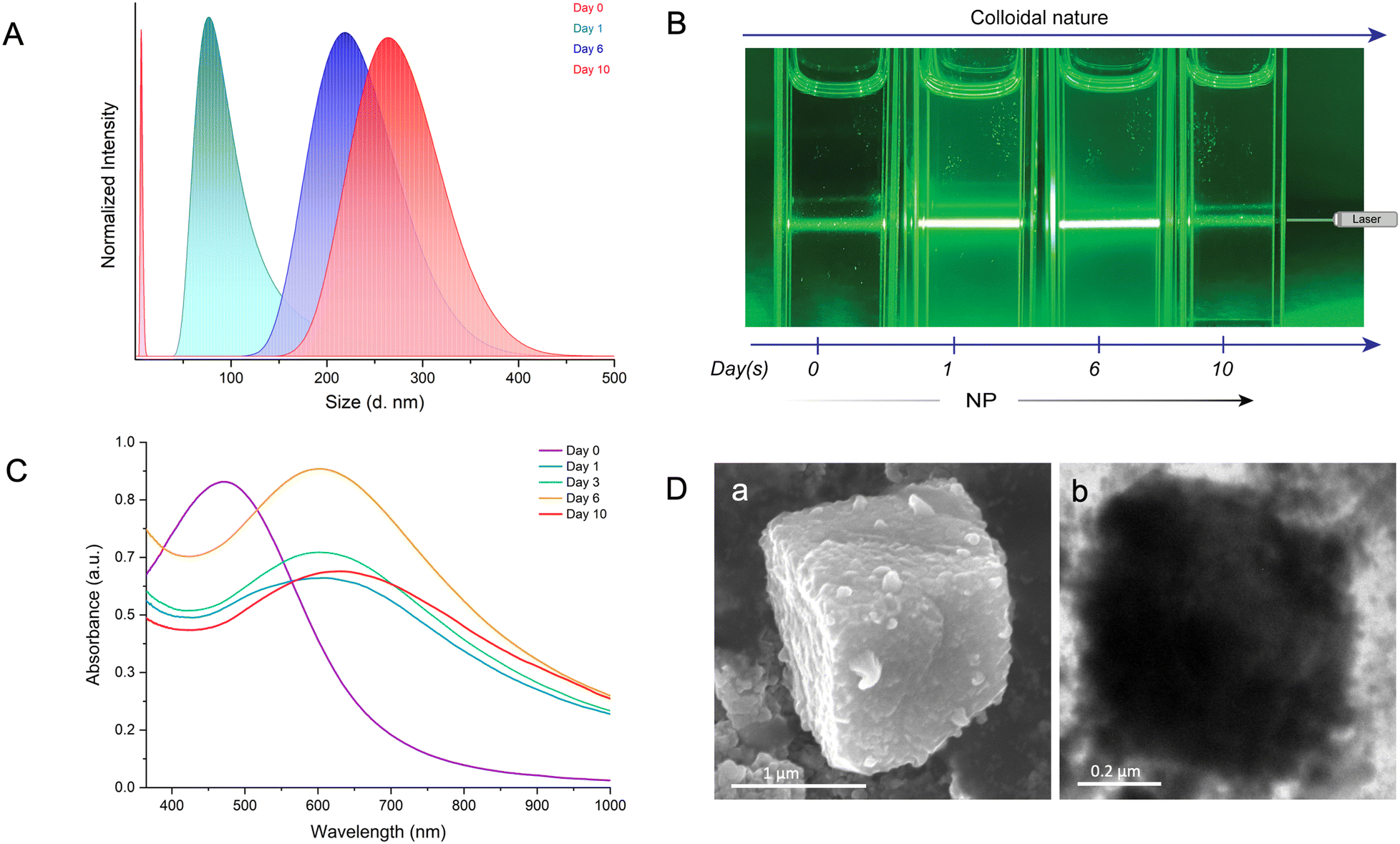 | ||
| Fig. 4 Time-dependent evolution of colloidal particles into crystalline mesostructures as monitored using (A) dynamic light scattering, (B) the Tyndall effect under irradiation with a 630 nm green laser, and (C) optical absorption spectroscopy. (D) FESEM (a) and TEM (b) images of the final mesocrystals as-obtained after 10 days of reaction. | ||
Based on the above discussion, the interparticle reaction in a non-polar solvent and at elevated temperature results in the in situ formation of mesostructures with well-packed bimetallic AgAu@PET nanocrystals. The nucleation and growth rate, and thus the formation of the mesostructures, is known to be influenced by various reaction conditions such as temperature, pressure, pH, concentration, and solvent.96 We carried out the same interparticle reaction at room temperature while keeping the other parameters, such as solvent, concentration, and RPM, constant (refer to the Experimental section in ESI for details‡). Under the previous reaction conditions, the process of mesocrystallization took approximately 100 days when performed at 45 °C, and nearly no change was observed when the process was conducted at room temperature. Next, we studied the same interparticle reaction at room temperature in DCM while maintaining other parameters constant (schematic illustration in Fig. 5A). The particle mixture formed a stable dispersion in DCM, which was continuously stirred at 250 RPM under ambient conditions. The reaction took ∼50 days for the complete precipitation to occur, and the product was analyzed using electron microscopy (Fig. 5B). In order to mitigate solvent loss over time, the reaction was conducted in the same leakproof container (as shown in Fig. S5‡). The FESEM image shows the formation of much larger cuboidal mesostructures of 51 nm × 47 nm (l × b; height information is unavailable) as a product (Fig. 5B a). The image was acquired at 3500× magnification using the same 10 keV accelerating voltage and a secondary electron detector in field-free mode. The mesostructure exhibited visible signs of NP nucleation, resulting in layer-by-layer structural growth at intermediate phases, and, finally, densely packed structures were formed at the end of the process (Fig. S9‡). The rough surface morphology of the as-obtained mesostructure can be attributed to the random packing of the NPs. The TEM image further confirmed the aggregation of the NPs within the precise boundaries of the mesostructures while retaining their original dimension (Fig. 5B b). To verify the retention of the NP characteristics in the final mesostructure, we degraded the sample by sonication in DCM. Post-sonication, the TEM image revealed the disintegration of the mesostructure into the NPs resembling the original AgAu@PET NPs (Fig. S10‡). AFM analysis of the selected mesostructure revealed a maximum height of 3.19 μm and a root mean square (RMS) roughness of 22.05 nm, indicating a highly disordered structure with significant surface irregularities (Fig. 5B c, and further details are provided in Fig. S11‡). Higher surface irregularity also suggests a random assembly of the NPs during the growth stage. A time-dependent optical absorption spectroscopic analysis of the interparticle reaction in DCM at ambient temperature is presented in Fig. 5C. Over time, we observed that the characteristic SPR peak corresponding to the parent Ag@PET NPs (corresponding spectrum on the 0th day) underwent a gradual red-shift and broadening upon reaction with [Au25(PET)18]− NCs. According to the FESEM EDS study, the mesostructures are made up of about 25% gold (Au), 30% silver (Ag), 14% sulfur (S), and 31% carbon (C) (Fig. S12,‡ based on the intensities of the respective EDS features). Mapping data confirmed a uniform distribution of Au, Ag, S, and C across the mesostructure (Fig. 5D a), indicating the AgAu@PET NPs as the building blocks of such structures (Fig. 5D b–f). Thus, we can infer that the parent Ag@PET NPs react to form alloy AgAu@PET NPs and eventually aggregate into higher-order assemblies with time. The continuous rise in the baseline of the optical absorption spectrum with the progress of the reaction can be related to the gradual nucleation of particles to a larger structure in solution.
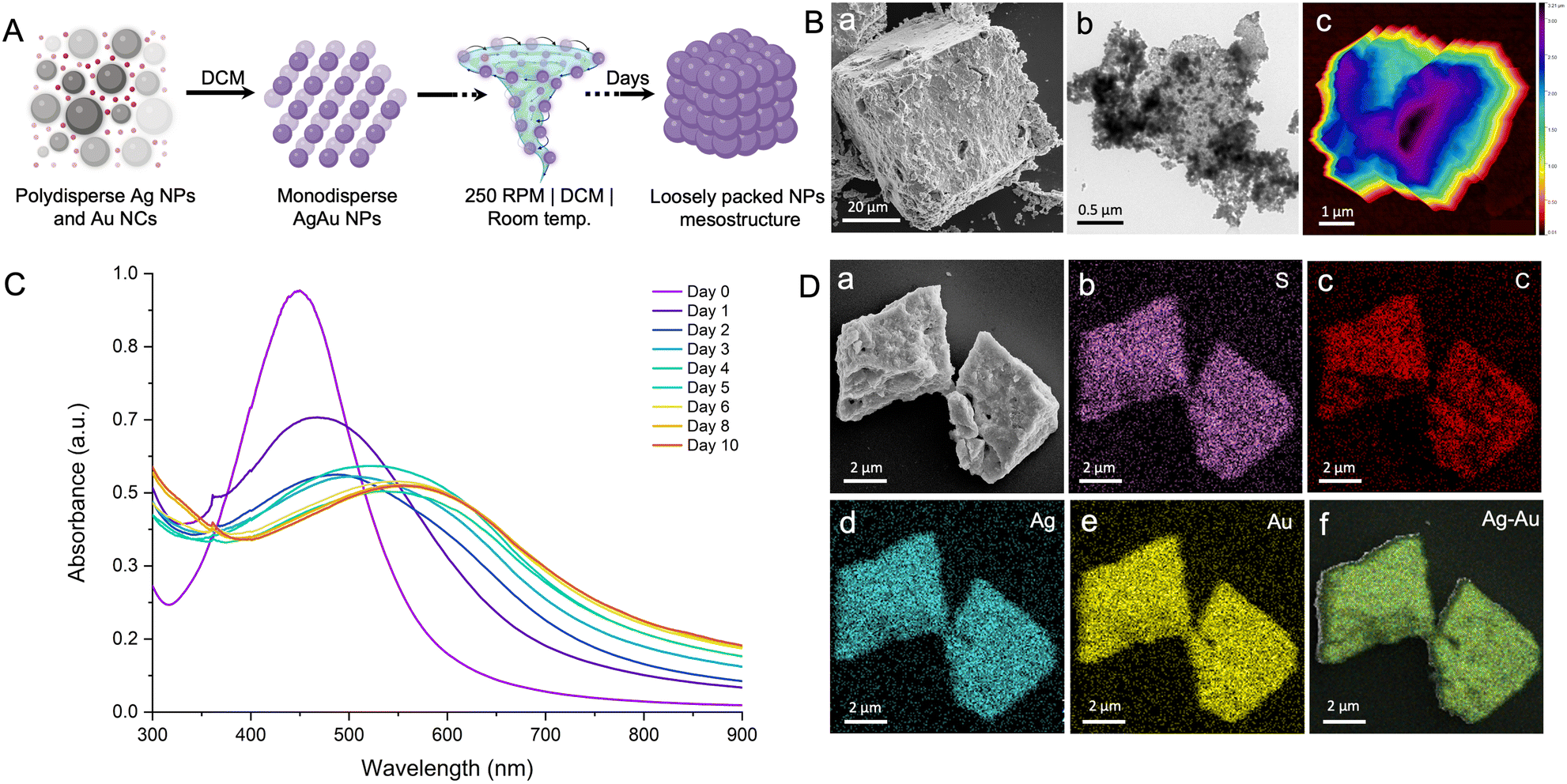 | ||
| Fig. 5 (A) Schematic representation of a room temperature interparticle reaction carried out in DCM with constant stirring at 250 RPM, resulting in larger mesostructures with lower packing. (B) FESEM (a), TEM (b), and AFM (c) images of the mesostructures. (C) Time-dependent evolution of colloidal particles into mesostructures as monitored using optical absorption spectroscopy. (D) FESEM image of the mesostructures (a) and the corresponding EDS maps of S (b), C (c), Ag (d), Au (e), and Ag–Au overlay (f). Illustrations are created with a licensed version of https://www.biorender.com/. | ||
For control experiments, we checked the stability of the parent particles under identical reaction conditions. Over the course of ∼05 days, a steady decline in the absorbance of the parent Ag@PET NPs was evident in the optical absorption spectra, indicating particle degradation (Fig. S13A‡). The [Au25(PET)18]− NCs showed an increased sensitivity to heat, and the NCs degraded rapidly (Fig. S13B‡). To understand the in situ nucleation and growth process, we studied the evolution of the reacted NPs in solution with TEM at different intervals as the mesocrystallization process continued (Fig. S14‡). The time-dependent series of TEM images indicated a significant difference in the patterns of nucleation and growth of the AgAu@PET NPs under two reaction conditions. For instance, the AgAu@PET NPs in toluene nucleate under an elevated temperature to form platelet-like nanocrystals, which eventually assemble and grow layer-by-layer until the final mesocrystal is formed (Fig. S14A‡). On the other hand, the AgAu@PET NPs in DCM randomize and then assemble under ambient conditions, followed by a layered growth until the final mesostructure is formed (Fig. S14B‡). From the above observation, we can understand that local chemical microenvironments involving temperature, solvent polarity, RPM, and interparticle forces play a crucial role in the morphogenesis of the mesocrystals.78,96 However, understanding such complex dynamics of nucleation and growth of the NPs requires in situ microscopic studies and advanced mesoscale modeling.
Inter-ligand interactions are known to play a prominent role in the self-assembly of metallic NPs.21,38,40,51 We performed reactions for various intermetallic (for example, Au NP–Ag NC) and intrametallic interparticle (Au NP–Au NC and Ag NP–Ag NC) systems to verify the role of ligands in such a reaction-driven in situ mesocrystallization process. Similar experimental conditions for the PET-capped particle system were used for the subsequent reactions (refer to the Experimental section in ESI‡). We studied the same Ag–Au intermetallic system using the reaction between the 2,4-dimethylbenzenethiol (DMBT)-protected particles, such as ∼4.5 nm Au@DMBT NPs and atomically precise [Ag25(DMBT)18]− NCs (refer to the ESI for details, Fig. S15‡). We recently reported a detailed study on the interparticle chemistry of Au@DMBT NPs and [Ag25(DMBT)18]− NCs, and showed that the Ag NCs mediate the alloying process via an interparticle metal–ligand exchange pathway.46 As per the literature, the insertion of Ag atoms into an Au NP is known to strongly alter the SPR feature in terms of peak position and shape depending on the size and composition of the alloy NP.97–99 Within a day of reaction, we observed a strong enhancement in the SPR feature, and a slight shift of ∼9 nm in the peak position of the reacted NPs, indicating the insertion of Ag atoms in the parent Au@DMBT NPs. Subsequently, the reacted NPs steadily degraded with time. However, the reacted NPs underwent stable aggregation within a day in DCM at room temperature. In the following experiment, we utilized the 2-phenylethanethiol (PET)-protected Au@PET NPs and [Au25(PET)18]− NCs (intrametallic particle system) to study the reaction (refer to the ESI for details, Fig. S16‡). The PET-protected particle mixture, however, displayed time-dependent optical absorption spectra that suggested a reaction over time. Over time, the reacted NPs in the solution self-assembled into a 2D capsid-like superstructure (Fig. S16A,‡ further details are provided in the ESI‡). The optical absorption spectra of the parent Au@PET NP dispersion in toluene showed stability over a span of a few days at an elevated temperature (Fig. S17‡). Next, we carried out a similar intrametallic interparticle reaction with the Ag@PET NPs and [Ag25(DMBT)18]− NCs (Fig. S18‡). However, in this case, the reaction product underwent rapid degradation (reaction at 45 °C in toluene). However, the product in solution showed a tendency to self-assemble into a 2D superlattice when the reaction was carried out at room temperature in DCM. A study of intrametallic systems is intriguing but goes beyond the scope of this paper.
3. Conclusions
In summary, we demonstrated a simple in situ reaction-driven technique for fabricating self-assembled bimetallic mesostructures. An interparticle reaction between PET-protected plasmonic Ag NPs and atomically precise Au NCs results in highly monodisperse bimetallic AgAu NPs with enhanced thermal stability and assembling properties. At an elevated temperature of 45 °C, the interparticle dispersion in toluene results in reaction-driven in situ nucleation and growth of AgAu@PET nanocrystals. These nanocrystals eventually self-assemble to create cuboidal mesocrystals. Under ambient conditions, however, the same interparticle reaction in DCM produces mesostructures of random close-packed AgAu@PET NPs. Electron microscopy coupled with EDS and light scattering experiments was used to examine the growth of NPs into mesostructures. An AFM study suggests that the reaction at 45 °C in toluene produces mesocrystals with reduced structural irregularity. Our studies also indicate that PET-capped intermetallic and intrametallic systems are capable of creating unique thermally stable NP-assembled nanoarchitectures. Tracing the origins of such an assembly event requires mesoscale modelling, which will be addressed in the future. This interparticle chemistry can be an alternative non-classical mesocrystallization method that offers flexibility to engineer bimetallic 3D nanomaterials. Using atomically precise noble metal NC reaction-driven phenomena with well-chosen NP–NC systems, it is now possible to achieve alloy particles with control over the size dispersity, leading to self-assembly, all in one pot.Data availability
The data supporting the study are included in the main article and its ESI,‡ uploaded as separate files. Any additional and raw data that were used during the preparation of the manuscript and ESI‡ are available from the corresponding author upon request. Schematic illustrations are prepared using Mercury Cambridge Structural Database and https://www.biorender.com/. The BioRender license files are compiled and uploaded separately with submission. The figure preparation includes professional tools like Adobe Illustrator and Microsoft PowerPoint 2021. Bibliographic references were prepared using the citing software Zotero.For further inquiries or access to the data, please contact E-mail: pradeep@iitm.ac.in.
Author contributions
P. B. performed the experiments and analyzed the results. P. S. carried out the FESEM imaging. M. P. carried out the AFM imaging. N. carried out the STEM imaging. A. R. K. contributed to the conceptualization of the experimental setup. A. S. carried out the TEM imaging. T. P. designed the project objective and supervised the progress. Every author contributed to the drafting of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Department of Science and Technology (DST), Government of India, and the Centre of Excellence Program of the Indian Institute of Technology Madras on the theme of Molecular Materials and Functions, under the Institutes of Eminence of Ministry of Education, India, for supporting our research. We thank the Academy of Finland for project funding (No. 352900), the Photonics Research and Innovation (PREIN) flagship, and the Tampere Microscopy Centre for imaging facilities. We thank Bhanupriya S. for her technical assistance with the transmission electron microscopy imaging at the DST Unit of Nanoscience, Indian Institute of Technology Madras. T. P. thanks the Science and Engineering Research Board (SERB), India, for funding through the SPR/2021/000439 research grant and a JC Bose Fellowship.References
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed
.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443 RSC
- Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Xie, Chem. Sci., 2021, 12, 99–127 RSC
-
P. Bose, G. Natarajan and T. Pradeep, in Atomically Precise Metal Nanoclusters, Elsevier, 2023, pp. 313–343 Search PubMed
-
T. Pradeep, Atomically precise metal clusters, Elsevier, 1st edn, 2023 Search PubMed
- Nonappa, T. Lahtinen, J. S. Haataja, T.-R. Tero, H. Häkkinen and O. Ikkala, Angew. Chem., Int. Ed., 2016, 55, 16035–16038 CrossRef CAS PubMed
- Nonappa, J. S. Haataja, J. V. I. Timonen, S. Malola, P. Engelhardt, N. Houbenov, M. Lahtinen, H. Häkkinen and O. Ikkala, Angew. Chem., Int. Ed., 2017, 56, 6473–6477 CrossRef CAS PubMed
- J. V. Rival, Nonappa and E. S. Shibu, ACS Appl. Mater. Interfaces, 2020, 12, 14569–14577 CrossRef CAS PubMed
- M. Jash, A. Jana, A. K. Poonia, E. Khatun, P. Chakraborty, A. Nagar, T. Ahuja, K. V. Adarsh and T. Pradeep, Chem. Mater., 2023, 35, 313–326 CrossRef CAS
- A. Chakraborty, A. C. Fernandez, A. Som, B. Mondal, G. Natarajan, G. Paramasivam, T. Lahtinen, H. Häkkinen, Nonappa and T. Pradeep, Angew. Chem., Int. Ed., 2018, 57, 6522–6526 CrossRef CAS PubMed
- A. Chakraborty, H. Dave, B. Mondal, Nonappa, E. Khatun and T. Pradeep, J. Phys. Chem. B, 2022, 126, 1842–1851 CrossRef CAS
- A. Som, A. Griffo, I. Chakraborty, H. Hähl, B. Mondal, A. Chakraborty, K. Jacobs, P. Laaksonen, O. Ikkala, T. Pradeep and Nonappa, Small, 2022, 18, 2201707 CrossRef CAS PubMed
- S. Chandra, Nonappa, G. Beaune, A. Som, S. Zhou, J. Lahtinen, H. Jiang, J. V. I. Timonen, O. Ikkala and R. H. A. Ras, Adv. Opt. Mater., 2019, 7, 1900620 CrossRef CAS
- A. Ghosh, T. Pradeep and J. Chakrabarti, J. Phys. Chem. C, 2014, 118, 13959–13964 CrossRef CAS
- A. Som, A. K. Samal, T. Udayabhaskararao, M. S. Bootharaju and T. Pradeep, Chem. Mater., 2014, 26, 3049–3056 CrossRef CAS
- A. Som, I. Chakraborty, T. A. Maark, S. Bhat and T. Pradeep, Adv. Mater., 2016, 28, 2827–2833 CrossRef CAS
- Nonappa and O. Ikkala, Adv. Funct. Mater., 2018, 28, 1704328 CrossRef
- P. Chakraborty, A. Nag, A. Chakraborty and T. Pradeep, Acc. Chem. Res., 2019, 52, 2–11 CrossRef CAS
- J. V. Rival, P. Mymoona, K. M. Lakshmi, T. Pradeep and E. S. Shibu, Small, 2021, 17, 2005718 CrossRef CAS
-
A. Nag and T. Pradeep, in Atomically Precise Metal Nanoclusters, ed. T. Pradeep, Elsevier, 2023, pp. 101–111 Search PubMed
- Nonappa, Chem. Commun., 2023, 59, 13800–13819 RSC
- R. Nakatani, S. Das and Y. Negishi, Nanoscale, 2024, 16, 9642–9658 RSC
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC
- J. Liu, J. Huang, W. Niu, C. Tan and H. Zhang, Chem. Rev., 2021, 121, 5830–5888 CrossRef CAS PubMed
-
A. Jana and T. Pradeep, in Atomically Precise Metal Nanoclusters, Elsevier, 2023, pp. 49–82 Search PubMed
- N. Alam, A. Kumar Das, P. Chandrashekar, P. Baidya and S. Mandal, Nanoscale, 2024, 16, 10087–10107 RSC
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed
- A. Nag, P. Chakraborty, M. Bodiuzzaman, T. Ahuja, S. Antharjanam and T. Pradeep, Nanoscale, 2018, 10, 9851–9855 RSC
- C. Cerretani, H. Kanazawa, T. Vosch and J. Kondo, Angew. Chem., Int. Ed., 2019, 58, 17153–17157 CrossRef CAS PubMed
- S. Antonello, T. Dainese, F. Pan, K. Rissanen and F. Maran, J. Am. Chem. Soc., 2017, 139, 4168–4174 CrossRef CAS PubMed
- Z. Wu, C. Dong, Y. Li, H. Hao, H. Zhang, Z. Lu and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 9952–9955 CrossRef CAS PubMed
- Z. Wu, J. Liu, Y. Li, Z. Cheng, T. Li, H. Zhang, Z. Lu and B. Yang, ACS Nano, 2015, 9, 6315–6323 CrossRef CAS PubMed
- A. M. Smith, K. A. Johnston, S. E. Crawford, L. E. Marbella and J. E. Millstone, Analyst, 2016, 142, 11–29 RSC
- V. Jain, S. Roy, P. Roy and P. P. Pillai, Chem. Mater., 2022, 34, 7579–7597 CrossRef CAS
- A. Rao, S. Roy, V. Jain and P. P. Pillai, ACS Appl. Mater. Interfaces, 2023, 15, 25248–25274 CrossRef CAS
- J. V. Rival, P. Mymoona, K. M. Lakshmi, Nonappa, T. Pradeep and E. S. Shibu, Small, 2021, 17, 2005718 CrossRef CAS
- A. Nag and T. Pradeep, ACS Nanosci. Au, 2022, 2, 160–178 CrossRef CAS
- A. Chakraborty, A. C. Fernandez, A. Som, B. Mondal, G. Natarajan, G. Paramasivam, T. Lahtinen, H. Häkkinen, Nonappa and T. Pradeep, Angew. Chem., Int. Ed., 2018, 57, 6522–6526 CrossRef CAS
- K. R. Krishnadas, A. Ghosh, A. Baksi, I. Chakraborty, G. Natarajan and T. Pradeep, J. Am. Chem. Soc., 2016, 138, 140–148 CrossRef CAS
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS PubMed
- P. Bose, K. K. Ramankutty, P. Chakraborty, E. Khatun and T. Pradeep, Nanoscale, 2024, 16, 1446–1470 RSC
- P. Bose, P. Chakraborty, J. S. Mohanty, Nonappa, A. Ray Chowdhuri, E. Khatun, T. Ahuja, A. Mahendranath and T. Pradeep, Nanoscale, 2020, 12, 22116–22128 RSC
- P. Bose, J. Roy, V. Khokhar, B. Mondal, G. Natarajan, S. Manna, V. Yadav, A. Nyayban, S. S. R. K. C. Yamijala, Nonappa and T. Pradeep, Chem. Mater., 2024, 36, 7581–7594 CrossRef CAS
- J. Roy, B. Mondal, G. Vishwakarma, Nonappa, N. V. Sridharan, P. Krishnamurthi and T. Pradeep, Nanoscale, 2023, 15, 8225–8234 RSC
- A. Chakraborty, M. M. Stanley, B. Mondal, Nonappa, M. Bodiuzzaman, P. Chakraborty, M. P. Kannan and T. Pradeep, Nanoscale, 2023, 15, 2690–2699 RSC
- M. S. Lee, D. W. Yee, M. Ye and R. J. Macfarlane, J. Am. Chem. Soc., 2022, 144, 3330–3346 CrossRef CAS
- R. K. Cersonsky, G. van Anders, P. M. Dodd and S. C. Glotzer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1439–1444 CrossRef CAS PubMed
- V. Linko, H. Zhang, Nonappa, M. A. Kostiainen and O. Ikkala, Acc. Chem. Res., 2022, 55, 1785–1795 CrossRef CAS PubMed
- C. L. Bassani, G. van Anders, U. Banin, D. Baranov, Q. Chen, M. Dijkstra, M. S. Dimitriyev, E. Efrati, J. Faraudo, O. Gang, N. Gaston, R. Golestanian, G. I. Guerrero-Garcia, M. Gruenwald, A. Haji-Akbari, M. Ibáñez, M. Karg, T. Kraus, B. Lee, R. C. Van Lehn, R. J. Macfarlane, B. M. Mognetti, A. Nikoubashman, S. Osat, O. V. Prezhdo, G. M. Rotskoff, L. Saiz, A.-C. Shi, S. Skrabalak, I. I. Smalyukh, M. Tagliazucchi, D. V. Talapin, A. V. Tkachenko, S. Tretiak, D. Vaknin, A. Widmer-Cooper, G. C. L. Wong, X. Ye, S. Zhou, E. Rabani, M. Engel and A. Travesset, ACS Nano, 2024, 18, 14791–15328 CrossRef CAS PubMed
- V. N. Manoharan, Science, 2015, 349, 1253751 CrossRef PubMed
- A. M. Kalsin, M. Fialkowski, M. Paszewski, S. K. Smoukov, K. J. M. Bishop and B. A. Grzybowski, Science, 2006, 312, 420–424 CrossRef CAS PubMed
- T. P. Bigioni, X. M. Lin, T. T. Nguyen, E. I. Corwin, T. A. Witten and H. M. Jaeger, Nat. Mater., 2006, 5, 265–270 CrossRef CAS PubMed
- K. E. Mueggenburg, X. M. Lin, R. H. Goldsmith and H. M. Jaeger, Nat. Mater., 2007, 6, 656–660 CrossRef CAS PubMed
- W. Cheng, M. J. Campolongo, J. J. Cha, S. J. Tan, C. C. Umbach, D. A. Muller and D. Luo, Nat. Mater., 2009, 8, 519–525 CrossRef CAS PubMed
- A. Dong, J. Chen, P. M. Vora, J. M. Kikkawa and C. B. Murray, Nature, 2010, 466, 474–477 CrossRef CAS PubMed
- T. Wen and S. A. Majetich, ACS Nano, 2011, 5, 8868–8876 CrossRef CAS
- C. Yu, X. Guo, M. Muzzio, C. T. Seto and S. Sun, ChemPhysChem, 2019, 20, 23–30 CrossRef CAS PubMed
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef CAS
- D. Liu, F. Zhou, C. Li, T. Zhang, H. Zhang, W. Cai and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 9596–9600 CrossRef CAS
- C. Pigliacelli, D. Maiolo, Nonappa, J. S. Haataja, H. Amenitsch, C. Michelet, P. Sánchez Moreno, I. Tirotta, P. Metrangolo and F. Baldelli Bombelli, Angew. Chem., Int. Ed., 2017, 56, 16186–16190 CrossRef CAS
- S. Li, J. Liu, N. S. Ramesar, H. Heinz, L. Xu, C. Xu and N. A. Kotov, Nat. Commun., 2019, 10, 4826 CrossRef PubMed
- C. Pigliacelli, K. B. Sanjeeva, Nonappa, A. Pizzi, A. Gori, F. B. Bombelli and P. Metrangolo, ACS Nano, 2019, 13, 2158–2166 CAS
- K. Kimura and T. Pradeep, Phys. Chem. Chem. Phys., 2011, 13, 19214 RSC
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS
- D. Frenkel, Phys. A, 2002, 313, 1–31 CrossRef CAS
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed
- J. Fang, B. Ding and H. Gleiter, Chem. Soc. Rev., 2011, 40, 5347–5360 RSC
- P. Qiu, B. Ma, C. T. Hung, W. Li and D. Zhao, Acc. Chem. Res., 2019, 52, 2928–2938 CrossRef CAS PubMed
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350–2365 CrossRef
- D. Gebauer and S. E. Wolf, J. Am. Chem. Soc., 2019, 141, 4490–4504 CrossRef CAS
- M. Jehannin, A. Rao and H. Cölfen, J. Am. Chem. Soc., 2019, 141, 10120–10136 CrossRef CAS
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed
- S. Karthika, T. K. Radhakrishnan and P. Kalaichelvi, Cryst. Growth Des., 2016, 16, 6663–6681 CrossRef CAS
- J. De Roo, Chem. Mater., 2022, 34, 5766–5779 CrossRef CAS
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson Jr., B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef PubMed
- N. Vogel, S. Goerres, K. Landfester and C. K. Weiss, Macromol. Chem. Phys., 2011, 212, 1719–1734 CrossRef CAS
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed
- C. Jenewein, S. M. Schupp, B. Ni, L. Schmidt-Mende and H. Cölfen, Small Sci., 2022, 2, 2200014 CrossRef CAS
- M. R. Jones, K. D. Osberg, R. J. Macfarlane, M. R. Langille and C. A. Mirkin, Chem. Rev., 2011, 111, 3736–3827 CrossRef CAS PubMed
- S. Sun, X. Yu, Q. Yang, Z. Yang and S. Liang, Nanoscale Adv., 2019, 1, 34–63 RSC
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS
- Y. Shi, Z. Lyu, M. Zhao, R. Chen, Q. N. Nguyen and Y. Xia, Chem. Rev., 2021, 121, 649–735 CrossRef CAS PubMed
- T. Tachikawa and T. Majima, NPG Asia Mater., 2014, 6, e100–e100 CrossRef CAS
- E. Uchaker and G. Cao, Nano Today, 2014, 9, 499–524 CrossRef CAS
- L. Zhou and P. O'Brien, J. Phys. Chem. Lett., 2012, 3, 620–628 CrossRef CAS PubMed
- B. Ni, G. Gonzalez-Rubio and H. Cölfen, Acc. Chem. Res., 2022, 55, 1599–1608 CrossRef CAS
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC
- P. Chakraborty, P. Bose, J. Roy, A. Nag, B. Mondal, A. Chakraborty and T. Pradeep, J. Phys. Chem. C, 2021, 125, 16110–16117 CAS
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-marza, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed
- K.-J. Wu, E. C. M. Tse, C. Shang and Z. Guo, Prog. Mater. Sci., 2022, 123, 100821 CrossRef CAS
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235–1237 CrossRef CAS
- S. Tokonami, N. Morita, K. Takasaki and N. Toshima, J. Phys. Chem. C, 2010, 114, 10336–10341 CrossRef CAS
- S. W. Verbruggen, M. Keulemans, J. A. Martens and S. Lenaerts, J. Phys. Chem. C, 2013, 117, 19142–19145 CrossRef CAS
Footnotes |
| † Dedicated to Parasuraman Selvam, a colleague and valued friend, to celebrate his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02820a |
| § Present address: Faculty of Engineering and Natural Sciences, Tampere University, Korkeakoulunkatu 3, FI-33720, Tampere, Finland |
| This journal is © The Royal Society of Chemistry 2025 |